

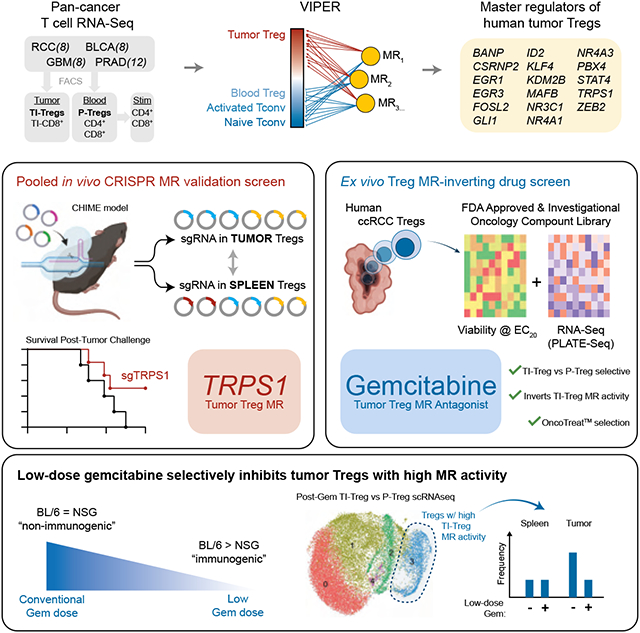
Summary
Due to their immunosuppressive role, tumor-infiltrating regulatory T cells (TI-Tregs) represent attractive immuno-oncology targets. Analysis of TI vs. peripheral Tregs (P-Tregs) from 36 patients, across four malignancies, identified 17 candidate Master Regulators (MRs) as mechanistic determinants of TI-Treg transcriptional state. Pooled CRISPR-Cas9 screening in vivo, using a chimeric hematopoietic stem cell transplant model, confirmed essentiality of 8 MRs in TI-Treg recruitment and/or retention, without affecting other T cell subtypes, and targeting one of the most significant MRs (Trps1) by CRISPR KO significantly reduced ectopic tumor growth. Analysis of drugs capable of inverting TI-Treg MR activity identified low-dose gemcitabine as the top prediction. Indeed, gemcitabine treatment inhibited tumor growth in immunocompetent but not immunocompromised allografts, increased anti-PD-1 efficacy, and depleted MR-expressing TI-Tregs in vivo. This study provides key insight into Treg signaling, specifically in the context of cancer, and a generalizable strategy to systematically elucidate and target MR proteins in immunosuppressive subpopulations.
Keywords: Tumor Immunology, Regulatory T Cells, Master Regulator Analysis, TRPS1, gemcitabine, Cancer Systems Biology
Graphical Abstract
eTOC Blurb
Obradovic et al. infer and functionally validate a set of master regulatory (MR) proteins specifically active in tumor-infiltrating regulatory T cells (TI-Tregs), including TRPS1. Inhibition of TRPS1 improves survival in mouse models. A drug screen revealed low-dose gemcitabine inhibits TI-Treg MRs and extends survival in immune-competent mice and is synergistic with aPD-1 immunotherapy.
Introduction
To manifest as clinically apparent disease, cancer must evade a complex repertoire of host-protective immune response mechanisms, the outcome of which is largely determined by the balance of inflammatory (anti-tumor) and tolerogenic (pro-tumor) immune cell function in the tumor microenvironment (TME)1. By contributing to a tolerogenic TME, the regulatory T cell (Treg) lineage—characterized by activation of the hallmark transcription factor FoxP3—promotes tumor growth and immunotherapy resistance. As such, increased Treg infiltration in the TME correlates with poor prognosis and increased resistance to immune checkpoint inhibitors across many human malignancies2,3,4,5,6,7. While this makes Tregs attractive therapeutic targets, several factors have prevented clinical translation. First, to avoid severe autoimmunity-mediated toxicity2,7, an optimal Treg-directed therapy should target tumor infiltrating Tregs (TI-Tregs) while sparing peripheral Tregs (P-Tregs). Second, the Treg transcriptional profile broadly recapitulates that of other activated T cells, thus complicating design of selective targeting strategies that would preserve anti-tumor cytotoxic CD8+ and CD4+ T cell function2,8. The majority of current Treg-targeting agents do not satisfy these criteria and, although effective in murine models, they have not effectively translated to human patients9,10,11. This highlights the need for elucidating the still elusive causal mechanisms that underlie Treg recruitment, retention, and/or function in the TME, thus leading to identification of more specific TI-Treg vulnerabilities.
To address this challenge several labs have profiled Tregs isolated from clinical tumor specimens to identify genes differentially expressed in TI vs. non-TI-Tregs and other T cells. However, differences identified so far have failed to provide tumor infiltration mechanisms that can be successfully targeted pharmacologically. For instance, several studies have successfully validated known TI-Treg biology, including high expression of IL2RA (CD25) and FOXP3 in conjunction with multiple T cell checkpoints (CTLA4, PDCD1 [PD-1], HAVCR2 [TIM-3], LAG3, TIGIT), TNF-family receptors (TNFRSF9 [4-1BB], TNFRSF18 [GITR], TNFRSF4 [OX-40]), wound-healing factors (ENTPD1 [CD39], IL1RL1 [ST2]), and proliferation programs8,12,13,14,15. In addition, a number of specific genes enriched in TI-Tregs have been observed across datasets, including LAYN, SAMSN1, IL1R2, MAGEH1, CD177, and the chemokine receptor CCR8. Of these, CCR8 is one leading candidate, showing preferential protein level expression in breast cancer14 and NSCLC TI-Tregs16, among others. Monoclonal antibodies targeting CCR8 and LAG-3 are currently in multiple clinical trials. However, while LAG-3 in combination with nivolumab improved progression free survival in metastatic melanoma by 5 months17,18, more recent data suggest CCR8 may be dispensable for Treg function16,19. Thus, despite these advancements, additional efforts are warranted to discover novel potential TI-Treg vulnerabilities via orthogonal approaches.
We have developed methodologies for the assembly and interrogation of lineage context-specific gene regulatory networks, including the Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe)20 and the Virtual Proteomics by Enriched Regulon analysis (VIPER) algorithm21, respectively (Figure 1A). These have been successful in nominating Master Regulator (MR) proteins, representing mechanistic drivers of both pathophysiologic and transformed transcriptional cell states22,23,24, which have been experimentally validated, including at the single cell level25,26,27. We thus sought to leverage these methodologies to interrogate a Treg-specific gene regulatory network with signatures of TI vs. P-Tregs, to first identify and then validate novel causal Master Regulators driving Treg infiltration to and retainment in the TME.
Figure 1: VIPER Enables Definition of Tumor vs Peripheral Treg Master Regulator Signature:

(A) Conceptual plot of ARACNe/VIPER protein activity inference process. (B) Principal Component Analysis (PCA) plot of Gene Expression colored by T cell , as indicated. P-Treg: peripheral Treg, TI-Treg: tumor-infiltrating Treg. (C) PCA plot of VIPER-inferred protein activity, colored as in B, showing spatial separation of T-cell sub-types. (D) PCA plot of VIPER-inferred protein activity separating TI-Tregs and P-Tregs only, colored as in B,C. (E) Heatmap of VIPER Protein Activity for Master Regulators Identified by Random Forest Feature Selection as best distinguishing TI-Tregs vs P-Tregs. (F) Heatmap of VIPER Protein Activity for Master Regulators Identified by Random Forest Feature Selection as best distinguishing TI-Tregs vs all peripheral controls (P-Tregs, Naïve Tconv, Activated Tconv). (G) Experimental design of over-expression screen, where the predicted TI-Treg MR ORFs (17 in total) are individually overexpressed in sorted P-Tregs and then 7 days later profiled by scRNA-seq. (H) LDA Plot showing unsupervised clustering of Treg phenotypes by scRNA-seq from experiment described in E. (I) Violinplot of cell-by-cell Gene Set Enrichment (GSEA) of 27 TI-Treg MRs for cells shown in F, such that cluster 3 cells are significantly enriched for TI-Treg signature. (J) Barplot of cluster 3 frequencies grouped by over-expressed gene, where negative controls (no gene overexpressed and EGFP overexpressed) are colored blue, and candidate MRs are colored red. *** indicates p<0.001 relative to negative control, ** indicates p<0.01, * indicates p<0.05, by Bonferroni-Adjusted Fisher’s Exact Test. (K) Heatmap of protein activity for inferred TI-Treg MR proteins at single-cell level, in the experiment described in G-J. Grouped by cluster as in H,I. Shows Co-upregulation of entire MR module in every cell from cluster 3, regardless of which individual MR was over-expressed.
See also Figures S1 and S2.
To generate tumor-agnostic signatures of TI-Treg vs. P-Treg state, we have collected patient-matched transcriptional profiles from multiple T cell subpopulations, isolated from the tumors and peripheral blood of 36 patients by FACS, using established antibody panels. We specifically focused on tumor types whose T cell repertoire is not well-represented in existing datasets, including prostate adenocarcinoma, bladder cancer, clear cell renal carcinoma, and glioblastoma. With this dataset, we identify transcriptional signatures differentially expressed in TI-Tregs, vs. patient-matched peripheral blood Tregs, conventional non-Treg CD4 (Tconv), and CD8+ T cells, across a set of highly diverse cancers. We leverage the VIPER algorithm to identify candidate MR proteins, whose transcriptional targets are most differentially expressed in TI- vs. P-Tregs, and thus most likely to comprise the protein module that mechanistically drives and homeostatically maintains the TI-Treg transcriptional state21. We have shown that this approach outperforms gene expression based analyses and compares favorably with single cell, antibody-based approaches25,28,29. Critically, while antibodies measure abundance, this approach measures the transcriptional activity of each regulatory protein, i.e., its ability to mechanistically regulate a transcriptional signature of interest.
To further assess whether candidate MR proteins are essential for TI-Treg infiltration and retention to the TME, we leverage two orthogonal yet complementary methodologies. First, we utilize a CHIME (Chimeric Immune Editing)30 model to perform a pooled, in vivo CRISPR-Cas9 screen to assess whether targeting of the candidate MRs by CRISPR KO would deplete TI-Tregs, without affecting P-Tregs, thus confirming their mechanistic role in mediating naïve Treg recruitment and/or TI-Treg retention to the tumor microenvironment. Second, we performed a systematic drug screen where patient-derived TI-Tregs were expanded ex vivo and their response to perturbations with a large compound library was assessed by RNA-seq profiling (perturbational profiles). Candidate MR-inverter compounds—capable of specifically inverting the activity of the TI-Treg MRs—were nominated using the NY/CA Dept. of Health approved, CLIA-compliant OncoTreat algorithm31 and validated. A critical value of this approach is its highly generalizable nature and potential for rapid translation of mechanism-based therapeutic strategies for modulating TI-Treg infiltration and retention to the TME, thus potentiating immunotherapy.
Results:
Isolating tumor (TI-Tregs) vs. blood derived (P-Tregs) Regulatory T Cells:
Tumor and patient-matched peripheral blood tissues were collected from 36 individuals, including 8 glioblastoma (GBM), 8 bladder adenocarcinoma (BLCA), 8 clear cell renal carcinoma (KIRC), and 12 prostate adenocarcinoma (PRAD) patients. Multiple T cell lineages were freshly sorted from each patient by antibody-based flow cytometry, including TI-Tregs, P-Tregs, peripheral blood CD4 T cells, and both tumor-infiltrating and peripheral blood CD8+ T cells, see Figure S1 for sorting strategies. Purity was assessed by flow and exceeded 95% for each population (Figure S1A-E). To provide additional controls for T cell activation, patient-matched flow sorted naive peripheral (i.e., blood-derived) CD4+ and CD8+ T cells from each of the 36 patients were stimulated for 72-hours with anti-CD3/anti-CD28 beads. Total RNA was purified from each of these seven distinct T cell subpopulations and RNA-seq profiles were generated by Illumina sequencing, for a total of 236 distinct RNA-seq profiles.
Nominating Candidate Master Regulators of TI-Treg Transcriptional State:
In order to maximize biological signal-to-noise we applied protein activity inference to the sorted RNA-Seq profiles (Figure 1A). Gene expression-based cluster analysis produced poor stratification of different T-cell subtypes (Figure 1B). This is likely due to inherent noise in transcriptional data, as RNA counts represent a proxy for the biological activity of proteins that determine cell state. After a protein is expressed, its activity is manifested only when it is effectively post-translationally modified, translated to the appropriate sub-cellular compartment, and forms complexes with critical cognate binding partners. By inferring a network of the downstream transcriptional targets for each TF, CoTF, and signaling protein using the ARACNe algorithm, we may effectively assess the activity of upstream proteins from the expression patterns of their targets using VIPER. Having shown that protein activity-based cluster analysis consistently outperforms expression-based analyses24,25, we proceeded to generate a tumor-infiltrating regulatory T cell-specific gene regulatory network by analyzing the 236 T cell-derived profiles using AP-ARACNe32 —the latest version of the ARACNe algorithm20 —followed by VIPER-based measurement of differential protein activity for each sample against the average of all samples (methods), as previously described in multiple publications21. Activity-based cluster analysis showed clear separation of naïve and activated T cells by 2D principal component analysis, with tumor-infiltrating cells comprising a distinct cluster (Figure 1C). Intriguingly, neither gene expression nor protein activity could stratify TI-Treg samples by tumor type, suggesting a relatively tumor-agnostic transcriptional state. However, while gene expression could not differentiate between TI and P-Tregs in PCA space, MR analysis nearly perfectly stratified the two subpopulations (Figure 1D).
Consistent with these findings, a Random Forest classifier for TI- vs. P-Treg state, independently trained on the statistically significant MRs (p ≤ 10−3), as assessed independently from samples of each tumor type (e.g., using GBM samples only), could precisely classify TI- vs. P-Tregs across all other tumor types (e.g., PRAD, BLCA, and KIRC), producing a perfect pairwise Area Under the Receiver Operating Curve metric (AUROC = 1.0) for all comparisons. Consistently, there was highly significant enrichment (ranging from p = 10−4 to p = 10−11) of candidate MRs inferred from a single tumor type—based on VIPER analysis of genes differentially expressed in tumor patient-specific TI- vs. P-Tregs (p ≤ 10−3)—in proteins differentially active in TI- vs. P-Tregs from each other tumor type, by Gene Set Enrichment Analysis (GSEA)33 (Table 1).
Table 1:
GSEA of TI vs. P-Treg MR proteins between tumor contexts, Bonferroni corrected.
| Candidate MRs |
Differentially Active Proteins |
P-value | Candidate MRs |
Differentially Active Proteins |
P-value |
|---|---|---|---|---|---|
| GBM | PRAD | 4.70E-07 | PRAD | GBM | 1.30E-11 |
| GBM | KIRC | 7.20E-07 | KIRC | GBM | 6.90E-11 |
| GBM | BLCA | 2.10E-05 | BLCA | GBM | 1.10E-06 |
| PRAD | KIRC | 3.40E-11 | KIRC | PRAD | 2.20E-04 |
| PRAD | BLCA | 8.10E-08 | BLCA | PRAD | 2.30E-07 |
| KIRC | BLCA | 2.10E-05 | BLCA | KIRC | 1.30E-08 |
To select the most discriminative candidate MRs, among those differentially active in TI-Tregs vs. other T cell populations—including P-Tregs, naïve CD4, and activated CD4 T cells—we used the Random Forest algorithm (see methods). Specifically, this analysis selected the minimal number of features (i.e., candidate MRs, starting from the most statistically significant one) that maximized the ratio between the AUROC from a Monte Carlo cross validation (MCCV) analysis, compared to the null hypothesis (i.e., equal number of randomly selected transcriptional regulators) (Figure 1E-F). The analysis yielded 15 candidate MR proteins significantly differentially active in TI- vs. P-Tregs, shown in Figure 1E (AUROC = 0.982 for TI vs. P-Treg classification by MCCV, Figure S2A). In addition, seven candidate MRs were found to optimally classify TI-Tregs vs. other control subpopulations, shown in Figure 1F (AUROC = 0.988, by MCCV, Figure S2A). Of these, only two were not included in the previous 15, yielding a total of 17 unique candidate MRs of Treg tumor infiltration, namely EGR1, NR3C1, PBX4, MAFB, ID2, STAT4, NR4A3, NR4A1, TRPS1, EGR3, BANP, ZEB2, KLF4, GLI1, CSRNP2, KDM2B, and FOSL2. Of these, the NR4A family of transcription factors34 as well as FOSL235 were previously reported as upstream regulators of FOXP3 expression in Tregs, the glucocorticoid receptor NR3C1 was shown to have Treg-specific function36, and EGR3 was reported as a negative regulator of T-cell activation37. However, none were previously reported as causal regulators of Treg tumor infiltration and none was significantly differentially expressed at the RNA level in TI-Tregs in our dataset.
