

Abstract
Gene silencing with siRNA nanoparticles (si-NPs) is promising but still clinically unrealized for inhibition of tumor driver genes. Ternary si-NPs containing siRNA, a single block NP core-forming polymer poly[(2-(dimethylamino)ethyl methacrylate)-co-(butyl methacrylate)] (DMAEMA-co-BMA, 50B), and an NP surface-forming diblock polymer 20kDa poly(ethylene glycol)-block-50B (20kPEG-50B) have the potential to improve silencing activity in tumors due to the participation of both 50B and 20kPEG-50B in siRNA electrostatic loading and endosome disruptive activity. Functionally, single block 50B provides more potent endosomolytic activity, while 20kPEG-50B colloidally stabilizes the si-NPs. Here, we systematically explored the role of the molecular weight (MW) of the core polymer and of the core:surface polymer ratio on ternary si-NP performance. A library of ternary si-NPs was formulated with variation in the MW of the 50B polymer and in the ratio of the core and surface forming polymeric components. Increasing 50B core polymer MW and ratio improved si-NP in vitro gene silencing potency, endosome disruptive activity, and stability, but these features also correlated with cytotoxicity. Concomitant optimization of 50B size and ratio resulted in the identification of lead ternary si-NPs 50B4-DP100, 50B8-DP100, 50B12-DP25 with potent activity and minimal toxicity. Following intravenous treatment in vivo, all lead si-NPs displayed negligible toxicological effects and enhanced pharmacokinetics and tumor gene silencing relative to more canonical binary si-NPs. Critically, a single 1 mg/kg intravenous injection of 50B8-DP100 si-NPs silenced the tumor driver gene Rictor at the protein level by 80% in an orthotopic breast tumor model. 50B8-DP100 si-NPs delivering siRictor were assessed for therapeutic efficacy in an orthotopic HCC70 mammary tumor model. This formulation significantly inhibited tumor growth compared to siControl-NP treatment. 50B8-DP100 si-NPs were also evaluated for long-term safety and were well-tolerated following a multi-dose treatment scheme. This work provides new insight on ternary si-NP structure-function relationships and identifies core polymer optimization strategies that can yield safe si-NP formulations with potent oncogene silencing.
Keywords: drug delivery, ternary nanoparticles, RNA interference, cancer
Graphical abstract.
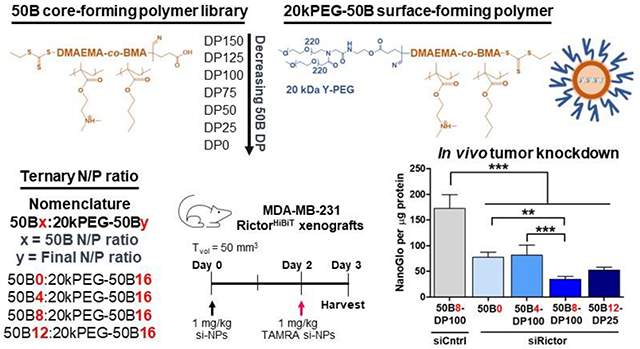
A library of si-NP formulations were screened to identify a lead formulation based on core polymer molecular weight and core:surface polymer ratio in order to overcome the multiple barriers that challenge siRNA cancer therapies.
Introduction
siRNA-based RNAi therapeutics are a promising strategy for the treatment of a variety of diseases that lack druggable targets. Systemic delivery of siRNA alone, however, is limited by rapid kidney clearance, inadequate inherent cellular uptake, and poor endosome escape. As a result, carrier free siRNAs have thus far required receptor-ligand targeting for success.[1] For a heterogeneous disease like cancer, where universal cellular receptors are lacking, nanoscale delivery vehicles can support target tissue siRNA delivery. Recent clinical success of nanocarriers for siRNA delivery was seen for Onpattro (patisiran), a lipid-based siRNA nanoparticle (LNP) formulation targeting hepatocytes for the treatment of transthyretin-mediated amyloidosis.[2, 3] While the FDA approval of Onpattro paves the way for the translation of other siRNA nano-systems, siRNA delivery to extrahepatic tissue, such as tumors, remains elusive.
LNPs such as Onpattro have been intensely investigated as systemic siRNA delivery systems.[4–8] In contrast, polymer-based siRNA nanoparticle (si-NP) systems are less studied. However, polymer-based si-NPs offer advantages in scalability, limitless chemical variations, and incorporation of “smart” components that respond to environmental stimuli or promote endosome escape. For instance, the high tunability of cationic polymer systems was recently employed to drive tissue-selective mRNA delivery to the spleen and lymph nodes.[9]
Nanoparticle chemical physical properties can be optimized to enhance passive tumor accumulation, independent of any specific receptor-ligand targeting. However, passive NP accumulation in tumor tissue requires a stable formulation that extends circulation time following intravenous (i.v.) delivery. Cationic polymer si-NPs are typically held together by electrostatic interactions between the cationic polymer and the negatively charged siRNA. Unfortunately, these electrostatic bonds are de-complexed through serum protein interactions in circulation, or through negatively charged proteoglycans and heparan sulfates of the kidney glomerular basement membrane (GBM), resulting in siRNA loss, primarily through renal filtration.[10] Circulating si-NPs must also circumvent other factors that reduce tumor bioavailability, such as phagocytosis by the macrophages of the reticuloendothelial system (RES).[11–14] Finally, nanoparticles, especially those with cationic components, have a narrow therapeutic index and can cause carrier-related off-target toxicities.[15, 16] There is a critical need to overcome siRNA carrier challenges and identify systems that provide potent silencing activity, circulation stability, and avoidance of carrier-associated toxicity.
Our lab and others have improved si-NP stability through a variety of strategies, particularly through enhancing core hydrophobic interactions in combination with canonical electrostatic interactions.[17] Shown by us and others, modification of the siRNA by conjugation with hydrophobic moieties such as palmitic acid[18, 19], cholesterol[20, 21], or other hydrophobes[22] increases carrier-cargo loading stability. Nanocarrier hydrophobicity can also be increased through ternary si-NP formulation strategies. Ternary si-NPs, to achieve a given N+/P− formulation ratio, substitute in a hydrophobic, RNA condensing core-forming polymer alongside the PEGylated diblock surface-forming polymer to enhance carrier stability.[23] Here, we created and tested a unique library of ternary si-NPs with variations in both the core polymer molecular weight and core:surface polymer ratio. This series of candidates was formulated to test the hypothesis that core polymer molecular weight and core:surface polymer ratio interact and should be simultaneously tuned for optimization of si-NP stability, activity, and safety.
The cationic and endosomolytic polymer block of both polymers of our ternary formulations have a 50:50 monomer composition of 2-(dimethylamino)ethyl methacrylate and butyl methacrylate (poly(DMAEMA-co-BMA), 50B). This monomer composition has been established as an ideal polymer containing a balance of cationic and hydrophobic monomer in a random copolymer composition that optimizes stability, endosome escape activity, and cytocompatibility.[17, 23] The core-forming polymer our of ternary si-NPs is a single block of 50B, and the surface-forming polymer is a diblock of 20kDa poly(ethylene glycol)-block-50B (20kPEG-50B). Previous studies comparing ternary si-NPs to “binary” si-NPs, containing solely a 5kPEG-bl-50B surface-forming polymer, revealed that the inclusion of a core-forming free 50B polymer endowed ternary si-NPs with greater activity and stability; one reason for the higher activity is that the core-forming 50B single block polymer both more stably complexes siRNA and has more potent endosome disruptive function versus 5kPEG-bl-50B because its siRNA and membrane interactions are not sterically hindered by junction with a PEG block.[23]
In other related work, we have shown that for binary formulations of PEG-bl-50B, the activity and toxicity of the formulation correlate with the 50B polymer block MW.[24] The current work, therefore, simultaneously tuned both the MW of the core-forming free 50B polymer and the core:surface polymer ratio with the hypothesis that si-NP potency could be enhanced in a manner that did not necessitate the use of exceedingly large 50B polymer blocks that would cause toxicity. MW of the core-forming 50B polymer is also a variable that has not been studied to our knowledge for this type of ternary system.
The design of ternary si-NPs comprised of core-forming and surface-forming polymers also presents the opportunity to optimize core-to-surface polymer ratio. Previous testing of the core-forming 50B polymer ratio revealed a tradeoff in si-NP performance. While a greater ratio of 50B improved activity, the consequent decrease in the surface-forming PEG polymer limited colloidal stability and charge shielding of the si-NP.[23] These studies, however, utilized a 5kPEG-bl-50B surface-forming polymer. Here, we use a 20kPEG-50B polymer with the hypothesis that a higher MW PEG will provide greater stabilization function,[25] even when it is incorporated at lower ratios in the si-NP. Previous work furthermore studied the effect of core-to-surface polymer ratio on si-NP activity solely at the in vitro level, and in vitro studies are not always predictive of in vivo performance. [26] In this work, we therefore screened multiple si-NP candidates in vivo rather than moving forward with a lead formulation identified through in vitro studies. In all, the library composed and tested here includes 18 ternary si-NPs that were screened for in vitro gene silencing, endosomolytic activity, and cargo loading stability, as well as a subset that were screened for in vivo si-NP tumor uptake and target gene silencing in an orthotopic breast tumor model.
Materials and Methods
Materials and reagents
Unless otherwise noted, chemicals and materials for biological assays were purchased from Sigma-Aldrich or Fisher Scientific. All oligonucleotides used in these studies were synthesized on a MerMade 12 Oligonucleotide Synthesizer (Bioautomation) using modified (2’-OMe and 2’-F) phosphoramidites. Sequences and modifications of all oligonucleotides can be found in Supplementary Table 1.
Polymer synthesis and characterization
All polymers were synthesized by RAFT polymerization using 4-cyano-4-(ethylsulfanylthiocarbonyl)sulfanylpentanoic acid (ECT) as the CTA, synthesized as previously described.[27] 50B polymers were synthesized at 50:50 molar ratios of 2-(dimethylamino)ethyl methacrylate (DMAEMA) and butyl methacrylate (BMA) using AIBN as an initiator and a 5:1 ratio of CTA:initiator, in 20% w/v dioxane for 24 hr. 50B polymers were purified by dialysis in water followed by lyophilization.
To synthesize 20kPEG-50B, 20kPEG-ECT was first synthesized by DIC/DMAP coupling of hydroxyl-terminated 20kDa PEG to ECT in dichloromethane for 48 hr, as previously described.[17, 25] A 10 molar excess of ECT, 10 molar excess of DIC, and 5 molar excess of DMAP to 20kDa PEG was used in a 10% w/v reaction. 20kPEG-ECT was purified by precipitation in diethyl ether and dried under vacuum. RAFT polymerization of 50:50 DMAEMA and BMA was performed as described above. 20kPEG-50B was purified by precipitation in 2:1 pentane:diethyl ether and dried under vacuum.
Polymers were characterized for degree of polymerization and MW using 1H nuclear magnetic resonance spectroscopy (H NMR, Bruker, 400 MHz) and dimethyl formamide mobile phase gel permeation chromatography (GPC, Agilent Technologies).
Formulation of lyoprotected si-NPs, characterization of size and surface charge
All ternary si-NPs were formulated using a 3 mg/mL polymer solution of 20kPEG-50B and 0.5 mg/mL polymer solution of 50B made up of 10% ethanol and 90% 0.1 M citrate buffer (pH 4). Binary si-NP 50B0 contained only the 20kPEG-50B polymer solution. si-NPs were formulated at a total polymer to nucleic acid ratio (N+/P−, calculated using the ratio of protonated amines on DMAEMA polymer to phosphates on the siRNA duplex) of 16. N/P of 50B was varied as 0, 4, 8, or 12 of the total polyplex N/P of 16. siRNA and polymer amounts were calculated as previously described[23], using the formulas below:
where Pol1 refers to the 50B polymer and Pol2 refers to the 20kPEG-50B polymer.
After mixing calculated amounts of polymer and siRNA, the solution was allowed to complex for 30 min. Following complexation, 5x v/v 0.2 M phosphate buffer (pH 8) was added to raise solution pH to 7.4.
For in vitro experiments, formulated si-NPs were added to Amicon centrifugal tubes (Millipore Sigma) with 50 kDa MW cut-off to concentrate si-NPs and remove excess levels of pH buffers. 7x v/v of 270 mM sucrose solution was added to concentrated si-NPs and si-NPs were once again spun to a final concentrated dose of 2000 nM. Concentrated si-NPs were flash-frozen, lyophilized overnight, and stored at −80 °C. To reconstitute lyophilized si-NPs, water was first added to si-NP cake at the volume that si-NPs were lyophilized in. si-NPs were allowed to sit for 20 min to rehydrate fully, and media or other buffers were then added to dilute si-NPs to their final working dose needed for respective assays.
For in vivo experiments, si-NPs were concentrated in pH buffers as described above and then washed with 7x 300 mM aqueous trehalose solution. si-NPs were spun down to a 1 mg/kg dose in a 100 uL injection volume and frozen and lyophilized as described above. In vivo lyophilized si-NPs were reconstituted with 100 uL sterile water to deliver si-NPs i.v. in an isotonic solution.
si-NP hydrodynamic diameter and zeta potential was measured by dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern Instruments).
