

Significance
We identified an AR ubiquitination site undergoing TRAF4-mediated nonclassical K27-linked ubiquitination under castration conditions. This ubiquitination enhances the interaction between AR and the pioneer factor FOXA1 and triggers a switch of AR genomic binding and AR-regulated transcriptome. Ubiquitinated AR subsequently hyperactivates E2F to promote CRPC. Our studies lay the foundation for identifying an appropriate CRPC patient population sensitive to CDK4/6 inhibitor treatment.
Keywords: androgen receptor, ubiquitination, CRPC, TRAF4, E2F
Abstract
Castration-resistant prostate cancer (CRPC) poses a major clinical challenge with the androgen receptor (AR) remaining to be a critical oncogenic player. Several lines of evidence indicate that AR induces a distinct transcriptional program after androgen deprivation in CRPCs. However, the mechanism triggering AR binding to a distinct set of genomic loci in CRPC and how it promotes CRPC development remain unclear. We demonstrate here that atypical ubiquitination of AR mediated by an E3 ubiquitin ligase TRAF4 plays an important role in this process. TRAF4 is highly expressed in CRPCs and promotes CRPC development. It mediates K27-linked ubiquitination at the C-terminal tail of AR and increases its association with the pioneer factor FOXA1. Consequently, AR binds to a distinct set of genomic loci enriched with FOXA1- and HOXB13-binding motifs to drive different transcriptional programs including an olfactory transduction pathway. Through the surprising upregulation of olfactory receptor gene transcription, TRAF4 increases intracellular cAMP levels and boosts E2F transcription factor activity to promote cell proliferation under androgen deprivation conditions. Altogether, these findings reveal a posttranslational mechanism driving AR-regulated transcriptional reprogramming to provide survival advantages for prostate cancer cells under castration conditions.
Prostate cancer (PCa) is the most commonly diagnosed malignancy and the second leading cause of cancer death among men in the United States (1). Androgen regulates normal and malignant prostate tissue growth via activation of androgen receptor (AR) signaling and plays an essential role in the initiation and progression of PCa. Androgen deprivation therapy (ADT) is the first-line treatment for locally advanced or metastatic PCa. Despite initial responses to ADT, nearly all patients eventually develop castration-resistant prostate cancer (CRPC) within a few years. It is now well recognized that CRPC continues to be dependent on AR signaling (2, 3). In CRPC, AR signaling is reactivated through different mechanisms including AR amplification or overexpression, AR point mutations, expression of constitutively active AR splice variants, and intratumoral androgen synthesis. Several lines of evidence indicate that AR regulates a distinct transcription program in CRPC to promote castration-resistant cancer growth (4–6). However, the underlying mechanisms triggering this AR-regulated transcription program switch are largely unknown.
AR belongs to a family of ligand-activated nuclear receptors. It consists of an N-terminal domain, which has a powerful and constitutively active activation function, a conserved central DNA-binding domain (DBD), an interdomain linker or hinge, and a C-terminal ligand-binding domain (LBD), which has a ligand-dependent activation function. Upon binding to androgen, AR is translocated into the nucleus and binds to specific DNA sequences known as androgen-responsive elements (AREs) at AR target gene promoters/enhancers, thereby regulating transcription of androgen-responsive genes. AR transcriptional activity can be regulated by posttranslational modifications such as phosphorylation, acetylation, sumoylation, and ubiquitination (7–9). Ubiquitination is an important posttranslational modification brought about by ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and ubiquitin ligases (E3s). Ubiquitin (Ub) is a highly conserved protein of 76 amino acids that becomes covalently attached to lysine residues of target proteins. Besides from proteasomal degradation, protein ubiquitination also can lead to various nonproteolytic cellular functions, including endocytosis, endosomal sorting, subcellular localization, kinase activation, and DNA repair (10).
AR ubiquitination has been shown to regulate AR activity and contribute to CRPC progression (9, 11). The mechanisms identified so far are mainly through ubiquitination-targeted inactive AR/corepressor complex degradation (11), or noncanonical ubiquitination-mediated recruitment of ubiquitin-binding domain containing AR coactivator (9). However, only a small subset of AR-targeted gene transcription is regulated by these mechanisms (9, 11). TRAF4, a RING finger domain E3 ubiquitin ligase, belongs to a family of TNF receptor (TNFR)-associated adaptor proteins. Unlike other TRAF family members, TRAF4 does not directly interact with TNFR. It is amplified and overexpressed in many types of cancers and plays an important role in promoting cancer development and metastasis (12, 13). We previously found that TRAF4 is highly expressed in metastatic CRPCs and promotes PCa metastasis (14). The role of TRAF4 in PCa castration resistance is not clear. Here, we demonstrate that TRAF4 mediates nonclassical K27-linked AR ubiquitination at its C-terminal end. This atypical ubiquitination increases AR interaction with the pioneer transcription factor FOXA1, and subsequently alters the AR genomic binding profile. Upon TRAF4 overexpression, ubiquitinated AR switches its transcription program and up-regulates a set of CRPC-associated genes to promote CRPC progression. Our study reveals an important role of posttranslation modification in governing the AR-regulated gene switch in CRPCs.
Results
TRAF4 Promotes Castration-Resistant PCa Growth.
