

Abstract

Pancreatic lipase is one of the crucial lipolytic enzymes of the gut that actively facilitates the digestion and absorption of the dietary triglycerides and cholesteryl esters. Although it has been deemed as one of the most reliable targets for the treatment of obesity and/or dyslipidemia, to date, orlistat is the only known FDA-approved, effective, oral pancreatic lipase inhibitor available for clinical use apart from the centrally acting antiobesity agents. However, it is known to be associated with adverse gastrointestinal and renal complications. In this study, we attempted to assess the antioxidant and porcine pancreatic lipase inhibitory potentials of Ziziphus oenoplia (L.)Mill. leaves through a systematic combination of in vitro and in silico approaches. Among the four different extracts including petroleum ether extract, ethyl acetate extract, ethanolic extract, and aqueous extract obtained through successive solvent extraction, the ethyl acetate extract has outperformed the other extracts and orderly displayed competent peroxide scavenging (IC50 value: 267.30 μg/mL) and porcine pancreatic lipase inhibitory (IC50 value: 444.44 μg/mL) potentials compared to the selected reference compounds: ascorbic acid (IC50 value: 251.50 μg/mL) and orlistat (IC50 value: 502.51 μg/mL) in the selected in vitro assay models. In addition, based on the molecular docking simulations of the six essential phytoconstituents of the leaves of Ziziphus oenoplia (L.)Mill. and their respective chemical analogues against the crystal structure of pancreatic lipase–colipase complex (PDB ID: 1LPB), four best-ranked molecules (PubChem CIDs: 15515703, 132582306, 11260294, and 44440845) have been proposed. Further, among these, the interaction potentials of the two top-ranked molecules (PubChem CIDs: 132582306 and 15515703) were analyzed through molecular dynamics (MD) simulations at a trajectory of 100 ns. Finally, absorption, distribution, metabolism, excretion, and toxicity (ADMET) parameters were theoretically predicted for all of the molecules using Swiss ADME and ADMET lab2.0. In conclusion, Ziziphus oenoplia (L.)Mill. leaves could become a prominent source for various potent bioactive compounds that may serve as prospective leads for the development of clinically cognizable pancreatic lipase inhibitors, provided their pharmacokinetic and in particular toxicity properties are thoroughly optimized.
1. Introduction
According to the World Health Organization (WHO), body mass index (BMI) values above 30.0 kg/m2 indicate obesity, whereas values between 25.0 and 29.9 kg/m2 indicate overweight.1 Despite the obscurity in the etiological background, genetic defects that affect peptide and nutrient signaling are known to be associated with obesity.2 Especially, leptin, peroxisome proliferator-activated receptor γ (PPAR γ), pro-opiomelanocortin, and agouti-related peptide are some of the principal examples.2 Besides, the modern lifestyle, which is indeed concerned with the sedentary habits, predisposes majority of the population to obesity irrespective of age and gender.2 Over the last 50 years, its prevalence has reached pandemic levels globally. Especially, regions including South Asia, Southeast Asia, Caribbean, and Southern Latin America experienced an accelerated increase in BMI.3 Obesity is also known to dramatically increase the risk for cardiovascular (myocardial infarction, stroke, and hypertension), neuropsychiatric (Alzheimer’s disease, dementia, and depression), and metabolic disorders (diabetes mellitus type II, cancers like breast, ovarian, colon, prostate, and liver).3
Apart from various endogenous hormones3 like leptin, leptin receptor, melanocortin 4 receptor, pro-opiomelanocortin, etc., lipases also play a substantial role in the lipid homeostasis and its related disorders like obesity.4 Physiologically, these facilitate the absorption of dietary fats by hydrolyzing them into simple glycerides and free fatty acids. Among different lipases, pancreatic lipase (classified as triacylglycerol esterase) is crucial for the fatty acid absorption in the intestine. The human pancreatic lipase is made up of 449 amino acids in which Ser-152 His-263 and Asp-176 constitute the typical catalytic triad, which is conserved.4 Its activity is dependent on another companion called colipase. Indeed, lipase inhibitors have been proven to be effective in both preclinical and clinical models of obesity.4
While healthy lifestyle and physical activity are the two fundamental and routine nonpharmacological approaches that influence energy imbalance in obesity, central appetite suppressants (like dexfenfluramine and phentermine) and inhibitors of intestinal fat absorption (orlistat) are the two commonly prescribed drug classes to manage obesity in the clinical practice.4 In addition, there are a wide number of plant species that are known to be promising in the treatment of obesity and its associated disorders. Some of the medicinal plants that have been recognized in the ayurvedic practice for the treatment of obesity include Artemisia indica,(5)Amaranthus spp.,(6)Salacia oblonga,(7)Salacia roxbhurgii,(7) and Garcinia indica.(7) In addition, certain phytochemical classes such as saponins (silphioside F and chikusetsusaponin), polyphenols (galangin and licochalcone A), terpenes (betulin and betulinic acid), and alkaloids (caffeine) are reported to exhibit reliable antiobesity/hypolipidemic activities.8
In addition, Ziziphus is one of the renowned genera of Rhamnaceae family with a vast number of plant species (around 135–170) in which majority of them are thorny shrubs or small trees, distributed globally in warm-temperate and subtropical regions.9 In addition, several species of Ziziphus have also been recognized to have diverse chemical and pharmacological profiles. About 165 different varieties of cyclopeptide-type alkaloids, 151 flavanoids, 31 saponins (triterpenoid and steroidal types), and 43 terpenoids were identified from various plants of this genus. Among these, cyclopeptide alkaloids are known to be predominantly distributed in a majority of the Ziziphus spp.9 Interestingly, among various parts of Ziziphus spp. that have been explored for the investigation of bioactive compounds, the leaves comprised the most common target.9 Reports from the traditional medicine revealed antipyretic, antimicrobial, antidepressant, antinociceptive, anticancer, hypolipidemic, antidiabetic, antioxidant, and anti-inflammatory properties9 with regard to this genus. Especially, Z. jujuba, Z. Nummularia, Z. Spina-christi, Z. Xylopyrus, and Z. mauritiana have been widely described in the literature.9 Despite the substantial medicinal significance of Ziziphus spp. for decades, their exact molecular mechanism, clinical utility, and toxicity profile have not been clearly delineated yet.9
Ziziphus oenoplia (L.)Mill. (Rhamnaceae) is one among those that is a straggling shrub found in the drier parts of Pakistan, Sri Lanka, India, Malaysia, and Tropical Asia. The folklore uses of its aerial parts and roots have been mentioned earlier in the era of ayurveda.10 In particular, its roots are used to cure ailments like fever, diarrhea, asthma, and ulcers. Its bark is especially useful for wound healing, and its fruits possess antidiabetic potential,11 and also used to treat gastric problems.12 The leaves exhibit wound healing,13 anti-inflammatory,13 and antihyperlipidemic10,14 activities, also known to contain some important bioactive principles including 6111-feruloylspinosin (flavanoid)15 and cyclopeptide alkaloids such as ziziphine A–F, amphibine-B, amphibine-F, and amphibine H.15
As a matter of fact, the currently marketed drugs for weight loss are known to pose a great deal of systemic adverse effects.4 Among those, the most commonly prescribed drug is orlistat,4 which is the only FDA-approved, blockbuster, oral pancreatic lipase inhibitor, that effectively opposes intestinal absorption of fats and eventually their accumulation in the adipose tissue. However, it has been reported to cause progressive gastrointestinal and renal complications,16−19 which may limit patient’s compliance. Hence, obviously there is a dire need for the development of more effective and especially safest drugs.
With the goal of promoting a healthier lifestyle, the use of herbal products has been strongly advocated in many regions of both developing and developed countries.20 It has been estimated that around 80% of the population in developing countries prefer to seek the practice of traditional medicine as a primary option for health care. In addition, herbal medicines constitute a substantial proportion of over-the-counter (OTC) products, which may be in part a reason for their demanding sales in the global market.20
In view of the above aspects and indeed lack of valid scientific reports on the lipase inhibitory properties of Ziziphus oenoplia, our study is primarily purposed to investigate and understand the pancreatic lipase inhibitory potential of Ziziphus oenoplia (L.)Mill. leaves using a combination of in vitro and in silico approaches. We hypothesize that this study shall certainly provide a strong proof-of-concept for the therapeutic significance of the selected Ziziphus species and facilitate identification of some prospective leads that could consecutively set a stage for the development of effective antiobesity drugs with improved efficacy and in particular safety.
2. Methodology
2.1. Plant Collection
Healthy leaves of Ziziphus oenoplia (L.)Mill. were collected in the month of April 2022 at a latitude of 13.630214° and a longitude of 79.398642° at Sri Venkateswara University (S V U) campus gardens in Tirupati, India. The plant material was identified and confirmed by the characteristic morphological features, and the concerned specimen bearing voucher no. SVUH-1557/1609 was deposited in the herbarium at the Department of Botany, S V U, Tirupati.
