

Key Points
Question
What are the genetic associations between modifiable risk factors and Alzheimer disease (AD)?
Findings
In this genetic association study using a mendelian randomization framework with the largest genomic data sets to date, including 39 106 participants with clinically diagnosed AD and 401 577 control participants without AD, genetically determined increased high-density lipoprotein cholesterol and increased systolic blood pressure were associated with higher risk of AD.
Meaning
These findings suggest that genetically determined increased high-density lipoprotein cholesterol and systolic blood pressure may be involved in AD pathogenesis, which may thus inspire new drug targeting and improved early dementia prevention.
This genetic association study uses mendelian randomization to examine potentially causal aspects of modifiable genetic risk factors for Alzheimer disease.
Abstract
Importance
An estimated 40% of dementia is potentially preventable by modifying 12 risk factors throughout the life course. However, robust evidence for most of these risk factors is lacking. Effective interventions should target risk factors in the causal pathway to dementia.
Objective
To comprehensively disentangle potentially causal aspects of modifiable risk factors for Alzheimer disease (AD) to inspire new drug targeting and improved prevention.
Design, Setting, and Participants
This genetic association study was conducted using 2-sample univariable and multivariable mendelian randomization. Independent genetic variants associated with modifiable risk factors were selected as instrumental variables from genomic consortia. Outcome data for AD were obtained from the European Alzheimer & Dementia Biobank (EADB), generated on August 31, 2021. Main analyses were conducted using the EADB clinically diagnosed end point data. All analyses were performed between April 12 and October 27, 2022.
Exposures
Genetically determined modifiable risk factors.
Main Outcomes and Measures
Odds ratios (ORs) and 95% CIs for AD were calculated per 1-unit change of genetically determined risk factors.
Results
The EADB-diagnosed cohort included 39 106 participants with clinically diagnosed AD and 401 577 control participants without AD. The mean age ranged from 72 to 83 years for participants with AD and 51 to 80 years for control participants. Among participants with AD, 54% to 75% were female, and among control participants, 48% to 60% were female. Genetically determined high-density lipoprotein (HDL) cholesterol concentrations were associated with increased odds of AD (OR per 1-SD increase, 1.10 [95% CI, 1.05-1.16]). Genetically determined high systolic blood pressure was associated with increased risk of AD after adjusting for diastolic blood pressure (OR per 10–mm Hg increase, 1.22 [95% CI, 1.02-1.46]). In a second analysis to minimize bias due to sample overlap, the entire UK Biobank was excluded from the EADB consortium; odds for AD were similar for HDL cholesterol (OR per 1-SD unit increase, 1.08 [95% CI, 1.02-1.15]) and systolic blood pressure after adjusting for diastolic blood pressure (OR per 10–mm Hg increase, 1.23 [95% CI, 1.01-1.50]).
Conclusions and Relevance
This genetic association study found novel genetic associations between high HDL cholesterol concentrations and high systolic blood pressure with higher risk of AD. These findings may inspire new drug targeting and improved prevention implementation.
Introduction
Dementia is a rapidly increasing health threat worldwide, affecting more than 50 million people and projected to triple in prevalence by 2050.1 A 2020 report by Livingston et al for the Lancet Commission for Dementia Prevention, Intervention and Care1 estimated that up to 40% of dementia could be prevented or delayed by modifying 12 risk factors throughout the life course.1 However, various degrees of inconsistency for these risk factors exist between observational studies and clinical trials, leading to mixed quality of evidence underpinning recommendations.2,3 Effective interventions should target ameliorating risk factors that lie in the causal pathways. Hence, thoroughly unfolding the genomic background for associations between modifiable risk factors and dementia might help to develop future efficacious preventive and therapeutic approaches.
Associations identified in observational studies are not equivalent to causality due to confounding and reverse causation; the latter may explain why associations between risk factors and dementia change across the lifespan, especially in late life.4,5 Although randomized clinical trials may demonstrate an unconfounded effect of a certain intervention on dementia, interventions after irreversible neuron damage or with a relatively short duration may have a negligible effect. The mendelian randomization (MR) design uses genetic variants associated with the exposure to investigate potential causal relationships between risk factors and outcomes. The exposure is thus lifelong, and the random allocation of variants at conception minimizes confounding and reverse causation.6 Thus, the MR approach may help establish causality and guide whether a comprehensive randomized clinical trial targeting the risk factor will be meaningful to perform. Several MR studies have been conducted to disentangle the associations between modifiable risk factors and Alzheimer disease (AD), the most common type of dementia and the only type of dementia with large-scale genomewide association studies (GWAS). Genetically, longer educational attainment has been well-established as associated with lower risk for AD, whereas other risk factors, including lipid traits, blood pressure (BP), body mass index (BMI), smoking, and alcohol consumption have shown inconclusive associations with AD. This may be due to lack of power, small number of genetic instruments, and other biases related to study design. Consequently, more powerful and state-of-the-art MR studies are warranted to examine genomic associations between modifiable risk factors and AD.
The new landmark paper on AD genetic etiology by the European Alzheimer & Dementia Biobank (EADB) provides new possibilities to disentangle potential causal aspects of modifiable risk factors for AD.7 Furthermore, the massively increasing availability of high-quality genotypic data in large consortia provides more powerful genetic instrumental variables. Collectively, this prompted us to scrutinize the genetic associations between modifiable risk factors and AD using complementary and up-to-date MR methods.
Methods
In this genetic association study, we implemented a 2-sample MR approach that uses genetic variants as instrumental variables for the exposure to investigate whether a lifetime exposure may be causally associated with an outcome (eFigure 1 in Supplement 1). Ethical approvals were obtained by each individual participating cohort; therefore, no additional ethical approvals or informed consents were required. Our study followed the Strengthening the Reporting of Genetic Association Studies (STREGA) reporting guideline and Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization (STROBE-MR) reporting guidelines. The schematic overview of the study design is presented in Figure 1, and previous main MR studies on modifiable risk factors and AD are listed in eTable 1 in Supplement 1. We used summary GWAS statistics for each exposure and AD.
Figure 1. Schematic Overview of the Study Design.