We recently found that TRAF4 is highly expressed in metastatic PCas and promotes PCa metastasis (14). Most of these metastatic patients were treated with androgen-deprivation therapy before developing metastasis (15–19). We also found that the TRAF4 protein level is higher in androgen-insensitive Lymph Node Carcinoma of the Prostate (LNCaP)-Abl cells compared to androgen-sensitive LNCaP PCa cells (SI Appendix, Fig. S1). To understand whether TRAF4 overexpression plays a role in the development of PCa castration-resistance, we examined the growth of TRAF4 stably overexpressed LNCaP cells or control LNCaP cells in the absence or presence of androgen dihydrotestosterone (DHT). LNCaP cells depend on androgen for cell growth; accordingly, DHT in the cell culture promoted control LNCaP cell growth (Fig. 1A). Overexpression of TRAF4 markedly increased cell growth in the absence of DHT while DHT had a minimal effect on TRAF4 overexpressing cell growth, suggesting that TRAF4 overexpression promotes androgen-independent cell growth. Similar results were also found in other AR-positive PCa cells, VCaP and 22Rv1 cells (SI Appendix, Fig. S2A). Since TRAF4 is a RING domain containing E3 ubiquitin ligase, we next investigated whether TRAF4-promoted androgen-independent cell growth requires its E3 ubiquitin ligase activity. We generated an LNCaP cell line ectopically expressing RING domain deletion mutant of TRAF4. It expresses comparable levels of the mutant TRAF4 compared to the wild-type (WT) TRAF4-expressing stable cell line (Fig. 1 A, Right) (14). Unlike the WT TRAF4-expressing stable cells, the mutant-expressing cells remained androgen-sensitive (Fig. 1A), suggesting that the ubiquitin ligase activity of TRAF4 is critical for its ability to promote androgen-independent growth.
Fig. 1.

TRAF4 overexpression promotes CRPC development. (A) Overexpression of TRAF4 WT but not its E3-ubiquitin ligase-defective mutant (ΔRING) promotes androgen-independent cell growth. Left Upper, schematic representation of WT TRAF4 and its mutant. Left Bottom, control or TRAF4-overexpressing LNCaP stable cell numbers in the absence or presence of 1 nM of androgen DHT at day 1 and day 6. Right, western blot analysis of TRAF4 expression levels in control, TRAF4 wild-type or ΔRING-expressing LNCaP cells. (B) TRAF4-overexpressing LNCaP xenograft tumors are castration resistant. Upper, control or TRAF4-overexpressing LNCaP xenograft (subcutaneously injected) tumor growth (n = 5). Bottom, time periods for xenograft tumor studies. * represents P < 0.05.
To substantiate the importance of TRAF4 in CRPC development in vivo, we injected TRAF4 overexpressing or control LNCaP cells into nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice subcutaneously. A comparable tumor growth rate was initially observed between the two cell lines. When tumors reached approximately 1 cm3 in volume, mice were castrated. The control tumors regressed and stopped growing in response to castration as expected (Fig. 1B). In contrast, TRAF4-overexpressing tumors continued to grow, suggesting that TRAF4 overexpression promotes castration-resistant growth of prostate tumors in vivo. We also injected doxycycline-inducible TRAF4 knockdown 22Rv-1 cells or control cells into castrated NOD/SCID mice subcutaneously and found that TRAF4 knockdown significantly reduced castration-resistant 22Rv-1 xenograft tumor growth (SI Appendix, Fig. S2B), confirming the role of TRAF4 in promoting CRPC growth.
TRAF4 Overexpression Promotes Upregulation of the E2F Pathway.
To understand how TRAF4 promotes CRPC, we performed a RNA sequencing on TRAF4-overexpressing and control LNCaP cells under androgen deprivation culture condition (Dataset S1). The volcano plot of the RNA seq and heat map of the top 20 altered genes are shown in SI Appendix, Fig. S3A. Gene set enrichment analysis revealed E2F- and myelocytomatosis oncogene (Myc)-regulated gene pathways as the top pathways enriched in TRAF4 overexpressing androgen-independent LNCaP cells (Fig. 2A). These pathways have been linked to CRPC (5, 14, 20–23), and have cross talks between them. Myc is known to modulate the cell cycle regulator E2F activity and its expression (24, 25). E2F also regulates the expression of Myc (26–28). E2F pathway activation has been found in multiple CRPC models and human CRPCs (20, 21, 29–31). In our RNA-Seq analysis, around 100 E2F target genes were up-regulated in TRAF4-overexpressing CRPC cells. We next performed qRT-PCR to validate the RNA-Seq results. As shown in SI Appendix, Fig. S3B, most of the E2F or Myc target genes we tested that are identified in RNA seq were up-regulated in TRAF4 overexpressing cells compared to control, confirming that TRAF4 indeed promotes these E2F and Myc target gene expressions. We also performed another RNA-Seq on TRAF4-overexpressing LNCaP xenograft tumors and the control castration sensitive tumors. E2F and Myc gene pathways were similarly found enriched in TRAF4 overexpressing CRPC tumors (SI Appendix, Fig. S3C).
Fig. 2.

