

In Brief
Using a DNA biorepository and the Utah Population Data Base, individuals with both a Chiari malformation and inclusion in a high-risk Chiari pedigree were identified and underwent whole-exome DNA sequencing. Sequence analysis of 24 individuals identified 38 rare candidate Chiari predisposition variants. The most interesting of these variants was HOXC4. All individuals with a HOXC4 mutation exhibited craniocervical kyphosis as part of their Chiari malformation, suggesting that a unique HOXC4 Chiari phenotype may exist.
Keywords: Chiari malformation, high-risk pedigree, predisposition, Utah Population Database, craniocervical kyphosis, protein prediction modeling
ABBREVIATIONS : CM = Chiari malformation, CXA = clivoaxial angle, C-C2SVA = condylar-C2 sagittal vertical alignment, GATK = Genome Analysis Toolkit, MAF = minor allele frequency, SNP = single nucleotide polymorphism, UPDB = Utah Population Database, VQSR = variant quality score recalibration, WES = whole-exome sequencing, WGS = whole-genome sequencing
Abstract
OBJECTIVE
Inherited variants predisposing patients to type 1 or 1.5 Chiari malformation (CM) have been hypothesized but have proven difficult to confirm. The authors used a unique high-risk pedigree population resource and approach to identify rare candidate variants that likely predispose individuals to CM and protein structure prediction tools to identify pathogenicity mechanisms.
METHODS
By using the Utah Population Database, the authors identified pedigrees with significantly increased numbers of members with CM diagnosis. From a separate DNA biorepository of 451 samples from CM patients and families, 32 CM patients belonging to 1 or more of 24 high-risk Chiari pedigrees were identified. Two high-risk pedigrees had 3 CM-affected relatives, and 22 pedigrees had 2 CM-affected relatives. To identify rare candidate predisposition gene variants, whole-exome sequence data from these 32 CM patients belonging to 24 CM-affected related pairs from high-risk pedigrees were analyzed. The I-TASSER package for protein structure prediction was used to predict the structures of both the wild-type and mutant proteins found here.
RESULTS
Sequence analysis of the 24 affected relative pairs identified 38 rare candidate Chiari predisposition gene variants that were shared by at least 1 CM-affected pair from a high-risk pedigree. The authors found a candidate variant in HOXC4 that was shared by 2 CM-affected patients in 2 independent pedigrees. All 4 of these CM cases, 2 in each pedigree, exhibited a specific craniocervical bony phenotype defined by a clivoaxial angle less than 125°. The protein structure prediction results suggested that the mutation considered here may reduce the binding affinity of HOXC4 to DNA.
CONCLUSIONS
Analysis of unique and powerful Utah genetic resources allowed identification of 38 strong candidate CM predisposition gene variants. These variants should be pursued in independent populations. One of the candidates, a rare HOXC4 variant, was identified in 2 high-risk CM pedigrees, with this variant possibly predisposing patients to a Chiari phenotype with craniocervical kyphosis.
Cerebellar tonsil ectopia, including types 1 and 1.5 Chiari malformation (CM), is a disorder defined by herniation of the cerebellar tonsils below the foramen magnum. CM causes a wide variety of neurological and musculoskeletal signs and symptoms, most of which result from abnormalities in cerebrospinal fluid flow or brainstem compression. The underlying cause of the malformation is unknown.
Although there is ample evidence that a genetic cause of CM exists,1–6 little evidence supports a direct link between a particular genetic variant and symptomatic CM in human patients. Several studies have implicated gene mutations in CHD8 alleles, resulting in insufficient gene product expression (haploinsufficiency), as a genetic driver of the CM phenotype. Zebrafish with CHD8 haploinsufficiency demonstrated macrocephaly and posterior hindbrain displacement reminiscent of CM.7,8 Mice with CHD8 haploinsufficiency developed cerebral megalencephaly with increased anterior-posterior length and cross-sectional area of the neocortex; however, this study did not note hindbrain displacement in the mice.9 A prospective analysis of humans with CHD8 haploinsufficiency revealed macrocephaly but not CM.7 Recently, Sadler et al.8 performed whole-exome sequencing (WES) on 668 individuals with CM and 232 of their family members in a specific search for de novo variants. Their findings implicated chromodomain and excessive brain growth genes in CM pathogenesis, including 3 de novo loss-of-function variants in the CHD8 genes. However, the vast majority of CM patients do not harbor CHD8 gene abnormalities, nor do they possess a pronounced increase in cortical tissue; thus, the genetic drivers of CM remain poorly characterized.
In 2018, Abbott et al.10 used the Utah Population Database (UPDB), a unique genealogical resource linking Utah genealogy records dating from the mid-1800s with medical data from the two largest healthcare providers in Utah, to describe significant familial clustering of CM. This study identified extended families with a significantly greater incidence of CM compared with the general population. Increased risks for CM were observed in both close and distant relatives of CM cases, supporting an inherited contribution to CM.