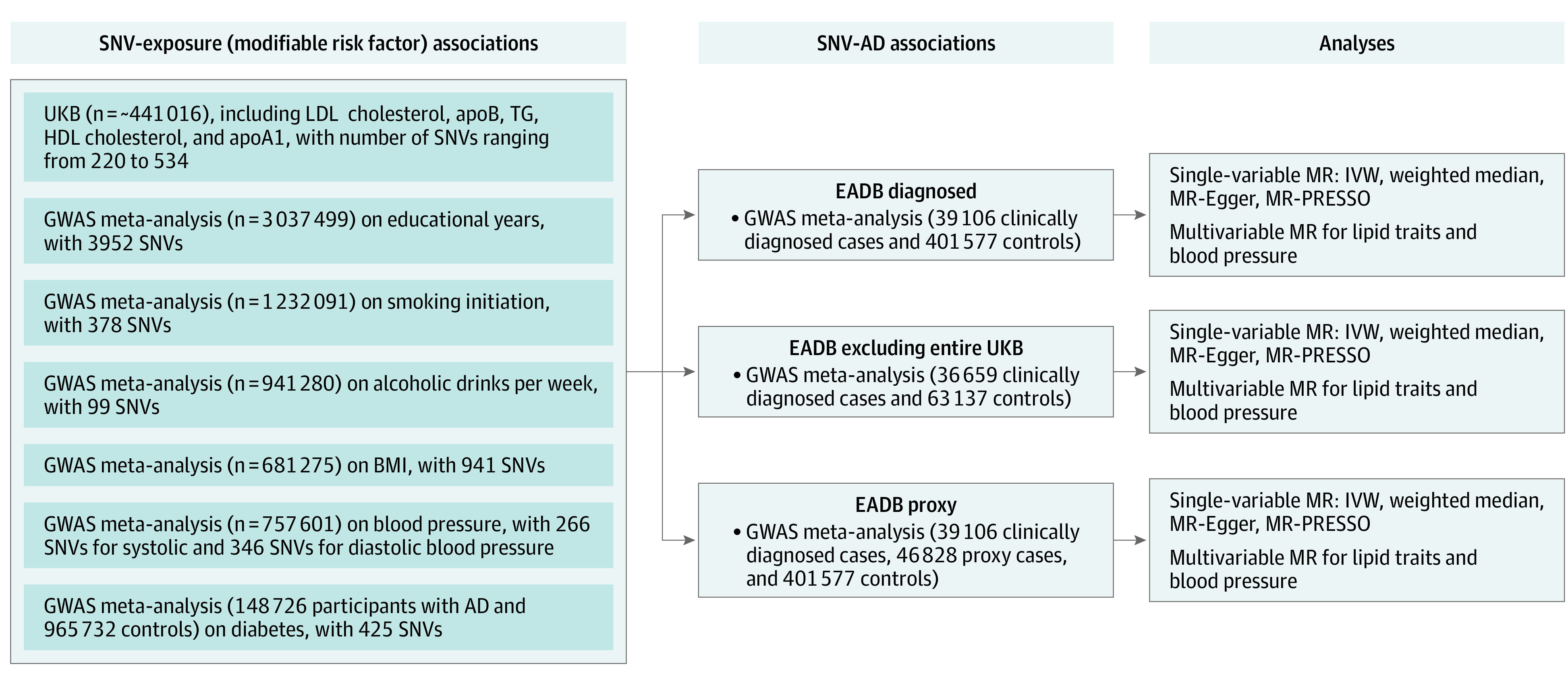
AD indicates Alzheimer disease; apoA1, apolipoprotein A1; apoB, apolipoprotein B; BMI, body mass index; EADB, European Alzheimer & Dementia Biobank; GWAS, genome-wide association study; HDL, high-density lipoprotein; IVW, inverse-variance weighted; LDL, low-density lipoprotein; MR, mendelian randomization; MR-PRESSO, mendelian randomization pleiotropy residual sum and outlier; SNV, single nucleotide variant; TG, triglycerides; UKB, UK Biobank.
Selection of Instrumental Variables
Modifiable risk factors include educational attainment,8 lipids and lipoproteins9 (low-density lipoprotein [LDL] cholesterol, triglycerides [TG], apolipoprotein B [apoB], high-density lipoprotein [HDL] cholesterol, and apolipoprotein A1 [apoA1]), BMI,10 alcohol consumption,11 smoking initiation,11 systolic BP (SBP) and diastolic BP (DBP),12 and type 2 diabetes.13 Details on the GWAS from which we obtained summary-level associations between genetic variants and risk factors are described in Table 1. Selection and summary information of the independent single nucleotide variants (SNVs) are in Table 2 and the eMethods in Supplement 1.
Table 1. GWAS Data Sources for Instrumental Variables Selection.
| Study | Risk factor | Consortium | Participantsa | Covariates |
|---|---|---|---|---|
| Okbay et al,8 2022 | Education attainment | SSGAC; UKB; 23andMe | 3 037 499 | Age, sex, age × sex, PCs |
| Richardson et al,9 2020 | Low-density lipoprotein cholesterol | UKB | 440 546 | Age, sex |
| High-density lipoprotein cholesterol | 403 943 | |||
| Triglycerides | 441 016 | |||
| Apolipoprotein A1 | 393 193 | |||
| Apolipoprotein B | 439 214 | |||
| Yengo et al,10 2018 | BMI | GIANT, UKB | 681 275 | Age, sex, PCs |
| Liu et al,11 2019 | Smoking initiation | GSCAN; 23andMe | 1 232 091 | Age, sex, age × sex, PCs |
| Alcohol consumption | 941 280 | |||
| Evangelou et al,12 2018 | Systolic blood pressure | UKB; ICBP | 757 601 | Age, sex, age2, BMI |
| Diastolic blood pressure | ||||
| Vujkovic et al,13 2020 | Type 2 diabetes | DIAMENTE | 148 726 with AD | Age, sex, PCs |
| 965 732 controls |
Abbreviations: AD, Alzheimer disease; BMI, body mass index; DIAMENTE, Diabetes Meta-analysis of Trans-ethnic Association Studies consortium; GIANT, Genetic Investigation of Anthropometric Traits consortium; GWAS, genome-wide association study; GSCAN, GWAS and Sequencing Consortium of Alcohol and Nicotine use; ICBP, International Consortium of Blood Pressure Genome Wide Association Studies; PCs, principal components; SSGAC, Social Science Genetic Association Consortium; UKB, UK Biobank.
Numbers are participants with European ancestry.
Table 2. Summary Information of Genetic Instruments for Modifiable Risk Factors.
| Risk factor | Unit | SNVs, No.a | LD thresholdb | Variation, % |
|---|---|---|---|---|
| Low-density lipoprotein cholesterol | SD in mmol/L | 220 | 0.001 | 7.7c |
| Triglycerides | SD in mmol/L | 440 | 0.001 | 10.3c |
| Apolipoprotein B | SD in g/L | 255 | 0.001 | 9.2c |
| High-density lipoprotein cholesterol | SD in mmol/L | 534 | 0.001 | 11.9c |
| Apolipoprotein A1 | SD in g/L | 440 | 0.001 | 10.1c |
| Educational attainment | years | 3952 | 0.1 | 12-16 |
| BMI | SD per 1 unit | 507 | 0.001 | 6 |
| Smoking initiation | Ever smoked regularly vs never smoke | 378 | 0.1 | 2.3 |
| Alcohol consumption | SD in alcoholic drinks per week | 99 | 0.1 | 0.7 |
| Systolic blood pressure | 10 mm Hg | 266 | 0.1 | 5.7 |
| Diastolic blood pressure | 10 mm Hg | 346 | 0.1 | 5.3 |
| Type 2 diabetes | Log odds | 425 | 0.05 | 19 |
Abbreviations: BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); LD, linkage disequilibrium; SNV, single nucleotide variant.
