

Abstract
Alzheimer's disease (AD) drug discovery has focused on a set of highly studied therapeutic hypotheses, with limited success. The heterogeneous nature of AD processes suggests that a more diverse, systems‐integrated strategy may identify new therapeutic hypotheses. Although many target hypotheses have arisen from systems‐level modeling of human disease, in practice and for many reasons, it has proven challenging to translate them into drug discovery pipelines. First, many hypotheses implicate protein targets and/or biological mechanisms that are under‐studied, meaning there is a paucity of evidence to inform experimental strategies as well as high‐quality reagents to perform them. Second, systems‐level targets are predicted to act in concert, requiring adaptations in how we characterize new drug targets. Here we posit that the development and open distribution of high‐quality experimental reagents and informatic outputs—termed target enabling packages (TEPs)—will catalyze rapid evaluation of emerging systems‐integrated targets in AD by enabling parallel, independent, and unencumbered research.
Keywords: Alzheimer's disease, drug development, drug targets, genomics, proteomics, systems biology, TEPs
1. INTRODUCTION
By 2050, Alzheimer's disease (AD) will affect 150 million people worldwide. Caring for these patients will exact a tremendous societal and economic toll, 1 especially because there is a paucity of efficacious disease‐modifying therapies approved for use in humans. The past two decades of drug discovery efforts in AD have been focused on a few therapeutic hypotheses. Although these were strongly supported by genetic evidence, none have been validated in patients. 2 There is, therefore, an urgent need to explore new target hypotheses, which will necessitate adopting a higher risk tolerance in our target selection and drug discovery strategies. To this end, the National Institute on Aging has launched an initiative, called TREAT‐AD (TaRget Enablement to Accelerate Therapy Development for AD). The goals of this program are to provide the community with a set of high‐quality core reagents that are needed to explore new targets or disease mechanisms, and to pursue the most promising by generating new high‐quality pharmacological tools that are active in animals (drug leads). TREAT‐AD comprises two centers. Each is a collective of institutions; one is led from the Indiana University School of Medicine and the other from Emory University (and includes Sage Bionetworks and the Structural Genomics Consortium [SGC]). Here we describe the strategy of the Emory‐Sage‐SGC TREAT‐AD Center to enable rapid diversification of the AD drug discovery pipeline and apply the strategy to targets suggested by systems‐level approaches.
Unbiased genome‐wide studies from genetics and systems biology have led to an abundance of novel disease‐associated proteins and pathways that have not been studied previously in relation to AD and may provide for new therapeutic hypotheses. This is both an advantage and a disadvantage: an advantage in that the approaches offer the potential for truly transformative ideas, but a disadvantage in that the research enterprise is reticent to work on previously understudied proteins. 3 In AD, this phenomenon is particularly true, and can be quantified by evaluating the publication intensity over time for the 184 genes reported as associated with AD risk (ie, “AD‐related genes”) in the National Human Genome Research Institute ‐ European Bioinformatics Institute (NHGRI‐EBI) Unbiased genome‐wide association studies (GWAS) catalog through 2015 4 (Figure 1). Specifically, we quantified the publications that mention both Alzheimer's and the gene or protein name in the keywords, abstract, or title in two time periods: before genetic association studies were published (1950–2000) and after these studies were published (2018–2020). As might be expected, prior to 2000 almost all of the AD gene‐based publications (93%) were focused on four genes (apolipoprotein E [APOE], amyloid precursor protein [APP], presenilin 1 [PSEN1], and presenilin 2 [PSEN2]) that were discovered through early‐onset familial AD studies. But despite strengthening evidence from clinical trials that some anti‐amyloid immunotherapies informed by these four genes/proteins have only small although significant benefit in at least a subset of AD cases, 5 clearly more robust therapeutic strategies are urgently needed. Nonetheless, these four genes remained the most studied genes in the 2018–2020 period, garnering fully 73% of the AD‐related gene publications. In contrast, the number of genes associated with AD risk continues to explode with larger well‐powered studies, with at least 75 gene associations confirmed—most with links to additional, understudied biology. 6
FIGURE 1.
