

Abstract
Rheumatoid arthritis (RA) is a risk factor for atherosclerotic cardiovascular diseases (CVD). Given the critical roles of the immune system and inflammatory signals in the pathogenesis of CVD, we hypothesized that interrogation of CVD-related proteins using integrative genomics might provide new insights into the pathophysiology of RA. We utilized two-sample Mendelian randomization (MR) for causal inference between circulating protein levels and RA by incorporating genetic variants, followed by colocalization to characterize the causal associations. Genetic variants from three sources were obtained: those associated with 71 CVD-related proteins measured in nearly 7000 Framingham Heart Study participants, a published genome-wide association study (GWAS) of RA (19 234 cases, 61 565 controls), and GWAS of rheumatoid factor (RF) levels from the UK Biobank (n = 30 565). We identified the soluble receptor for advanced glycation end products (sRAGE), a critical inflammatory pathway protein, as putatively causal and protective for both RA (odds ratio per 1-standard deviation increment in inverse-rank normalized sRAGE level = 0.364; 95% confidence interval 0.342–0.385; P = 6.40 × 10–241) and RF levels (β [change in RF level per sRAGE increment] = − 1.318; SE = 0.434; P = 0.002). Using an integrative genomic approach, we highlight the AGER/RAGE axis as a putatively causal and promising therapeutic target for RA.
Subject terms: Cardiovascular diseases, Rheumatoid arthritis, Data integration, Genome informatics, Proteome informatics, Computational models
Introduction
Rheumatoid arthritis (RA) is one of the most common chronic autoimmune diseases with a worldwide prevalence of 0.5–1% in adults1. Risk factors for RA include a strong genetic component2, prompting large-scale genome-wide association studies (GWAS) that have revealed more than 100 RA-associated genetic loci2.
RA is also a risk factor for cardiovascular disease (CVD) and multiple studies demonstrate a 1.5 to 2-fold risk of coronary artery disease in RA patients3–6. Currently hypothesized mechanisms for the predisposition to CVD among RA patients include shared genetic and environmental risk factors and dysregulation of inflammation and immune function4,7. Indeed, it was recently shown that baseline inflammatory markers such as erythrocyte sedimentation rate and C-reactive protein levels were associated with higher heart failure risk at 5- and 10-yr follow up8. There also have been recent early murine models and cross-sectional studies showing the link of IL-6 trans-signalling in the progression of atherosclerosis in RA9,10, as well as the role of annexin A1 in interrupting the progression of cardiomyopathy in arthritis models11. The exact immune-mediated and inflammatory mechanisms linking RA and CVD, however, are unknown and warrant elucidation.
Materials and methods
We analysed causal relations of 71 CVD-associated proteins to RA using protein quantitative trait loci (pQTL) from GWAS of plasma protein levels in 6,861 Framingham Heart Study (FHS) participants12 in conjunction with large-scale GWAS of RA13 and circulating rheumatoid factor (RF) levels14, which reflects anti-IgG immunoglobulins present in 80–90% of patients with RA15.
To assess the potential causal association between CVD-related proteins and RA, we employed Mendelian randomization16 (MR), a statistical approach to infer causality of an exposure for an outcome by mimicking randomized control trials using genetic variants as instrumental variables (IVs; Fig. S1), and colocalization, a Bayesian approach to assess shared genetic signals for two traits17. We applied two-sample MR16 to identify proteins causally associated with RA and RF and assessed the probability that the signals from MR are due to shared genetic variants18,19. While colocalization is not a test of causal inference between the exposure and the outcome, it identifies shared genetic variants, and when carried out in conjunction with Mendelian randomization can both provide additional insight into the mechanism of the causal association and reduce the probability of horizontal pleiotropy19,20. We further hypothesized that this integrative genomics approach might reveal CVD-related proteins that are causally linked to RA, thereby highlighting promising targets for the treatment of RA.
Study design
The study consisted of five steps (Fig. 1). First, from over 16,000 pQTL variants identified from GWAS of 71 CVD-related proteins measured in 6861 FHS participants12, we characterized pQTL variants that coincided with genetic variants from GWAS of RA13,16. Second, using cis-pQTL variants (i.e. residing within 1 Mb of the protein-coding gene) as IVs, we conducted MR testing to infer causal effects of proteins on RA (Fig. S1). Third, any causal protein from RA MR analysis was subject to MR analysis investigating its causal effect on RF levels. Fourth, colocalization analysis was performed on the putatively causal protein with RA to tease out potential loci that modulate the causal association. Fifth, MR analysis for a causal protein from the primary MR analysis was repeated with external replication with pQTLs from the INTERVAL study21 and a smaller GWAS of RA from the UK Biobank22.
Figure 1.

Study design. Flowchart of the study design. The study consisted of four steps: i. Identify pQTL variants overlapping with genetic variants for RA from GWAS, ii. and iii. Mendelian randomization analyses of the primary and secondary traits, iv. Colocalization analysis using the pQTL and GWAS, and v. Replication utilizing external pQTL and GWAS. The GWAS for RA was obtained via MRCIEU13,16 and the GWAS for RF14 was obtained via the UK Biobank. v. External replication was then achieved with the INTERVAL pQTL21 and the GWAS for RA22 obtained via the UK Biobank.
