

Abstract
Introduction
Aducanumab was approved in 2021 by the US Food and Drug Administration (FDA) under the accelerated approval pathway. Since then, there have been many misconceptions about the approval decision despite multiple publications from the FDA to explain the rationale.
Methods
Even though the FDA's final decision was accelerated approval, the Office of Clinical Pharmacology recommended regular/full approval based on its own analyses. Exposure–response analyses were conducted to quantify the relationship between aducanumab longitudinal exposure and responses (standardized uptake values ratios for amyloid beta and various clinical endpoints) in all clinical trials. To explain the difference between aducanumab and other compounds with negative results in the past, publicly available data were combined with the aducanumab data to demonstrate the relationship between amyloid reduction and clinical endpoint change across multiple compounds with similar mechanism of action. The probability to observe the overall positive findings in the aducanumab program was quantified under the assumption that aducanumab is ineffective.
Results
Positive exposure–response (disease progression) relationship for multiple clinical endpoints from all clinical trials was identified. Positive exposure–amyloid reduction relationship was established. Consistent amyloid reduction–clinical endpoint change relationship across multiple compounds was observed. If aducanumab is assumed to be ineffective, it is extremely unlikely we would observe the overall positive findings in the aducanumab program.
Conclusion
These results provided convincing evidence to support aducanumab's effectiveness. In addition, the observed effect size in the studied patient population represents a clinically meaningful benefit given the magnitude of disease progression within the trial duration.
Highlights
Totality of evidence supports the Food and Drug Administration (FDA)’s approval decision for aducanumab.
Different opinions were clearly explained in the FDA's public reviews from different disciplines.
Readers are encouraged to read the FDA's reviews to understand the FDA's rationale to approve aducanumab.
1. INTRODUCTION
It has been more than 1 year since the Food and Drug Administration (FDA)’s approval of aducanumab. 1 Many events have happened within this 1 year. Two of the aducanumab advisory committee members resigned. 2 Two federal investigations were initiated. 3 , 4 The Centers for Medicare & Medicaid Services (CMS) made the decision that Medicare will only cover aducanumab in the case of FDA‐ or National Institutes of Health (NIH)–approved clinical trials. 5 It was a historical record to quickly publish the key FDA reviews (medical review to support both regular/full approval and accelerated approval, statistical review to support no approval, and clinical pharmacology review to support regular/full approval) 6 on June 28, 2021, only 3 weeks after the FDA approval decision was announced on June 7, 2021. The purpose of the swift publication of key FDA reviews was to officially and comprehensively explain the rationale for the FDA's approval decision despite the negative recommendations (10 no, 1 uncertain for the voting question “In light of the understanding provided by the exploratory analyses of Study 301 and Study 302, along with the results of Study 103 and evidence of a pharmacodynamic effect on Alzheimer's disease pathophysiology, is it reasonable to consider Study 302 as primary evidence of effectiveness of aducanumab for the treatment of Alzheimer's disease”) 7 from the advisory committee meeting that was held on November 6, 2020. Unfortunately, the FDA staff's efforts to expedite the publication of these reviews did not lead to any meaningful impact as demonstrated by the continued publication of negative papers 8 , 9 that did not reflect some important points included in the clinical pharmacology review 10 almost 1 year after its publication. The length of the review (147 pages) or a strongly held negative view on aducanumab's approval decision may have contributed to the lack of interest in reading the full review. Even though multiple articles 11 , 12 , 13 were published by the FDA staff to explain the rationale for the accelerated approval decision, the recommendation from the Office of Clinical Pharmacology at the FDA was a regular/full approval, not an accelerated approval. Therefore, it is necessary to summarize the key points that were used to support the regular/full approval recommendation in a scientific journal article. It is impossible to thoroughly explain all the technical details of the review in a perspective article. Nevertheless, it is the author's hope that this perspective can stimulate more interest in the scientific community to read the full clinical pharmacology review and other key reviews for an in‐depth understanding of the different methods used for data analyses and conclusions reached by different disciplines within the FDA.