Cell culture
All cells used in this work were cultured in Dulbecco’s modified eagle’s medium (DMEM, Gibco Cell Culture), containing 4.5 g/L glucose, 10% FBS (Gibco), and 1% Antibiotic-Antimycotic (Gibco). All cells were tested for Mycoplasma contamination using MycoAlert Mycoplasma Detection Kit (Lonza).
In vitro assessment of si-NP target gene silencing and cell viability following si-NP treatment
Luciferase gene silencing and si-NP cell viability were assessed in Luciferase-expressing MDA-MB-231 cells that were transduced with the LUCIFERASE gene as previously described. [25] Cells were seeded in opaque 96-well plates at 4,000 cells/well. After adhering overnight, cells were treated with si-NPs encapsulating either siControl or siLuciferase siRNA. si-NP treatment was replaced at 24 hr. Luciferase gene knockdown was assessed at 48 and 72 hr through the addition of luciferin-containing media (150 ug/mL) and imaging of luminescence by IVIS (Caliper Life Sciences). Knockdown of luminescence signal was normalized against cells treated with siControl-NPs.
RICTOR gene silencing was assessed in RictorHiBiT-expressing MDA-MB-231 cells. HiBiT-tagged Rictor cells were generated by CRISPR-Cas9 mediated homology directed repair. A single-stranded oligo donor (ssODN) sandwiching the RICTOR STOP codon was used for HiBiT sequence insertion. HiBiT sequence insertion was confirmed by successful Nano-Luc complementation using a Nano-Glo HiBiT Lytic Detection assay (Promega) according to manufacturer’s instructions. Cells were dilution cloned as singlecell derived colonies to identify subpopulations with high levels of Nano-Luc complementation. Selected cells were plated and treated for si-NP silencing studies as described above. Rictor gene knockdown was assessed at 72 hr using the Nano-Glo HiBiT Lytic Detection assay, and luminescence was measured by plate reader (Tecan Infinite F500). Knockdown of luminescence signal was normalized against cells treated with siControl-NPs.
siControl-containing si-NPs were used for viability assessment at 24 hr. CellTiter-Glo assay (Promega) was used according to manufacturer’s instructions for relative viability assessment normalized to untreated wells.
In vitro cell uptake
si-NPs harboring fluorescent TAMRA-siRNA were used to assess uptake in MDA-MB-231 cells. 100,000 cells/well were seeded in 12-well plates and allowed to adhere overnight. Cells were treated with 100 nM si-NPs for 4, 8, and 24 hrs, and untreated cells were used as negative controls. At study endpoint, si-NPs were removed, and cells were trypsinized and washed with PBS three times. Cells were reconstituted a final time in PBS and assessed for TAMRA fluorescence using flow cytometry (Guava easyCyte SL System, Luminex). TAMRA fluorescence was monitored using the Yellow filter off the 488 nm Blue laser (YEL-B parameter). Quantification of geometric mean fluorescence intensity and percent positive cells was performed on FlowJo software.
In vitro assessment of si-NP endosome disruptive activity
Gal8-YFP measurement were carried out as previously described.[15, 24, 28] Gal8-YFP expressing MDA-MB-231 cells were seeded at 2,000 cells/well in opaque halfarea 96-well plates. After allowing cells to adhere for 24 h, cells were treated with 200 nM si-NPs. Cells were imaged for Gal8 recruitment by automated fluorescent microscopy (Nikon C1si + confocal microscope system on a Nikon Eclipse Ti-0E inverted microscope base, Plan Apo VC 20 × objective, Galvano scanner, and 408/488/543 dichroic mirror). Wells were imaged by a software-controlled motorized stage that moved the plate between images. Recruited Gal8 was identified by a MATLAB script recognizing Gal8-YFP fluorescent puncta, and intensity was normalized to total cell area.
Characterization of si-NP encapsulation, stability against heparin and serum
si-NP encapsulation was assessed using the Quant-iT Ribogreen assay kit (ThermoFisher). Assay reagents were prepared following manufacturer’s instructions and heparin salts were added at final concentrations of 0, 25, and 50 U/mL. si-NPs were prepared at a 100 nM final dose. Final well volumes in an opaque 96-well plate were made up of 50 μL si-NPs in TE buffer, 50 uL heparin in TE buffer, and 100 uL Ribogreen reagent. Fluorescence was measured on a plate reader (Tecan Infinite F500) at 520 nm over time, and encapsulation efficiency was calculated against a standard curve of siRNA-only wells containing known levels of siRNA.
si-NP stability in fetal bovine serum (FBS) and heparin salts was assessed using black hole quencher assay. si-NPs were co-loaded with TAMRA-tagged siRNA and Black hole quencher-tagged siRNA (BHQ2, capable of quenching TAMRA emission). To synthesize TAMRA and BHQ-tagged siRNAs, standard controlled pore glass synthesis columns containing the respective fluorophores were purchased from Glen Research (20-5910-41M, 20-5932-42M), and Control sense oligonucleotide strands were grown. 100 nM si-NPs and final FBS concentration of either 10% or 50% were added to opaque 96-well plates. TAMRA was excited at 546 nm and fluorescence was measured at 576 nm. As TAMRA signal was restored following si-NP cargo release and loss of Black hole quenching, rate of relative TAMRA fluorescence increase over time was plotted as a measure of si-NP destabilization kinetics.
In vivo si-NP toxicology studies
For acute toxicity studies, wild-type immunocompetent Balb/C mice (4-6 week old, Charles River) were injected i.v. in the tail vein with 1 mg/kg si-NPs. 300 mM aqueous trehalose was injected as a vehicle control. Mouse blood was collected via cardiac punch 30 min after injection to assess for acute platelet activating factor (PAF)-related si-NP toxicities. Mouse EDTA-blood samples were spun 1,000×g for 10 min at 4 °C to isolate plasma for analysis using a PAF acetyl hydrolase activity kit (Caymen Chemical). Plasma samples were assayed in duplicate according to the manufacturer’s protocol using 10 μL plasma samples in each well.
For multi-dose toxicity studies, wild-type immunocompetent Balb/C mice (4-6 week old, Charles River) were injected i.v. in the tail vein with 1 mg/kg si-NPs bearing siRNA targeting Rictor in the mouse genome (simRictor-NPs) on Days 0, 3, and 7. Mouse blood was collected via cardiac punch on Day 8, 24 hr after the final injection. Plasma biochemistry analysis for systemic markers (AST, ALT, BUN, glucose) was performed by Antech GLP through the Vanderbilt Translational Pathology Shared Resource (TPSR). Biochemistry markers were compared against a normal value range for Balb/C mice, and ranges were referenced from the Mouse Phenome Database by The Jackson Laboratory.
Collected blood was also analyzed for complete blood counts (including hematocrit and red blood cell measures) by TPSR. Mouse organs were harvested (liver, kidney, spleen) and fixed in 10% formalin for H&E histologic analysis of si-NP toxicity.
Western analysis for Rictor protein knockdown was performed on liver, kidney, and spleen tissues. Organs were homogenized in ice-cold lysis buffer (RIPA lysis buffer supplemented with 2% NP-40, 1X protease inhibitor cocktail (Roche), and 1x phosphatase inhibitor cocktail (Roche)). Protein concentration was determined by BCA assay (Pierce), and samples were resolved by SDS-PAGE. Separated proteins were transferred to nitrocellulose membranes, blocked, and probed with primary antibodies Rictor (Sigma-Aldrich), and β-actin (Cell Signaling Technologies).
Intravital microscopy and biodistribution
Anesthetized CD-1 mice were immobilized on a heated stage for fluorescence scanning of the ear vasculature by confocal microscopy. Microscopy was performed using a Nikon Czsi+ system with a Nikon Eclipse Ti-oE inverted microscopy base, Plan ApoVC 20× differential interference contrast N2 objective, 0.75 NA, Galvano scanner, and 543 dichroic mirror. Prior to imaging, mouse ears were depilated and positioned on a glass coverslip using immersion oil. Light microscopy was used to focus upon a prominent ear vein where flowing red blood cells could be easily visualized. Once a vein was in focus, confocal microscopy was initiated, and mice were tail vein injected with si-NPs harboring fluorescent, Cy5-tagged siRNA. Intravital fluorescence decay via laser scanning was monitored as one image per second, with a laser gain of 98 throughout. All image analysis and acquisition were done using Nikon NIS-Elements AR version 4.30.01. For image analysis, a region of interest (ROI) was highlighted within the mouse ear vasculature, and a fluorescence decay curve from this ROI was generated. Background fluorescence was first subtracted, and fluorescence values were normalized to a maximum initial fluorescence intensity. Fluorescence decay curves were fit to a one-compartment intravenous bolus model in PK Solver[29] to determine pharmacokinetic parameters. A one-compartment model was chosen because it had the greatest fit (R2 value) compared to other models tested in PK Solver.
In vivo si-NP tumor studies
Athymic nude Balb/C mice (4-6 weeks, Envigo) were injected with 1e6 HiBiT-tagged Rictor MDA-MB-231 cells in 50:50 Matrigel:serum-free DMEM in the inguinal mammary fat pad. Once tumors reached 50 mm3 in volume (Day 0), mice were divided into treatment groups and injected i.v. with 1 mg/kg si-NPs bearing siControl or siRNA targeting RICTOR in the human genome (sihRictor). Mice injected with 300 mM aqueous trehalose were used as a vehicle control. si-NPs bearing fluorescent TAMRA-tagged siRNAs were injected i.v. on Day 2. On Day 3 (72 hr after siRictor-NP treatment and 24 hr after siTAMRA-NP treatment) tumors were harvested for downstream analysis.
A portion of harvested tumors were assessed for si-NP uptake by flow cytometry (Amnis CellStream, Luminex). Tumors were processed for flow analysis as previously described[25] and quantified for TAMRA fluorescence using a 583 nm filter off the 532 nm laser (D4 laser parameter). Uptake analysis was performed on FlowJo software.
A portion of harvested tumors were assessed for gene silencing by Nano-Luciferase complementation of HiBiT-tagged Rictor. Tumors were lysed and quantified for total protein using BCA Assay (Pierce). HiBiT-tagged Rictor knockdown was assessed in lysates using Nano-Glo HiBiT Lytic Detection assay (Promega) according to manufacturer’s instructions.
For therapeutic tumor growth inhibition studies, athymic nude Balb/C mice (4-6 weeks, Envigo) were injected with 1e6 HCC70 cells in 50:50 Matrigel:serum-free RPMI in the inguinal mammary fat pad. Once tumors reached 50 mm3 in volume (Day 0), mice were divided into treatment groups and injected i.v. with 1 mg/kg si-NPs bearing siControl or sihRictor. Mice were again injected with si-NPs on Day 3. Tumor volumes were monitored by digital caliper measurements (Tvol = length x width2 / 2).
Statistical Analyses
Data were analyzed using GraphPad Prism 8 software (Graphpad Software, Inc.). In most cases, si-NP performance was compared by one-way ANOVA analysis. Specific statistical tests used for data can be found in corresponding figure legends. Data is plotted as average ± SEM. For all figures, * p≤0.05; ** p≤0.01; *** p≤0.001; ns, not significant.
Results and Discussion
Polymer synthesis and characterization
A library of six core-forming polymers was synthesized by reversible addition-fragmentation chain-transfer (RAFT) to create 50:50 copolymers of DMAEMA and BMA (50B). While the ratio of DMAEMA to BMA was kept equimolar (50:50), the total size was varied by iterating the degree of polymerization (DP) of 50B from 150 (DP150) to 25 (DP25) (Figure 1A). The DP of the 50B block in the surface-forming 20kPEG-50B polymer was kept constant (Figure 1B). DP150 was synthesized as the largest 50B polymer in our library to match previously published 50B lengths used in binary si-NP (PEG-bl-50B diblock polymer) formulations.[18, 25] The DP of 50B was then decreased with the hypothesis that smaller 50B polymers can provide similar levels of si-NP activity while mitigating toxicity.
Figure 1. Ternary si-NP polymer chemistry.