The 71 plasma proteins were selected based on their relation to CVD as described previously12. Protein levels in FHS participants were measured using Luminex bead-based assays (Luminex, Inc, Austin, TX)23. Genotyping of participants was performed using Affymetrix genotyping arrays (Affymetrix, Inc, Santa Clara, CA) as well as the Illumina Exome Chip array (Illumina, Inc. San Diego, CA). GWAS of inverse-rank normalized protein levels was performed in R and SAS using genotype dosages based on 1000 Genomes Project imputation (Affymetrix genotypes) or observed genotypes (Exome Chip) in linear mixed-effects models12.
Our primary analysis used the UK Medical Research Council Integrative Epidemiology Unit’s (MRCIEU) summary statistics for the trans-ethnic GWAS of RA (19 234 cases, 61 565 controls) by Okada et al.13,16. All RA cases fulfilled diagnostic criteria of the American College of Rheumatology or were diagnosed by a rheumatologist13. Secondary analysis was conducted on the UK Biobank GWAS of circulating RF levels (n = 30 565)14. The protocols for measuring serum RF and genotyping are described elsewhere14. Replication analyses were conducted with the INTERVAL pQTLs (n = 3301)21 for the exposure, and the UK Biobank GWAS of RA (4412 cases, 365 085 controls)22 for the outcome, with protocols for serum protein measurements, case definitions, and genotyping defined elsewhere21,22,24.
MR for causal inference
Two-sample MR16 was used to infer causal association, with protein level (using cis-pQTL variants as IVs) as the exposure, and RA (or RF) as the outcome. For the exposure, 40 (of 71) proteins with cis-pQTLs shared by the outcome GWAS were used12. For each outcome, summary statistics were obtained from the corresponding GWAS13,14,16.
MR requires three assumptions to be fulfilled (Fig. S1). First, the genetic variants should be associated with the exposure. Second, the genetic variants should not be associated with a confounder. Third, the genetic variants should be associated with the outcome only through the exposure; violation of this assumption is referred to as horizontal pleiotropy9. The first assumption was fulfilled by utilizing cis-pQTLs that reflect association with the exposure (plasma protein levels). The second and third assumptions were tested using sensitivity analyses including horizontal pleiotropy analyses that utilize the intercept term in MR Egger regression as an indicator, leave-one-out analyses to determine if a single SNP is driving the association and colocalization analyses as described below25,26.
Pruned cis-pQTL variants with linkage disequilibrium (LD) r2 < 0.01 for each protein were used as IVs to minimize the chances of a single nucleotide polymorphism (SNP) in LD being a confounder for the MR analysis. For proteins with only one independent pQTL variant after LD pruning, causal effect was determined using the Wald test, i.e., a ratio of effect per risk allele on RA to effect per risk allele on inverse-rank normalized protein levels. When multiple non-redundant pQTL variants were present, multi-SNP MR was conducted using fixed-effect inverse-variance weighted estimates. All MR analyses were conducted using the TwoSampleMR package in R16.
Colocalization analysis
Colocalization analysis was conducted as an additional characterization of the inferred causal association in which shared genetic loci were identified between an exposure and an outcome26. The loci of proteins that were identified as putatively causal from the MR analyses were tested for colocalization with the loci of the outcomes to further explore the MR result and to consider potential confounders.
We first identified sentinel cis- and trans-pQTL variants for each protein. A locus was defined as within 1 Mb upstream or downstream (total span of 2 Mb) of each sentinel SNP. We then identified the SNPs within each locus that overlapped with the RA GWAS at P < 5.13 × 10–9 (0.05/9,739,304 variants)13. To estimate the probability that the overlapping locus reflects the same sentinel variant for both the protein and RA, we conducted a Bayesian test for colocalization of all SNPs in each locus using the coloc package in R. This method requires specifying a prior probability for a SNP being associated with RA only (p1), protein levels only (p2), and with both traits (p12). We applied the default values for p1 and p2 of 1 × 10–4 and p12 was specified as 1 × 10–6. We prioritized the analysis of the posterior probability (PP) of hypothesis H4, where one shared SNP is associated with both trait 1 and 2. Significant colocalization was defined as H4 > 0.9027.
Results
Table 1 summarizes MR results for two proteins (P < 0.05), and Table S1 presents the comprehensive MR results for all 40 proteins. Statistical significance was defined as P < 0.00125 (0.05/40). sRAGE was causally implicated (odds ratio [OR] per 1 standard deviation [SD] increment in inverse rank-normalized sRAGE levels = 0.364; 95% confidence interval [CI] 0.342–0.385; P = 6.40 × 10–241) with a protective effect (OR/ΔSD < 1) on RA with the sentinel cis-variant rs2070600 having the most replicable effect on the causal relationship passing the sensitivity analyses (Figs. 2, 3).
Table 1.