Candidate MR Validation by In Vitro Overexpression Assay:
To further assess whether candidate MRs play a mechanistic role in controlling Treg state, we tested whether ectopic expression of any of the 17 computationally predicted individual TI-Treg MRs was sufficient to convert naïve Tregs to a TI-Treg-like state. For this, we performed an arrayed (one open reading frame (ORF) / well) overexpression screen in human peripheral Tregs, where we lentivirally overexpressed each of the 17 predicted TI-Treg MR ORFs, with one MR over-expressed per cell (Figure 1G). EGFP-ORF and non-transduced naïve Tregs were used as assay negative controls. 7 days after the lentiviral transductions, we performed single-cell RNA-Seq (scRNA-seq) profiling of the resulting cells.
We performed unsupervised clustering of Treg phenotypes for all the single cells within our assay (Figure 1H). In this analysis, one cluster (C3) emerged as significantly enriched in the TI-Treg gene signature (the set of 17 genes identified as TI-Treg MRs in our analysis above) by GSEA (p < 0.05) (Figure 1I). Confirming our predictions, within this C3 / TI-Treg cluster, the ORFs of 11 / 17 candidate MRs were significantly enriched, meaning that by individually overexpressing any of these 11 MRs caused a concurrent change in the entire MR module and overall shift in transcriptional signature closer to TI-Treg cell state. The strongest enrichments were seen with overexpression of FOSL2 (32%, p = 1.3e-15) and TRPS1 (25%, p = 0.0062), compared to only 13% of Tregs harboring negative control that spontaneously acquired this phenotype via anti-CD3/anti-CD28 bead-based stimulation in culture (Figure 1J). Critically, the entire set of TI-Treg MRs was concurrently active in C3 at a single-cell level, regardless of which individual one had been over-expressed (Figure 1K). This result shows that several of the VIPER-inferred MRs play a causal, mechanistic role in reprogramming Treg cell state and suggests that activation or inhibition of the entire MR module may induce even more significant effects.
Candidate MR Validation by In Vivo Pooled CRISPR KO Screen:
To functionally validate whether the computationally predicted MRs are essential for TI-Treg recruitment and/or retention to the TME, we performed an in vivo pooled CRISPR knockout screen using the CHIME (Chimeric Immune Editing) system30. Briefly, we sorted Lin−Sca-1+c-Kit− cells enriched for hematopoietic stem cells (HSCs) from constitutive Cas9-expressing mice, and lentivirally transduced them with a sgRNA library targeting 34 genes, for a total of 102 guides (i.e., 3 guides/gene). Target genes included the 17 MRs described above, 6 randomly selected negative control genes with low baseline expression in T-cells, and 4 positive controls known to be essential in Tregs or in all cells, including Cd4 (CD4 T-cell essential), Foxp3 (Treg essential), and Plk1 and Cdk1 (globally essential) (Fig 2A-B). Guides were cloned in the pXPR_053 vector (see methods), which includes a VexGFP (Vex) fluorophore for transduced cell selection. HSCs were then implanted into irradiated Cas9-tolerized recipients, allowing the immune system to reconstitute de novo over at least 10 weeks, such that all Vex+ immune lineage cells, including Tregs, harbored co-expression of a given guide RNA and Cas9. Syngeneic MC38 colon carcinoma tumors, chosen for their well-established reliance on an intact TI-Treg compartment for in vivo growth38, were implanted and allowed to grow for approximately three weeks. Finally, Vex+ Tregs as well as CD4 Tconv were flow-sorted from the tumor and spleen (control) of each mouse (Fig 2C). The latter was selected as an effective reservoir of P-Tregs—such that differential sgRNA barcode abundance could be compared in TI-Tregs vs spleen P-Tregs. Since this screen was intended for validation rather than discovery, measures were taken to minimize false positive rate, such that negative controls consisted of randomly sampled non-MR genes rather than non-targeting guides, and guide frequencies were compared by permutation-based non-parametric empirical p-value.
Figure 2: Chimeric Immune Editing Mouse Model Enables Validation of Treg Tumor-Infiltration Master Regulators:

(A) Experimental design for in vivo CRISPR KO Validation of TI-Treg MRs (B) List of sgRNAs targeting 17 TI-Treg MRs, 6 negative control genes, and 4 positive control genes. (C) Representative flow cytometry gating for Vex+ CRISPR-transduced Tregs, CD4nonTregs, and CD8+ T cells in spleen and tumor. (D) Correlation of sgDNA frequency distribution between replicates of spleen and tumor Tregs in experimental cohorts 1 (top) and 2 (bottom). Within each cohort, samples of spleen and tumor represent technical replicates of pooled tissue, while the two cohorts are themselves independent biological replicate experiments. (E) Plot of -log10(Bonferroni-corrected p-value) versus p-value rank for gene depletion in P-Tregs versus input plasmid library, where blue indicates positive control genes, red indicates candidate TI-Treg MRs, and grey indicates negative controls. Horizontal dashed line indicates p=0.05 (F) Plot of -log10(Bonferroni-corrected p-value) versus p-value rank for gene depletion in TI-Tregs versus P-Tregs, with color-coding and dashed line as in E. (G) Plot of −log10(Bonferroni-corrected p-value) versus p-value rank for gene depletion in TI-Tregs versus Tumor-Infiltrating CD4nonTregs, with color-coding and dashed line as in E. (H) Tumor growth curves of MCA205 (8x105 implanted subcutaneously) in mice bearing single gene Trps1 sgRNAs (red) versus scrambled sgRNAs (black) in the hematopoietic lineage. Data shown as average across mice (I) Individual growth curves of mice in H, with numbers of mice tumor free (TF) noted. (J) Kaplan-Meier plot for overall survival time of mice in I, showing significant difference in tumor growth (p=0.002 by logrank test).
See also Figure S2.
Upon engraftment and reconstitution of the hematopoietic system, roughly 25-40% of immune cells expressed VexGFP-fluorophore, indicating that most transduced Treg cells harbored a single sgRNA perturbation (Figure S2B). To assess reproducibility, two separate CHIME chimera cohorts were implanted with syngeneic MC38 tumors, the second being implanted with Lin−Sca-1+c-Kit− hematopoietic stem cells from the bone marrow of the first. Tumors in the 2nd cohort were grown for 18 days before CD4+CD25+ Tregs and CD4+CD25− Tconv were sorted from the tumor and spleen of each animal (Figure 2C) and sequenced to assess differences in sgRNA representation. Confirming reproducibility, differential representation of individual sgRNAs in TI- vs. P-Tregs was significantly correlated in the two cohorts (p < 0.01, Figure 2D). All four positive control genes were depleted as expected in Tregs relative to the starting plasmid library (Figure 2E). Of the 17 candidate MR proteins, 8 presented significantly depleted sgRNAs in TI-Tregs vs. spleen P-Tregs—including Trps1, Mafb, Fosl2, Egr3, Gli1, Kdm2b, Nr3c1, and Klf4 (Figure 2F)—suggesting a causal role in Treg tumor infiltration and/or retention to the TME. Frequency distribution of sgRNAs in P-Tregs and TI-Tregs for both experimental cohorts are shown in Figure S2E-F. Critically, 5 of the 8 validated MR candidates—including Trps1, Mafb, Fosl2, Klf4, and Nr3c1—were also significantly depleted in TI-Tregs relative to tumor CD4 Tconv, (Figure 2G), thus supporting their TI-Treg-specific rather than T cell-specific function. Of note, the positive control Foxp3, which is Treg essential but not CD4+ Tconv essential, was significantly depleted in Tregs relative to CD4+ Tconv (Figure 2G). The most statistically significant protein emerging from the comparison of TI-Tregs vs P-Tregs was Trps1 (p = 2.21×10−13), a protein with previously unknown function in T-cells, including Tregs (Figure 2F).
CRISPR KO targeting of Trps1 in Hematopoietic Lineages Inhibits Tumor Growth:
Based on these findings, we further interrogated the phenotype induced by CRISPR KO targeting of Trps1, the MR whose guide RNAs were most significantly depleted in TI-Tregs vs. P-Tregs, and whose expression was sufficient to induce the TI-Treg cell state in naïve human P-Tregs. Specifically, two guide RNAs targeting the encoding gene, Tprs1, were transduced into Cas9-expressing LSKs that were then used to reconstitute the lethally-irradiated bone marrow of thirteen chimeras across two experimental cohorts. As negative controls, we reconstituted the bone marrow of nine mice with LSKs transduced with two non-targeting (scramble) guides. To assess the tumor-agnostic nature of TI-Treg infiltration MRs, we implanted these mice with a complementary syngeneic tumor model, MCA205, representing a well-studied, poorly immunogenic fibrosarcoma39. In initial studies, we found that the MC38 tumor model ultimately experiences spontaneous tumor regression in CHIME mice on longer time-scales even with non-targeting guides, motivating the use of MCA205 as an orthogonal and more immune-resistant tumor, and higher bar for survival effect of targeting Trps1. Confirming the MR’s functional relevance, we observed a significant survival advantage in sgTrps1 mice vs. sgControl mice. In particular, we observed spontaneous, durable tumor rejection (> 60 days) in seven of the thirteen sgTrps1 (54%) but none of the sgControl animals (Figure 2H-I), and overall survival comparison p-value of 0.002 (Figure 2J).
In conjunction with the above-described CRISPR KO and overexpression screens, these data suggest that TRPS1 activity is essential for Tregs to acquire and maintain their infiltrating, immunosuppressive potential in the TME. Supporting the tumor context-specific role for TRPS1, we observed no statistically significant decrease in ex vivo suppressive capacity of Tregs containing Trps1-sgRNAs vs scramble sgRNAs (Figure S2C) and no consistent signs of autoimmunity or immunopathology across peripheral tissues (Skin, Colon, Small Intestine, Liver, and Kidney) in these mice upon histological review by a trained pathologist blinded to sample group (Figure S2D). These data support that the observed immuno-modulatory effects of sgTrps1-ko on Tregs are restricted to the tumor microenvironment.
Systematic Identification of TI-Treg-specific MR-inverter Drugs:
To identify drugs that could specifically inhibit Treg infiltration/retention to the TME by targeting the TI-Treg MR proteins identified by our study such as TRPS1, we generated RNA-seq profiles of sorted human TI-Tregs at 24h following treatment with a library of clinically relevant compounds. To reduce study complexity and cost, we first assessed the effect of a library of 1,554 FDA-approved and investigational compounds on human-derived P-Treg viability at a single, relatively large concentration (5μM). For this screen, human P-Tregs were flow sorted, expanded ex vivo, and drug treated in a 96-well plate format (Figure 3A). We then selected 195 bioactive compounds that inhibited P-Treg viability ≥ 60% (Figure 3B). To further reduce the number of candidate drugs, we then generated 10-point drug response curves to identify the 48h EC20 concentration of the 195 compounds and selected the 86 compounds with the lowest EC20 for efficient perturbational profile analysis in a 96-well format, also considering inclusion of vehicle controls (DMSO). As previously reported40,41, the 48h EC20 (maximum sublethal) concentration was selected to effectively assess each drug’s mechanism of action at 24h, while reducing confounding effects arising from activation of cell stress or death pathways. Finally, the 48h EC20 concentration of each compound was used to perturb TI-Tregs flow-sorted from a treatment-naïve human clear cell carcinoma specimen and expanded, ex vivo, into 96-well plates, followed by RNA-seq profiling using the fully automated PLATE-Seq technology40,41.
Figure 3: High-Throughput Drug Screening Platform Identifies Potential Drug Candidates with Tumor-Treg-Directed Toxicity:

(A) Experimental design of High-Throughput Treg-Directed Drug Toxicity Screen. (B) Results from initial set of 1,554 FDA-approved and investigational oncology compounds screened at single-dose for peripheral Treg growth inhibition, with 195 compounds showing >60% inhibition at 5μM. (C) Viability results of the PLATE-Seq screen, where human tumor Tregs were assessed for growth inhibition on sorted Tumor Tregs at peripheral-Treg EC20 dose, resulting in 7 drugs with higher toxicity in TI-Tregs relative to P-Tregs. Data shown as % viability for each drug vs. DMSO control (D) Heatmap of VIPER protein activity for Tumor vs Peripheral Treg MRs defined in 1E, 1F comparing transcriptional effect of drugs in (C) vs untreated control, with downregulation of nearly all identified Master Regulators by these drugs. (E) Patient-by-Patient Drug predictions according to inversion of patient Tumor Treg vs Peripheral Treg protein activity signature by drug-treatment protein activity signature. Each drug predicted to invert Tumor Treg signature with - log10(Bonferroni-Corrected p-value) < 0.01 in a particular patient is colored red. Patients are grouped by tumor type. Subset to drugs identified by tumor Treg growth screen in (C), with columns colored by tumor type and clustered by unsupervised hierarchical clustering.
See also Figures S3 and S4.
Viability data, as well as perturbational RNA-seq profiles of TI-Tregs were collected (Figure 3C-D). Based on the differential protein activity signature in drug- vs. vehicle control-treated TI-Tregs, we identified compounds capable of inducing statistically significant inactivation of TI-Treg-specific MR proteins, using the OncoTreat algorithm31. From this analysis, 32 compounds were nominated as statistically significant inhibitors of the 17-MR TI-Treg signature (p < 1E-5), which includes TRPS1 (Figure S3A). Of these, seven preferentially depleted TI-Treg vs. P-Treg viability in vitro (Figure 3C-D) and were also predicted by OncoTreat to inhibit the TI-Treg vs P-Treg MRs signature—on a patient-by-patient basis—across all tumor types and nearly all patients (Figure 3E). Of these, three (i.e., gemcitabine, triapine, and floxuridine) were among the seven most significant TI-Treg MR activity inhibitors—as predicted across all 36 patients in the study (Figure S3B)—and also among the top 6 inducing the most significant differential TI- vs. P-Treg viability reduction in vitro (Figure 3C).
Dose response curves of these three drugs revealed that only gemcitabine had a gradual dose-dependent effect on Treg viability reduction, as a function of its concentration, while the other two had sharp elbows consistent with a. threshold effect that would challenge appropriate concentration selection for in vivo studies (Figure S4A-C). In addition, floxuridine had cytostatic rather than cytotoxic, activity even at high concentration, and neither floxuridine nor triapine were confirmed to affect overall survival in MC38 mouse model (Figure S4D). Surprisingly, gemcitabine was predicted to drive TI- to P-Treg signature conversion, including TRPS1 inhibition, at a remarkably low concentration in vitro (10nM) (Figure S3B), which is much lower than the concentration achieved at clinical doses. As a result, we focused on this drug for in vivo validation purposes.
