

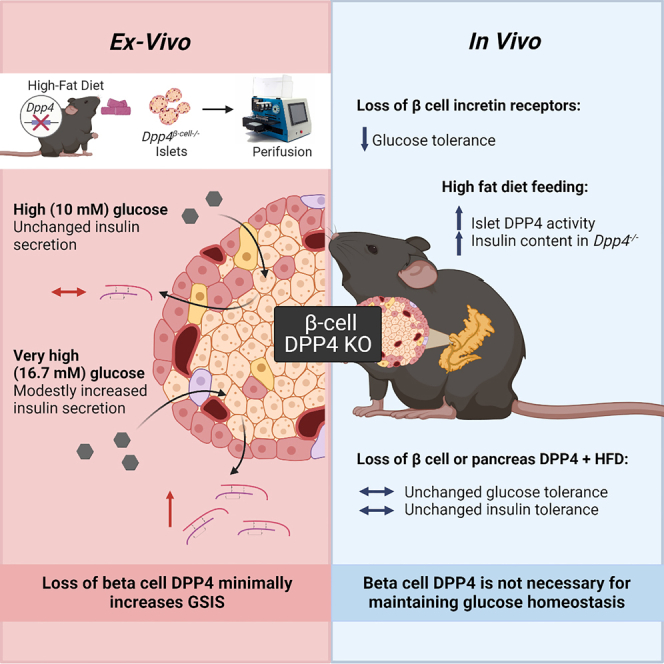
Summary
Mice systemically lacking dipeptidyl peptidase-4 (DPP4) have improved islet health, glucoregulation, and reduced obesity with high-fat diet (HFD) feeding compared to wild-type mice. Some, but not all, of this improvement can be linked to the loss of DPP4 in endothelial cells (ECs), pointing to the contribution of non-EC types. The importance of intra-islet signaling mediated by α to β cell communication is becoming increasingly clear; thus, our objective was to determine if β cell DPP4 regulates insulin secretion and glucose tolerance in HFD-fed mice by regulating the local concentrations of insulinotropic peptides. Using β cell double incretin receptor knockout mice, β cell- and pancreas-specific Dpp4−/− mice, we reveal that β cell incretin receptors are necessary for DPP4 inhibitor effects. However, although β cell DPP4 modestly contributes to high glucose (16.7 mM)-stimulated insulin secretion in isolated islets, it does not regulate whole-body glucose homeostasis.
Subject areas: Biological sciences, Physiology, Molecular biology, Cell biology
Graphical abstract
Highlights
-
•
β cell incretin receptors are required for DPP4i-mediated glucose lowering
-
•
Dpp4β-cell−/− increases glucose-stimulated (16.7 mM) insulin secretion in isolated islets
-
•
Glucose tolerance is not improved in HFD-fed Dpp4β-cell−/− or Dpp4Pan−/− mice
-
•
Insulin secretion is unchanged in HFD-fed Dpp4β-cell−/− or Dpp4Pan−/− mice
Biological sciences; Physiology; Molecular biology; Cell biology
Introduction
Hormone secretion from the islets in the pancreas is tightly linked to nutrient ingestion through the action of the gut-derived incretin hormones, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP).1 Biologically active incretins regulate glucagon secretion and potentiate insulin secretion to tailor postprandial glucose uptake and utilization.2,3 Together, GLP-1 and GIP action account for 50–70% of the postprandial rise in insulin in healthy individuals,4 but their collective effect is greatly diminished in patients with type 2 diabetes (T2D), even during early disease pathogenesis where reliable glycemic control is maintained.5,6 Active incretins are rapidly cleaved by dipeptidyl peptidase-4 (DPP4), a widely expressed serine protease, which limits the half-life of GLP-1 and GIP to minutes and eliminates their glucoregulatory action.7,8 Three classes of drugs based on enhancing incretin signaling are current treatments for T2D: DPP4 inhibitors (DPP4i) that selectively inhibit the catalytic action of DPP4, GLP-1 receptor agonists (GLP-1RAs) and structural variants of endogenous human GLP-1 or lizard exendin-4 that are resistant to degradation by DPP4, as well as the recently approved GLP-1R/GIPR (GIP receptor) dual agonist, tirzepatide.9,10,11
Although the role of DPP4 in the regulation of incretin hormone action and glucose homeostasis is well documented, the cell and tissue types essential for incretin degradation have remained elusive. Mice with whole-body Dpp4 knockout are protected against HFD feeding-induced obesity and insulin resistance; they display improved glycemic excursions, elevated plasma GLP-1 and insulin, reduced islet hypertrophy, and protection from streptozotocin-induced β cell death.12 Ablation of Dpp4 in endothelial cells (ECs), but not in adipocytes, or enterocytes, leads to a significant increase in glucose-induced active GLP-1 and GIP levels and partially recapitulates the improved glucose tolerance seen in global Dpp4 knockout.13,14 Hepatocyte-derived DPP4 regulates portal GLP-1 bioactivity and hepatic glucose production.15 Still, glucose control is improved further with systemic DPP4 inhibition,16 revealing that ECs contribute most significantly to systemic DPP4 activity and that additional unknown cell types also robustly contribute to DPP4-mediated postprandial glucoregulation.
There is now increasing evidence of local production and paracrine action of GLP-1 within the islet due to the presence of prohormone convertase (PC) ⅓, an enzyme that cleaves islet-derived preproglucagon into GLP-1 and several other peptides.17,18,19,20,21,22 The expression of intra-islet PC1/3 is increased under the conditions of hyperglycemia, hyperlipidemia, or metabolic stress17,19,23 and can be induced pharmacologically by activation of the β cell GLP-1R with the GLP-1RA, liraglutide.24 GLP-1RA treatment has been shown to increase the expression of prohormone convertase subtilisin/kexin type 1 (PCSK1), as well as the abundance of GLP-1+ signal in a subset of human α cells,24 raising the possibility that the pool of intra-islet GLP-1 can be pharmacologically manipulated. While colonic L cells are a bona fide source of glucoregulatory GLP-1,25 several studies have supported that pancreatic islets synthesize a distinct pool of glucoregulatory GLP-1.26,27 Indeed, the hyperglycemic effects of the GLP-1R antagonist, exendin 9-39, cease to exist in mice with loss of glucagon (Gcg) expression in α cells, but not L cells,26 suggesting the glucoregulatory actions of proglucagon peptides originate from α cells. Consistent with this, the islet-derived proglucagon pool is required for maintaining glucose homeostasis during metabolic stress induced by high-fat feeding.26,27,28 However, these studies must be interpreted in the context of the ability of glucagon to act as a GLP-1RA, inducing an insulin response from β cells in a paracrine manner,29 even in the absence of a functional Gcg receptor30 through mechanisms that primarily rely on activation of the GLP-1R. Still, the emerging evidence that α cells are a meaningful source of active GLP-1 and insulin secretion forces the consideration of local DPP4 expression within the islet as a key regulator of this local paracrine system. In support of this, intra-islet DPP4 has been suggested to modulate islet function via a paracrine mechanism due to its direct co-localization with β cells in the mouse islet.31 However, this has yet to be empirically tested. Therefore, we aimed to understand intra-islet DPP4 in the paracrine regulation of insulin secretion and glucose metabolism by studying mice with selective deletion of Dpp4 in β cells.
Results
GLP-1R and GIPR signaling within the β cell is required for regulation of glucose by systemic DPP4 inhibition
Previous studies in whole-body double incretin receptor knockout (DIRKO) and β cell-specific Glp1r knockout mice have demonstrated the importance of GLP-1R and GIPR signaling for proper glucose homeostasis, both of which are required for the glucoregulatory effects of the DPP4i, sitagliptin.32,33,34 GLP-1R and GIPR action within the portal circulation and enteric or central nervous system has also been proposed to contribute to their net glucoregulatory effect.35,36,37,38,39,40 To determine if incretin receptor activation on the β cell alone underlies the entirety of their glucose-lowering effects and eliminates the ability for one incretin receptor to compensate for lack of the other,32,34 we generated a mouse model with selected elimination of both Glp1r and Gipr within β cells (β cell double incretin receptor knockout; DIRKOβ-cell−/− mice). Gipr and Glp1r mRNA transcripts were significantly reduced in islets from DIRKOβ-cell−/− mice compared to controls (mixed group of mouse insulin (Ins1) promoter with mouse estrogen receptor (ERT), MIP-Cre and Gipr/Glp1rfl/fl) (Figure 1A), with no differences detected in other tissues (Figures 1B and 1C ), strongly suggesting the specific deletion of the incretin hormone receptors in the most abundant islet cell type, the β cell. We then investigated if the β cell incretin receptors were required for the DPP4i-dependent glucose-lowering in response to either a dose of sitagliptin that provides selective enteral inhibition (14 μg) and systemic inhibition (10 mg/kg).14,32 Both doses of sitagliptin improved oral glucose tolerance in control mice (Figures 1D and 1E ) but failed to elicit a glucose-lowering effect in DIRKOβ-cell−/− mice (Figures 1F and 1G ). As anticipated, the area under the curve (AUC) from the glucose tolerance test (GTT) was greater in DIRKOβ-cell−/− compared to control mice (Figure 1H). Together, these findings demonstrate that β cell incretin receptors are essential for DPP4i-mediated regulation of glucose control.