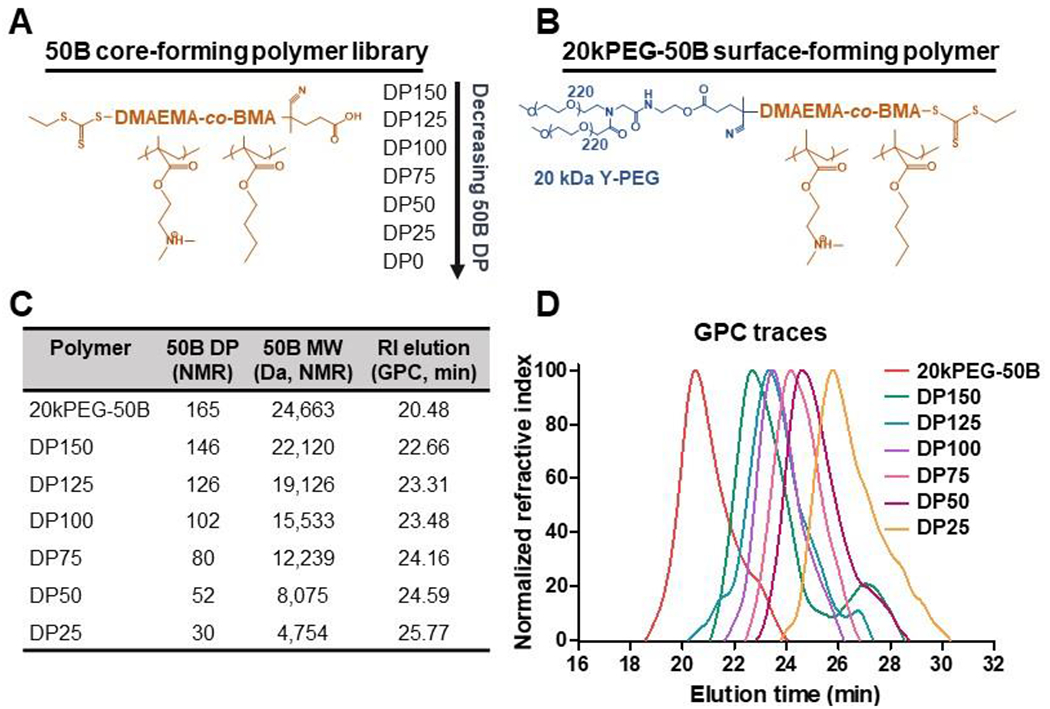
(A) The degree of polymerization (DP) was varied for the si-NP core-forming 50B polymer from DP150 to DP25. (B) si-NP surface-forming 20kPEG-50B polymer had a constant chemical structure. (C) Table of polymer size characteristics. (D) Gel permeation chromatography confirms synthesis of 20kPEG-50B and 50B polymers with varied molecular weight.
20kPEG-50B was synthesized as previously described where the 20 kDa PEG was conjugated to a RAFT chain transfer agent (CTA) and chain extended to add the 50B block.[17, 25] RAFT polymerization allowed for tight control of 50B DP, calculated by monomer consumption using 1H nuclear magnetic resonance (NMR) spectroscopy (Figure 1C, Supplementary Figure 1). A unimodal polymer population for the 50B series and 20kPEG-50B was confirmed by gel permeation chromatography (Figure 1D). Unlike early-generation ternary si-NPs that contained a surface-forming polymer made up of a 5 kDa PEG,[23] the novel si-NPs described here use a high MW (20 kDa) Y-shape PEG, shown to improve in vitro stability in serum, decrease protein adsorption, and enhance in vivo pharmacokinetics when compared to coronas of smaller MW.[25, 30] Furthermore, 20 kDa Y-shape PEG is an FDA-approved surface-forming polymer for enhancing drug pharmacokinetics and was used here as a standard to allow us to rigorously study effects of the 50B core-forming polymer.[31–33]
Sucrose containing ternary si-NP formulations retain structure and silencing activity upon reconstitution following lyophilization
To generate the library of ternary si-NPs, siRNA was complexed with our panel of core-forming 50B polymers (DP150 to DP25) and mixed in different ratios with the 20kPEG-50B surface-forming polymer (Figure 2A). The relative amount of 50B and 20kPEG-50B polymers comprising the si-NP was defined based on their contribution to the overall ratio of cationic amine (N) to anionic phosphate (P) in the formulation. The final N/P of all si-NPs was kept constant at 16, with the N contribution calculated based on the DMAEMA monomer in both the 50B core-forming polymer and the 50B block of the 20kPEG-50B polymer. We progressively increased the 50B polymer content in the si-NP ternary library from 0 to 12 (50B0, 50B4, 50B8, and 50B12) (Figure 2B). Notably, this formulation strategy results in an inverse correlation between the 50B:20kPEG-50B ratio and the total PEGylation on the final ternary si-NP (Figure 2C). The respective N/P contribution from 20KPEG-50B polymer in those formulations was therefore 16, 12, 8, and 4, respectively.
Figure 2. Ternary si-NP library design.
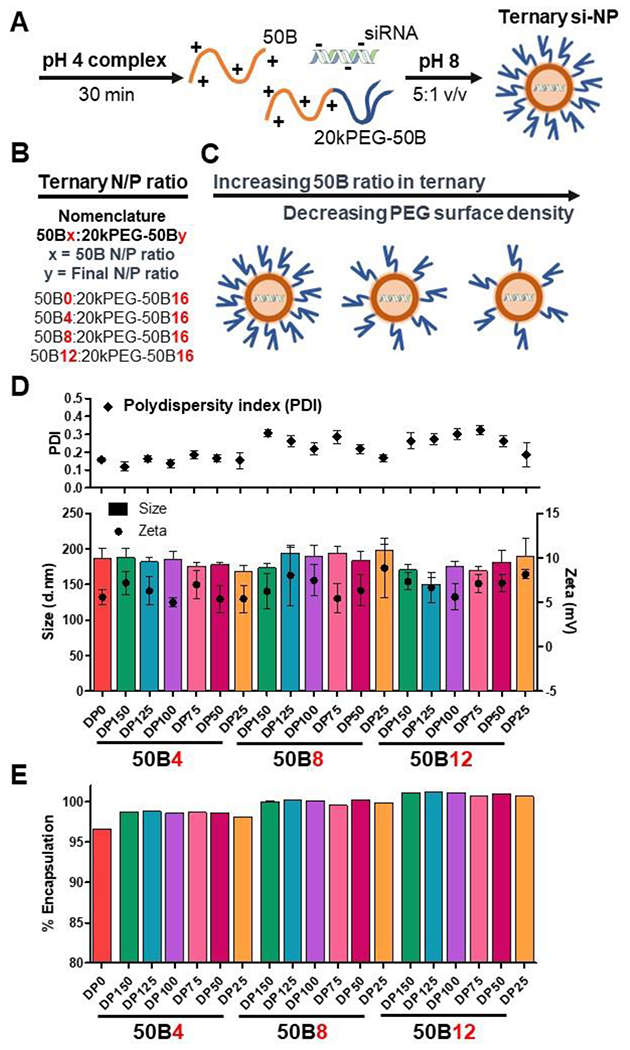
(A) Ternary si-NPs were formulated by complexing 50B and 20kPEG-50B polymers with siRNA in pH 4 buffer. The solution was brought to physiologic pH through the addition of pH 8 buffer, to stabilize the resultant ternary si-NPs. (B) si-NPs were formulated at a total N+/P− of 16, while ratio of 50B polymer was increased from N/P 4, 8, and 12. (C) Increasing 50B ratio in the si-NP results in lower amounts of 20kPEG-50B surface-forming polymer available for si-NP surface shielding. (D) All si-NPs displayed similar hydrodynamic diameter (d.nm) and surface charge (N = 3). (E) All si-NPs achieved greater than 95% siRNA encapsulation efficiency (N = 3).
To support large batch synthesis of si-NPs with consistent formulations across all experiments, lyophilized storage conditions were optimized for our ternary system. Lyophilized formulations also address major limitations of the siRNA nanomedicine field in that they allow for longer-term, stable storage and greater accessibility to underdeveloped areas in which requirement of a cold chain is a limitation.[34, 35] Prior to freezing, salt-containing buffers used during siRNA complexation and si-NP formation were exchanged for isotonic sucrose solution (Supplementary Figure 2A). Sucrose, and other sugars such as trehalose and glucose, are commonly used as cryo- and lyoprotectant agents during the freeze-drying process to prevent NP aggregation and promote cargo stabilization.[36, 37] The resulting si-NPs in sucrose were concentrated by centrifugation and freeze-dried (Supplementary Figure 2A). Following lyophilization, si-NPs were easily rehydrated by reconstitution in water.
Size characterization revealed lower size dispersity of lyophilized si-NPs than fresh, while also keeping the hydrodynamic diameter under 200 nm (Supplementary Figure 2B-C). Lyophilized si-NPs were assessed for siRNA encapsulation and retention of siRNA over time and displayed virtually identical siRNA encapsulation to freshly formulated si-NPs (Supplementary Figure 3A). Lyophilized si-NPs also maintained hydrodynamic size following multiple freeze-thaw cycles, demonstrating the integrity of these lyophilized formulations (Supplementary Figure 3B). To determine if lyophilization-reconstitution of si-NPs affects silencing activity, we generated ternary si-NPs harboring an siRNA sequence against the model gene LUCIFERASE (siLuc) or a non-targeting siRNA control (siControl). Fresh si-NPs or lyophilized-reconstituted si-NPs were transfected into MDA-MB-231 cells stably expressing Luc. At 48 hours after transfection, Luc activity was measured, revealing that both fresh and previously lyophilized siLuc-NPs diminished Luc activity to <35% compared to siControl-NPs in the DP150 formulation. (Supplementary Figure 3C). After confirming that lyophilized si-NPs had similar properties to freshly formulated si-NPs, all studies described in this work were completed using si-NPs that underwent the described lyophilization protocol.
The full ternary library, varying 50B size and ratio, was first assessed for si-NP size and zeta potential. All formulations had similar hydrodynamic diameters ranging from 150-200 nm with relatively narrow dispersity (Figure 2D). All si-NPs were slightly positive in surface charge ranging from 4 to 9 mV when assessed in deionized water (Figure 2D). si-NPs displayed a near neutral surface charge when measured in physiologic saline (Supplementary Figure 4). As 50B content was increased, there was no discernable increase in zeta potential for 50B8 and 50B12 si-NPs compared to the 50B0 and 50B4 si-NPs. This suggests that even the 50B12 formulations have effective PEG surface coverage contributed from the 4:1 ratio of the 20kPEG-50B polymer. Branched Y-shape PEG surface-forming polymers like the one used here are known to provide an “umbrella-like” covering that can shield larger portions of the NP surface, compared to linear PEGs of the same MW.[38] si-NPs were furthermore assessed for siRNA encapsulation, and all formulations displayed greater than 95% cargo loading (Figure 2E). As core-forming single block 50B polymer content was increased, there was an appreciable increase in siRNA encapsulation, perhaps due to lower PEG steric hindrance for siRNA complexation.
In vitro si-NP activity and toxicity is correlated to 50B polymer MW and ratio
Through tuning 50B core-forming polymer size and ratio in ternary si-NP formulations, we aim to find a balance between si-NP activity and toxicity. Gene silencing activity was assessed by treating Luc-expressing MDA-MB-231 cells with 100 nM si-NPs harboring siLuc and measuring bioluminescence at 48 hr (Figure 3A) and 72 hr (Supplementary Figure 5). Overall, treatment with si-NPs harboring larger 50B core polymer sizes resulted in greater silencing activity. Within the 50B4 si-NP group, treatment with DP150 resulted in 28% remaining Luc activity at 48 hr while treatment with DP25 had greater than double remaining Luc activity at 71%. Similar trends in activity were seen in the 50B8 and 50B12 si-NP groups. Level of knockdown did plateau with increasing 50B size. Compared to treatment with si-NPs harboring DP100, si-NPs harboring DP125 and DP150 did not significantly improve silencing activity. Silencing activity also improved with increasing 50B ratio, where treatment with 50B12 si-NPs resulted in the greatest Luc knockdown (Figure 3A). In all cases except 50B4-DP25 si-NPs, addition of the core 50B polymer resulted in improved silencing activity compared to binary 50B0 si-NPs (Supplementary Table 2).
Figure 3. Increasing 50B polymer MW and ratio improves gene silencing activity but reduces cell viability.