2. EVIDENCE USED TO SUPPORT REGULAR/FULL APPROVAL
Table 1 outlines the key milestones of aducanumab's development and regulatory process. It was certainly not a typical case of new drug development and regulatory interactions. The early termination of the two phase 3 clinical studies (301 and 302) led to the following series of events that were further complicated by the apparent inconsistency of the primary efficacy results between the two studies. The usual (and unusual) statistical arguments against the approval of aducanumab can be found in the FDA statistical review. 14 However, a different approach of analyzing the same data led to the opposite conclusion. From a quantitative clinical pharmacology (pharmacometrics) perspective, the following four points (referred to as Points 1, 2, 3, and 4 later in the paper), which will be expanded further below, support the efficacy of aducanumab:
1. Clear exposure–efficacy and dose–response relationships for aducanumab.
2. Consistent pharmacodynamic effect (amyloid beta [Aβ] plaque reduction) in clinical studies and a clear relationship between Aβ plaque reduction and clinical endpoint Clinical Dementia Rating Sum of Boxes (CDR‐SB) for aducanumab.
3. Similar relationship (group level) between Aβ plaque reduction and clinical endpoint CDR‐SB reported with other candidate drugs targeting Aβ pathways.
4. Clinical trial simulations showed very low probability of a false positive finding for the high‐dose group in Study 302, strongly suggesting that the Study 301 result could be a chance finding driven by a subgroup in the high‐dose group.
TABLE 1.
| Date | Event |
|---|---|
| December 16, 2014 | A Type B End of Phase 2 Meeting was held. The meeting included preliminary discussion regarding study population, endpoints, and dosing for the applicant's two proposed phase 3 studies, and the Division suggested a SPA be requested for an in‐depth review of the protocols. The Division and applicant agreed to the safety monitoring plan and the safety database. |
| September 28, 2015 | SPA agreements for Study 301 and Study 302 were reached with the Division and the trials were initiated. During the trials, the target dose for all apolipoprotein E ε4 carriers in the high‐dose arm was increased from 6 to 10 mg/kg. This protocol change was called Protocol Version 4 (PV4). The rationale for this change can be found on page 121 of the FDA's medical review. 15 |
| March 21, 2019 | Biogen announces the termination of the Phase 3 program (Studies 301 and 302) based on results of a prespecified interim futility analysis (data generated up to December 26, 2018). The first formal request from Biogen to the FDA regarding a discussion of the decision to terminate the studies came on May 15, 2019, in the form of a Type C Meeting Request. |
| June 14, 2019 | A Type C Meeting was held to discuss the applicant's analysis of the intent to treat populations of Study 301 and Study 302 including all data prior to the March 21, 2019, announcement of the termination of the studies. The Division advised the applicant that the development of aducanumab should not be abandoned as the available clinical data suggested the drug may be clinically active and did not provide convincing evidence that the drug is ineffective. The Division recommended that further analyses of the available data should be conducted to understand the effect of early termination of the studies on the interpretability of the data and to address the partially conflicting results for Study 301 compared to those for Study 302. |
| October 21, 2019 | A Type C Meeting was held to discuss the additional analyses proposed at the June 14, 2019, meeting. Based on these analyses, the Division agreed that the results of Study 301 and Study 302 are interpretable and suitable for additional consideration. The Division further advised that planning for submission of a marketing application was a reasonable option. |