Immunomodulatory effects of low dose gemcitabine contribute to its efficacy and synergy with immunotherapy:
To validate the preferential TI-Treg targeting of gemcitabine in vivo, we implanted C57BL/6J mice subcutaneously with MC38 syngeneic tumors, and initiated therapy 12 days later, a “late stage” of growth when MC38 tumors are resistant to anti-PD-1 immunotherapy42. Low-dose gemcitabine was administered intra-peritoneally (IP) on days 12, 15, and 18, at 12 mg/kg, representing ~1/10th of the lowest conventional clinically-relevant dose in mice (120 mg/kg)43,44 In an additional treatment arm, mice received gemcitabine in combination with anti-PD-1 administered IP on days 12, 15, and 18 (Figure 4A). As expected, late stage MC38 tumors failed to respond to anti-PD-1. However, single agent low-dose gemcitabine temporarily controlled MC38 progression, conferring a significant reduction in growth kinetics (p = 0.003) and prolongation of survival (p = 0.006) relative to vehicle-treated mice (Figure 4A-D). In combination, low-dose gemcitabine sensitized late stage MC38 tumors to anti-PD-1, achieving complete responses in 50% of animals, translating to a significant survival advantage compared to gemcitabine alone (p = 0.009) (Figure 4A-D).
Figure 4: Low-Dose Gemcitabine is Immunogenic and Potentiates anti-PD-1 Therapy:

(A) Schematic of in vivo validation studies. Experiment consists of 6 mice per group. (B) Tumor growth curves for each treatment group, (C) Kaplan-Meier survival curves, and (D) forest-plot showing the result of multiple cox regression assessing treatment effect on time-to-death for each of the treatments described in (A). Hazard ratios are shown with 95% confidence interval and p-value. Results are representative of two independent experiments. (E) Tumor growth and Kaplan Meier survival curves of NSG mice, C57BL/6J mice, and C57BL/6J mice exposed to anti-PD-1 therapy receiving the indicated dose of gemcitabine between 1-10 mg/kg. Statistical significance for survival was calculated by Mantel-cox log rank test. (F) Experimental design of flow cytometry experiment. (G) Overall flow cytometry clustering of tumor immune cells. (H) Stacked barplot of frequencies for clusters shown in G, split by timepoint and gemcitabine dose. (I) Stacked barplot of frequencies for clusters shown in G, split by timepoint and gemcitabine dose. (J) Violinplot of Treg absolute numbers per mg of tumor, split by timepoint and gemcitabine treatment dose, where * indicates p<0.05 and * indicates p<0.01 by two-way ANOVA with multiple testing correction. (K) Violinplot of Treg absolute numbers per mg of spleen, split by timepoint and gemcitabine treatment dose. (L) Violinplot of Helios+CD103+ TI-Treg cluster absolute numbers per mg tumor 24 hours post-treatment, split by gemcitabine dose. (M) Tumor growth and Kaplan Meier survival curves of NSG mice, RAG1-KO mice, C57BL/6J mice, and C57BL/6J mice exposed to anti-PD-1 therapy receiving the indicated dose of gemcitabine between 0-120 mg/kg. Statistical significance for survival was calculated by Wilcoxon test. All significant pairwise comparisons (p < 0.05) are shown.
See also Figures S4 and S5.
To assess whether low-dose gemcitabine effects were immune-mediated, we performed parallel dose titrations in immune-competent C57BL/6J mice and severely immune-deficient NSG (NOD.Cg-Prkdcscid I12rgtm1Wjl/SzJ) mice, lacking both innate and adaptive immunity. At a clinically-relevant dose (120 mg/kg)43,44 gemcitabine inhibited tumor growth in both C57BL/6J and NSG mice relative to vehicle control (p < 0.001, by Cox regression analysis) with no significant difference between the two strains (p = 0.19, Figure S4E-F). We found efficacy was lost in both strains between the range of 12 mg/kg and 1.2 mg/kg, with a modest advantage in C57BL/6 mice at 12 mg/kg (p = 0.012) and a trending advantage at 1.2 mg/kg (p = 0.09 Figure S4F). Though modest, these data are in line with previous observations in immunodeficient nude mice45, suggesting the therapeutic effects of low-dose gemcitabine are at least in part due to its immunomodulatory properties.
To test whether immune-dependent activity could be observed in this range of concentrations, we dosed cohorts of mice with 1-10mg/kg gemcitabine, with an additional cohort of C57BL/6J mice receiving anti-PD-1 in combination. We found that doses as low as 3 mg/kg, which lack any activity in NSG mice (p = 0.84), reveal sensitivity to anti-PD-1 via tumor growth kinetic reduction (p = 0.01) and enhanced survival (p = 0.0029) in the combination group (Figure 4E). As above, the benefit observed in C57BL/6J vs. NSG strains is modest (p = 0.253) however in immune-competent mice this dose is sufficient to augment anti-PD-1 therapy to achieve curative responses and a significant enhancement of survival relative to gemcitabine or anti-PD-1 alone (p = 0.048, p = 0.005, respectively; Figure 4A-D). Taken together, these data show that low-dose gemcitabine is in part dependent upon host immunity and effectively, sensitizes anti-PD-1 resistant MC38 tumors to immune checkpoint blockade therapy.
Low-dose gemcitabine selectively targets TI-Tregs in vivo:
To better understand the immunomodulatory effects of low dose gemcitabine and evaluate whether they are restricted to TI-Tregs, we performed high-parameter flow cytometry analysis of tumors and spleens from MC38 tumor-bearing C57BL/6J mice post-gemcitabine treatment, utilizing a 34-parameter spectral flow cytometry panel. We evaluated tissues from mice receiving high-dose gemcitabine (120 mg/kg), low-dose gemcitabine (12 mg/kg) or a minimally effective dose of gemcitabine (3 mg/kg), as well as vehicle-treated controls, and evaluated immediate effects 24 hours post-treatment, as well as delayed/secondary effects at 48- and 72-hour post-treatment (Figure 4F-I, S5A-C). Though gemcitabine elicits compositional changes throughout the CD45+ infiltrate (Figure 4G-H), Tregs are the only immune subset significantly reduced in absolute number in tumors by low-dose gemcitabine (Figure 4J; S5D). Splenic Tregs are not reduced in number by gemcitabine at any dose, confirming the inhibitory effects of low-dose gemcitabine are restricted to Tregs infiltrating the tumor (Fig 4K). Our data suggest this tumor-specific effect is not solely based on greater Treg proliferation within tumors, as gemcitabine inhibited Ki67 staining in Tregs in both the tumor and the periphery (Fig S5E-F). Rather, low-dose gemcitabine inhibited a specific phenotypic subset revealed by unsupervised clustering on 15 phenotypic and functional Treg markers, primarily defined by expression of Helios and CD103 (Figure 4L, S5G) Prior studies showed that Helios+CD103+GITR+ TI-Tregs are the most potently suppressive Treg subset in tumors72. With respect to compositional effects of gemcitabine on other immune populations, we noted a putative transient recruitment of polymorphonuclear cells (PMNs) from spleen to tumor (Figure S5H), transient reduction in tumor-infiltrating NK cells at maximum dose (Figure S5I), and induced differentiation of monocytes to macrophages in the tumor (Figure S5J-N;). Notably, none of these populations exhibited differential effects from low-dose gemcitabine, as was observed for TI-Tregs in this system.
To test whether gemcitabine-mediated myeloid modulation, as observed in our flow cytometry data, may confer therapeutic benefit independent of Tregs, we compared effects of low- vs high-dose gemcitabine on MC38 growth in Rag1−/− vs. NSG and C57BL/6J mouse strains. We found identical MC38 growth kinetics in Rag1−/− vs. NSG mice, supporting the notion that the effects of low-dose gemcitabine are largely T cell mediated, in line with a prior report (Figure 4M)45. Taken together, these data suggest that the ability of low-dose gemcitabine to augment responses to immunotherapy in this preclinical model correlate broadly with its effect on TI-Tregs.
Low-Dose gemcitabine targets the TI-Treg MR signature:
To delineate the mechanism by which low-dose gemcitabine modulates TI-Treg frequency and acquisition of the TI-Treg transcriptional phenotype, we generated single cell RNA-seq (scRNA-seq) profiles from MC38 tumor- and spleen-derived Tregs, at 24 hours after treatment with a single 12 mg/kg dose of either gemcitabine or vehicle control (Figure 5A). For this study, we implanted FoxP3Yfp-Cre mice with MC38 tumor cells to facilitate specific flow-sorting of CD4+ FoxP3+ Tregs from tumor and spleen using YFP as a FoxP3 expression marker. Using 5-mouse per group, we obtained high quality profiles from ~10,000 spleen-derived and ~1,000 tumor-derived Tregs from each group (Figure S6A). While raw gene expression data were noisy (Figure S6B), Protein activity-based cluster analysis stratified the cells into five clusters (TRC1 – TRC5) (Fig 5B, S6C), with cluster TRC3 highly enriched for human TI-Treg MRs including TRPS1 (Figure 5C-D). Notably, this cluster also had highest expression of IKZF2 (Helios), concordant with the gemcitabine-sensitive population of Tregs observed by flow cytometry (Figure 5E). In vehicle-treated control animals, the TRC3 cluster comprised 7.8% of splenic Tregs vs. 30.1% of TI-Tregs (p = 1.8×10−84). Gemcitabine treatment reduced TRC3 frequency by ~50%, to 14.9% of the TI-Treg cells, while inducing virtually no change in the spleen population (Figure 5F-G). Furthermore, treatment resulted in a proportional increase in TRC1 occupancy, which exhibits signs of interferon exposure (high IFI16 activity). These data suggest low-dose gemcitabine has antagonistic effects on TI-Tregs and prevents TI-Treg MR activity in vivo.
Figure 5: Single-Cell RNA-Sequencing Suggests Low-Dose Gemcitabine Depletes TI-Tregs.

Exhibiting high TI-Treg Master-Regulator Activity: (A) Schematic of experimental workflow. (B) UMAP plot and unsupervised clustering by VIPER-inferred protein activity of Tregs from untreated and gemcitabine-treated tumor and spleen. (C) Heatmap of cell-by-cell protein activity for each Tumor-Treg MR identified by scRNA-seq, grouped by cluster. (D) Distribution of the 17-gene TI-Treg MR signature normalized enrichment score by Gene Set Enrichment Analysis (GSEA), grouped by cluster, such that cluster TRC3 is most enriched for the TI-Treg MR signature. (E) Distribution of IKZF2 (Helios) Normalized Gene Expression, grouped by cluster, such that the cluster TRC3 has highest expression. (F) Barplot of cluster frequencies in each sample, such that cluster TRC3 has a baseline frequency of 7.8% in spleen of vehicle-control sample and 30.1% in tumor (p = 1.78e-84), with frequency of only 14.9% in tumor of gemcitabine-treated sample (p = 1.51e-20). (G) Cox proportional hazard ratios of cluster TRC3 frequencies in vehicle vs gemcitabine treated mice in tumor (OR = 0.407 [95% CI: 0.334-0.494]) and spleen (p = 0.242, OR = 1.063 [95% CI: 0.958-1.17]).
See also Figure S6.
Discussion
Treg immunosuppression in the TME is a major barrier to antitumor immunity and undermines efficacy of checkpoint blockade immunotherapy, which is effective only in a minority of cancer patients2,46. To address the critical need for more effective agents to counteract human TI-Treg number or function, we harnessed new tools to identify and validate previously unappreciated regulators of TI- vs. P-Treg transcriptional state. Protein activity analysis—using the VIPER algorithm on a novel dataset of TI-Tregs, P-Tregs, and additional CD4+ and CD8+ non-Treg controls across 36 patients—identified a set of TI-Treg MRs functionally validated by a pooled in vivo CRISPR screen. Most significant among the validated targets was TRPS1, a transcription factor not previously studied in the context of Treg biology. In parallel, we conducted a systematic ex vivo drug screen and found gemcitabine possesses preferential cytotoxic capacity against TI-Tregs and inhibits transcriptional activity of TI-Treg MRs, including TRPS1, across multiple tumor types. In vivo validation studies confirmed that sub-clinical doses of gemcitabine, lacking activity in immune deficient animals, effectively potentiated immune checkpoint blockade-mediated control of established, anti-PD-1 resistant MC38 tumors. These findings have implications for both basic understanding of TI-Treg biology as well as clinical use of available chemotherapeutics for the purpose of modulating TI-Treg activity. Critically, they provide a highly generalizable integrative framework, both computational and experimental, to identify critical, pharmacologically actionable dependencies of other tolerogenic subpopulations in the TME.
While our findings showing immune-modulating properties for gemcitabine are broadly consistent with prior reports, they provide critical mechanism-based insight into these effects by elucidating TI-Treg-specific activity of previously unreported MR proteins, especially TRPS1, whose activity is inhibited by gemcitabine. While in agreement with prior observations that gemcitabine antagonizes Tregs in mouse and man, our findings also clarify that, at low doses, gemcitabine is differentially toxic in TI- vs. P-Tregs, which was not fully investigated in prior studies.
Specifically, early studies showed clinically equivalent doses of gemcitabine systemically decrease MDSC and B cell numbers without substantial effects on T cells, and in fact promote T cell trafficking into tumors47,48,49. In multiple pre-clinical models, tumor growth in T cell-deficient nude mice or specific CD8+ T cell depletion rendered gemcitabine less effective, suggesting that gemcitabine exhibits T cell-dependent immunogenic activity in addition to direct tumoricidal killing45. Informed by prior investigation into immunogenic effects of low dose or metronomic dosing of other chemotherapeutic agents such as cyclophosphamide50,51,52 or oxaliplatin53, more recent studies have shown that sub-clinical “low” doses of gemcitabine are immunomodulatory in various ways, with effects on NK cell function54, myeloid polarization55,56 and Tregs57,58,59,60,61. However, the mechanisms and effector proteins underlying manifestation of these immunomodulatory effects were not previously identified.
Additional studies are required to more fully understand how gemcitabine selectively modulates TI-Treg MRs. Furthermore, our gemcitabine titration studies in immunocompetent C57BL/6 versus severely immunodeficient NSG mice defined a more narrow “low dose” range at which gemcitabine is primarily immunomodulatory, building upon previous studies in nude mice that were confounded by the presence of functional NK and myeloid cells, as these are also known to be modulated by gemcitabine45,54. Our flow cytometry profiling and therapeutic studies further inform these prior observations and confirm a T cell-mediated immunomodulatory effect by showing lack of response to low-dose gemcitabine in RAG1−/− mice. We found the range between 3-10 mg/kg of gemcitabine dosed Q3D to be immunogenic, which represents 2.5-8.3% of the standard murine maximum tolerated dose of 120 mg/kg, and roughly translates to a human equivalent dose62 of 9-30 mg/m2, as compared to the standard clinical dose of 1,000 mg/m2. Although we fully acknowledge the challenges of translating dosing strategies between species, our studies support the development of dose-finding studies of gemcitabine in combination with immune modulating agents such as anti-PD-1, particularly in settings where the benefit of anti-PD-1 monotherapy is sub-optimal or in long-term maintenance therapy.
A major finding of our study was the discovery and validation of TRPS1 as a putative master regulator of TI-Tregs, such that CRISPR KO targeting of Trps1 inhibits tumor Treg infiltration without depleting peripheral Tregs, preferentially inhibits TI-Tregs relative to tumor CD4+ Tconv, and inhibits overall tumor growth, with a 54% cure rate in MCA205 tumor model without exogenous intervention. Conversely, over-expression of TRPS1 induces the entire module of TI-Treg MRs and drives P-Tregs toward a TI-Treg transcriptional phenotype. TRPS1 is a transcription factor classically linked to skeletal development, as subjects with germline alterations in the Trps1 gene suffer from autosomal dominant trichorhinophalangeal syndromes with characteristic craniofacial abnormalities63,64. More recently, TRPS1 has been implicated in tumorigenesis in breast cancer65,66 and osteosarcoma67 potentially through promotion of dysregulated cell replication resulting in accumulation of genomic aberrations68. Functionally, TRPS1 is thought to function uniquely as a transcriptional repressor via its GATA domain69, although notably TRPS1 contains two Ikaros-like domains whose specific functions are poorly characterized. Other Ikaros family proteins including Helios and Aiolos are expressed in hematopoietic tissues with important functions in Treg differentiation and function70,71,72, thus it is tempting to speculate that TRPS1 governs TI-Treg activities via its Ikaros domain. At this point, the specific functions of TRPS1 in Tregs remain to be described and additional future work is warranted to understand mechanisms of TRPS1 regulation in Tregs both within and outside of the tumor microenvironment. Additionally, our results support the design of specific inhibitors of TRPS1 activity. As compared to Treg-targeting agents in clinical translation such as CCR8, which is relatively TI-Treg-specific but not required for Treg function16,19, TRPS1 as a putative target has the benefit of being TI-Treg specific, functionally required for TI-Treg recruitment and/or retention, present in TI-Tregs across multiple cancer types, and in certain cases also present in malignant cells65,66,67.