2.2. Successive Solvent Extraction and Qualitative Phytochemical Screening
The leaves were air-dried for a period of 7 days, coarsely blended into powder, and sieved. The processed powder was then successively extracted (Figure 1) with four different solvents in increasing order of their polarity (petroleum ether, ethyl acetate, ethanol, and water) through continuous active reflux for 5 h at a temperature range not exceeding the boiling point of the respective solvent. Further, the extracts were carefully collected, thoroughly dried, and stored under anhydrous conditions. The yields of all of the extracts were recorded, and they were eventually subjected to preliminary, qualitative phytochemical screening13 and in vitro biological assays.
Figure 1.
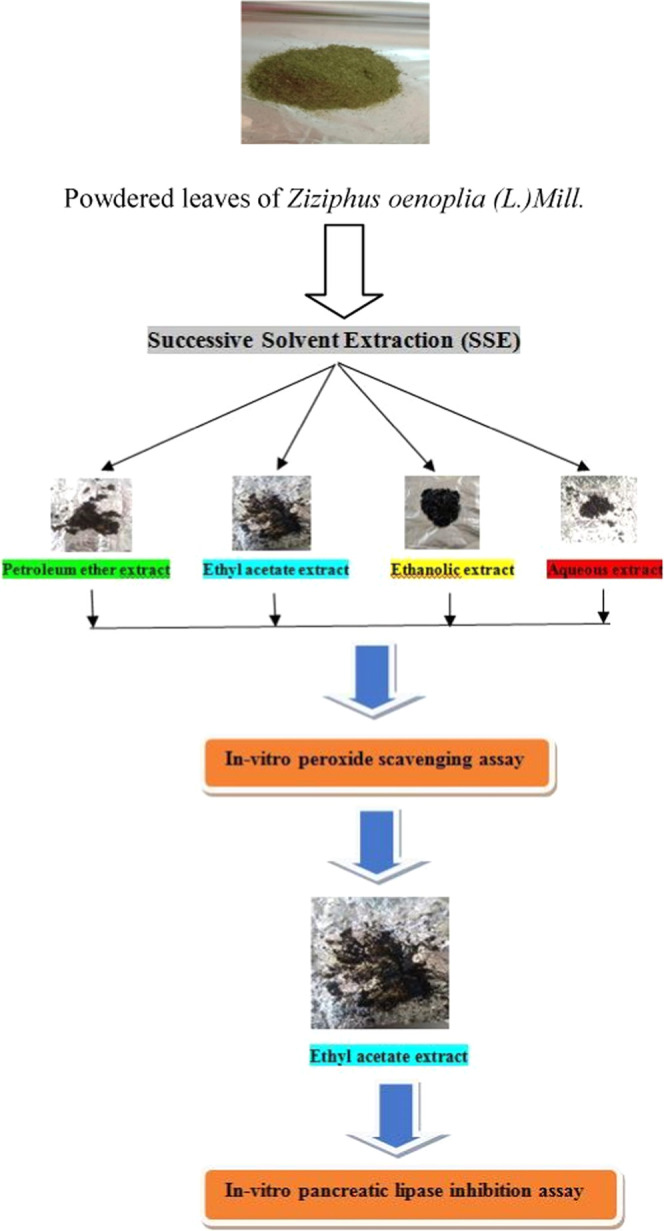
Pictorial overview of the experimental design for the assessment of in vitro pancreatic lipase inhibitory potential of Ziziphus oenoplia (L.)Mill. leaves.
2.3. In Vitro Peroxide Scavenging Assay
The assay was performed as per Bhatti et al.21 with modifications. Aliquots of different selected concentrations (100–500 μg/mL) of the extracts were transferred into the Eppendorf tubes and made up to the required volume with the phosphate buffer (pH 7.4). Further, 0.6 mL of freshly prepared hydrogen peroxide (H2O2) solution was added to the sample tubes and incubated for 10 min. Finally, the absorbance of the reaction mixture was measured at 230 nm, using the ascorbic acid as a positive control or standard. The experiment was performed in triplicate, and the percent (%) peroxide radical scavenging ability was calculated using the following formula:
where A0 is the absorbance of the control and A1 is the absorbance of the sample.
2.4. In Vitro Pancreatic Lipase Inhibition Assay
The porcine pancreatic lipase inhibitory activity was estimated using p-nitrophenyl palmitate (p-NPP) as a substrate with slight modifications of the reported method.22 The assay principle deals with the enzymatic hydrolysis of p-NPP in the reaction medium to yield p-nitrophenol, a colored end product that can be spectroscopically measured at 410 nm. The lipase (0.1 mg) was dissolved in tris-buffer (50 mM, pH 8), stirred thoroughly for 15 min, centrifuged at 2000 rpm for 10 min, and the clear supernatant was recovered and used for the study. The selected concentrations of the ethyl extract (100, 200, 400, and 800 μg/mL) were treated with 0.5 mL of the enzyme solution and incubated for 30 min at 37 °C. Subsequently, 1 mL of p-NPP (3 mM in 2-propanol) was added, and the absorbance was recorded at 410 nm. The experiment was performed in triplicate, and the percent (%) enzyme inhibition was calculated using the following formula by comparing with the positive control or standard, orlistat.
where Ac and As are the absorbance of the control and the sample, respectively.
2.5. In Silico studies
2.5.1. Selection of the Target Protein and the Ligands
The findings of the in vitro pancreatic lipase inhibition assay were further supported by in silico studies in which the crystal structure of the pancreatic lipase–colipase complex bound to the inhibitor C11 alkyl phosphonate (PDB ID: 1LPB) and having a good resolution (2.46 Å) was retrieved from the RCSB Protein Data Bank.23,24 This enzyme belongs to the class hydrolases and is composed of 449 amino acids wherein amino acids serine-152, aspartate-176, and histidine-263 (Ser–Asp–His) constitute the crucial catalytic triad that actively takes part in the breakdown of complex triglycerides into simple fatty acids.4,25
In addition, a total of six important chemical constituents (Table 1) of Ziziphus oenoplia (L.)Mill. have been identified from the literature15 and considered as lead-like molecules (natural ligands) in the present study. The three-dimensional (3D) structures of jujubogenin, amphibine H, and ziziphine N were retrieved from the PubChem database.26 However, as the 3D structures of spinosin-6111-(E)-P-coumarate, 6111-feruloylspinosin, and mucronine D are not available in the PubChem database, they were constructed manually using the Chem3D Pro 14 computational tool.27 Also, respective chemical analogues (comprising the analogue data set) of the above lead-like molecules were searched and retrieved from the PubChem database on the basis of one of Lipinski’s Ro5 criteria,28 i.e., molecular weight < 500. The 3D structure of the Orlistat (refer Table 1) was used as a positive control or standard.
Table 1. Two-Dimensional Chemical Structures of Six Important Phytochemicals (1–6) from the Leaves of Ziziphus oenoplia (L.)Mill. and Orlistat Control (7).

2.5.2. Molecular Docking Simulations
To computationally estimate the binding affinity of the selected lead-like molecules and their respective analogues, they were docked with the protein 1LPB using the PyRx Autodock vina29 tool. By means of the CASTp server,30 the active site of 1LPB was located, and the grid box of 30.8561, 30.3628, and 31.0593 Å was used, fixing the grid center at −4.6058, 21.1572, and 29.3771. For maximizing the conformational search of the docking strategy and reliably sampling the available search space, a total of eight docking solutions were generated for each molecule. Further, UCSF chimera 1.1631 and BIOVIA discovery studio 21.1.0.0 were used to visualize the protein and the key protein–ligand interactions. The binding affinity scores (in kcal/mol) of all of the test molecules were compared with the orlistat.
2.5.3. Molecular Dynamics Simulations
Molecular dynamics simulations help to ascertain the nature of the molecular interactions and favorable conformations (poses) with regard to the protein–ligand complexes.32 To analyze the physical dynamic motions of the crucial active-site protein atoms, the molecular topology and force-field parameters were generated using Amber Tools 21. GROMACS-version 2021.333 was used to perform the MDS on the protein structure 1LPB and its top-ranked two complexes. For energetically mapping the macromolecules through a 100 ns simulation, the GROMACS-AMBER99 force field was employed. Through the TIP3P water model as a solvent over the dodecahedron simulation box, the structures were neutralized by adding 0.15 M of sodium chloride. The steepest descent method was used to energetically relax the system, and using 100000 frames per simulation, the macromolecules were simulated at 300 K. Lastly, the molecular trajectories of the protein and its complexes were individually integrated using the leap-frog algorithm and were analyzed using the encoded scoring measures, viz. radius of gyration (Rg), overall and per-residue solvent accessible surface area (SASA), ligand root-mean-square deviations (RMSD), structural RMSD, and root-mean-square fluctuations (RMSFs) across the residues. These molecules have been graphically represented using Pymol and VMD.34
2.5.4. Absorption, Distribution, Metabolism, Elimination, and Toxicity (ADMET) Predictions
Several significant pharmacokinetic and toxicity parameters were theoretically estimated for all of the molecules using Swiss ADME35 and ADMETlab 2.0,36 while taking into account the likelihood of susceptibility to the undesired pharmacokinetics and toxicity. In addition, the drug-likeness was appraised by considering a set of six important physicochemical properties including lipophilicity as (LIPO), size (SIZE), polarity as topological polar surface area (POLAR), water solubility as per logS scale (INSOLU), flexibility based on the number of the rotatable bonds (FLEX), and saturation as per the fraction of SP3 carbons (INSATU) and plotted as bioavailability radars.