| February 20, 2020 | The applicant opened BLA 761178 and submitted nonclinical information. |
| July 7, 2020 | BLA submission complete. |
| November 6, 2020 | AC meeting (Yes: 0; No: 10; Uncertain: 1). |
| January 28, 2021 | The Division notified the applicant that their January 27, 2021, submission constituted a major amendment to the application. PDUFA date was changed to June 7, 2021. |
| March 31, 2021, and April 7, 2021 (additional internal Agency meetings to discuss the application and differences of opinion on approvability) | MPPRC meetings. This meeting provided an opportunity to discuss the reviews conducted by the review team and receive input and feedback from the members of the committee. At the MPPRC meetings, many different perspectives were discussed extensively as to whether there is substantial evidence of effectiveness for aducanumab's intended use to treat Alzheimer's disease. |
| April 26, 2021 (additional internal Agency meeting to discuss the application and differences of opinion on approvability) | Center director briefing meeting. The meeting included CDER senior leaders, as well as Dr. Peter Marks, Director of the CBER, and Dr. Rick Pazdur, Director of the OCE. The meeting reviewed the approach to approval the Office of Neuroscience and the Office of New Drugs supported, specifically, using the accelerated approval pathway based on substantial evidence of the effect of aducanumab on reduction in brain amyloid plaques, an effect that is reasonably likely to predict clinical benefit. This approach was supported by Dr. Cavazzoni, Dr. Marks, Dr. Pazdur, and the Office Directors for Clinical Pharmacology (OCP; Dr. Issam Zineh) and Medical Policy (Dr. Jacqueline Corrigan‐Curay). The Director of the Office of Translational Sciences (which includes both the Office of Biostatistics and the Office of Clinical Pharmacology), Dr. Shahvree Buckman‐Garner, commented that she understood the arguments for and against approval. Dr. Sylva Collins, Director of the Office of Biostatistics (OB), dissented on the approach, stating her belief that there is insufficient evidence to support accelerated approval or any other type of approval. |
| June 7, 2021 | Accelerated approval by the FDA. |
Abbreviations: AC, Advisory Committee; BLA, Biologics License Application; CBER, Center for Biologics Evaluation and Research; CDER, Center for Drug Evaluation and Research; FDA, US Food and Drug Administration; MPPRC, Medical Policy and Program Review Council; OCE, Oncology Center of Excellence; PDUFA, Prescription Drug User Fee Act; SPA, Special Protocol Assessment.
Detailed analyses to support each point can be found in the FDA clinical pharmacology review (Section 3.3.1.2 on pages 19 to 22, Section 3.3.1.3 on pages 23 to 24, Section 3.3.1.4 on pages 25 to 33, and Section 3.3.1.5 on pages 34 to 40). The collection of individual longitudinal drug concentration (exposure) data from almost all patients in both studies made it possible to conduct exposure–response (efficacy) analyses to fully take advantage of the rich individual longitudinal exposure–response data by incorporating the dose titration during the trial and the dose increase for the high dose after protocol version 4 amendment (Post‐PV4). During the trials, the target dose for all apolipoprotein E ε4 carriers in the high‐dose arm was increased from 6 to 10 mg/kg. This protocol change was called Protocol Version 4 (PV4, see details in Table 1). Another advantage of the model‐based exposure–response analyses is the increased power of detecting a positive relationship between drug exposure and the efficacy endpoint after including a parametric model to quantify the