Figure 1.

Activation of the GLP-1R and GIPR within the β cell is required for regulation of glucose by DPP4i
(A–E) Abundance of Gipr and Glp1r mRNA in islets of male and female control and DIRKOβ-cell−/− mice normalized to Tbp. (B and C) Expression of Gipr (B) and Glp1r (C) mRNA transcripts in whole extracts of jejunum, epididymal white adipose tissue (eWAT), and lung tissue normalized to Tbp. (D-E) Oral glucose tolerance test (2 g/kg) glycemia (D) and AUC (E) in male and female control (MIP-Cre and Dpp4fl/fl) mice administered sitagliptin (14 μg/mouse or 10 mg/kg) by oral gavage 30 min prior to gavage of glucose.
(F and G) Oral glucose tolerance test (2 g/kg) glycemia (F) and AUC (G) in DIRKOβ-cell−/− mice administered sitagliptin (14 μg/mouse or 10 mg/kg) by oral gavage 30 min prior to gavage of glucose.
(H) Oral glucose tolerance test AUC in male and female control compared to DIRKOβ-cell−/− mice without sitagliptin. Statistical differences between groups were analyzed by an unpaired t test (A–C, H) or ANOVA (1 way or mixed-effects analysis) with post-hoc Tukey test (D–G). Data are represented as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01 (control compared to sita 10 mg/kg), ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Insulin secretion is enhanced in islets lacking DPP4 in the β cell
Next, we investigated the role of endogenous DPP4 in islet composition and function. First, we found that islet Dpp4 mRNA levels trended higher in wild-type (WT) C57BL6/J mice fed an HFD compared with those fed a standard laboratory chow diet (chow) (p = 0.0754; Figure 2A). Additionally, DPP4 activity was not detectable in islets isolated from female mice fed a chow diet; however, three out of four age-matched female mice on HFD for 5 weeks had islet DPP4 activity for the same amount of islet protein (Figure 2B). We then evaluated GLP-1 secretion in islets during dynamic perifusion from WT male and female mice fed chow or a high-fat high-cholesterol (HFHC) diet. Islets from most mice fed both diets had higher active GLP-1 secretion at 16.7 mM glucose than 10 mM glucose which was associated with significantly higher insulin secretion in both diets (Figures 2C and 2D ). Dpp4 knockout mice (Dpp4−/−) had marginally, but insignificantly, higher pancreatic insulin content compared to WT controls (p = 0.114) when fed chow (Figure 2E). However, insulin content was significantly greater in Dpp4−/− mice following 4 weeks of HFD feeding compared to WT controls and chow-fed Dpp4−/− mice (Figure 2E).
Figure 2.

Islets from HFD-fed Dpp4β-cell−/− mice have significantly reduced islet DPP4 activity and expression
(A) mRNA abundance of Dpp4 in islets isolated from 52-week-old wild-type C57BL/6J mice fed chow or a high-fat diet.
(B) DPP4 activity normalized to total protein in isolated islets from young female mice fed chow or high-fat diet (5 weeks).
(C and D) Active GLP-1 and insulin in perfusate from islets of WT male and female mice fed chow or HFHC (12 weeks) diets exposed to 16.7 mM and 10 mM glucose in dynamic perifusion.
(E) Insulin content measured in the pancreas of 12-week-old Dpp4+/+ or Dpp4−/− littermate controls fed chow or HFD (4 weeks).
(F) DPP4 activity normalized to total protein measured in whole extracts of tissues (liver, spleen, gut, heart, pancreas, islets) control (WT, MIP-Cre, Dpp4fl/fl) and Dpp4β-cell−/− male mice.
(G and H) Plasma DPP4 activity (G) and concentration (H) in plasma of HFD-fed control and Dpp4β-cell−/− male mice.
(I) Immunofluorescence staining of glucagon (purple) and DPP4 (green) and insulin (purple) and DPP4 (green) in HFD-fed male and female mouse pancreatic sections (20X) (WT – top, Dpp4−/− – middle, Dpp4β-cell−/− – bottom). DAPI staining (blue) was used to identify nuclei. DPP4 staining in liver sections of WT – top, Dpp4−/− – middle, Dpp4β-cell−/− – bottom mice.
(J) Magnified Dpp4β-cell−/− islet cells showing presence of lack of double glucagon and DPP4 or insulin and DPP4 stain.
(K–M) Glucose-stimulated insulin secretion (GSIS) and area under the curve (AUC) measured in islets isolated from control (WT, Dpp4fl/fl, and MIP-Cre) and Dpp4β-cell−/− (n = 3–5) 40-week-old HFD-fed male mice in response to perifusion with buffer containing 2.8 mM glucose, 16.7 mM glucose, or 2.8 mM glucose + KCl (30 mM). (L,M) GSIS measured in islets isolated from 40-week-old HFD-fed control (n = 9) (L) or Dpp4β-cell−/− (n = 6) (M) male mice in response to perifusion with and without DPP4 inhibitor sitagliptin. Graphs were analyzed using mixed-effects analysis and Sidak’s multiple comparisons (C-E, K-M) or unpaired t test (A, B, F, G, H and AUCs). Data are represented as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Scale bars represent 50 μm (I) and 10 μm (J).
Based on previous findings showing that HFD-fed Dpp4−/− mice have stronger glycemic control than Dpp4 EC knockout (Dpp4EC−/−) mice14 and that dysregulated glucose is associated with upregulation of DPP4 activity,41 we hypothesized that the increased intra-islet DPP4 may contribute to whole-body glucose dysregulation in response to the high metabolic demand induced by HFD feeding. To determine this, we generated a β cell-specific Dpp4 knockout model (Dpp4β-cell−/−) and compared it to controls (Dpp4+/+ MIP-Cre+, denoted as MIP-Cre). We confirmed that Dpp4β-cell−/− mice had a 40% reduction in DPP4 activity within islets compared to controls with no difference in other tissues including the liver (Figure 2F). Other dipeptidyl peptidases in the DPP4 family, fibroblast activation protein (FAP), DPP8, and DPP9, may contribute to the remaining protease activity in Dpp4β-cell−/− islets. Plasma DPP4 activity was not different between Dpp4β-cell−/− mice and controls, and DPP4 protein in plasma was insignificantly increased in Dpp4β-cell−/− mice (Figures 2G and 2H ). β cell-derived DPP4 protein was detected by fluorescence microscopy using a protocol previously described42 whereby DPP4 was co-stained with insulin or glucagon. While HFD-fed WT mouse islets had uniform DPP4 signal, no detectable DPP4 was found in the islets or liver of the Dpp4−/− group used as a negative control (Figure 2I). In the Dpp4β-cell−/− liver, DPP4 signal was similar to WT controls, confirming the specificity of the knockout (Figure 2I). Consistent with reduced DPP4 activity, islets of HFD-fed Dpp4β-cell−/− demonstrated a nearly complete loss of DPP4 signal (Figure 2I), of which the remaining signal colocalizes with glucagon+ and insulin− islet cells (Figure 2J). Additionally, we performed flow cytometry in dissociated islets, defined α, β, and δ cells as previously described,43 and quantified DPP4 levels in these populations. In accordance with the histology, DPP4 expression was reduced by 79% in the β cell population (Figure S1A). Alpha (21%) and delta (3.1%) cell populations had much less reduction in DPP4 expression by comparison (Figure S1A). However, the overall recovery of β cells was lower than expected in both control and Dpp4β-cell−/− mice; this may be due to the effects of MIP-Cre expression or tamoxifen treatment (Figure S1B).44,45
We then evaluated the role of intra-islet DPP4 on islet secretory function ex vivo. Consistent with relatively low immuno-detectable DPP4 in chow-fed mice, DPP4 inhibition in perifusion of islets isolated from WT mice fed chow had similar glucose-stimulated insulin secretion (GSIS) as control treated islets (Figures S2A and S2B). While basal glucose and KCl-induced insulin release remained similar between all groups, higher GSIS was observed in Dpp4β-cell−/− islets isolated from mice fed an HFD for 15 weeks and sacrificed at 40 weeks of age compared to controls after perifusion with 16.7 mM glucose (incremental AUC (iAUC), p = 0.055) (Figure 2K). To further investigate these differences, we compared GSIS in islets from another set of mice from these two groups with and without the addition of 200 nM sitagliptin. Here, sitagliptin significantly increased insulin response to 16.7 mM glucose in islets from control mice (Figure 2L) but had no effect in islets from Dpp4β-cell−/− mice (Figure 2M). Taken together, the increase in GSIS in Dpp4β-cell−/− islets vs. controls (Figure 2K) and the marked increase in GSIS following the sitagliptin treatment in control islets (Figure 2L), but not Dpp4β-cell−/− (Figure 2M), suggest that the catalytic action of β cell DPP4 is a regulator of GSIS ex vivo. Although not detectable in all perifusion samples, similar to our studies in WT mice (Figure 2C), we were able to measure GLP-1 from a subset of high glucose-stimulated effluents (16.7 mM), with Dpp4β-cell−/− islets releasing ∼4-fold more active GLP-1 than control islets (Dpp4β-cell−/− islets [n = 8 mice], 0.692 ± 0.119 pM vs. MIP-Cre islets [n = 4 mice], 0.164 ± 0.065 pM, p = 0.014), consistent with increased glucose-stimulated local GLP-1 (data not shown). To determine if the increase in GSIS was the result of enhanced signaling through the GLP-1R, islets from MIP-Cre and Dpp4β-cell−/− mice were treated with 100 nM of the GLP-1R antagonist exendin 9-39. Blockade of the GLP-1R resulted in a 25% reduction in GSIS in islets isolated from both MIP-Cre (Figure S3A) and Dpp4β-cell−/− mice (Figure S3B) indicating a similar reliance in both groups on GLP-1R signaling.