TRAF4 overexpression drives E2F pathway activation to promote CRPC. (A) Gene set enrichment analysis (GSEA) of the genes associated with TRAF4 overexpression in LNCaP cells under androgen-deprived culture condition. Genes enriched in E2F, and MYC pathways with the normalized enrichment scores (NES), nominal P-value and false discovery rate (FDR) q-value are shown. The green line plots show the enrichment of genes associated with TRAF4 overexpression (Left). The normalized enrichment score indicates the ranking list of gene expression differences of TRAF4 overexpression from control cells. The genes associated with the leading edge of the enrichment score are shown in the heat map (Right). The color gradient matches the expression status of individual genes, as shown in the color index. (B) TRAF4 increases E2F transcriptional activity. Shown is an E2F-driven luciferase reporter assay in LNCaP cells transiently transfected with the luciferase reporter and a TRAF4-expressing plasmid. (C) Inhibition of E2F activation abolishes TRAF4-promoted androgen-independent growth. Control or TRAF4-overexpressing LNCaP cells were grown in an androgen-deprived culture medium treated with the CDK4/6 inhibitor Palbociclib (1 µM) or vehicle. The cell growth was monitored through the MTS assay. * represents P < 0.05.
To test whether TRAF4 regulates E2F activity, we monitored an E2F-driven luciferase reporter activity in the presence of transfected TRAF4. As shown in Fig. 2B, increasing concentrations of TRAF4 markedly increased E2F-Luc activity, suggesting that TRAF4 indeed enhances E2F activity.
We next examined whether inhibition of E2F activity could abolish TRAF4-promoted androgen-independent cell growth. Palbociclib is a selective cyclin-dependent kinase CDK4/6 inhibitor. It inhibits CDK4/6-mediated Rb phosphorylation, thereby preventing E2F activation. As shown in Fig. 2C, TRAF4 overexpressing LNCaP cells grow faster in an androgen-deprived medium compared to control cells. Treatment of Palbociclib reduced TRAF4 stable cell growth to the level of control cells. Similar effects were also found with E2F1 knockdown (SI Appendix, Fig. S3D). We next examined the effects of Palbociclib on TRAF4-promoted E2F target gene expressions and found that treatment of Palbociclib completely abolished TRAF4-mediated gene upregulation (SI Appendix, Fig. S3E). These results substantiate that E2F activation is an important downstream effector regulating TRAF4-promoted androgen-independent growth.
TRAF4 Promotes AR Ubiquitination through the Nonclassical K27 Ubiquitin Linkage.
Since TRAF4 E3 ubiquitin ligase activity is essential for promoting androgen-independent cell growth, we tried to identify TRAF4 ubiquitin substrates that are important for mediating this function. AR is a well-known driver in CRPC (32–34). We found that knockdown of AR abolished TRAF4-enhanced E2F transcriptional activity (Fig. 3A), suggesting that AR mediates the effect of TRAF4 on E2F activity.
Fig. 3.
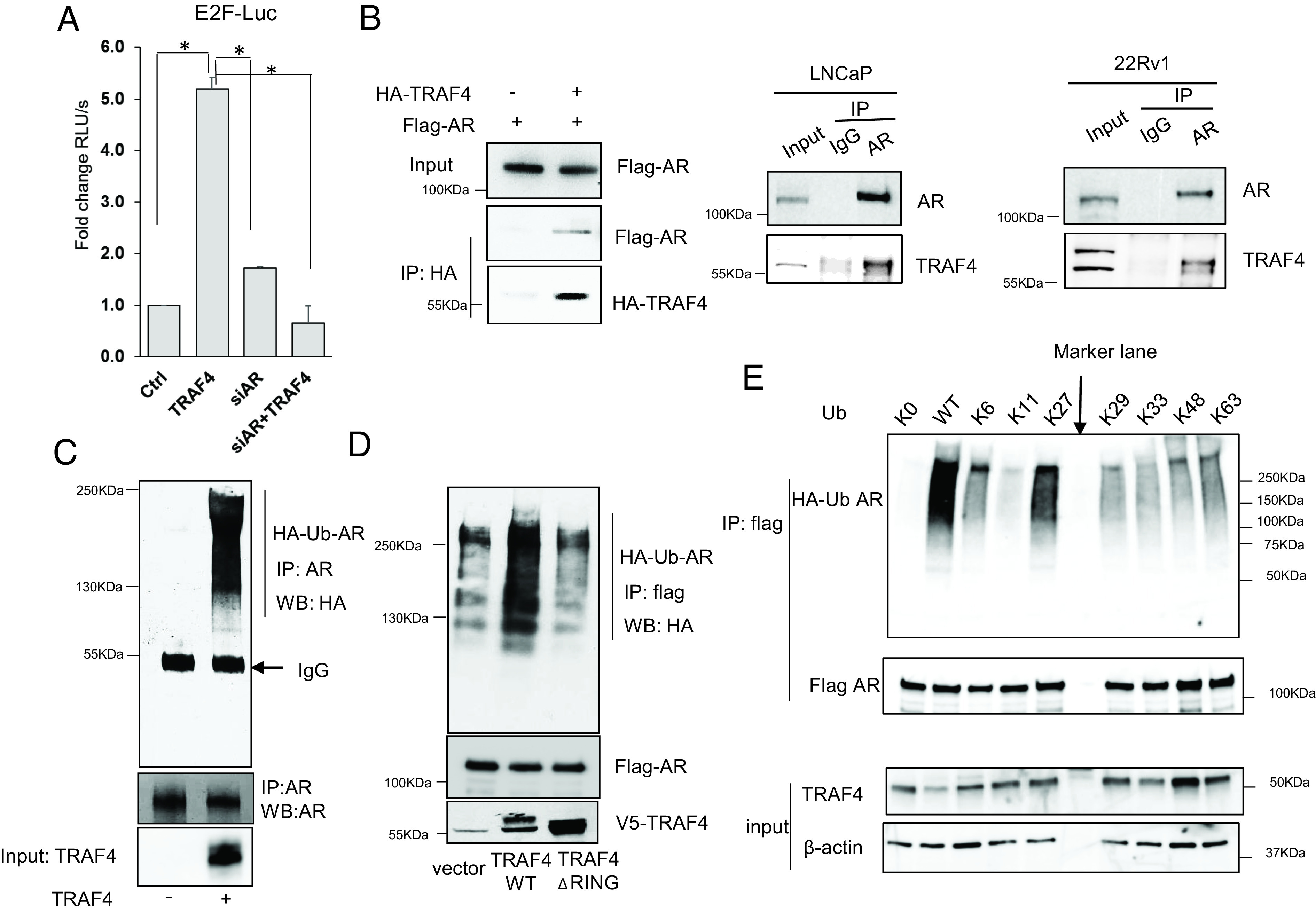