disease progression of the efficacy endpoints over time. No single parametric model was assumed. Instead, multiple possible structure models (such as linear model and various non‐linear models) were tested based on prespecified criteria and the optimal model (linear model) was selected based on the observed data. The drug effect was evaluated on the slope of the disease progression (how fast the clinical endpoint deteriorated over time). This parametric mixed‐effect modeling approach including individual level exposure–response data that minimized the impact of the missing data at later visits. Compared to the primary statistical analysis method (mixed‐model with repeated measurements [MMRM], using time as a categorical variable without any time pattern assumption), the parametric longitudinal modeling approach was less influenced by the missing data at later visits, giving appropriate weight to the data from the subgroup recruited after PV4 amendment (Figure 1). As a result, even the failed Study 301 showed an overall positive exposure–response relationship (Figure 1A) although the magnitude of exposure–response slope is smaller in Study 301 than Study 302 and the subgroup of Pre‐PV4 in Study 301 did not show the expected exposure–response relationship. More importantly, the Post‐PV4 subgroup in Study 301 showed the expected positive exposure–response relationship, completely consistent with the results from Study 302 (both Pre‐PV4 and Post‐PV4, Figure 1B). The failure of the MMRM analysis for the high dose in Study 301 was mainly due to the unusual pattern (the high‐dose group performed numerically worse than both the low‐dose and the placebo groups) of exposure/dose–response in the Pre‐PV4 subgroup. Even though almost half of the overall patients were recruited after Post‐PV4, there were more missing data at later visits (especially week 78, the primary efficacy visit) for these patients due to the early termination decision. As a result, the overall average result was mainly driven by the Pre‐PV4 subgroup when the MMRM analysis was used, giving very little weight to the data from the Post‐PV4 subgroup. This is a key difference between the primary statistical analysis and the model‐based pharmacometric analysis. The typical challenges of confounded exposure–response analysis (either time‐independent or time‐dependent) were not identified in this case. 15 , 16 The outlier nature of the high dose in Study 301 was further highlighted in the standardized uptake value ratios (SUVR, a quantitative measure of Aβ) versus CDR‐SB (the clinical endpoint) relationship (Figure 2). It should be noted that only a subgroup of patients provided the paired SUVR and CDR‐SB data (roughly N = 100 in each arm) and the CDR‐SB values in Figure 2 are not identical to those from the overall randomized groups. Even though this is the second point that was presented at the advisory committee meeting to support the efficacy of aducanumab, it was clearly not convincing enough for any of the committee members. It should be emphasized that the last two points (Points 3 and 4) were based on additional analyses conducted after the advisory committee meeting.
FIGURE 1.
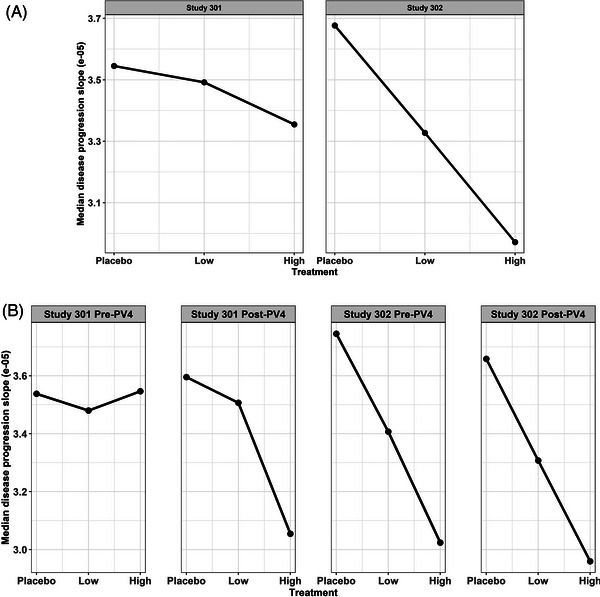
Exposure (aducanumab concentration)–CDR‐SB response (disease progression slope quantifying how fast the clinical endpoint deteriorated over time) relationship. A, Overall groups. B, Subgroups based on Pre‐PV4 and Post‐PV4. CDR‐SB, Clinical Dementia Rating Sum of Boxes; Low, low dose; High, high dose; PV4, Protocol Version 4.
FIGURE 2.
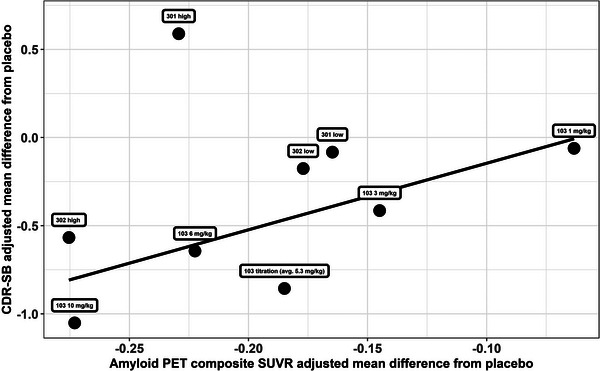
Relationship between mean SUVR and CDR‐SB difference from placebo for aducanumab only. CDR‐SB, Clinical Dementia Rating Sum of Boxes; PET, positron emission tomography; SUVR, standardized uptake value ratio.