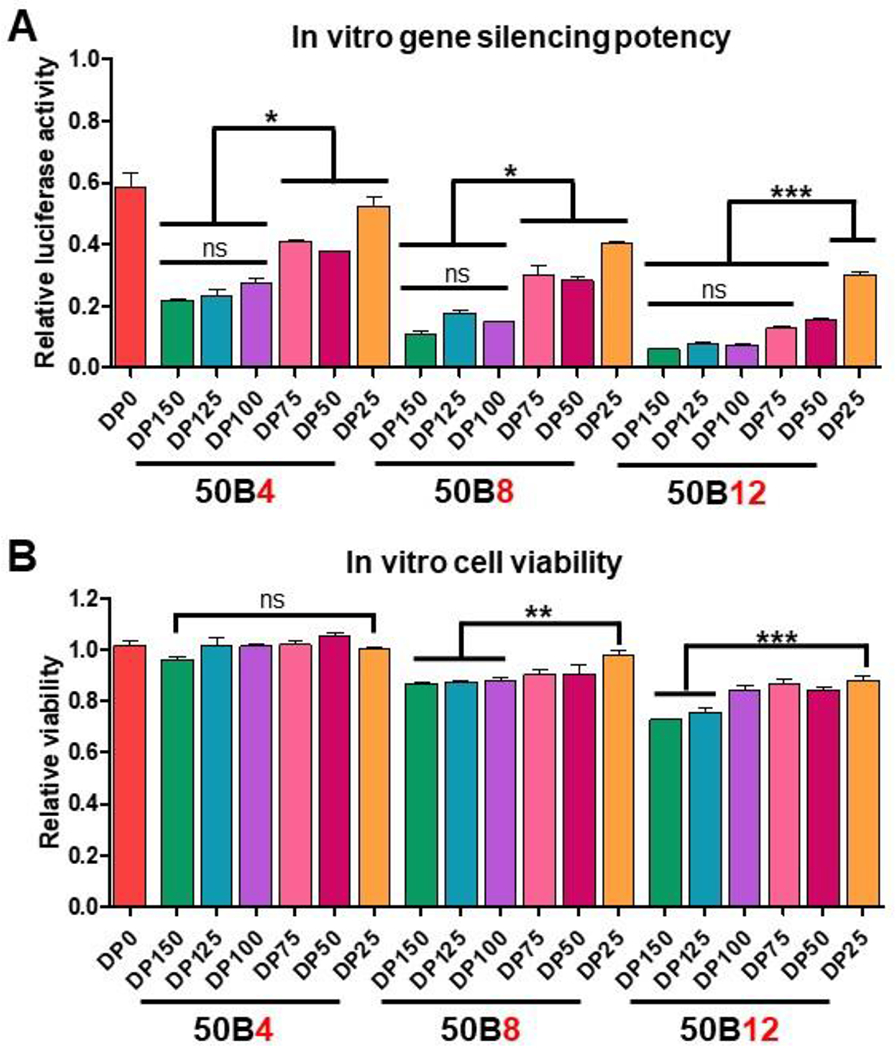
(A) Luciferase activity 48 hr after siLuciferase treatment using si-NP library (N = 3). Activity assessed relative to cells treated with each si-NP formulation loaded with siControl. (B) Cell viability 24 hr after treatment with si-NP library (N = 3). Viability assessed relative to non-si-NP treated cells. One-way ANOVA analysis with Tukey’s multiple comparison test was used to compare differences (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Though 50B quantity correlated with higher silencing activity, addition of 50B core-forming polymer also trended with cytotoxicity in relation to binary 50B0 si-NPs (Supplementary Table 3). In ternary si-NPs, these trends were apparent both with increasing 50B size and ratio. Within the 50B4 ratio group, treatment with all 50B DPs resulted in similar levels of cell viability. However, in the 50B8 and 50B12 ratio groups, cytotoxicity resulting from increasing 50B DP was apparent (Figure 3B). Independent of 50B size, increasing 50B quantity also increased si-NP toxicity. While all si-NPs in the 50B4 ratio group displayed cell viability above 95%, viability fell to approximately 85% for 50B8-DP150 si-NPs and approximately 75% for 50B12-DP150 si-NPs (Figure 3B). Together, these data indicate that 50B content, both in terms of MW and contribution to the N/P ratio, is a critical variable in ternary si-NP tuning for maximum silencing activity and minimal toxicity. For example, though 50B12-DP25 si-NPs were the least active compared to other 50B12 formulations, these si-NPs still outperformed 50B4-DP75 and 50B8-DP75 si-NPs while having greater than 85% cell viability.
To ultimately achieve gene silencing activity, cell internalized si-NPs must trigger endosomal siRNA escape to the cytosol, which can be enhanced by carriers that respond to the acidic environment of endolysosomal vesicles.[4, 39] Endosome disruption causes diffusely dispersed Galectin 8 (Gal8) to translocate and accumulate at the inner endolysosomal leaflet. Thus, yellow fluorescent protein (YFP) fused with Gal8 serves as a reporter for endosomal disruption,[24, 28] appearing as bright puncta.[40] Endosome disruption after treatment with si-NPs was quantified as average Gal8 puncta intensity per cell area (Figure 4A, Supplementary Figure 6). Processed images show Gal8 puncta recognition based on fluorescent intensity (Figure 4B). Overall, level of endosome disruption increased with both 50B size and ratio. si-NPs comprised of DP150 50B displayed significantly enhanced endosome disruption compared to si-NPs harboring smaller DPs; these trends are consistent with previous work correlating endosomolytic capability to polymer MW.[24] Interestingly in the 50B12 ratio group, there were no significant differences between DP150, DP125, and DP100 si-NPs, indicating that the larger 50B MW benefit may saturate at around DP100. The overall increase in endosome escape activity for DPs of 100 or larger correlates with higher gene silencing activity within our library, where si-NPs containing larger 50B core polymers induced greater silencing (Figure 3A). Greater levels of endosome escape, which will consequently allow for greater siRNA delivery to the cytosol, is likely a major mechanism for the increased silencing activity seen from si-NPs containing larger 50B DPs. Increasing 50B ratio also increased endosomolytic activity. As an example, 50B8-DP100 si-NPs had >2-fold greater disruptive activity (3.46) over 50B4-DP100 (1.66), while 50B12-DP100 si-NPs had >8-fold greater activity (14.71) compared to 50B4-DP100. In comparison, our binary 50B0 si-NP, comprised solely of the 20kPEG-50B polymer, had less endosome disruptive activity (0.79). These data further suggest that 50B is more potent than 20kPEG-50B at endosome escape and that, even for a given quantity of 50B used in si-NP formulation, endosome escape potency correlates with the 50B MW.
Figure 4. Ternary si-NP endosomal disruptive activity is dependent on 50B size and ratio.
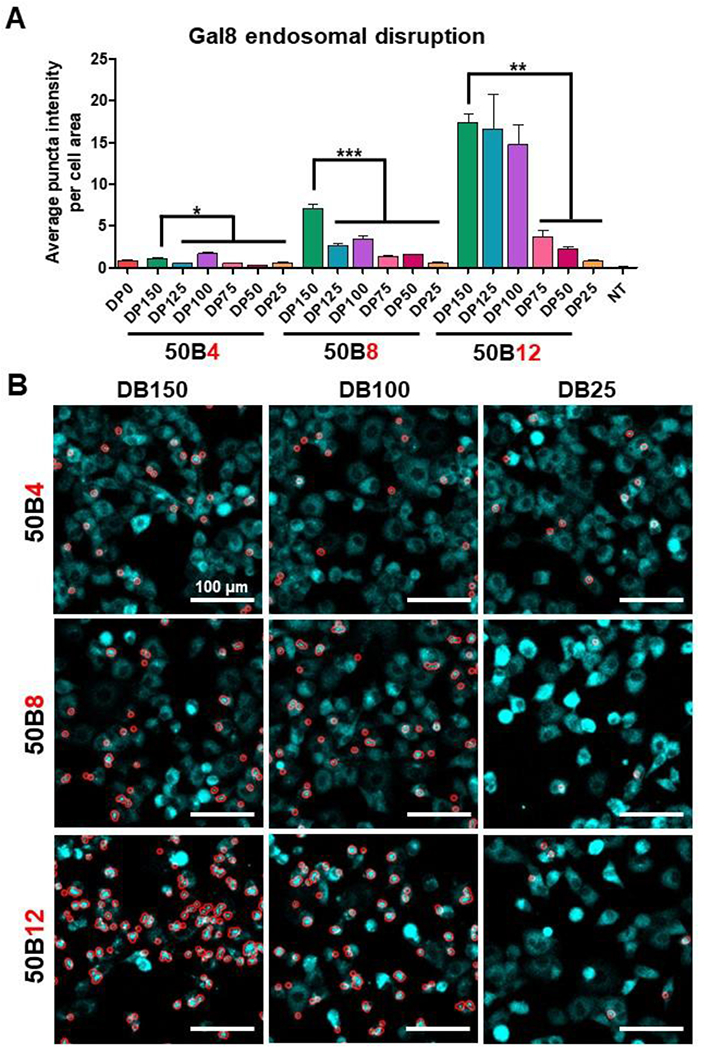
(A) Gal8-YFP expressing cells were tracked for Gal8 redistribution and puncta formation following si-NP treatment. Average puncta intensity per cell area was calculated for 4 hr after si-NP treatment (N = 3). One-way ANOVA analysis with Tukey’s multiple comparison test was used to compare activity differences (*, p < 0.05; **, p < 0.01; ***, p < 0.001). (B) Representative micrographs show automated puncta recognition (encircled in red) by MATLAB script.
50B polymer MW and ratio are critical regulators of ternary si-NP stability
To assess stability of the different si-NPs, siRNA encapsulation was quantified in the presence of heparin salt over time. Heparan sulfate is a negatively charged protein found in the GBM that can electrostatically interact with positively charged polymers that make up the si-NP carrier.[10, 41] Though si-NPs such as ours are designed with hydrodynamic diameters that bypass the ~10 nm GBM filtration barrier, si-NP disassembly would make the siRNA cargo vulnerable to renal filtration.[10, 42] si-NPs were therefore challenged with either 0 U/mL, 25 U/mL, or 50 U/mL heparin, and siRNA encapsulation was assessed as a measure of si-NP stability (Figure 5A-C). Overall, increasing 50B size and ratio in ternary si-NPs improved si-NP stability in heparin. At 0 U/mL heparin, all si-NPs showed slow siRNA release, and 50B0 si-NPs were fully disassembled by 9 hr (Figure 5A). However, incorporation of core-forming 50B polymer vastly improved stability, and even the minimally stable 50B4 si-NPs retained at least 37% of their siRNA cargo by 9 hr. We observed similar trends when challenging si-NPs with 25 U/mL or 50 U/mL heparin. By 120 min of si-NPs challenged with 50 U/mL heparin, all 50B4 si-NPs were disassembled, whereas all 50B12 si-NPs still retained 35% or more of their cargo (Figure 5C). To better quantify these data, the area under the curve (AUC) at each heparin condition was calculated (Figure 5D-G). We also calculated the slope of disassembly at each heparin condition and plotted the resulting rate constants (Supplementary Figure 7). 50B0 si-NPs had the lowest AUC, matching its data showing quick loss of encapsulated siRNA (Figure 5D). AUC increased with increasing 50B ratio (Figure 5E-G), indicating greater siRNA retention. While one may anticipate that 50B12 si-NPs would be more susceptible to heparin-mediated disassembly due to their lower surface PEGylation, this effect was not apparent. These data therefore suggest that free 50B polymer can complex more tightly with siRNA than 20kPEG-50B and that the single block 50 core-forming polymer content is more critical than the PEG content for cargo loading stability across the range of formulations conditions used here.
Figure 5. Ternary si-NP stability in heparin improves with increasing 50B size and ratio.
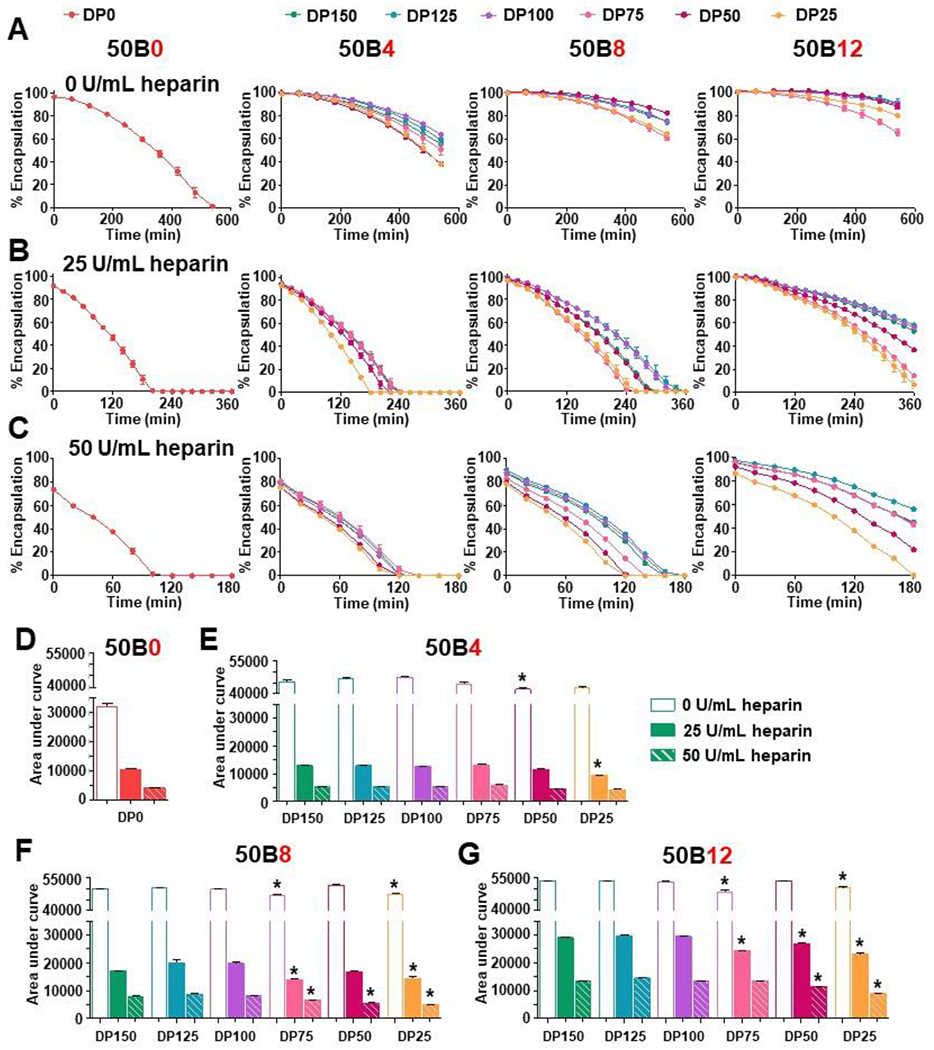
(A) si-NP stability was assessed by siRNA encapsulation in the presence of 0 u/mL heparin, (B) 25 U/mL heparin, and (C) 50 U/mL heparin over time (N = 3). (D) Area under the curve of plots of %siRNA encapsulation vs. time were calculated for various heparin concentrations for 50B0, (E) 50B4, (F) 50B8, and (G) 50B12 si-NPs. Hollow, solid, and dashed bars represent 0, 25, and 50 U/mL heparin conditions, respectively. One-way ANOVA analysis with Tukey’s multiple comparison test was used to compare stability differences; * show si-NPs with AUC significantly lower (p < 0.05) from DP150 si-NP within the same heparin condition.