Mendelian randomization results for rheumatoid arthritis (p < 0.05) and the corresponding Mendelian randomization for rheumatoid factor.
| A. | ||||||||
|---|---|---|---|---|---|---|---|---|
| Rheumatoid arthritis | Rheumatoid factor | |||||||
| Exposure | nsnp | Effect size | 95% CI | P value | nsnp | Effect size | SE | P value |
| sRAGE | nsnp | 0.364 | [0.342; 0.385] | 6.40E-241* | 3 | -1.318 | 0.43 | 0.02† |
| sICAM1 | 3 | 1.200 | [1.026; 1.403] | 0.02† | 1 | 1.235 | 0.96 | 0.198 |
| B. | ||||||||
|---|---|---|---|---|---|---|---|---|
| Rheumatoid arthritis | Rheumatoid factor | |||||||
| Exposure | SNP | Effect size | 95% CI | P value | Effect size | SE | P value | |
| sRAGE | rs2070600 (6: 32151443T < C) | 0.475 | [0.446; 0.505 ] | 5.25E–122* | − 1.262 | 0.48 | 0.008† | |
| rs9266529 (6: 31342029A < G) | 1.365 | [0.829; 2.249] | 0.222 | − 1.877 | 1.35 | 0.164 | ||
| rs6923504 (6: 32428186G < C) | 0.004 | [0.003; 0.005 ] | < E308* | − 1.150 | 1.62 | 0.478 | ||
| sICAM1 | rs5498 (19:10395683G < A) | 1.200 | [1.026; 1.403 ] | 0.02† | 1.235 | 0.96 | 0.198 | |
*Denotes Bonferroni-corrected significance at P < 0.00125 (0.05/40).
†Denotes Bonferroni-corrected significance at P < 0.025 (0.05/2) for sRAGE and sICAM1.
Effect size for RA is odds ratio per 1 standard deviation [SD] increment in inverse rank-normalized sRAGE levels.
Effect size for RF is change in RF level (IU/mL) per 1 standard deviation [SD] increment in inverse rank-normalized sRAGE levels.
Figure 2.

Mendelian randomization sensitivity analysis of sRAGE in relation to rheumatoid arthritis. (A): Forest plot of individual sRAGE cis-pQTL variant’s effect size (odds ratio per 1-standard deviation increment in inverse-rank normalized sRAGE level) in relation to RA. The fixed-effect inverse-variance weighted average effect of the SNPs were highly influenced by both rs2070600 and rs6923504 compared to rs9266529. (B): Leave-one-out analysis. Leaving rs2070600 out affected the confidence interval of the overall MR the most, while leaving rs6923504 did not affect the significance of the results. (C): Scatter plot of SNP effect size (odds ratio) in exposure GWAS (x-axis) and SNP effect size (standard deviation increment in inverse-rank normalized sRAGE level) in outcome GWAS (y-axis) for each cis-pQTL. rs2070600 is the driver of the causal association between sRAGE and RA. While rs2070600 is inversely associated with circulating sRAGE levels and positively associated with RA in their respective GWAS, note that the plotting algorithm transforms (x,y) to (− x,− y) if the x value is negative for plotting purposes (the slope and y-intercept remain the same).
Figure 3.

Mendelian randomization sensitivity analysis of sRAGE in relation to rheumatoid factor levels. (A): Forest plot of individual sRAGE cis-pQTL variant’s effect size (odds ratio per 1-standard deviation increment in inverse-rank normalized sRAGE level) in relation to plasma RF levels. rs2070600 had the narrowest confidence interval and contributed the most to the fixed-effect inverse-variance weighted effect of sRAGE on the outcome. (B): Leave-one-out analysis. Leaving rs2070600 out affected the confidence interval of the overall MR the most. (C): Scatter plot shows rs2070600 having the strongest contribution to the putatively causal association.
Single-SNP MR was performed using rs2070600 to further characterize the effect of rs2070600, which demonstrated a causal effect of sRAGE on RA (Wald test OR = 0.474; 95% CI 0.446–0.505; P = 5.25 × 10–122). While rs6923504 also contributed to the protective effect of sRAGE with a statistically significant p value, its effect size on RA was only 0.004, and its effect on RF was not statistically significant.sRAGE also was significant in MR analysis of RF levels as a secondary outcome (β, RF level change per sRAGE increment = − 1.318; SE = 0.434; P = 0.002). rs2070600 had the most substantial contribution to the RF causal relationship (Fig. 3), and single-SNP MR using rs2070600 revealed evidence of causality (Wald test β = − 1.263; SE = 0.477; P = 0.008).
Horizontal pleiotropy sensitivity analysis using MR Egger intercepts showed no significant horizontal pleiotropy between sRAGE and RA (Ppleiotropy = 0.336) or RF (Ppleiotropy = 0.843). Leave-one-out sensitivity analysis revealed that leaving rs2070600 out affected the confidence interval of the overall MR association more than any other pQTL SNPs for sRAGE.
In MR of both RA and RF, sRAGE was the only protein biomarker that passed the multiple testing corrected significance threshold. sRAGE was putatively casual and protective (OR/ΔSD < 1; Beta/ΔSD < 0) in relation to RA and RF.
The putatively causal protective relation of sRAGE to RA and RF was externally replicated, using the INTERVAL pQTL (rs2070600) for sRAGE as the exposure and the UK Biobank GWAS of RA as the outcome (Table 2; Fig. S2). The net protective effect of sRAGE was recapitulated both using the original FHS pQTL as the exposure and UK Biobank of RA as the outcome, as well as using the INTERVAL pQTL as the exposure and original MRC-IEU GWAS of RA was the outcome. The net inverse causal relation of sRAGE levels to RF levels was replicated with the INTERVAL pQTL.