In this study, we aimed to extend the findings of Abbott et al.10 by taking advantage of two repositories of genomic DNA for Utah CM cases and their close relatives, as well as statewide genealogy data, to identify pairs of related CM cases from pedigrees exhibiting a significant excess of CM cases. We performed WES on 32 CM patients who were members of 24 high-risk CM pedigrees and accessed whole-genome sequencing (WGS) data previously generated for a separate group of 7 high-risk CM nuclear families, with some small overlap between the CM-affected individuals included in the two sets of pedigrees. We identified the rare variants shared by the affected pairs who belonged to high-risk pedigrees as representing a set of candidate predisposition gene variants for CM. We used 3D protein structure prediction methods to examine potential structural changes that may provide insights on mechanisms of pathogenicity.11
Methods
Utah Population Database
The UPDB consists of genealogy data from Utah’s early immigrants in the mid-1800s and their modern-day descendants that have been linked to medical data from the two largest health care providers in Utah (University of Utah Health and Intermountain Healthcare) since the mid-1990s.12 Approximately 2 million individuals with at least 3 generations of genealogy and linked Utah medical data were analyzed here. Among those individuals with at least 3 generations of genealogy data in UPDB, a total of 2871 patients at either University of Utah Health or Intermountain Healthcare had an International Classification of Diseases code indicating a diagnosis of CM. The cohort-specific population rates of CM used to test each pedigree for a statistical excess of CM cases were estimated from these 2 million individuals.
Each pedigree was tested for a significant excess of CM cases among the descendants as follows. All individuals in UPDB with linked genealogy and medical data were assigned to a cohort based on their biological sex, 5-year birth-year range, and birth state (Utah or not). Cohort-specific rates of CM were estimated as the total number of CM cases in each cohort divided by the total number of individuals with genealogy and linked medical data in each cohort. The observed number of CM cases among the descendants within a pedigree was compared with the expected number of CM cases on the basis of the cohort-specific rates for each pedigree in UPDB. The expected number of CM cases in each pedigree was estimated as the sum of the cohort-specific rates for CM of each descendant who had linked medical data. An excess of observed to expected CM cases (p < 0.05) was used to identify those pedigrees with a significant excess of CM; these were classified as high-risk pedigrees. Twenty-two of the high-risk pedigrees identified included a pair of sampled CM-affected relatives, and 2 of the high-risk pedigrees included 3 related cases diagnosed with CM.
Utah CM Cases and High-Risk Pedigrees
An IRB-approved project in the Genetics Research Program at the University of Utah has prospectively gathered whole blood and extracted DNA from volunteer pediatric CM patients and their parents since 2013. In all cases, the CM status of the parents was unknown. We included a total of 451 stored samples from 186 individuals with at least 3 generations of linked genealogy data, 61 of whom had a diagnosis of CM (Fig. 1). Thirty-two of these 61 sampled CM cases with linked genealogy in UPDB were found to be related to others in 24 identified high-risk pedigrees. No pedigrees were completely overlapping, but individuals could belong to more than 1 high-risk CM pedigree through different ancestors. Because only a small number of Utah CM cases were sampled, the relationships of the pairs of related CM cases were typically distant, varying in genetic distance from fourth cousins to more distant tenth cousins (Fig. 2), with the amount of shared genetic material among these distant cousins varying from 0.002 for fourth cousins to 5 × 10−7 for tenth cousins. The strength of our high-risk pedigree approach depends on the hypothesis that the related CM cases were members of pedigrees with a significant excess of CM cases and thus were likely to share an inherited predisposition for CM from a common ancestor. Data are available from us with the approval and permission of UPDB.
FIG. 1.
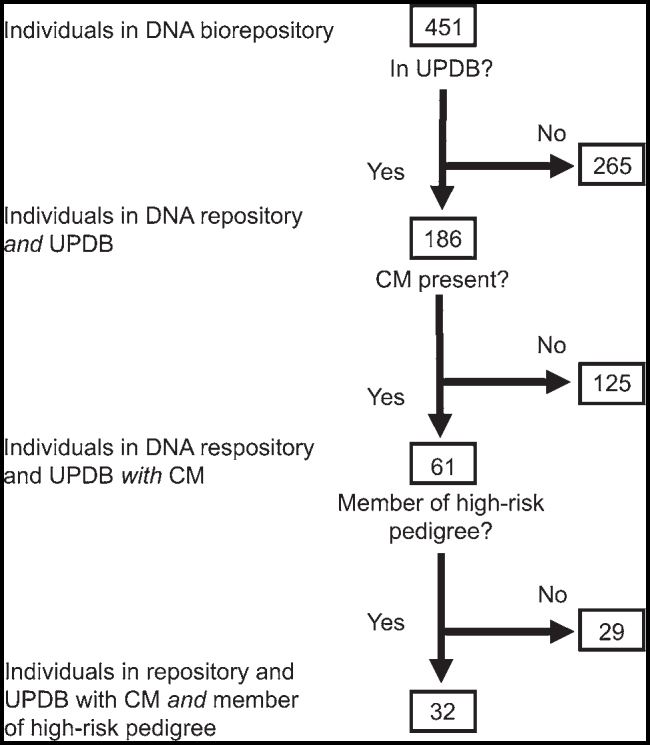
Flowchart showing patient identification.
FIG. 2.

High-risk CM pedigree. A: HOXC4 variant–carrying, high-risk CM pedigree, including a pair of distantly related, affected cases who both have the HOXC4 variant. CM cases are fully shaded. Sequenced HOXC4 carriers (+) are noted. Slashes indicate deceased individuals. B: Midline sagittal cervical T2-weighted MR images showing CM and craniocervical kyphosis in the 2 affected individuals included in panel A. The CXA values are shown for each patient. C: Radiological measurements of the 2 affected individuals. CXA is shown in degrees, and pBC2, tonsillar ectopia, and C-C2SVA are shown in millimeters. pBC2 = maximum perpendicular distance to the basion-inferoposterior point of the C2 body. Figure is available in color online only.