Within each 50B ratio group, si-NPs harboring larger 50B sizes retained their siRNA cargo for a longer period while DP50 and DP25 si-NPs tended to disassemble the fastest (Figure 5A-C). AUC data calculated from these kinetic graphs show that increasing 50B size decreased rate of siRNA release. However, DP150 si-NPs do not confer strikingly greater stability over DP125 or DP100 si-NPs. This is reflected in the AUC calculated for the 50 U/mL heparin condition. 50B8-DP100 si-NPs increased AUC appreciably from 50B8-DP75 si-NPs (8300 %min 50B8-DP100 si-NPs vs. 6573 %min 50B8-DP75 si-NPs); however, 50B8-DP100 si-NPs have a comparable AUC to 50B8-DP150 si-NPs (7929 %min) (Figure 5F). Similar trends in 50B size were seen in rate constants calculated for each si-NP (Supplementary Figure 7), further supporting these AUC trends.
Serum proteins can also factor into si-NP destabilization following in vivo systemic delivery. In circulation, serum proteins such as albumin, IgGs, and lipoproteins can create a “corona” on the nanoparticles that can trigger recognition and clearance by the RES.[43] To assess stability of our si-NPs in serum, we utilized quencher-based methods where ternary si-NPs were co-loaded with tetramethylrhodamine (TAMRA)-tagged siRNA and non-fluorescent acceptor Black Hole Quencher (BHQ2)-tagged siRNA and challenged with 10% (Supplementary Figure 8A) or 50% (Figure 6) fetal bovine serum (FBS). When excited TAMRA is in close proximity to BHQ2 (e.g., co-loaded into an si-NP), its emission is absorbed by the quencher, suppressing the measurable fluorescent signal.[44] However, as the si-NPs disassemble and distance is increased between the two tagged siRNAs, TAMRA emission signal is restored (Figure 6A). TAMRA fluorescence in the presence of FBS was plotted over time for 50B0 si-NPs and the ternary library (Figure 6B-E). Like the trends seen in the presence of heparin (Supplementary Figure 8B), si-NP stability in FBS was increased both by increasing 50B MW and increasing 50B contribution to the N/P ratio. We calculated the half-time to TAMRA signal plateauing as a measure of si-NP stability, where more stable si-NPs had longer half-times to fluorescence restoration (Figure 6F). In accordance with stability trends seen following heparin challenge, si-NPs containing DP150, DP125, and DP100 did not significantly differ in half-time, suggesting that these si-NPs conferred similar levels of stability. Furthermore, the effect of 50B MW on si-NP serum stability was more apparent as 50B ratio was increased. For instance, the half-time for 50B8-DP150 si-NPs (63.9 min) was 2-fold greater than 50B8-DP25 (29.1 min) si-NPs, but the half-time for 50B12-DP150 si-NPs (100.4 min) was >3-fold compared to 50B12-DP25 counterparts (32.3 min). For in vivo delivery, this may suggest that the effects of 50B size and 50B ratio are additive and tuning one parameter could compensate for the other. For example, the half-time for 50B12-DP50 si-NPs (65.9 min) may be significantly reduced compared to 50B12-DP150, but it is virtually equivalent to the half-time displayed by 50B8-DP150 si-NPs (64.0 min). These collective data on our si-NP library indicate that 50B4, 50B8, and 50B12 si-NPs containing DP100 are lead candidates most worth of further in vivo studies, as they exhibit near maximal gene silencing activity, endosome disruptive ability, and stability, while also limiting toxicity.
Figure 6. Ternary si-NP stability in serum improves with increasing 50B size and ratio.
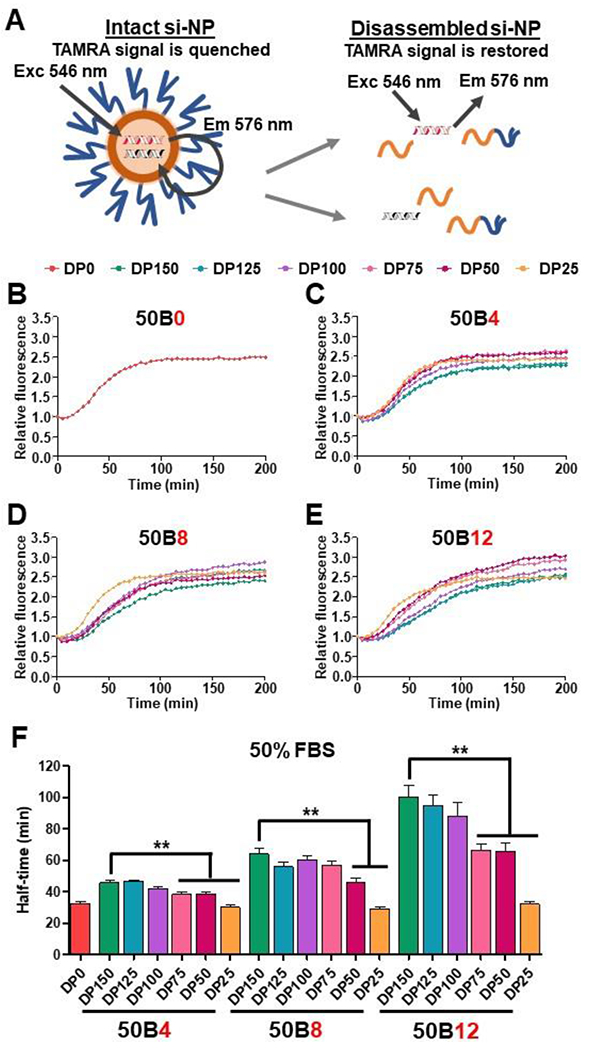
(A) si-NPs were co-loaded with fluorescent TAMRA-siRNA and quencher BHQ2-siRNA. Fluorescence de-quenching was measured to detect cargo release from si-NP destabilization. (B) Relative fluorescence was plotted for 50B0 si-NPs and (C) 50B4, (D) 50B8, and (E) 50B12 ternary si-NPs challenged with 50% FBS over time (N = 6). (F) Half-time to fluorescence signal plateau was calculated for each si-NP as a measure of relative stability. One-way ANOVA analysis with Tukey’s multiple comparison test was used to compare stability differences (**, p < 0.01).
Lead ternary si-NPs display a desirable safety response in vivo
Toxicology studies are an important facet of interrogation toward understanding the impact of 50B size and ratio on the in vivo use of ternary si-NPs. The above activity and stability studies suggest that 50B4-DP100, 50B8-DP100, and 50B12-DP100 si-NPs may be lead candidates for subsequent in vivo studies. However, due to the higher cytotoxicity displayed by 50B12 si-NPs in vitro (Figure 3B), preliminary in vivo toxicity studies were performed using 50B12 si-NPs to assess mouse survival following treatment. A 1 mg/kg dose of 50B12 si-NPs were injected i.v. into wild-type mice, and mice were observed for any adverse events or succumbing to treatment (Supplementary Figure 9). si-NPs harboring smaller 50B sizes were injected, and mice responded well to 1 mg/kg treatment with 50B12-DP25 si-NPs. Though 50B12-DP100 si-NPs displayed minimal in vitro toxicity and enhanced gene silencing activity (Figure 3), we found 50B12-DP25 si-NPs to be the more optimal formulation based on greater safety with i.v. administration in vivo. Based on these pilot studies, we moved forward with more rigorous in vitro toxicity studies using 50B0 si-NPs as a binary si-NP control and 50B4-DP100, 50B8-DP100, and 50B12-DP25 si-NPs as our lead ternary si-NP candidates. An initial in vitro viability screen using our lead si-NPs revealed that toxicity was 50B ratio-dependent at high doses; however, si-NPs displayed minimal cytotoxicity overall, with >85% viability for 50 nM doses and lower (Supplementary Figure 10).
Wild-type, immune-competent Balb/C mice were i.v. injected with lead si-NPs and resulting toxicity markers were compared to vehicle control at 30 min following injection. We and others have previously shown that acute dose-limiting nanocarrier and viral toxicities—occurring within 1 hr post-injection—are driven by the release and downstream effects of lipid mediator platelet activating factor (PAF).[15, 45] PAF was therefore used as a marker of acute si-NP toxicities in this work. Because PAF is extremely unstable and difficult to measure, it’s levels can be indirectly estimated by measurement of plasma PAF acetyl hydrolase (PAF-AH) activity. PAF-AH hydrolyzes PAF into an inactive form and its activity is negatively correlated with PAF levels.[46, 47] We evaluated mouse plasma for PAF-AH activity 30 minutes after si-NP injection (Figure 7A). None of the lead si-NPs had significantly decreased PAF-AH activity compared to trehalose vehicle control. This suggests that our lead si-NPs did not induce PAF related nanocarrier toxicities and were well-tolerated. To further confirm these results, we assessed mouse whole blood for elevated hematocrit and red blood cell concentration compared to vehicle. As a major mediator of anaphylaxis, PAF induces shock-like effects such as vascular permeability and hemoconcentration when it is present in the bloodstream.[48] There was no apparent vascular congestion following si-NP injection (Figure 7B-C). We also examined liver histology following acute si-NP delivery and observed no difference compared to vehicle (Figure 7D). Other primary si-NP clearance organs, kidneys and spleen, were also observed and displayed normal histology (Supplementary Figure 11).
Figure 7. Lead ternary si-NPs display minimal acute toxicity and improved in vivo pharmacokinetics following intravenous si-NP treatment.
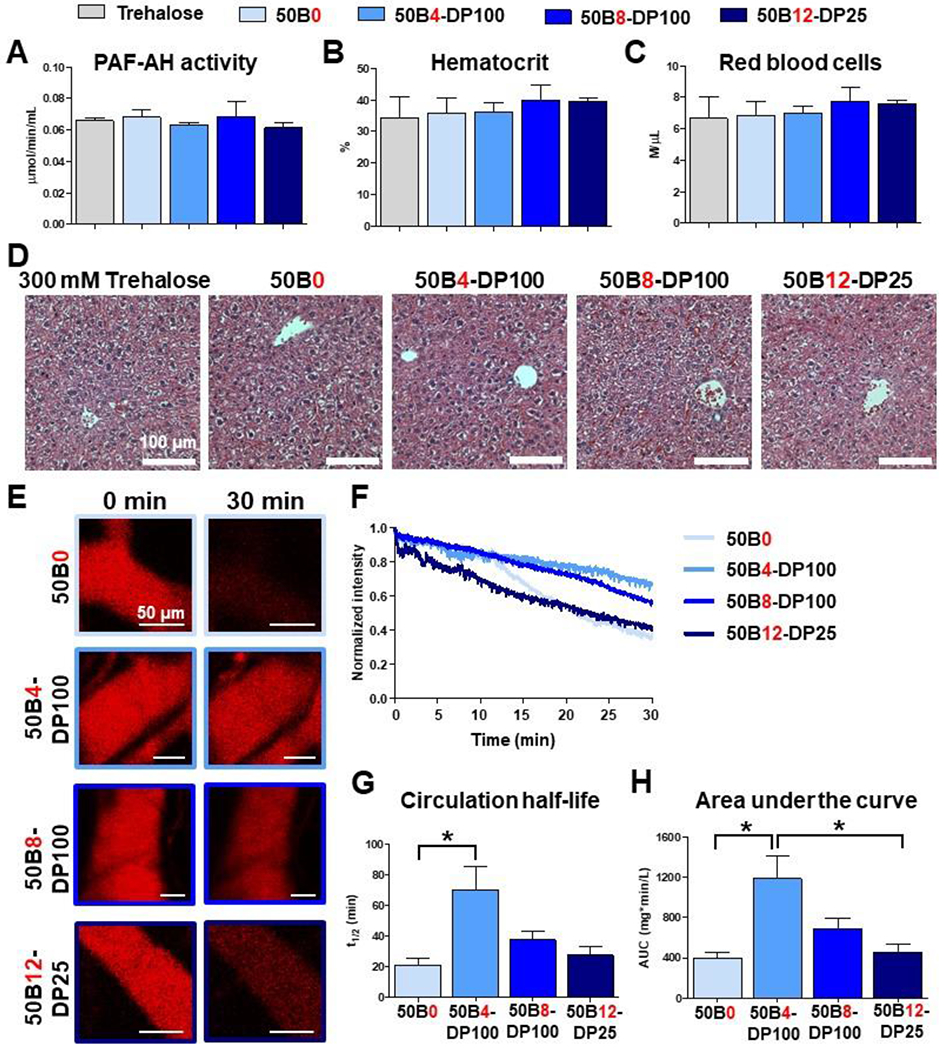
(A-D) Mice were i.v. treated with either trehalose vehicle or si-NPs and assessed after 30 min. Plasma PAF-AH activity was measured (A) as an indirect measure of plasma PAF levels, a biomarker for acute liver Kupffer cell toxicity (N = 4-5). Mouse blood was assessed for signs of PAF-induced hemoconcentration by measuring % hematocrit (B) and red blood cell level (C, N = 7-9). Mouse livers were evaluated for histological signs of toxicity, in particular vascular congestion, by H&E staining (D). (E) Representative intravital microscopy images of fluorescent Cy5 si-NPs in mouse ear vasculature (N = 3-4). (F) Average pharmacokinetic curves of each si-NP formulation. (G) Plasma half-life and (H) area under curve was calculated for each si-NP from intravital microscopy data. One-way ANOVA analysis with Tukey’s multiple comparison test was used to compare differences in pharmacokinetic parameters (*, p < 0.05).
