

Significance
The outcome of viral infection in cells is dependent on the balance between host restriction factors and viral countermeasures. BST2/tetherin is a restriction factor that reduces HIV-1 dissemination by tethering virions at the cell surface. Its action is counteracted by the viral protein Vpu through multiple mechanisms. Here, we provide a description of the initial step of a non-canonical autophagic pathway, called LC3C-associated pathway, subverted by Vpu to counteract BST2 antiviral activities. We found that ATG5 acts as a transducer by targeting phosphorylated and dimerized virus-tethered BST2 from cell surface to degradation. Our discovery opens an avenue in the discovery of unconventional functions of ATG5, as an adaptor for receptor at the plasma membrane initiating an unconventional autophagy process.
Keywords: ATG5, BST2/tetherin, HIV-1, LC3-associated pathway, non-canonical autophagy
Abstract
Bone marrow stromal antigen 2 (BST2)/tetherin is a restriction factor that reduces HIV-1 dissemination by tethering virus at the cell surface. BST2 also acts as a sensor of HIV-1 budding, establishing a cellular antiviral state. The HIV-1 Vpu protein antagonizes BST2 antiviral functions via multiple mechanisms, including the subversion of an LC3C-associated pathway, a key cell intrinsic antimicrobial mechanism. Here, we describe the first step of this viral-induced LC3C-associated process. This process is initiated at the plasma membrane through the recognition and internalization of virus-tethered BST2 by ATG5, an autophagy protein. ATG5 and BST2 assemble as a complex, independently of the viral protein Vpu and ahead of the recruitment of the ATG protein LC3C. The conjugation of ATG5 with ATG12 is dispensable for this interaction. ATG5 recognizes cysteine-linked homodimerized BST2 and specifically engages phosphorylated BST2 tethering viruses at the plasma membrane, in an LC3C-associated pathway. We also found that this LC3C-associated pathway is used by Vpu to attenuate the inflammatory responses mediated by virion retention. Overall, we highlight that by targeting BST2 tethering viruses, ATG5 acts as a signaling scaffold to trigger an LC3C-associated pathway induced by HIV-1 infection.
Host restriction factors, a set of proteins with direct or indirect antiviral activity, are an important facet of the innate immune response controlling infection in a cell-intrinsic manner (1). Among them, the restriction factor bone marrow stromal antigen 2 (BST2)/tetherin interferes with the release of diverse mammalian enveloped viruses including the HIV-1 by physically trapping de novo formed viral particles at the surface of infected cells (2, 3). Tethered virions can be endocytosed and then targeted for lysosomal degradation (4). The antiviral activity of BST2 relies on its presence at the viral budding site and on its ability to be incorporated into budding virions, bridging virions and cellular membranes (5). Prior to its identification as an antiviral factor, BST2 was identified as an inducer of proinflammatory genes expression via NFĸB activation (6). Coupled to its viral tether activity, BST2 acts as a sensor of HIV-1 budding, establishing a cellular antiviral state, and thus making infected cells more visible to innate and adaptative immunity (7, 8).
Viruses have thus adopted diverse strategies to antagonize this restriction and further optimize viral release and attenuate the cellular antiviral response. They notably encode counteracting viral proteins, such as the HIV-1 accessory protein Vpu. Vpu countermeasures against BST2 depend on the direct interaction between Vpu transmembrane domain and that of BST2 (5, 9, 10). Vpu downregulates BST2 cell surface and global expression (2, 3), by modifying BST2 intracellular trafficking (11–15) and accelerating ESCRT-mediated sorting of BST2 for degradation (16–19). Vpu also displaces BST2 from viral budding sites. This activity is crucial to overcome BST2 restriction on HIV-1 release. It involves hijacking of clathrin adaptor protein complexes (AP)-dependent endocytic pathways (11–13) and is independent of Vpu effect on BST2 cell surface expression (2, 3, 9, 13, 20). Moreover, Vpu attenuates the NFĸB-dependent pro-inflammatory response mediated by viral-induced BST2 signaling (7, 21). Interestingly, we revealed an additional way by which Vpu counteracts BST2 innate barrier. We reported that Vpu acts with LC3C, an autophagy protein (ATG) to favor the removal of BST2 tethering virus at the plasma membrane and their targeting in a single-membrane compartment for degradation (22). Two ATG proteins, ATG5 and Beclin 1/ATG6, but not all the components of the autophagy pathway, are also implicated in this process, supporting the view that a non-canonical autophagy process reminiscent of LC3-associated pathway contributes to Vpu-induced removal and degradation of the fraction of BST2 molecules tethering virus at the plasma membrane (22).
The LC3-associated pathway is a non-canonical autophagy process controlled by a subset of ATG proteins, linking activation of a surface receptor with phagocytosis (LC3-associated phagocytosis, LAP) (23) or endocytosis (LC3-associated endocytosis, LANDO) (24). LC3-associated pathway has a prominent anti-inflammatory function and represents a key cell-intrinsic restriction mechanism against infections (25–27). As expected for such an antimicrobial pathway, pathogens have acquired the ability to subvert it to attenuate the host responses or establish a proliferative niche in the host (22, 27). LC3-associated pathway differs from canonical autophagy by the formation of a single-membrane structure from invagination of the plasma membrane (28). The engagement of the surface receptor triggers the recruitment of several components of the canonical autophagy process to conjugate LC3 at the phagosomal or endosomal membrane (26, 29). This recruitment accelerates the fusion of these single membrane compartments with the lysosomes and thus the degradation of their contents. In this process, ATG5 in conjugation with ATG12 is part of the LC3 conjugation system which catalyzes the conjugation of LC3 on the single membrane vesicle. Although the molecular players involved in the initiation and regulation of LC3-associated phagocytosis are becoming better characterized, the molecular mechanism driving the selective engagement of BST2 molecules in the LC3-associated pathway induced by HIV-1 infection is not elucidated (29–31). In this study, we dissected the initial events driving BST2 in the LC3C-associated pathway subverted by Vpu and analyzed the contribution of this pathway on Vpu countermeasure to pro-inflammatory signaling induced by BST2 sensing.
Here, we identify a role for ATG5 as a key player in the LC3C-associated pathway subverted by Vpu. We show that the formation of an ATG5 and BST2 complex at the cell surface is the first step of this viral-induced LC3C-associated process. It occurs independently of Vpu, ahead of the recruitment of LC3C, and requires cysteine-linked homodimerization of BST2. We revealed that the unconjugated form of ATG5 can efficiently recruit the dimerized form of BST2. We also provide evidence that ATG5 preferentially targets phosphorylated virus-tethered BST2, the specific form triggering BST2 signaling. Finally, we found that this LC3C-associated pathway is used by Vpu to attenuate the inflammatory responses mediated by BST2 sensing of virion retention. In summary, we propose that by targeting BST2 tethering viruses, ATG5 acts as a transducer of the LC3C-associated pathway induced by HIV-1 infection.
Results
ATG5 and BST2 Assemble as a Complex Independently of Vpu and Ahead of the Recruitment of LC3C.
We previously described a non-canonical autophagy pathway reminiscent of LC3-associated phagocytosis contributing to HIV-1 Vpu antagonism of the BST2 innate barrier. We highlighted that ATG5 and Beclin 1, but not all the components of the autophagic pathway, act with LC3C in this process. We revealed a selective interaction between HIV-1 Vpu and LC3C (Fig. 1A) (22).
Fig. 1.

ATG5–BST2 complex forms independently of Vpu and ahead of LC3C Recruitment. (A) Overview of the interaction between BST2, Vpu, LC3C and ATG5. (B) Immunoprecipitation of endogenous BST2 in HeLa cells extracts co-transfected with plasmids expressing Flag-ATG5, Flag-LC3C, and Flag-Beclin 1. Immunoprecipitated proteins were detected by western blotting using rabbit anti-BST2 and anti-Flag HRP antibodies. (C) Immunoprecipitation of endogenous BST2 in HeLa cells extracts co-transfected with plasmids expressing Flag-ATG5 and GFP or Vpu-GFP. Immunoprecipitated proteins were detected by western blotting using rabbit anti-BST2 and anti-Flag HRP antibodies. All western blots presented are representative of at least three independent experiments. Immunoprecipitation of endogenous BST2 were done with mouse anti-BST2 antibodies in all experiments.