Number of independent SNVs at genome-wide significance level (P < 5 × 10−8).
LD refers to the degree to which an allele of 1 genetic variant is inherited or correlated with an allele of a nearby genetic variant within a given population. The threshold to prune for LD was obtained in the original genome-wide association studies.
Variation explained by genetic instrumental variables were calculated based on the formula: β2 × 2 × MAF × (1 − MAF), where MAF denotes mean minor allele frequency from European populations, obtained through Phenoscanner V2. Calculation of the remaining percentages are given in the original articles.
Alzheimer Disease Data Sources
The associations between SNVs and late-onset AD were retrieved from EADB, the largest AD genomic consortia. EADB brings together a range of European cohorts and GWAS consortia, and summary estimates were based on 39 106 participants with clinically diagnosed AD, 46 828 participants with proxy AD, and 401 577 control participants without AD7 (generated in August 31, 2021). Proxy AD was only identified from the UK Biobank via questionnaire data asking if parents of the participants had AD (“Has/did your father or mother ever suffer from Alzheimer’s disease/dementia?”). Participants were categorized into proxy AD if the answer was yes, otherwise they were controls. Three summary data sets were used: (1) the EADB-diagnosed data set in which only participants who had been clinically diagnosed with AD were included in the summary data; (2) the EADB data set, excluding the entire UK Biobank (UKB) (generated on September 19, 2022); and (3) the EADB-proxy data set, which included both participants who had been clinically diagnosed with AD and those with proxy AD (eMethods in Supplement 1) from the UKB were included (generated on February 10, 2022).
Statistical Analysis
All analyses were performed between April 12 to October 27, 2022. The MR results are given as odds ratios (ORs) with corresponding 95% CIs of log odds of AD per unit increase in genetically determined risk factors. The estimates are scaled by year of education completed, ever smoked regularly vs never smoked, 10–mm Hg increase of BP, SD for consumption of alcoholic drinks per week, and SD for the other continuous risk factors; for diabetes, the estimates represent the OR of AD per 1-unit higher log odds of diabetes. In the reverse direction, the results represent the relative increase in the odds of AD per 1-unit change in each behavioral risk factor. Statistical power for MR analyses were calculated using the power calculation tool.14 We have 80% power to detect a minimum of 3% change in log odds of AD (eFigure 2 in Supplement 1).
To maintain statistical power while still limiting the number of false-positive conclusions, we corrected for multiple testing per MR-method using false discovery rate proposed by Benjamini and Hochberg.15 Two-sided P < .05 indicated statistical significance. All the analyses were undertaken using R version 4.0.2 (R Project for Statistical Computing).
Associations between genetic variants and risk factors and AD were harmonized to ensure that estimates were aligned on the same allele. Ambiguous genetic variants with palindromic genotypes were excluded. We used the inverse-variance weighted (IVW) method as the primary analysis, which combines SNV-specific estimates calculated by Wald ratios through dividing the genetic association with AD by the genetic association with each risk factor. When a genetic variant affects other traits that influence the outcome independently of the hypothesized exposure, known as horizontal pleiotropy, this may violate 1 of the key MR assumptions of exclusion restriction. IVW assumes no violation of MR assumptions, particularly no directional pleiotropic effect of each instrumental variable, and constrains intercepts to zero. Furthermore, we performed several MR sensitivity analyses to address invalid instruments, unbalanced pleiotropy, outliers, and correlated risk factors. The weighted median estimator and MR-Egger allows the inclusion of pleiotropic genetic variants and were used to investigate whether bias in IVW estimates were present due to invalid instruments.16,17 The regression slope from MR-Egger represents the estimated effect of an exposure on the outcome, and the freely estimated intercept additionally provides a mean magnitude of the pleiotropic effects across all genetic variants if it deviates from zero. MR-Egger is statistically less efficient (ie, with wider CIs) but provides a causal estimate that accounts for horizontal pleiotropy. Therefore, the point estimates from these 2 methods might be close to null even if a strong association is observed through IVW; however, the CIs should largely overlap. To assess the distortions of the IVW estimate from any heterogeneity or horizontal pleiotropy, MR-PRESSO was applied to detect and correct for outliers, giving an unbiased estimate.18
For correlated risk factors, we performed multivariable MR, an extension of the basic MR design and estimates the effects of 2 or more related exposures on an outcome simultaneously. Subsequently, the direct effect, ie, the effect not confounded or mediated by other factors, of each exposure in the model is obtained.19 For lipids, apoA1 and apoB are highly correlated with HDL and LDL cholesterol, respectively. To avoid multicollinearity in the multivariable MR model, we adjusted HDL and TG for LDL and apoB, HDL and LDL for TG, and LDL and TG for HDL and apoA1.
Four additional sensitivity analyses were conducted and are described in detail in eMethods in Supplement 1. First, we used the Causal Analysis using Summary Effect Estimates) method, accounting for correlated pleiotropy.20 A second analysis excluded SNVs on the entire chromosome 19, addressing the independence of the strong apolipoprotein E (APOE) locus. Third, we used cross-trait linkage disequilibrium-score regression, accounting for sample overlap.21 Fourth, we evaluated possible reverse causation of AD on behavioral risk factors.