Together, the integrative systems biology approach proposed here—combining CRISPR validation of putative regulatory proteins in an in vivo functional genomics system with ex vivo drug screening and transcriptional profiling of treatment response—provides a highly generalizable framework for the systematic discovery of Treg-directed immunotherapy targets. Of note, this platform could in theory be extended to other tolerogenic cell types in the TME, opening up additional possibilities for target identification and validation across the field of immune-oncology. Furthermore, our PLATE-Seq screening method can be feasibly extended to a significantly larger compound libraries, thus supporting discovery of additional TI-Treg modulating compounds. While the development of TRPS1-directed therapeutics will require additional effort, our findings on low-dose gemcitabine are readily translatable to human studies aimed at improving the clinical activity of anti-PD(L)-1 agents in the clinic.
Limitations of the study
Future follow-up studies may further clarify the mechanism of TRPS1 function in TI-Tregs in vivo by generating Treg-lineage-specific TRPS1 KO mice and performing further scRNA-seq and immunophenotyping in these mice across tissue contexts. The loss of TRPS1 across all immune lineages in our present model represents a limitation of the study such that potential contribution of TRPS1 KO in other immune cell types to improved overall survival cannot entirely be ruled out. Relatedly, evidence for Tregs mediating response to low-dose gemcitabine in this study is correlative based on observed depletion of intratumoral Tregs by flow cytometry, and of the TI-Treg transcriptional sub-phenotype in particular by scRNA-seq. We cannot fully rule out contribution of effects on other immune cell subtypes, downstream of or in parallel to the observed effect on TI-Tregs, as contributing to overall treatment response. Finally, differences in engraftment rate of sgRNA-bearing stem cells in our Chimeric mouse model result in lower statistical power for certain proteins in the pooled screen. As a result, while we have functionally validated 8 of the predicted TI-Treg MRs, including TRPS1, further repeat cohorts may validate additional predicted MRs which had lower baseline engraftment rate in our study.
STAR Methods
Resource Availability
Lead contact
Requests for further information and reagents should be directed to the lead contact, Dr. Andrea Califano (ac2248@cumc.columbia.edu).
Materials availability:
Plasmids from this article will be available from Addgene following publication.
Data and code availability:
All data reported on in this manuscript are available in a public Mendeley Data repository at DOI: 10.17632/vnrsbb4gk9.1 as of the date of publication, grouped by experiment. VIPER algorithm used for data analysis is publicly available as an R package on Bioconductor and single-cell VIPER helper functions are available as part of previously published workflow on github All original code has been deposited on Mendeley. DOIs in Key Resources Table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse TCR-β BUV395 (clone H57-597) | BD | Cat# 742485 |
| Anti-Mouse CD103 BUV496 (clone M290) | BD | Cat# 741083 |
| Anti-Mouse CD44 BUV563 (clone IM7) | BD | Cat# 741227 |
| Anti-Mouse PD-1 BUV615 (clone J43) | BD | Cat# 752299 |
| Anti-Mouse Nrp1 BUV661 (clone V46-1954) | BD | Cat# 752461 |
| Anti-Mouse/Human Ki67 BUV737 (clone B56) | BD | Cat# 567130 |
| Anti-Mouse CD4 BUV805 (clone GK1.5) | BD | Cat# 612900 |
| Anti-Mouse CD39 BV421 (clone Y23-1185) | BD | Cat# 567105 |
| Anti-Mouse IA-IE Pacific Blue (clone M5/114.15.2) | BioLegend | Cat# 107620 |
| Anti-Mouse ST2 BV480 (clone U29-93) | BD | Cat# 746701 |
| Anti-Mouse CD8 Pacific Orange (clone 5H10) | ThermoFisher Scientific | Cat# MCD0830 |
| Anti-Mouse CD62L BV570 (clone MEL-14) | BioLegend | Cat# 104433 |
| Anti-Mouse CD11c BV605 (clone N418) | BioLegend | Cat# 117334 |
| Anti-Mouse ICOS BV650 (clone C398.4A) | BioLegend | Cat# 313550 |
| Anti-Mouse KLRG1 BV750 (clone 2F1) | BD | Cat# 746972 |
| Anti-Mouse PD-L1 BV785 (clone MIH5) | BD | Cat# 741014 |
| Anti-Mouse iNOS FITC (clone REA982) | Miltenyi Biotec | Cat# 130-116-357 |
| Anti-Mouse CD45 A532 (clone 30-F11) | ThermoFisher Scientific | Cat# 58-0451-82 |
| Anti-Mouse CD19 NB610 (clone 1D3) | ThermoFisher Scientific | Cat# M004T02B06 |
| Anti-Mouse Ly6C PerCP-Cy5.5 (clone HK1.4) | BioLegend | Cat# 128012 |
| Anti-Mouse CD206 PerCPeF710 (clone MR6F3) | ThermoFisher Scientific | Cat# 46-2069-42 |
| Anti-Mouse/Human TOX PE (clone REA473) | Miltenyi Biotec | Cat# 130-120-716 |
| Anti-Mouse Ly6G SYG563 (clone 1A8) | BioLegend | Cat# 127668 |
| Anti-Mouse Helios PE-Dazzle 594 (clone 22F6) | BioLegend | Cat# 137232 |
| Anti-Mouse CD80 PE-Cy5 (clone 16-10A1) | BioLegend | Cat# 104712 |
| Anti-Mouse FoxP3 PE-Cy7 (clone FJK-16s) | ThermoFisher Scientific | Cat# 25-5773-82 |
| Anti-Mouse/Human B220 PE/Fire 810 (clone RA3-6B2) | BioLegend | Cat# 103287 |
| Anti-Mouse/Human TCF-1 APC (clone C63D9) | Cell Signaling Technologies | Cat# 37636S |
| Anti-Mouse CD69 SNIR685 (clone H1.2F3) | BioLegend | Cat# 104558 |
| Anti-Mouse/Human CD11b A700 (clone M1/70) | BioLegend | Cat# 101222 |
| Anti-Mouse F4/80 APC/Fire 750 (clone BM8) | BioLegend | Cat# 123152 |
| Anti-Mouse NK1.1 APC/Fire 810 (clone S17016D) | BioLegend | Cat# 156519 |
| TruStain FcX PLUS (clone S17011E) | BioLegend | Cat# 156604 |
| Anti-Mouse Ter-119 PE (clone TER-119) | BioLegend | Cat# 116208 |
| Anti-Mouse/Human CD11b PE (clone M1/70) | BioLegend | Cat# 101208 |
| Anti-Mouse Gr-1 PE (clone RBC-8C5) | BioLegend | Cat# 108408 |
| Anti-Mouse CD3ε PE (clone 145-2C11) | BioLegend | Cat# 100308 |
| Anti-Mouse CD5 PE (clone 53-7.3) | BioLegend | Cat# 100608 |
| Anti-Mouse/Human B220 PE (clone RA3-6B2) | BioLegend | Cat# 103208 |
| Anti-Mouse CD117 (c-kit) APC (clone 2B8) | BioLegend | Cat# 105812 |
| Anti-Mouse Sca-1 BV421 (clone D7) | BioLegend | Cat# 108127 |
| Anti-Human CD4 BV421 (clone OKT-4) | BioLegend | Cat# 317434 |
| Anti-Human CD25 APC (clone M-A251) | BioLegend | Cat# 356110 |
| Anti-Human CD127 PE (clone A019D5) | BioLegend | Cat# 351304 |
| Anti-Mouse CD25 BV421 (clone PC61) | BioLegend | Cat# 102043 |
| Anti-Mouse CD3 BV711 (clone 145-2C11) | BioLegend | Cat# 100349 |
| Anti-Mouse CD8 PE (clone 53-6.7) | BioLegend | Cat# 100708 |
| Anti-Mouse CD127 PE-Cy7 (clone SB/199) | BioLegend | Cat# 121119 |
| Anti-Mouse CD4 APC (clone GK1.5) | BioLegend | Cat# 100411 |
| InVivoMAb Anti-Mouse PD-1 (clone RMP1-14) | BioXCell | Cat# BE0146 |
| Bacterial and virus strains | ||
| NEBstable Competent E. coli | NEB | Cat# C3040H |
| Endura Electrocompetent cells | Lucigen | Cat# 60242-1 |
| Biological samples | ||
| Healthy donor PBMC buffy coats | New York Blood Center | https://www.nybc.org/ |
| ccRCC Nephrectomy Specimens | Columbia University Irving Medical Center | |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human IL-2 | PeproTech | Cat# 200-02 |
| Recombinant Mouse TPO | PeproTech | Cat# 315-14 |
| Recombinant Mouse SCF | PeproTech | Cat# 250-03 |
| Recombinant Mouse Flt3-L | PeproTech | Cat# 250-31L |
| Recombinant Mouse IL-7 | PeproTech | Cat# 217-17 |
| RetroNectin Recombinant Human Fibronectin | Takara Bio | Cat# T100B |
| FDA-approved and Investigational Oncology Drug Compound Library | Selleckchem | Cat# N/A (custom) |
| Gemcitabine (Ly-188011) | Selleckchem | Cat# S1714 |
| Triapine | Selleckchem | Cat# S7470 |
| Floxuridine (NSC 27640) | Selleckchem | Cat# S1299 |
| SYTOX Green Ready Flow Reagent | ThermoFisher Scientific | Cat# R37168 |
| CellTrace Violet Proliferation Kit | ThermoFisher Scientific | Cat# C34557 |
| Ficoll-Paque PLUS (1.077 g/mL) | GE Healthcare | Cat# 17144003 |
| Mitomycin C | Millipore Sigma | Cat# 10107409001 |
| DNase I | Roche/Sigma | Cat# 10104159001 |
| Collagenase D | Roche/Sigma | Cat# 11088866001 |
| LiveDead Fixable Blue Dye | ThermoFisher Scientific | Cat# L34962 |
| BD Brilliant Stain Buffer Plus | BD | Cat# 566385 |
| FoxP3 Fixation/Permeabilization Kit | eBioscience/Thermo | Cat# 00-55214-00 |
| TruStain Monocyte Blocker | BioLegend | Cat# 426103 |
| Fugene HD | Pro mega | Cat# E2312 |
| RNAseA | Qiagen | Cat# 19101 |
| Biotium EVAGREEN DYE 20X IN WATER 1 ML | Fisher Scientific | Cat# NC0521178 |
| GeneJET Gel Extraction Kit | Fisher Scientific | Cat# FERK0692 |
| Gibson Assembly® Master Mix | NEB | Cat# E2611L |
| KAPA HiFi HotStart PCR Kit, with dNTPs | Kapa Biosystems | Cat# KK2502 |
| NucleoBond® Xtra Midi EF (50 preps) | Macherey-Nagel | Cat# 740420.50 |
| Puradisc 25 mm PES Syringe Filters | Cytiva | Cat# 6780-2504 |
| RIPA BUFFER | Teknova | Cat# R3792 |
| Phenol:Chloroform:Isoamyl Alcohol (25:24:1, v/v) | Invitrogen | Cat# 15593031 |
| KAPA HiFi HotStart ReadyMix (2X) | Kapa Biosystems | Cat# KK2612 |
| Critical commercial assays | ||
| CD4+ T cell Isolation Kit, Human | Miltenyi Biotec | Cat# 130-096-533 |
| Human Treg Expander DynaBeads | Gibco | Cat# 11129D |
| Human Tumor Dissociation Kit | Miltenyi Biotec | Cat# 130-095-929 |
| Mouse Tumor Dissociation Kit | Miltenyi Biotec | Cat# 130-096-730 |
| 10x Chromium Next GEM Single Cell 3’ Kit v3.1 | 10x Genomics | Cat# 1000269 |
| Deposited data | ||
| Sorted T-cell Populations RNA-Seq From Patient Blood & Tumor | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| Overexpression Screen Single-Cell RNA-Seq Data | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| Gemcitabine Treatment Single-Cell RNA-Sequencing Data | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| sgRNA counts from CRISPR KO experiment | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| Drug Screen PLATE-Seq Data | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| T-cell ARACNe Network | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| Experimental models: Cell lines | ||
| MC38 | Kerafast | Cat# ENH204-FP |
| MCA205 | Millipore Sigma | Cat# SCC173 |
| HEK293T | ATCC | Cat# CRL-11268 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain# 000664 |
| Mouse: H11-Cas9 (Igs2tm1.1(CAG-cas9*)Mmw/J) | The Jackson Laboratory | Strain# 027650 |
| Mouse: NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Strain# 005557 |
| Mouse: Rag1-KO (B6.129S7-Rag1tm1Mom/J | The Jackson Laboratory | Strain# 002216 |
| Mouse FoxP3-YFP/Cre (B6.129(Cg)-Foxp3tm4(YFP/icre)Ayr/J | The Jackson Laboratory | Strain# 016959 |
| Recombinant DNA | ||
| pXPR_053 | Addgene | Cat# 113591 (PubMed 30971695 |
| PsPAX2 | Addgene | Cat# 12260 (gift from Didier Trono) |
| pMD2.G | Addgene | Cat# 12259 (gift from Didier Trono) |
| Tet-O-FUW-EGFP | Addgene | Cat# 30130 (PubMed 20107439) |
| Tet-O-FUW-EGFP-P2A-mCherry | This paper | N/A |
| FUW-M2rtTA | Addgene | Cat# 20342 (PubMed 18786421) |
| Human CSRNP2 ORF clone | GenScript | Cat# OHu04521 |
| Human TRPS1 ORF clone | GenScript | Cat# OHu21177 |
| Human BANP ORF clone | GenScript | Cat# OHu10822 |
| Human MAFB ORF clone | GenScript | Cat# OHu25119 |
| pFUW-tetO-NR4A3 | Addgene | Cat# 139818 (PubMed 30530727) |
| GFP-FBXL10 (KDM2B) | Addgene | Cat# 126542 (PubMed 29985131) |
| KLF4 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| STAT4 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| NR4A1 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| EGR1 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| ZEB2 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| EGR3 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| PBX4 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| ID2 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| FOSL2 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| NR3C1 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| GLI1 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| Tet-O-FUW-CSRNP2-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-TRPS1-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-BANP-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-MAFB-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-NR4A3-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-KDM2B-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-KLF4-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-STAT4-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-NR4A1-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-EGR1-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-ZEB2-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-EGR3-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-PBX4-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-ID2-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-FOSL2-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-NR3C1-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-GLI1-P2A-mCherry | This paper | N/A |
| Software and algorithms | ||
| R v3.6.2 | https://cran.r-project.org/bin/macosx/ | |
| ARACNe | Margolin, et al. | http://califano.c2b2.columbia.edu/aracne |
| VIPER | Alvarez, et al. | http://califano.c2b2.columbia.edu/viper |
| GraphPad Prism v9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo v10.8.1 | BD | https://www.flowjo.com/ |
| Single-Cell VIPER | Obradovic, et al. | https://github.com/Aleksobrad/single-cell-rcc-pipeline |
| Custom Analysis Scripts | This paper | DOI: 10.17632/vnrsbb4gk9.1 |
Experimental model and subject details
Cell lines
The HEK293T cell line (female) was mycoplasma tested before the lentiviral viral production. Cells were maintained in a 5% CO2, 95% air, humidified incubator at 37°C, in DMEM supplemented with 1X penicillin-streptomycin and 10% FBS (Sigma, F2442). MC38 colon carcinoma cells (female) were purchased from Kerafast and maintained in a 5% CO2, 95% air, humidified incubator at 37°C in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin. MCA205 fibrosarcoma cells (unknown sex) were purchased from Millipore Sigma and maintained in a 5% CO2, 95% air, humidified incubator at 37°C in RPMI supplemented with 10% FBS, 100 U/mL penicillin, 100 mg/mL streptomycin, 1 mM sodium pyruvate, 100 μM non-essential amino acids, and 0.055 nM 2-mercaptoethanol. MC38 and MCA205 cells utilized in in vivo experiments were under 5 passages from purchase.