The above tools especially facilitate the theoretical estimation of physicochemical properties, drug-likeness, medicinal chemistry, pharmacokinetics, and toxicity parameters with a better input efficiency along with accuracy and robustness in the interpretation.
3. Results and Discussion
Based on the literature evidence, various species of Ziziphus are known to have a great deal of therapeutic benefits owing to the presence of a diverse array of bioactive phytochemicals.37 Around 431 compounds have been identified from various plant parts (including seeds, fruits, leaves, stems, and root bark) of Ziziphus spp., and most of them are alkaloids and flavonoids.37,38Ziziphus oenoplia (L.)Mill. (Rhamnaceae) is one of the important medicinal plants of Ziziphus genera with an incredible pharmacological profile exhibiting antimicrobial, antidiabetic, hypolipidemic,anti-inflammatory, immunomodulatory, anticancer, hepatoprotective, wound healing, and antiulcer activities.39 In the present study, the petroleum ether, ethyl acetate, ethanolic, and water were used as solvents to prepare respective extracts of Ziziphus oenoplia (L.)Mill. leaves, and the proposed bioactivity was successfully assessed at a preliminary level using a combination of in vitro and in silico approaches,with a credible outcome.
3.1. Successive Solvent Extraction and Qualitative Phytochemical Screening
The yields of the extracts including petroleum ether (0.840%), ethyl acetate (0.890%), ethanolic (1.536%), and aqueous (0.906%) were expressed as % total dry weight of the crude plant material. The preliminary, qualitative phytochemical screening of the extracts revealed the presence of alkaloids, glycosides, flavonoids, saponins, and triterpenoids, as enlisted in Table 2.
Table 2. Preliminary Qualitative Phytochemical Tests Performed on the Extracts Obtained Through Successive Solvent Extraction of the Powdered Leaves of Ziziphus oenoplia (L.)Mill.a.
| S. no. | Test | Petroleum ether extract | Ethyl acetate extract | Ethanolic extract | Aqueous extract |
|---|---|---|---|---|---|
| 1. | Test for Alkaloids: | +++ | +++ | +++ | +++ |
| 2. | Test for Glycosides: | – – – | – – – | +++ | +++ |
| 3. | Test for Flavonoids: | – – – | – – – | +++ | +++ |
| 4. | Test for Saponins: | – – – | – – – | +++ | +++ |
| 5. | Test for Steroids/ Triterpenoids: | +++ | +++ | – – – | – – – |
The symbol “+++” represents a positive reaction and “– – –“represents a negative reaction.
3.2. In Vitro Peroxide Scavenging Assay
The antioxidant activity of all of the extracts was evaluated in vitro by hydrogen peroxide scavenging assay. In this study, different extracts of Ziziphus oenoplia (L.)Mill. were tested for the peroxide scavenging ability at varying concentrations (100–500 μg/mL). All of the extracts displayed a concentration-dependent peroxide scavenging ability, as evident from Figure 2. At the maximum selected concentration (500 μg/mL), the petroleum ether, ethyl acetate, ethanolic, and aqueous extracts orderly showed 75.12 ± 0.0017, 93.52 ± 0.0053, 65.92 ± 0.0039, and 86.18 ± 0.1475% scavenging abilities, respectively. Ascorbic acid was used as a standard whose % scavenging ability has been found to be 99.36 ± 0.2101% at 500 μg/mL with an IC50 value of 251.50 μg/mL, whereas the IC50 values of petroleum ether, ethyl acetate, ethanolic, and aqueous extracts are found to be 332.8, 267.3, 379.2, and 290 μg/mL, respectively. Among all of the extracts, ethyl acetate extract exhibited a significant scavenging ability at the maximum concentration (93.52 ± 0.00532% at 500 μg/mL) with an IC50 of 267.3 μg/mL, competing very closely with the ascorbic acid.
Figure 2.

The percent (%) peroxide radical scavenging ability of the different extracts of Ziziphus oenoplia (L.)Mill. leaves in comparison with ascorbic acid as a standard. Data represent the results of the experiment done in triplicate, n = 3. The values indicate the mean ± standard deviation of the three independent experiments.
Figure 2 represents the scavenging ability of the extracts, which could be ranked as follows: ethyl acetate > aqueous extract > petroleum extract > ethanolic extract.
3.3. In Vitro Pancreatic Lipase Inhibition Assay
Owing to the potency of ethyl acetate extract over the other extracts in the in vitro peroxide scavenging assay, it was selected and tested further for understanding the porcine pancreatic lipase inhibitory potential. The enzyme inhibition was calculated at various selected concentrations, i.e., 100, 200, 400, and 800 μg/mL, and the extract clearly shows a concentration-dependent inhibition as illustrated in Figure 3. Interestingly, the inhibitory potential of the extract at all of the tested concentrations has been observed to be exceptionally good (82.20 ± 1.14, 87.00 ± 0.60, 88.50 ± 0.14, and 90.00 ± 1.07%, respectively) compared to that of the orlistat (75.25 ± 0.09, 77.19 ± 0.14, 78.59 ± 0.07, and 79.60 ± 0.08%, respectively). At a maximum concentration of 800 μg/mL, the extract showed 90.00 ± 1.07% inhibition with an IC50 value of 444.44 μg/mL, whereas orlistat showed only 79.60 ± 0.08% inhibition with an IC50 value of 502.51 μg/mL. This unequivocally corroborates the potency of the ethyl acetate extract over the orlistat.
Figure 3.

Effect of different concentrations of ethyl acetate extract of Ziziphus oenoplia (L.)Mill. leaves on porcine pancreatic lipase (% enzyme inhibition) in comparison with the orlistat as a standard. Data represent the results of the experiment done in triplicate, n = 3. The values indicate the mean ± standard deviation of the three independent experiments.
3.4. In Silico studies
3.4.1. Molecular Docking Simulations
Molecular docking simulations were successfully carried out using the PyRx Autodock tool for both lead-like molecules and their respective chemical analogues against the selected protein target 1LPB (Figure 4A). Among a total of eight solutions (poses) that were generated for each molecule, the poses with the lowest conformational energy and highest binding affinity score (indicated by the most negative value) were considered. Based on the binding affinity scores (in kcal/mol) of the lead-like molecules (Table 3), the 3D structures of a total of 44 chemical analogues concerning the best five lead-like molecules (including Pubchem CIDs 15515703, 51029223, 44258335, 5373023, and 21597353) were retrieved successfully from the PubChem database search. The binding affinity scores of these chemical analogues are enlisted in descending order as shown in Table S1. However, since the binding affinity score of Ziziphine N is relatively poor (−7.7 kcal/mol) when compared to the remaining lead-like molecules, the chemical analogues of Ziziphine N have not been considered and included in the present study. The difference among the binding affinity scores of most of the analogues can be ruled out as they fall approximately in the same range (−8.8 to 8.0). However, there are a few molecules whose scores fall above (Pubchem CID 132582306) and below (Pubchem CIDs 21603474, 10454451, 131752176, 6451798, 5318659, 44257871, 5370466, 162907626, 85362951, 162901393, 44258317, 16037498, 1102158311, 24806298, 4419068, and 53589131) this range (refer Table S1).
Figure 4.


(A) Three-dimensional (3D) representation of the pancreatic lipase–colipase complex (PDB ID: 1LPB) where the blue region encompasses the catalytic triad. (B) and (C) Three-dimensional images of the two top-ranked molecules (represented in a stick/framework model) 132582306 and 15515703 that are bound at a site different and distant (allosteric) from the catalytic triad comprising serine-152, aspartate-176, and histidine-263. There is a slight variation in the distance (in Å) between the two ligands and the catalytic triad, where the distance between the 132582306-catalytic triad is found to be 26.303 Å, while the 15515703-catalytic triad is found to be 21.934 Å. (D) and (E) Two-dimensional interaction maps illustrating the crucial atomic contacts between the top-ranked molecules (132582306 and 15515703) and the protein residues. The colored broken lines represent the conventional hydrogen-bonding interactions.