Results
EADB-Diagnosed Data Set
The EADB-diagnosed cohort included 39 106 participants with clinically diagnosed AD and 401 577 control participants without AD. In the EADB-diagnosed data set, the mean age ranged from 72 to 83 years among participants with AD and 51 to 80 years among control participants without AD. Among participants with AD, 54% to 75% were female, and among control participants, 48% to 60% were female. A detailed description of the demographic characteristic is given in the original literature.7
The results from the EADB-diagnosed data set are presented in eTable 1 in Supplement 2 and visualized in Figure 2. Increased HDL cholesterol was associated with increased odds of AD (OR per 1-SD increase, 1.07 [95% CI, 1.01-1.13]). The point estimate for HDL cholesterol was enhanced on correction for outliers using MR-PRESSO (OR, 1.10 [95% CI, 1.05-1.16]; P < .001) and remained similar to the IVW estimate in other MR sensitivity methods (eTable 1 in Supplement 2). After adjusting for DBP, SBP was associated with increased risk of AD in multivariable MR (OR per 10–mm Hg increase, 1.22 [95% CI, 1.02-1.46]). Analyses using different univariable MR methods were not statistically significant (OR per 10–mm Hg increase, 1.02 [95% CI, 0.96-1.09]), but analysis using other MR sensitivity methods remained significant. A 10–mm Hg genetically determined higher level of DBP was associated with lower risk of AD across different MR methods. Genetic predisposition to longer educational attainment was associated with lower odds of AD in all analyses (IVW OR, 0.83 [95% CI, 0.79-0.87]). The estimates for apoA1, smoking, and BMI were inconclusive. LDL cholesterol, apoB, TG, alcohol consumption, and diabetes were consistently not associated with the odds of AD in all MR methods. A detailed list of SNVs involved in the HDL cholesterol signal is given in eTable 2 in Supplement 2.
Figure 2. Associations of Genetically Determined Modifiable Risk Factors and Alzheimer Disease (AD) in the European Alzheimer & Dementia Biobank Data Set of Participants With Clinically Diagnosed AD.
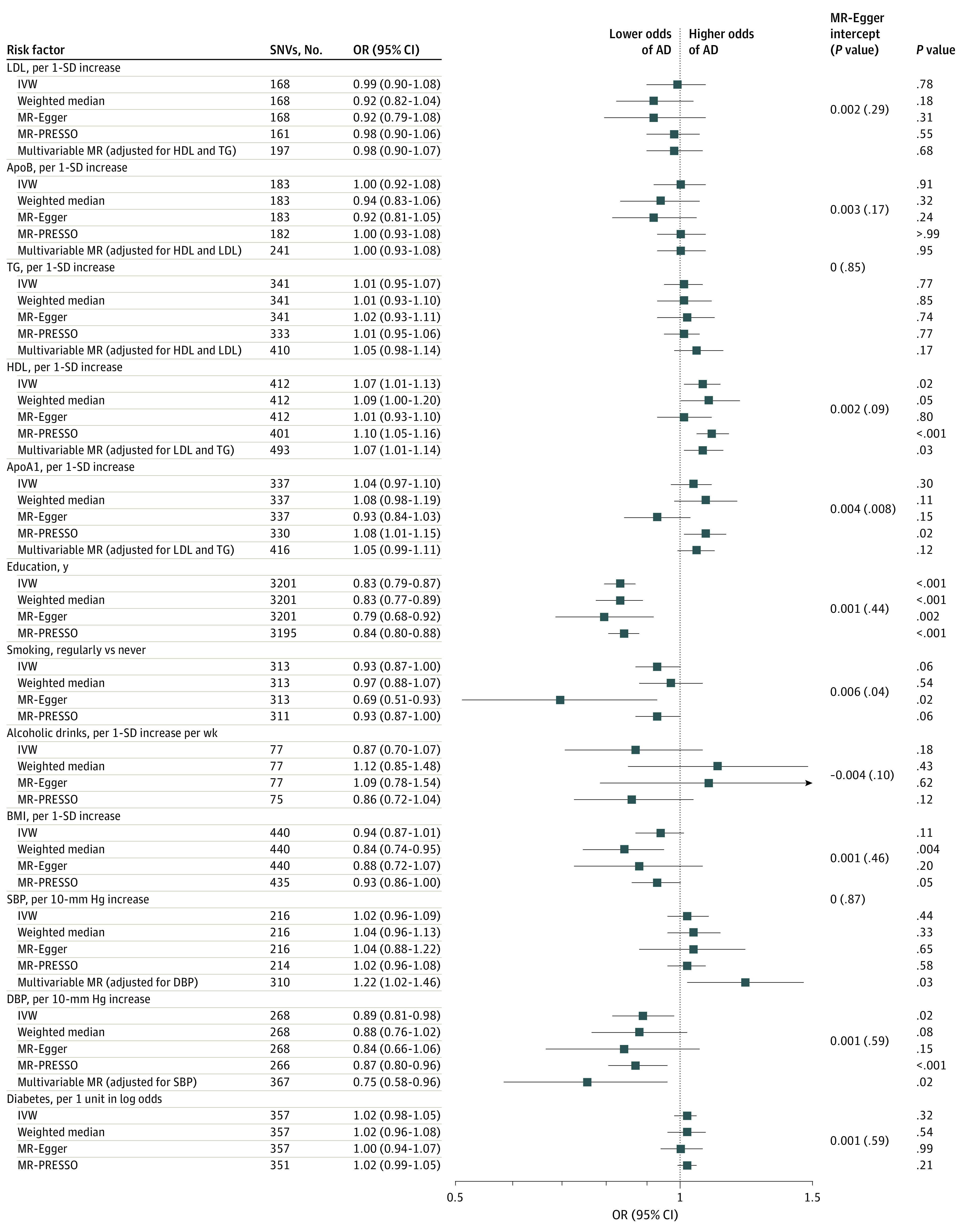
Multivariable mendelian randomization was performed for correlated phenotypes only (lipid traits and blood pressure). ApoA1 indicates apolipoprotein A1; apoB, apolipoprotein B; BMI, body mass index; DBP, diastolic blood pressure; HDL, high-density lipoprotein; IVW, inverse-variance weighted; LDL, low-density lipoprotein; MR-PRESSO, mendelian randomization pleiotropy residual sum and outlier; OR, odds ratio; SBP, systolic blood pressure; SNV, single nucleotide variant; TG, triglycerides.
EADB Excluding the Entire UKB
The results from the EADB data set excluding UKB generally resemble those from the EADB diagnosed data set (Figure 3; eTable 3 in Supplement 2). For HDL cholesterol concentrations, the IVW and multivariable MR analyses found the same results (ORs for both, 1.08 [95% CI, 1.02-1.15]). After adjusting for DBP, increased SBP was associated with increased risk of AD (OR per 10–mm Hg increase, 1.23 [95% CI, 1.01-1.50]). The analyses corrected for multiple testing remained significant in both IVW and MR-PRESSO. Higher DBP was associated with lower risk of AD in the IVW analysis (OR per 10–mm Hg increase, 0.87 [95% CI, 0.79-0.97]) and remained similar using other sensitivity methods. Genetic predisposition to longer educational attainment was associated with lower odds of AD in all analyses (IVW OR, 0.85 [95% CI, 0.81-0.90]). Smoking initiation and higher BMI were associated with lower odds of AD (smoking: IVW OR, 0.90 [95% CI, 0.83-0.98]; BMI: IVW OR, 0.91 [95% CI, 0.84-0.99]). No associations were found between other modifiable risk factors and odds of AD.