To determine the initial step driving the engagement of BST2 molecules present at the budding site in the LC3C-associated pathway induced by HIV-1 infection, we examined whether BST2 interacts with ATG5, Beclin 1, or LC3C. We found through co-immunoprecipitation experiments that BST2 interacts with Beclin 1 and LC3C albeit with low affinity (Fig. 1B, compare lanes 7 and 8 to lane 5). By contrast, we revealed a strong interaction between BST2 and both the unconjugated form of ATG5 (ATG5) and ATG5 conjugated to ATG12 (ATG5-ATG12) (Fig. 1B, compare lanes 5 to 6). Moreover, we observed that Vpu expression did not modify this interaction (Fig. 1 C, Right, compare lanes 3 to 4), suggesting that ATG5 interacts with BST2 independently of Vpu and of its conjugation to ATG12.
We previously showed that, upon infection, Vpu binds specifically to the ATG protein LC3C. This interaction favors the targeting of BST2 molecules present at the budding site to an LC3C-associated pathway (22). The complex made by LC3C, Vpu, and BST2 plays a central part in this process (Fig. 1A). We thus analyzed the contribution of LC3C in the interaction between BST2 and ATG5. Using two LC3C knockout (KO) cell lines previously described in ref. 22, we observed that the presence of LC3C is not required for ATG5 to bind BST2 (SI Appendix, Fig. S1 A, Right panel, compare lanes 5 and 6 to 4). Conversely, we examined whether the formation of the complex made by LC3C, Vpu, and BST2 relies on the expression of ATG5. For that, we established two different ATG5−/− HeLa clones (ATG5−/− 129#9 and 92#3) using the CRISPR-Cas9 system. As expected, the KO of ATG5 gene blocked the lipidation of LC3 proteins and only the nonlipidated form of LC3B (LC3B-I) was observed even upon autophagy induction by amino-acid starvation (ES) (SI Appendix, Fig. S1B). Using co-immunoprecipitation, we showed that only the lipidated form of LC3C (LC3C-II) binds weakly to BST2 in presence of ATG5 and this binding is strongly increased in presence of Vpu, correlating with a Vpu-dependent co-immunoprecipitation of LC3C with BST2 (SI Appendix, Fig. S1 C, Right panel, compare lanes 4 to 10) (22). In contrast, the KO of ATG5 resulted in a loss of binding of LC3C to BST2 even upon Vpu expression (SI Appendix, Fig. S1 C, Right panel, compare lanes 11 and 12 to 10), indicating that ATG5 is a key determinant for the formation of this 3-partner complex.
Altogether, these data support the view that ATG5 and BST2 assemble as a complex independently of Vpu and ahead of the recruitment of LC3C.
ATG5 Takes in Charge BST2 Molecules Tethering Viral Particles at the Cell Surface.
To confirm the implication of ATG5 in the engagement of BST2 molecules in the LC3C-associated pathway induced by HIV-1 infection, we assessed the impact of ATG5 depletion on the distribution of BST2 and viral components at the surface of HIV-1-infected cells. Confocal analysis of cell surface BST2 staining revealed rare small BST2 puncta at the surface of control-infected cells (Fig. 2 A, Upper) compared to BST2 staining in non-infected cells, illustrating the well-documented BST2 downregulation mediated by Vpu (2). Interestingly, in ATG5-depleted cells, large puncta of BST2 were still observed in specific area of the plasma membrane, colocalizing with HIV structural components, the tagged HIV-1 Gag product MA/YFP and the HIV-1 Envelope glycoprotein (Env) (Fig. 2A), despite an overall downregulation of cell surface BST2 in infected cells compared to neighboring non-infected cells. Indeed, the measurement of BST2 cell surface level by flow cytometry showed that Vpu-mediated downregulation of cell surface BST2 is still efficient in ATG5-depleted cells infected with HIV-1 wild type (WT) (CAp24-positive cells) compared to non-infected cells or cells infected with HIV-1 that do not express Vpu (HIV-1 Udel) (SI Appendix, Fig. S2).
Fig. 2.
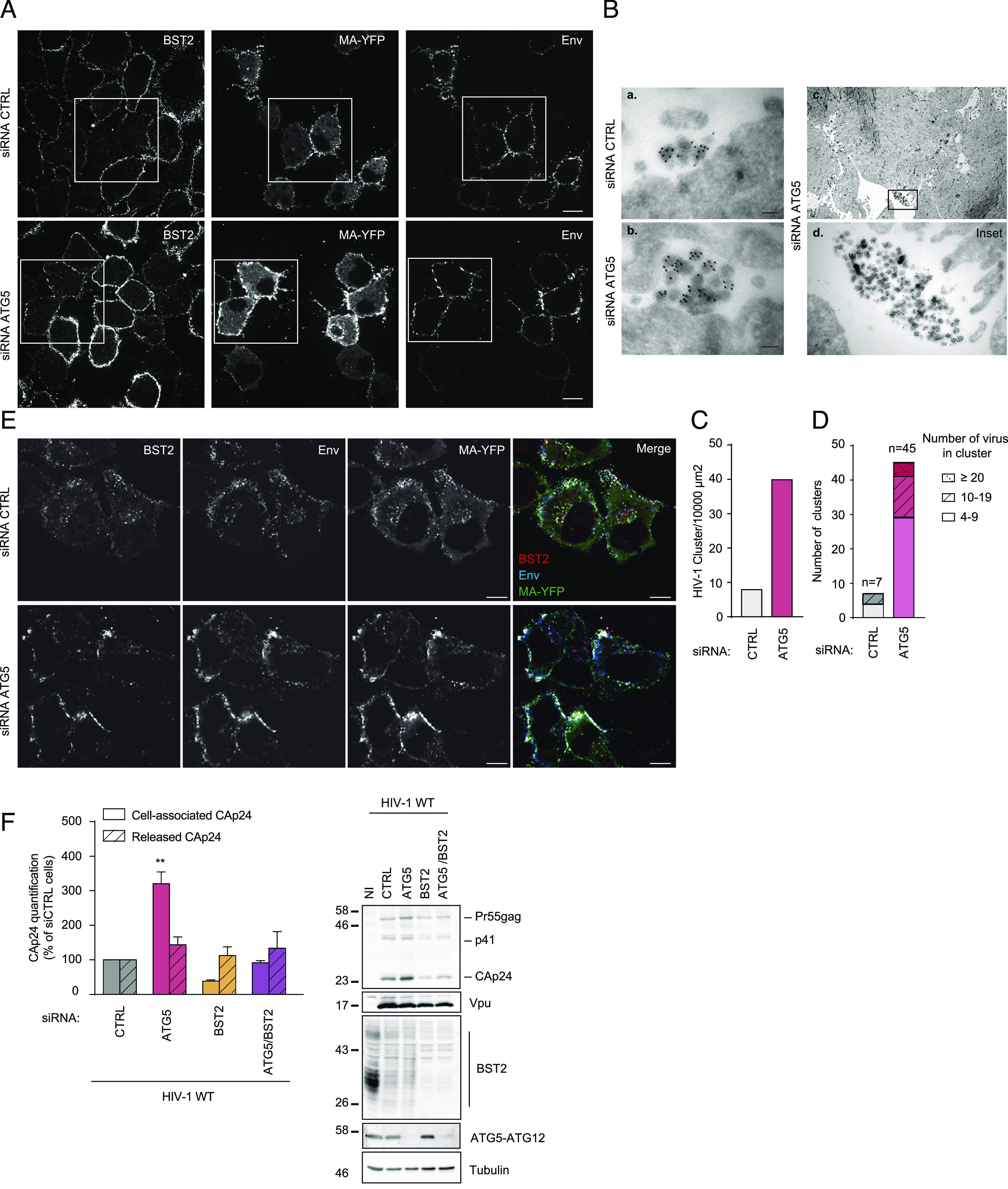
ATG5 depletion leads to virions retention at the plasma membrane in a BST2-dependent manner. (A) Confocal fluorescence microscopy of HeLa cells transfected with indicated siRNA and infected with VSV-G-pseudotyped HIV-1 WT MA-YFP at a MOI of 0.5 for 24 h following cell surface staining with human anti-Env (SUgp120) and rabbit anti-BST2 antibodies. (Scale bar: 10 μm). Immunofluorescence images presented are representative of at least three independent experiments. (B) Cryosections of HeLa cells treated with CTRL (a) or ATG5 (b) siRNA for 72 h and then infected with VSV-G pseudotyped HIV-1 WT for 48 h before labelling with antibodies against CAp24 and 10-nm protein A-gold. (Scale bar, 100 nm.) (c) Clusters of viral particles at the cell surface in ATG5 depleted cells. (d) Enlargement of the region indicated in the upper panel (c). (C) Quantification of the number of clusters of viral particles per 10,000 µm2 analyzed in (B). The number of clusters of viral particles in control and ATG5-depleted cells was determined by scanning 8,815 and 11,540 µm2, respectively. (D) Relative distribution of HIV-1 particles in cluster presented in (A). The total number of clusters of HIV-1 particles counted in control and ATG5-depleted cells was 7 and 45, respectively. (E) Confocal fluorescence microscopy of HeLa cells transfected with indicated siRNA and infected with VSV-G-pseudotyped HIV-1 Udel MA-YFP at a MOI of 0.5 for 24 h following antibody-feeding assay using human anti-Env (SUgp120) and rabbit anti-BST2 antibodies. (Scale bar: 10 μm). Immunofluorescence images presented are representative of at least three independent experiments. (F) HeLa cells transfected with indicated siRNA were infected with a VSV-G pseudotyped HIV-1 NL4-3 WT at a MOI of 0.5 for 48 h. Cell-associated CAp24 and released CAp24 were quantified by Elisa. Statistical analysis using two-way ANOVA with Holm-Sidak’s multiple comparisons test, mean ± SEM, n = 3 experiments; **P ≤ 0.01. Western blot analysis of HIV-1 Gag and CAp24 products, Vpu, BST2, ATG5 and Tubulin in infected siRNA-treated cells. All western blot are representative of at least three independent experiments.