Table 3. Details of the Selected Lead-like Molecules and Orlistat in Descending Order of Their Binding Affinity Scores (in kcal/mol) Confirmed Through Docking Analysis.
| S. no | PubChem CID | Binding affinity (in kcal/mol) |
|---|---|---|
| 1 | 15515703 (Jujubogenin) | –9.0 |
| 2 | 51029223 (Amphibine H) | –8.5 |
| 3 | 44258335 (Spinosin-6111-(E)-P-coumarate) | –8.5 |
| 4 | 5373023 (Mucronine D) | –8.3 |
| 5 | 21597353 (6111-feruloylspinosin) | –8.1 |
| 6 | 5273923 (Ziziphine N) | –7.7 |
| 7 | Orlistat | –5.7 |
The molecules 132582306 (amphibine H analogue) and 15515703 (jujubogenin) have found to exhibit the highest binding affinity scores (−9.3 and −9.0 kcal/mol) among the chemical analogues and the lead-like molecules.
Figures 4B, and C apparently reveal that these two molecules are bound at an allosteric site that is 26.303 Å (Pubchem CID 132582306) and 21.934 Å (Pubchem CID 15515703) away from the catalytic site principally via conventional hydrogen bonding (as depicted in Figure 4D,E). This scenario might eventually lead to negative modulation of the catalytic property to a considerable extent. Surprisingly, the binding affinity scores of most of the molecules are relatively predominant compared to the orlistat (−5.7 kcal/mol). As per Table S1, the best three molecules from the analogue data set are Pubchem CID 132582306 (Amphibine H analogue), Pubchem CID 11260294 (Jujubogenin analogue), and Pubchem CID 44440845 (6111-feruloylspinosin analogue) respectively.
3.4.2. Molecular Dynamics Simulations
When a molecule is forced to fit into a target protein, the conformational changes that take place can be studied using a reliably accurate method called molecular dynamics simulations. For the simulated protein/protein–ligand complex, the conformational stability is examined across the simulation trajectory in nanoseconds (ns). The conformational stability against any physicochemical strain is estimated using Rg or the average deviation between the center of mass and the rotating axis.40 Here, the two top-ranked molecules (Pubchem CIDs 132582306 and 15515703) have successfully converged within the molecular dynamics simulations window of 100 ns. Besides few residues, the average Rg score for the 1LPB-15515703 complex is 2.696 Å as opposed to 2.721Å for the 1LPB-132582306 complex (Figure 5A), which is quite similar to the value of 2.705 Å for 1LPB. It indicates that 1LPB becomes a bit more compact structure after binding with these molecules.
Figure 5.

Molecular dynamics simulations analysis of 1LPB and its 15515703 and 132582306 complexes for (A) radius of gyration, (B) ligand-RMSD, (C) ligand RMSF, (D) number of hydrogen bonds, and (E) internal energy scores.
Another useful metric for determining the overall conformational divergence and structural stability at a given temperature is the RMSD.41 The average RMSD score for the native 1LPB is 0.18 Å between 0 and 0.324 Å, as opposed to the two complexes with respective scores of 0.196 Å between 0 and 0.29 Å and 0.203 Å from 0 to 0.313 Å (Figure 5B). This indicates a slightly higher stability of the 1LPB-15515703 complex. The macromolecular stability can also be estimated reliably using RMSF-scoring undulations, and its lower score indicates a greater stability.41 The protein and its two complexes show an equivalent RMSF variation across the trajectory (Figure 5C). While the RMSF of the native protein lies within 0.043–0.425Å, variations for the two complexes orderly lie within 0.04–0.338 and 0.04–0.458 Å, with a corresponding average score of 0.109, 0.099, and 0.106 Å. This probably implies that the former complex formation stabilizes the atomic fluctuations of 1LPB, showing that it could be a potentially better substrate for pancreatic lipase.
Further, the number of hydrogen bonds was plotted for the native protein and its complexes to note down their variations across the trajectory.41 The two molecules orderly showed a variation within 0–4 and 0–3, with a corresponding average of 1.298 and 0.596. It further affirms a higher credibility of the molecule 15515703 to be a prospective pancreatic lipase inhibitor (Figure 5D). The plot clearly indicates a more stable interaction of this molecule with 1LPB throughout the trajectory. Since appropriate anchoring of the molecule within the active site is predominantly driven by hydrogen and hydrophobic bonds, this might have resulted in a stronger interaction.41
To excavate these interactions, the free interaction energies of the binding of 1LPB to the selected molecules were estimated using the Parrinello–Rahman parameter, and it has been observed that the 1LPB-15515703 complex shows higher stability throughout the period of 100 ns simulation (Figure 5E).
In contrast to the molecule (Pubchem CID132582306), which has an interaction energy score of −20.058 KJ, the molecule (Pubchem CID 15515703) has been found to interact (with 1LPB) with an average interaction energy of −54.614 KJ. The detailed chemical interactions between various amino acid residues of 1LPB and each of the two molecules (Pubchem CIDs 15515703 and 132582306) at their binding region have been tabulated (Table 4).
Table 4. Details of the Chemical Interactions between Each of the Two Top-Ranked Molecules and the Amino Acid Residues at the Binding Site of 1LPB Highlighting the Bond Distance and the Bond Category.
| Amino acid residue: chain involved in the interaction | Bond distance (in Å) | Bond Category |
|---|---|---|
| PubChem CID: 15515703 | ||
| His 30: A | 3.3837 | Conventional hydrogen bond |
| Arg 38: A | 4.7492 | Hydrophobic |
| Ala 40: A | 5.4679 | Hydrophobic |
| Ile 248: B | 4.9569 | Hydrophobic |
| Leu 41: A | 5.0606 | Hydrophobic |
| Ile 248: B | 5.4502 | Hydrophobic |
| Arg 38: A | 3.7957 | Hydrophobic |
| PubChem CID: 132582306 | ||
| Lys 24: A | 2.1324 | conventional hydrogen bond |
| Arg 65: A | 4.0454 | electrostatic (Pi-cation) |
| Tyr 369: B | 4.9796 | hydrophobic (Pi- Pi, T-shaped) |
| Lys 367: B | 4.4472 | hydrophobic |
| Leu 41: A | 4.5674 | hydrophobic |
| Lys 42: A | 5.2725 | hydrophobic |
| Lys 42: A | 4.5527 | hydrophobic |
| Arg 65: A | 5.2355 | hydrophobic |
3.4.3. In Silico Absorption, Distribution, Metabolism, Elimination, and Toxicity (ADMET) Predictions
The selected best five lead-like molecules and 44 respective chemical analogues are fed to the Swiss ADME tool for predicting various pharmacokinetic parameters in silico (Tables 5–7). The parameters predicted include molecular weight, hydrogen bonding, molar refractivity (M R), consensus log Po/w (C log Po/w), gastrointestinal absorption (GIA), blood–brain barrier permeability (B3P), Cytochrome P450 3A4 (CYP3A4) inhibitor, P-glycoprotein substrate (P-gp s), Lipinski’s and Veber’s criteria, and bioavailability score (B.S). Except the top-ranked lead-like molecule 15515703 (jujubogenin) with a molecular weight of 472.70 g/mol, molecular weights of the remaining four lead-like molecules fall beyond 500 g/mol. The M R values of all of the five molecules are beyond the acceptable range (20–70). The C log Po/w is one of the important physicochemical parameters expressed as the logarithm of partition coefficient between n-octanol and water. It indicates the compound’s lipophilicity and also provides an estimate of intestinal absorption and/or permeability. In general, Lipinski’s rule (Ro5)28 emphasizes that the ideal range of log P is ≤ 5 to ≥ 0, which is most desirable for the bioavailability of orally administered drugs. Here, majority of the molecules are found to lie within this range with only a few exceptions (including molecules 5748594, 11546834, 14887606, and 5318659). The C log Po/w42 is predicted as the arithmetic mean of ilogP,43 XlogP3,44 MlogP,45,46 WlogP,47 and SILICOS-IT,48 respectively. The values of the four lead-like molecules excluding jujubogenin (whose value is 5.29) are within the range of 5, and this could be imputable to the presence of polar groups like amide (in Pubchem CIDs51029223 and 5373023) and polyhydroxy types (in Pubchem CIDs 44258335 and 21597353) that consequently impart polarity to them over 15515703. However, their solubility is moderate (Pubchem CIDs 44258335 and 21597353) to poor (Pubchem CIDs 5373023 and 51029223) due to their higher molecular weights (exceeding 500 g/mol) compared to jujubogenin (472.70 g/mol), which is nonpolar with poor solubility. The solubility is predicted as LogS by considering three important models including ESOL,49 Ali et al.,50 and SILICOS-IT.48 Actually, it is one of the important physicochemical properties that significantly affect the absorption process and enables the formulation of parenteral dosage forms. The passive gastrointestinal absorption and B3P were predicted based on brain or intestinal estimated permeation method (BOILED-Egg),51 wherein three (Pubchem CIDs 15515703, 5373023, and 51029223) of them showed high gastrointestinal absorption and none of them has B3P. Out of the six crucial metabolic isozymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP2E1, and CYP3A4) of the cytochrome P450 system, CYP3A4 has a significant role in the biotransformation of several xenobiotics and drug–drug interaction mechanisms.52 These are widely distributed in the liver and other extra-hepatic tissues like kidney, skin, intestine, and lungs.52 Swiss ADME facilitates theoretically predicting whether the test molecules could act as CYP and P-gp substrates or not through a support vector machine (SVM) algorithm.53 The output generates as either “yes” or “no” based on the tendency of the test molecules to act as substrates or nonsubstrates of various CYP isozymes and P-gp.