Table 2.
Mendelian randomization external replication for sRAGE in relation to rheumatoid arthritis and rheumatoid factor.
| A. | |||||
|---|---|---|---|---|---|
| Exposure | Outcome | nsnp/SNP | Effect size | 95% CI | P value |
| FHS pQTL for sRAGE* | MRCIEU RA GWAS* | 3 | 0.364 | [0.342; 0.385] | 6.40E−241 |
| rs2070600 | 0.475 | [0.446; 0.505] | 5.25E−122 | ||
| rs9266529 | 1.365 | [0.829; 2.249] | 0.222 | ||
| rs6923504 | 0.004 | [0.003; 0.005] | < E308 | ||
| UK Biobank RA GWAS | 3 | 0.420 | [0.191; 0.924] | 0.031 | |
| rs2070600 | 0.475 | [0.419; 0.539] | 1.03E−30 | ||
| rs9266529 | 0.607 | [0.431; 0.854] | 0.004 | ||
| rs6923504 | 0.053 | [0.034; 0.081] | 8.77E−40 | ||
| INTERVAL pQTL for sRAGE | MRCIEU RA GWAS* | 1 | 0.394 | [0.317; 0.472] | 5.25E−122 |
| rs2070600 | |||||
| UK Biobank RA GWAS | 1 | 0.395 | [0.236; 0.553] | 1.03E−30 | |
| rs2070600 | |||||
| B. | |||||
|---|---|---|---|---|---|
| Exposure | Outcome | nsnp/SNP | Effect size | SE | P value |
| FHS pQTL for sRAGE* | UK Biobank RF GWAS* | 3 | − 1.318 | 0.43 | 0.002 |
| rs2070600 | − 1.262 | 0.48 | 0.008 | ||
| rs9266529 | − 1.877 | 1.35 | 0.164 | ||
| rs9266529 | − 1.150 | 1.62 | 0.478 | ||
| INTERVAL 1 pQTL for sRAGE | UK Biobank RF GWAS* | 1 | − 1.58 | 0.60 | 0.008 |
| rs2070600 | |||||
Effect size for RA is odds ratio per 1 standard deviation [SD] increment in inverse rank-normalized sRAGE levels.
Effect size for RF is the change in RF level per 1 standard deviation [SD] increment in inverse rank-normalized sRAGE levels.
* = dataset used for primary analysis.
External MR replication using the INTERVAL pQTL (n = 3301)21 and a smaller RA GWAS from the UK Biobank (4412 cases, 365 085 controls)22 was consistent with the primary finding. MR recapitulated the net protective effect of sRAGE. Replication with INTERVAL for the UK Biobank RF GWAS was also consistent with the primary MR analysis.
The minor T allele for rs2070600 was associated with 20–50% lower sRAGE levels in FHS participants (Table S2). This minor allele was associated with increased risk of RA and higher RF levels in the corresponding GWAS13,16 (OR [per risk allele] = 1.700; 95% CI 1.626–1.777; P = 3.60 × 10–127, and β [RF change per risk allele] = 0.899; SE = 0.340; P = 0.008, respectively).
Various other clinical characteristics such as mean age, percent women, body mass index, smoking status, history of diabetes, history of cardiovascular disease, and mean IL6 and CRP levels were also investigated by sRAGE levels. Quartile tabulation by sRAGE levels showed a significant association between sRAGE levels and BMI, current smoking status, and mean CRP levels (Tabe S3A). Cross-sectional multivariable regression model between sRAGE levels and clinical traits (history of diabetes and cardiovascular diseases) and inflammatory biomarker levels (Il-6 and CRP) revealed a significant inverse association between circulating sRAGE levels and CRP levels (Table S3B).
Colocalization analysis for sRAGE was conducted for three sentinel loci: rs4253272 (trans, Chromosome 4), rs116653040 (trans determined to be long-range cis, Chromosome 6), and rs2070600 (cis, Chromosome 6). At the posterior probability of > 0.90, only the rs116653040 locus (1 Mb window) significantly colocalized, reflecting an association of sRAGE levels with RA (PP.H4 = 1.00; Table S4). While the rs2070600 locus did not significantly colocalize (PP.H4 = 5.08 × 10–48), rs116653040 is in significant linkage disequilibrium with rs2070600 (R2 = 0.3041, P < 0.0001), indicating that rs116653040 is associated with sRAGE level and RA while acting as a long-range cis-locus for sRAGE. Thus, the causal association of sRAGE with RA from MR is strongly driven by rs2070600, although it may confer effects in conjunction with other SNPs in LD (e.g. with rs9266529 [the other cis-pQTL used as an IV for sRAGE]; r2 = 0.0438, P = 0.0032)28 that modulate sRAGE levels.