We then investigated the nature of the large patches of viral products accumulated at the periphery of ATG5-depleted cells by electron microscopy. Labeling of cryosections of HIV-1 WT-infected cells with antibody targeting the viral Gag subproduct CAp24 (Capsid) mainly showed mature HIV-1 particles accumulating at the plasma membrane in ATG5-depleted cells (Fig. 2 B, b). The analysis of HIV-1 particle distribution revealed a higher number of HIV-1 particle clusters per 10,000 µm2 found at the cell surface in ATG5-depleted cells compared to infected control cells (Figs. 2 B, c and d and C). Furthermore, the average number of HIV-1 particles per cluster upon ATG5 depletion was superior to that in control cells (Fig. 2D). Interestingly, while we saw numerous intracellular structures containing mature HIV-1 particles in LC3C-depleted cells in our previous study (22), we rarely observed HIV-1 particles sequestered in intracellular compartments in ATG5-depleted cells. These data provide evidence that, upon HIV-1 infection, ATG5 acts at the cell surface level and upstream of LC3C.
BST2-tethered viruses were shown to undergo endocytosis (4). We thus assessed the impact of ATG5 depletion on the internalization of BST2 molecules tethering viruses using an antibody-feeding assay. The analysis was performed on cells infected with Vpu-defective HIV-1 to avoid any misinterpretation of our results due to Vpu downregulation of cell surface BST2. SiRNA-treated cells infected with Vpu-defective HIV-1 were incubated at 37 °C with anti-Env (2G12) and anti-BST2 antibody to label surface-exposed proteins and viruses. In CTRL cells, we observed intracellular BST2 staining colocalizing with HIV structural components, the tagged HIV-1 Gag product MA/YFP, and the labeled HIV-1 envelope glycoprotein (Env), indicating an endocytosis of virus-tethered BST2 from the cell surface (Fig. 2 E, Upper). In ATG5-depleted cells, an accumulation of HIV structural components (MA/YFP and labeled Env) colocalizing with labeled BST2 was mostly seen at the cell surface (Fig. 2 E, Lower), indicating that virus-tethered BST2 remain at the cell surface and are not endocytosed, compared to CTRL cells. These data suggest that ATG5 is necessary for the internalization of virion tethered by BST2.
We finally checked whether the effect of ATG5 depletion on HIV-1 particle retention at the cell surface was related to BST2 expression. Upon HIV-1 infection, ATG5 depletion induced a strong accumulation of the cell-associated viral CAp24 protein without impacting the amount of CAp24 released found in the supernatant. This effect can also be detected by western blot in ATG5-depleted infected cells (Fig. 2F). Interestingly, ATG5 knockdown did not induce an accumulation of cell-associated CAp24 nor affected the amount of CAp24 released in BST2-depleted cells suggesting, along with the cell microscopy data, that the effect of ATG5 silencing on viral retention is linked to BST2.
Together, these results suggest that ATG5 takes in charge BST2 molecules tethering viral particles at the cell surface.
The Interaction Between ATG5 and BST2 Is Strengthened in HIV-1–Producing Cells and Independent of ATG5 Conjugation Status.
Based on the above observations (Fig. 2), we hypothesized that ATG5 acts as an adaptor to trigger the targeting of virus-tethered BST2 onto the LC3C-associated pathway subverted by Vpu. We thus explored the binding of ATG5 on BST2 in HIV-1–producing cells. We found through co-immunoprecipitation that the interaction between ATG5 and BST2 is strengthened upon HIV-1 production (Fig. 3A and SI Appendix, Fig. S3A). Interestingly, we noticed that HIV-1 productive infection favors the interaction of unconjugated form of ATG5 with BST2, independently of Vpu expression (Fig. 3 A, Right panel, compare lanes 4 and 6 to 2). This correlates with the Vpu-independent binding of ATG5 to BST2 shown in Fig. 1C.
Fig. 3.

Productive infection favors ATG5–BST2 interaction independently on ATG5 conjugation with ATG12. (A) HEK293T cells were co-transfected with p3XFlag-ATG5, either pcDNA or pcDNA-BST2 WT and the provirus HIV-1 WT or Vpu-deleted (Udel) for 24 h. BST2 was immunoprecipitated with mouse anti-BST2 antibodies and immunoprecipitated proteins were detected by western blotting using rabbit anti-BST2 and anti-Flag-HRP antibodies. (B) HeLa cells were co-transfected with vector encoding for Flag-ATG5 and the provirus HIV-1 WT or Vpu-deleted (Udel) for 24 h. Proteins were detected by western blotting using anti-Flag-HRP and anti-GAPDH antibodies. Quantification of the relative amount of unconjugated ATG5 after normalization to the ATG5-ATG12 conjugate form upon infection in five experiments. Statistical analysis using one-way ANOVA with Turkey's multiple comparisons test, mean ± SEM, n = 5 experiments; ***P ≤ 0.001. (C) Immunoprecipitation of GFP in HeLa cells extracts transfected with vectors expressing GFP, GFP-ATG5 or GFP-ATG5 K130R. Immunoprecipitated proteins were detected by western blotting using rabbit anti-BST2 and anti-GFP HRP antibodies.
As unconjugated ATG5 binds BST2, we next studied the conjugation pattern of ATG5 upon infection. Intriguingly, we observed that independently of Vpu expression, the amount of ATG5-ATG12 conjugate is markedly reduced in infected cells, whereas the amount of free ATG5 increased significantly suggesting that the infection reduces the formation of ATG5-ATG12 conjugate, making free ATG5 available (Fig. 3B).
Finally, we evaluated whether the binding of ATG5 to BST2 relies on its conjugation status with ATG12, and thus on its function in autophagy. We performed a green fluorescent protein affinity immunoprecipitation (GFP-Trap) in cells expressing GFP-ATG5 or GFP-ATG5 K130R, an autophagy-incompetent mutant bearing a point mutation that prevents the formation of the ATG5-ATG12 conjugate essential in autophagy. The results showed that GFP-ATG5 WT or GFP-ATG5 K130R similarly immunoprecipitate BST2. Interestingly, both constructs bound predominantly the dimeric form of BST2 (Fig. 3C).
Thus, these data suggest that ATG5-ATG12 conjugation is dispensable for the interaction between ATG5 and BST2 and that ATG5 could bind preferentially dimeric forms of BST2.
Binding of ATG5 to the Cytosolic Domain of BST2 Requires BST2 Dimerization.