TRAF4 mediates atypical K27-linked AR polyubiquitination. (A) AR is important in mediating the activation effect of TRAF4 on E2F activity. LNCaP cells were treated with control or siAR, and then transiently transfected with an E2F-driven luciferase reporter and a TRAF4 expression plasmid. Shown is the luciferase reporter activity. (B) TRAF4 interacts with AR in cells. Left, Flag-AR was transiently transfected into 293T cells in the absence or presence of HA-TRAF4 cotransfection. A coimmunoprecipitation (co-IP) experiment was then carried out using an HA-specific antibody. Middle and Right, endogenous AR interacts with endogenous TRAF4 in LNCaP and 22Rv1 cells, respectively. AR was immunoprecipitated using an AR-specific Ab or IgG control followed by western blot analysis. (C) TRAF4 promotes AR in vitro ubiquitination. Purified recombinant AR protein was incubated with purified UBE1, UbcH5a, HA-ubiquitin in the absence or presence of purified TRAF4 protein. An IP was then carried out using an AR-specific antibody followed by western blot analysis using an HA antibody to detect AR ubiquitination. (D) TRAF4 WT but not its RING domain deletion mutant promotes AR ubiquitination in cells. Flag-AR and HA-ubiquitin, V5-WT TRAF4, ΔRING mutant or vector control were transiently transfected into 293T cells followed by IP using a flag antibody. The levels of AR ubiquitination were measured using an HA antibody in a western blot analysis. (E) TRAF4-mediated AR ubiquitination mainly occurs through the K27-linkage. K0 represents all lysine residues in ubiquitin were mutated. K6 to K63 represents the ubiquitin mutant with all lysine mutations except the depicted number of lysine. Flag AR, V5-TRAF4, and different HA-ubiquitin (WT or mutants) were transiently transfected into 293T cells. An IP experiment was carried out using a flag antibody followed by a western blot analysis to detect AR ubiquitination with an HA antibody. * represents P < 0.05.
To investigate whether TRAF4 plays a key role in regulating AR function, we performed a co-IP experiment in 293T cells transfected with Flag-AR with or without HA-TRAF4 cotransfection. As shown in Fig. 3 B, Left, an association of Flag-AR and HA-TRAF4 was detected. We also detected the interaction between endogenous AR and endogenous TRAF4 in LNCaP and 22Rv1 cells through co-IP experiments using an AR-specific antibody for IP (Fig. 3 B, Middle and Right, respectively). A reciprocal IP using a TRAF4-specific antibody also detected the association of endogenous AR (SI Appendix, Fig. S4A). In contrast, we did not detect a direct interaction between E2F1 and TRAF4 in this co-IP experiment (SI Appendix, Fig. S4A). We next performed an in vitro ubiquitination assay to determine whether AR is a TRAF4-mediated ubiquitination substrate using purified recombinant proteins. As shown in Fig. 3C, the presence of TRAF4 substantially increased AR ubiquitination in vitro, suggesting that AR is a TRAF4-targeted substrate. We further confirmed that TRAF4 WT but not the ΔRING mutant promoted AR ubiquitination in cells (Fig. 3D). However, AR protein levels were not significantly changed in TRAF4 WT or ΔRING overexpressing cells (SI Appendix, Fig. S4B), suggesting that TRAF4-mediated AR ubiquitination does not promote AR protein turnover. We also did not detect any significant changes in E2F1 protein levels in these cells (SI Appendix, Fig. S4B).