Primary cell cultures
Human PBMC-derived regulatory T cells were freshly sorted from healthy donor whole buffy coats obtained from the New York Blood Center, or from treatment-naïve clear cell renal cell carcinoma tissues received from patients undergoing standard of care nephrectomy at Columbia University Irving Medical Center. Demographic information relating to donors was kept blinded to researchers, however all donors underwent routine pathogen screening and were found negative. Flow sorted cells were cultured in a 5% CO2, 95% air, humidified incubator at 37°C in X-VIVO 15 (Lonza) supplemented with 10-500 U/mL recombinant human IL-2 (PeproTech) at a density of 35,000 – 200,000 cells/well in 96-well plates.
Animals
Male and female C57BL/6J (Strain #000664), H11-Cas9 (Igs2tm1.1[CAG-cas9*]Mmw/J; Strain #027650), RAG1-KO (B6.129ST-Rag1tm1Mom/J; Strain #002216), NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; Strain #005557), and FoxP3YFP/Cre (B6.129(Cg)-Foxp3tm4(YFP/icre)Ayr/J; Strain #016959) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were 6-8 weeks old at time of use. All animals were housed and bred in strict accordance with NIH and American Association of Laboratory Animal Care regulations. All experiments and procedures for this study were approved by the Columbia University Medical Center Institutional Animal Care and Use Committee (IACUC).
Method details
Clinical Sample Collection, Sorting, and RNA-Sequencing:
These methods relate to data as shown in Figure 1. Tissue was collected from treatment-naïve resected tumors across patients with four tumor types, including 8 patients with glioblastoma multiforme, 8 patients with clear cell renal carcinoma, 8 patients with bladder cancer, and 12 patients with prostate cancer (from radical prostastectomies). For prostate cancers, since it can be difficult to identify tumor from freshly sectioned prostatectomies, for each case, prostates were inked and sliced fresh, 8 mm punches were taken, and a thin slice was taken for frozen section to ensure the presence of tumor in adjacent tissues used for cell dissociation and flow cytometry. For each patient, 50ml of peripheral blood was drawn same day as tumor resection. Tumors were dissociated with the GentleMACS OctoDissociator following manufacturer’s instruction, and subsequently Tregs and CD8+ T-cells were flow-sorted from tumor along with Tregs, naïve CD4nonTregs, and naïve CD8+ T cells from peripheral blood. An aliquot of flow sorted naïve CD8+ and CD4+ non-Treg were stimulated ex vivo with anti-CD3/anti-CD28 beads for 72 hours to induce T-cell activation. Flow-sorted and ex-vivo-stimulated populations were processed to prepare RNA-Seq libraries. RNA-Seq libraries were generated using the Nugen Ovation RNA-Seq System v2 kit (Nugen). Libraries were sequenced on an Illumina HiSeq 2500 with paired end 2 × 100 bp reads. RSEM (v1.2.8–1.2.9) was used with bowtie2 to derive gene-level expression measures, represented as posterior transcripts per million (pmeTPM).
Gene Expression and VIPER Analysis:
Gene Expression was combined across all samples and scaled to log10(Transcripts Per Million + 1). Gene Expression was subsequently scaled across rows by z-score transformation and used as input for Principal Component Analysis (Figure 1B) and differential gene expression.
Log10(TPM+1) matrix was separately used to infer gene regulatory network structure by the ARACNe algorithm. ARACNe was run with 100 bootstrap iterations using 1785 transcription factors (genes annotated in gene ontology molecular function database as GO:0003700, “transcription factor activity”, or as GO:0003677, “DNA binding” and GO:0030528, “transcription regulator activity”, or as GO:0003677 and GO:0045449, “regulation of transcription”), 668 transcriptional cofactors (a manually curated list, not overlapping with the transcription factor list, built upon genes annotated as GO:0003712, “transcription cofactor activity”, or GO:0030528 or GO:0045449), 3455 signaling pathway related genes (annotated in GO biological process database as GO:0007165, “signal transduction” and in GO cellular component database as GO:0005622, “intracellular” or GO:0005886, “plasma membrane”), and 3620 surface markers (annotated as GO:0005886 or as GO:0009986, “cell surface”). ARACNe is only run on these gene sets so as to limit protein activity inference to proteins with biologically meaningful downstream regulatory targets, and we do not apply ARACNe to infer regulatory networks for proteins with no known signaling or transcriptional activity for which protein activity may be difficult to biologically interpret. Parameters were set to zero DPI (Data Processing Inequality) tolerance and MI (Mutual Information) p-value threshold of 10−8, computed by permuting the original dataset as a null model.
Using the ARACNe gene regulatory network structure, VIPER protein activity inference was performed on gene expression signature. First directly on z-score-scaled gene expression signature for all T-cell subtypes, used for Principal Component Analysis and clustering (Figure 1C,1D). Then separately scaling Tumor and Peripheral Tregs against naïve CD4nonTregs by viperSignature command in Rstudio for comparison of Tumor Treg vs Peripheral Treg (Figure 1E), and scaling all Tregs and CD4nonTregs against naïve CD8+ T-cells by viperSignature for comparison of Tumor Treg vs all Treg and CD4nonTreg controls (Figure 1F).
Random Forest Feature Selection:
The full dataset was randomly split into 75% training data and 25% testing data. On training data, a Random Forest Model was built with VIPER-inferred protein activity to classify Tumor Treg vs Peripheral Treg (Figure 1E) or Tumor Treg vs all Controls (Figure 1F), taking the list of all differentially active proteins (t-test p-value < 0.01) as an initial feature set. Features were ranked by mean decrease in model accuracy and included one-by-one to construct random forest models with feature selection. Predictive power was assessed by Area-Under-ROC-Curve (AUC) in the held-out testing data, and a null model of AUC was constructed from random sampling of the same number of genes (from the set of genes with differential activity p-value = 1.0) 1000 times. For each comparison, the maximum number of discriminative genes was selected for which AUC vs null model remained statistically significant (Figure S2A). These genes are shown in Figure 1C and 1D and aggregated into a combined list of 17 putative Tumor Treg vs Peripheral Treg Master Regulators with Activity specifically upregulated in Tumor Tregs.
CRISPR KO library design:
For in vivo CRISPR KO screening we designed the target gene list to include 34 genes, which consisted of 17 predicted Tumor Treg MRs, 13 randomly sampled negative control genes (genes with p = 1.0 comparing Tumor Tregs to Peripheral Tregs: In the final analysis, 6 of these genes with negligible baseline expression in Tregs were utilized as true negative controls), Treg context-specific positive controls Foxp3 and Cd4, and core-essential genes Cdk1 and Plk1. All these genes were targeted with 3 sgRNAs. For guide design, we used the Broad Institute Genetic perturbation platform (GPP) sgRNA designer-tool73. The guide sequences are found in (Table S1).
CRISPR KO oligo synthesis and library cloning:
Oligo libraries (102 oligos) were ordered from Twist-biosciences (Table S1):
From the initial oligo pool, this TREG sub-library was amplified first with KAPA polymerase (KK2502) with the following TREG_1F and TREG_1R PCR primers (see Supplementary Table 2) and with the following settings:
| DNA (oligo pool 1ng/ul) | 2ul | |
| 5xHF-buffer | 5 | |
| dNTPs | 0.75ul | |
| TREG_1F(10uM) | 0.75ul | |
| TREG_1R(10uM) | 0.75ul | |
| KAPA pol | 0.5ul | |
| SYBR | 1.25ul | |
| H2O | to 25ul | |
| PCR 1 Protocol: | ||
| 95C | 3min | |
| 98C | 20s | |
| 55C | 15s | |
| 72C | 15s | |
| 72C | 1min | |
| 4C | --- | |
The PCR product from PCR1 was gel purified with GeneJet gel purification-kit.
The 2nd PCR prior to the Gibson cloning-step was done with the TREG_2F and TREG_2R primers and the following settings:
| DNA (product from 1st PCR) | 3ng | |
| 5xHF-buffer | 5ul | |
| dNTPs | 0.75ul | |
| TREG_2F(10uM) | 0.75ul | |
| TREG_2R(10uM) | 0.75ul | |
| KAPA pol | 0.5ul | |
| SYBR | 1.25ul | |
| H2O | to 25ul | |
| PCR 2 Protocol: | ||
| 95C | 3min | |
| 98C | 20s | |
| 64C | 15s | |
| 72C | 15s | |
| 72C | 1min | |
| 4C | --- | |
Both of these amplifications were done with qPCR and the PCR program was stopped before the amplification started to plateau. After PCR the insert was gel purified (GeneJet) and Gibson cloned into BsmBI-digested pXPR_053 (Addgene# 113591). Gibson cloned insert and vector was column purified (GeneJet) and large-scale electroporated into Lucigen Enduro competent cells. The bacterial colonies were scraped from 24,5cm x 24,5cm agar plates, so that the estimated library complexity was > 1000 colonies / sgRNA. Library-plasmid DNA was extracted with NucleoBond Xtra Midi EF-kit (Macherey-Nagel).
Lentiviral packaging of the sgRNA library:
13 million HEK293T cells were seeded for each 15cm dish the night before transfection. The following morning, viral transfections were conducted with the following components:
22.1ug sgRNA containing pXPR_053 (Addgene 113591).
16.6ug PsPAX2 (Addgene 12260)
5.5ug PMD2G (Addgene 12259).
1660ul of sterile H2O.
After mixing the plasmids and H2O, 110,6ul of Fugene HD (Promega) was added to the mix. The transfection mixture was vortexed, then incubated for 10 minutes before adding dropwise to 293T cells. The transfection mixture was removed the following day and fresh media was added to the cells. Virus was collected at 48h and 72h after initial transfections. To remove cellular debris, the virus-containing supernatant was centrifuged 500 x g for 5min and filtered with 0.45um PES filters (Millipore), followed by ultracentrifugation (25,000rpm for 2h), dissolving the viral pellet into PBS, aliquoting the virus and storing the aliquots at −80C. Viral titer was measured with 293T cells by using FACS and violet-excited GFP in the pXPR_053-plasmid.
sgRNA library transductions into hematopoietic LSK cells:
Confirmatory evidence that the predicted proteins regulate tumor Treg infiltration was generated in murine models in a pooled CRISPR KO screen (Figure 2); by comparing the differential representation of Tregs containing MR targeting sgRNAs in tumor versus non-tumor tissue (spleen, as a control).
LSKs from donor Cas9+ mice mice were sorted into 96-well plate (100k LSKs/well) and incubated overnight in SFEM media supplemented with 100 ng/mL of the following cytokines: SCF, TPO, Flt3-Ligand, and IL-7. Pen/Strep was also used in all in vitro cultures. The following day, LSK cells were transferred into Retronectin (Takara)-coated 24-well plate and sgRNA library-containing Lentiviruses were added to the wells with MOI 30 (based on viral titering in 293T cells, similarly as in LaFleur et. al, 2019). The final volume was adjusted to 400ul / well by adding cytokine supplemented SFEM stem cell media. The cells were centrifuged at 650 x g for 1.5 hours at 37°C with an acceleration of 2 and a brake of 1. After centrifugation, the plate was placed into 37C incubator for 1h, before adding 500 microliters of prewarmed stem cell media on top of the LSKs followed by overnight incubation. Next day the transduced LSKs were implanted into donor mice irradiated with two doses of 600rads, spaced four hours apart, by intravenous tail vein injection immediately following the second irradiation.
CRISPR Validation in Chimeric Immune Editing Model:
The transduced stem cells were reimplanted into lethally irradiated (6 Gy x2) recipient H11-Cas9 mice in two cohorts (six replicate mice for cohort 1 and three replicate mice for cohort 2), allowing reconstitution of the entire immune system, including Tregs, with a unique pool of MR sgRNAs and control sgRNAs in place. Subsequent subcutaneous implantation of 1x106 MC38 murine colon adenocarcinoma cells allowed direct observation of differential infiltration of tumors by Tregs receiving selected CRISPR guides, in a single, high-throughput experimental screen. For both cohorts, the stem cells were separately implanted and harvested, and Vex+ sgRNA-bearing Tregs and CD4nonTregs were flow-sorted from Tumor and spleen, separately.
Genomic DNA extraction and Preparation of NGS libraries:
Since the number of Vex+ tumor Tregs was very low in any individual mouse and because the mice all share the same genetic background, we decided to pool all tumor Tregs and tumor CD4s together across all mice before the gDNA extraction step in order to reliably purify gDNA with sufficient yield. Before pooling the tumor TREGs or tumor CD4s, the TREG / CD4 cell numbers coming from each individual tumor were carefully counted during FACS sorting.
First the pooled cells (all tumor TREGs or all tumor CD4s) were lysed with 400ul of RIPA-buffer (Teknova) + RNAseA (Qiagen), followed by 1h incubation in 65C. After this, 400ul of Phenol/Chloroform/Isoamyl alcohol (Invitrogen) was added, followed by 6 min centrifugation at room temperature. Finally, the gDNA was recovered by Isopropanol precipitation.
For spleen Tregs and spleen CD4s all the gDNA extractions were done individually for each mouse-sample (not pooled together before the lysis-stage as was done with tumor Tregs and tumor CD4s), since the number of Spleen extracted Vex+ cells was much higher than with tumor Tregs / tumor CD4s. After the gDNAs of spleen Tregs and spleen CD4s samples were individually purified, spleen Tregs and spleen CD4s gDNAs were pooled before the NGS library prep PCRs. This was done by pooling Spleen Tregs gDNAs and Spleen CD4s gDNAs in the same ratio as earlier Tumor Tregs and Tumor CD4s were pooled prior to gDNA extractions (as measured by Vex+ FACS cell count).
For the NGS library preparations, the extracted gDNA was split evenly into 8 (for cohort 1) or 4 (for cohort 2) separate technical replicates. Library prep PCRs (2-step PCR protocol with 2 x KAPA Mastermix (KK2612, KAPA Biosystems)) and NGS were done individually to all these technical replicates. Both 1st (with TREG_NGS_1F and TREG_NGS_1R) and the 2nd PCRs (with TREG_NGS_2F and TREG_NGS_2R) were run in qPCR machine and stopped before amplification started to saturate in order to avoid biases in the library coverage. The following primers and PCR programs were used for the NGS library preps:
| 1st PCR: | ||
|---|---|---|
| gDNA | 12.5 - 25% of pooled material (depending on the cohort) | |
| 2 x KAPA mastermix | 12.5ul | |
| TREG_NGS_1F(10uM) | 1ul | |
| TREG_NGS_1R(10uM) | 1ul | |
| SYBR | 1.25ul | |
| H2O | to 25ul | |
| PCR protocol TREGS_NGS_1: | ||
| 95C | 3min | |
| 98C | 20s | |
| 60C | 15s | |
| 72C | 20s | |
| 72C | 1min | |
| 2nd PCR: | ||
|---|---|---|
| 1:50 diluted DNA template from PCR 1 | 8ul | |
| 2 x KAPA mastermix | 12.5ul | |
| TREG_NGS_2F(10uM) | lul | |
| TREG_NGS_2R(10uM) | lul | |
| SYBR | 1.25ul | |
| H2O | to 25ul | |
| PCR protocol TREGS_NGS_2: | ||
| 95C | 3min | |
| 98C | 30s | |
| 52.5C | 15s | |
| 72C | 20s | |
| 72C | 1min | |
After the 2nd PCR, samples were gel-purified (GenJet), pooled and sequenced with Illumina.
Critically, this experiment would not have been possible on a genome-wide level without initial narrowing of candidate master regulators by VIPER protein activity analysis, due to fundamental limitations in achieving a sufficient number of tumor-infiltrating Tregs harboring guide DNAs for the full set of mouse genes. This is because we typically find fewer than 10,000 tumor-infiltrating Tregs in MC38 tumor model.