To further understand the initial event that governs the recruitment of ATG5 on BST2, we investigated which domain of BST2 interacts with ATG5 and spotted that ATG5 binds to the cytoplasmic domain of BST2, since the truncation of the first 21 amino-acids of BST2 strongly decreases ATG5-BST2 interaction (Fig. 4A, compare lanes 2 to 3). To confirm our finding, we performed an immunoprecipitation assay using the short isoform of BST2 lacking the first 12 amino acids of the cytoplasmic tail (BST2 M1A) (32) (SI Appendix, Fig. S3B). We observed that the interaction between ATG5 and the short isoform of BST2 (BST2 M1A) is less efficient than with BST2 WT, indicating that the integrity of the cytoplasmic tail of BST2 is necessary for its interaction with ATG5. We next performed an alanine scanning mutagenesis along the cytoplasmic domain of BST2 to determine the minimal sequence required for the binding of ATG5 to BST2 (SI Appendix, Fig. S3 C and D). Interestingly, none of the mutants affected the binding of ATG5 to BST2, implying that current mutations are not enough to alter the motif or structure essential for the ATG5-BST2 interaction.
Fig. 4.

BST2 dimerization is an essential for efficient ATG5 Binding. (A) HeLa cells were transfected with plasmids expressing HA-ATG5 and either Flag, Flag-BST2 or Flag-BST2 truncated for its cytosolic domain (BST2 Delta CT). Flag-tagged proteins were immunoprecipitated with anti-Flag antibodies. Immunoprecipitated proteins were detected by western blotting using anti-HA-HRP and anti-Flag-HRP antibodies. (B) Schematic representation of human BST2 showing the GPI anchor, the coiled-coil and the transmembrane domains as well as the cytoplasmic tail. Indicated in blue, red and green respectively were the amino acids essential for the N-linked glycosylation (N65 and N92), dimerization (C53, C63, and C91) and phosphorylation (Y6, Y8) of BST2. (C) HEK293T cells were co-transfected with vectors expressing Flag-ATG5, either pcDNA-BST2 WT or BST2 Y6Y8 (double tyrosine phosphorylation defective mutant) or BST2 C3A (triple cysteine dimerization defective mutant) and the provirus WT or Vpu-deleted (Udel) NL4-3 HIV-1 for 24 h. BST2 was immunoprecipitated with mouse anti-BST2 antibodies and immunoprecipitated proteins were detected by western blotting using rabbit anti-BST2 and anti-Flag-HRP antibodies. All Western blots presented are representative of at least three independent experiments.
Upon infection, BST2 acts as a viral tether via multimerization by bridging viral particles to the cells as well as a viral sensor capable of initiating the recruitment of proteins and signaling cascades upon infection (7, 21). We thus investigated whether mutations that disrupt the sensing and tethering function of BST2 affect the binding of ATG5. To alter the sensing function of BST2, we mutated the two conserved tyrosine residues (Y6 and Y8) that are phosphorylated upon retroviral particle retention (8). This BST2 Y6Y8-AA mutant (BST2 Y6Y8) is unable to activate the downstream NFĸB pathway (Fig. 4B) (7, 21, 32). Accordingly, BST2 Y6Y8 was not recognized by an antibody that we developed, targeting specifically phosphorylated forms of BST2 (pBST2) (SI Appendix, Fig. S3E, compare lanes 2 to 3). For the tethering function of BST2, we focused on BST2 C3A mutant in which three cysteines (C53, C63, and C91) of BST2 ectodomain required for dimerization have been mutated to alanine (Fig. 4B). This mutant is unable to dimerize and to tether virions at the cell surface (33). Migration of this mutant under non-reducing condition confirmed its inability to dimerize (SI Appendix, Fig. S3F, compare lanes 2 to 4). Co-immunoprecipitation experiments showed that the substitution of Y6Y8 residues to alanine residues (BST2 Y6Y8) did not alter ATG5-BST2 interaction, indicating that phosphorylation of the cytoplasmic tail is not required for ATG5 interaction (Fig. 4C, compare lanes 2 to 3, lanes 6 to 7 and lanes 10 to 11 and SI Appendix, Fig. S3G). Interestingly, we found that the mutation of the 3 cysteines of BST2 ectodomain (BST2 C3A) decreases the binding of ATG5 to BST2 (Fig. 4 C, Right panel, compare lanes 2 to 4, lanes 6 to 8 and lanes 10 to 12).
Altogether, these results indicate that ATG5 binding to BST2 requires the integrity of the cytoplasmic tail of BST2 as well as the cysteine residues implicated in BST2 dimerization and virus tethering.
ATG5 Targets Phosphorylated Dimerized Virus-Tethered BST2.
To further define the form of BST2 targeted by ATG5 upon tethering of viral particles, we determined the impact of ATG5 depletion on the N-linked glycosylation pattern of BST2 upon infection. BST2 is a N-linked glycosylated protein on asparagine 65 and 92 (N65; N92) (Fig. 4B) that can be found in a monomeric (mBST2) and dimeric form (dBST2). Following crosslinking with formaldehyde, allowing us to study simultaneously monomeric and dimeric status of BST2, mBST2 migrates as a smear of heterogeneously glycosylated forms with apparent molecular weights between 26 to 43 kDa in control cells, whereas dBST2 migrates between 43 to 95 kDa (SI Appendix, Fig. S4A, lane 1). Several glycosylated forms of BST2 can be detected: the non-glycosylated species, the high mannose modification, and complex-type carbohydrates at either or both positions. Interestingly, ATG5 depletion induced an upward shift of BST2 smear for both mBST2 and dBST2, compared to control cells or LC3C depleted cells (SI Appendix, Fig. S4A, lanes 2, 5 and 8 to lanes 1, 3 and 4, and to lanes 6, 7 and 9), suggesting that the N-linked glycosylated forms of BST2 are enriched in absence of ATG5. As similar results were observed in cells infected with HIV-1 WT and Udel compared to non-infected cells, this phenotype was independent of the infection and the expression of Vpu (SI Appendix, Fig. S4A, compare lanes 5 and 8 to lane 2; see also SI Appendix, Fig. S4B, line scan HIV-1 WT vs. HIV-1 Udel).
To go further, we assessed the impact of ATG5 depletion on the dimerization of BST2. A PNGase treatment, which removes all oligosaccharides on BST2 molecules enriched after immunoprecipitation, resulted in deglycosylated mBST2 with a molecular weight of 17kDa and a deglycosylated dBST2 with a molecular weight of 38kDa (SI Appendix, Fig. S4C, compare Lane 1 to lane 4). By evaluating the ratio between dBST2 and mBST2 to assess the variations of BST2 expression in each condition, we revealed an increased amount of dBST2 in ATG5 depleted cells compared to control cells, in both infected and non-infected cells (Fig. 5 A, Upper panel, compare lanes 2, 5, and 8 to lanes 1, 4, and 7 and SI Appendix, Fig. S4D). These results suggest that, upon infection, ATG5 targets virus-tethered BST2 which is mainly N-linked glycosylated and under its dimeric form, confirming the results shown in Figs. 3C and 4C.
Fig. 5.