WES Data Generation
We sequenced the protein-coding regions of the genome (using WES) at the Huntsman Cancer Institute High-Throughput Genomics Core by using the stored germline DNA from the 32 CM cases belonging to the 24 high-risk pedigrees. DNA libraries were prepared from 1.5 μg of DNA using the IDT xGEN Human Exome v2 capture kit. Samples were run on the Illumina NextSeq instrument. Reads were mapped to the human genome GRCh37 reference using the BWA-MEM algorithm for alignment, and variants were called using Genome Analysis Toolkit (GATK) version 4.1.3.0 software according to the Broad Institute’s Best Practices Guidelines.13 Exome capture resulted in an average of 95% of the target bases covered by greater than 10× coverage across the exome, with an average depth of 232×. Variants occurring outside the exon capture kit’s intended coverage area were removed. Variants were called and filtered using standard GATK variant quality score recalibration (VQSR) filters (QD < 2.0, QUAL < 30, MQRankSum < –12.5, ReadPosRankSum < –8.0, FS > 60.0, SOR > 3.0, maximum Gaussian variables 2, and truth-sensitivity-filter-level 99.0). Variants were annotated with ANNOVAR, which contains predicted pathogenicity scores from 15 in silico functional prediction algorithms. The sequence data generated for young, related cases have not been submitted to a public database because of the risk of patient identifiability; however, these data are available after communication with us and with appropriate confidentiality agreements.
Previously Sequenced Utah CM Nuclear Families
In a previous unpublished effort, 7 nuclear CM families, each including at least 2 CM-affected siblings who had been sampled as part of the Genetics Research Program, had WGS data generated. The DNA of most of these cases were no longer available for the current study, but their DNA sequence data were accessed for this study. Some previously sequenced CM-affected patients were related to members of the current study, thus slightly extending some of the high-risk pedigrees sequenced here.
Protein Prediction Methods
To perform 3D structure prediction with I-TASSER structure prediction software using full homology modeling,14–17 we used the canonical sequence of HXC4_Human (reference number P09017 in UniProt) for the wild-type species. The sequence was manually changed to obtain the mutated sequence using replacement N178S. All obtained structures were visualized and analyzed using Chimera.18 Variant pathogenicity characterization was obtained with the PolyPhen-2 predictor.19
Results
The DNA sequencing of the 32 CM-affected individuals in 24 high-risk CM pedigrees identified a total of approximately 18,000 exon-specific coding variants (synonymous, nonsynonymous, frameshift indels, nonframeshift indels, start-loss, stop-gain, stop-loss, and splicing) with minor allele frequencies (MAFs) < 0.005 in gnomAD 2.1 that occurred in at least 1 sequenced CM case. Removal of synonymous and poor-quality variants that failed the VQSR tranche filters resulted in approximately 9000 rare variants. Removal of singleton variants (i.e., allele count of 1) resulted in the identification of 693 rare variants. Of these, we identified 38 rare candidates that were shared by both CM cases in at least 1 CM-affected pair from a high-risk pedigree (Table 1).
TABLE 1.
Details for 38 strong candidate variants identified as rare and shared by at least 2 individuals belonging to 24 sequenced high-risk pedigrees
| Chromosome | Start | End | Reference* | Alternate* | Function | Gene | Exonic Function† | Allele Frequency† | Pedigree‡ |
|---|---|---|---|---|---|---|---|---|---|
| 1 |
6727803 |
6727804 |
TC |
— |
Exonic |
DNAJC11
|
Frameshift deletion |
0.0001 |
16 & 17 |
| 1 |
53793511 |
53793512 |
TG |
— |
Exonic |
LRP8
|
Frameshift deletion |
0.0018 |
11 |
| 1 |
53793514 |
53793514 |
A |
— |
Exonic |
LRP8
|
Frameshift deletion |
0.001 |
11 |
| 1 |
1.04 × 108 |
1.04 × 108 |
— |
A |
Exonic |
RNPC3
|
Frameshift insertion |
0.0018 |
21 |
| 1 |
1.84 × 108 |
1.84 × 108 |
C |
T |
Exonic |
COLGALT2
|
Nonsynonymous SNV |
0.0005 |
21 |
| 2 |
9989571 |
9989571 |
A |
— |
Exonic |
TAF1B
|
Frameshift deletion |
0.0008 |
8 |