Figure 3. Associations of Genetically Determined Modifiable Risk Factors and Alzheimer Disease (AD) in the European Alzheimer & Dementia Biobank Data Set Excluding the Entire UK Biobank.
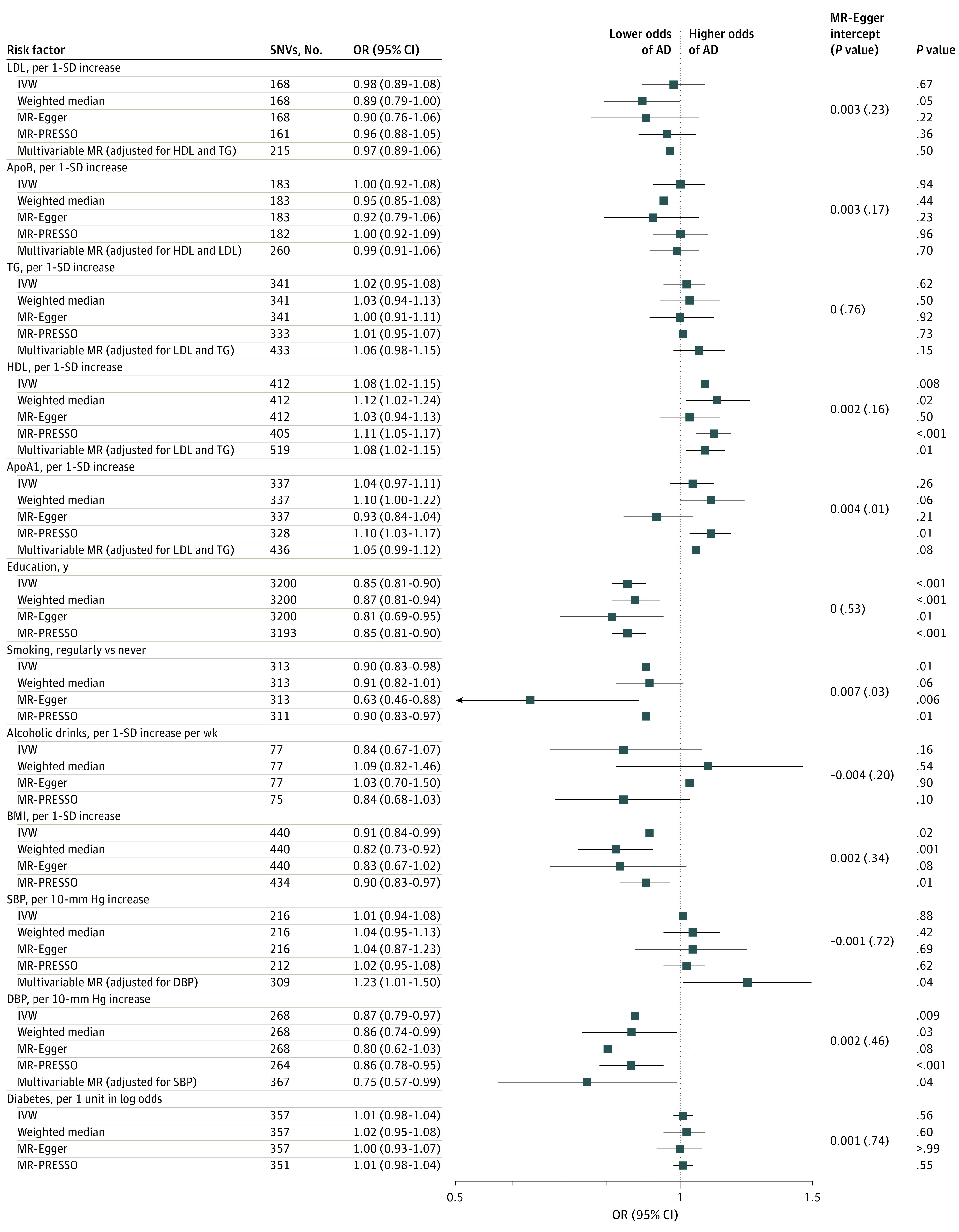
Multivariable mendelian randomization was performed for correlated phenotypes only (lipid traits and blood pressure). Abbreviations: apoA1, apolipoprotein A1; apoB, apolipoprotein B; BMI, body mass index; DBP, diastolic blood pressure; HDL, high-density lipoprotein; IVW, inverse-variance weighted; LDL, low-density lipoprotein cholesterol; MR-PRESSO, mendelian randomization pleiotropy residual sum and outlier; OR, odds ratio; SBP, systolic blood pressure; SNV, single nucleotide variant; TG, triglycerides.
EADB-Proxy Data Set
Results using EADB-proxy data set are shown in eFigure 3 in Supplement 1. Longer educational attainment was associated with higher odds of AD (IVW OR, 1.06 [95% CI, 1.01-1.10]). A detailed illustration leading to this counterintuitive finding is provided in eFigure 4 in Supplement 1. In IVW analyses, higher HDL cholesterol (OR, 1.10 [95% CI, 1.04-1.15]) and higher apoA1 (OR, 1.07 [95% CI, 1.00-1.13]) were associated with higher odds of AD, whereas smoking (OR, 0.88 [95% CI, 0.82-0.94]), higher BMI (OR, 0.89 [95% CI, 0.83-0.95]), and higher DBP (OR, 0.85 [95% CI, 0.78-0.92]) were associated with lower odds of AD. LDL, apoB TG, alcohol consumption, SBP, and diabetes showed no association (eFigure 3 in Supplement 1).
Other Sensitivity Analyses
The various sensitivity analyses examining the association between genetically determined modifiable risk factors and AD generally showed similar results (eAppendix 1, eTable 2, eFigure 5, and eFigure 6 in Supplement 1). Furthermore, genetic predisposition to high odds of AD were not associated with educational attainment, smoking, alcohol consumption, or BMI (eTable 3 in Supplement 1).
Discussion
This genetic association study using 2-sample mendelian randomization based on the largest genomic consortia found that genetically determined high HDL cholesterol and high SBP were associated with higher odds of AD. There was no consistent evidence supporting genetic associations of other lipid traits, BMI, alcohol consumption, smoking initiation, or diabetes with odds of AD. Moreover, our study suggested that meticulous care should be taken when using individuals with proxy AD from the UKB in 2-sample MR studies, as the results can be seriously biased.