Table 5. In Silico ADME Prediction of the Best Five Lead-like Molecules.
| s. no. | PubChem CID | M.W (g/mol) | no. of H-bond acceptors and donors | M.R | C log Po/w | solubility | GIA | B3P | CYP3A4 inhb | P-gpS | Lipinski’s (violations) | Veber’s (violations) | B.S | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 15515703 | 472.70 | 04 | 02 | 136.70 | 5.29 | poor | high | no | no | no | yes(1) | yes | 0.55 |
| 2 | 5373023 | 661.83 | 07 | 03 | 197.47 | 3.41 | poor | high | no | yes | yes | no(2) | no(1) | 0.17 |
| 3 | 44258335 | 754.69 | 17 | 09 | 184.83 | 0.26 | moderate | low | no | no | yes | no(3) | no(2) | 0.17 |
| 4 | 51029223 | 605.70 | 07 | 03 | 178.24 | 2.21 | poor | high | no | yes | yes | no(2) | yes | 0.17 |
| 5 | 21597353 | 784.71 | 18 | 09 | 191.32 | 0.42 | moderate | low | no | no | yes | no(3) | no(2) | 0.17 |
Table 7. In Silico ADME Prediction of the Remaining Chemical Analogues.
| s. no | PubChem CID | M.W (g/mol) | no. of H-bond acceptors and donors | M.R | C log Po/w | solubility | GIA | B3P | CYP3A4 inhb | P-gpS | Lipinski’s (violations) | Veber’s (violations) | B.S | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 22288010 | 474.41 | 11 | 05 | 115.85 | 0.68 | soluble | low | no | no | yes | yes(1) | no(1) | 0.55 |
| 2 | 74977829 | 430.40 | 09 | 04 | 109.42 | 1.20 | soluble | high | no | yes | yes | yes | yes | 0.55 |
| 3 | 10454451 | 414.41 | 08 | 04 | 108.75 | 1.80 | moderate | high | no | yes | yes | yes | yes | 0.55 |
| 4 | 163020300 | 418.52 | 06 | 02 | 109.66 | 2.34 | soluble | high | no | no | yes | yes | yes | 0.55 |
| 5 | 5748594 | 464.38 | 12 | 08 | 110.16 | –0.27 | soluble | low | no | no | no | no (2) | no (1) | 0.55 |
| 6 | 12443368 | 432.38 | 10 | 06 | 106.11 | 0.31 | soluble | low | no | no | no | yes (1) | no (1) | 0.55 |
| 7 | 73981696 | 478.40 | 12 | 07 | 114.63 | 0.11 | soluble | low | no | yes | yes | no (2) | no (1) | 0.17 |
| 8 | 10095770 | 460.43 | 10 | 04 | 115.05 | 1.00 | soluble | low | no | yes | yes | yes | no (1) | 0.55 |
| 9 | 11546834 | 448.38 | 11 | 07 | 108.13 | –0.11 | soluble | low | no | no | no | no (2) | no (1) | 0.17 |
| 10 | 5321398 | 432.38 | 10 | 06 | 106.11 | 0.45 | soluble | low | no | no | no | yes (1) | no (1) | 0.55 |
| 11 | 53398699 | 458.40 | 10 | 04 | 113.83 | 0.89 | soluble | low | no | yes | yes | yes | no(1) | 0.55 |
| 12 | 44147684 | 482.44 | 10 | 05 | 120.55 | 1.59 | moderate | low | no | yes | no | yes | no (1) | 0.55 |
| 13 | 131751472 | 478.40 | 12 | 07 | 115.49 | 0.13 | soluble | low | no | yes | no | no (2) | no (1) | 0.17 |
| 14 | 14887606 | 492.39 | 13 | 07 | 115.24 | –0.29 | soluble | low | no | no | yes | no (2) | no (1) | 0.11 |
| 15 | 442725 | 492.43 | 12 | 06 | 115.89 | 0.10 | soluble | low | no | no | yes | no (2) | no (1) | 0.17 |
| 16 | 14681458 | 490.41 | 12 | 07 | 118.37 | 0.26 | soluble | low | no | no | no | no (2) | no (1) | 0.17 |
| 17 | 5918474 | 446.40 | 10 | 05 | 110.58 | 0.79 | soluble | low | no | yes | yes | yes | no (1) | 0.55 |
| 18 | 11248556 | 476.57 | 05 | 03 | 144.52 | 1.89 | moderate | high | no | yes | yes | yes | yes | 0.55 |
| 19 | 44257871 | 446.40 | 10 | 06 | 111.08 | 0.37 | soluble | low | no | no | no | yes(1) | no(1) | 0.55 |
| 20 | 53589131 | 268.35 | 05 | 01 | 80.69 | 0.89 | soluble | high | no | no | no | yes | yes | 0.55 |
| 21 | 6451798 | 436.41 | 10 | 07 | 106.14 | 0.05 | soluble | low | no | no | yes | yes (1) | no (1) | 0.55 |
| 22 | 12302035 | 478.40 | 12 | 07 | 114.63 | 0.29 | soluble | low | no | no | yes | no (2) | no (1) | 0.17 |
| 23 | 5318659 | 434.39 | 10 | 07 | 106.46 | –0.02 | soluble | low | no | no | no | yes (1) | no (1) | 0.55 |
| 24 | 14861226 | 474.41 | 11 | 05 | 116.71 | 1.16 | soluble | low | no | no | yes | yes (1) | no (1) | 0.55 |
| 25 | 159460 | 476.43 | 11 | 05 | 117.07 | 0.90 | moderate | low | no | yes | yes | yes (1) | no (1) | 0.55 |
| 26 | 21603474 | 438.43 | 10 | 06 | 108.76 | 0.56 | soluble | low | no | no | yes | yes (1) | no (1) | 0.55 |
| 27 | 44258317 | 446.40 | 10 | 06 | 111.08 | 0.26 | soluble | low | no | yes | no | yes (1) | no (1) | 0.17 |
| 28 | 85362951 | 392.36 | 09 | 06 | 94.51 | 0.11 | soluble | low | no | no | no | yes (1) | no (1) | 0.55 |
| 29 | 131752855 | 448.42 | 10 | 05 | 108.16 | 0.36 | soluble | low | no | no | yes | yes | no (1) | 0.55 |
| 30 | 162941227 | 490.59 | 05 | 03 | 149.42 | 2.20 | moderate | high | no | yes | yes | yes | yes | 0.55 |
| 31 | 102353416 | 490.60 | 05 | 03 | 149.42 | 2.20 | moderate | high | no | yes | yes | yes | yes | 0.55 |
| 32 | 134926872 | 470.56 | 06 | 02 | 138.93 | 1.34 | soluble | high | no | no | no | yes | yes | 0.55 |
| 33 | 131752176 | 448.38 | 11 | 06 | 108.66 | 0.11 | soluble | low | no | yes | no | no (2) | no (1) | 0.17 |
| 34 | 102158311 | 373.45 | 05 | 03 | 113.91 | 1.56 | soluble | high | no | no | yes | yes | yes | 0.55 |
| 35 | 14861229 | 474.41 | 11 | 05 | 116.71 | 1.11 | soluble | low | no | no | yes | yes (1) | no (1) | 0.55 |
| 36 | 44191068 | 357.40 | 05 | 02 | 107.08 | 0.91 | soluble | high | no | no | yes | yes | yes | 0.55 |
| 37 | 162901393 | 428.52 | 05 | 02 | 129.84 | 1.32 | soluble | high | no | no | yes | yes | yes | 0.55 |
| 38 | 24806298 | 475.58 | 06 | 02 | 133.94 | 2.84 | moderate | high | no | yes | yes | yes | no (1) | 0.55 |
| 39 | 5370466 | 478.58 | 05 | 03 | 142.81 | 2.32 | moderate | high | no | yes | yes | yes | yes | 0.55 |
| 40 | 162907626 | 428.48 | 06 | 02 | 124.51 | 0.26 | soluble | high | no | no | yes | yes | yes | 0.55 |
| 41 | 16037498 | 473.56 | 06 | 02 | 139.75 | 2.41 | moderate | high | no | yes | yes | yes | yes | 0.55 |
Table 6. In Silico ADME Prediction of the Best Three Chemical Analogues.