There are 7 lysine residues in the ubiquitin. Different ubiquitin lysine linkages could mediate different functions, including protein turnover, protein–protein interaction, and cellular trafficking. TRAF4 was previously reported to mediate K48- or K63-linked ubiquitination (35, 36). We recently also found that TRAF4 is able to promote protein ubiquitination through nonclassical K27 and K29-linkages (14, 37). To determine the type of TRAF4-mediated ubiquitin linkage on AR, we cotransfected Flag-AR and TRAF4, WT HA-ubiquitin or different ubiquitin mutants. These ubiquitin mutants either have all seven lysine residues mutated to arginine (K0), or only contain one lysine while all other six lysine residues were mutated to arginine residues (K6, K11, K27, K29, K33, K48, or K63). As shown in Fig. 3E, AR is polyubiquitinated mainly in the presence of WT or K27 ubiquitin, suggesting that TRAF4 mediates AR ubiquitination mainly through the K27 linkage.
Identification of TRAF4-Targeted AR Ubiquitination Site.
To identify TRAF4-targeted AR ubiquitination sites, we first determined the functional domain at which the ubiquitination sites are located. A flag-tagged full-length WT AR, an AR splicing variant ARV7 that lacks the LBD, or two AR truncation mutants (Mut 1-669 that lacks the LBD and Mut 669-920 that only contains the LBD) were transiently cotransfected with TRAF4 and HA ubiquitin into cells. ARV7 or the LBD deletion mutant of AR (1-669) had barely detectable levels of ubiquitination compared to full-length or the LBD containing AR mutants (Fig. 4A), suggesting that TRAF4-targeted AR ubiquitination occurs at its LBD. An in vitro TRAF4 pull-down experiment using Ni beads bound-purified recombinant 6×His taggedTRAF4 to pull-down Flag-tagged full length or different deletion mutants of AR also demonstrated a direct binding between TRAF4 and AR LBD (SI Appendix, Fig. S5). We further narrowed down the ubiquitination target region and generated a series of C-terminal deletion mutants. We found that deletion of the last 53 amino acid residues at the C terminus of AR abolished TRAF4-mediated ubiquitination (Fig. 4B). There are four lysine residues located in this region. We generated each individual lysine to arginine mutants and found that a single K to R mutation at K913 is sufficient to abolish AR ubiquitination, suggesting that this residue is the TRAF4-targeted ubiquitination site (Fig. 4C).
Fig. 4.

TRAF4 mediates AR ubiquitination at the K913 residue. (A) TRAF4 mainly targets AR LBD for ubiquitination. Upper, schematic representation of WT, splicing variant (V7) or deletion mutants of AR. Bottom (from the top to bottom), the ubiquitination levels of flag-AR WT or its mutant in cells (Shown is a western blot analysis using an HA-specific Ab to detect HA-ubiquitinated AR); the level of flag-AR or its mutants following an IP with a flag Ab; the levels of TRAF4 from the inputs and β-actin as a loading control of the input. (B) Deletion of the last 53aa in AR abolished TRAF4-mediated ubiquitination. 293T cells were transiently transfected with flag-AR WT or the deletion mutant (1-867), TRAF4 WT, ΔRING, or vector control, together with HA-ubiquitin, V5-TRAF4. An IP was carried out using a flag Ab followed by western blot analysis. Bottom, the amino acid sequence of the last 53aa, which contains 4 lysine residues. (C) K913R mutation abolished TRAF4-mediated AR ubiquitination.
To confirm that K913 is a bona fide ubiquitination target site, we performed a mass spectrometry to determine ubiquitination modification of AR protein. 6×His- ubiquitin, purified TRAF4, UBE1, and UbcH5a were incubated with recombinant AR protein purified from baculovirus in the in vitro ubiquitination assay. His- ubiquitinated AR was then captured using a nickel affinity column followed by mass spectrometry analysis. Although this method cannot distinguish the AR ubiquitination that preexisted in cells or was modified in vitro, we did find K913 among a total of eight ubiquitinated lysine residues (SI Appendix, Fig. S6 and Dataset S2). These results substantiate that K913 is a TRAF4-targeted ubiquitination site.
TRAF4 Overexpression Selectively Up-regulates CRPC-Associated AR Target Gene Transcription.
To investigate the potential role of TRAF4 on AR function, we generated two TRAF4 stable LNCaP cells with low (T1) and high (T2) TRAF4 expression (SI Appendix, Fig. S7). Since TRAF4 overexpression did not alter the AR protein level (SI Appendix, Fig. S4B), we next examined whether TRAF4 regulates AR-targeted gene transcription in these cells in either a complete culture medium containing androgen, or in an androgen-deprived medium. Unexpectedly, we did not find any major effects of TRAF4 overexpression on the expression of canonical AR target genes TMPRSS2 and KLK3/prostate-specific antigen (PSA) (Fig. 5A). We observed an inhibitory effect of TRAF4 overexpression on another AR target gene (Serum/Glucocorticoid Regulated Kinase or SGK) expression (Fig. 5 A, Right). Upon androgen deprivation, TRAF4 overexpression appears to reduce these canonical AR target gene expressions. It was previously reported that AR regulates a distinct transcriptional program in androgen-independent compared to androgen-dependent PCa cells (4). Several M-phase cell cycle genes, such as UBE2C and CDC20 which are important for CRPC development, are regulated by AR selectively in androgen-independent PCa cells. We found that UBE2C and CDC20 were significantly up-regulated in TRAF4 overexpressing cells in both complete culture medium and androgen-deprived medium (Fig. 5B and SI Appendix, Fig. S8A). The levels of these gene transcriptions were higher in high TRAF4-expressing T2 cells compared to low TRAF4-expressing T1 cells, suggesting that TRAF4 expression levels influence the transcription of UBE2C and CDC20. TRAF4 overexpression also increased UBE2C and CDC20 levels in VCaP and 22Rv1 cells (SI Appendix, Fig. S8B), further confirming this notion.