Conflicting results for modifiable risk factors and AD have been reported in previous MR studies. For HDL cholesterol in particular, genetic studies have found no association22,23,24,25,26,27,28,29 or an association of high concentration of extra-large particles with lower risk of AD.30,31 These inconsistencies may be attributed to insufficient power and other biases, including pleiotropy. To our knowledge, our study is the first to identify an association between high HDL cholesterol concentrations and higher AD risk in a comprehensive range of complementary analyses. The genetic instruments for HDL cholesterol are marking well-known genes in HDL cholesterol biology, including ATP binding cassette A and G transporters, cholesteryl ester transfer protein, endothelial lipase, hepatic lipase, lipoprotein lipase, and scavenger receptor B1, further strengthening the validity of our findings. Although the underlying mechanisms remain unclear, there are a few biologically plausible explanations. HDL particles are complex, comprising a wide spectrum of sizes, compositions, and functionality. Small, but not large, HDL particles exchange lipids between plasma and cerebrospinal fluid compartments and form apoE and apoA1 small HDL particles through the interaction between plasma-derived apoA1 and brain-derived apoE.32 These particles subsequently promote neuronal membrane lipid remodeling and synaptic plasticity, limit apoE self-aggregation, and increase receptor binding and amyloid-β clearance.33 Indeed, the concentration of small particles in cerebrospinal fluid is highly correlated with the concentrations in plasma and is positively associated with cognitive function.34 However, high HDL cholesterol concentrations in plasma lead to a shift toward large HDL particles and significant increases in apoA1.35 Therefore, high plasma HDL cholesterol concentrations characterizing large buoyant HDL particles may play a role in dementia pathogenesis by disrupting the homeostasis between plasmatic particles and the beneficial apoE and apoA1 small HDL particles in cerebrospinal fluid.
Observationally, hypertension in midlife has been suggested as an independent risk factor for AD, whereas hypertension in late life showed null or reverse associations with AD, particularly for DBP.36,37 Sustained hypertension from midlife to late life, compared with midlife and late-life BP within reference ranges, was associated with increased risk of dementia.38 Nevertheless, most studies have BP measured at 1 or few separate time points, which may not fully capture the longitudinal changes and their cumulative effect. Results from individual antihypertensive randomized clinical trials are inconclusive. A meta-analysis combining 12 trials (baseline BP 154/83.3 mm Hg) concluded that antihypertensive treatment is associated with significantly decreased dementia risk through decreasing SBP.39 Similarly, a pooled individual-participant data analysis of 5 randomized clinical trials provided evidence supporting benefits associated with antihypertensive treatment in late midlife and later life to lower the risk of dementia.40 However, these effects last only for the duration of the trials. Our findings of genetically determined and thus lifelong high SBP and low DBP independently associated with high AD risk were partly in line with previous MR findings.41,42 These associations are reinforced by a study in which a long-term cumulative SBP increase was associated with subsequently higher dementia risk, whereas a cumulative DBP increase was associated with lower risk.43 There are several hypothesized explanations, which are discussed in detail in eAppendix 2 in Supplement 1.
We observed associations of high BMI and smoking initiation with lower risk of AD. Individual-level data analyses have suggested that the BMI association might be restricted to older age groups only.44 The association between smoking initiation or lifetime smoking and AD were mixed in previous MR studies using summary data; individual-level data from a genetically homogenous Danish population in a 1-sample MR study observed a higher risk of AD with high smoking quantity.45 Nevertheless, a meta-analysis that pooled the results from 2 summary statistics-based MR studies found no associations either for smoking initiation or quantity46; however, the estimates from the included studies showed opposite directions, resulting in significant heterogeneity. The mechanisms behind these findings need further investigation. Finally, no genetic associations of LDL cholesterol, apoB, TG, alcohol consumption, or diabetes with risk of AD were observed.
Despite the confirmation of the association between longer educational attainment and low AD risk in EADB participants with clinically diagnosed AD, the association counterintuitively reversed when including participants with proxy AD. There are significant differences and genetic heterogeneity in the associations between education and clinically diagnosed AD and a self-reported proxy phenotype.47 A possible explanation may be that the status of a proxy AD diagnosis may be associated with the person’s educational level, which most likely is influenced by their parental educational level. Thus, the selected instrumental variables were associated with parental educational attainment through the genetic variants for parental education, violating the independence assumption of the MR design. This may also apply to other modifiable factors that are associated with education, such as LDL cholesterol and BMI, as manifested by their associations with AD becoming stronger when including proxy AD diagnoses.
The main strength of this study is the use of the largest genomic consortia to date, yielding ample statistical power and instrumental variables explaining much phenotypic variation. The mixed definition of AD in the consortia allowed us to explore the potential influence of proxy AD on the associations between behavioral risk factors and AD. Furthermore, several MR sensitivity analyses were performed to account for bias related to study design. The statistical correction for sample overlap between exposure and end point data, as well as the possibility to use the EADB data excluding the entire UKB, enabled us to produce robust findings.
Limitations
This study has some limitations. An inherent limitation of our study is that the included genetic studies predominantly consist of individuals of European ancestry, which limits the extrapolation of our findings to individuals of other ethnicities. Moreover, to our knowledge, all MR studies performed to date take advantage of genetic variants that are associated with 1-time measurement of the exposure. The associations of some risk factors at midlife and late life, such as BMI, have been contradictory. This may also explain the associations of high BMI with lower AD risk both in previous MR studies and in our MR study. Several observational studies have examined risk factor trajectories throughout the life course, capturing a more complete picture and representing the associations of time-varying factors, which might be more relevant than a single point measurement in examining risk of AD. However, no additional analyses could be performed due to the lack of trajectory GWAS.
Conclusions
This genetic association study found novel genetic associations between high HDL cholesterol concentrations and high SBP with higher risk of AD. These findings may inspire new drug targeting and improved early dementia prevention.
eMethods.