Elimination of β cell-derived Dpp4 fails to improve glucose tolerance or prevent incretin degradation in male mice on an HFD
Mice were fed an HFD to assess the impact of DPP4 in the context of metabolic stress. After five weeks of HFD feeding, both Dpp4β-cell−/− and control mice exhibited similar glucose tolerance in both oral and intraperitoneal (i.p.) glucose administration (Figures 3A and 3E). Sitagliptin reduced glucose excursion with oral, but not i.p. glucose challenge, in both groups (Figures 3A and 3E ). Consistent with previous findings,14 systemic inhibition with sitagliptin prior to GTTs increased the concentration of active circulating GIP (Figures 3B and 3F ) and active GLP-1 (Figures 3C) 15 min post-glucose challenge; however, no increase in plasma insulin was observed in Dpp4β-cell−/− compared to MIP-Cre HFD-fed mice (Figures 3D and 3G ). Insulin response to glucose was insignificantly increased with sitagliptin, likely due to HFD-induced metabolic dysfunction (Figures 3D and 3G ). Dpp4β-cell−/− mice demonstrated similar insulin sensitivity compared to control mice regardless of glycemia being represented in raw units (mmol/L) or expressed as a percentage of the fasting glucose (Figures 3H and 3I ).
Figure 3.

Elimination of β cell-derived DPP4 does not improve glucose tolerance and insulin tolerance or prevent incretin degradation in male HFD-fed mice
(A–D) Oral glucose tolerance (A) and plasma active GIP (B), active GLP-1 (C), and insulin (D) in male HFD-fed Dpp4β-cell−/− and MIP-Cre control mice in response to oral glucose gavage ± sitagliptin.
(E–G) Intraperitoneal (i.p.) glucose tolerance (E) and plasma active GIP (F), and insulin (G) secreted in response to i.p. glucose injection. Sitagliptin was given by oral gavage 30 min prior to glucose tolerance tests (2 g/kg body weight).
(H) Glucose levels and AUC during an insulin (0.6 IU/kg) tolerance test in male, HFD-fed mice.
(I) Insulin tolerance test glycemia as a percentage of fasted glucose and AUC.
(J) Insulin secretion measured in perifusion of islets from 65-week-old HFD-fed Dpp4β-cell−/− (n = 4) and MIP-Cre control (n = 4) mice with arginine (1 mM), GLP-1 (0.3 μm) and GIP (100 nM). Graphs were analyzed by one-way ANOVA (A, E AUCs), unpaired t test (H, I AUCs), or mixed-effects analysis with Tukey’s multiple comparisons. All data are represented as the mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Finally, to assess islet function ex vivo, male mice were sacrificed at 63–65 weeks of age after HFD feeding for 38–40 weeks. Islets were sequentially perifused with Krebs-Ringer Bicarbonate HEPES (KRBH) buffer containing (in order) 2.8 mM glucose and 10 mM glucose with arginine (1 mM), GLP-1 (0.3 μM), and GIP (100 nM). Unlike when given 16.7 mM glucose (Figure 2K) no trends or differences in GSIS were observed in islets from Dpp4β-cell−/− (n = 4) or MIP-Cre (n = 4) mice at 10 mM glucose (Figure 3J). This may be due to higher islet GLP-1 contribution at 16.7 mM versus 10 mM glucose exposure as we have seen in islet from WT male and female, chow- and HFHC-fed mice (Figure 2C). Taken together, these data demonstrate that, in male mice, elimination of DPP4 from pancreatic β cells is not sufficient to improve glucose tolerance achieved by the whole-body Dpp4−/− mice or systemic DPP4 inhibition with sitagliptin.14
Dpp4 deletion in β cells does not affect glucose tolerance, incretin levels, or GSIS in HFD-fed female mice
Following HFD feeding, female Dpp4β-cell−/− mice demonstrated an overall glycemic response similar to the MIP-Cre controls in response to an oral glucose tolerance test (oGTT) (Figure 4A), including levels of plasma active GIP and insulin (Figures 4B and 4C ). We also observed no change in i.p. glucose tolerance with and without sitagliptin (Figure 4D), plasma active GIP levels (Figure 4E), or insulin levels (Figure 4F) following glucose administration between control and Dpp4β-cell−/− mice. Insulin tolerance was also unchanged (Figures 4G and 4H ). Finally, islets isolated from female Dpp4β-cell−/− and MIP-Cre control mice exhibited similar GSIS in perifusion, with no significant differences observed after stimulation with arginine, GLP-1, or GIP (Figure 4I), indicating that β cell-specific DPP4 plays an insignificant role in physiological glycemic regulation in HFD-fed male and female mice.
Figure 4.

Elimination of β cell-derived DPP4 does not improve glucose tolerance, incretin levels, or GSIS in HFD-fed female mice
(A–C) Oral glucose tolerance (A), active GIP (B), and insulin secreted (C) in female HFD-fed Dpp4β-cell−/− and MIP-Cre control mice in response to oral glucose gavage ± sitagliptin.
(D–F) Intraperitoneal (i.p.) glucose tolerance (D), active GIP (E), and insulin secreted (F) in female HFD-fed Dpp4β-cell−/− and MIP-Cre control mice in response to i.p. glucose injection. Sitagliptin was given by oral gavage 30 min prior to glucose tolerance tests (2 g/kg body weight for oGTT and at 1.2 g/kg for ipGTT) (n = 7–10 per group).
(G) Glucose levels and AUC during an insulin (0.6 IU/kg) tolerance test in female HFD-fed Dpp4β-cell−/− and MIP-Cre control mice.
(H) Insulin tolerance test glycemia as a percentage of fasted glucose and AUC.
(I) GSIS measured during perifusion of islets isolated from 65-week-old HFD-fed Dpp4β-cell−/− (n = 4) and MIP-Cre control (n = 4) mice with arginine (1 mM), GLP-1 (0.3 μm) and GIP (100 nM). AUC graphs represent area under the curve, analyzed by ANOVA with post-hoc Tukey test (A, D) or unpaired t test (G, H). For non-AUC graphs we used mixed-effects analysis with Tukey’s multiple comparisons. All data are represented as the mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Loss of whole pancreas Dpp4 does not improve glucose tolerance or GSIS in HFD-fed mice
Since we found no differences in glucose tolerance in vivo with Dpp4β-cell−/− mice but did detect DPP4 signal in other islet and pancreas cells (Figures 2I and 2J ), we decided to probe if a more global deletion of DPP4 within the pancreas would impact whole-body glucose homeostasis. We generated Dpp4Pan−/− mice and pancreatic and duodenal homeobox 1 (PDX)-Cre littermate controls and fed them an HFD. We confirmed DPP4 elimination with flow cytometry (Figures S4A and S4B), immunostaining of pancreatic sections, and RT-qPCR in islets (Figures 5A and 5B). Male and female HFD-fed control and Dpp4Pan−/− mice were challenged with oral and i.p. GTTs, and their islets were isolated for perifusion. There was no difference in oral and i.p. glucose tolerance between male HFD-fed Dpp4Pan−/− and control mice (Figures 5C and 5D ). In isolated islets from male Dpp4Pan−/− and control mice, GSIS was not different (Figure 5E). Interestingly, treatment with GLP1-R antagonist exendin 9-39 only reduced GSIS in the Dpp4Pan−/− male mice (trend, p = 0.0607), indicating that consistent with the Dpp4β-cell−/− mice they are more reliant on GLP-1R signaling for maximal insulin secretion in response to high glucose (Figure 5E). In females, no differences in glucose tolerance were detected in control and Dpp4Pan−/− HFD-fed mice (Figures 5F and 5G ). In islets isolated from female mice, loss of pancreas Dpp4 did not impact GSIS with and without exendin 9-39 treatment (Figure 5H).
Figure 5.

Ablation of whole pancreas Dpp4 does not improve glucose tolerance or GSIS
(A) Immunofluorescence staining of glucagon (purple) and DPP4 (green) and insulin (purple) and DPP4 (green) in HFD-fed mouse pancreatic sections of PDX-Cre and Dpp4Pan−/−. DAPI staining (blue) was used to identify nuclei.
(B) Absolute mRNA abundance of Dpp4 in islets isolated from one-year-old control (Dpp4(fl/fl)) and Dpp4Pan−/− male and female mice.
(C and D) Glycemia and AUC glucose during (C) oral and (D) i.p. glucose tolerance tests in HFD-fed control and Dpp4Panl−/− male mice.