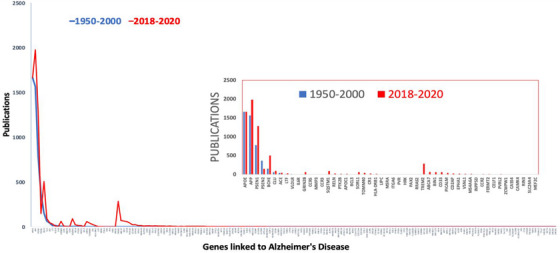
Publication intensity over time for 184 genes reported as associated with Alzheimer's disease (AD) risk.
RESEARCH IN CONTEXT
SYSTEMATIC REVIEW: The authors reviewed the scientific literature using standard approaches (GoogleScholar, PubMed) to develop our perspective on open drug discovery in Alzheimer's disease (AD).
INTERPRETATION: We present an overview of a novel approach to target validation in AD, which utilizes open science practices to accelerate drugs through the drug discovery pipeline.
FUTURE DIRECTIONS: Our center is generating openly available resources to enable target validation and drug discovery efforts in AD. Future directions based on our work include the adoption of these specific resources in experimental programs. For example, with an in vivo chemical or molecular probe inhibitor to a target of interest, a researcher could design experiments to examine a role for that target in relevant AD‐related phenotypes in human induced pluripotent stem cells (iPSCs) and relevant AD mouse models.
In our center, we hypothesize that researchers will be more willing to devote attention to understudied genes and proteins if provided with open access to high‐quality molecular and cellular research tools, 3 and our aim is to generate these tools for under‐studied proteins linked to AD.
1.1. The target enabling packages (TEPs): Open reagents for target evaluation
Our center is committed to providing a core set of data and reagents (target‐enabling packages [TEPs]) for a range of protein targets nominated through systems approaches and otherwise by the community. 7 As we define it, the minimal TEP comprises the purified protein, validated renewable or recombinant antibodies for a range of applications, and a verified gene knockout cell line. These minimal TEP components are the basic and validated experimental tools needed to start probing target biology. Each TEP component must meet our quality criteria prior to its release and open distribution. The quality criteria are listed in Table 1. For prioritized targets, we are generating additional TEP components that might include crystal structures, biochemical and biophysical assays, and cell‐based assays that would facilitate the identification of drug candidates. For each protein target, we pre‐define the composition of the TEPs, based on its function and localization, and with input from the wider community. All TEP components are being made available through trusted commercial and/or non‐profit distributors.
TABLE 1.
Quality acceptance criteria for target enabling package components.
| TEP component | Quality criteria |
|---|---|
| Genetic tools | |
| CRISPR‐deleted in selected cell lines |
|
| CRISPRa, CRISPRi, or induced‐degradation in relevant cell lines (or iPSCs) or RNAi |
|
| CRISPR‐edited cell lines containing disease‐linked mutations |
|
| Validated antibodies | |
| General | Preferably monoclonal or recombinant and with registered Research Resource Identifiers (RRIDs) |
| WB |
|
| IF |
|
| IP |
|
| IHC |
|
| Protein expression | |
| Purified protein/functional domain |
|
| Modified versions of the target protein if required for assays |
|
| If relevant, assembly into relevant protein complex |
|
| Fundamental assays | |
Biophysical assays:
|
|
In vitro biochemical assays:
|
|
|
|
Intracellular target engagement:
|
|
Cellular phenotypes
|
|
| Low‐throughput assays (additional criterion) |
|
| High‐throughput assays (additional criterion) |
|
| Structures | |
| Crystal structures |
|
| Chemical probes | |
Potency refers to EC50/IC50
|
|
| Biological probes | |
|
Abbreviations: BID, twice daily; BRET, Bioluminescence Resonance Energy Transfer; CETSA, cellular thermal shift assay; CNS, central nervous system; CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) ‐ deleted; CRISPRa (CRISPRactivation); CRISPRi (CRISPRinterference); HTS, high‐throughput screening; IF, immunofluorescence; IHC, immunistochemistry; IP, immunoprecipitation; IP‐MS, IP‐mass spectrometry; KO, knock‐out; PK, pharmacokinetic; QD, once daily; SAR, structure activity relationship; Tm, melting point, WB, western blot.