Due to the inconsistency between the FDA's final decision and the advisory committee members’ recommendation, there has been a general perception that the FDA ignored advisory committee members’ opinions. In fact, the additional analyses to support Points 3 and 4 were based on the recommendations provided by the advisory committee members. It was entirely reasonable to question why all historical drug candidates targeting Aβ were negative while only aducanumab could be successful (if we believe the positive Study 302 represents the truth). In addition, when there were so many negative trials in the past and the current data for aducanumab are partially inconsistent between two phase 3 studies, it is difficult to be convinced that the negative CDR‐SB result of the high dose in Study 301 was due to a random chance (type 2 error) and other factors (dose and fast progressors’ imbalance) while the positive results in Study 302 and other supporting evidence in Study 301 represent the truth. We took these concerns seriously and conducted additional analyses to address them. We collected all other antibodies targeting Aβ to understand the reason for the historical failures and the difference between the recently developed antibodies (aducanumab, lecanemab/BAN2401, donanemab) and the past ones that reported negative findings. The combined data (Figure 3) clearly revealed the reason for the past negative trials. Despite some impressive P‐values for SUVR reduction under a large sample size, the magnitudes of SUVR reduction for all past negative trials are quite small (< = 0.1 unit) compared to aducanumab, lecanemab, and donanemab. A large enough magnitude of SUVR reduction is required to achieve a meaningful clinical endpoint change. This hypothesis can be further tested with more accumulated data. Figure 3 includes three additional small molecule compounds that also reported negative findings in large phase 3 trials in addition to the seven antibodies included in the FDA original review. The readers are encouraged to keep adding more data points to Figure 3 when new data (such as Clarity AD, GRADUATE I and II) become available to evaluate whether the observed pattern holds. The FDA was criticized for assuming the Study 302 result represented the truth and trying to find reasons to explain why the result in Study 301 should not have happened at the advisory committee meeting 17 (page 303 of the advisory meeting transcript). One committee member recommended the FDA should do the opposite: assume aducanumab does not work and evaluate the chance of observing the positive results in Study 302. That's what we did after the advisory committee meeting. The results actually showed an extremely low chance of observing the overall positive results in Study 302 (< 1 in 10,000,000) if aducanumab was assumed to be ineffective. These additional analyses significantly increased our confidence in the evidence to support the regular/full approval recommendation. Our medical colleagues reached a similar conclusion while accelerated approval was also recommended as a potential pathway.
FIGURE 3.
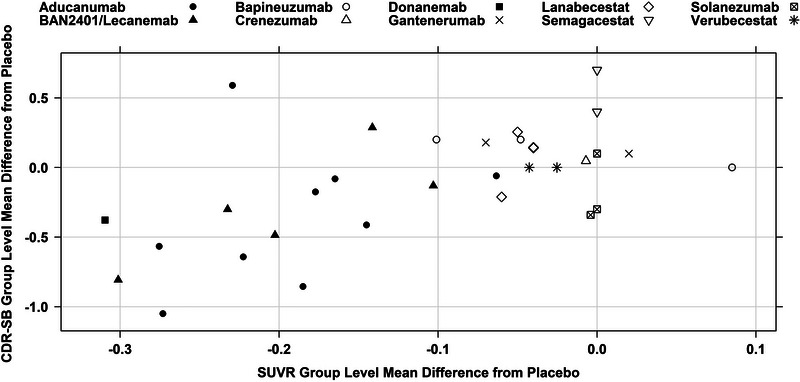
Relationship between mean SUVR and CDR‐SB difference from placebo for multiple compounds. CDR‐SB, Clinical Dementia Rating Sum of Boxes; SUVR, standardized uptake value ratio.
Even though it was never our intention to proactively search for errors in our statistical colleagues’ review, we accidentally identified multiple errors or inappropriate analyses during our additional analyses after the advisory committee meeting. The following two key points were presented to the committee members at the advisory meeting.