(E) Perifusion GSIS in male mice fed HFD for 25–30 weeks, with and without Exendin 9-39.
(F and G) Glycemia and AUC glucose during (F) oral and (G) i.p. glucose tolerance tests in HFD-fed control and Dpp4Pan−/− female mice.
(H) Perifusion GSIS in female mice fed HFD for 25–30 weeks, with and without Exendin 9-39 (100 nM). Statistical analysis was done with mixed-effects analysis and Tukey’s multiple comparisons (C–H), one-way ANOVA (AUC E and H) or unpaired t test (B, AUC C, D, F and G). All data are represented as the mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Scale bars represent 50 μm.
GLP-1R signaling has been proposed to be an important link between the α and β cell.28,46 To probe this relationship more closely, we performed arginine tolerance tests and meal tolerance tests in male and female Dpp4Pan−/− and control mice fed a long-term HFD. When challenged with an i.p. arginine injection, glycemia was not different between Dpp4Pan−/− and controls, neither in males (Figure S4C) nor in females (Figure S4D). Post-arginine injection glucagon (Figure S4E-F) levels were not different between Dpp4Pan−/− and controls in males or females. To evaluate the response to a mixed meal, mice were fasted and gavaged with Ensure meal replacement. Dpp4Pan−/−, and control mice had similar glucose before and up to 90 min after gavage in both sexes (Figures S4G–S4J). Similarly, active GLP-1 was not different between groups in either sex (Figures S4K and S4L).
Discussion
Previous work has identified vascular ECs as a principal site of action for the acute glucoregulatory actions of DPP4 inhibitors.14,32 However, despite a significant improvement in glucoregulation of HFD-fed Dpp4EC−/− mice, they did not achieve the same degree of improvement in glucose tolerance than observed in Dpp4−/− mice, which indicated that inhibition of DPP4 in other cell types is contributing to these beneficial glycemic effects.14 In this report, we evaluated the role of intra-islet DPP4 by deleting Dpp4 in MIP+ or PDX+ cells in the regulation of glycemia, insulin resistance, and islet hormone release.
Whole-body deletion of the incretin receptors impairs glucose tolerance through suppressed insulin response to oral glucose in mice.32,47 Mice in which only the β cell Glp1r is deleted still respond to DPP4i pointing to the importance of GIPR signaling and compensation.34 Our study found that limiting the genetic deletion of Gipr and Glp1r to the islet β cell compartment also leads to elevated glucose excursion during an oGTT. Moreover, this increase could not be improved with a systemic dose of sitagliptin, demonstrating that the activation of the incretin receptors on the β cell is essential for the glucose-lowering action of sitagliptin.
To further explore the link between metabolic dysregulation, islet-derived DPP4, and insulin secretion, we generated a line of β cell-specific Dpp4 knockout mice (Dpp4β-cell−/−). In human islets, direct inhibition of islet DPP4 with the DPP4 inhibitors MK-062626,27 or sitagliptin22 has been shown to improve β cell function, survival, and insulin secretion. Consistent with this, we show that Dpp4−/− mice have a significant increase in pancreatic insulin content with HFD feeding. We also found that islets from HFD-fed Dpp4β-cell−/− mice secreted more insulin in perifusion experiments with very high glucose (16.7 mM) compared to controls. However, KCl-stimulated membrane depolarization resulted in comparable insulin release in both groups, suggesting the difference is dependent on augmented signaling in response to glucose and not just improved islet function. Further studies would be needed to determine if loss of islet DPP4 improves islet health or survival through prolonged α cell GLP-1 activity. Furthermore, pharmacological inhibition of DPP4 significantly increased GSIS in control HFD-fed DPP4-positive islets but had no further effects on the Dpp4β-cell−/− islets. Our data are consistent with previous findings in human islets treated with a DPP4i48 and the increase in glucose-stimulated insulin granule exocytosis observed with the DPP4i (MK-0626).49
The production of active GLP-1 has been observed in both mouse and human islet α cells, but whether it is essential for glucose regulation in vivo remains controversial.20,22,23,26,27,50,51,52 The importance of intra-islet communication between α cell-derived products and β cell signaling to regulate insulin secretion has been recently established.28,29,30 Deciphering the contribution of glucagon from other proglucagon-derived peptides is challenging because it is a more abundant53 but weaker GLP-1RA28 than other proglucagon-derived peptides. Studies in α cell-ablated mice have identified pancreatic GLP-1 as required for glucose homeostasis during metabolic stress and shown that a deficiency in α cell-derived GLP-1 could be partially rescued by inhibition of DPP4i with sitagliptin.27 While GLP-1 levels in mouse pancreas effluents are frequently undetectable,30 we were able to detect increased GLP-1 secreted from islet perifusion chambers from HFD-fed Dpp4β-cell−/− mice at higher levels than controls. This is consistent with recent detection of low levels of GLP-1 (7–36 amide or 7–37) in mouse islets using product ion monitoring approaches.53 Together with the 25% reduction in GSIS with exendin 9-39 following Dpp4 deletion, our data indicate that islet-derived GLP-1R signaling contributes to the increase in GSIS50 but only in response to very high-glucose treatment (16.7 mM). Studies incubating islets from healthy human donors or donors with T2D in more intermediate concentrations of glucose (11.1 mM) reported sitagliptin to increase active GLP-1 levels with no meaningful effect on GSIS.23 Our findings also suggest that glucose concentrations must be sufficiently high to stimulate robust paracrine effects from the α cell to the β cells and that enzymatic cleavage by DPP4 and GLP-1R signaling is at best a very modest regulator of GSIS in islets from HFD-fed mice in vitro.
Although GIP is also a DPP4 substrate and regulator of GSIS, recent proteomic analysis performed in islets from mice on a chow or HFD did not detect the presence of intra-islet GIP,53 and our in vivo studies did not demonstrate any differences in GIP bioactivity in the circulation of Dpp4β-cell−/− mice. However, our study could not rule out intra-islet contributions from other DPP4 substrates, including but not limited to protein YY (PYY), stromal-derived growth factor 1 (SDF-1), and insulin growth factor 1 (IGF-1).54,55,56 While SDF-1 may increase GLP-1 secretion from α cells after stress and IGF-1 may be important in maintaining post-streptozotocin (STZ) insulin secretion, the impact of truncation by DPP4 on these effects is unknown.55,56 On the other hand, PYY may differentially impact GSIS in its cleaved and uncleaved forms.54,57 Additionally, the effects of paracrine signaling in mouse islets may contribute less than in human islets because of reduced contact between cell types with mouse islets having a mostly β cell core and an α and δ cell mantle.23
Diet-induced weight gain and metabolic stress may induce increased islet PC1/3 expression,51,52 which has been proposed to increase intra-islet GLP-1 production, amplifying the potential differences in glucose metabolism between groups fed an HFD. However, no differences in oral glucose, i.p. glucose, or insulin tolerance were observed between male or female Dpp4β-cell−/− and HFD-fed control mice. Our data demonstrate that intra-islet DPP4 does not significantly contribute to circulating DPP4 concentration or activity, incretin secretion, incretin cleavage, or insulin secretion following glucose gavage or injection in HFD-fed mice. DPP4 inhibition with sitagliptin improved glycemic excursion to an oral glucose gavage of both groups but had no effects during an i.p. injection in either group regardless of the diet, providing further evidence for a gut-dominant incretin response in mice.
Taken together, our data indicate that regulation of GLP-1 bioactivity in the mouse islet by DPP4 is dispensable for regulation of whole-body glucose homeostasis but under conditions of metabolic stress may have some control of intra-islet insulin release.