| s. no. | PubChem CID | M.W (g/mol) | no. of H-bond acceptors and donors | M.R | C log Po/w | solubility | GIA | B3P | CYP3A4 inhb | P-gpS | Lipinski’s (violations) | Veber’s (violations) | B.S | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 132582306 | 487.55 | 05 | 01 | 146.60 | 2.24 | moderate | high | no | yes | yes | yes | yes | 0.55 |
| 2 | 11260294 | 486.70 | 04 | 02 | 141.24 | 2.22 | poor | high | no | no | no | yes (1) | yes | 0.55 |
| 3 | 44440845 | 448.42 | 10 | 05 | 107.72 | 1.87 | soluble | low | no | no | yes | yes | no (1) | 0.55 |
P-gp54,55 is a vital efflux pump that protects various tissues and especially the central nervous system from the entry of harmful xenobiotics, metabolites, and toxins. As per these predictions, jujubogenin (Pubchem CID15515703) along with the two other molecules (Pubchem CIDs 44258335 and 21597353) are poor substrates for CYP3A4. Except jujubogenin, all of the four molecules are P-gp substrates. Above all, so as to empirically judge the drug-likeness on the basis of a combination of physicochemical and pharmacokinetic properties, Lipinski’s Ro528,56 and Veber’s57 criteria have been introduced. The molecules that violate these criteria (with more than one violation) are considered as noncompliant and ineligible with regard to drug-likeness and oral administration. Here, the number of violations in the case of Lipinski’s criteria for Pubchem CID 15515703 is only 1, for Pubchem CIDs 5373023 and 51029223, it is 2, and for the remaining two molecules (Pubchem CIDs 44258335 and 21597353), it is 3. In the case of Veber’s criteria, Pubchem CIDs 15515703 and 51029223 show no violations compared to the other three molecules. In addition, the bioavailability score58 has been calculated on the basis of a combination of total charge, topological polar surface area (TPSA), and Lipinski’s rule, and the ranges are classified into four categories as 11, 17, 56, or 85%. It serves as a probability predictor of at least 10% oral bioavailability in rat or measurable Caco 2 permeability. Here, jujubogenin showed a better score of 0.55 (55%) compared to the other four with a common score of 0.17 (17%).
Further, the bioavailability radar59,60 is a typical graphical representation that helps to assess the drug-likeness of molecules by taking six important physicochemical parameters (lipophilicity, flexibility, polarity, size, solubility, and saturation) into account.
The illustrative bioavailability radars in Figure 6 clarify that Pubchem CID15515703 (jujubogenin) is in better compliance with the drug-likeness criteria compared to the other four molecules due to its optimum physicochemical parameters.
Figure 6.

Bioavailability radars of the selected lead-like molecules indicating the properties related to drug-likeness. The pink section in the radars indicates the optimum range necessary to comply with the drug-likeness criteria. The images are retrieved from the Swiss ADME tool.
The M R values of all of the 44 analogues are found to be above 100, which is beyond the acceptable limit as described above (i.e., 20–70). The C log Po/w values of all of the 44 analogues fall within 5. Moreover, 25 molecules have shown values less than 1, and among them, four have exhibited negative values (Pubchem CIDs 5748594, 11546834, 14887606, and 5318659), which could be due to the exceeding polarity. Except 11260294 (poor solubility), all of the remaining molecules have shown moderate to better solubility. Around 27 analogues have shown poor GIA. A good number of analogues have been found to act as CYP3A4 inhibitors and P-gp substrates. The top-ranked analogue (Pubchem CID 132583206) has been predicted to act as a substrate for both CYP3A4 and P-gp, and 10 of them (Pubchem CIDs 11260294, 5748594, 12443368, 11546834, 14681458, 44257871, 53589131, 5318659, 85362951, and 134926872) are not CYP3A4 and P-gp substrates. In addition, none of them displayed B3P. In general, the violations of Lipinski’s and Veber’s criteria did not exceed more than 2. However, 16 molecules (Pubchem CIDs 132582306, 4440845, 74977829, 10454451, 163020300, 11248556, 53589131, 162941227, 102353416, 134926872, 102158311, 44191068, 162901393, 162907626, 5370466, and 16037498) have shown no violations for both the criteria. The B.S of Pubchem CID 14887606 is only 0.11 (11%), the score of eight molecules (Pubchem CIDs 73981696, 11546834, 131751472, 442725, 14681458, 12302035, 44258317, and131752176) is 0.17 (17%), and the score of all of the remaining molecules is 0.55 (55%).
Further, the illustrative bioavailability radar images of Figure 7 substantiate that the top-ranked analogue 132582306 shows a better compliance in terms of drug-likeness.
Figure 7.

Bioavailability radars of the best three chemical analogues indicating the properties related to drug-likeness.
The in silico toxicity predictions for all of the lead-like molecules and their analogues were performed using ADMET lab2.0 to evaluate certain toxicity-associated parameters like hERG blockage ability, AMES toxicity, rat oral acute toxicity, carcinogenicity, and respiratory toxicity. The intensity of the respective toxicity has been scored as depicted in Tables 8 and 9. The overall scores range between 0 and 1, and a score of 0.9–1.0 indicates maximum toxicity. Most of the molecules lack hERG blockage ability and show moderate-to-weak AMES toxicity indicating some tendency of mutagenicity. However, six molecules 163020300, 134926872, 162901393, 24806298, 5370466, and 16037498 lack AMES toxicity.
Table 8. In Silico Toxicity Prediction of the Top-Ranked Lead-like Molecule and the Best Three Chemical Analoguesa.
| s. no | PubChem CID | hERG blockers | AMES toxicity | Rat oral acute toxicity | Carcinogenicity | Respiratory toxicity |
|---|---|---|---|---|---|---|
| 1 | 15515703 | – – – | – – – | ++ | – – – | +++ |
| 2 | 132582306 | – | – – | +++ | +++ | – – |
| 3 | 11260294 | – – – | – – | – – | – – – | +++ |
| 4 | 44440845 | – – – | – | – – – | ++ | – – – |
The symbols represent the corresponding range of the toxicity scores 0–0.1(− – –), 0.1–0.3(− –), 0.3–0.5(−), 0.5–0.7(+), 0.7–0.9(++), and 0.9–1.0(+++).
Table 9. In Silico Toxicity Prediction of the Remaining Chemical Analoguesa.
| S. no | PubChem CID | hERG blockers | AMES toxicity | Rat oral acute toxicity | Carcinogenicity | Respiratory toxicity |
|---|---|---|---|---|---|---|
| 1 | 22288010 | – – – | – | – – – | ++ | – – – |
| 2 | 74977829 | – – – | + | – – – | – | – – – |
| 3 | 10454451 | – – – | ++ | – | – | – |
| 4 | 163020300 | – – – | – – – | +++ | + | ++ |
| 5 | 5748594 | – – | ++ | – – – | – – – | – – – |
| 6 | 44257871 | – – – | + | – – – | – – – | – – |
| 7 | 73981696 | – – – | ++ | – – – | – – – | – – – |
| 8 | 10095770 | – – | – | – – – | – – | – – – |
| 9 | 11546834 | – – | + | – – – | – – – | – – – |
| 10 | 5321398 | – – | + | – – – | – | – – – |
| 11 | 53398699 | – – – | – | – – | ++ | – – – |
| 12 | 44147684 | – – – | – | – – | + | – – – |
| 13 | 131751472 | – – – | ++ | – – – | – – | – – – |
| 14 | 14887606 | – – – | – | – – – | – – – | – – – |
| 15 | 442725 | – – | ++ | – – – | – – – | – – – |
| 16 | 14681458 | – – – | + | – – – | – – – | – – – |
| 17 | 5918474 | – – – | + | – – – | ++ | – – – |
| 18 | 11248556 | + | ++ | ++ | + | – – |
| 19 | 53589131 | – – – | + | – – – | ++ | – – – |
| 20 | 6451798 | – – | – | – – | + | – – – |
| 21 | 12302035 | – – – | ++ | – – – | – – | – – – |
| 22 | 5318659 | – – | ++ | – | + | – – – |
| 23 | 14861226 | – – – | ++ | – – – | – – | – – – |
| 24 | 159460 | – | + | – – – | – – | – – – |
| 25 | 21603474 | – – – | + | – – – | ++ | – – – |
| 26 | 44258317 | – – – | ++ | – – | – – – | – – |
| 27 | 85362951 | – – – | ++ | – – – | – | – – – |
| 28 | 131752855 | – – – | – | – – – | +++ | – – – |
| 29 | 162941227 | – – – | – | – – | ++ | – |
| 30 | 102353416 | + | ++ | ++ | + | – – |
| 31 | 134926872 | – – – | – – – | ++ | +++ | – – |
| 32 | 131752176 | – – – | ++ | – – – | – – – | – – – |
| 33 | 102158311 | + | – | + | + | ++ |
| 34 | 14861229 | – – – | + | – – – | – – | – – – |
| 35 | 12443368 | – – – | + | – – – | – – | – – – |
| 36 | 44191068 | – – – | + | + | +++ | – |
| 37 | 162901393 | – – | – – – | – | + | – – |
| 38 | 24806298 | – – – | – – – | – – | ++ | – – – |
| 39 | 5370466 | + | – – – | – – | – – – | + |
| 40 | 162907626 | – – – | ++ | – – – | – – – | – – – |
| 41 | 16037498 | – – – | – – – | – – | +++ | – – – |
The symbols represent the corresponding range of the toxicity scores 0–0.1(− – −), 0.1–0.3(− –), 0.3–0.5(−), 0.5–0.7(+), 0.7–0.9(++), and 0.9–1.0(+++).