In practice, the first step in assembling a TEP is to secure and assay existing commercial reagents (such as antibodies and cell lines) to determine if they meet our quality criteria. If any do, we will make this information freely available. If we identify reagents that are missing, our center will produce and characterize them. To date our center has generated TEPs for seven AD‐relevant targets. These TEPs are distributed through the AD Knowledge Portal. 8
The generation of reagents for the chromosome 9 open reading frame 72 (C9ORF72) protein provides an excellent example of the potential impact of TEPs, and of our approach. The C9orf72 gene is one of the most commonly mutated genes in individuals with frontotemporal dementia or amyotrophic lateral sclerosis (ALS). This link was discovered almost 10 years ago, and in the immediate years after there was significant interest in its molecular and cellular biology. Many of the studies on C9ORF72 conflicted; however, different studies localized the protein to different subcellular components. The situation was clarified by focusing on the quality of the reagents. In 2018, McPherson et al. launched a TEP project to create or identify existing tools for the C9ORF72 proteins. 9 Focusing on antibodies to start, they used knockout cells as controls to screen commercially available antibodies for those that recognized the protein specifically and selectively. They found a high‐performing antibody, made the results publicly available, and used the antibody to localize C9ORF72 to the lysosomal membrane. Their work also revealed that many of the antibodies used previously to localize the protein to other compartments did not in fact even recognize the protein. The trajectory of C9ORF72 cell biology research has now changed in response to the availability of high‐quality, specific, and selective reagents.
1.2. TEP targets from AD systems biology
The current target portfolio in our center (Table 2) was assembled by evaluating prioritized proteins that were nominated as promising, yet understudied, targets for AD through the Accelerating Medicines Partnership in AD (AMP‐AD) program. 10 , 11 , 12 , 13 , 14 , 15 We select targets using an iterative process in which we evaluate lists of targets from AMP‐AD studies by calculating an unbiased target risk score, summarizing existing literature, and assessing the tractability and therapeutic intervention potential of each target. Our full target scoring pipeline is detailed in Cary et al., 16 but briefly, all targets are scored across a hierarchy of criteria that is broadly organized into genetic, multiomic, neuropathological, and literature‐based metrics (Figure 2). Following this initial scoring, targets are evaluated for therapeutic potential, with an emphasis on tractability. If targets are judged to not be tractable, the starting gene lists are expanded to incorporate other genes that are in the same network (using co‐expression or pathway annotation information). 16
TABLE 2.
TREAT‐AD emerging gene targets in Alzheimer's disease for TEP development.
| Target genes | |
|---|---|
| APOE | NDUFS2 |
| APP | NTN1 |
| ARHGEF2 | NTN3 |
| BDH2 | NXPH1 |
| C4A | OLFML3 |
| CAPN2 | PAK1 |
| CD44 | PLEC |
| CNN3 | PRDX1 |
| COL11A1 | PRDX6 |
| COL25A1 | PTN |
| CTHRC1 | QPRT |
| CTSH | RABEP1 |
| DAG1 | RENBP |
| DDX1 | SDC4 |
| DHX58 | SFRP1 |
| ECE1 | SLIT1 |
| EPHX2 | SLIT2 |
| FCER1G | SMOC1 |
| FLT1 | SNX32 |
| FRZB | SPOCK1 |
| GPC5 | SPOCK2 |
| GPNMB | SPOCK3 |
| HTRA1 | SPON1 |
| IFIH1 | STX4 |
| LRP1 | SYK |
| MDK | TICAM1 |
| MSN | TMEFF2 |
FIGURE 2.