1. Placebo worsening explains the apparent high dose effect in Study 302.
2. There is no correlation between week 78 CDR‐SB changes and SUVR week 78 changes for high dose.
However, these two conclusions were derived from inadvertent errors and inappropriate analyses. The detailed explanation can be found in the FDA clinical pharmacology review 8 (pages 134–143). The different methods and conclusions from the FDA's Office of Clinical Pharmacology and Office of Biostatistics were acknowledged and highlighted in Dr. Peter Stein's (Director of Office of New Drugs at FDA) official memorandum. 18 Also, the reason the Office of Clinical Pharmacology's analyses were considered more appropriate and used to support the FDA's final decision was also clearly explained and justified in that memorandum.
3. WHAT IS CONSIDERED CLINICALLY MEANINGFUL?
Even if there is sufficient evidence to demonstrate that aducanumab is effective in lowering Aβ and improving the clinical endpoint, is the effect size on CDR‐SB clinically meaningful? A commonly raised question is that the 0.39 unit change relative to placebo may be too small when we consider the range of CDR‐SB endpoint (0–18). Such a general statement seems reasonable without considering the specific design of the two phase 3 studies and the included patient population. Many journalists or scientists became so confident in their scientific judgement that some concluded “one doesn't have to be a statistician to understand that aducanumab's highest (and least safe) dose slows cognitive decline by just 0.39 on an 18 point scale (i.e., < 2.2%). That could never be considered an important effect, almost regardless of sample size and concerns for statistical significance.” 19 Another argument is that at least 1 to 2 units of CDR‐SB change should be achieved to be clinically meaningful. 20 These numbers even became one of the major reasons to support CMS’ decision to not cover aducanumab for the general patient population despite FDA's approval decision.
At the design stage for both studies 301 and 302, FDA and Biogen reached a consensus on the expected effect size and this consensus was captured in the Special Protocol Assessment (SPA) agreement. “The sample size was planned to have approximately 90% power to detect a true mean difference of 0.5 in change from baseline CDR‐SB at Week 78. The mean difference of 0.5 represents an approximately 25% reduction assuming the placebo mean change in CDR‐SB is 2 at Week 78.” 21 In the actual trials, the disease progression quantified by the mean change in CDR‐SB of the placebo group was even slower than the assumed 2 units at Week 78. It was 1.56 in Study 301 and 1.74 in Study 302 with an average of 1.65. Therefore, the effect margin for any potentially effective treatment to improve is only 1.65 units instead of the full 18‐unit range. As a result, even a change of 0.39 unit represents 24% reduction of the disease progression relative to the placebo group, almost the same as the assumed effect size at the design stage. When we apply 1 to 2 units change against the 1.65 units of placebo disease progression, that represents 61% to 121% reduction of the disease progression. One hundred percent reduction means a complete stop of the disease progression, an effect size expected from a cure for mild cognitive impairment (MCI) and mild Alzheimer's disease (AD) patients, the target patient population that was included in the two phase 3 studies. More than 100% reduction means a treatment that can reverse these MCI and mild AD patients back to almost normal subjects. It is certainly a good wish to have such a highly effective treatment in the future. However, given the challenges we witnessed in AD drug development during the last two decades, expecting the first treatment addressing the underlying disease to be a cure for MCI and mild AD is totally unrealistic. If requiring a treatment to be a cure was not the intention, it is not clear whether those who cited 1 to 2 units of change fully understood the implication of this request within the trial design of studies 301 and 302. Then is 24% reduction of the disease progression clinically meaningful? Both the FDA and Biogen certainly agreed that this magnitude of change was clinically meaningful because 25% change was assumed to plan the trials and calculate the sample size at the design stage. Despite many opinions or articles from those who argued against the clinical meaningfulness of aducanumab, some experts concluded that aducanumab produced a clinically meaningful benefit in association with amyloid lowering. 22 If the observed linear disease progression in the placebo group and the relative magnitude of change under aducanumab treatment can be extrapolated to a longer time, the 24% reduction of disease progression will translate to a larger magnitude of absolute units.