ATG5 selectively engages phosphorylated virus-tethered BST2 in an LC3-associated pathway controling viral-induced BST2 signaling. (A) HeLa cells transfected with either control siRNA (CTRL) or siRNA targeting ATG5 or LC3C were infected with a VSV-G pseudotyped WT or Udel NL4-3 HIV-1 at a MOI of 0.5 for 24 h. Endogenous BST2 was immunoprecipitated with a mouse anti-BST2 antibody and deglycosylated precipitates were analyzed by western blot using rabbit antiphosphorylated BST2 (pBST2) and rabbit anti-BST2 antibodies. dBST2: dimer of BST2; mBST2: monomer of BST2. (B) Quantification of the relative amount of the phosphorylated monomeric form of BST2 (pBST2) after normalization to immunoprecipitated monomeric BST2. Statistical analysis using two-tailed unpaired Student’s t test, mean ± SEM, n = 3 experiments, *P ≤ 0.05. (C) Confocal fluorescence microscopy of HeLa cells infected with VSV-G-pseudotyped WT HIV-1 MA-YFP for 24 h following total staining with rabbit anti-pBST2 antibodies. (Scale bar: 20 μM). All western blots and immunofluorescence images presented are representative of at least three independent experiments. (D) HeLa BST2 WT and HeLa BST2 −/− cells transfected with either control siRNA (CTRL) or siRNA targeting ATG5 were transfected with the WT or Vpu-deleted (Udel) NL4-3HIV-1 provirus for 28 h. Total RNA was analyzed for TNFα mRNA levels relative to KDSR by qRT-PCR. Statistical analysis using two-tailed unpaired Student’s t test, mean ± SEM, n = 3 experiments, *P ≤ 0.05, ns: non-significant. (E) HeLa BST2 WT and HeLa BST2 −/− cells transfected with either control siRNA (CTRL) or siRNA targeting LC3C were transfected with the provirus HIV-1 NL4-3 WT or Udel for 28 h. Total RNA was analyzed for TNFα mRNA levels relative to KDSR by qRT-PCR. Statistical analysis using two-tailed unpaired Student’s t test, mean ± SEM, n = 3 experiments, *P ≤ 0.05, ns: non-significant. (F) Purified CD4+T cells from three independent donors were nucleofected with the indicated gRNA and then infected with a VSV-G pseudotyped HIV-1 NL4-3 WT or Udel at a MOI of 0.5 for 48 h. Total RNA was analyzed for CXCL10 and IFNβ mRNA levels relative to FKBP4 by qRT-PCR. (G) HeLa cells transfected with either control siRNA (CTRL) or siRNA targeting ATG5 or LC3C were infected with a VSV-G pseudotyped WT or Udel NL4-3 HIV-1 at a MOI of 0.5 for 24 h. Endogenous BST2 was immunoprecipitated with a mouse anti-BST2 antibody and deglycosylated precipitates were analyzed by western blot using rabbit antiphosphorylated Syk (pSyk) and rabbit anti-BST2 antibodies.
Multiples studies correlate the multimerization state of BST2 upon viral tether and its phosphorylation on tyrosine residues present in its cytosolic domain (Y6 and Y8) (7, 21, 32). It has also been previously documented that the sensing function of BST2 is interrelated to its phosphorylation status and leads to the activation of the NFĸB pathway. We thus explored the phosphorylation status of the virus-tethered BST2 in cells depleted for ATG5. In accordance with previous studies (8), we confirmed an increase of the level of pBST2 upon infection, notably in cells infected with HIV-1 Udel (Fig. 5 A, Lower panel, compare lanes 4 to 7 to lane 1, and Fig. 5B). Interestingly, depletion of ATG5 and LC3C strongly enriched the monomeric and dimeric forms of phosphorylated BST2 in cells infected with HIV-1 WT and Udel, compared to control cells (Fig. 5 A, Lower panel, compare lanes 5-6 and 8-9 to 2-3, and Fig. 5B). Accordingly, confocal analysis of pBST2 staining revealed an accumulation of pBST2 in HIV-1 WT-infected cells upon ATG5 depletion compared to control cells (Fig. 5C). Furthermore, these patches of pBST2 were distributed in the vicinity of HIV-1 Gag product, MA/YFP at or near the plasma membrane (Fig. 5 C, Lower panel, Inset).
Altogether, these data support the view that ATG5 as well as LC3C might promote the degradation of phosphorylated forms of BST2 induced by virions retention through an LC3C-associated pathway
LC3C-Associated Process Regulates BST2-Mediated Signaling Induced by Virions Retention.
Phosphorylated BST2 tethering viruses have been regarded as signaling complexes that induced NFĸB-dependent pro-inflammatory responses. We thus speculated that ATG5 as well as LC3C, via an LC3C-associated pathway, exert an effect on BST2-mediated signaling. To examine the involvement of these two ATG proteins, parental and BST2 KO HeLa cells (BST2−/−) (34) were co-transfected with control (CTRL) siRNA or siRNA targeting ATG5 or LC3C along with the WT (HIV-1 WT) or Vpu-defective (HIV-1 Udel) HIV-1 provirus. The outcome of these extinctions on the NFĸB-dependent target gene expression TNFα was evaluated. To solely analyze the contribution of ATG5 and LC3C in the control of BST2-induced pro-inflammatory responses by Vpu, we normalized values obtained to the non-infected (NI) condition of each cell line. As previously reported (21), the expression of HIV-1 Udel provirus in parental control cells leads to a higher level of mRNA for the proinflammatory cytokine TNFα than those observed upon expression of HIV-1 WT provirus (Fig. 5 D and E). This increase was lost in BST2−/− cells, underlying the BST2 dependency of the enhanced proinflammatory response observed in these cell lines upon HIV-1 Udel expression (Fig. 5 D and E and SI Appendix, Fig. S5A). Surprisingly, the depletion of ATG5 decreased TNFα mRNA induction observed upon HIV-1 Udel provirus expression at a level similar to the one obtained with HIV-1 WT provirus expression (Fig. 5D and SI Appendix, Fig. S5A), suggesting that the enhanced proinflammatory response observed upon HIV-1 Udel expression is dependent on both ATG5 and BST2 expression. Interestingly, the knockdown of LC3C led to an increased level of TNFα mRNA upon expression of HIV-1 WT and Udel provirus in parental cells (Fig. 5E and SI Appendix, Fig. S5B), which was completely lost in BST2−/− cells. This suggests that LC3C is implicated in the attenuation of BST2-induced proinflammatory gene expression mediated by Vpu upon HIV-1 infection.
We next assessed the implication of ATG5 in BST2-induced proinflammatory response upon viral infection in primary CD4+ T cells. Purified CD4+ T cells from three healthy donors were depleted for ATG5 using CRISPR-Cas9 system and then infected with HIV-1 WT or Udel (SI Appendix, Fig. S5C). The level of infection was monitored by analyzing mRNA level of HIV-1 Nef (SI Appendix, Fig. S5C). NF-ĸB activity was measured indirectly by analyzing mRNA level for the downstream targets CXCL10 and IFNβ. Depending on the donors, HIV-1 Udel infection of CD4+ T cells induced a 1.8 to 15.4 fold increase in CXCL10 mRNA level and a 1.3- to 13.9-fold increase in IFNβ mRNA level (Fig. 5F and SI Appendix, Fig. S5C), compared to cells infected with HIV-1 WT. These results are consistent with the clustering of Vpu-defective virus at the cell surface mediated by BST2 in absence of Vpu countermeasure. Interestingly, ATG5 depletion of CD4+ T cells strongly limited the induction of CXCL10 and IFNβ mRNA, detected upon HIV-1 Udel infection (CXCL10: 1.1 to 1.7-fold increase; IFNβ: 0.9 to 1.9-fold increase between HIV Udel vs. WT; Fig. 5F) These results indicate that ATG5 is critical to transduce BST2-mediated signaling induced by virions retention.
Previous study showed that blockade of tethered virion endocytosis potentiates signaling, suggesting that the clustering of BST2 at the plasma membrane in assembling virions is the primary trigger for BST2-mediated signaling (21). Moreover, BST2 phosphorylation induced by viral retention leads to the recruitment of the kinase Syk and its activation by phosphorylation, consequently promoting NFĸB activation (8). Thus, we examined whether ATG5 is essential for the recruitment of the phosphorylated form of Syk, pSyk on BST2. For that, BST2 immunoprecipitation was performed on WT and Udel HIV-1-infected cells depleted or not for ATG5. We found that, though pSyk is detected in HIV-1 WT- and Udel-infected cells lysates, activated pSyk is mainly recruited on immunoprecipitated BST2 from HIV-1 Udel-infected cells, compared to non-infected cells or HIV-1 WT-infected cells (Fig. 5G). These corroborated with previous results showing that BST2-clustering induced by viral retention is the initial trigger for BST2 signal transduction. Moreover, we observed that depletion of ATG5 alters the recruitment of pSyk on BST2 in HIV-1 Udel-infected cells, even though pSyk could be detected in the corresponding cell lysate (Fig. 5G). These results are consistent with the signaling phenotype observed in ATG5-depleted cells and suggest that ATG5 may contribute to BST2-mediated signalling through regulating the recruitment of pSyk on BST2.
Altogether, these results indicate that ATG5 is critical to initiate BST2-mediated signaling cascade, while LC3C and Vpu attenuate the inflammatory response induced by BST2.