Discussion
Using an integrative genomic strategy, we identified sRAGE as putatively causal and protective protein against both RA and RF. sRAGE is a soluble form of RAGE, a transmembrane protein coded by the AGER gene (Fig. 4). AGER is located in the human leukocyte antigen (HLA) class III locus, near HLA-DRB1 and the HLA class II locus, both of which have been reported to be associated with RA2. Ligands that bind to membrane-bound RAGE, including advanced glycation end products, S100 proteins, and high mobility group box-1 protein (HMGB1), trigger proinflammatory pathways29. Circulating sRAGE is derived from proteolytic cleavage of membrane-bound RAGE (mRAGE) or via endogenous secretion of an alternatively spliced isoform (esRAGE) that lacks the trans-membrane domain of the RAGE protein. sRAGE acts as a decoy receptor and binds to RAGE ligands without inciting RAGE-mediated inflammatory signalling, explaining its protective effect. Indeed, a recent study30 found that sRAGE-overexpressing mesenchymal stem cells (MSCs) had reduced proinflammatory molecule production and increased immunomodulatory molecule expression. Similarly, IL-1Ra-knockout mice transplanted with sRAGE-overproducing MSCs demonstrated a reduction in inflammatory arthritis30. Of note, methotrexate, a first-line RA treatment, acts in part by directly binding to the RAGE ligand HMGB1 to inhibit the HMGB1/RAGE pathway31.
Figure 4.
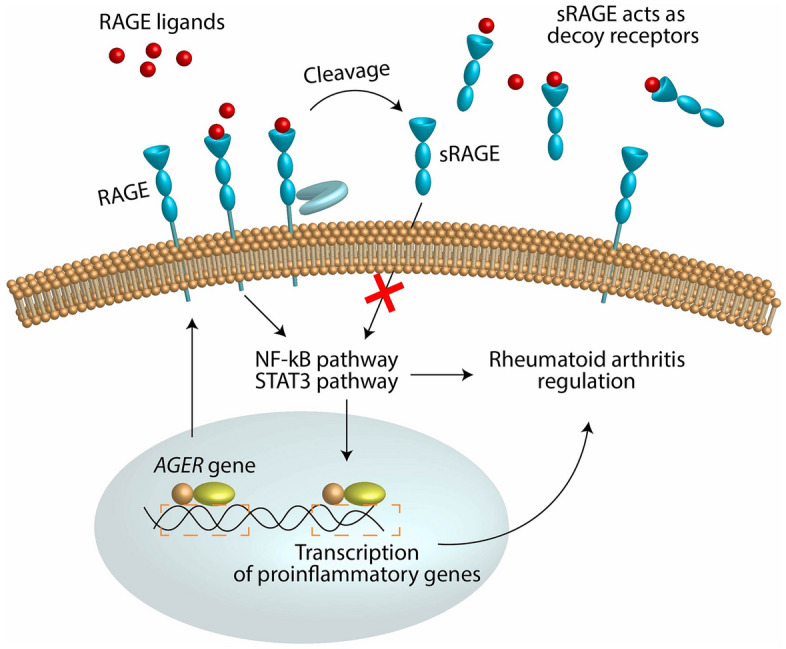
The protective role of sRAGE in relation to rheumatoid arthritis. Depiction of the protective mechanism of sRAGE in relation to RA. RAGE is a membrane-bound receptor that triggers pro-inflammatory pathways implicated with RA. sRAGE, a circulating form of RAGE, acts as a decoy receptor for RAGE ligands and therefore downregulates pro-inflammatory pathways.
Of the genetic variants driving the observed causal effect of sRAGE, we decided to focus our analysis on rs2070600 based on the sensitivity analysis ruling rs6923504 out as a significant contribution to the association. rs2070600 is a missense variant in AGER exon 3 with higher prevalence in RA patients29. The amino acid substitution (Gly82Ser) resides at the ligand-binding domain and increases the affinity for RAGE ligands29, enhancing proinflammatory signalling. This polymorphism is thought to simultaneously make RAGE less susceptible to cell surface RAGE cleavage29, reducing the generation of sRAGE. This has the dual effects of increasing RAGE proinflammatory ligand-binding and decreasing availability of sRAGE to act as a decoy receptor for ligands. Consistent with these joint effects, we found that the rs2070600 Ser (versus Gly) substitution was associated with lower circulating sRAGE levels in FHS participants12 and positively associated with both RA and RF levels in GWAS13,16.
Previous proteomic MR studies of RA have reported IL-632, CRP33, and sex hormone-binding globulin34 as causal biomarkers of RA. While IL-6 and sex hormone-binding globulin were not in our panel of proteins, we found that CRP was not causal for RA (P = 0.512; Table S1). We posit that the 22 SNPs used for CRP as the exposure in the prior MR study did not distinguish cis- or trans- pQTLs and spanned multiple chromosomes—and therefore may have contributed to horizontal pleiotropy from other genes remote from the CRP locus.
The AGER gene is located in the human leukocyte antigen (HLA) class III locus on chromosome 6, between the HLA class I and the HLA class II locus, which has been reported to be associated with RA2. Colocalization analysis hinted at the presence of other long-range effects on sRAGE and RA in addition to the modulation by missense variant rs2070600. Since the HLA locus is highly polymorphic and with sizable LD across the region35–37, the relationship at the HLA class II locus (e.g. rs116653040) was interrogated.