| 3 |
42674218 |
42674218 |
C |
T |
Exonic |
NKTR
|
Nonsynonymous SNV |
0.0009 |
23 |
| 3 |
47452833 |
47452833 |
G |
A |
Exonic |
PTPN23
|
Nonsynonymous SNV |
0.0023 |
23 |
| 3 |
52521865 |
52521865 |
A |
G |
Exonic |
NISCH
|
Nonsynonymous SNV |
0.004 |
23 |
| 3 |
52548768 |
52548768 |
C |
T |
Exonic |
STAB1
|
Nonsynonymous SNV |
0.0036 |
23 |
| 3 |
88184224 |
88184224 |
A |
G |
Exonic |
ZNF654
|
Nonsynonymous SNV |
0.0031 |
23 |
| 3 |
1.85 × 108 |
1.85 × 108 |
A |
C |
Exonic |
VPS8
|
Nonsynonymous SNV |
0.0031 |
14 |
| 4 |
1360144 |
1360144 |
G |
A |
Exonic |
UVSSA
|
Nonsynonymous SNV |
0.0019 |
8 |
| 4 |
3076654 |
3076654 |
G |
0 |
Exonic |
HTT
|
|
|
21 |
| 6 |
51656129 |
51656129 |
C |
G |
Exonic |
PKHD1
|
Nonsynonymous SNV |
0.0022 |
23 |
| 6 |
76596587 |
76596587 |
C |
T |
Exonic |
MYO6
|
Nonsynonymous SNV |
0.0029 |
10 |
| 7 |
1.39 × 108 |
1.39 × 108 |
AG |
— |
Exonic |
FMC1-LUC7L2 & FMC1-LUC7L2 |
Frameshift deletion |
0.0004 |
8 |
| 8 |
10467637 |
10467637 |
T |
C |
Exonic |
RP1L1
|
Nonsynonymous SNV |
|
5 |
| 8 |
1.45 × 108 |
1.45 × 108 |
— |
AA |
UTR3 |
EPPK1
|
|
|
4 |
| 10 |
35495901 |
35495901 |
C |
T |
Exonic |
CREM
|
Nonsynonymous SNV |
0.0027 |
14 |
| 11 |
694968 |
694968 |
G |
0 |
Exonic |
DEAF1
|
|
|
10 |
| 11 |
4842839 |
4842839 |
A |
G |
Exonic |
OR51F2
|
Nonsynonymous SNV |
0.0012 |
23 |
| 11 |
6411934 |
6411934 |
G |
0 |
Exonic |
SMPD1
|
|
|
18 |
| 12 |
54448727 |
54448727 |
A |
G |
Exonic |
HOXC4
|
Nonsynonymous SNV |
0.0046 |
7 |
| 12 |
1.32 × 108 |
1.32 × 108 |
C |
T |
Exonic |
ADGRD1
|
Nonsynonymous SNV |
0.0033 |
16 |
| 13 |
1.13 × 108 |
1.13 × 108 |
ATG… |
0 |
Exonic |
TUBGCP3
|
|
|
16 |
| 14 |
45716018 |
45716018 |
— |
T |
Exonic |
MIS18BP1
|
Frameshift insertion |
0.0003 |
14 |
| 15 |
72190309 |
72190309 |
A |
G |
Exonic |
MYO9A
|
Nonsynonymous SNV |
0.0012 |
10 |
| 17 |
17697101 |
17697105 |
AGCAG |
0 |
Exonic |
RAI1
|
|
|
19 |
| 19 |
2425415 |
2425415 |
G |
A |
Exonic |
TMPRSS9
|
Nonsynonymous SNV |
0.0017 |
1 |
| 19 |
13875437 |
13875437 |
C |
T |
Exonic |
MRI1
|
Nonsynonymous SNV |
0.0014 |
21 |
| 19 |
17417057 |
17417057 |
G |
A |
Exonic |
MRPL34
|
Nonsynonymous SNV |
|
2 |
| 20 |
307202 |
307202 |
G |
0 |
Exonic |
SOX12
|
|
|
6 |
| 20 |
1212260 |
1212260 |
T |
C |
Exonic |
RAD21L1
|
Nonsynonymous SNV |
0.0025 |
6 |
| 20 |
18446005 |
18446006 |
TC |
— |
Exonic |
DZANK1
|
Frameshift deletion |
0.0011 |
8 |
| 20 |
48808076 |
48808076 |
C |
T |
Exonic |
CEBPB
|
Nonsynonymous SNV |
0.0002 |
8 |
| 21 |
40569102 |
40569102 |
G |
A |
Exonic |
BRWD1
|
Nonsynonymous SNV |
0.0013 |
21 |
| X | 50037948 | 50037948 | C | T | Exonic | CCNB3 | Nonsynonymous SNV | 0.0008 | 17 |
SNV = single nucleotide variant.
0 indicates multiple discrete alleles at this position in the genome and in the variant call format; dashes (—) indicate an insertion or deletion.
Determined with gnomAD 2.1. Empty cells indicate no known allele frequency at this position, implying a novel variant.
The pedigree column shows in which of the 24 high-risk pedigrees the variant was observed.
Two of the 38 rare candidate variants identified may be of particular interest: DNAJC11 and HOXC4. The variant in DNAJC11 was the only concordant variant found in more than 1 relative pair (it was observed in 2 relative pairs in 2 independent pedigrees); the DNAJC11 variant was also observed in 3 single CM cases who were members of pairs in other sequenced high-risk pedigrees. This variant has not yet been pursued.
The HOXC4 variant is perhaps of more significant interest. This variant is a nonsynonymous single nucleotide variant, in which the mutation results in the substitution of an asparagine amino acid for a serine amino acid within an exon encoding the nonspecific DNA interaction loop. It was predicted to be damaging by 12 of 15 prediction algorithms. Figures 2 and 3 show the presence of the HOXC4 variant in the 2 Utah high-risk pedigrees in which it was observed. Figure 2A shows a high-risk pedigree with 2 distantly related CM-affected cousins, both of whom carry the HOXC4 variant and demonstrate abnormal craniocervical kyphosis. The affected male had a clivoaxial angle (CXA) of 122°, whereas the affected female had a CXA of 125° (Fig. 2B).