In these studies, 50B0 was used as a benchmark si-NP that has served as a historical standard in our lab and has a desirable safety profile.[15, 25] Though all of our lead ternary si-NPs were safe upon treatment, there was a slight trend towards decreased plasma PAF-AH activity as 50B core polymer ratio was increased (Figure 7A). Treatment with our ternary si-NPs, however, did not result in significant differences in PAF-AH activity when compared to mice treated with binary 50B0 si-NPs. We previously showed that PAF related nanocarrier toxicities are driven by the level of uptake and endosome disruptive activity by liver Kupffer cells.[15] This is contrary to other cationic carriers, such as the “gold standard” PEI, which can induce toxicity via aggregation with serum proteins and excessive cell membrane disruption upon binding.[49, 50] 50B core polymer size and ratio can both regulate endosome disruptive activity (Figure 4) which may in turn promote PAF related toxicities. It was found here that balancing the MW and quantity of the 50B component allows development of promising, safe si-NP formulations.
Lead ternary si-NPs enhance in vivo pharmacokinetics
We next assessed our lead si-NPs for in vivo pharmacokinetics and biodistribution. si-NPs were loaded with Cy5-tagged siRNA, and fluorescence of the injected si-NPs was tracked by laser scanning confocal intravital microscopy (IVM) imaging of the mouse ear vasculature (Figure 7E-F). This longitudinal fluorescence tracking on a per-second basis enables a robust quantification of pharmacokinetic parameters, revealing increased circulation half-life and area under curve (AUC) upon inclusion of a 50B core-forming polymer in ternary si-NPs compared to binary 50B0 si-NPs (Figure 7G-H). This finding is consistent with previous studies showing that ternary si-NPs have greater in vivo tumor bioavailability than binary si-NPs.[23] Plasma half-lives for 50B0, 50B4-DP100, 50B8-DP100, and 50B12-DP25 si-NPs were 21.2, 70.1, 37.5, and 27.6 min, respectively. Similar relative trends were seen in AUC values calculated for each si-NP, where 50B4-DP100 si-NPs had close to a 3-fold increase in AUC compared to 50B0 si-NPs. The relative benefit of the core-forming 50B polymer decreases as its ratio within the si-NP is increased. While 50B4-DP100 si-NPs exhibited maximal blood circulation pharmacokinetics, 50B8-DP100 si-NPs had a 1.72-fold increase and 50B12-DP25 si-NPs only had a 1.13-fold increase in AUC over 50B0 si-NPs. Similar trends were seen in si-NP clearance, where 50B4-DP100 si-NPs had the lowest values, and 50B0 si-NPs had the greatest clearance (Table 1). As 50B core polymer ratio is increased, there is a consequent decrease in the 20kPEG-50B surface-forming polymer, necessary for si-NP charge shielding. These data suggest that, in the in vivo setting, a balance in core:surface polymer ratio is required for optimal blood circulation and that the lack of a core-forming polymer (50B0) or too much core polymer (50B12) can diminish in vivo bioavailability. To optimize for pharmacokinetics, our ternary si-NPs ideally balance two main competing factors: (1) higher 50B core polymer ratios promote stronger siRNA complexation whereas the 20kPEG-based surface-forming polymer likely sterically hinders electrostatic interactions between the cationic polymer blocks and siRNA. For example, higher packaging stability in the presence of serum is expected to contribute to improved pharmacokinetics, and we indeed observed increased stability in FBS for our ternary si-NPs as we increased 50B ratio (Figure 6). (2) lower 20kPEG-50B surface polymer ratios, resulting from higher 50B core polymer ratios, minimize stealth shielding and increase si-NP zeta potential and protein adsorption, properties that contribute to rapid clearance and poorer in vivo pharmacokinetics.
Table 1.
Pharmacokinetic parameters quantified from intravital imaging of si-NPs. Parameters include half-life (T1/2), area under the curve (AUC), and clearance (Cl).
| Formulation | T1/2 (min) | AUC0-t (mg*min/L) | AUC0-inf (mg*min/L) | Cl (mL/min) |
|---|---|---|---|---|
| 50B0 | 21.2 ± 4.15 | 264 ± 28 | 398 ± 56 | 0.049 ± 0.006 |
| 50B4-DP100 | 70.1 ± 15.67 | 440 ± 48 | 1187 ± 224 | 0.018 ± 0.004 |
| 50B8-DP100 | 37.5 ± 5.8 | 358 ± 18 | 688 ± 104 | 0.029 ± 0.005 |
| 50B12-DP25 | 27.6 ± 5.78 | 285 ± 39 | 451 ± 85 | 0.045 ± 0.010 |
Organ siRNA biodistribution was also assessed 1 hour after si-NP delivery, revealing similar biodistribution profiles for all si-NPs tested (Supplementary Figure 12). Greatest si-NP accumulation occurred in the liver (>45%), with minimal heart accumulation (<2.5%). Biodistribution to kidneys was ≤20% for all si-NPs, suggesting limited si-NP disassembly in the GBM. 50B12-DP25 si-NPs had greater localization to the liver and spleen, major clearance organs making up the RES, compared to other si-NPs. Since 50B12-DP25 si-NPs have the lowest ratio of surface-forming PEG polymer, this may suggest that they are more vulnerable to recognition and clearance by phagocytic cells. This underscores the importance of maintaining a threshold level of surface forming PEG polymer in ternary si-NP formulations, in order to minimize recognition and clearance by phagocytic cells.
Ternary si-NP formulations enable robust knock down of the oncogenic protein Rictor
The favorable toxicity profiles and pharmacokinetic performance displayed by our lead si-NPs justified advancement into studies on in vivo tumor target gene silencing activity. In vitro data on our full ternary library indicates that si-NP activity improves with increasing 50B size and ratio. We therefore hypothesized that increasing 50B ratio within our lead si-NPs will enhance in vivo tumor gene silencing potency, a parameter that has not been studied in our ternary si-NPs. Our previous silencing studies, however, were performed on cells exogenously expressing model gene LUC. Moving forward, we assessed si-NP silencing ability on an endogenously expressed tumor driver, choosing specifically to target the known tumor-driver gene RICTOR.[51, 52] The protein Rictor has established roles in tumor formation and tumor cell survival. To enable high throughput, protein-level screening of endogenous Rictor expression, CRISPR/Cas9 gene editing was used to introduce a HiBiT peptide tag in frame with Rictor (RictorHiBiT) at the endogenous RICTOR gene locus of MDA-MB-231 breast cancer cells (Supplementary Figure 13) [53]. Through split Nano-Luciferase (Nano-Luc) complementation, RictorHiBiT produces a bioluminescent signal, acting as a quantitative reporter for endogenous Rictor protein levels.
RictorHiBiT cells were treated with increasing doses of si-NPs loaded with siRICTOR (200 nM to 0.0976 nM) (Figure 8A). After 72 hours, relative Nano-Luc activity measurements revealed greater endogenous Rictor knockdown in cells treated with 50B8-DP100 and 50B12-DP25 si-NPs. For example, 50B8-DP100 and 50B12-DP25 si-NPs induced >80% Rictor knockdown at a 12.5 nM dose, versus 50B4-DP100 (60%) and 50B0 (34%). Notably, the EC50 values for 50B8-DP100 and 50B12-DP25 si-NPs were both subnanomolar and similar to each other, recorded at >10-fold lower than those seen in 50B4-DP100 si-NPs (Figure 8B). At high doses, all si-NPs had similar levels of activity, with greater than 80% Rictor knockdown at a 200 nM dose.
Figure 8. 50B8-DP100 si-NPs are the lead formulation for in vivo tumor uptake and tumor gene silencing.
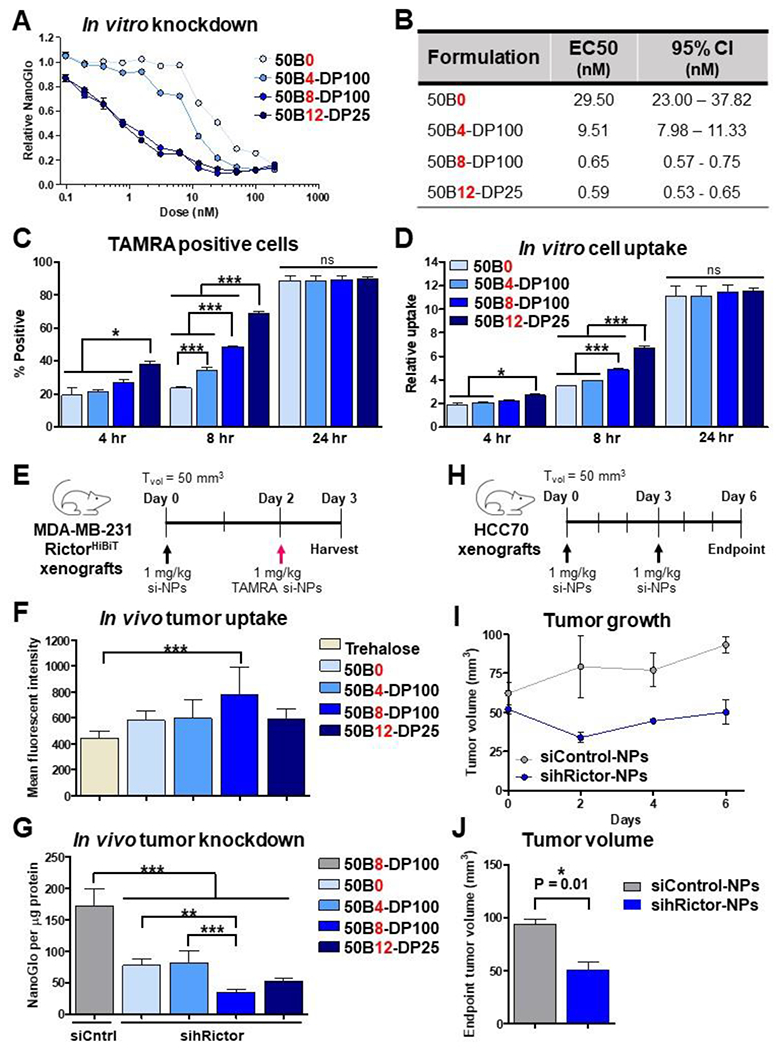
(A) Rictor-HiBiT cells were treated with a subset of si-NPs at a range of siRNA doses, and relative Rictor silencing was assessed by NanoGlo detection of HiBiT (N = 6). (B) EC50 values for each si-NP were calculated as a measure of si-NP silencing potency. (C-D) si-NPs loaded with fluorescent TAMRA-siRNA were used to assess in vitro uptake at 4, 8, and 24 hr of treatment (N = 3). (E) Mice bearing Rictor-HiBiT tumors were injected with trehalose vehicle, 1 mg/kg siControl-NPs, or 1 mg/kg sihRictor-NPs once tumors reached 50 mm3 on Day 0. Mice were injected with 1 mg/kg fluorescent TAMRA si-NPs on Day 2. Tumors were harvested for analysis on Day 3. (F) si-NP in vivo tumor uptake at 24 hr after si-NP treatment was measured by flow cytometric quantification of TAMRA fluorescence (N = 5-6 mice). One-way ANOVA analysis with Dunnett’s Multiple Comparison Test was used to compare uptake differences to trehalose vehicle-treated mice (***, p < 0.001). (G) si-NP in vivo tumor silencing of Rictor at 72 hr after si-NP treatment was quantified by NanoGlo detection of HiBiT levels (N = 4-6 mice). One-way ANOVA analysis with Tukey’s multiple comparison test was used to compare silencing differences (**, p < 0.01; ***, p < 0.001). (H) Mice bearing HCC70 tumors were injected with 1 mg/kg 50B8-DP100 siControl-NPs or sihRictor-NPs once tumors reached 50 mm3 on Day 0 and again on Day 3. (I-J) Tumor volumes of treated mice were monitored until Day 6 (N = 3). Unpaired t-test analysis was used to compare tumor volume differences on Day 6.