eAppendix 1. Supplementary Results
eAppendix 2. Supplementary Discussion
eFigure 1. The Concept of Mendelian Randomization Design
eFigure 2. Statistical Power of Each Exposure in the EADB Consortium
eFigure 3. Associations of Genetically Determined Modifiable Risk Factors and AD in the EADB-Proxy Data Set
eFigure 4. Directed Acyclic Graph Illustration on the Association Between Educational Attainment and Proxy-AD
eFigure 5. Associations of Genetically Determined Modifiable Risk Factors and AD in the EADB-Diagnosed Data Set Excluding SNVs From Chromosome 19
eFigure 6. Associations of Genetically Determined Modifiable Risk Factors and AD in the EADB-Proxy Data Set Excluding SNVs From Chromosome 19
eTable 1. Previous Main Mendelian Randomization Studies on Modifiable Risk Factors and AD Based on Summary Statistics
eTable 2. Results From CAUSE Mendelian Randomization
eTable 3. Association of Genetic Predisposition to High Risk of AD and Behavioral Risk Factors
eAppendix 3. Acknowledgements for EADB Cohorts
eReferences
eTable 1. Mendelian Randomization Results From EABD-Diagnosed Data Set
eTable 2. Information of Genetic Instrumental Variables for High-Density Lipoprotein Cholesterol
eTable 3. Mendelian Randomization Results From EADB Dataset Excluding the Entire UKB
Data Sharing Statement
References
- 1.Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396(10248):413-446. doi: 10.1016/S0140-6736(20)30367-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guidelines WHO; Approved by the Guidelines Review Committee . Risk Reduction of Cognitive Decline and Dementia: WHO Guidelines. World Health Organization; 2019. [PubMed] [Google Scholar]
- 3.Chowdhary N, Barbui C, Anstey KJ, et al. Reducing the risk of cognitive decline and dementia: WHO recommendations. Front Neurol. 2022;12:765584. doi: 10.3389/fneur.2021.765584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh-Manoux A, Dugravot A, Shipley M, et al. Obesity trajectories and risk of dementia: 28 years of follow-up in the Whitehall II Study. Alzheimers Dement. 2018;14(2):178-186. doi: 10.1016/j.jalz.2017.06.2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abell JG, Kivimäki M, Dugravot A, et al. Association between systolic blood pressure and dementia in the Whitehall II cohort study: role of age, duration, and threshold used to define hypertension. Eur Heart J. 2018;39(33):3119-3125. doi: 10.1093/eurheartj/ehy288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richmond RC, Davey Smith G. Mendelian randomization: concepts and scope. Cold Spring Harb Perspect Med. 2022;12(1):a040501. doi: 10.1101/cshperspect.a040501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellenguez C, Küçükali F, Jansen IE, et al. ; EADB; GR@ACE; DEGESCO; EADI; GERAD; Demgene; FinnGen; ADGC; CHARGE . New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54(4):412-436. doi: 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okbay A, Wu Y, Wang N, et al. ; 23andMe Research Team; Social Science Genetic Association Consortium . Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals. Nat Genet. 2022;54(4):437-449. doi: 10.1038/s41588-022-01016-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson TG, Sanderson E, Palmer TM, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable mendelian randomisation analysis. PLoS Med. 2020;17(3):e1003062. doi: 10.1371/journal.pmed.1003062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yengo L, Sidorenko J, Kemper KE, et al. ; GIANT Consortium . Meta-analysis of genome-wide association studies for height and body mass index in ~700000 individuals of European ancestry. Hum Mol Genet. 2018;27(20):3641-3649. doi: 10.1093/hmg/ddy271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu M, Jiang Y, Wedow R, et al. ; 23andMe Research Team; HUNT All-In Psychiatry . Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51(2):237-244. doi: 10.1038/s41588-018-0307-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evangelou E, Warren HR, Mosen-Ansorena D, et al. ; Million Veteran Program . Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50(10):1412-1425. doi: 10.1038/s41588-018-0205-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vujkovic M, Keaton JM, Lynch JA, et al. ; HPAP Consortium; Regeneron Genetics Center; VA Million Veteran Program . Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet. 2020;52(7):680-691. doi: 10.1038/s41588-020-0637-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burgess S. Sample size and power calculations in mendelian randomization with a single instrumental variable and a binary outcome. Int J Epidemiol. 2014;43(3):922-929. doi: 10.1093/ije/dyu005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1-2):279-284. doi: 10.1016/S0166-4328(01)00297-2 [DOI] [PubMed] [Google Scholar]
- 16.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304-314. doi: 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512-525. doi: 10.1093/ije/dyv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693-698. doi: 10.1038/s41588-018-0099-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burgess S, Thompson SG. Multivariable mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181(4):251-260. doi: 10.1093/aje/kwu283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrison J, Knoblauch N, Marcus JH, Stephens M, He X. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat Genet. 2020;52(7):740-747. doi: 10.1038/s41588-020-0631-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mounier N, Kutalik Z. Bias correction for inverse variance weighting mendelian randomization. bioRxiv. Preprint posted online September 20, 2022. doi: 10.1101/2021.03.26.437168 [DOI] [PubMed]
- 22.Nordestgaard LT, Christoffersen M, Lauridsen BK, et al. Long-term benefits and harms associated with genetic cholesteryl ester transfer protein deficiency in the general population. JAMA Cardiol. 2022;7(1):55-64. doi: 10.1001/jamacardio.2021.3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peloso GM, van der Lee SJ, Destefano AL, Seshardi S; International Genomics of Alzheimer’s Project (IGAP) . Genetically elevated high-density lipoprotein cholesterol through the cholesteryl ester transfer protein gene does not associate with risk of Alzheimer’s disease. Alzheimers Dement (Amst). 2018;10:595-598. doi: 10.1016/j.dadm.2018.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrews SJ, Goate A, Anstey KJ. Association between alcohol consumption and Alzheimer’s disease: a mendelian randomization study. Alzheimers Dement. 2020;16(2):345-353. doi: 10.1016/j.jalz.2019.09.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang SY, Yang YX, Zhang YR, et al. Investigating causal relations between circulating metabolites and Alzheimer’s disease: a mendelian randomization study. J Alzheimers Dis. 2022;87(1):463-477. doi: 10.3233/JAD-220050 [DOI] [PubMed] [Google Scholar]
- 26.Kjeldsen EW, Thomassen JQ, Juul Rasmussen I, Nordestgaard BG, Tybjærg-Hansen A, Frikke-Schmidt R. Plasma high-density lipoprotein cholesterol and risk of dementia: observational and genetic studies. Cardiovasc Res. 2022;118(5):1330-1343. doi: 10.1093/cvr/cvab164 [DOI] [PubMed] [Google Scholar]