Few molecules have been found to display moderate-to-high rat acute oral toxicity (Pubchem CIDs 132582306 and 16302030), carcinogenicity (Pubchem CIDs 132582306, 131752855, 134926872, 44191068, and 16037498), and respiratory toxicity (Pubchem CIDs 15515703, 11260294, and 163020300).
4. Conclusions and Future Recommendations
Orlistat is the one and only FDA-approved, blockbuster oral pancreatic lipase inhibitors commonly used for the treatment of typical obesity or hyperlipidemia. However, the likelihood of risk for patient noncompliance may increase as a result of rebound weight gain, gastrointestinal, and renal complications. This condition may certainly necessitate the development of therapeutic agents with the utmost safety. In addition, a vast majority of the world’s population have been relying on traditional medicine for decades in order to get cured of a variety of ailments, including obesity-associated disorders. In this study, we sought to comprehend the antioxidant and pancreatic lipase inhibitory potential of Ziziphus oenoplia (L.)Mill. by a combination of in vitro and in silico methods.
The literature findings substantiated the richness of Ziziphus oenoplia (L.)Mill. in terms of the presence of several bioactive phytochemicals, especially alkaloids, flavonoids, and terpenoids, that are known to be distributed in various parts of the plant. Particularly, leaves, stem bark, roots, and the fruits have credible therapeutic qualities. In light of this, we successfully prepared crude petroleum ether, ethyl acetate, ethanol, and aqueous extracts of the dried leaves of Ziziphus oenoplia (L.)Mill. in good yields through successive solvent extraction. All of the extracts were tested for in vitro peroxide scavenging ability among which the ethyl acetate extract has demonstrated convincing scavenging ability by nearly competing with the ascorbic acid (standard). In addition, it has also displayed exceptionally potent inhibitory effect against porcine pancreatic lipase in vitro when compared to the orlistat (standard).
Furthermore, , out of the six lead-like molecules (Pubchem CIDs 15515703, 5373023, 44258335, 51029223, 21597353, and 5273923), the best five molecules (Pubchem CIDs 15515703, 5373023, 44258335, 51029223, and 21597353) were selected on the basis of the binding affinity scores (in kcal/mol) and their respective chemical analogues (a total of 44) comprising the analogue dataset were also retrieved from the PubChem database. The binding affinities of most of the molecules in the analogue data set against the crystal structure of the selected target protein (PDB ID: 1LPB) seem to be good. Among the lead-like molecules and chemical analogues, Pubchem CIDs 15515703 and 132582306 are found to be the top-ranked molecules. In addition, the molecular dynamics simulations confirmed that the interactions of these two molecules with the selected target protein were found to be reasonably stable throughout the trajectory in which the stability of the 15515703–1LPB complex is relatively more. The binding site of these molecules is different and distant from the active catalytic triad, which is evident from Figure 4B,C.
Based on this observation, we presume that these molecules might modulate the enzymatic activity by a typical mechanism of allosteric inhibition. From the in silico ADMET predictions, it can be inferred that both molecules have optimum physicochemical properties that are required for drug-likeness. However, they have been predicted to exhibit respiratory toxicity and carcinogenicity. Apart from this, most of the remaining molecules in the data set have acceptable physicochemical properties with moderate-to-poor toxicity profiles. In conclusion, our study was successful in understanding the significant inhibitory potential of Ziziphus oenoplia (L.)Mill. leaves against porcine pancreatic lipase. We strongly believe that these preliminary findings could be further successfully validated by a systematic combination of chemical, genomic, and proteomic approaches. In fact, these studies may also facilitate the development of certain innovative Ziziphus oenoplia-based polyherbal formulations (as such types of formulations are known to be rare to date) for the treatment of obesity-related disorders with intended potency, efficacy, and safety. Moreover, the two top-ranked molecules Pubchem CIDs 15515703 and 132582306 along with the other less toxic analogues of the data set, if, diligently optimized in the laboratory settings by modern chemical approaches, could certainly set a stage for the development of distinguishable chemical libraries with promising and improved drug-like properties.
Acknowledgments
The authors are extremely grateful to the fraternity of the S V University College of Pharmaceutical Sciences, S V University, Tirupati, India, and Department of Pharmacology, Raghavendra Institute of Pharmaceutical Education and Research (RIPER), Anantapur, India, for providing the necessary laboratory facilities for our research work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c07361.
Details of the respective chemical analogues of the best five lead-like molecules in descending order of their binding affinity scores confirmed by docking analysis (Table S1) (PDF)
Author Contributions
S.R.V. has contributed to the conceptualization, data analysis, data interpretation (in vitro studies, in silico molecular docking simulations and ADMET predictions), manuscript writing and critical revision of the finalized manuscript. A.R. has performed the in silico molecular docking simulations, data analysis, data interpretation (in silico molecular dynamics simulations), and critically reviewed the finalized manuscript. S.K.R. and R.S.P.Y. have performed in vitro peroxide scavenging assay and collected the experimental data and also retrieved the target protein and PubChem compounds required for the in silico studies. P.S. has carried out the molecular dynamics simulations. P.V.L.S.B. and N.S. have interpreted the outcome of simulations. S.A. and S.R. have performed the in vitro enzyme inhibition assay and collected the experimental data. A.R.C. and S.D.S.M. have supervised the work and critically reviewed the finalized manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Mehrzad R.Definition and Introduction to Epidemiology of Obesity. In Obesity: Global Impact and Epidemiology, 1st ed.; Academic Press (Elsevier), 2020. [Google Scholar]
- George A.; Bray M. D. Etiology and pathogenesis of obesity. Clin. Cornerstone 1999, 2, 1–15. 10.1016/S1098-3597(99)90001-7. [DOI] [PubMed] [Google Scholar]
- Blüher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. 2019, 15, 288–298. 10.1038/s41574-019-0176-8. [DOI] [PubMed] [Google Scholar]
- Liu T. T.; Liu X. T.; Chen Q. X.; Shi Y. Lipase inhibitors for obesity: A Review. Biomed. Pharmacother. 2020, 128, 110314 10.1016/j.biopha.2020.110314. [DOI] [PubMed] [Google Scholar]
- Ahmad W.; Khan I.; Khan M. A.; Ahmad M.; Subhan F.; Karim N. Evaluation of antidiabetic and antihyperlipidemic activity of Artemisia indica linn (aeriel parts) in Streptozotocin induced diabetic rats. J Ethnopharmacol. 2014, 151, 618–623. 10.1016/j.jep.2013.11.012. [DOI] [PubMed] [Google Scholar]
- Girija K.; Lakshman K. Anti-hyperlipidemic activity of methanol extracts of three plants of Amaranthus in triton-WR 1339 induced hyperlipidemic rats. Asian. Pac. J. Trop. Biomed. 2011, 1, S62–S65. 10.1016/S2221-1691(11)60125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subhasree N.; Kamella A.; Kaliappan I.; Agrawal A.; Dubey G. P. Antidiabetic and antihyperlipidemic activities of a novel polyherbal formulation in high fat diet/streptozotocin induced diabetic rat model. Indian J. Pharmacol. 2015, 47, 509–513. 10.4103/0253-7613.165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunagariya N. A.; Patel N. K.; Jagtap S. C.; Bhutani K. K. Inhibitors of pancreatic lipase: state of the art and clinical perspectives. EXCLI J. 2014, 13, 897–921. [PMC free article] [PubMed] [Google Scholar]
- Maaiden E. E.; Kharrassi Y. E.; Qarah N. A. S.; Essamadi A. K.; et al. Genus Ziziphus: A comprehensive review on ethnopharmacological, phytochemical, and pharmacological properties. J. Ethnopharmacol. 2020, 259, 112950 10.1016/j.jep.2020.112950. [DOI] [PubMed] [Google Scholar]
- Shukla A.; Garg A.; Mourya P.; Jain C. P. Zizyphus oenoplia Mill: A review on Pharmacological aspects. Adv. Pharm. J. 2016, 1, 8–12. [Google Scholar]