In vitro si-NP uptake was quantified in MDA-MB-231 cells using fluorescent TAMRA-tagged siRNA at 4, 8, and 24 hr of treatment. While the percentage of TAMRA+ cells increased over time in all groups (90% TAMRA+ by 24 hr), (Figure 8C) a greater percentage of 50B12-DP25 treated cells were TAMRA+ at earlier timepoints (Figure 8D). This result is consistent with previous studies revealing an inverse correlation between PEG coating density and cell uptake. Since 50B12-DP25 si-NPs contain the highest ratio of core 50B polymer, and consequently lowest amounts of PEG shielding (Figure 2C), it is possible that their decreased PEG coating facilitates rapid cell entry. Overall, these data suggest that all lead formulations tested in vivo had robust siRNA delivery to tumor cells.
Optimized ternary si-NP core polymer content enhances tumor cell uptake in vivo and oncogene targeting in an orthotopic breast cancer model
To assess in vivo si-NP gene silencing activity by our lead si-NP candidates, we established orthotopic mammary tumors using MDA-MB-231.RictorHiBiT cells. Once tumors reached a volume of 50 mm3 [Day 0] mice were randomized into groups for treatment with ternary si-NPs (50B0, 50B4-DP100, 50B8-DP100, and 50B12-DP25) loaded with siRNA against RICTOR (siRictor) or with non-targeting siRNA (siControl, 50B8-DP100) at 1 mg/kg (Figure 8E). Treatment with trehalose vehicle served as a control. A second treatment on Day 2 with si-NPs harboring fluorescent non-targeting siRNA (TAMRA si-NPs) enabled measurements of si-NP uptake by tumor cells on Day 3, when tumors were dissociated and analyzed by flow cytometry for mean fluorescent intensity (MFI). These studies revealed that 50B8-DP100 si-NPs exhibited the greatest MH compared to any other si-NPs (Figure 8F). This is in contrast to our findings performed in cell culture demonstrating highest cell uptake of 50B12-DP25 si-NPs. This suggests that tumor biodistribution and cell uptake of 50B8-DP100 si-NPs benefit from greater PEG shielding in vivo in ways that were not detected from in vitro experiments.
Tumors assessed on Day 3 post-treatment for protein levels of RictorHiBiT revealed that, as compared to tumors from mice treated with vehicle or with siControl-loaded si-NPs, tumors treated with si-NPs loaded with human Rictor-targeting siRNAs (sihRictor-NPs) exhibited Nano-Luc activity that was decreased by >50%, reflecting siRNA-mediated knockdown of endogenous Rictor (Figure 8G). Consistent with in vivo tumor cell uptake data, tumors treated with 50B8-DP100 sihRictor-NP displayed the greatest Rictor knockdown (approx. 80%) relative to tumors treated with siControl-NPs and were significantly more potent than binary 50B0 sihRictor-NPs containing no 50B core polymer. Interestingly, gene silencing activity by 50B8-DP100 si-NPs was modestly, albeit not significantly, greater than 50B12-DP25 si-NPs (70% knockdown), potentially due to greater in vivo circulation time (Figure 7G–7H), in vivo tumor cell uptake (Figure 8F), and endosome disruptive activity (Figure 4).The 50B8-DP100 ternary si-NP formulation therefore exhibits characteristics that overcome multiple siRNA delivery barriers, positioning it as a frontrunner ternary formulation for future exploration of si-NP use in a therapeutic setting. The treatment efficacy of 50B8-DP100 si-NPs was, therefore, next tested in an orthotopic mammary tumor model using HCC70 cells. Once tumors surpassed a volume of 50 mm3 [Day 0], mice were randomized into groups for i.v. treatment with 50B8-DP100 si-NPs bearing non-targeting siRNA (siControl-NPs) or siRNA targeting the oncogene Rictor in the human genome (sihRictor-NPs) at 1 mg/kg on Days 0 and 3 (Figure 8H). Tumor volume was monitored throughout the study. Mice treated with sihRictor-NPs had significantly diminished tumor volume (50.22 mm3) compared to mice treated with siControl-NPs (93.27 mm3) by the study endpoint (Figure 8I-J), indicating that 50B8-DP100 si-NPs have high potential for future application in oncological therapy.
Optimized 50B8-DP100 si-NPs are well-tolerated in a muti-dose treatment setting
After establishing the therapeutic potential of 50B8-DP100 si-NPs in the cancer setting, we next assessed the long-term safety of this formulation following multiple i.v. treatments. Healthy, wild-type Balb/c mice were treated with three injections (Days 0, 3, and 7) of trehalose vehicle or 50B8-DP100 si-NPs harboring siControl or siRNA targeting Rictor in the mouse genome (simRictor) (Figure 9A, Supplementary Figure 14A-B). Mice were treated with either siControl- or simRictor-NPs to assess for potential carrier related toxicities of si-NPs or potential toxicities resulting from Rictor silencing in off-target organs. Mice were assessed on Day 8 for kidney and liver toxicity markers, blood urea nitrogen (BUN), alanine aminotransferase (ALT), and aspartate aminotransferase (AST). Both groups of si-NP treated mice possessed plasma marker levels within the normal ranges for healthy mice (Figure 9B-D). Complete blood count levels following si-NP treatment were also similar to levels seen in vehicle treated mice (Supplementary Figure 14C-J). Treatment with simRictor-NPs has the potential to produce off-target Rictor knockdown in the liver, which could result in hyperglycemia.[54] Mice treated with si-NPs were, therefore, assessed for glucose levels at the study endpoint, and baseline levels were similar to vehicle treated mice (Figure 9E). We furthermore probed the si-NP treated mice for Rictor knockdown within the liver, kidney, and spleen by western analysis (Figure 9F). While our si-NPs displayed the greatest biodistribution to the liver (Supplementary Figure 12), simRictor-NPs mediated a relatively low level of Rictor protein knockdown (~15%) in this organ. We did not observe Rictor protein knockdown within the kidney, and similar to liver, spleens had a relatively low (~20%) protein knockdown. Finally, histological assessment of the liver, kidney, and spleen produced no signs of tissue damage following si-NP treatment (Figure 9G). Together, these data indicate that 50B8-DP100 si-NPs display minimal toxicological effects following multiple treatments while also limiting knockdown activity in off-target organs. In the therapeutic setting, efficacious delivery of siRNA is likely to involve repeat si-NP administration. These studies therefore suggest that our optimized 50B8-DP100 ternary formulation would be well-tolerated in a long-term clinical scenario.
Figure 9. 50B8-DP100 si-NPs display minimal toxicological effects following multi-dose treatments.
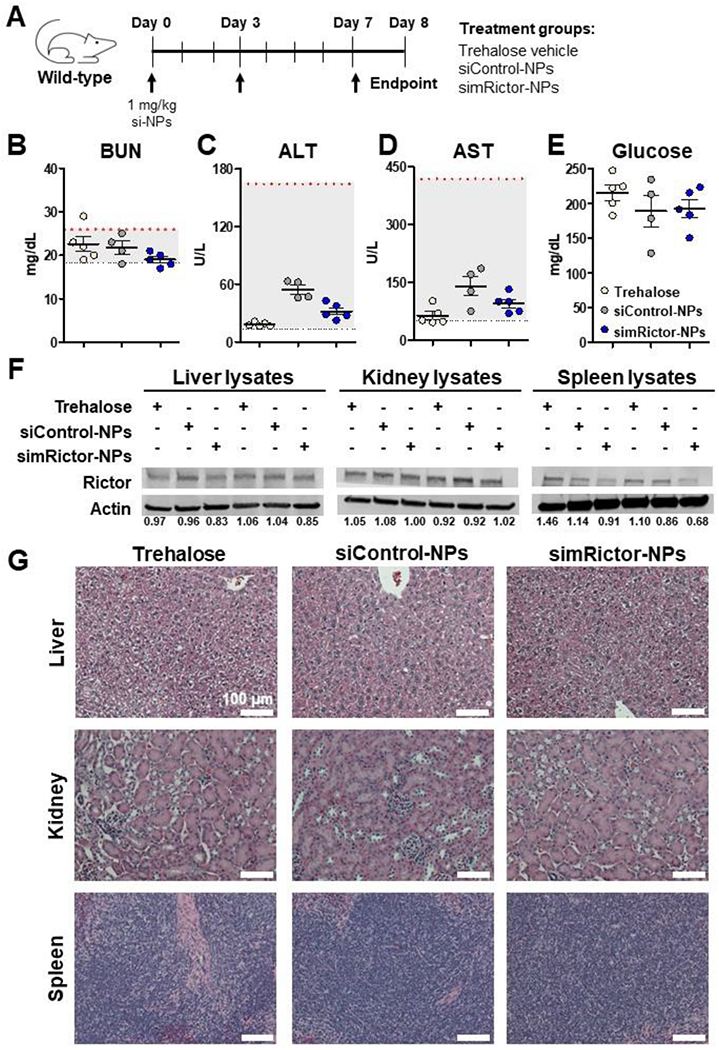
(A) Healthy, wild-type mice were treated with three injections over the course of a week and analyzed for safety and off-target activity on Day 8, 24 hr after the final injection. (B-D) Plasma BUN, ALT, and AST levels were within the normal ranges following multiple si-NP injections. Normal ranges for each marker are indicated by dotted y-axis lines. (E) simRictor-NP treatment did not result in elevated baseline glucose levels compared to vehicle treatment. (F) Livers, kidneys, and spleens were probed for Rictor protein knockdown by western analysis. Densiometric analysis of bands was calculated relative to average of siControl-NP bands. (G) Livers, kidneys, and spleens were assessed for tissue damage by H&E staining.
Ternary si-NP designs have been used in various delivery scenarios, including siRNA delivery to tumors, likely due to the versatility and multiple functionalities that they can offer. Notably, the clinically approved Onpattro comprises multiple components, including a cationic DLin-MC3-DMA core lipid and 2k PEG-lipid surface-forming unit, as well as cholesterol and DSPC phospholipids. While others have reported use of PEG-lipid amphiphiles as NP surfactants, we find that our 20kPEG-50B diblock surface-forming polymer provides comparable charge shielding, while advantageously also contributing to both siRNA complexation and endosome escape. The relatively simple design of our si-NP, comprised of siRNA and two polymers, also offers additional flexibility for future work to separately vary the cationic block composition of the core and surface-forming polymers.
Novel ternary si-NP formulations are being intensely pursued, including si-NPs containing a cationic polyamine core and hyaluronic acid coating for siRNA therapy against pancreatic cancer.[55] Similar to our work, the introduction of hydrophobic modifications to the core—in this case, through the addition of perfluoroalkyl moieties in the prodrug—improved si-NP stability. Yet, other groups utilizing ternary si-NP designs for tumor delivery have shown successful tumor gene silencing with their systems, often demonstrating ~60% or more tumor gene silencing by si-NP treatment.[23, 51, 56–58] We have built upon these advancements by using a comprehensive approach to optimize the ternary si-NP delivery platform, mitigating the need for high treatment doses and multiple injections regimes that have been used in the past. The systematic study of different formulations in vivo allowed us to achieve potent tumor gene silencing with a single dose given at 1 mg/kg. Studies presented herein advance the goal of siRNA delivery for cancer therapeutics through a comprehensive understanding of the structure-activity relationships of ternary si-NP components and how they govern in vivo efficacy.
Conclusions
Here, we aimed to understand the effect of core polymer content on ternary si-NP function both at the in vitro and in vivo level. We identified lead si-NP candidates that displayed maximal activity and stability without sacrificing safety in vivo and demonstrated that careful variation of core polymer size and ratio can balance these characteristics for efficacious si-NP tumor delivery. Addition of a core 50B polymer improved si-NP stability and activity in comparison to binary si-NPs across a very broad range of 50B MWs and formulation conditions. However, si-NP library screening also revealed trends among the ternary si-NPs, consistently showing that increasing 50B MW and ratio improved gene silencing potency, endosome disruptive ability, and stability when challenged with heparin salts or serum. However, increasing 50B content also came at the cost of heightened cytotoxicity. By simultaneously balancing 50B size and ratio, we identified ternary si-NP formulations that enhanced the si-NP therapeutic index, improved in vivo pharmacokinetics, and conferred potent tumor gene silencing in an orthotopic mammary tumor model. These studies underline the flexibility of ternary si-NP designs, as they can be easily tuned to overcome multiple delivery barriers. Our findings furthermore generate critical knowledge on the structure-activity relationships driven by ternary si-NP core polymer MW and content. These insights will guide future siRNA nanoparticle design for improved in vivo efficacy following systemic delivery.