Discussion
BST2 is one of the main restriction factors for mammalian enveloped viruses release. It considerably reduces viral dissemination by physically trapping de novo formed viral particles at the surface of infected cells. Studies demonstrate that BST2 forms clusters at the HIV-1 budding sites (35, 36) and that virus-tethered BST2 can be endocytosed and targeted for lysosomal degradation (4). Moreover, coupled with its viral tethering function, BST2 acts as a sensor of HIV-1 budding. By tethering viruses, BST2 activates the NF-ĸB signaling pathway leading to the expression of pro-inflammatory genes, hence establishing an antiviral state of infected cells (21). To counteract this restriction, HIV-1, notably through the viral protein Vpu, engages multiple mechanisms (3, 16, 37). Among them, an LC3C-associated pathway is usurped by Vpu to favor the removal of virus-tethered BST2 from the budding site and their targeting into single membrane compartments to promote their degradation (22). Indeed, the attachment of LC3 proteins to the single membrane vesicles results in an increased acidification and degradation of its content (23).
LC3-associated pathways are initiated by the recognition of a surface receptor by immune complexes, exogenous particles, or pathogens (23, 26, 29). The binding of these ligands to their receptors triggers the invagination of the plasma membrane in a single membrane structure decorated by LC3. Several receptors have been reported to initiate this process, such as TLR1/2, TLR2/6, TLR4, TIM4, and FcR, resulting in the recruitment of some, but not all, members of the autophagic machinery to a single membrane compartment such as phagosome or endosome. These pathways are orchestrated by the concerted action of specific ATG proteins (e.g., LC3, ATG5, and Beclin-1), independently of the autophagy preinitiation complex (38). The work presented here reveals a new unexpected role for ATG5 as a key determinant in i) the recognition of plasma membrane virus-tethered BST2 and ii) the initiation of the LC3C-associated process subverted by Vpu. Indeed, i) ATG5 and BST2 assemble as a complex, independently of Vpu and LC3C (Fig. 1), ii) ATG5 takes in charge BST2 molecules tethering viral particles at the cell surface for their internalization (Fig. 2) and iii) binding of ATG5 to BST2 is strengthened by HIV-1 production (Fig. 3A). Moreover, BST2 preferentially binds to the unconjugated form of ATG5 (Fig. 3A) and HIV-1 expression reduces the formation of ATG5–ATG12 conjugate, making free ATG5 available (Fig. 3B). ATG5 preferentially interacts with dimerized BST2 and ATG5–ATG12 conjugation is dispensable for this interaction (Fig. 3C). These results support a contribution of ATG5 in the selection at the cell surface and the engagement of virus-tethered BST2 molecules in an LC3C-associated pathway that does not require its conjugation to ATG12.
LC3-associated pathway has been initially described in phagocytic cells as LC3B-associated phagocytosis, leading to engulfment and degradation of exogenous cargos (pathogens, cells…)(23, 29). However, recent studies have revealed other pathways linked to LC3. An LC3-associated pathway has been connected to endocytic pathway with LC3-associated endocytosis (LANDO) to favor the degradation of β-amyloid protein and the recycling of its receptor to the cell surface (24). The LC3C-endocytic-associated-pathway (LEAP), is another LC3-associated pathway that has been recently described (39). This pathway involves the targeting of cell surface receptors or plasma membrane-derived proteins via the endocytic machinery to ATG9-containing vesicles and ultimately autophagosome. In contrast to the LAP or LANDO, involving the lipidation of LC3B, the LEAP pathway does not rely on direct LC3C lipidation to vesicular membranes, but instead on LC3C localization to endocytic compartment. While the stimuli, represented by the engagement of a plasma membrane receptor and the fate, implying the recycling or the degradation of these engaged receptors, are known, intracellular adaptors that activate these pathways are not yet known. In our study, we reveal that via its interaction with viral-tethered BST2, ATG5 engages an alternative route for the internalization of cell surface HIV-1 clusters, an LC3C-associated process, targeting them to degradation. Multiple studies show that ATG5 in its conjugated and unconjugated forms can modulate cellular signaling pathways in addition to its traditional role in autophagy (40). This unconventional role for ATG5 at the plasma membrane in the LC3C-associated process induced by HIV-1 infection has never been described and could open a new avenue in the discovery of autophagy-independent functions of this protein, as an adaptor for receptor at the plasma membrane initiating an LC3-associated process.
We thus propose a model in which (1) the recognition of virus-tethered BST2 by ATG5 initiates the LC3C-associated pathway (2). This engagement induces their endocytosis in a single membrane vesicle (3). Then, through its interaction with the transmembrane domain of BST2, Vpu recruits selectively LC3C onto this vesicle membrane (22) (4). The presence of ATG5 at the level of this vesicle, via its interaction with BST2, will favor, after its conjugation with ATG12, the lipidation of LC3C and thus its anchoring to the vesicular membrane. Next, (5) the LC3C recruitment will accelerate the docking of this compartment to the lysosomes leading to the degradation of virus-tethered BST2 (Fig. 6, Left). Conversely, in absence of ATG5, the virus-tethered BST2 remain docked at the plasma membrane and virions accumulate in large clusters that are not endocytosed (Fig. 6, Right).
Fig. 6.

Model for the action of ATG5 in the selection and engagement of virus-tethered BST2 in Vpu-subverted LC3-associated pathway. (Left) (1) ATG5 initiated the LC3-associated pathway by recognizing and targeting phosphorylated virus-tethered BST2. (2) The interaction induces the endocytosis via regulators of clathrin-mediated vesicular trafficking of viral-tethered pBST2 in a single membrane compartment. (3) LC3C will be recruited to the BST2-ATG5 complex through its interaction with Vpu. (4) The presence of ATG5 at the level of the compartment will favor the lipidation of LC3C and thus its anchoring to this compartment sequestering virus-tethered pBST2. (5) The LC3C involvement will accelerate the docking of this compartment to the lysosomes leading to the degradation of virus-tethered pBST2. Upon Vpu expression, the degradation of virus-tethered pBST2 will attenuate the signaling mediated by BST2. Without Vpu expression, viral-tethered pBST2 will still be endocytosed. However, in absence of LC3C recruitment onto this compartment, the fusion with lysosome will be delayed and BST2-mediated signaling will persist. (Right) Without ATG5, virus-tethered BST2 will not be endocytosed and degraded. The virion retention mediated by BST2 will not induce the activation of NF-ĸB signaling.
Our model hypothesizes that ATG5 engages specific forms of BST2 tethered to virions to an LC3C-associated pathway, in which endosomal structures are customized by some ATG proteins. Interestingly, recent proteomic approaches described a strong link between ATG5 and clathrin adaptor protein complexes (AP)-dependent endocytic pathway (41). Moreover, several studies reported that Vpu/BST2 complex can interact with the AP complexes and that these interactions are important for BST2 antagonism by Vpu (11). It is thus tempting to speculate that ATG5 and regulators of clathrin-mediated vesicular trafficking acts in concert to target phosphorylated and dimerized forms of BST2 tethering virions for the degradation in an LC3C-associated pathway. Indeed, analysis of ATG5-BST2 interaction showed that ATG5 selectively engages phosphorylated BST2 (pBST2) in the LC3C-associated pathway induced by HIV-1 infection. ATG5 depletion leads to a marked accumulation of pBST2 (Fig. 5 A and B), implying that BST2 acts like a sensor for the LC3C-associated pathway. Multiple studies showed that the viral sensing function of BST2, after phosphorylation of its tyrosine residues Y6Y8, induces the activation of NFĸB signaling (7, 8, 21, 32). This induction of the NFĸB signaling pathway requires the internalization of BST2 (7) and Vpu is able to attenuate BST2-induced pro-inflammatory responses (21). LC3-associated pathways as well as the membrane trafficking pathway, in regulating transport of internalized receptors, are known to be an important signaling regulatory network. Here, we found that BST2-mediated signaling induced by HIV-1 infection is impaired in ATG5-depleted HeLa and CD4+ T cells (Fig. 5 D and F), whereas this signaling is increased in LC3C-knockdown cells (Fig. 5E). Moreover, we found that ATG5 may be required in the early step of the BST2-mediated signaling cascade by either i) participating to the recruitment of Syk and/or to its activation (pSyk) on BST2 (Fig. 5G) or ii) by favoring the recruitment of other cellular actors required for Syk activation, a crucial step for the establishment of the BST2 signalosome (8).