Limitations of the study
A major limitation of the findings presented in this report is the use of CreERT2/loxP system to induce genetic recombination. The use of the MIP-Cre model for genetic deletion has established caveats,58 including the presence of human growth hormone that becomes apparent with STZ treatment in addition to HFD feeding.45,59 However, multiple metabolic comparisons between WT and MIP-Cre have been performed, and no significant differences are observed with the presence of the transgene or tamoxifen treatment in the absence of STZ.60 Also, PDX-Cre lines also have established limitations with regards to cell specificity.58 Additionally, Dpp4β-cell−/− mice may have adapted following gene deletion to compensate for the loss of pancreatic DPP4, influencing the impact of the deletion, although measurements of related family member fibroblast activation protein within the pancreas were very low.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Polyclonal goat anti-mouse DPP4 | R&D Systems | Cat#AF954; RRID:AB_355739 |
| Alexa Fluor488 donkey anti-goat | Thermo Fisher | Cat#A32814; RRID:AB_2762838 |
| Recombinant rabbit monoclonal anti-insulin | Abcam | Cat#ab181547; RRID:AB_2716761 |
| Recombinant anti-glucagon | Abcam | Cat#ab92517; RRID:AB_10561971 |
| Alexa Fluor Plus 647 donkey anti-rabbit | Thermo Fisher | Cat#A32795; RRID:AB_2762835 |
| TER-119/Erythroid Cells | BioLegend | Cat#133307; RRID:AB_11124348 |
| Biotin anti-mouse CD45 Antibody | BioLegend | Cat#103103; RRID:AB_312968 |
| Biotin anti-mouse CD31 Antibody | BioLegend | Cat#102503; RRID:AB_312910 |
| APC/Fire750 anti-mouse CD24 Antibody | BioLegend | Cat#101839; RRID:AB_2650875 |
| FITC anti-mouse CD71 Antibody | BioLegend | Cat#113805; RRID:AB_313566 |
| PerCP/Cyanine5.5 anti-mouse CD326 (Ep-CAM) Antibody | BioLegend | Cat#118219; RRID:AB_2098647 |
| PE anti-human/Mouse CD49f Antibody | BioLegend | Cat#313611; RRID:AB_893374 |
| Anti-CD26 Rat Monoclonal Antibody | BioLegend | Cat#137807; RRID:AB_10663403 |
| Streptavidin-BV421 | BioLegend | Cat#405226 |
| Chemicals, peptides, and recombinant proteins | ||
| Sitagliptin (Januvia 100mg tablets) | Merck Laboratories | Not Applicable |
| Sitagliptin Phosphate Monohydrate (for perifusion) | BioVision | Cat#1757 |
| Humalog (insulin lispro injection) | Lilly | DIN#02229704 |
| GIP-(D-Ala2)-GIP | Bachem | Cat#4054476 |
| GLP-1 7-36 | Bachem | Cat#4030663 |
| Exendin 9-39 | Bachem | Cat#4017799 |
| Collagenase from Clostridium histolyticum | Sigma | Cat# C7657 |
| Histopaque(R)-1077,sterile-filtered, density: 1.077 g/mL | Sigma-Aldrich | Cat#10771 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| Critical commercial assays | ||
| Ultrasensitive Mouse Insulin ELISA | ALPCO | Cat#80-INSMSU-E01 |
| High Range Mouse Insulin ELISA | ALPCO | Cat#80-INSMSH |
| U-PLEX Mouse GLP-1 (active) Assay | Mesoscale | Cat#K1526LK |
| Mouse Active GIP Elisa Kit | CrystalChem | Cat#81511 |
| Mouse Glucagon Elisa Kit | CrystalChem | Cat#81518 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| Pierce Rapid Gold BCA Protein Assay Kit | Thermo Fisher | Cat#A53225 |
| Mouse DPPIV/CD26 DuoSet ELISA | R&D Systems | Cat#DY954 |
| H-Gly-Pro-AMC HBr (for DPP4 activity assay) | Bachem | Cat#I-1225 |
| AMC (for DPP4 activity assay) | Bachem | Cat#Q-1025 |
| Experimental models: Organisms/strains | ||
| MIP-Cre mice (B6.Cg-Tg(Ins1-cre/ERT)1Lphi/J) | The Jackson Laboratory | Stock #024709; RRID:IMSR_JAX:024709 |
| Glp1rfl/fl mice | R. Seeley lab | Not applicable |
| Giprfl/fl mice | D. Drucker lab | Not applicable |
| Dpp4fl/fl mice (C57BL/6-Dpp4tm1.1Mrl) | Merck Laboratories; (Distributed by Taconic) | Model#10935 |
| Dpp4-/- mice | Generously provided by Didier Marguet | Not applicable |
| PDX-Cre mice (B6.FVB-Tg(Pdx1-cre)6Tuv/J) | The Jackson Laboratory | Stock #014647; RRID:IMSR_JAX:014647 |
| Oligonucleotides | ||
| Tbp | Applied Biosystems | Mm01277042_m1 |
| Hprt | Applied Biosystems | Mm03024075_m1 |
| Glp1r | Applied Biosystems | Mm00445292_m1 |
| Gipr | Applied Biosystems | Mm01316344_m1 |
| Dpp4 | Applied Biosystems | Mm00494538_m1 |
| Software and algorithms | ||
| Graph Pad Prism 9 | Graphpad Software | https://www.graphpad.com |
| Other | ||
| Regular Chow Diet | Harlan Teklad | Cat#2018 |
| High Fat Diet | Research Diets | Cat#D12451 |
| High Fat High Cholesterol Diet | Envigo | Cat#TD88137 |
Resource availability
Lead contact
Requests for related resources or information will be addressed by the lead contact, Dr. Erin Mulvihill (emulvihi@uottawa.ca).
Materials availability
No unique reagents were generated in this study.
Experimental model and subject details
Experimental animals
Experiments were performed in adult, male and female mice, aged 12 to 65 weeks old as described in figure legends. Male and female mice were analyzed separately apart from the verification of the knockout models (Figures 1, 3I, 3, and 5B). Gender as a social construct was not applicable to our study.
Dpp4-/- mice were re-derived from a colony described previously.61 Dpp4fl/fl mice were obtained from Merck Research Laboratories.14 B6.Cg-Tg(Ins1-cre/ERT)1Lphi/J (MIP-Cre) (stock # 024709) mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Animals were cared for in accordance with the Canadian Guide for the Care and Use of Laboratory animals (CCAC). All experimental procedures were approved under AUP#2909 and AUP#2029 by the University of Ottawa Animal Care and Veterinary Service. Dr. Daniel Drucker (Lunenfeld Tanenbaum Research Institute, Toronto Centre for Phenogenomics) kindly provided Glp1r fl/fl/Gipr fl/fl MIP-Cre (β cell Double Incretin Knockout; DIRKO) mice and littermate controls. Dpp4+/+ MIP-Cre+ mice were crossed with Dpp4fl/fl mice, producing Dpp4fl/+MIP-Cre+/- offspring. Intercrossing Dpp4fl/+MIP-Cre+/- with Dpp4fl/+ generated four experimental groups: Dpp4+/+ (WT), Dpp4+/+ WT with MIP-Cre expression (MIP-Cre), Dpp4 floxed without Cre (Dpp4fl/fl), and Dpp4fl/fl MIP-Cre+ (Dpp4β-cell-/-). Controls for Dpp4β-cell-/- experiments are combined WT, MIP-Cre control and Dpp4fl/fl as no significant differences were observed among groups. In our hands, MIP-Cre and WT mice have identical glucose tolerance and islet function under high-fat fed conditions.60 For pancreas specific Dpp4 knockout mice (Dpp4Pan-/-), PDX-Cre-expressing mice (JAX stock #014647)62 were crossed with Dpp4fl/fl mice and resulting Dpp4fl/+ offspring with and without PDX-Cre were then crossed. This generated four genotypes: Dpp4+/+ (WT), Dpp4 WT with Cre expression (PDX-Cre), Dpp4 floxed without PDX-Cre (Dpp4fl/fl), and Dpp4 floxed with Cre expression (Dpp4Pan-/-). Controls for Dpp4Pan-/- experiments are combined WT, PDX-Cre, and Dpp4fl/f unless indicated as Cre control where only PDX-Cre mice are used.
Mice were housed under a 12-hour light/dark cycle at the University of Ottawa Heart Institute, and maintained on regular chow (Harlan Teklad, 2018, 18% kcal from fat) prior to HFD (Research Diets #D12451, 45% kcal from fat) with free access to food and water, unless otherwise noted. The HFHC diet has 0.2% per weight cholesterol, 42% kcal from fat, and 42.7 % kcal from carbohydrate (Teklad, TD88137). For metabolic tests, food was removed at 8 am for a five hour fast. Cre activity was induced by administering tamoxifen (Sigma-Aldrich, #T5648) in corn oil (50 mg/mL, Sigma-Aldrich) by oral gavage in all Dpp4β-cell-/- and control genotypes (5 mg/mouse/day) for 5 consecutive days.63 Tamoxifen was not administered to PDX-Cre mice.62
Method details
Glucose, insulin, meal, and arginine tolerance tests
Oral glucose (2 g/kg), intraperitoneal (i.p.) glucose (1.2 g/kg females, 2 g/kg males), intraperitoneal arginine (2 g/kg), and insulin (0.6 IU/kg) tolerance tests were performed as previously described where glucose is measured for 90 minutes after gavage or i.p. injection.14 For the meal tolerance test, mice were bled before and after 200 μL Ensure meal replacement gavage. Blood for active GLP-1 (K1526LK, Mesoscale), GIP (CrystalChem, USA), insulin (Mouse Ultrasensitive Insulin ELISA, Alpco, 80-INSMSU-E01), and glucagon (CrystalChem, USA) measurements was collected in K2EDTA-coated tubes and blood was mixed with 10% TED (5000 KIU/mL Trasylol, 1.2 mg/mL EDTA, 0.1 nmol/L Diprotin A). Plasma was stored at -80°C until analysis. Terminal blood samples were collected via cardiac puncture. Tissues were flash frozen in liquid nitrogen and stored at -80°C. Tissues for histological analyses were fixed in 4% paraformaldehyde for 24 hours prior to transferring to 70% ethanol, processing, and embedding in paraffin wax.
Gene Expression
RNA was isolated using TRIzol reagent (Life Technologies, Canada) and cDNA was synthesized from total RNA using a high-capacity cDNA reverse transcription kit (Applied biosystems). Specific mRNA levels were measured by quantitative real-time PCR on a QuantStudio 5. mRNA abundance was determined using the standard curve method and expression was normalized to housekeeping gene Tbp or Hprt.