Supplementary Material
Acknowledgements
The authors acknowledge the assistance of the Vanderbilt Translational Pathology Shared Resource (TPSR), supported by NCI/NIH Vanderbilt Cancer Center Support Grant P30CA068485. DLS nanoparticle characterization was conducted at the Vanderbilt Insitute of Nanoscale Science and Engineering (VINSE).
Funding
This work was supported by the National Institutes of Health (NIH R01 CA224241) and the National Science Foundation (NSF GRF 1937963).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The raw/processed data required to reproduce these findings are available upon request.
References
- 1.Nair JK, et al. , Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res, 2017. 45(19): p. 10969–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akinc A, et al. , The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nature Nanotechnology, 2019. 14(12): p. 1084–1087. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, et al. , Patisiran Pharmacokinetics, Pharmacodynamics, and Exposure-Response Analyses in the Phase 3 APOLLO Trial in Patients With Hereditary Transthyretin-Mediated (hATTR) Amyloidosis. Journal of clinical pharmacology, 2020. 60(1): p. 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilleron J, et al. , Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nature Biotechnology, 2013. 31(7): p. 638–646. [DOI] [PubMed] [Google Scholar]
- 5.Miao L, et al. , Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nature Biotechnology, 2019. 37(10): p. 1174–1185. [DOI] [PubMed] [Google Scholar]
- 6.Kulkarni JA, et al. , Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics. Accounts of Chemical Research, 2019. 52(9): p. 2435–2444. [DOI] [PubMed] [Google Scholar]
- 7.Zhu X, et al. , Long-circulating siRNA nanoparticles for validating Prohibitin1-targeted non-small cell lung cancer treatment. Proceedings of the National Academy of Sciences, 2015. 112(25): p. 7779–7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gujrati M, et al. , Multifunctional Cationic Lipid-Based Nanoparticles Facilitate Endosomal Escape and Reduction-Triggered Cytosolic siRNA Release. Molecular Pharmaceutics, 2014. 11(8): p. 2734–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu S, et al. , Zwitterionic Phospholipidation of Cationic Polymers Facilitates Systemic mRNA Delivery to Spleen and Lymph Nodes. Journal of the American Chemical Society, 2021. 143(50): p. 21321–21330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zuckerman JE, et al. , Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane. Proceedings of the National Academy of Sciences of the United States of America, 2012. 109(8): p. 3137–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moghimi SM and Patel HM, Serum-mediated recognition of liposomes by phagocytic cells of the reticuloendothelial system – The concept of tissue specificity. Advanced Drug Delivery Reviews, 1998. 32(1): p. 45–60. [DOI] [PubMed] [Google Scholar]
- 12.Moghimi SM, Hunter AC, and Murray JC, Long-Circulating and Target-Specific Nanoparticles: Theory to Practice. Pharmacological Reviews, 2001. 53(2): p. 283. [PubMed] [Google Scholar]
- 13.Leu D, et al. , Distribution and Elimination of Coated Polymethyl [2-14C]Methacrylate Nanoparticles After Intravenous Injection in Rats. Journal of Pharmaceutical Sciences, 1984. 73(10): p. 1433–1437. [DOI] [PubMed] [Google Scholar]
- 14.Göppert TM and Müller RH, Polysorbate-stabilized solid lipid nanoparticles as colloidal carriers for intravenous targeting of drugs to the brain: Comparison of plasma protein adsorption patterns. Journal of Drug Targeting, 2005. 13(3): p. 179–187. [DOI] [PubMed] [Google Scholar]
- 15.Jackson MA, et al. , Kupffer cell release of platelet activating factor drives dose limiting toxicities of nucleic acid nanocarriers. Biomaterials, 2021. 268: p. 120528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urits I, et al. , A Review of Patisiran (ONPATTRO®) forthe Treatment of Polyneuropathy in People with Hereditary Transthyretin Amyloidosis. Neurology and therapy, 2020. 9(2): p. 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson CE, et al. , Balancing Cationic and Hydrophobic Content of PEGylated siRNA Polyplexes Enhances Endosome Escape, Stability, Blood Circulation Time, and Bioactivity in Vivo. ACS Nano, 2013. 7(10): p. 8870–8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson MA, et al. , Dual carrier-cargo hydrophobization and charge ratio optimization improve the systemic circulation and safety of zwitterionic nano-polyplexes. Biomaterials, 2019. 192: p. 245–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarett SM, et al. , Hydrophobic interactions between polymeric carrier and palmitic acid-conjugated siRNA improve PEGylated polyplex stability and enhance in vivo pharmacokinetics and tumor gene silencing. Biomaterials, 2016. 97: p. 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oe Y, et al. , Actively-targeted polyion complex micelles stabilized by cholesterol and disulfide cross-linking for systemic delivery of siRNA to solid tumors. Biomaterials, 2014. 35(27): p. 7887–7895. [DOI] [PubMed] [Google Scholar]
- 21.Ambardekar VV, et al. , The modification of siRNA with 3’ cholesterol to increase nuclease protection and suppression of native mRNA by select siRNA polyplexes. Biomaterials, 2011. 32(5): p. 1404–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishina K, et al. , Efficient In Vivo Delivery of siRNA to the Liver by Conjugation of α-Tocopherol. Molecular Therapy, 2008. 16(4): p. 734–740. [DOI] [PubMed] [Google Scholar]
- 23.Werfel TA, et al. , Combinatorial optimization of PEG architecture and hydrophobic content improves ternary siRNA polyplex stability, pharmacokinetics, and potency in vivo. Journal of Controlled Release, 2017. 255: p. 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kilchrist KV, et al. , Gal8 Visualization of Endosome Disruption Predicts Carrier-Mediated Biologic Drug Intracellular Bioavailability. ACS nano, 2019. 13(2): p. 1136–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson MA, et al. , Zwitterionic Nanocarrier Surface Chemistry Improves siRNA Tumor Delivery and Silencing Activity Relative to Polyethylene Glycol. ACS Nano, 2017. 11(6): p. 5680–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paunovska K, et al. , A Direct Comparison of in Vitro and in Vivo Nucleic Acid Delivery Mediated by Hundreds of Nanoparticles Reveals a Weak Correlation. Nano Lett, 2018. 18(3): p. 2148–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Convertine AJ, et al. , Development of a novel endosomolytic diblock copolymer for siRNA delivery. Journal of controlled release : official journal of the Controlled Release Society, 2009. 133(3): p. 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kilchrist KV, Tierney JW, and Duvall CL, Genetically Encoded Split-Luciferase Biosensors to Measure Endosome Disruption Rapidly in Live Cells. ACS Sensors, 2020. 5(7): p. 1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, et al. , PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed, 2010. 99(3): p. 306–14. [DOI] [PubMed] [Google Scholar]
- 30.Miteva M, et al. , Tuning PEGylation of mixed micelles to overcome intracellular and systemic siRNA delivery barriers. Biomaterials, 2015. 38: p. 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mishra P, Nayak B, and Dey RK, PEGylation in anti-cancer therapy: An overview. Asian Journal of Pharmaceutical Sciences, 2016. 11(3): p. 337–348. [Google Scholar]
- 32.Caliceti P and Veronese FM, Pharmacokinetic and biodistribution properties of poly(ethylene glycol)–protein conjugates. Advanced Drug Delivery Reviews, 2003. 55(10): p. 1261–1277. [DOI] [PubMed] [Google Scholar]
- 33.Yamaoka T, Tabata Y, and Ikada Y, Distribution and Tissue Uptake of Poly(ethylene glycol) with Different Molecular Weights after Intravenous Administration to Mice. Journal of Pharmaceutical Sciences, 1994. 83(4): p. 601–606. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y and Grainger DW, Lyophilized liposome-based parenteral drug development: Reviewing complex product design strategies and current regulatory environments. Advanced Drug Delivery Reviews, 2019. 151-152: p. 56–71. [DOI] [PubMed] [Google Scholar]
- 35.Cavallaro G, et al. , Polymeric nanoparticles for siRNA delivery: Production and applications. International Journal of Pharmaceutics, 2017. 525(2): p. 313–333. [DOI] [PubMed] [Google Scholar]
- 36.Radmanovic N, et al. , Understanding the Freezing of Biopharmaceuticals: First-Principle Modeling of the Process and Evaluation of Its Effect on Product Quality. Journal of Pharmaceutical Sciences, 2013. 102(8): p. 2495–2507. [DOI] [PubMed] [Google Scholar]
- 37.Fonte P, et al. , Effect of cryoprotectants on the porosity and stability of insulin-loaded PLGA nanoparticles after freeze-drying. Biomatter, 2012. 2(4): p. 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veronese FM, Caliceti P, and Schiavon O, Branched and Linear Poly(Ethylene Glycol): Influence of the Polymer Structure on Enzymological, Pharmacokinetic, and Immunological Properties of Protein Conjugates. Journal of Bioactive and Compatible Polymers, 1997. 12(3): p. 196–207. [Google Scholar]
- 39.Sahay G, et al. , Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nature biotechnology, 2013. 31(7): p. 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kilchrist KV, et al. , Mechanism of Enhanced Cellular Uptake and Cytosolic Retention of MK2 Inhibitory Peptide Nano-polyplexes. Cellular and molecular bioengineering, 2016. 9(3): p. 368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanwar YS and Farquhar MG, Presence of heparan sulfate in the glomerular basement membrane. Proceedings of the National Academy of Sciences, 1979. 76(3): p. 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi HS, et al. , Renal clearance of quantum dots. Nature biotechnology, 2007. 25(10): p. 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rampado R, et al. , Recent Advances in Understanding the Protein Corona of Nanoparticles and in the Formulation of “Stealthy” Nanomaterials. Frontiers in Bioengineering and Biotechnology, 2020. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J, et al. , Nonfluorescent quenchers to correlate single-molecule conformational and compositional dynamics. Journal of the American Chemical Society, 2012. 134(13): p. 5734–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Z, et al. , Induction of Shock After Intravenous Injection of Adenovirus Vectors: A Critical Role for Platelet-activating Factor. Molecular Therapy, 2010. 18(3): p. 609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohnishi T, et al. , Serum platelet-activating factor acetylhydrolase activity in patients with atopic dermatitis. Journal of Dermatological Science, 2003. 33(1): p. 70–72. [DOI] [PubMed] [Google Scholar]
- 47.Graham RM, et al. , Plasma degradation of platelet-activating factor in severely ill patients with clinical sepsis. Critical Care Medicine, 1994. 22(2). [DOI] [PubMed] [Google Scholar]
- 48.Kelefiotis D and Vakirtzi-Lemonias C, In vivo responses of mouse blood cells to plateletactivating factor (PAF): role of the mediators of anaphylaxis. Agents Actions, 1993. 40(3-4): p. 150–6. [DOI] [PubMed] [Google Scholar]
- 49.Jäger M, et al. , Branched and linear poly(ethylene imine)-based conjugates: synthetic modification, characterization, and application. Chem Soc Rev, 2012. 41(13): p. 4755–67. [DOI] [PubMed] [Google Scholar]
- 50.Godbey WT, Wu KK, and Mikos AG, Poly(ethylenimine)-mediated gene delivery affects endothelial cell function and viability. Biomaterials, 2001. 22(5): p. 471–80. [DOI] [PubMed] [Google Scholar]
- 51.Werfel TA, et al. , Selective mTORC2 Inhibitor Therapeutically Blocks Breast Cancer Cell Growth and Survival. Cancer research, 2018. 78(7): p. 1845–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morrison Joly M, et al. , Rictor/mTORC2 Drives Progression and Therapeutic Resistance of HER2-Amplified Breast Cancers. Cancer research, 2016. 76(16): p. 4752–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwinn MK, et al. , CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chemical Biology, 2018. 13(2): p. 467–474. [DOI] [PubMed] [Google Scholar]
- 54.Hagiwara A, et al. , Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab, 2012. 15(5): p. 725–38. [DOI] [PubMed] [Google Scholar]
- 55.Yu A, et al. , Hyaluronate-coated perfluoroalkyl polyamine prodrugs as bioactive siRNA delivery systems for the treatment of peritoneal cancers. Biomaterials Advances, 2022: p. 212755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang X-Z, et al. , Sheddable Ternary Nanoparticles for Tumor Acidity-Targeted siRNA Delivery. ACS Nano, 2012. 6(1): p. 771–781. [DOI] [PubMed] [Google Scholar]
- 57.Cao S, et al. , Reduction-responsive RNAi nanoplatform to reprogram tumor lipid metabolism and repolarize macrophage for combination pancreatic cancer therapy. Biomaterials, 2022. 280: p. 121264. [DOI] [PubMed] [Google Scholar]
- 58.Ohyama A, et al. , Ternary complexes of folate-PEG-appended dendrimer (G4)/α-cyclodextrin conjugate, siRNA and low-molecular-weight polysaccharide sacran as a novel tumor-selective siRNA delivery system. International Journal of Biological Macromolecules, 2017. 99: p. 21–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw/processed data required to reproduce these findings are available upon request.