Correlation between replicates by gDNA frequency was assessed in each cohort and for each set of replicates following library sequencing (Figure 2D).
In vivo CRISPR KO-screen analysis:
Sequencing reads were aligned to a reference of sgRNA template sequences by kallisto to determine a counts matrix of reads per guide for each sample. Differential frequency of guides in Tumor Treg vs Peripheral Treg and Tumor Treg vs Tumor CD4nonTreg was assessed by empiric p-values from normal distribution fitted to a permutation-based null model for each guide (1000 permutations per sgRNA), with Bonferroni correction, and p-values for guides targeting the same gene integrated by Stouffer’s Method. P-values across the two replicate experiments were then also integrated by Stouffer’s Method (Figure 2F-G). For the final analysis 6 /13 of the randomly sampled negative controls with low baseline expression in T cells were ultimately selected as negative controls for the assay. Positive control sgRNAs were significantly depleted post-transduction, indicating successful gene-editing.
CRISPR KO-based Tumor Growth Experiments:
Tumor growth was assessed in single-gene CHIME chimeras generated by the CHIME protocol described above. Two unique guide RNAs per gene were pooled to create either Trps1-targeted chimeras (sgTrps1_1 and sgTrps1_2), or non-targeting control chimeras (sgNon-targeting guide 1 and sgNon-targeting guide 2)30. For sgRNA sequences see Supplementary Table 2. LSKs were transduced with sgTrps1 or sgControl RNA pools at MOI 50 based on 293T cell line titering. Two cohorts of 5-10 mice were generated, with FACS-sorted Vex+ (sgRNA-containing) LSK cells from cohort 1 used to generate cohort 2 chimeras.
Due to variability in growth and spontaneous regression of MC38 tumors in these chimeras, mice were subcutaneously implanted with MCA205 (Millipore Sigma). In cohort 1, male and female mice were implanted with 8x105 MCA205 cells on the opposite flank from which spontaneous MC38 regression occurred. In cohort 2, mice were only implanted with 8x105 MCA205. Growth kinetics and survival outcomes were highly reproducible across cohorts. Tumor volumes were measured every 2-3 days by electronic caliper, and mice were euthanized when tumor volume exceeded 1,000mm3 or when ulceration exceeded 5mm in diameter. Differential survival between groups was assessed by Kaplan-Meier test. At endpoint, tissues were harvested from cohort 2 mice for downstream assays as described below.
ORF cloning for Transcription factor overexpression / reprogramming assay:
Full-length open reading frame (ORF) clones for the top 17 predicted TI-TREG-MRs (EGR1, NR3C1, PBX4, MAFB, ID2, STAT4, NR4A3, NR4A1, TRPS1, EGR3, BANP, ZEB2, KLF4, GLI1, CSRNP2, FOSL2 and KDM2b) and EGFP (as assay control) were cloned into modified Tet-O-FUW lentiviral expression plasmid (Addgene #30130), which include P2A and mCherry-selection marker. All cloned ORFs were sequence verified. For each ORF-construct we introduced an ORF specific 22nt barcode sequence and 10nt random UMI sequence (see Supplementary Table 2) located approx. 200 bp upstream of the lentiviral 3’-long terminal repeat (LTR) region of the plasmid. This produces a polyadenylated transcript, which contains the ORF-specific barcode proximal to its 3’ end. To increase the 10nt random UMI diversity for each ORF construct, the final step of the cloning procedure (cloning in the ORF specific barcode and ORF UMI into modified Tet-O-FUW (which is already containing all full length ORFs)) was done in pooled fashion with Lucigen Endura competent cells and electroporation for each ORF-construct separately.
For all other cloning-related transformation steps we used NEB Stable competent cells (NEB).
Transcription factor overexpression / reprogramming assay (scRNA-Seq):
All lentiviruses were produced, similarly as with “Lentiviral packaging of the sgRNA library”, and viral titers were measured individually for each virus. P-Tregs were collected from healthy donor PBMC. Briefly, healthy donor buffy coats (New York Blood Center) were diluted 1:1 in PBS and layered over ficoll (GE Healthcare; 1.077 g/mL) and spun for 30 min at 400xg with the brake off. PBMC were isolated from the interface and pooled, RBC lysed (ACK buffer; Quality Biological), then washed with PBS. CD4+ T cells were enriched with the Miltenyi CD4+ T cell Isolation kit according to manufacturer’s instructions, then stained for flow cytometry sorting with antibodies targeting CD4, CD127, and CD25. Viable Tregs were sorted (Sytox Green− CD4+CD25+CD127lo) using a FACSAria II sorter into X-VIVO 15 medium (Lonza) supplemented with 10 U/mL human recombinant IL-2 (PeproTech). Approximately 50k cells were seeded per well in flat-bottom 96-well plates in Treg expansion media (X-VIVO 15 + 500 U/mL human IL-2), and activated with Human Treg Expander beads (Dynabeads; Gibco) according to manufacturer’s protocol. The next day, ORF containing lentiviruses were transduced into human P-Tregs by spin-infection (930 x g, 2h, +30C) in arrayed fashion +/− M2RTTA (FUW-M2rtTA, Addgene #20342), a tetracycline-inducible transcriptional amplifier allowing monitoring of MR overexpression at higher and lower levels74. The total viral dose for each ORF-virus was optimized to maximize the transduction efficiency for each clone without ill-effects on the TREG viability (data not shown). One day after the transductions, the media was changed and doxycycline (0.5ug/ml) was added to wells in order to activate the M2RTTA-driven tet-promoter. 7 days after the spin-transduction, the arrayed cells were FACS sorted into 2 pools (+M2RTTA and −M2RTTA) in approx. equal cell numbers for each ORF and control. This was followed by 10x chromium run and NGS.
After the Chromium-run, and cDNA amplification, the ORF barcode transcripts were specifically enriched by amplifying with ORF_BC_amplif_oligo_F and ORF_BC_amplif_R-oligos (see Supplementary Table 2).
This amplified product was spiked in spiked in the final NGS library at 10% total amounts respectively.
High-Throughput Treg-Directed Drug Screening:
From an initial library of 1,554 FDA-approved or investigational oncology compounds (SelleckChem), single-dose viability screening was performed in vitro on human Tregs sorted from buffy coats as described above. 195 compounds were identified which reduced peripheral Treg growth by at least 60% relative to DMSO control at 5uM. For these, dose-response titrations were performed to identify the IC20 dose at which peripheral Treg growth is inhibited by 20%, either by direct toxicity to Tregs or inhibition of Treg cell division. Subsequently, Tumor-Infiltrating Tregs were sorted from a large clear cell renal carcinoma tumor and plated with Treg-expansion beads in culture for 5 days, resulting in 5x106 TI-Tregs. These were suspended at 160,000cells/mL and divided among 2 replicate plates for downstream RNA sequencing (PLATE-Seq) and 1 plate for viability testing in comparison to peripheral Tregs at the P-Treg IC20 dose. Seven drugs with significantly greater toxicity to tumor Tregs vs peripheral Tregs were identified (Figure 3C).
Wells of drug-treated Tregs were RNA-Sequenced and each normalized with viperSignature against the internal DMSO-control wells on the same PLATE. VIPER was run on the normalized gene expression using the T-cell ARACNe network inferred from sorted bulk-RNA-Sequencing clinical data. Drugs were ranked on their overall inversion across patients of the 17-gene Master Regulator signature previously identified and validated by CRISPR (Figure S3B), as well as on their patient-by-patient inversion of Tumor-Treg vs Peripheral-Treg protein activity signature by OncoTreat (Figure 3E).
Drug-based Tumor Growth Experiments:
5-10 female mice per treatment arm were implanted with subcutaneous MC38 (Kerafast) tumor cells. Treatment was initiated after 8-12 days of initial tumor growth when average tumor volume reached 150mm3 (12 days for C57BL/6J, 10 days for Rag1-KO, 8 days for NSG). Mice were randomized prior to treatment to equalize mean tumor volume and size distribution between groups. Tumors were measured by electronic caliper every 2-3 days, and mice were euthanized when tumor volume exceeded 1000mm3 or ulceration exceeded a diameter of 5mm. Gemcitabine was administered IP in 100ul sterile PBS for 3 total doses (q3D) at indicated dose. Floxuridine and triapine were injected IP daily for 9 doses at 1mg/kg and 5mg/kg, respectively. Mice received 200ug anti-PD-1 (RMP1-14; BioXCell) IP in sterile PBS for 3 total doses (q3D). Treatment response outcomes were assessed by cox proportional hazards model, Kaplan-Meier curve, and computation of mean tumor growth slope over time. Tumor growth curves display the average tumor volume over time with standard deviation represented by error bars or shading, with each average growth curve terminating when the first animal in each treatment condition reached end stage.
In vitro Treg Suppression Assay:
At tumor growth endpoint, spleens from Trps1-targeted CHIME chimeras were harvested, dissociated into single cell suspensions, RBC lysed, removed of B cells (Miltenyi CD19+ Microbeads; per manufacturer’s protocol), and frozen in 90% FBS + 10% DMSO. Later, cells were thawed and rested overnight in T cell media (RPMI + 10% FBS + 100U/mL penicillin + 100mg/mL streptomycin + 1mL sodium pyruvate + 100uM non-essential amino acids + 5mM HEPES + 0.055nM 2-mercaptoethanol) at 2x106 cells/mL. Tregs were flow sorted on a BD FACSAria II cytometer (Sytox Green−CD3+CD4+CD25+), and Tregs containing Trps1-sgRNAs were separated from non-perturbed Trps1-WT Tregs by Vex fluorescence. Spleens from naive C57BL/6J mice were harvested to isolate responder T cells and APCs for the suppression assay. Briefly, spleens were mechanically mashed through a 70um filter, RBC lysed, and resuspended in MACS buffer (PBS + 0.5% BSA + 2mM EDTA). Splenocytes were split in half for CD4 T cell isolation (Miltenyi mouse CD4+ T cell isolation kit) and APC enrichment (Miltenyi CD3+ microbeads) according to manufacturer’s protocols. APCs were fixed by incubation with 25ug/mL mitomycin C (Millipore Sigma) in T cell media for 30 min at 37o C. CD4+ T cells were stained with 10uM CellTrace Violet in 1mL T cell media for 5 min at room temperature protected from light. For the assay, Tregs containing Trps1-sgRNAs and Trps1-WT Tregs were serially diluted in triplicate, targeting 20k Tregs for the 1:1 ratio wells. 20k CTV+ CD4+ responder T cells were added to all wells, then 20k fixed APCs were added, followed by soluble anti-CD3 antibody (clone 145-2C11; BioLegend; final concentration of 1ug/mL). Cells were incubated at 37° C for ~80 hours, then CTV dilution was assayed in responder CD4+ T cells using a Cytek Aurora full spectrum flow cytometer.
Histology:
At endpoint, shaved skin, colon, small intestine, liver, and kidney tissues were harvested from TRPS1-targeted and Scrambled control CHIME chimeras. Intestinal organs were flushed with PBS. All tissues were fixed in 15mL 10% formalin for 24-36 hours, then were transferred to 70% ethanol before routine FFPE embedding, slide sectioning, and H&E staining at the Columbia Molecular Pathology Shared Resource core. Tissue slides were scanned up to 40X magnification and imaged in QuPath-0.3.2. Pathology scores were provided by independent expert pathologist review, blinded to sample group status.
High Dimensional Spectral Flow Cytometry Immune Profiling:
Tumors and spleens were collected from 12-day MC38 tumor bearing mice 24, 48, or 72 hours post treatment with Gemcitabine (120mg/kg, 12mg/kg, or 3mg/kg; 5-10 mice per group per time point). Tumors and spleens were minced and digested by 30 min incubation with shaking at 37° in X-VIVO 15 media containing 0.16ug/mL DNaseI (Roche) and 1mg/mL Collagenase D (Roche). Digest reactions were quenched by addition of RPMI + 10%FBS and vortexing for 30 seconds. Spleen samples were RBC lysed in 1mL ACK buffer for 1-2 minutes, followed by quenching with 9mL RPMI + 10%FBS. Samples and cells for single stain controls were plated in U bottom 96-well plates, then were washed with PBS followed by staining with LiveDead Fixable Blue dye (ThermoFisher) for 30 minutes at room temperature on a shaker. Cells were washed, blocked with Tru-Stain anti-mouse FcX Plus (BioLegend), then stained with surface marker antibodies supplemented with BD Brilliant Stain buffer and TruStain Monocyte Blocker (BioLegend) for 30 minutes at room temperature on a shaker. Samples were washed, then fixed and permeabilized using the FoxP3 Fixation kit (eBioscience) for 30 minutes at room temperature on a shaker. Cells were washed in 1X PermWash (eBioscience) then stained with intracellular antibodies in 1X PermWash overnight at 40 C on a shaker. Cells were then washed with 1X PermWash and FACS buffer then acquired on a 5-laser Cytek Aurora full spectrum flow cytometer. High dimensional data analysis was completed in FlowJo v10.8.1 (BD) using UMAP v3.1 and FlowSOM v3.0.18 plugins downloaded from FlowJo Exchange.
Single-Cell RNA-Seq Profiling of Gemcitabine Effect on TI-Tregs:
To test the hypothesis that low-dose Gem modulates TI-Tregs, we performed single cell RNA sequencing of MC38 tumor- and spleen-derived Tregs 24 hours after exposure to a single dose of 12 mg/kg Gem as well as 24 hours after vehicle control. For this study, we implanted FoxP3Yfp-Cre mice with 1x106 MC38 subcutaneously to facilitate flow-sorting of TCR-β+ CD4+ FoxP3+ Tregs from tumor and spleen specifically by the YFP marker. Tissue was harvested at day 14 post tumor-implantation, and fresh tissue was minced to 2-4 mm sized pieces in a 6-cm dish and subsequently digested to single cell suspension using Mouse Tumor Dissociation Kit (Miltenyi Biotec) and a gentleMACS OctoDissociator (Miltenyi Biotec) according to the manufacturer’s instructions.
Dissociated cells were flow-sorted for YFP+ Tregs and processed for single-cell gene expression capture (scRNASeq) using the 10X Chromium 3’ Library and Gel Bead Kit (10x Genomics), following the manufacturer’s user guide at the Columbia University Genome Center. After GelBead in-Emulsion reverse transcription (GEM-RT) reaction, 12-15 cycles of polymerase chain reaction (PCR) amplification were performed to obtain cDNAs used for RNA-seq library generation. Libraries were prepared following the manufacturer’s user guide and sequenced on Illumina NovaSeq 6000 Sequencing System. Single-cell RNASeq data were processed with Cell Ranger software at the Columbia University Single Cell Analysis Core. Illumina base call files were converted to FASTQ files with the command “cellranger mkfastq.” Expression data were processed with “cellranger count” on pre-built mouse reference. Cell Ranger performed default filtering for quality control, and produced a barcodes.tsv, genes.tsv, and matrix.mts file containing transcript counts for each cell, such that expression of each gene is in terms of the number of unique molecular identifiers (UMIs) tagged to cDNA molecules corresponding to that gene.
These data were loaded into the R version 3.6.1 programming environment, where the publicly available Seurat package was used to further quality-control filter cells to those with fewer than 25% mitochondrial RNA content, more than 1,000 unique UMI counts, and fewer than 15,000 unique UMI counts. Pooled distribution of UMI counts, unique gene counts, and percentage of mitochondrial DNA after QC-filtering is shown in Figure S5A. Gene Expression UMI count matrix was processed in R using the Seurat SCTransform command followed by Seurat Anchor-Integration. The sample was clustered on gene expression by a Resolution-Optimized Louvain Algorithm25,75. Protein activity was inferred for all cells by VIPER using the SCTransform gene expression signature and the T-cell ARACNe network derived from sorted T-cell bulk-RNA-Seq. The single-cell data were then re-clustered on VIPER protein activity (Figure S5B). Top 5 most differentially upregulated proteins per cluster were assessed by t-test (Figure S5C). Enrichment of the TI-Treg MRs was assessed by Gene Set Enrichment Analysis (GSEA) on a cell-by-cell basis, with normalized enrichment scores shown in Figure 5E and protein activity of the individual MRs shown in Figure 5D. Cluster frequencies were plotted for each sample (Vehicle-Treated Tumor, Vehicle-Treated Spleen, Gem-Treated Tumor, Gem-Treated Spleen), with pairwise comparisons in frequency assessed by Fisher’s Exact test and cox proportional hazards model (Figure 5G).