Fig. 5.

TRAF4 promotes upregulation of selective AR-targeted CRPC-associated genes. (A) TRAF4 overexpression does not activate AR activity on classical AR targets under androgen-deprived condition. T1, low TRAF4-expressing LNCaP cells. T2, high TRAF4-expressing LNCaP cells. (B) TRAF4 overexpression up-regulates UBE2C gene expression. (C) AR depletion abolishes TRAF4-up-regulated UBE2C expression. (D) TRAF4 depletion in CRPC cells decreases UBE2C expression. LNCaP-C4-2 cells were treated with control siRNA or siTRAF4. The levels of TRAF4 and UBE2C mRNA were measured through qRT-PCR. (E) Depletion of TRAF4 or UBE2C inhibits CRPC Lymph Node Carcinoma of the Prostate-androgen ablation (LNCaP-Abl) cell growth. LNCaP-Abl cell growth was measured by counting cell numbers at 0, 7, and 10 d after siRNA transfection. *represents P < 0.05.
To determine whether increased transcription of UBE2C and CDC20 was dependent on AR, we knocked-down AR or TRAF4 in control LNCaP or T2 cells. In control cells, neither AR nor TRAF4 knockdown had a noticeable effect on UBE2C transcription (Fig. 5C). This is consistent with the previous report that AR does not regulate UBE2C transcription in androgen-sensitive PCa cells (4). In TRAF4-overexpressing cells, however, upregulation of UBE2C transcription was abolished upon AR or TRAF4 knockdown. A similar result also was observed for CDC20 transcription (SI Appendix, Fig. S8C). In contrast to UBE2C and CDC20 transcription, KLK3 and TMPRSS2 transcriptions were not differentially regulated by siTRAF4 in control and T2 cells. Their transcriptions were significantly reduced upon siAR treatment in both cells (SI Appendix, Fig. S9). In addition to UBE2C and CDC20, we found TRAF4 overexpression also similarly up-regulates several other AR-regulated CRPC signature genes (SI Appendix, Fig. S8F). These genes were identified as a set of core CRPC genes from a distinct transcriptional program downstream of the AR targets in human CRPC tissue (5). We found AR knockdown similarly abolished TRAF4-mediated upregulation of these genes (SI Appendix, Fig. S8F). TRAF4 expression levels were also significantly correlated with these gene expressions in prostate adenocarcinomas (SI Appendix, Fig. S8G). These results suggest that TRAF4 overexpression switches on AR function by activating a set of CRPC-associated gene transcriptions.
To further confirm that TRAF4 plays a role in regulating UBE2C and CDC20 transcription in CRPC cells, we knocked down TRAF4 in androgen-independent C4-2 cells. As shown in Fig. 5D and SI Appendix, Fig. S8D, siRNA-mediated TRAF4 knockdown (siTRAF4) indeed reduced UBE2C and CDC20 transcription. It was previously reported that AR binds to the UBE2C distal enhancer region in LNCaP-Abl cells but not in LNCaP cells (4). We found that TRAF4 depletion significantly reduced AR recruitment to the UBE2C enhancer but not the KLK3 (PSA) enhancer in Lymph Node Carcinoma of the Prostate-androgen ablation (LNCaP-Abl) cells (SI Appendix, Fig. S10 A and B, respectively). These results suggest that TRAF4 plays a role in regulating AR genomic binding to CRPC-associated gene enhancers. Accordingly, TRAF4 depletion markedly decreased the growth of CRPC LNCaP-Abl and C4-2 cells (Fig. 5E and SI Appendix, Fig. S8E, respectively).
TRAF4 Overexpression Alters AR Genomic Binding Profile.
Since TRAF4 promotes the ability of AR to turn on genes that are normally not regulated by AR in androgen-sensitive PCa cells, it is likely that TRAF4 overexpression alters AR genomic binding profile. To examine this, we performed an AR chromatin immunoprecipitation sequencing (ChIP-Seq) analysis in control and TRAF4-overexpressing LNCaP cells under androgen-deprived condition. The AR-binding peaks in the two cell lines clearly did not completely overlap (Fig. 6A). TRAF4 overexpression caused a loss of about 50 to 60% AR-binding peaks and a gain of 40 to 50% new AR genomic binding sites. We analyzed the top gained AR-binding peaks upon TRAF4 overexpression (log2 TRAF4/vector >1) and found that the majority of them are located 5 to 500 kb away from transcriptional start sites (SI Appendix, Fig. S11A), suggesting that these binding sites are not resident in proximal promoter regions but are instead likely located at enhancer regions. The top transcription factor-binding motifs among these sites are not the classical AR-binding motifs (ARE) but the motifs associated with transcription factors from Forkhead domain family and homeodomain family (Fig. 6B). FOXA1 and HOXB13 are known pioneer factors that colocalize with AR and facilitate AR cistrome reprogramming in prostate tumors (38–40). Although shared AR-binding peaks between control and TRAF4 stable cells contain significant numbers of FOXA1-binding motifs, we noticed that even stronger FOXA1-binding sites were colocalized with AR-binding sites in TRAF4 stable cells compared to the control cells (Fig. 6C and SI Appendix, Fig. S11B). A similar result also was found for the HOXB13-binding sites (SI Appendix, Fig. S11C). These unexpected results suggest that TRAF4 overexpression alters the AR genomic binding profile and increases AR binding to FOXA1- and HOXB13-binding sites.