- Goyal P. K.; Jeyabalan G.; Singh Y. In-vitro free radical scavenging and hypoglycemic evaluation of fruit extract and solvent fractions of Ziziphus oenoplia mill (Rhamnaceae). Int. J Pharmacogn. 2021, 8, 216–223. [Google Scholar]
- Eswari M. L.; Bharathi R. V.; Jayashree N. Preliminary Phytochemical Screening and Heavy Metal Analysis of Leaf Extracts of Ziziphus oenoplia (L) Mill. Gard. IJPSDR. 2013, 5, 38–40. [Google Scholar]
- Potaraju A.; Samanthula K. S.; Gadda K. Y. Evaluation of Wound Healing and Anti-inflammatory Activities of Leaves of Ziziphus oenoplia. Int. J. Pharm.Biol. Sci. 2019, 9, 1502–1512. [Google Scholar]
- Mourya P.; Shukla A.; Rai G.; Lodhi S. Hypoglycemic and hypolipidemic effects of ethanolic and aqueous extracts from Ziziphus oenoplia (L) Mill on alloxan-induced diabetic rats. BJBAS. 2017, 6, 1–9. 10.1016/j.bjbas.2016.12.002. [DOI] [Google Scholar]
- Alam M.; Chakrabarty N.; Majumder M.; Khan M. F.; Faruk M. O.; et al. Assessment of Antioxidant, Antihelmintic, and Cytotoxic activities of Zizyphus oenoplia (L.) leaves and identification of potential lead compounds through molecular docking analysis. Pharmacologyonline. 2020, 1, 55–67. [Google Scholar]
- Chaudhari D.; Crisostomo C.; Ganote C.; Youngberg G. Acute Oxalate Nephropathy Associated with Orlistat: A Case Report with a Review of the Literature. Case. Rep. Nephrol. 2013, 2013, 124604 10.1155/2013/124604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho A. K.; Glancey G. R. Orlistat, an under-recognised cause of progressive renal impairment. Nephrol. Dial. Transplant. 2013, 28, 172–174. 10.1093/ndt/gft066. [DOI] [PubMed] [Google Scholar]
- Filippatos T. D.; Derdemezis C. S.; Gazi I. F.; et al. Orlistat-associated adverse effects and drug interactions: a critical review. Drug Saf. 2008, 31, 53–65. 10.2165/00002018-200831010-00005. [DOI] [PubMed] [Google Scholar]
- Singh A.; Sarkar S. R.; Gaber L. W.; Perazella M. A. Acute Oxalate Nephropathy Associated with Orlistat, a Gastrointestinal Lipase Inhibitor. Am. J. Kidney Dis. 2007, 49, 153–157. 10.1053/j.ajkd.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Ekor M. The growing use of herbal medicines: issues relating to adverse reactions and challenges in monitoring safety. Front Pharmacol. 2014, 4, 177. 10.3389/fphar.2013.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatti M. Z.; Ali A.; Ahmad A.; Saeed A.; Malik S. A. Antioxidant and phytochemical analysis of Ranunculus arvensis L. extracts. BMC Res Notes 2015, 8, 279. 10.1186/s13104-015-1228-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maqsood M.; Ahmed D.; Atique I.; Malik W. Lipase inhibitory activity of Lagenaria siceraria fruit as a strategy to treat obesity. Asian Pac. J. Trop. Med. 2017, 10, 305–310. 10.1016/j.apjtm.2017.03.010. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley S. K.; Bhikadiya C.; Bi C.; Bittrich S.; Chen L. RCSB Protein Data Bank: powerful new tool for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering, and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. 10.1093/nar/gkaa1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler F. K.; Arcy A. D.; Hunziker W. Structure of human pancreatic lipase. Nature 1990, 343, 771–774. 10.1038/343771a0. [DOI] [PubMed] [Google Scholar]
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2019, 49, D1388–D1395. 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemdraw (RRID: SCR_016768). http://www.perkinelmer.co.uk/category/chemdraw.
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Dallakyan S.; Olson A. J.. Small-molecule library screening by docking with PyRx. In Methods in Molecular Biology, 2015; Vol. 1263, pp 243–250. [DOI] [PubMed] [Google Scholar]
- Tian W.; Chen C.; Lei X.; Zhao J.; Liang J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petterson E. F.; Goddard T. D.; Huang C. C.; Couch S. G.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Hu X.; Zeng Z.; Zhang J.; Wu D.; Li H.; Geng F. Molecular dynamics simulation of the interaction of food proteins with small molecules. Food Chem. 2023, 405, 134824 10.1016/j.foodchem.2022.134824. [DOI] [PubMed] [Google Scholar]
- GROMACS Development Team. 10.5281/zenodo.5053201. [DOI]
- Humphrey W.; Dalke A.; Schulten K. VMD – Visual Molecular Dynamics. J. Molec. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017, 7, 42717 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong G.; Wu Z.; Yi J.; Fu L.; Yang Z.; Hsieh C.; Yin M.; Zeng X.; Wu C.; Lu A.; Chen X.; Hou T.; Cao D. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. 10.1093/nar/gkab255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsadig Karar M. G.; Quiet L.; Rezk A.; Jaiswal R.; Rehders M.; et al. Phenolic profile and in vitro assessment of cytotoxicity and antibacterial activity of Ziziphus spina-christi leaf extracts. Med. Chem. 2016, 6, 143–156. 10.4172/2161-0444.1000339. [DOI] [Google Scholar]
- Alsayari A.; Wahab S. Genus Ziziphus for the treatment of chronic inflammatory diseases. Saudi J. Biol. Sci. 2021, 28, 6897–6914. 10.1016/j.sjbs.2021.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrin A.; Junaid Md.; Afrose S. S.; Barua A.; Akter Y.; et al. Ziziphus oenoplia Mill.: A systematic review on ethnopharmacology, phytochemistry, and pharmacology of an important traditional medicinal plant. Mini. Rev. Med. Chem. 2022, 22, 640–660. 10.2174/1389557521666210810153311. [DOI] [PubMed] [Google Scholar]
- Lobanov M. Y.; Bogatyreva N. S.; Galzitskaya O. V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. 10.1134/S0026893308040195. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Hess A. R.; Buuren R. V.; Apol E.; Meulenhoff P. J.; Tieleman D. P.; Sijbers A. L.; Feenstra K. A.; Drunen R. V.; Berendsen H. J.. Gromacs User Manual, version 4.5.6, 2010.
- Mannhold R.; Poda G. I.; Ostermann C. Calculation of molecular lipophilicity: State-of-the-art comparison of log P methods on more than 96,000 compounds. J Pharm. Sci. 2009, 98, 861–893. 10.1002/jps.21494. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- Cheng T.; et al. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. 10.1021/ci700257y. [DOI] [PubMed] [Google Scholar]
- Moriguchi I.; Shuichi H.; Liu Q.; Nakagome I.; Matsushita Y. Simple method of calculating octanol/ water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. 10.1248/cpb.40.127. [DOI] [Google Scholar]
- Moriguchi I.; Shuichi H.; Nakagome I.; Hirano H. Comparison of reliability of log P values for drugs calculated by several methods. Chem. Pharm. Bull. 1994, 42, 976–978. 10.1248/cpb.42.976. [DOI] [Google Scholar]
- Wildman S. A.; Crippen G. M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Model. 1999, 39, 868–873. 10.1021/ci990307l. [DOI] [Google Scholar]
- http://silicos-it.be.s3-website-eu-west-1.amazonaws.com/software/filter-it/1.0.2/filter-it.html. accessed June 2016.
- Delaney J. S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Model. 2004, 44, 1000–1005. 10.1021/ci034243x. [DOI] [PubMed] [Google Scholar]
- Ali J.; Camilleri P.; Brown M. B.; Hutt A. J.; Kirton S. B. Revisiting the general solubility equation: in silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 2012, 52, 420–428. 10.1021/ci200387c. [DOI] [PubMed] [Google Scholar]
- Daina A.; Zoete V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem. MedChem. 2016, 11, 1117–1121. 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogu C. C.; Maxa J. L. Drug interactions due to cytochrome P450.. BUMC Proceedings 2000, 13, 421–423. 10.1080/08998280.2000.11927719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes C.; Vapnik V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. 10.1007/BF00994018. [DOI] [Google Scholar]
- Szakács G.; Váradi A.; Ozvegy-Laczka C.; Sarkadi B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discovery Today 2008, 13, 379–393. 10.1016/j.drudis.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Montanari F.; Ecker G. F. Prediction of drug-ABC-transporter interaction–Recent advances and future challenges. Adv. Drug Delivery Rev. 2015, 86, 17–26. 10.1016/j.addr.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Martin Y. C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. 10.1021/jm0492002. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Ritchie T. J.; Ertl P.; Lewis R. The graphical representation of ADME- related molecule properties for medicinal chemists. Drug Discovery Today 2011, 16, 65–72. 10.1016/j.drudis.2010.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.