Quantification and statistical analysis
All analyses and statistics were performed in R version 3.6.2 or GraphPad Prism v9. P-values were considered statistically significant at less than 0.05, with multiple testing correction by Benjamini-Hochberg method where appropriate. All data are available in a Mendeley Data repository (Reserved DOI: 10.17632/vnrsbb4gk9.1).
Highlights
Discovery of proteins specifically active in human TI-Tregs vs P-Tregs
TRPS1 validated as the most significant regulator of TI-Treg phenotype
TRPS1-knockout in hematopoietic cells associates with improved survival in mice
Drug screen identifies low-dose gemcitabine as differential inhibitor of TI-Tregs
Supplementary Material
Supplementary Table S1, related to STAR Methods: sgRNA Sequences used for pooled CRISPR KO experiment.
Acknowledgements
This research was supported by National Institutes of Health (NIH) grants R01 CA127153, 1P50CA58236-15 and P30CA006973, the Prostate Cancer Foundation Challenge Grant, and CUMC institutional funds to Charles Drake, by NIH grants R35CA197745, U01DA217858, S10 OD012351 and S10 OD021764 to Andrea Califano, by NIH grant F30CA260765-01 to Aleksandar Obradovic, and by NIH grants UL1TR001873 and TL1TR001875 to Casey Ager. This study was also supported by Sigrid Juselius Foundation (MT). We thank the Sidney Kimmel Comprehensive Cancer Center Experimental and Computational Genomics Core at Johns Hopkins, supported by NIH/NCI grant P30CA006973, for support in performing the bulk-RNA sequencing on sorted cells from human cancer subjects. We also thank Drs. A.Jolma and J.Taipale for kindly providing some of the full-length cDNA of the TI-TREG MRs. This study was also supported by the NIH/NCI SPORE in Prostate Cancer (P50CA58236) and the U.S. Department of Defense Prostate Cancer Research Program (PCRP) Prostate Cancer Biospecimen Network Site (W81XWH-18-2-0015).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Dr. Drake is a co-inventor on patents licensed from JHU to BMS and Janssen, has served as a paid consultant to AZ Medimmune, BMS, Pfizer, Roche, Sanofi Aventis, Genentech, Merck, and Janssen, and has received sponsored research funding to his institution from BMS IioN and Janssen. Dr. Califano is founder, equity holder, consultant, and director of DarwinHealth Inc., which has licensed IP related to these algorithms from Columbia University. Columbia University is an equity holder in DarwinHealth Inc. S.Y. has received sponsored research support to his institution from Celgene/BMS, Janssen, and Cepheid/Danaher, and has served as a paid consultant to Cepheid/Danaher. Dr. Obradovic, Dr. Ager, Dr. Drake, and Dr. Califano are co-inventors on U.S. provisional patent No. 63/188,970 “Therapeutic modulation of regulatory T cells through master regulatory protein targeting” which relates to the work described here.
References
- 1.Schreiber RD, Old LJ, and Smyth MJ (2011). Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 331, 1565–1570. [DOI] [PubMed] [Google Scholar]
- 2.Chao JL, and Savage PA (2018). Unlocking the Complexities of Tumor-Associated Regulatory T Cells. J Immunol 200, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flammiger A, Weisbach L, Huland H, Tennstedt P, Simon R, Minner S, Bokemeyer C, Sauter G, Schlomm T, and Trepel M (2013). High tissue density of FOXP3+ T cells is associated with clinical outcome in prostate cancer. European Journal of Cancer 49, 1273–1279. [DOI] [PubMed] [Google Scholar]
- 4.Muroyama Y, Nirschl TR, Kochel CM, Lopez-Bujanda Z, Theodros D, Mao W, Carrera-Haro MA, Ghasemzadeh A, Marciscano AE, Velarde E, et al. (2017). Stereotactic Radiotherapy Increases Functionally Suppressive Regulatory T Cells in the Tumor Microenvironment. Cancer immunology research 5, 992–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Obradovic AZ, Dallos MC, Zahurak ML, Partin AW, Schaeffer EM, Ross AE, Allaf ME, Nirschl TR, Liu D, Chapman CG, et al. (2020). T-Cell Infiltration and Adaptive Treg Resistance in Response to Androgen Deprivation With or Wthout Vaccination in Localized Prostate Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 26, 3182–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shang B, Liu Y, Jiang S.-j., and Liu Y (2015). Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep 5, 15179–15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bos PD, Plitas G, Rudra D, Lee SY, and Rudensky AY (2013). Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med 210, 2435–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freeman ZT, Nirschl TR, Hovelson DH, Johnston RJ, Engelhardt JJ, Selby MJ, Kochel CM, Lan RY, Zhai J, Ghasemzadeh A, et al. (2020). A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. The Journal of clinical investigation 130, 1405–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, and Korman AJ (2013). Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer Immunology Research 1, 32–42. [DOI] [PubMed] [Google Scholar]
- 10.Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, Allison JP, Ribas A, and Sharma P (2019). Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research 25, 1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. (2013). Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 210, 1695–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJP, Provasi E, Sarnicola ML, Panzeri I, et al. (2016). Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity 45, 1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magnuson AM, Kiner E, Ergun A, Park JS, Asinovski N, Ortiz-Lopez A, Kilcoyne A, Paoluzzi-Tomada E, Weissleder R, Mathis D, et al. (2018). Identification and validation of a tumor-infiltrating Treg transcriptional signature conserved across species and tumor types. Proc Natl Acad Sci U S A 115, E10672–E10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, and Rudensky AY (2016). Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 45, 1122–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng C, Zheng L, Yoo J-K, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. (2017). Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356.e1316. [DOI] [PubMed] [Google Scholar]
- 16.Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, Kancheva D, Martens L, Murgaski A, Bardet PMR, et al. (2021). Therapeutic depletion of CCR8(+) tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. Journal for immunotherapy of cancer 9, e001749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrews LP, Marciscano AE, Drake CG, and Vignali DA (2017). LAG3 (CD223) as a cancer immunotherapy target. Immunological reviews 276, 80–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E, Rutkowski P, Gogas HJ, Lao CD, De Menezes JJ, et al. (2022). Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. The New England journal of medicine 386, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whiteside SK, Grant FM, Gyori DS, Conti AG, Imianowski CJ, Kuo P, Nasrallah R, Sadiyah F, Lira SA, Tacke F, et al. (2021). CCR8 marks highly suppressive Treg cells within tumours but is dispensable for their accumulation and suppressive function. Immunology 163, 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, and Califano A (2005). Reverse engineering of regulatory networks in human B cells. Nature Genetics 37, 382–390. [DOI] [PubMed] [Google Scholar]
- 21.Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, and Califano A (2016). Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nature genetics 48, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aytes A, Mitrofanova A, Lefebvre C, Alvarez MJ, Castillo-Martin M, Zheng T, Eastham JA, Gopalan A, Pienta KJ, Shen MM, et al. (2014). Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell 25, 638–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. (2010). The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paull EO, Aytes A, Jones SJ, Subramaniam PS, Giorgi FM, Douglass EF, Tagore S, Chu B, Vasciaveo A, Zheng S, et al. (2021). A modular master regulator landscape controls cancer transcriptional identity. Cell 184, 334–351.e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Obradovic A, Chowdhury N, Haake SM, Ager C, Wang V, Vlahos L, Guo XV, Aggen DH, Rathmell WK, Jonasch E, et al. (2021). Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 184, 2988–3005.e2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Son J, Ding H, Farb TB, Efanov AM, Sun J, Gore JL, Syed SK, Lei Z, Wang Q, Accili D, et al. (2021). BACH2 inhibition reverses β cell failure in type 2 diabetes models. The Journal of clinical investigation 131, e153876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Califano A, and Alvarez MJ (2017). The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat Rev Cancer 17, 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 9, 1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Obradovic A, Graves D, Korrer M, Wang Y, Roy S, Naveed A, Xu Y, Luginbuhl A, Curry J, Gibson M, et al. (2022). Immunostimulatory Cancer-Associated Fibroblast Subpopulations Can Predict Immunotherapy Response in Head and Neck Cancer. Clin Cancer Res 28, 2094–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaFleur MW, Nguyen TH, Coxe MA, Yates KB, Trombley JD, Weiss SA, Brown FD, Gillis JE, Coxe DJ, Doench JG, et al. (2019). A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nat Commun 10, 1668–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez MJ, Subramaniam PS, Tang LH, Grunn A, Aburi M, Rieckhof G, Komissarova EV, Hagan EA, Bodei L, Clemons PA, et al. (2018). A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nature genetics 50, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lachmann A, Giorgi FM, Lopez G, and Califano A (2016). ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 32, 2233–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bandukwala HS, and Rao A (2013). 'Nurr'ishing Treg cells: Nr4a transcription factors control Foxp3 expression. Nat Immunol 14, 201–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Renoux F, Stellato M, Haftmann C, Vogetseder A, Huang R, Subramaniam A, Becker MO, Blyszczuk P, Becher B, Distler JHW, et al. (2020). The AP1 Transcription Factor Fosl2 Promotes Systemic Autoimmunity and Inflammation by Repressing Treg Development. Cell Reports 31, 107826. [DOI] [PubMed] [Google Scholar]
- 36.Rocamora-Reverte L, Tuzlak S, von Raffay L, Tisch M, Fiegl H, Drach M, Reichardt HM, Villunger A, Tischner D, and Wiegers GJ (2019). Glucocorticoid Receptor-Deficient Foxp3(+) Regulatory T Cells Fail to Control Experimental Inflammatory Bowel Disease. Front Immunol 10, 472–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morita K, Okamura T, Inoue M, Komai T, Teruya S, Iwasaki Y, Sumitomo S, Shoda H, Yamamoto K, and Fujio K (2016). Egr2 and Egr3 in regulatory T cells cooperatively control systemic autoimmunity through Ltbp3-mediated TGF-β3 production. Proc Natl Acad Sci U S A 113, E8131–E8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, Solomon I, Lesko MH, Ruef N, Roddie C, et al. (2018). Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 33, 649–663.e644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. (2016). Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 44, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bush EC, Ray F, Alvarez MJ, Realubit R, Li H, Karan C, Califano A, and Sims PA (2017). PLATE-Seq for genome-wide regulatory network analysis of high-throughput screens. Nat Commun 8, 105–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Douglass EF, Allaway RJ, Szalai B, Wang W, Tian T, Fernández-Torras A, Realubit R, Karan C, Zheng S, Pessia A, et al. (2020). A Community Challenge for Pancancer Drug Mechanism of Action Inference from Perturbational Profile Data (Cold Spring Harbor Laboratory). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor MA, Hughes AM, Walton J, Coenen-Stass AML, Magiera L, Mooney L, Bell S, Staniszewska AD, Sandin LC, Barry ST, et al. (2019). Longitudinal immune characterization of syngeneic tumor models to enable model selection for immune oncology drug discovery. Journal for immunotherapy of cancer 7, 328–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ager CR, Boda A, Rajapakshe K, Lea ST, Di Francesco ME, Jayaprakash P, Slay RB, Morrow B, Prasad R, Dean MA, et al. (2021). High potency STING agonists engage unique myeloid pathways to reverse pancreatic cancer immune privilege. Journal for immunotherapy of cancer 9, e003246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. (2011). CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suzuki E, Sun J, Kapoor V, Jassar AS, and Albelda SM (2007). Gemcitabine has significant immunomodulatory activity in murine tumor models independent of its cytotoxic effects. Cancer Biology & Therapy 6, 880–885. [DOI] [PubMed] [Google Scholar]
- 46.Zappasodi R, Merghoub T, and Wolchok JD (2018). Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 33, 581–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daikeler T, Maas K, Weiss B, Hartmann J, Knobloch A, Arning M, Kanz L, and Bokemeyer C (1997). The influence of gemcitabine on the CD4/CD8 ratio in patients with solid tumours. Oncology Reports. [DOI] [PubMed] [Google Scholar]
- 48.Nowak AK, Robinson BW, and Lake RA (2002). Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemoimmunotherapy. Cancer Res 62, 2353–2358. [PubMed] [Google Scholar]
- 49.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, and Albelda SM (2005). Gemcitabine Selectively Eliminates Splenic Gr-1+/CD11b+ Myeloid Suppressor Cells in Tumor-Bearing Animals and Enhances Antitumor Immune Activity. Clinical Cancer Research 11, 6713–6721. [DOI] [PubMed] [Google Scholar]
- 50.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, and Chauffert B (2006). Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunology, Immunotherapy 56, 641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lutsiak MEC, Semnani RT, De Pascalis R, Kashmiri SVS, Schlom J, and Sabzevari H (2005). Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 105, 2862–2868. [DOI] [PubMed] [Google Scholar]
- 52.Wada S, Yoshimura K, Hipkiss EL, Harris TJ, Yen H-R, Goldberg MV, Grosso JF, Getnet D, Demarzo AM, Netto GJ, et al. (2009). Cyclophosphamide augments antitumor immunity: studies in an autochthonous prostate cancer model. Cancer Res 69, 4309–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, Willimsky G, Ammirante M, Strasner A, Hansel DE, et al. (2015). Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 521, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang X, Wang D, Li Z, Jiao D, Jin L, Cong J, Zheng X, and Xu L (2020). Low-Dose Gemcitabine Treatment Enhances Immunogenicity and Natural Killer Cell-Driven Tumor Immunity in Lung Cancer. Front Immunol 11, 331–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deshmukh SK, Tyagi N, Khan MA, Srivastava SK, Al-Ghadhban A, Dugger K, Carter JE, Singh S, and Singh AP (2018). Gemcitabine treatment promotes immunosuppressive microenvironment in pancreatic tumors by supporting the infiltration, growth, and polarization of macrophages. Sci Rep 8, 12000–12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, Capretti G, Mineri R, Todoric J, Zerbi A, et al. (2015). Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut 65, 1710–1720. [DOI] [PubMed] [Google Scholar]
- 57.Homma Y, Taniguchi K, Nakazawa M, Matsuyama R, Mori R, Takeda K, Ichikawa Y, Tanaka K, and Endo I (2013). Changes in the immune cell population and cell proliferation in peripheral blood after gemcitabine-based chemotherapy for pancreatic cancer. Clinical and Translational Oncology 16, 330–335. [DOI] [PubMed] [Google Scholar]
- 58.Rettig L, Seidenberg S, Parvanova I, Samaras P, Knuth A, and Pascolo S (2011). Gemcitabine depletes regulatory T-cells in human and mice and enhances triggering of vaccine-specific cytotoxic T-cells. International Journal of Cancer 129, 832–838. [DOI] [PubMed] [Google Scholar]
- 59.Shevchenko I, Karakhanova S, Soltek S, Link J, Bayry J, Werner J, Umansky V, and Bazhin AV (2013). Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. International Journal of Cancer 133, 98–107. [DOI] [PubMed] [Google Scholar]
- 60.Skavatsou E, Semitekolou M, Morianos I, Karampelas T, Lougiakis N, Xanthou G, and Tamvakopoulos C (2021). Immunotherapy Combined with Metronomic Dosing: An Effective Approach for the Treatment of NSCLC. Cancers (Basel) 13, 1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tongu M, Harashima N, Monma H, Inao T, Yamada T, Kawauchi H, and Harada M (2012). Metronomic chemotherapy with low-dose cyclophosphamide plus gemcitabine can induce anti-tumor T cell immunity in vivo. Cancer Immunology, Immunotherapy 62, 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nair AB, and Jacob S (2016). A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 7, 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lüdecke HJ, Schaper J, Meinecke P, Momeni P, Gross S, von Holtum D, Hirche H, Abramowicz MJ, Albrecht B, Apacik C, et al. (2001). Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet 68, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Momeni P, Glöckner G, Schmidt O, von Holtum D, Albrecht B, Gillessen-Kaesbach G, Hennekam R, Meinecke P, Zabel B, Rosenthal A, et al. (2000). Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nature Genetics 24, 71–74. [DOI] [PubMed] [Google Scholar]
- 65.Ai D, Yao J, Yang F, Huo L, Chen H, Lu W, Soto LMS, Jiang M, Raso MG, Wang S, et al. (2020). TRPS1: a highly sensitive and specific marker for breast carcinoma, especially for triple-negative breast cancer. Modern Pathology 34, 710–719. [DOI] [PubMed] [Google Scholar]
- 66.Cornelissen LM, Drenth AP, van der Burg E, de Bruijn R, Pritchard CEJ, Huijbers IJ, Zwart W, and Jonkers J (2020). TRPS1 acts as a context-dependent regulator of mammary epithelial cell growth/differentiation and breast cancer development. Genes Dev 34, 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Z, Jia M, Wu X, Cui J, Pan A, and Li L (2015). Overexpression of Trps1 contributes to tumor angiogenesis and poor prognosis of human osteosarcoma. Diagn Pathol 10, 167–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang J, Liu X, Huang Y, He L, Zhang W, Ren J, Wang Y, Wu J, Wu X, Shan L, et al. (2021). TRPS1 drives heterochromatic origin refiring and cancer genome evolution. Cell Reports 34, 108814. [DOI] [PubMed] [Google Scholar]
- 69.Malik TH, Von Stechow D, Bronson RT, and Shivdasani RA (2002). Deletion of the GATA domain of TRPS1 causes an absence of facial hair and provides new insights into the bone disorder in inherited tricho-rhino-phalangeal syndromes. Mol Cell Biol 22, 8592–8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Getnet D, Grosso JF, Goldberg MV, Harris TJ, Yen H-R, Bruno TC, Durham NM, Hipkiss EL, Pyle KJ, Wada S, et al. (2010). A role for the transcription factor Helios in human CD4(+)CD25(+) regulatory T cells. Mol Immunol 47, 1595–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, and Shevach EM (2010). Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol 184, 3433–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zabransky DJ, Nirschl CJ, Durham NM, Park BV, Ceccato CM, Bruno TC, Tam AJ, Getnet D, and Drake CG (2012). Phenotypic and functional properties of Helios+ regulatory T cells. PLoS One 7, e34547–e34547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016. Feb;34(2):184–191. doi: 10.1038/nbt.3437. Epub 2016 Jan 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hockemeyer D, Soldner F, Cook EG, Gao Q, Mitalipova M, and Jaenisch R (2008). A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell stem cell 3, 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1, related to STAR Methods: sgRNA Sequences used for pooled CRISPR KO experiment.