FIG. 3.

High-risk CM pedigree. A: HOXC4 variant–carrying, high-risk CM pedigree in which 1 girl (arrow) who carried the HOXC4 variant was identified with UPDB pedigree analysis. The carrier status of her mother, father, and CM-affected sister identified in a previous WGS CM analysis of this nuclear family are also shown. Both sisters and the nonpedigree-member mother carry the variant. CM cases are fully shaded. Sequenced HOXC4 carriers (+) and noncarriers (–) are noted. Slashes indicate deceased individuals. B: Radiological measurements of the sequenced patient and her CM-affected sister are shown. CXA is shown in degrees, and pBC2, tonsillar ectopia, and C-C2SVA are shown in millimeters. C: Midline sagittal cervical T2-weighted MR images showing CM and craniocervical kyphosis in the case identified with UPDB pedigree analysis. D: Midline sagittal cervical T2-weighted MR images of the CM-affected sister. OCF = occipitocervical fusion; SODD = suboccipital decompression; TOR = transoral odontoid resection; TPX = tonsillopexy. Figure is available in color online only.
Figure 3A shows another high-risk pedigree harboring 2 distantly related, CM-affected cousins. In the pedigree shown, only 1 case carried the HOXC4 variant; however, the HOXC4 variant carrier was identified as 1 of 2 sisters who also underwent the previously described WGS effort (this WGS portion of the original high-risk pedigree is shown in Fig. 3B). Both CM-affected sisters carried the rare HOXC4 variant and had abnormal craniocervical kyphosis. One sister had a CXA of 117°, and the other had a CXA of 115° (Fig. 3C). The 2 affected sisters shared a total of 3 rare variants that were observed in our WES data (in HOXC4, TNFRSF21, and COL8A2), but the HOXC4 variant was the only one of these 3 variants that was also shared by a CM-affected pair from an additional independent high-risk pedigree (Fig. 2). Of note, the 2 affected sisters also shared a HOXD13 variant inherited from their father (unknown CM phenotype), whose genome was sequenced in the WGS project.
Independent Variant Analysis in a Russian Tartar Cohort
The 38 rare candidate variants were analyzed in an independent WES data set consisting of 9 Russian Tartar CM pedigrees studied by Musolf et al.1 Only 2 of the 38 candidate variants identified in the Utah high-risk pedigrees were also observed in these Russian CM pedigrees (variants in NKTR and RP1L1), and both were primarily identified in unaffected pedigree members (who were used as controls). The NKTR variant was observed in only 1 control. The RP1L1 variant was enriched in this data set (MAF 0.41) but was observed in 24 unaffected controls and 20 cases.
Protein Prediction
HOXC4-expressed protein HXC4_HUMAN functions as a sequence-specific transcription factor that is part of a developmental regulatory system that provides cells with specific positional identities on the anterior-posterior axis (UniProt P09017). The homeobox region, residues 156–215, has been reported as a DNA-binding domain. The mutation reported here is in this region (Fig. 4).
FIG. 4.

Wild-type homeobox sequence of HOXC4. The site of the mutation reported here is shown in red and underlined. Figure is available in color online only.
The variant reported here has been previously reported (rs35406888) but has not been associated with any disorder (UniProt P09017). Analysis of the variant with PolyPhen19 reported that this variant is "probably damaging."
In Fig. 5, we depict the superpositions of the wild-type (yellow/cyan) and mutant (red/green) predicted structures (model 1) of the homeobox domain with their relationships to the predicted DNA binding. Although it is apparent that there were no significant structural changes to the homeobox region upon mutation, careful observation of the coiled region of the mutation under consideration (residues 161–178) shows that the mutated structure has a shorter coil than that of the wild type. This observation led us to hypothesize that this minor structural change may lead to a change in the binding affinity of DNA to the homeobox region of the protein.
FIG. 5.

Superpositions of the wild-type (yellow/cyan) and mutant (red/green) predicted structures (model 1) of the homeobox domain with their relationships to the predicted DNA binding.
Discussion
By using an extensive Utah genealogic resource combined with a DNA biorepository of CM cases, we have performed a unique and powerful analysis of sequence data from related CM cases belonging to high-risk CM pedigrees. The analysis identified a set of 38 rare candidate predisposition variants with strong evidence for a role in CM predisposition.
Two of the 38 candidate CM predisposition variants identified here were also observed in an independent set of CM pedigrees,1 but neither variant showed associations with CM risk in those data. This finding may be due to the low overall frequencies of these variants, as well as differences in genetic makeup between the Russian Tartar pedigrees and the Utah–Northern European pedigrees. Two of the remaining candidate variants may be of particular interest: DNAJC11 and HOXC4. One candidate variant, HOXC4, was observed in 2 distinct Utah pedigrees and appeared to be associated with the development of a CM subtype characterized by craniocervical kyphosis.