Altogether, our data support the view that the LC3C-associated pathway subverted by Vpu is implicated in the control of the inflammatory response mediated by BST2. Therefore, to complement our model, we propose that the recruitment by ATG5 of the phosphorylated virus-tethered BST2 at the plasma membrane and the engagement of these complexes onto an LC3C-associated pathway, can additionally lead to the activation of NF-ĸB signaling (Fig. 6). Vpu, by recruiting LC3C on the vesicles that sequestered virus-tethered pBST2, enhances the degradation of these complexes and thus attenuates the proinflammatory response mediated by viral tethering (Fig. 6, Left, compare the model with and without Vpu). Without ATG5, virion retention mediated by BST2 does not trigger the activation of NF-ĸB signaling (Fig. 6, Right).
It has been documented that Vpu inhibits stimulation of NF-ĸB activity induced by viral infection, cytokine, MAVS or BST2/tetherin overexpression (7, 21, 42–44). Previous reports showed that this inhibitory effect of Vpu is driven by its capacity to bind and sequester β-TrCP, an E3 ubiquitin ligase responsible for the ubiquitination and degradation of phosphorylated IĸB (7, 42). Further analyses of determinants responsible of Vpu inhibitory function also revealed that Vpu effect on BST2-mediated NFĸB signaling could not solely result from its binding to and sequestration of β-TrCP (43, 44). Here, we propose that the subversion of the LC3C-associated pathway by Vpu could be an additional mechanism by which Vpu attenuates NFκB activation mediated by BST2-induced virions retention, independently of its action on β-TrCP. Indeed, this mechanism involves a selective interaction between Vpu and LC3C, implicating the helix 2 of Vpu, and not the DS52GNES56 binding motif to β-TrCP (22). Furthermore, this mechanism mainly relies on the ability of Vpu to perturb the intracellular trafficking: By accelerating the targeting of the « BST2 signalosome » induced by cell surface virions retention to the degradation (Fig. 2E), Vpu specifically attenuates the inflammatory responses mediated by BST2 sensing of virion retention. Whether this pathway contributes as well on Vpu inihibitory action on other stimuli-induced NFĸB signalling pathway would require further investigations. Vpu-mediated antagonism of BST2 restriction is a multifaceted process involving so far, the AP1/2-mediated mistrafficking of BST2, the β-TrCP-mediated ubiquitination and the ESCRT machinery. The discovery of an LC3-associated process implicated in the targeting of virus-tethered BST2 from the viral budding site to the degradation adds another layer of complexity. How these pathways might work together need to be elucidated.
BST2 has broad activity against diverse families of enveloped viruses (45). It will be thus interesting to assess whether the LC3-associated pathway could be subverted by other viruses to counteract BST2 restriction and/or attenuate the cellular antiviral state established by BST2. Importantly, the discovery of a novel autophagic-independent function of ATG5, as an adaptor for receptor at the plasma membrane will open new path to decipher the contribution of ATG5 in the engagement of a surface receptor in non-canonical autophagic pathway.
Materials and Methods
For detailed protocols, see SI Appendix online on the PNAS Website.
Cell Lines and CRISPR-Cas9 KO in Hela Cells.
Cells lines used in this study and generation CRISPR-Cas9 KO in HeLa cells are described in SI Appendix, Supplemental Materials and Methods.
Small Interfering RNA and Transfection.
Cells were transfected with relevant small interfering RNA (siRNA) oligonucleotide using Lipofectamine RNAiMAX (Life TechnologiesSpecific). The siRNA targeting ATG5, LC3C, and BST2 are previously described (22, 46). The siRNA (D-001810-01 from Dharmacon) was used as negative control (referred as siRNA Control).
Mammalian Expression Vectors and Transfection.
All mammalian expression vectors used in this study are described in SI Appendix, Supplemental Materials and Methods. Transfections of HeLa cells or HEK293T cells with mammalian expression vectors were performed using Lipofectamine LTX with PLUS Reagent (Life technologies).
Viral Stocks.
Stocks of VSV-G pseudotyped WT NL4-3 HIV-1, NL4-3 Udel HIV-1, WT NL4-3 (MA/YFP) HIV-1 and Udel NL4-3 (MA/YFP) HIV-1 were obtained by transfection of HEK293T cells with HIV-1 proviral DNA along with a VSV-G expression vector (pMD.G) and polyethylenimine (Polysciences).
Antibodies.
The antibodies used in this study for immunoblotting and/or immunofluorescence are described in SI Appendix, Supplemental Materials and Methods.
Immunoprecipitation Assay.
To observe pBST2 after immunoprecipitation, cells were incubated for 5 min in DMEM containing 100 µM of pervanadate before lysis. Immunoprecipitations were performed on equal amounts of protein for each sample by incubating whole cell extracts overnight at 4 °C with monoclonal mouse anti-Flag or monoclonal mouse anti-BST2 antibody, or mouse IgG CTRL coupled to Dynabeads protein G (Life Technologies). The beads were washed four times with lysis buffer, and proteins were eluted in 2X Laemmli (Sigma-Aldrich) or deglycosylated by a 1-h treatment with PNGase F (NEB) then eluted in 2X Laemmli as previously described.
GFP-tagged proteins were immunoprecipitated using GFP-TRAP beads (Chromotek) using TNTE buffer (20mM Tris-HCL pH 7.4, 150 mM NaCl, 5mM EDTA, 0.5% Triton X-100) with complete protease inhibitor cocktail. Immunoprecipitations were performed by incubating indicated whole cell extracts for 1 h30 at 4 °C with GFP-TRAP beads. The beads were washed and proteins were eluted as previously described.
Western Blotting and Formaldehyde Crosslinking of BST2.
The experimental procedure for western blotting and formaldehyde crosslinking of BST2 are described in SI Appendix, Supplemental Materials and Methods.
Quantitative RT-PCR.
Total cellular RNA was extracted using the Reliaprep RNA Cell Miniprep System kit (Promega; Z6012). For each sample, 500 ng to 2 µg of total RNA was subjected to DNase I treatment and cDNA synthesis was performed using the High-capacity cDNA Reverse Transcription Kit (Applied Biosystems; 4368814). The different mRNA levels were assayed using SYBR Green Supermix (Bio-Rad; 1725275) in a real-time PCR detection system (LightCycler® 480).
Fluorescence Microscopy and Electron Microscopy.
Cells were grown on coverslips, transfected with siRNA, infected, and then fixed with 4% paraformaldehyde in PBS for 20 min. For staining of pBST2, cells were incubated for 5 min in DMEM containing 100 µM of pervanadate before fixation with 4% paraformaldehyde. Cells were permeabilized with 0.2% Triton X-100 for 10 min. Coverslips were blocked with 5% BSA for 1 h, incubated with primary antibody in 1% BSA for 1 h, washed with PBS, and incubated with secondary antibody in 1% BSA for 30 min, before final washing with PBS and MilliQ water.
For extracellular staining of BST2, living cells were incubated for 1 h at 4 °C with rabbit polyclonal anti-BST2 (NIH) together with mouse anti-Env SUgp120 (110H, Hybridolab). Then, cells were fixed with 4% PFA in PBS for 20 min and labelled with appropriate fluorophore-conjugated secondary antibodies.
The method used for electron microscopy is those described previously (16, 47).
HIV-1 Production Assay.
For a HIV-1 production assay, HeLa BST2 WT and HeLa BST2 KO cells were treated with siRNA (7.5 to 30 nM) as described above and transfected with HIV-1 proviral DNA NL4-3 (WT or Udel). Twenty-eight hours after transfection, cell lysates were analyzed by qRT-PCR and western blotting.