Data Availability Statement
All data reported on in this manuscript are available in a public Mendeley Data repository at DOI: 10.17632/vnrsbb4gk9.1 as of the date of publication, grouped by experiment. VIPER algorithm used for data analysis is publicly available as an R package on Bioconductor and single-cell VIPER helper functions are available as part of previously published workflow on github All original code has been deposited on Mendeley. DOIs in Key Resources Table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse TCR-β BUV395 (clone H57-597) | BD | Cat# 742485 |
| Anti-Mouse CD103 BUV496 (clone M290) | BD | Cat# 741083 |
| Anti-Mouse CD44 BUV563 (clone IM7) | BD | Cat# 741227 |
| Anti-Mouse PD-1 BUV615 (clone J43) | BD | Cat# 752299 |
| Anti-Mouse Nrp1 BUV661 (clone V46-1954) | BD | Cat# 752461 |
| Anti-Mouse/Human Ki67 BUV737 (clone B56) | BD | Cat# 567130 |
| Anti-Mouse CD4 BUV805 (clone GK1.5) | BD | Cat# 612900 |
| Anti-Mouse CD39 BV421 (clone Y23-1185) | BD | Cat# 567105 |
| Anti-Mouse IA-IE Pacific Blue (clone M5/114.15.2) | BioLegend | Cat# 107620 |
| Anti-Mouse ST2 BV480 (clone U29-93) | BD | Cat# 746701 |
| Anti-Mouse CD8 Pacific Orange (clone 5H10) | ThermoFisher Scientific | Cat# MCD0830 |
| Anti-Mouse CD62L BV570 (clone MEL-14) | BioLegend | Cat# 104433 |
| Anti-Mouse CD11c BV605 (clone N418) | BioLegend | Cat# 117334 |
| Anti-Mouse ICOS BV650 (clone C398.4A) | BioLegend | Cat# 313550 |
| Anti-Mouse KLRG1 BV750 (clone 2F1) | BD | Cat# 746972 |
| Anti-Mouse PD-L1 BV785 (clone MIH5) | BD | Cat# 741014 |
| Anti-Mouse iNOS FITC (clone REA982) | Miltenyi Biotec | Cat# 130-116-357 |
| Anti-Mouse CD45 A532 (clone 30-F11) | ThermoFisher Scientific | Cat# 58-0451-82 |
| Anti-Mouse CD19 NB610 (clone 1D3) | ThermoFisher Scientific | Cat# M004T02B06 |
| Anti-Mouse Ly6C PerCP-Cy5.5 (clone HK1.4) | BioLegend | Cat# 128012 |
| Anti-Mouse CD206 PerCPeF710 (clone MR6F3) | ThermoFisher Scientific | Cat# 46-2069-42 |
| Anti-Mouse/Human TOX PE (clone REA473) | Miltenyi Biotec | Cat# 130-120-716 |
| Anti-Mouse Ly6G SYG563 (clone 1A8) | BioLegend | Cat# 127668 |
| Anti-Mouse Helios PE-Dazzle 594 (clone 22F6) | BioLegend | Cat# 137232 |
| Anti-Mouse CD80 PE-Cy5 (clone 16-10A1) | BioLegend | Cat# 104712 |
| Anti-Mouse FoxP3 PE-Cy7 (clone FJK-16s) | ThermoFisher Scientific | Cat# 25-5773-82 |
| Anti-Mouse/Human B220 PE/Fire 810 (clone RA3-6B2) | BioLegend | Cat# 103287 |
| Anti-Mouse/Human TCF-1 APC (clone C63D9) | Cell Signaling Technologies | Cat# 37636S |
| Anti-Mouse CD69 SNIR685 (clone H1.2F3) | BioLegend | Cat# 104558 |
| Anti-Mouse/Human CD11b A700 (clone M1/70) | BioLegend | Cat# 101222 |
| Anti-Mouse F4/80 APC/Fire 750 (clone BM8) | BioLegend | Cat# 123152 |
| Anti-Mouse NK1.1 APC/Fire 810 (clone S17016D) | BioLegend | Cat# 156519 |
| TruStain FcX PLUS (clone S17011E) | BioLegend | Cat# 156604 |
| Anti-Mouse Ter-119 PE (clone TER-119) | BioLegend | Cat# 116208 |
| Anti-Mouse/Human CD11b PE (clone M1/70) | BioLegend | Cat# 101208 |
| Anti-Mouse Gr-1 PE (clone RBC-8C5) | BioLegend | Cat# 108408 |
| Anti-Mouse CD3ε PE (clone 145-2C11) | BioLegend | Cat# 100308 |
| Anti-Mouse CD5 PE (clone 53-7.3) | BioLegend | Cat# 100608 |
| Anti-Mouse/Human B220 PE (clone RA3-6B2) | BioLegend | Cat# 103208 |
| Anti-Mouse CD117 (c-kit) APC (clone 2B8) | BioLegend | Cat# 105812 |
| Anti-Mouse Sca-1 BV421 (clone D7) | BioLegend | Cat# 108127 |
| Anti-Human CD4 BV421 (clone OKT-4) | BioLegend | Cat# 317434 |
| Anti-Human CD25 APC (clone M-A251) | BioLegend | Cat# 356110 |
| Anti-Human CD127 PE (clone A019D5) | BioLegend | Cat# 351304 |
| Anti-Mouse CD25 BV421 (clone PC61) | BioLegend | Cat# 102043 |
| Anti-Mouse CD3 BV711 (clone 145-2C11) | BioLegend | Cat# 100349 |
| Anti-Mouse CD8 PE (clone 53-6.7) | BioLegend | Cat# 100708 |
| Anti-Mouse CD127 PE-Cy7 (clone SB/199) | BioLegend | Cat# 121119 |
| Anti-Mouse CD4 APC (clone GK1.5) | BioLegend | Cat# 100411 |
| InVivoMAb Anti-Mouse PD-1 (clone RMP1-14) | BioXCell | Cat# BE0146 |
| Bacterial and virus strains | ||
| NEBstable Competent E. coli | NEB | Cat# C3040H |
| Endura Electrocompetent cells | Lucigen | Cat# 60242-1 |
| Biological samples | ||
| Healthy donor PBMC buffy coats | New York Blood Center | https://www.nybc.org/ |
| ccRCC Nephrectomy Specimens | Columbia University Irving Medical Center | |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human IL-2 | PeproTech | Cat# 200-02 |
| Recombinant Mouse TPO | PeproTech | Cat# 315-14 |
| Recombinant Mouse SCF | PeproTech | Cat# 250-03 |
| Recombinant Mouse Flt3-L | PeproTech | Cat# 250-31L |
| Recombinant Mouse IL-7 | PeproTech | Cat# 217-17 |
| RetroNectin Recombinant Human Fibronectin | Takara Bio | Cat# T100B |
| FDA-approved and Investigational Oncology Drug Compound Library | Selleckchem | Cat# N/A (custom) |
| Gemcitabine (Ly-188011) | Selleckchem | Cat# S1714 |
| Triapine | Selleckchem | Cat# S7470 |
| Floxuridine (NSC 27640) | Selleckchem | Cat# S1299 |
| SYTOX Green Ready Flow Reagent | ThermoFisher Scientific | Cat# R37168 |
| CellTrace Violet Proliferation Kit | ThermoFisher Scientific | Cat# C34557 |
| Ficoll-Paque PLUS (1.077 g/mL) | GE Healthcare | Cat# 17144003 |
| Mitomycin C | Millipore Sigma | Cat# 10107409001 |
| DNase I | Roche/Sigma | Cat# 10104159001 |
| Collagenase D | Roche/Sigma | Cat# 11088866001 |
| LiveDead Fixable Blue Dye | ThermoFisher Scientific | Cat# L34962 |
| BD Brilliant Stain Buffer Plus | BD | Cat# 566385 |
| FoxP3 Fixation/Permeabilization Kit | eBioscience/Thermo | Cat# 00-55214-00 |
| TruStain Monocyte Blocker | BioLegend | Cat# 426103 |
| Fugene HD | Pro mega | Cat# E2312 |
| RNAseA | Qiagen | Cat# 19101 |
| Biotium EVAGREEN DYE 20X IN WATER 1 ML | Fisher Scientific | Cat# NC0521178 |
| GeneJET Gel Extraction Kit | Fisher Scientific | Cat# FERK0692 |
| Gibson Assembly® Master Mix | NEB | Cat# E2611L |
| KAPA HiFi HotStart PCR Kit, with dNTPs | Kapa Biosystems | Cat# KK2502 |
| NucleoBond® Xtra Midi EF (50 preps) | Macherey-Nagel | Cat# 740420.50 |
| Puradisc 25 mm PES Syringe Filters | Cytiva | Cat# 6780-2504 |
| RIPA BUFFER | Teknova | Cat# R3792 |
| Phenol:Chloroform:Isoamyl Alcohol (25:24:1, v/v) | Invitrogen | Cat# 15593031 |
| KAPA HiFi HotStart ReadyMix (2X) | Kapa Biosystems | Cat# KK2612 |
| Critical commercial assays | ||
| CD4+ T cell Isolation Kit, Human | Miltenyi Biotec | Cat# 130-096-533 |
| Human Treg Expander DynaBeads | Gibco | Cat# 11129D |
| Human Tumor Dissociation Kit | Miltenyi Biotec | Cat# 130-095-929 |
| Mouse Tumor Dissociation Kit | Miltenyi Biotec | Cat# 130-096-730 |
| 10x Chromium Next GEM Single Cell 3’ Kit v3.1 | 10x Genomics | Cat# 1000269 |
| Deposited data | ||
| Sorted T-cell Populations RNA-Seq From Patient Blood & Tumor | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| Overexpression Screen Single-Cell RNA-Seq Data | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| Gemcitabine Treatment Single-Cell RNA-Sequencing Data | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| sgRNA counts from CRISPR KO experiment | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| Drug Screen PLATE-Seq Data | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| T-cell ARACNe Network | This Manuscript | DOI: 10.17632/vnrsbb4gk9.1 |
| Experimental models: Cell lines | ||
| MC38 | Kerafast | Cat# ENH204-FP |
| MCA205 | Millipore Sigma | Cat# SCC173 |
| HEK293T | ATCC | Cat# CRL-11268 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain# 000664 |
| Mouse: H11-Cas9 (Igs2tm1.1(CAG-cas9*)Mmw/J) | The Jackson Laboratory | Strain# 027650 |
| Mouse: NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Strain# 005557 |
| Mouse: Rag1-KO (B6.129S7-Rag1tm1Mom/J | The Jackson Laboratory | Strain# 002216 |
| Mouse FoxP3-YFP/Cre (B6.129(Cg)-Foxp3tm4(YFP/icre)Ayr/J | The Jackson Laboratory | Strain# 016959 |
| Recombinant DNA | ||
| pXPR_053 | Addgene | Cat# 113591 (PubMed 30971695 |
| PsPAX2 | Addgene | Cat# 12260 (gift from Didier Trono) |
| pMD2.G | Addgene | Cat# 12259 (gift from Didier Trono) |
| Tet-O-FUW-EGFP | Addgene | Cat# 30130 (PubMed 20107439) |
| Tet-O-FUW-EGFP-P2A-mCherry | This paper | N/A |
| FUW-M2rtTA | Addgene | Cat# 20342 (PubMed 18786421) |
| Human CSRNP2 ORF clone | GenScript | Cat# OHu04521 |
| Human TRPS1 ORF clone | GenScript | Cat# OHu21177 |
| Human BANP ORF clone | GenScript | Cat# OHu10822 |
| Human MAFB ORF clone | GenScript | Cat# OHu25119 |
| pFUW-tetO-NR4A3 | Addgene | Cat# 139818 (PubMed 30530727) |
| GFP-FBXL10 (KDM2B) | Addgene | Cat# 126542 (PubMed 29985131) |
| KLF4 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| STAT4 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| NR4A1 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| EGR1 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| ZEB2 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| EGR3 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| PBX4 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| ID2 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| FOSL2 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| NR3C1 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| GLI1 in pENTR223.1 | Jussi Taipale lab | PubMed 23332764 |
| Tet-O-FUW-CSRNP2-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-TRPS1-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-BANP-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-MAFB-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-NR4A3-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-KDM2B-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-KLF4-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-STAT4-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-NR4A1-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-EGR1-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-ZEB2-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-EGR3-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-PBX4-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-ID2-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-FOSL2-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-NR3C1-P2A-mCherry | This paper | N/A |
| Tet-O-FUW-GLI1-P2A-mCherry | This paper | N/A |
| Software and algorithms | ||
| R v3.6.2 | https://cran.r-project.org/bin/macosx/ | |
| ARACNe | Margolin, et al. | http://califano.c2b2.columbia.edu/aracne |
| VIPER | Alvarez, et al. | http://califano.c2b2.columbia.edu/viper |
| GraphPad Prism v9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo v10.8.1 | BD | https://www.flowjo.com/ |
| Single-Cell VIPER | Obradovic, et al. | https://github.com/Aleksobrad/single-cell-rcc-pipeline |
| Custom Analysis Scripts | This paper | DOI: 10.17632/vnrsbb4gk9.1 |