The known functions and patterns of expression of HOX genes and the severe phenotype that we observed in the CM individuals harboring the HOXC4 mutation make this mutation an attractive candidate for investigations of a causal relationship between mutation and phenotype. Mammals possess 4 similar Hox gene clusters (A–D) comprising 9 to 11 tandemly arranged genes, with each cluster located on different chromosomes. All HOX genes encode transcription factors expressed in a wide variety of tissues. They play a central role in specifying regions of the body plan along the caudal-rostral axis during the embryological development of all animals. Hox genes are critical regulators of the organization and morphology of the vertebral elements in vertebrates.20 For example, inactivation of Hoxd3 in mice is known to produce occiput-axis assimilation, whereas rostral extension of the expression domain of Hoxd4 leads to premature disjunction of the occipital bone, with subsequent "cervicalization of the clivus."20 Previous work in mice by Boulet and Capecchi21 demonstrated that homozygous disruption of Hoxc4 creates abnormal vertebral kyphosis in the high thoracic spine area in a process described by Matsuoka et al.22 as follows: "Neural crest anchors the head onto the anterior lining of the shoulder girdle, while a Hox gene-controlled mesoderm links trunk muscles to the posterior neck and shoulder skeleton." Other work by Saegusa et al.23 found that in mice with homozygous Hoxc4 mutations, "various abnormalities" were seen "in the cervical and thoracic regions. The most frequent abnormality was a partial posterior homeotic transformation of the seventh cervical vertebra."
Although some of the abovementioned mutations lead to abnormalities in areas other than the craniocervical region, it is clear that HOX genes regulate the tissue types (i.e., bone, cartilage, and muscle) of the paraxial mesoderm that are relevant to the CM phenotype. For instance, a case-control association study of single nucleotide polymorphisms (SNPs) across 58 candidate genes involved in early paraxial mesoderm development in 415 CM patients and 524 controls found that HOXC4 had the highest number of SNPs relative to the other HOX genes analyzed (22 SNPs, with the next greatest being 11 SNPs in HOXD3).24 Thus, a mutation in HOXC4 could lead to the morphological changes seen in the bones of the posterior fossa and upper cervical spinal cord, resulting in the observed CM phenotype. The concept of the primary problem being within the skeletal axis in CM patients is supported by the often observed phenomenon that the anatomy of the neural components of the CM phenotype (e.g., herniated tonsils, cord syrinx) can normalize with bony decompression alone.
Another candidate variant of interest found in 2 high-risk pedigrees in this study—DNAJC11 frameshift mutation—does not have an obvious causal relationship with the CM phenotype. DNAJC11 encodes a chaperone protein required for mitochondrial inner membrane organization via its interactions with the mitochondrial MICOS (mitochondrial contact site and cristae organizing system) and SAM (sorting and machinery) complexes.25 In a previous study, a splicing mutation in a mouse model led to motor neuron pathology associated with cristae disorganization and lymphocyte abnormalities. However, a query of the Human Protein Atlas (https://www.proteinatlas.org/ENSG00000007923-DNAJC11/tissue) revealed the expression of DNAJC11 transcripts in all tissue types. Thus, the specific DNAJC11 mutation seen in our patients may manifest in the specific tissues that lead to the observed CM phenotype.
Despite the strong likelihood that our candidate genes have a causal relationship to CM, we need more studies to ascertain the precise mechanisms of how these mutations manifest into the phenotype. WGS or WES analysis of greater numbers of CM patients may help to reveal which of our candidate gene mutations exist in other patients and to find new candidate gene mutations. Scientists can study these mutations using the powerful experimental systems of modern developmental biological research. For instance, high-throughput next-generation sequencing of gene expression and protein expression array profiling of the affected tissues in CM patients versus controls may provide insights into the dysregulation of the developmental pathways involved in CM. Given that epigenetic factors play an obvious and important role in CM and that transcription factors regulate the accessibility of genes to the transcriptional machinery, array-based methylation phenotyping would also be of tremendous value. Numerous in vitro genetic and biochemical studies can be performed to determine how these gene mutations affect gene expression, biological pathway activation, proliferation, and survival and development of bone, cartilage, and muscle cells. Finally, experimental manipulation of model animal systems such as zebrafish and mice may help determine whether candidate genes identified in this and other studies lead to phenotypic changes similar to those of human CM. The expression levels of mutated candidate genes and targeted disruption of these genes confined to critical tissue types (e.g., posterior skull base, upper cervical bone and cartilage, cervical medullary dura mater) using advanced techniques such as CRISPR/Cas9 and site-specific promoters may lead to essential findings that could not have been determined if the candidate gene was simply disrupted in the entire organism. Further studies at the atomic level that use empirical methods to determine the binding affinity between wild-type and mutant HXC4_HUMAN proteins and DNA may provide more solid evidence of the molecular hypothesis postulated here.
Clinical Implications
The clinical implications of these findings are intriguing given recent knowledge about CM-related craniocervical morphology. In 2012, Bollo et al.26 published a retrospective single-center series of 101 CM patients that demonstrated that craniocervical kyphosis (defined as a CXA < 125°) and CM type 1.5 (defined as herniation of the obex through the foramen magnum) predicted need for craniocervical fusion and transoral odontoid resection. Patients in this population were described as having "complex Chiari" malformation, implying that craniocervical bony geometry and biomechanics may play a role in the natural history of that particular subset of CM patients. This study led to a series of publications further investigating this concept. Specifically, a recent paper by Ravindra et al.27 described the concept of condylar-C2 sagittal vertical alignment (C-C2SVA) measurement and its relationship to craniocervical kyphosis, along with its influence on the clinical behavior of CM. The subsequent validation of C-C2SVA measurement in a large cohort of patients from the Park-Reeves Syringomyelia Research Consortium28 further strengthened the argument that bony geometry and sagittal craniocervical alignment are important aspects of CM clinical behavior.