Fig. 6.

TRAF4 overexpression alters AR genomic binding profile and increases its interaction with FOXA1. (A) Venn diagram and distribution of AR genomic binding peaks in TRAF4-overexpressing cells or control LNCaP cells. Cells were cultured in an androgen-deprived medium for 2 d followed by ChIP using an AR antibody. ChIP seq analysis was then performed. (B) Top gained AR-binding peaks in TRAF4-overexpressing cells are enriched with forkhead domain family and homeodomain family-binding motifs. Left panel, the heatmap of AR-binding intensity in TRAF4-overexpressing and control cells. Right panel, top enriched forkhead domain family and homeodomain family transcription factor-binding motifs. (C) AR gained more binding at the FOXA1-binding sites in TRAF4-overexpressing cells compared to vector control cells (blue peak vs. green peak). The red peak represents shared AR-binding sites between TRAF4- and vector-expressing cells. (D) TRAF4 overexpression enhances the association between AR and FOXA1. LNCaP cells were infected with adenovirus expressing TRAF4 or GFP control. A co-IP experiment was then carried out using an AR antibody followed by a western blot analysis using a FOXA1 antibody to detect AR-associated FOXA1. (E) AR ubiquitination mutation abolished the TRAF4-promoted AR-FOXA1 interaction. Shown is a co-IP experiment in flag-AR WT or ubiquitination mutant (K913/911R)-expressing cells. Cells were treated with siAR to reduce the levels of endogenous AR.
FOXA1 protein previously was reported to physically interact with AR (41). To examine whether TRAF4 overexpression could regulate the interaction between endogenous AR and FOXA1, we performed a co-IP experiment using an AR antibody in LNCaP cells infected with GFP or TRAF4-expressing adenovirus. As shown in Fig. 6D, more FOXA1 was associated with AR when TRAF4 was overexpressed, suggesting that TRAF4 increased the AR–FOXA1 interaction. We next determined whether TRAF4-mediated AR ubiquitination is important for TRAF4-promoted AR–FOXA1 interaction. A Flag-AR WT or AR ubiquitination mutant (K913/911R)-expressing LNCaP stable cell line was generated. Cells were treated with siRNA against AR to minimize the effect of endogenous AR followed by GFP- or TRAF4-expressing adenovirus infection. A co-IP experiment was then performed using a flag antibody (Fig. 6E). TRAF4 overexpression significantly increased the association of FOXA1 with Flag-AR WT but not the mutant, suggesting that AR ubiquitination is important for TRAF4-enhanced AR–FOXA1 interaction.
TRAF4 Promotes AR Binding to Olfactory Receptors (ORs) and Regulates cAMP Signaling.
To understand how alteration of the AR genomic binding profile affects androgen-independent cell growth, we performed KEGG pathway analysis on top gained AR-binding sites in TRAF4 stable cells (log2 TRAF4/vector > 2). Interestingly, OR transduction is the most enriched pathway (Fig. 7A). The UCSC genome browser view of AR binding to OR gene chromatin locations in LNCaP control or TRAF4-overexpressing cells is shown in SI Appendix, Fig. S12. ORs are the largest family of G-protein-coupled receptors, which stimulate intracellular cyclic adenosine monophosphate (cAMP) signaling upon activation (42–44). Using a lower threshold (log2 TRAF4/vector > 1) for gene ontology analysis with the same dataset, we found renin secretion and cAMP signaling as the topmost enriched pathways (SI Appendix, Fig. S13). The cAMP signaling is also the central and stimulatory pathway for renin secretion (45). Both analyses suggest that the OR-cAMP signaling pathway gained the most AR binding upon TRAF4 overexpression. In addition to the olfactory epithelium, ORs are expressed in a number of nonolfactory tissues and cancerous tissues including PCa (46–51). Ectopic overexpression of ORs can promote PCa progression, metastasis, and neuroendocrine differentiation (52–55).
Fig. 7.