4. SUMMARY
There is no doubt that the aducanumab program was riddled with complexity and mishaps. However, from both the clinical pharmacology and medical perspectives at the FDA, the review teams supported the approval decision after an extensive review of all the evidence including both data from the aducanumab program and data from other clinical programs. Novel analyses were required in this complex case. These included: exposure–response analysis including individual longitudinal pharmacokinetic data and multiple efficacy endpoints from all relevant clinical studies, meta‐analysis including other failed or ongoing clinical programs to understand the consistency/difference across multiple compounds, and clinical trial simulation to assess the possibility of false positive findings under the null assumption (aducanumab is ineffective). These novel analyses were necessary to maximize the understanding of a complex case when the traditional prespecified statistical method could not deliver a clear answer based on the incomplete data due to an interim decision to prematurely terminate the two phase 3 studies.
Challenging disease areas such as AD require an incremental progress in new drug development to maintain the momentum until we may eventually find a cure. Scientists with realistic expectation and thorough understanding of the disease and clinical trial design are urgently needed to speak out and publish their findings and opinions to balance those non‐scientific articles or seemingly scientific articles based on incomplete understanding of AD drug development. Clarifying some commonly held misconceptions or misunderstandings is necessary to advance the drug development for the AD field.
CONFLICT OF INTEREST STATEMENT
I am one of the decision makers who supported the FDA's decision to approve aducanumab. I was the Director of Division of Pharmacometrics, Office of Clinical Pharmacology, CDER, FDA. I conducted some of the additional analyses after the advisory committee meeting in November 2020, served as the supervisory review of all analyses included in the FDA clinical pharmacology review, and supported the regular/full approval recommendation.Author disclosures are available in the supporting information.
CONSENT STATEMENT
Consent was not necessary.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The author would like to acknowledge the entire FDA aducanumab review team for their action of approving aducanumab by applying the best science and listening to patients’ voice. There was no funding involved.
Wang Y. An insider's perspective on FDA approval of aducanumab. Alzheimer's Dement. 2023;9:e12382. 10.1002/trc2.12382
REFERENCES
- 1.accessed on July 18, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/761178Orig1s000ltr.pdf
- 2. Block Jonathan, Two more FDA advisory panel members resign over Biogen aducanumab approval. accessed on July 18, 2022. https://seekingalpha.com/news/3705638‐two‐more‐fda‐advisory‐panel‐members‐resign‐over‐biogen‐aducanumab‐approval
- 3. Review of the FDA's Accelerated Approval Pathway. accessed on July 18, 2022. https://oig.hhs.gov/reports‐and‐publications/workplan/summary/wp‐summary‐0000608.asp
- 4. Chairs Pallone and Maloney Request Answers from FDA on the Approval Process for Alzheimer's Drug Aduhelm. accessed on July 18, 2022. https://oversight.house.gov/news/press‐releases/chairs‐pallone‐and‐maloney‐request‐answers‐from‐fda‐on‐the‐approval‐process‐for
- 5. CMS Finalizes Medicare Coverage Policy for Monoclonal Antibodies Directed Against Amyloid for the Treatment of Alzheimer's Disease. accessed on July 18, 2022. https://www.cms.gov/newsroom/press‐releases/cms‐finalizes‐medicare‐coverage‐policy‐monoclonal‐antibodies‐directed‐against‐amyloid‐treatment