In a single round of infection, HeLa cells were treated with siRNA (7.5 to 30 nM) as described above. Forty-eight hours after, siRNA-treated Hela cells were infected with VSV-G pseudotyped NL4-3 (WT or Udel) HIV-1 for 2h30 at a multiplicity of infection (M.O.I.) of 0.5. Forty-eight hours after infection, Supernatants were then collected, 0.45-µm–filtered and used for HIV-1 CAp24 quantification by ELISA (released CAp24) (Perkin Elmer). Cell lysates were analyzed by western blotting.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Stéphane Emiliani, Florence Margottin-Goguet, and Mark Scott for helpful discussions and for critical reading of the manuscript, Annegret Pelchen-Mathews for IEM images. We thank all the members of the “Interactions hôte-virus” Laboratory for comments and helpful discussions. We thank the Imaging photonic Facility IMAG’IC and the Cytometry and Immunobiology Facility CYBIO of the Cochin Institute for technical assistance. The following reagents were obtained through the National Institute for Biological Standards and Control Centralised Facility for AIDS Reagents, which is supported by the EU Program EVA and the UK Medical Research Council: HIV-1 gp120 monoclonal antibody (2G12) from Dr. H. Katinger and mouse antibodies against CAp24 (EVA365) from B. Wahren. The following reagents were obtained through the AIDS research and reference reagent program, Division of AIDS, NIAID, NIH: antihuman BST2 from Drs. K. Strebel and A. Andrew, HIV-1 NL4-3 Vpu antiserum from Drs. K. Strebel and F. Maldarelli and pNL4-3 from Dr. M. Martin. We thank Dr. K. Strebel for the gift of pNL4-3/Udel proviral DNA and Dr. P. Bieniasz for the gift of pNL4-3(MA/YFP) proviral DNA. We thank Dr. S. Frémont for the gift of BST2 KO cell line. D.J. holds a fellowship from ANRS and then from SIDACTION, A.K., F.B., and P.V. from ANRS. M.P. and L.L. hold a fellowship from the “Ministère Français de l’enseignement supérieur et de la Recherche”. This work is funded by ANRS and SIDACTION.
Author contributions
D.J. and C.B.-T. designed research; D.J., M.V., F.B., M.P., P.V., A.K., L.L., and C.B.-T. performed research; D.J., A.K., S.G.-M., K.J., and C.B.-T. contributed new reagents/analytic tools; D.J., M.V., F.B., A.K., and C.B.-T. analyzed data; P.V., S.G.-M., and K.J. editing paper; and D.J. and C.B.-T. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Delphine Judith, Email: delphine.judith@inserm.fr.
Clarisse Berlioz-Torrent, Email: clarisse.berlioz@inserm.fr.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Malim M. H., Bieniasz P. D., HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2, a006940 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neil S. J., Zang T., Bieniasz P. D., Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Van Damme N., et al. , The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host. Microbe 3, 245–252 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neil S. J., Sandrin V., Sundquist W. I., Bieniasz P. D., An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host. Microbe. 2, 193–203 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez-Caballero D., et al. , Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139, 499–511 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuda A., et al. , Large-scale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene 22, 3307–3318 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Tokarev A., et al. , Stimulation of NF-kappaB activity by the HIV restriction factor BST2. J. Virol. 87, 2046–2057 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galao R. P., Pickering S., Curnock R., Neil S. J., Retroviral retention activates a Syk-dependent HemITAM in human tetherin. Cell Host. Microbe. 16, 291–303 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McNatt M. W., Zang T., Bieniasz P. D., Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 9, e1003299 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vigan R., Neil S. J., Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J. Virol. 84, 12958–12970 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia X., et al. , Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. Elife 3, e02362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kueck T., et al. , Serine Phosphorylation of HIV-1 Vpu and Its Binding to Tetherin Regulates Interaction with Clathrin Adaptors. PLoS Pathog. 11, e1005141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pujol F. M., et al. , HIV-1 Vpu Antagonizes CD317/Tetherin by Adaptor Protein-1-Mediated Exclusion from Virus Assembly Sites. J. Virol. 90, 6709–6723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt S., Fritz J. V., Bitzegeio J., Fackler O. T., Keppler O. T., HIV-1 Vpu blocks recycling and biosynthetic transport of the intrinsic immunity factor CD317/tetherin to overcome the virion release restriction. mBio 2, e00036–00011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dube M., et al. , HIV-1 Vpu antagonizes BST-2 by interfering mainly with the trafficking of newly synthesized BST-2 to the cell surface. Traffic 12, 1714–1729 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janvier K., et al. , The ESCRT-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 7, e1001265 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell R. S., et al. , Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 5, e1000450 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dube M., et al. , Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 6, e1000856 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lau D., Kwan W., Guatelli J., Role of the endocytic pathway in the counteraction of BST-2 by human lentiviral pathogens. J. Virol. 85, 9834–9846 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goffinet C., et al. , Antagonism of CD317 restriction of human immunodeficiency virus type 1 (HIV-1) particle release and depletion of CD317 are separable activities of HIV-1 Vpu. J. Virol. 84, 4089–4094 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galao R. P., Le Tortorec A., Pickering S., Kueck T., Neil S. J., Innate sensing of HIV-1 assembly by Tetherin induces NFkappaB-dependent proinflammatory responses. Cell Host. Microbe. 12, 633–644 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madjo U., et al. , LC3C Contributes to Vpu-Mediated Antagonism of BST2/Tetherin Restriction on HIV-1 Release through a Non-canonical Autophagy Pathway. Cell Rep. 17, 2221–2233 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Sanjuan M. A., et al. , Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Heckmann B. L., et al. , LC3-Associated Endocytosis Facilitates beta-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 178, 536–551 e514 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munz C., Autophagy proteins influence endocytosis for MHC restricted antigen presentation. Semin. Cancer Biol. 66, 110–115 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Florey O., Kim S. E., Sandoval C. P., Haynes C. M., Overholtzer M., Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 13, 1335–1343 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schille S., et al. , LC3-associated phagocytosis in microbial pathogenesis. Int. J. Med. Microbiol. 308, 228–236 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Galluzzi L., Green D. R., Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez J., et al. , Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 17, 893–906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Hooper K. M., et al. , V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J. Cell Biol. 221, e202105112 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ligeon L. A., et al. , Oxidation inhibits autophagy protein deconjugation from phagosomes to sustain MHC class II restricted antigen presentation. Nat. Commun. 12, 1508 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cocka L. J., Bates P., Identification of alternatively translated Tetherin isoforms with differing antiviral and signaling activities. PLoS Pathog. 8, e1002931 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrew A. J., Miyagi E., Kao S., Strebel K., The formation of cysteine-linked dimers of BST-2/tetherin is important for inhibition of HIV-1 virus release but not for sensitivity to Vpu. Retrovirology 6, 80 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Presle A., et al. , The viral restriction factor tetherin/BST2 tethers cytokinetic midbody remnants to the cell surface. Curr. Biol. 31, 2203–2213 e2205 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Habermann A., et al. , CD317/tetherin is enriched in the HIV-1 envelope and downregulated from the plasma membrane upon virus infection. J. Virol. 84, 4646–4658 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hammonds J., Wang J. J., Yi H., Spearman P., Immunoelectron microscopic evidence for Tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog. 6, e1000749 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neil S. J., Eastman S. W., Jouvenet N., Bieniasz P. D., HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2, e39 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez J., et al. , Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. U.S.A. 108, 17396–17401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coelho P. P., et al. , Endosomal LC3C-pathway selectively targets plasma membrane cargo for autophagic degradation. Nat. Commun. 13, 3812 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramani S., Malhotra V., Non-autophagic roles of autophagy-related proteins. EMBO Rep. 14, 143–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baines K., Yoshioka K., Takuwa Y., Lane J. D., The ATG5 interactome links clathrin-mediated vesicular trafficking with the autophagosome assembly machinery. Autophagy Rep. 1, 88–118 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bour S., Perrin C., Akari H., Strebel K., The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J. Biol. Chem. 276, 15920–15928 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Sauter D., et al. , Differential regulation of NF-kappaB-mediated proviral and antiviral host gene expression by primate lentiviral Nef and Vpu proteins. Cell Rep. 10, 586–599 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pickering S., et al. , Preservation of tetherin and CD4 counter-activities in circulating Vpu alleles despite extensive sequence variation within HIV-1 infected individuals. PLoS Pathog. 10, e1003895 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le Tortorec A., Willey S., Neil S. J., Antiviral inhibition of enveloped virus release by tetherin/BST-2: action and counteraction. Viruses 3, 520–540 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Muhlinen N., et al. , LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol. Cell 48, 329–342 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caillet M., et al. , Rab7A is required for efficient production of infectious HIV-1. PLoS Pathog. 7, e1002347 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.