- 27.Larsson SC, Traylor M, Malik R, Dichgans M, Burgess S, Markus HS; CoSTREAM Consortium, on behalf of the International Genomics of Alzheimer’s Project . Modifiable pathways in Alzheimer’s disease: mendelian randomisation analysis. BMJ. 2017;359:j5375. doi: 10.1136/bmj.j5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Østergaard SD, Mukherjee S, Sharp SJ, et al. ; Alzheimer’s Disease Genetics Consortium; GERAD1 Consortium; EPIC-InterAct Consortium . Associations between potentially modifiable risk factors and Alzheimer disease: a mendelian randomization study. PLoS Med. 2015;12(6):e1001841. doi: 10.1371/journal.pmed.1001841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Proitsi P, Lupton MK, Velayudhan L, et al. ; Alzheimer’s Disease Neuroimaging Initiative; GERAD1 Consortium . Genetic predisposition to increased blood cholesterol and triglyceride lipid levels and risk of Alzheimer disease: a mendelian randomization analysis. PLoS Med. 2014;11(9):e1001713. doi: 10.1371/journal.pmed.1001713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lord J, Jermy B, Green R, et al. Mendelian randomization identifies blood metabolites previously linked to midlife cognition as causal candidates in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2021;118(16):e2009808118. doi: 10.1073/pnas.2009808118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q, Xu F, Wang L, Zhang WD, Sun CQ, Deng HW. Detecting potential causal relationship between multiple risk factors and Alzheimer’s disease using multivariable mendelian randomization. Aging (Albany NY). 2020;12(21):21747-21757. doi: 10.18632/aging.103983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koch S, Donarski N, Goetze K, et al. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res. 2001;42(7):1143-1151. doi: 10.1016/S0022-2275(20)31605-9 [DOI] [PubMed] [Google Scholar]
- 33.Hubin E, Verghese PB, van Nuland N, Broersen K. Apolipoprotein E associated with reconstituted high-density lipoprotein-like particles is protected from aggregation. FEBS Lett. 2019;593(11):1144-1153. doi: 10.1002/1873-3468.13428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez AE, Weissberger G, Kuklenyik Z, et al. The small HDL particle hypothesis of Alzheimer’s disease. Alzheimers Dement. 2023;19(2):391-404. doi: 10.1002/alz.12649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zanoni P, Khetarpal SA, Larach DB, et al. ; CHD Exome+ Consortium; CARDIoGRAM Exome Consortium; Global Lipids Genetics Consortium . Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science. 2016;351(6278):1166-1171. doi: 10.1126/science.aad3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuller LH, Snitz BE, Hughes TM, et al. Low untreated systolic blood pressure over 18 years is associated with survival free of dementia age 90. Alzheimers Dement. 2022;18(11):2176-2187. doi: 10.1002/alz.12493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiu C, von Strauss E, Fastbom J, Winblad B, Fratiglioni L. Low blood pressure and risk of dementia in the Kungsholmen project: a 6-year follow-up study. Arch Neurol. 2003;60(2):223-228. doi: 10.1001/archneur.60.2.223 [DOI] [PubMed] [Google Scholar]
- 38.Walker KA, Sharrett AR, Wu A, et al. Association of midlife to late-life blood pressure patterns with incident dementia. JAMA. 2019;322(6):535-545. doi: 10.1001/jama.2019.10575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hughes D, Judge C, Murphy R, et al. Association of blood pressure lowering with incident dementia or cognitive impairment: a systematic review and meta-analysis. JAMA. 2020;323(19):1934-1944. doi: 10.1001/jama.2020.4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peters R, Xu Y, Fitzgerald O, et al. ; Dementia Risk Reduction (DIRECT) Collaboration . Blood pressure lowering and prevention of dementia: an individual patient data meta-analysis. Eur Heart J. 2022;43(48):4980-4990. doi: 10.1093/eurheartj/ehac584 [DOI] [PubMed] [Google Scholar]
- 41.Andrews SJ, Fulton-Howard B, O’Reilly P, Marcora E, Goate AM; Collaborators of the Alzheimer’s Disease Genetics Consortium . Causal associations between modifiable risk factors and the Alzheimer’s phenome. Ann Neurol. 2021;89(1):54-65. doi: 10.1002/ana.25918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sproviero W, Winchester L, Newby D, et al. High blood pressure and risk of dementia: a two-sample mendelian randomization study in the UK Biobank. Biol Psychiatry. 2021;89(8):817-824. doi: 10.1016/j.biopsych.2020.12.015 [DOI] [PubMed] [Google Scholar]
- 43.Li C, Zhu Y, Ma Y, Hua R, Zhong B, Xie W. Association of cumulative blood pressure with cognitive decline, dementia, and mortality. J Am Coll Cardiol. 2022;79(14):1321-1335. doi: 10.1016/j.jacc.2022.01.045 [DOI] [PubMed] [Google Scholar]
- 44.Brenowitz WD, Zimmerman SC, Filshtein TJ, et al. Extension of mendelian randomization to identify earliest manifestations of Alzheimer disease: association of genetic risk score for Alzheimer disease with lower body mass index by age 50 years. Am J Epidemiol. 2021;190(10):2163-2171. doi: 10.1093/aje/kwab103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nordestgaard AT, Nordestgaard BG, Frikke-Schmidt R, Juul Rasmussen I, Bojesen SE. Self-reported and genetically predicted coffee consumption and smoking in dementia: a mendelian randomization study. Atherosclerosis. 2022;348:36-43. doi: 10.1016/j.atherosclerosis.2022.03.022 [DOI] [PubMed] [Google Scholar]
- 46.Larsson SC, Burgess S. Appraising the causal role of smoking in multiple diseases: a systematic review and meta-analysis of mendelian randomization studies. EBioMedicine. 2022;82:104154. doi: 10.1016/j.ebiom.2022.104154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu H, Hu Y, Zhang Y, et al. Mendelian randomization highlights significant difference and genetic heterogeneity in clinically diagnosed Alzheimer’s disease GWAS and self-report proxy phenotype GWAX. Alzheimers Res Ther. 2022;14(1):17. doi: 10.1186/s13195-022-00963-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods.
eAppendix 1. Supplementary Results
eAppendix 2. Supplementary Discussion
eFigure 1. The Concept of Mendelian Randomization Design
eFigure 2. Statistical Power of Each Exposure in the EADB Consortium
eFigure 3. Associations of Genetically Determined Modifiable Risk Factors and AD in the EADB-Proxy Data Set
eFigure 4. Directed Acyclic Graph Illustration on the Association Between Educational Attainment and Proxy-AD
eFigure 5. Associations of Genetically Determined Modifiable Risk Factors and AD in the EADB-Diagnosed Data Set Excluding SNVs From Chromosome 19
eFigure 6. Associations of Genetically Determined Modifiable Risk Factors and AD in the EADB-Proxy Data Set Excluding SNVs From Chromosome 19
eTable 1. Previous Main Mendelian Randomization Studies on Modifiable Risk Factors and AD Based on Summary Statistics
eTable 2. Results From CAUSE Mendelian Randomization
eTable 3. Association of Genetic Predisposition to High Risk of AD and Behavioral Risk Factors
eAppendix 3. Acknowledgements for EADB Cohorts
eReferences
eTable 1. Mendelian Randomization Results From EABD-Diagnosed Data Set
eTable 2. Information of Genetic Instrumental Variables for High-Density Lipoprotein Cholesterol
eTable 3. Mendelian Randomization Results From EADB Dataset Excluding the Entire UKB
Data Sharing Statement