A query of National Cancer Institute's LDtrait Tool38 for variants in LD with rs2070600 or rs116653040 that have been reported to be associated with RA in Europeans revealed a signal for rs6910071 (OR [95% CI] = 2.73–3.03, P = 1 × 10–299 from a GWAS with 5,539 cases and 20,169 controls)39. rs6910071 is a tag SNP for the HLA-DRB1*0401 allele near C6orf1040. rs6910071 is in LD with rs2070600 with r2 = 0.1081 (P < 0.0001).
Based on reported whole blood expression quantitative loci (eQTL) in GTEx41, rs2070600 was found to be significantly associated with expression of HLA-DQA2 (p = 1.1 × 10–13) and HLA-DRB1 (P = 2.2 × 10–5). rs116653040, the trans-pQTL for sRAGE in LD with rs2070600 (r2 = 0.3041, P < 0.0001), was associated with HLA-C in whole blood (p = 4.9E−11). rs6910071, the RA-associated HLA-DRB1 tag-SNP in LD with rs2070600, was associated with HLA-DQA2 (P = 2.4 × 10–21) and HLA-DRB1 (P = 1.5 × 10–7). All three genes, HLA-DQA2, HLA-DRB1, and HLA-C, have been reported to be associated with RA2,42. The degree to which the causal association of sRAGE with RA is driven by the RAGE-mediated inflammatory pathways, by interaction with HLA class II genes, or both, warrants further investigation.
Additionally, rs2070600 is associated with other phenotypes including asthma43, lung function44, and celiac disease45. While the diverse role of missense variant rs2070600 raises the spectre of horizontal pleiotropy, we posit that the effects are likely explained by sRAGE and its effect on inflammatory signalling. Therefore, another consideration of this missense variant is vertical pleiotropy, whereby the additional traits associated with rs2070600 represent the downstream effects of the exposure and do not violate MR assumptions and premises25,46.
Our study has several limitations. First, while we utilized the FHS pQTL variants identified from 71 CVD-related proteins, they are not representative of the entire human plasma proteome. While we rationalize the use of the FHS pQTLs associated with CVD-related proteins due to the purported link between CVD and RA, a similar analysis with more comprehensive pQTLs could reveal additional significant pathways and potential confounders. There remains a need for a pan-protein pQTL resource with sufficient sample size and SNP associations to run reliable Mendelian randomization analyses. The FHS pQTL dataset based on GWAS of nearly 7000 individuals allowed us to conduct a statistically powerful MR with a sufficient number of SNPs. Second, the proteins were measured in plasma, which may yield conclusions not translatable to tissue-specific protein effects. While circulating sRAGE levels are correlated with synovial fluid sRAGE levels (rs = 0.48, P = 0.0002)47, our findings should be confirmed in tissue-specific settings. Third, since the SNPs accounting for the causal association between sRAGE and RA are in close proximity with HLA class II, the interaction between RAGE-mediated effects on RA with HLA genes should be delineated further. Fourth, while MR testing allowed inference of causal effects of protein levels on RA and RF, further cell and animal studies are warranted. If our findings are confirmed, modulation of AGER/RAGE to reduce inflammatory signalling, by altering sRAGE production, may lead to novel therapies for RA.
Conclusions
Through Mendelian randomization using pQTLs of CVD-related proteins along with GWAS of RA and RF, sRAGE was identified as putatively protective for both RA and RF levels. Given that sRAGE was previously identified as a potential inhibitor of RAGE-mediated inflammation related to RA, we hypothesize that the AGER/RAGE axis is a promising therapeutic target for RA.
Supplementary Information
Acknowledgements
We would like to thank the participants and volunteers for the FHS and the UK Biobank for their contribution to the data. We also are grateful to the staff at the Framingham Heart Study for helping with the medical chart review of participants that allowed us to begin our inquiry into the relationship between RA and CVD.
Disclaimer
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
Author contributions
G.L. designed the project, executed the statistical analyses, and wrote the manuscript. S.H., C.Y., D.H.L., and J.M. helped with troubleshooting, validating, and interpreting the analyses. S.H. and R.J. helped with accessing, cleaning, and analysing the FHS GWAS and data on plasma protein levels based on genotype. R.C.E., L.L.M., and C.L. provided feedback and guidance throughout the course of the project and helped with the interrogation of RA status of FHS participants with the chart review from the LifeHealth initiative. D.L. was the principal investigator and helped refine the project design, interpret the analyses, acquire funding, and organize the manuscript.
Funding
This work was supported by the National Institutes of Health (grant ref. N01-HC-25195) and the Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD (D. Levy, Principal Investigator).
Data availability
The study utilized de-identified human data consisting of summary statistics of genome-wide association studies and de-identified aggregate clinical data from the Framingham Heart Study. The datasets generated and/or analysed during the current study are available in the dbGaP and BioLINCC repositories. The data used for this study are all publicly available12–14,16,21,22. The FHS pQTL resource can be found at https://www.nature.com/articles/s41467-018-05512-x12 and the INTERVAL pQTL can be found at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6093935/21. The RA GWAS from Okada et al. accessed via MRCIEU can be found at https://gwas.mrcieu.ac.uk/datasets/ieu-a-833/13,16, the UK Biobank GWAS of RA can be found at http://pheweb.sph.umich.edu/SAIGE-UKB/pheno/714.122,24, and the UK Biobank GWAS of RF can be found at https://gwas.mrcieu.ac.uk/datasets/ukb-d-30820_raw/16,48. The results of the Mendelian randomization analysis from this study are available in full in Supplementary Table S1.