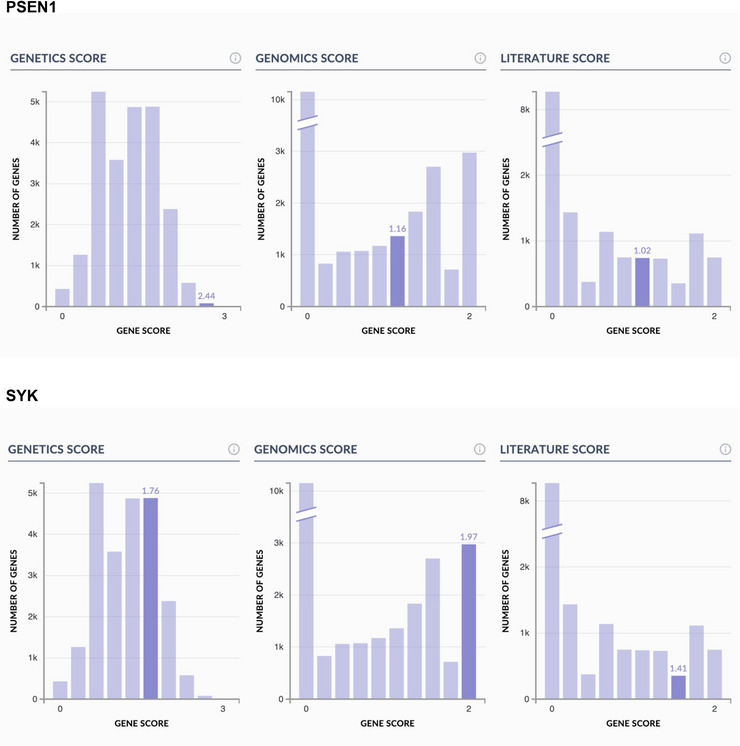
Representative histograms of TaRget Enablement to Accelerate Therapy Development for AD (TREAT‐AD) consortium target‐ranking scores encompassing genomics, genetics, and literature evidence for a classical Alzheimer's disease (AD) example target, presenilin1 (PSEN1), as well as a target prioritized by our TREAT‐AD center, spleen‐associated tyrosine kinase (SYK).
In addition to selection of individual understudied targets, we are developing resources to interrogate a set of functionally connected targets, by developing TEPS for multiple targets within one specific protein module. Deep proteomic profiling study of AD and control brains from AMP‐AD revealed new AD‐related protein co‐expression modules that were highly preserved across cohorts and brain regions. 15 One of these modules, Module 42, ontologically linked to the matrisome, was highly associated with the global burden of pathology in the brain, with a correlation coefficient of 0.75. Strikingly, the small group of 32 proteins co‐expressed in this module was enriched in AD risk genes and included APP and apoE. Evaluation of the potential therapeutic relevance of matrisome module proteins is a considerable challenge given the paucity of understanding of most of the proteins in the module, and the many possible mechanisms by which they influence disease pathology individually or collectively. By developing TEPs for multiple matrisome module targets, we aim to provide resources that can be used to dissect the function of this module and understand the relevance of individual module members to AD.
1.3. TEPs and open science
One of the anticipated major public benefits of this program is our commitment to share all reagents, including chemical inhibitors, without restriction or patent protection. This policy is designed to promote rapid and broad scientific discovery so that target validation can be robustly and rapidly performed across multiple independent research groups. Emerging evidence indicates that scientific discovery is slowed by the cautionary actions required to protect potential future intellectual property, such as imposing restrictive legal agreements on potential collaborators. 17 In further support of this, scientific discovery—as measured by citations—has been shown to be greater for openly available small molecule inhibitors than for analogous inhibitors that are encumbered by restrictions on use. 18 Several avenues exist to advance an unpatented TREAT‐AD drug candidate toward a therapy, including through the National Institute on Aging (NIA) drug discovery pipeline. Companies such as M4ND Pharma 19 do so by practicing open science business models. Alternatively, TREAT‐AD assets can be used to enable others to invent new patentable chemistry for a target. Our model offers the promise of stimulating new approaches to drug discovery by removing intellectual property restrictions as a barrier to research and openly sharing all tools that we generate.
2. CONCLUSION
The primary aim of this work is to increase the numbers of new targets considered in AD drug discovery pipelines by providing the community with the TEPs and drug‐like tools necessary to carry out robust target characterization. The TREAT‐AD program is the early translational partner of the NIA Translational Research Program and, so, operates on target priorities identified within this ecosystem. Reagents developed through TREAT‐AD are designed to uncover new biology, invalidate some target hypotheses, and increase the scientific attention on lesser‐studied human proteins and other targets. This alone will be a tremendous contribution to AD research. If one of the targets proves promising and advances into drug discovery and development, then its impact would extend from basic understanding to translational medicine.
Our center is designed as a community resource. As such, we actively solicit input on experimental prioritization from domain experts and promote the use of center outputs, in particular to test emergent therapeutic hypotheses. To encourage use by others, all available TREAT‐AD resources, including TEP reports, are cataloged in the AD Knowledge Portal 8 (Figure 3). An overview of the full TREAT‐AD portfolio, including resources currently in development, can be found on the TREAT‐AD website. 20
FIGURE 3.