But how does craniocervical kyphosis occur? What are the genetic determinants of skull base and upper cervical spine development? And what is its relationship to CM? Skull base development is a highly complex process primarily driven by genetic factors, with the HOX gene complex responsible for the positional identity and morphology of the embryonic craniocervical sclerotome elements. Other genes and developmental factors, such as those controlling cranial vault skull growth, brain growth, and cerebrospinal fluid flow, undoubtedly contribute to the development of CM during the critical period of early embryonic development. The fact that a percentage of CM cases exhibit craniocervical kyphosis with HOXC4 mutations and tend to occur in high-risk pedigrees, as shown here, is a compelling argument that a distinct genetic phenotype of CM may exist.
Strengths and Limitations
This study has both strengths and limitations. The strengths include physician-confirmed CM diagnoses with detailed characteristics for all sequenced cases. There was no recall or ascertainment bias because statewide medical diagnosis data were used and cases were distantly related. The existence of linked genealogic data for Utah allowed the identification of the high-risk status of each pedigree studied.
The limitations of the study include the small number of available CM cases and the homogeneous population analyzed. Utah genealogical data primarily represent the Northern European ancestry of the state;29 validation in other populations is needed. Our analysis identified 5 overlapping candidate genes that were found in the data from Sadler et al., which noted evidence of de novo predisposition variants (HTT, MYO6, ADGRD1, MIS18BP1, and DZANK1 [Table 1]).8 However, the variants identified for each gene were different. Furthermore, the sampled CM cases accessed for this study consisted of samples conveniently available rather than those representative of a more population-based sample. The CM cases available were distantly related, which increases the likelihood of heterogeneity and the likelihood that some pedigrees may have been incorrectly identified as high risk; nevertheless, the genetic distances of the cases can also be considered a strength in that the sharing of rare variants between distant relatives provides possible evidence of causality. Again, given the rarity of the phenotype and lack of samples for additional related cases in the pedigrees of interest (i.e., the only available sampled CM cases were from the Genetics Research Program), validation of segregation of the variants with CM in additional affected relatives was not possible.
Conclusions
This analysis of CM pedigrees displays the power of data from related cases who are members of high-risk pedigrees and was used to identify a small set of candidate predisposition variants for a rare disorder. Intriguing evidence for HOXC4 was observed and should be pursued in both pedigrees in which it was observed in Utah as well as in other populations. Furthermore, each candidate variant identified in this study has potential and should be pursued further using additional resources.
Acknowledgments
We thank Kristin Kraus for editorial assistance.
Partial support for all datasets within the Utah Population Database is provided by the University of Utah, Huntsman Cancer Institute (HCI), and the HCI Cancer Center Support grant no. P30 CA42014 from the National Cancer Institute (NCI). L.A.C.A. is partially funded by NCI grant no. P30 CA42014. This project was funded by a pilot grant from the Primary Children’s Center for Personalized Medicine, Primary Children’s Hospital, Intermountain Healthcare, Salt Lake City, Utah. This work was partially supported by the Utah Clinical and Translational Science Institute funded by the National Center for Advancing Translational Sciences award no. 1ULTR002538. Computer resources were provided by the University of Utah Center for High-Performance Computing, which has been partially funded by the NIH Shared Instrumentation grant no. 1S10OD02164401A1.
Disclosures
The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.
Author Contributions
Conception and design: Brockmeyer, Cheshier, Cannon-Albright. Acquisition of data: Brockmeyer, Stevens, Rowe, Heiss, Musolf, Viskochil, Cannon-Albright. Analysis and interpretation of data: Brockmeyer, Cheshier, Stevens, Facelli, Heiss, Musolf, Allen-Brady, Cannon-Albright. Drafting the article: Brockmeyer, Cheshier, Stevens, Heiss, Musolf, Cannon-Albright. Critically revising the article: Brockmeyer, Cheshier, Facelli, Heiss, Musolf, Cannon-Albright. Reviewed submitted version of manuscript: Brockmeyer, Cheshier, Cannon-Albright. Approved the final version of the manuscript on behalf of all authors: Brockmeyer. Administrative/technical/material support: Viskochil, Allen-Brady, Cannon-Albright. Study supervision: Brockmeyer.