Islet isolation and perifusion
The pancreas was inflated through the pancreatic duct with collagenase (7.5 mg/mL, Sigma C7657) in HBSS (5 mM glucose, 1 mM MgCl2), excised and incubated at 37°C for 12 minutes. The digestion was quenched with ice-cold HBSS (5 mM glucose, 1 mM MgCl2, 1 mM CaCl2). The digested pancreas was washed with HBSS, and islets were separated using a Histopaque gradient (Sigma, 10771). Islets were handpicked to achieve final purity of 95-99% and incubated in complete RPMI (10% FBS, 1% penicillin/streptomycin) overnight at 37°C and 5% O2 before perifusion.
A Biorep perifusion system (Biorep Technologies) was used to stimulate islet hormone secretion in vitro. Solutions were prepared in KRBH buffer (115 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 24 mM NaHCO3, 10 mM HEPES, and 1% BSA; pH = 7.4). Islets were equilibrated for 48 minutes with 2.8 mM glucose and then perifused in intervals in the indicated experimental conditions. Flow rate (100 μl/min) and temperature (37°C) remained constant, and the type of treatment is indicated at the top of each figure (Arginine, Sigma, #A5006; GLP-1 7-36, Bachem, 4030663; GIP-(D-Ala2)-GIP, Bachem #4054476; Exendin 9-39, Bachem #4017799). Effluent fractions collected at 2-minute intervals were stored at -80°C for further assays. Insulin concentration was measured using a Mouse Ultrasensitive or High Range Insulin ELISA (ALPCO, 80-INSMSU-E01 or 80-INSMSH).
DPP4 activity and protein measurements
Mouse DPP4 ELISA (R&D Systems) was used to measure plasma DPP4 concentrations following the manufacturer’s protocol. DPP4 activity was measured by a fluorometric assay as previously described.14 Tissue DPP4 activity was normalized to total protein concentration measured by BCA assay (Pierce Rapid Gold BCA Protein Assay Kit, Thermo Fisher).
Immunofluorescent analysis
Five μm thick sections of paraffin embedded pancreas and liver tissues were deparaffinized in toluene and rehydrated. Antigen retrieval was done in the microwave with sodium citrate buffer (0.1 M, pH 6, with 0.05% Tween-20) and slides were washed with water and PBS before blocking with 10% donkey serum in PBS for 30 minutes at room temperature. Sections were incubated with polyclonal goat anti-mouse DPP4 (1/40, R&D Systems, AF954), recombinant rabbit monoclonal anti-insulin (1/250; Abcam ab181547, clone number EPR17359), and recombinant anti-glucagon (1/500; Abcam ab92517, clone number EP3070) primary antibodies overnight at 4°C. Sections were incubated with Alexa Fluor488 donkey anti-goat (1/500; Thermo Fisher Scientific A32814) and Alexa Fluor Plus 647 donkey anti-rabbit (1/500; Thermo Fisher Scientific A32795) secondary antibodies for 45 minutes and with DAPI for 5 minutes protected from light at room temperature. Images were captured with a Zeiss Axioscope AxioImagerZ1 fluorescent microscope using the 20X objective or 63X oil objective.
Flow cytometry
Following islet isolation from pancreas tissue, islets from 3 mice were pooled and dissociated in 0.25% trypsin EDTA for 15 minutes at 37°C. Trypsin was quenched with FBS and PBS. Dispersed islet cells were centrifuged at 400 x g for 5 min and resuspended in 1% FBS in PBS. Cells were incubated with biotin-conjugated lineage (TER-119, CD45, CD31; BioLegend) and surface antibodies (EpCAM-PerCP/Cy5.5, CD49f-PE, CD71-FITC, CD24-APC/Fire750, DPP4-APC) for 20 min at room temperature and washed with 1% FBS in PBS. Cells were then incubated with streptavidin-BV421 for 20 min at room temperature and washed with 1% FBS in PBS. Cells were resuspended in 1% FBS/2mM EDTA/0.5 μg/mL DAPI in PBS and acquired on an LSRFortessa cytometer (BD). Analysis was done in FlowJo v10.7 (BD).
Quantification and statistical analysis
Data are expressed as the mean ± SEM. Comparisons between groups were performed using one-way ANOVA or a t-test, depending on the number of variables (GraphPad Prism9), with a post-hoc Tukey test as specified in figure legends. All time course and perifusion glycemic curves were analyzed using 2-way ANOVA with mixed model analysis and Sidak’s or Tukey’s multiple comparisons and AUC calculations were performed. Statistically significant differences are indicated as ∗p<0.05, ∗∗p<0.01, ∗∗∗p<0.001, ∗∗∗∗p<0.0001.
Acknowledgments
We would like to thank the Stewart Whitman Histopathology Core and Xiaoling Zhao for helping with sample preparation, sectioning, and immunohistochemistry. The graphical abstract was created with Biorender.com (publication license: TR258QWXV0). E.E.M is the guarantor of this work and, as such, has full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. This work was supported in part by a research grant from the Investigator Initiated Studies Program (IISP) by MERCK, by Canadian Institutes of Health Research Grants, ARJ-162628 and Project Grant 156136 to E.E.M and 148634 to M.D.F. In addition, M.D.F. was supported by a Diabetes Canada operating grant (OG-3-22-5657-MF). J.E.C.’s work is supported by funding from the NIH (DK123075, DK125353, DK046492). C.A.A.L. has previously received funding from Queen Elizabeth II Graduate Scholarship in Science and Technology (QEII-GSST), an Ontario Graduate Scholarship, and is the current recipient of an NSERC CREATE CIRTN-R2FIC award. N.A.T is the recipient of a UOHI Cardiac Endowment Scholarship. M.E.C. is supported by NIH funding (DK129417). N.M.M. is the recipient of a Canadian Institutes of Health Doctoral Research Award. E.M.V. was the recipient of a Diabetes Canada Post-Doctoral Fellowship. E.E.M. is the recipient of a Heart and Stroke New Investigator Award. The opinions expressed in this paper are those of the authors and do not reflect those of MERCK, its affiliates, or other companies.
Author contributions
Study concept and design: E.F., J.E.C., and E.E.M.; Acquisition of data: E.F., C.A.A.L., N.A.T., M.A.N, M.E.C., B.V., N.M.M., I.LS., A.A,H., P.G., E.M.V., M.A.D., R.S., J.E.C., and E.E.M.; Analysis and interpretation of data: E.F., C.A.A.L., M.D.F., J.E.C., and E.E.M.; Drafting of the manuscript E.F., C.A.A.L., and E.E.M.; Obtained funding: E.E.M. All authors provided critical revision of the manuscript for important intellectual content.
Declaration of interests
The Mulvihill lab received funding from the Merck IISP program for pre-clinical studies directly related to this work. While she performed experiments as a post-doctoral research fellow for these studies, E.M.V. currently works for Eli Lilly Canada. The Campbell lab receives funding for basic science from Eli Lilly and Novo Nordisk.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: April 26, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106748.
Supplemental information
Data and code availability
-
•
All data will be shared upon request to the lead contact. No standardized datatype data were generated in this study.
-
•
This study did not generate new code.
-
•
Any additional analysis information for this work is available by request to the lead contact.