Competing interests
The National Heart, Lung, and Blood Institute and Ionis Pharmaceuticals entered into a Cooperative Research and Development Agreement (CRADA) to conduct research targeting the AGER gene, which encodes sRAGE, based in part, on the results of this research. Dr. Levy is the NHLBI principal investigator on the CRADA. Neither Dr. Levy nor the NHLBI received any support from Ionis in relation to the CRADA. All other authors do not have any conflict of interest.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-023-35098-4.
References
- 1.Myasoedova E, Crowson CS, Kremers HM, Therneau TM, Gabriel SE. Is the incidence of rheumatoid arthritis rising?: Results from Olmsted County, Minnesota, 1955–2007. Arthritis Rheum. 2010;62:1576–1582. doi: 10.1002/art.27425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okada Y, Eyre S, Suzuki A, Kochi Y, Yamamoto K. Genetics of rheumatoid arthritis: 2018 status. Ann. Rheum. Dis. 2019;78:446–453. doi: 10.1136/annrheumdis-2018-213678. [DOI] [PubMed] [Google Scholar]
- 3.Solomon DH, et al. Patterns of cardiovascular risk in rheumatoid arthritis. Ann. Rheum. Dis. 2006;65:1608–1612. doi: 10.1136/ard.2005.050377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skeoch S, Bruce IN. Atherosclerosis in rheumatoid arthritis: Is it all about inflammation? Nat. Rev. Rheumatol. 2015;11:390–400. doi: 10.1038/nrrheum.2015.40. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, et al. The association between inflammatory markers, serum lipids and the risk of cardiovascular events in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2014;73:1301–1308. doi: 10.1136/annrheumdis-2013-204715. [DOI] [PubMed] [Google Scholar]
- 6.Liao KP, Liu J, Lu B, Solomon DH, Kim SC. Association between lipid levels and major adverse cardiovascular events in rheumatoid arthritis compared to non-rheumatoid arthritis patients. Arthritis Rheumatol. 2015;67:2004–2010. doi: 10.1002/art.39165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J. Intern. Med. 2015;278:483–493. doi: 10.1111/joim.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang S, et al. The association between inflammation, incident heart failure, and heart failure subtypes in patients with rheumatoid arthritis. Arthritis Care Res. 2021 doi: 10.1002/acr.24804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi: 10.1136/bmj.k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies R, et al. The role of interleukin-6 trans-signalling on cardiovascular dysfunction in inflammatory arthritis. Rheumatology. 2021;60:2852–2861. doi: 10.1093/rheumatology/keaa725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, et al. Annexin A1 attenuates cardiac diastolic dysfunction in mice with inflammatory arthritis. Proc. Natl. Acad. Sci. U. S. A. 2021 doi: 10.1073/pnas.2020385118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao C, et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat. Commun. 2018;9:3268. doi: 10.1038/s41467-018-05512-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okada Y, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neale, B. GWAS of UK Biobank Biomarker Measurements (2019).
- 15.Walker DJ, Pound JD, Griffiths ID, Powell RJ. Rheumatoid factor tests in the diagnosis and prediction of rheumatoid arthritis. Ann. Rheum. Dis. 1986;45:684–690. doi: 10.1136/ard.45.8.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemani G, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. doi: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giambartolomei C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGowan LM, Davey Smith G, Gaunt TR, Richardson TG. Integrating Mendelian randomization and multiple-trait colocalization to uncover cell-specific inflammatory drivers of autoimmune and atopic disease. Hum. Mol. Genet. 2019;28:3293–3300. doi: 10.1093/hmg/ddz155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Q, Pan J, Berzuini C, Rutter MK, Guo H. Integrative analysis of Mendelian randomization and Bayesian colocalization highlights four genes with putative BMI-mediated causal pathways to diabetes. Sci. Rep. 2020;10:7476. doi: 10.1038/s41598-020-64493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burgess S, Foley CN, Zuber V. Inferring causal relationships between risk factors and outcomes from genome-wide association study data. Annu. Rev. Genomics Hum. Genet. 2018;19:303–327. doi: 10.1146/annurev-genom-083117-021731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore C, et al. The INTERVAL trial to determine whether intervals between blood donations can be safely and acceptably decreased to optimise blood supply: Study protocol for a randomised controlled trial. Trials. 2014;15:363. doi: 10.1186/1745-6215-15-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gagliano Taliun SA, et al. Exploring and visualizing large-scale genetic associations by using PheWeb. Nat. Genet. 2020;52:550–552. doi: 10.1038/s41588-020-0622-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho JE, et al. Protein biomarkers of cardiovascular disease and mortality in the community. J. Am. Heart Assoc. 2018;7:e008108. doi: 10.1161/JAHA.117.008108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bycroft C, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng J, et al. Recent developments in Mendelian randomization studies. Curr. Epidemiol. Rep. 2017;4:330–345. doi: 10.1007/s40471-017-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hemani G, Bowden J, Davey Smith G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum. Mol. Genet. 