References
- 1. Musolf AM, Ho WSC, Long KA, et al. Small posterior fossa in Chiari I malformation affected families is significantly linked to 1q43-44 and 12q23-24.11 using whole exome sequencing. Eur J Hum Genet. 2019;27(10):1599–1610. doi: 10.1038/s41431-019-0457-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Speer MC, George TM, Enterline DS, Franklin A, Wolpert CM, Milhorat TH. A genetic hypothesis for Chiari I malformation with or without syringomyelia. Neurosurg Focus. 2000;8(3):E12. doi: 10.3171/foc.2000.8.3.12. [DOI] [PubMed] [Google Scholar]
- 3. Boyles AL, Enterline DS, Hammock PH, et al. Phenotypic definition of Chiari type I malformation coupled with high-density SNP genome screen shows significant evidence for linkage to regions on chromosomes 9 and 15. Am J Med Genet A. 2006;140(24):2776–2785. doi: 10.1002/ajmg.a.31546. [DOI] [PubMed] [Google Scholar]
- 4. Markunas CA, Enterline DS, Dunlap K, et al. Genetic evaluation and application of posterior cranial fossa traits as endophenotypes for Chiari type I malformation. Ann Hum Genet. 2014;78(1):1–12. doi: 10.1111/ahg.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Markunas CA, Lock E, Soldano K, et al. Identification of Chiari type I malformation subtypes using whole genome expression profiles and cranial base morphometrics. BMC Med Genomics. 2014;7:39. doi: 10.1186/1755-8794-7-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Markunas CA, Soldano K, Dunlap K, et al. Stratified whole genome linkage analysis of Chiari type I malformation implicates known Klippel-Feil syndrome genes as putative disease candidates. PLoS One. 2013;8(4):e61521. doi: 10.1371/journal.pone.0061521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bernier R, Golzio C, Xiong B, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158(2):263–276. doi: 10.1016/j.cell.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sadler B, Wilborn J, Antunes L, et al. Rare and de novo coding variants in chromodomain genes in Chiari I malformation. Am J Hum Genet. 2021;108(3):530–531. doi: 10.1016/j.ajhg.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gompers AL, Su-Feher L, Ellegood J, et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat Neurosci. 2017;20(8):1062–1073. doi: 10.1038/nn.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abbott D, Brockmeyer D, Neklason DW, Teerlink C, Cannon-Albright LA. Population-based description of familial clustering of Chiari malformation Type I. J Neurosurg. 2018;128(2):460–465. doi: 10.3171/2016.9.JNS161274. [DOI] [PubMed] [Google Scholar]
- 11. Hernandez R, Facelli JC. Understanding protein structural changes for oncogenic missense variants. Heliyon. 2021;7(1):e06013. doi: 10.1016/j.heliyon.2021.e06013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cannon Albright LA. Utah family-based analysis: past, present and future. Hum Hered. 2008;65(4):209–220. doi: 10.1159/000112368. [DOI] [PubMed] [Google Scholar]
- 13. Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high-confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43(1):11.10.11–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y. I-TASSER: fully automated protein structure prediction in CASP8. Proteins. 2009;77(suppl 9):100–113. doi: 10.1002/prot.22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 19. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pang D, Thompson DN. Embryology and bony malformations of the craniovertebral junction. Childs Nerv Syst. 2011;27(4):523–564. doi: 10.1007/s00381-010-1358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boulet AM, Capecchi MR. Targeted disruption of hoxc-4 causes esophageal defects and vertebral transformations. Dev Biol. 1996;177(1):232–249. doi: 10.1006/dbio.1996.0159. [DOI] [PubMed] [Google Scholar]
- 22. Matsuoka T, Ahlberg PE, Kessaris N, et al. Neural crest origins of the neck and shoulder. Nature. 2005;436(7049):347–355. doi: 10.1038/nature03837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saegusa H, Takahashi N, Noguchi S, Suemori H. Targeted disruption in the mouse Hoxc-4 locus results in axial skeleton homeosis and malformation of the xiphoid process. Dev Biol. 1996;174(1):55–64. doi: 10.1006/dbio.1996.0051. [DOI] [PubMed] [Google Scholar]
- 24. Urbizu A, Toma C, Poca MA, et al. Chiari malformation type I: a case-control association study of 58 developmental genes. PLoS One. 2013;8(2):e57241. doi: 10.1371/journal.pone.0057241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ioakeimidis F, Ott C, Kozjak-Pavlovic V, et al. A splicing mutation in the novel mitochondrial protein DNAJC11 causes motor neuron pathology associated with cristae disorganization, and lymphoid abnormalities in mice. PLoS One. 2014;9(8):e104237. doi: 10.1371/journal.pone.0104237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bollo RJ, Riva-Cambrin J, Brockmeyer MM, Brockmeyer DL. Complex Chiari malformations in children: an analysis of preoperative risk factors for occipitocervical fusion. J Neurosurg Pediatr. 2012;10(2):134–141. doi: 10.3171/2012.3.PEDS11340. [DOI] [PubMed] [Google Scholar]
- 27. Ravindra VM, Iyer RR, Awad AW, Bollo RJ, Zhu H, Brockmeyer DL. Defining the role of the condylar-C2 sagittal vertical alignment in Chiari malformation type I. J Neurosurg Pediatr. 2020;26(4):439–444. doi: 10.3171/2020.4.PEDS20113. [DOI] [PubMed] [Google Scholar]
- 28. Ravindra VM, Iyer RR, Yahanda AT, et al. A multicenter validation of the condylar-C2 sagittal vertical alignment in Chiari malformation type I: a study using the Park-Reeves Syringomyelia Research Consortium. J Neurosurg Pediatr. 2021;28(2):176–182. doi: 10.3171/2020.12.PEDS20809. [DOI] [PubMed] [Google Scholar]
- 29. Cannon-Albright LA, Farnham JM, Thomas A, Camp NJ. Identification and study of Utah pseudo-isolate populations-prospects for gene identification. Am J Med Genet A. 2005;137A(3):269–275. doi: 10.1002/ajmg.a.30893. [DOI] [PubMed] [Google Scholar]