- 6. Drug Approval Package: Aduhelm (aducanumab‐avwa). accessed on July 18, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761178Orig1s000TOC.cfm
- 7. Final Summary Minutes of the Peripheral and Central Nervous System Drugs Advisory Committee Meeting for aducanumab, accessed on July 18, 2022. https://www.fda.gov/media/145690/download
- 8. Ebell MH, Barry HC. Why physicians should not prescribe aducanumab for Alzheimer disease. Am Fam Physician. 2022;105(4):353‐354. PMID: 35426626. [PubMed] [Google Scholar]
- 9. Karlawish J. Fix the process that led to Alzheimer's drug fiasco. Nature. 2022;606(7912):9. doi: 10.1038/d41586-022-01507-3. PMID: 35641673. [DOI] [PubMed] [Google Scholar]
- 10. FDA Integrated Clinical Pharmacology Review. accessed on July 18, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761178Orig1s000ClinPharm_Redacted.pdf
- 11. Dunn B, Stein P, Temple R, Cavazzoni P. An appropriate use of accelerated approval ‐ aducanumab for Alzheimer's disease. N Engl J Med. 2021;385(9):856‐857. doi: 10.1056/NEJMc2111960. Epub 2021 Jul 28. PMID: 34320283. [DOI] [PubMed] [Google Scholar]
- 12. Dunn B, Stein P, Cavazzoni P. Approval of aducanumab for Alzheimer disease‐The FDA's perspective. JAMA Intern Med. 2021;181(10):1276‐1278. doi: 10.1001/jamainternmed.2021.4607. PMID: 34254984. [DOI] [PubMed] [Google Scholar]
- 13. Zhu H, Mehta M, Huang SM, Wang Y. Toward bridging unmet medical need in early Alzheimer's disease: an evaluation of beta‐amyloid (Aβ) plaque burden as a potential drug development tool. Clin Pharmacol Ther. 2022;111(4):728‐731. 10.1002/cpt.2536. Epub 2022 Feb 20. PMID: 35187648. [DOI] [PubMed] [Google Scholar]
- 14. FDA Statistical Review. accessed on July 18, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761178Orig1s000StatR_Redacted.pdf
- 15. Yang J, Zhao H, Garnett C, et al. The combination of exposure‐response and case‐control analyses in regulatory decision making. J Clin Pharmacol. 2013;53(2):160‐6. doi: 10.1177/0091270012445206. Epub 2013 Jan 24. PMID: 23436261. [DOI] [PubMed] [Google Scholar]
- 16. Liu C, Yu J, Li H, et al. Association of time‐varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin Pharmacol Ther. 2017;101(5):657‐666. doi: 10.1002/cpt.656. Epub 2017 Mar 22. PMID: 28182273. [DOI] [PubMed] [Google Scholar]
- 17. Transcript for the November 6, 2020 Meeting of the Peripheral and Central Nervous System Drugs Advisory Committee. accessed on July 18, 2022. https://www.fda.gov/media/145691/download
- 18. Concurrence Memorandum from Peter Stein, MD, Director, Office of New Drugs. accessed on January 23, 2023. www.accessdata.fda.gov/drugsatfda_docs/nda/2021/Aducanumab_BLA761178_Stein_2021_06_07.pdf
- 19. Gortler David, Unforgivable Hypocrisy From FDA's Career CDER Leadership. https://www.forbes.com/sites/davidgortler/2021/07/19/unforgivable‐hypocrisy‐from‐fdas‐career‐cder‐leadership/?sh=70a9e61f3418
- 20. Andrews JS, Desai U, Kirson NY, Zichlin ML, Ball DE, Matthews BR. Disease severity and minimal clinically important differences in clinical outcome assessments for Alzheimer's disease clinical trials. Alzheimers Dement (N Y). 2019;5:354‐363. doi: 10.1016/j.trci.2019.06.005. PMID: 31417957; PMCID: PMC6690415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. FDA Medical Review. accessed on July 18, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761178Orig1s000MedR_Redacted.pdf,
- 22. Cummings J, Aisen P, Lemere C, Atri A, Sabbagh M, Salloway S. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimers Res Ther. 2021;13(1):98. doi: 10.1186/s13195-021-00838-z. PMID: 33971962; PMCID: PMC8111757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. FDA Office of Neurology's Summary Review Memorandum. accessed on January 23, 2023. www.accessdata.fda.gov/drugsatfda_docs/nda/2021/Aducanumab_BLA761178_Dunn_2021_06_07.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information