Screenshot depicting the Target Enabling Resources page within the AD Knowledge Portal, found at https://adknowledgeportal.synapse.org/Explore/Target%20Enabling%20Resources.
We are committed to generating high‐quality probes that can be applied alongside an appropriate suite of reagents to enable discoveries about fundamental biology and validate the roles of these and other potential emerging AD targets. We will continue to make resources available and to collaborate widely with the research community—and we call on the community to use these reagents and engage with us to guide future reagent development.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the Supporting Information.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors would like to acknowledge the support of Drs. Benjamin A. Logsdon, Stephen V. Frye, and Larsson Omberg for insightful discussions and suggestions during the development of the center strategy. The Target Enablement to Accelerate Therapy Development for Alzheimer's Disease (TREAT‐AD) Consortium was established by the National Institute on Aging (NIA). The research reported in this manuscript was led by the Emory‐Sage‐SGC TREAT center and supported by grant U54AG065187 from the NIA.
Axtman AD, Brennan PE, Frappier‐Brinton T, et al. Open drug discovery in Alzheimer's disease. Alzheimer's Dement. 2023;9:e12394. 10.1002/trc2.12394
REFERENCES
- 1. WHO . Global action plan on the public health response to dementia. Geneva: WHO. 2017;2017‐2025. [Google Scholar]
- 2. Liu KY, Howard R. Can we learn lessons from the FDA's approval of aducanumab? Nat Rev Neurol. 2021;17:715‐722. [DOI] [PubMed] [Google Scholar]
- 3. Edwards AM, Isserlin R, Bader GD, Frye SV, Willson TM, Yu FH. Too many roads not taken. Nature. 2011;470:163‐165. [DOI] [PubMed] [Google Scholar]
- 4. GWAS Catalog. https://www.ebi.ac.uk/gwas/efotraits/EFO_0000249
- 5. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2023;388:9‐21. [DOI] [PubMed] [Google Scholar]
- 6. Bellenguez C, Kucukali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54:412‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bradley AR, Echalier A, Fairhead M, et al. The SGC beyond structural genomics: redefining the role of 3D structures by coupling genomic stratification with fragment‐based discovery. Essays Biochem. 2017;61:495‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. AD knowledge portal. https://adknowledgeportal.synapse.org/
- 9. Laflamme C, McKeever PM, Kumar R, et al. Implementation of an antibody characterization procedure and application to the major ALS/FTD disease gene C9ORF72. eLife. 8:2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Higginbotham L, Ping L, Dammer EB, et al. Integrated proteomics reveals brain‐based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer's disease. Sci Adv. 2020;6:eaaz9360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnson ECB, Dammer EB, Duong DM, et al. Large‐scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020;26:769‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wingo AP , Fan W, Duong DM, et al. Shared proteomic effects of cerebral atherosclerosis and Alzheimer's disease on the human brain. Nat Neurosci. 2020;23:696‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wingo AP, Liu Y, Gerasimov ES, et al. Integrating human brain proteomes with genome‐wide association data implicates new proteins in Alzheimer's disease pathogenesis. Nat Genet. 2021;53:143‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu L. Boyle PA, Wingo A, et al. Neuropathologic correlates of human cortical proteins in Alzheimer disease and related dementias. Neurology. 2022;98:e1031‐e1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson ECB, Carter EK, Dammer EB, et al. Large‐scale deep multi‐layer analysis of Alzheimer's disease brain reveals strong proteomic disease‐related changes not observed at the RNA level. Nat Neurosci. 2022;25:213‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cary GA, et al. Genetic and multi‐omic risk assessment of Alzheimer's disease implicates core associated biological domains. 2022. Preprint at 10.1101/2022.12.15.22283478 [DOI] [PMC free article] [PubMed]
- 17. Gold ER. The fall of the innovation empire and its possible rise through open science. Research Policy. 2021. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arshad Z, Smith J, Roberts M, et al. Open access could transform drug discovery: a case study of JQ1. Expert Opin Drug Discov. 2016;11:321‐332. [DOI] [PubMed] [Google Scholar]
- 19. M4K Pharma – Open Science for Children's Health. https://m4kpharma.com/
- 20. TREAT‐AD. Home. Treat‐AD. https://treatad.org/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information