References
- 1.Campbell J.E., Drucker D.J. Pharmacology physiology and mechanisms of incretin hormone action. Cell Metabol. 2013;17:819–837. doi: 10.1016/j.cmet.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Baggio L.L., Drucker D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 3.El K., Campbell J.E. The role of GIP in alpha-cells and glucagon secretion. Peptides. 2020;125:170213. doi: 10.1016/j.peptides.2019.170213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nauck M.A., Homberger E., Siegel E.G., Allen R.C., Eaton R.P., Ebert R., Creutzfeldt W. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J. Clin. Endocrinol. Metab. 1986;63:492–498. doi: 10.1210/jcem-63-2-492. [DOI] [PubMed] [Google Scholar]
- 5.Knop F.K., Aaboe K., Vilsbøll T., Vølund A., Holst J.J., Krarup T., Madsbad S. Impaired incretin effect and fasting hyperglucagonaemia characterizing type 2 diabetic subjects are early signs of dysmetabolism in obesity. Diabetes Obes. Metabol. 2012;14:500–510. doi: 10.1111/j.1463-1326.2011.01549.x. [DOI] [PubMed] [Google Scholar]
- 6.Michaliszyn S.F., Mari A., Lee S., Bacha F., Tfayli H., Farchoukh L., Ferrannini E., Arslanian S. beta-cell function, incretin effect, and incretin hormones in obese youth along the span of glucose tolerance from normal to prediabetes to type 2 diabetes. Diabetes. 2014;63:3846–3855. doi: 10.2337/db13-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deacon C.F., Nauck M.A., Toft-Nielsen M., Pridal L., Willms B., Holst J.J. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes. 1995;44:1126–1131. doi: 10.2337/diab.44.9.1126. [DOI] [PubMed] [Google Scholar]
- 8.Knudsen L.B., Pridal L. Glucagon-like peptide-1-(9-36) amide is a major metabolite of glucagon-like peptide-1-(7-36)amide after in vivo administration to dogs and it acts as an antagonist on the pancreatic receptor. Eur. J. Pharmacol. 1996;318:429–435. doi: 10.1016/s0014-2999(96)00795-9. [DOI] [PubMed] [Google Scholar]
- 9.Willard F.S., Douros J.D., Gabe M.B., Showalter A.D., Wainscott D.B., Suter T.M., Capozzi M.E., van der Velden W.J., Stutsman C., Cardona G.R., et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight. 2020;5:e140532. doi: 10.1172/jci.insight.140532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenstock J., Wysham C., Frías J.P., Kaneko S., Lee C.J., Fernández Landó L., Mao H., Cui X., Karanikas C.A., Thieu V.T. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021;398:143–155. doi: 10.1016/S0140-6736(21)01324-6. [DOI] [PubMed] [Google Scholar]
- 11.Coskun T., Sloop K.W., Loghin C., Alsina-Fernandez J., Urva S., Bokvist K.B., Cui X., Briere D.A., Cabrera O., Roell W.C., et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol. Metabol. 2018;18:3–14. doi: 10.1016/j.molmet.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conarello S.L., Li Z., Ronan J., Roy R.S., Zhu L., Jiang G., Liu F., Woods J., Zycband E., Moller D.E., et al. Mice lacking dipeptidyl peptidase IV are protected against obesity and insulin resistance. Proc. Natl. Acad. Sci. USA. 2003;100:6825–6830. doi: 10.1073/pnas.0631828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varin E.M., Mulvihill E.E., Beaudry J.L., Pujadas G., Fuchs S., Tanti J.F., Fazio S., Kaur K., Cao X., Baggio L.L., et al. Circulating levels of soluble dipeptidyl peptidase-4 are dissociated from inflammation and induced by enzymatic DPP4 inhibition. Cell Metabol. 2019;29:320–334.e5. doi: 10.1016/j.cmet.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Mulvihill E.E., Varin E.M., Gladanac B., Campbell J.E., Ussher J.R., Baggio L.L., Yusta B., Ayala J., Burmeister M.A., Matthews D., et al. Cellular sites and mechanisms linking reduction of dipeptidyl peptidase-4 activity to control of incretin hormone action and glucose homeostasis. Cell Metabol. 2017;25:152–165. doi: 10.1016/j.cmet.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 15.Trzaskalski N.A., Vulesevic B., Nguyen M.A., Jeraj N., Fadzeyeva E., Morrow N.M., Locatelli C.A., Travis N., Hanson A.A., Nunes J.R., et al. Hepatocyte-derived DPP4 regulates portal GLP-1 bioactivity, modulates glucose production, and when absent influences NAFLD progression. JCI Insight. 2023;8:e154314. doi: 10.1172/jci.insight.154314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varin E.M., Hanson A.A., Beaudry J.L., Nguyen M.A., Cao X., Baggio L.L., Mulvihill E.E., Drucker D.J. Hematopoietic cell- versus enterocyte-derived dipeptidyl peptidase-4 differentially regulates triglyceride excursion in mice. JCI Insight. 2020;5:e140418. doi: 10.1172/jci.insight.140418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Souza A.H., Tang J., Yadev A.K., Saghafi S.T., Kibbe C.R., Linnemann A.K., Merrins M.J., Davis D.B. Intra-islet GLP-1, but not CCK, is necessary for beta-cell function in mouse and human islets. Sci. Rep. 2020;10:2823. doi: 10.1038/s41598-020-59799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellingsgaard H., Hauselmann I., Schuler B., Habib A.M., Baggio L.L., Meier D.T., Eppler E., Bouzakri K., Wueest S., Muller Y.D., et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. 2011;17:1481–1489. doi: 10.1038/nm.2513nm.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kilimnik G., Kim A., Steiner D.F., Friedman T.C., Hara M. Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic alpha-cells in mouse models of ss-cell regeneration. Islets. 2010;2:149–155. doi: 10.4161/isl.2.3.11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandoval D.A., D'Alessio D.A. Physiology of proglucagon peptides: role of glucagon and GLP-1 in health and disease. Physiol. Rev. 2015;95:513–548. doi: 10.1152/physrev.00013.2014. [DOI] [PubMed] [Google Scholar]
- 21.Teitelman G. Heterogeneous expression of proinsulin processing enzymes in beta cells of non-diabetic and type 2 diabetic humans. J. Histochem. Cytochem. 2019;67:385–400. doi: 10.1369/0022155419831641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell S.A., Johnson J., Salamon N., Light P.E. The DPP4 inhibitor sitagliptin increases active GLP-1 levels from human islets and may increase islet cell survival prior to transplantation. OBM Transplantation. 2019;3:14. doi: 10.21926/obm.transplant.1902069. [DOI] [Google Scholar]
- 23.Campbell S.A., Golec D.P., Hubert M., Johnson J., Salamon N., Barr A., MacDonald P.E., Philippaert K., Light P.E. Human islets contain a subpopulation of glucagon-like peptide-1 secreting alpha cells that is increased in type 2 diabetes. Mol. Metabol. 2020;39:101014. doi: 10.1016/j.molmet.2020.101014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saikia M., Holter M.M., Donahue L.R., Lee I.S., Zheng Q.C., Wise J.L., Todero J.E., Phuong D.J., Garibay D., Coch R., et al. GLP-1 receptor signaling increases PCSK1 and beta cell features in human alpha cells. JCI Insight. 2021;6:e141851. doi: 10.1172/jci.insight.141851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song Y., Koehler J.A., Baggio L.L., Powers A.C., Sandoval D.A., Drucker D.J. Gut-proglucagon-derived peptides are essential for regulating glucose homeostasis in mice. Cell Metabol. 2019;30:976–986.e3. doi: 10.1016/j.cmet.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chambers A.P., Sorrell J.E., Haller A., Roelofs K., Hutch C.R., Kim K.S., Gutierrez-Aguilar R., Li B., Drucker D.J., D'Alessio D.A., et al. The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metabol. 2017;25:927–934.e3. doi: 10.1016/j.cmet.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traub S., Meier D.T., Schulze F., Dror E., Nordmann T.M., Goetz N., Koch N., Dalmas E., Stawiski M., Makshana V., et al. Pancreatic alpha cell-derived glucagon-related peptides are required for beta cell adaptation and glucose homeostasis. Cell Rep. 2017;18:3192–3203. doi: 10.1016/j.celrep.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 28.Capozzi M.E., Svendsen B., Encisco S.E., Lewandowski S.L., Martin M.D., Lin H., Jaffe J.L., Coch R.W., Haldeman J.M., MacDonald P.E., et al. Beta Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight. 2019;4:e126742. doi: 10.1172/jci.insight.126742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu L., Dattaroy D., Pham J., Wang L., Barella L.F., Cui Y., Wilkins K.J., Roth B.L., Hochgeschwender U., Matschinsky F.M., et al. Intra-islet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight. 2019;5:e127994. doi: 10.1172/jci.insight.127994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Svendsen B., Larsen O., Gabe M.B.N., Christiansen C.B., Rosenkilde M.M., Drucker D.J., Holst J.J. Insulin secretion depends on intra-islet glucagon signaling. Cell Rep. 2018;25:1127–1134.e2. doi: 10.1016/j.celrep.2018.10.018. [DOI] [PubMed] [Google Scholar]
- 31.Liu L., Omar B., Marchetti P., Ahrén B. Dipeptidyl peptidase-4 (DPP-4): localization and activity in human and rodent islets. Biochem. Biophys. Res. Commun. 2014;453:398–404. doi: 10.1016/j.bbrc.2014.09.096. [DOI] [PubMed] [Google Scholar]
- 32.Waget A., Cabou C., Masseboeuf M., Cattan P., Armanet M., Karaca M., Castel J., Garret C., Payros G., Maida A., et al. Physiological and pharmacological mechanisms through which the DPP-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology. 2011;152:3018–3029. doi: 10.1210/en.2011-0286. [DOI] [PubMed] [Google Scholar]