2018;27:R195–r208. doi: 10.1093/hmg/ddy163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet. 2021;17:e1009440. doi: 10.1371/journal.pgen.1009440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Machiela MJ, Chanock SJ. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Serveaux-Dancer M, et al. Pathological implications of receptor for advanced glycation end-product (AGER) gene polymorphism. Dis. Markers. 2019;2019:2067353. doi: 10.1155/2019/2067353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park MJ, et al. Overexpression of soluble RAGE in mesenchymal stem cells enhances their immunoregulatory potential for cellular therapy in autoimmune arthritis. Sci. Rep. 2016;6:35933. doi: 10.1038/srep35933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bedoui Y, et al. Methotrexate an old drug with new tricks. Int. J. Mol. Sci. 2019 doi: 10.3390/ijms20205023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li B, Xiao Y, Xing D, Ma XL, Liu J. Circulating interleukin-6 and rheumatoid arthritis: A Mendelian randomization meta-analysis. Medicine. 2016;95:e3855. doi: 10.1097/md.0000000000003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prins BP, et al. Investigating the causal relationship of C-reactive protein with 32 complex somatic and psychiatric outcomes: A large-scale cross-consortium Mendelian randomization study. PLoS Med. 2016;13:e1001976. doi: 10.1371/journal.pmed.1001976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qu Z, et al. Sex hormone-binding globulin and arthritis: A Mendelian randomization study. Arthritis Res. Ther. 2020;22:118. doi: 10.1186/s13075-020-02202-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen N, et al. The distributions of HLA-A, HLA-B, HLA-C, HLA-DRB1 and HLA-DQB1 allele and haplotype at high-resolution level in Zhejiang Han population of China. Int. J. Immunogenet. 2019;46:7–16. doi: 10.1111/iji.12411. [DOI] [PubMed] [Google Scholar]
- 36.Evseeva I, Nicodemus KK, Bonilla C, Tonks S, Bodmer WF. Linkage disequilibrium and age of HLA region SNPs in relation to classic HLA gene alleles within Europe. Eur. J. Hum. Genet. 2010;18:924–932. doi: 10.1038/ejhg.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matern BM, Olieslagers TI, Voorter CEM, Groeneweg M, Tilanus MGJ. Insights into the polymorphism in HLA-DRA and its evolutionary relationship with HLA haplotypes. Hla. 2020;95:117–127. doi: 10.1111/tan.13730. [DOI] [PubMed] [Google Scholar]
- 38.Lin SH, Brown DW, Machiela MJ. LDtrait: An online tool for identifying published phenotype associations in linkage disequilibrium. Cancer Res. 2020;80:3443–3446. doi: 10.1158/0008-5472.Can-20-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stahl EA, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denny JC, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 2013;31:1102–1110. doi: 10.1038/nbt.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.GTEx Consortium The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vignal C, et al. Genetic association of the major histocompatibility complex with rheumatoid arthritis implicates two non-DRB1 loci. Arthritis Rheum. 2009;60:53–62. doi: 10.1002/art.24138. [DOI] [PubMed] [Google Scholar]
- 43.Bui H, et al. A genomic approach identifies sRAGE as a putatively causal protein for asthma. J. Allergy Clin. Immunol. 2022;149:1992–1997.e1912. doi: 10.1016/j.jaci.2021.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keefe J, et al. An integrative genomic strategy identifies sRAGE as a causal and protective biomarker of lung function. Chest. 2022;161:76–84. doi: 10.1016/j.chest.2021.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamat MA, et al. PhenoScanner V2: An expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35:4851–4853. doi: 10.1093/bioinformatics/btz469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davey Smith G, Hemani G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014;23:R89–R98. doi: 10.1093/hmg/ddu328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pullerits R, Bokarewa M, Dahlberg L, Tarkowski A. Decreased levels of soluble receptor for advanced glycation end products in patients with rheumatoid arthritis indicating deficient inflammatory control. Arthritis Res. Ther. 2005;7:R817–R824. doi: 10.1186/ar1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sinnott-Armstrong, N. et al.bioRxiv (2019).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The study utilized de-identified human data consisting of summary statistics of genome-wide association studies and de-identified aggregate clinical data from the Framingham Heart Study. The datasets generated and/or analysed during the current study are available in the dbGaP and BioLINCC repositories. The data used for this study are all publicly available12–14,16,21,22. The FHS pQTL resource can be found at https://www.nature.com/articles/s41467-018-05512-x12 and the INTERVAL pQTL can be found at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6093935/21. The RA GWAS from Okada et al. accessed via MRCIEU can be found at https://gwas.mrcieu.ac.uk/datasets/ieu-a-833/13,16, the UK Biobank GWAS of RA can be found at http://pheweb.sph.umich.edu/SAIGE-UKB/pheno/714.122,24, and the UK Biobank GWAS of RF can be found at https://gwas.mrcieu.ac.uk/datasets/ukb-d-30820_raw/16,48. The results of the Mendelian randomization analysis from this study are available in full in Supplementary Table S1.