- 33.Ahrén B., Yamada Y., Seino Y. The incretin effect in female mice with double deletion of GLP-1 and GIP receptors. J. Endocr. Soc. 2020;4:bvz036. doi: 10.1210/jendso/bvz036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith E.P., An Z., Wagner C., Lewis A.G., Cohen E.B., Li B., Mahbod P., Sandoval D., Perez-Tilve D., Tamarina N., et al. The role of beta cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs. Cell Metabol. 2014;19:1050–1057. doi: 10.1016/j.cmet.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aulinger B.A., Perabo M., Seeley R.J., Parhofer K.G., D'Alessio D.A. Rapid hepatic metabolism blunts the endocrine action of portally infused GLP-1 in male rats. Am. J. Physiol. Endocrinol. Metab. 2020;318:E189–E197. doi: 10.1152/ajpendo.00298.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balkan B., Li X. Portal GLP-1 administration in rats augments the insulin response to glucose via neuronal mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000;279:R1449–R1454. doi: 10.1152/ajpregu.2000.279.4.R1449. [DOI] [PubMed] [Google Scholar]
- 37.Malbert C.H., Chauvin A., Horowitz M., Jones K.L. Glucose sensing mediated by portal glucagon-like peptide 1 receptor is markedly impaired in insulin-resistant obese animals. Diabetes. 2021;70:99–110. doi: 10.2337/db20-0361. [DOI] [PubMed] [Google Scholar]
- 38.Nishizawa M., Moore M.C., Shiota M., Gustavson S.M., Snead W.L., Neal D.W., Cherrington A.D. Effect of intraportal glucagon-like peptide-1 on glucose metabolism in conscious dogs. Am. J. Physiol. Endocrinol. Metab. 2003;284:E1027–E1036. doi: 10.1152/ajpendo.00503.2002. [DOI] [PubMed] [Google Scholar]
- 39.Nishizawa M., Nakabayashi H., Kawai K., Ito T., Kawakami S., Nakagawa A., Niijima A., Uchida K. The hepatic vagal reception of intraportal GLP-1 is via receptor different from the pancreatic GLP-1 receptor. J. Auton. Nerv. Syst. 2000;80:14–21. doi: 10.1016/s0165-1838(99)00086-7. [DOI] [PubMed] [Google Scholar]
- 40.Burcelin R., Da Costa A., Drucker D., Thorens B. Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes. 2001;50:1720–1728. doi: 10.2337/diabetes.50.8.1720. [DOI] [PubMed] [Google Scholar]
- 41.Mannucci E., Pala L., Ciani S., Bardini G., Pezzatini A., Sposato I., Cremasco F., Ognibene A., Rotella C.M. Hyperglycaemia increases dipeptidyl peptidase IV activity in diabetes mellitus. Diabetologia. 2005;48:1168–1172. doi: 10.1007/s00125-005-1749-8. [DOI] [PubMed] [Google Scholar]
- 42.Omar B.A., Liehua L., Yamada Y., Seino Y., Marchetti P., Ahrén B. Dipeptidyl peptidase 4 (DPP-4) is expressed in mouse and human islets and its activity is decreased in human islets from individuals with type 2 diabetes. Diabetologia. 2014;57:1876–1883. doi: 10.1007/s00125-014-3299-4. [DOI] [PubMed] [Google Scholar]
- 43.Berthault C., Staels W., Scharfmann R. Purification of pancreatic endocrine subsets reveals increased iron metabolism in beta cells. Mol. Metabol. 2020;42:101060. doi: 10.1016/j.molmet.2020.101060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahn S.H., Granger A., Rankin M.M., Lam C.J., Cox A.R., Kushner J.A. Tamoxifen suppresses pancreatic beta-cell proliferation in mice. PLoS One. 2019;14:e0214829. doi: 10.1371/journal.pone.0214829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oropeza D., Jouvet N., Budry L., Campbell J.E., Bouyakdan K., Lacombe J., Perron G., Bergeron V., Neuman J.C., Brar H.K., et al. Phenotypic characterization of MIP-CreERT1Lphi mice with transgene-driven islet expression of human growth hormone. Diabetes. 2015;64:3798–3807. doi: 10.2337/db15-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin H.V., Wang J., Wang J., Li W., Wang X., Alston J.T., Thomas M.K., Briere D.A., Syed S.K., Efanov A.M. GPR142 prompts glucagon-like Peptide-1 release from islets to improve beta cell function. Mol. Metabol. 2018;11:205–211. doi: 10.1016/j.molmet.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansotia T., Baggio L.L., Delmeire D., Hinke S.A., Yamada Y., Tsukiyama K., Seino Y., Holst J.J., Schuit F., Drucker D.J. Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP-IV inhibitors. Diabetes. 2004;53:1326–1335. doi: 10.2337/diabetes.53.5.1326. [DOI] [PubMed] [Google Scholar]
- 48.Bugliani M., Syed F., Paula F.M.M., Omar B.A., Suleiman M., Mossuto S., Grano F., Cardarelli F., Boggi U., Vistoli F., et al. DPP-4 is expressed in human pancreatic beta cells and its direct inhibition improves beta cell function and survival in type 2 diabetes. Mol. Cell. Endocrinol. 2018;473:186–193. doi: 10.1016/j.mce.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 49.Ferdaoussi M., Smith N., Lin H., Bautista A., Spigelman A.F., Lyon J., Dai X., Manning Fox J.E., MacDonald P.E. Improved glucose tolerance with DPPIV inhibition requires beta-cell SENP1 amplification of glucose-stimulated insulin secretion. Phys. Rep. 2020;8:e14420. doi: 10.14814/phy2.14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marchetti P., Lupi R., Bugliani M., Kirkpatrick C.L., Sebastiani G., Grieco F.A., Del Guerra S., D'Aleo V., Piro S., Marselli L., et al. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia. 2012;55:3262–3272. doi: 10.1007/s00125-012-2716-9. [DOI] [PubMed] [Google Scholar]
- 51.Sancho V., Daniele G., Lucchesi D., Lupi R., Ciccarone A., Penno G., Bianchi C., Dardano A., Miccoli R., Del Prato S. Metabolic regulation of GLP-1 and PC1/3 in pancreatic alpha-cell line. PLoS One. 2017;12:e0187836. doi: 10.1371/journal.pone.0187836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wideman R.D., Covey S.D., Webb G.C., Drucker D.J., Kieffer T.J. A switch from prohormone convertase (PC)-2 to PC1/3 expression in transplanted alpha-cells is accompanied by differential processing of proglucagon and improved glucose homeostasis in mice. Diabetes. 2007;56:2744–2752. doi: 10.2337/db07-0563. [DOI] [PubMed] [Google Scholar]
- 53.Galvin S.G., Kay R.G., Foreman R., Larraufie P., Meek C.L., Biggs E., Ravn P., Jermutus L., Reimann F., Gribble F.M. The human and mouse islet peptidome: effects of obesity and type 2 diabetes, and assessment of intraislet production of glucagon-like peptide-1. J. Proteome Res. 2021;20:4507–4517. doi: 10.1021/acs.jproteome.1c00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lafferty R.A., Gault V.A., Flatt P.R., Irwin N. Effects of 2 novel PYY(1-36) analogues, (P(3)L(31)P(34))PYY(1-36) and PYY(1-36)(lys(12)PAL), on pancreatic beta-cell function, growth, and survival. Clin. Med. Insights Endocrinol. Diabetes. 2019;12 doi: 10.1177/1179551419855626. 1179551419855626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Z., Stanojevic V., Avadhani S., Yano T., Habener J.F. Stromal cell-derived factor-1 (SDF-1)/chemokine (C-X-C motif) receptor 4 (CXCR4) axis activation induces intra-islet glucagon-like peptide-1 (GLP-1) production and enhances beta cell survival. Diabetologia. 2011;54:2067–2076. doi: 10.1007/s00125-011-2181-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nackiewicz D., Dan M., Speck M., Chow S.Z., Chen Y.C., Pospisilik J.A., Verchere C.B., Ehses J.A. Islet macrophages shift to a reparative state following pancreatic beta-cell death and are a major source of islet insulin-like growth factor-1. iScience. 2020;23:100775. doi: 10.1016/j.isci.2019.100775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi Y.C., Loh K., Bensellam M., Lee K., Zhai L., Lau J., Cantley J., Luzuriaga J., Laybutt D.R., Herzog H. Pancreatic PYY is critical in the control of insulin secretion and glucose homeostasis in female mice. Endocrinology. 2015;156:3122–3136. doi: 10.1210/en.2015-1168. [DOI] [PubMed] [Google Scholar]
- 58.Wicksteed B., Brissova M., Yan W., Opland D.M., Plank J.L., Reinert R.B., Dickson L.M., Tamarina N.A., Philipson L.H., Shostak A., et al. Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes. 2010;59:3090–3098. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Estall J.L., Screaton R.A. Of mice and men, redux: modern challenges in beta cell gene targeting. Endocrinology. 2020;161:bqaa078. doi: 10.1210/endocr/bqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Douros J.D., Lewis A.G., Smith E.P., Niu J., Capozzi M., Wittmann A., Campbell J., Tong J., Wagner C., Mahbod P., et al. Enhanced glucose control following vertical sleeve gastrectomy does not require a beta-cell glucagon-like peptide 1 receptor. Diabetes. 2018;67:1504–1511. doi: 10.2337/db18-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marguet D., Baggio L., Kobayashi T., Bernard A.M., Pierres M., Nielsen P.F., Ribel U., Watanabe T., Drucker D.J., Wagtmann N. Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. Proc. Natl. Acad. Sci. USA. 2000;97:6874–6879. doi: 10.1073/pnas.120069197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hingorani S.R., Petricoin E.F., Maitra A., Rajapakse V., King C., Jacobetz M.A., Ross S., Conrads T.P., Veenstra T.D., Hitt B.A., et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 63.Campbell J.E., Ussher J.R., Mulvihill E.E., Kolic J., Baggio L.L., Cao X., Liu Y., Lamont B.J., Morii T., Streutker C.J., et al. TCF1 links GIPR signaling to the control of beta cell function and survival. Nat. Med. 2016;22:84–90. doi: 10.1038/nm.3997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data will be shared upon request to the lead contact. No standardized datatype data were generated in this study.
-
•
This study did not generate new code.
-
•
Any additional analysis information for this work is available by request to the lead contact.