TRAF4 promotes AR binding to OR enhancers, up-regulates OR gene expression and intracellular cAMP levels. (A) Olfactory transduction is the top enriched pathway in AR-gained genomic binding loci upon TRAF4 overexpression. Shown is the KEGG pathway analysis in top AR-gained peaks (log2 TRAF4/vector > 2). (B) TRAF4 promotes AR binding to several OR gene enhancers. ChIP experiments were performed in TRAF4-overexpressing LNCaP cells or control cells using an AR antibody or IgG control to independently validate ChIP seq results. (C) The AR ubiquitination mutation inhibits AR binding to OR gene enhancers. Left, a ChIP experiment in LNCaP-TRAF4 stable cells with knockdown of endogenous AR followed by overexpression of either WT AR or AR ubiquitination mutant K913R. Right, expression levels of AR and TRAF4 in LNCaP cells expressing AR WT or K913R. *P < 0.05 by 2-way ANOVA with Tukey’s multiple comparisons test. Data are presented as mean ± SEM. (D) TRAF4 overexpression up-regulates several OR gene expression. Shown are the mRNA levels of ORs through real-time qPCR analysis in control or TRAF4-overexpressing cells. (E) AR depletion reduced OR gene expression in TRAF4 stable LNCaP cells. LNCaP-TRAF4 cells were treated with control siRNA or siAR. The levels of OR gene expression were detected by qRT-PCR. (F) TRAF4 overexpression significantly increases intracellular cAMP levels. Shown is the cAMP level in LNCaP control and TRAF4 stable cells with and without OR agonist β-ionone treatment. (G) OR activation enhances E2F activity in TRAF4 overexpressing cells. Shown are E2F-driven luciferase reporter activities in LNCaP control and TRAF4 stable cells with and without β-ionone treatment. (H) Inhibition of protein kinase A (PKA) significantly reduced TRAF4-promoted androgen-independent growth. The growth of LNCaP cells with or without TRAF4 overexpression under androgen-deprivation condition was monitored in the absence or presence of a PKA inhibitor H89 (10 µM). *P < 0.05 by 1-way ANOVA with Tukey’s multiple comparisons test. Data are presented as mean ± SEM, (n = 3).
To determine whether TRAF4 can regulate OR expression via AR, we first performed a ChIP experiment to validate the binding of AR to the enhancer/promoter region of several OR genes in control or TRAF4-overexpressing LNCaP cells. Strong AR binding to these OR gene enhancer/promoter regions was observed in TRAF4-overexpressing cells but not in control cells (Fig. 7B), confirming that TRAF4 overexpression indeed increased AR binding to these regions. In contrast, AR binding to KLK3 (PSA) enhancer was not significantly changed in control or TRAF4 overexpressing cells (SI Appendix, Fig. S14A). Similar results were also found in VCaP and 22Rv1 cells (SI Appendix, Fig. S14B). To ascertain whether TRAF4-mediated AR ubiquitination is required for AR binding to the enhancer/promoter region of OR genes, we performed a ChIP experiment in LNCaP-TRAF4 stable cells with knockdown of endogenous AR followed by overexpression of either WT or K913R-mutated AR. We observed strong binding of WT but not K913R-mutated AR to OR gene enhancer/promoter regions in LNCaP-TRAF4 cells (Fig. 7C). In contrast, K913R mutation did not affect AR binding to the KLK3 (PSA) enhancer/promoter region (SI Appendix, Fig S14C). Collectively, these data indicate that TRAF4-mediated AR ubiquitination is critical for AR binding to the enhancer/promoter regions of OR genes.
We next examined whether the transcriptions of these OR genes are indeed altered upon TRAF4 overexpression. Significantly higher expression levels of these genes were found in TRAF4-overexpressing LNCaP, VCaP, and 22Rv1 cells compared to the control cells (Fig. 7D and SI Appendix, Fig. S14B). Importantly, knockdown of AR reduced OR gene expression levels in TRAF4 cells (Fig. 7E). These results suggest that the TRAF4-promoted AR binding to OR gene enhancer regions up-regulates their transcriptions. We also found that knockdown of FOXA1 significantly reduced AR binding to these OR gene enhancers in TRAF4-overexpressing cells (SI Appendix, Fig S15), suggesting an important role of FOXA1 in directing AR binding to these regions.
To investigate the functional impact of OR gene upregulation, we examined the levels of intracellular cAMP in control or TRAF4-overexpressing LNCaPcells with or without OR agonist β-ionone stimulation. β-ionone treatment did not have a significant impact on cAMP levels in control cells, likely due to the low expression levels of ORs in LNCaP cells. In contrast, it dramatically increased the cAMP levels in TRAF4 stable cells (Fig. 7F). These results confirm that TRAF4 overexpression up-regulates OR gene expression and imply a role of TRAF4 in regulating intracellular cAMP signaling.
OR Activation Regulates E2F Transcriptional Activity and Androgen-Independent Growth.
Since we found that the E2F pathway is important for TRAF4-promoted androgen-independent cell growth (Fig. 2C) and it is AR-dependent (Fig. 3A), we next determined whether upregulation of OR signaling upon TRAF4 overexpression can regulate E2F activation. To this end, we examined the effects of β-ionone treatment-induced cAMP signaling on E2F transcriptional activity in control and TRAF4 stable LNCaP cells. As shown in Fig. 7G, β-ionone treatment caused a modest increase of E2F-driven luciferase reporter activity in control cells but it markedly increased E2F activity in TRAF4 stable cells, consistent with the levels of cAMP observed upon β-ionone treatment in these cells (Fig. 7F). These results suggest that OR activation enhances E2F activity when TRAF4 is overexpressed.
Since K913R mutation reduced AR binding to OR gene enhancers (Fig. 7C), we next examined whether TRAF4-mediated AR ubiquitination is important for regulating E2F activity. TRAF4-overexpressing LNCaP cells were infected with doxycycline-inducible AR WT or K913R-expressing lentivirus to generate stable cell lines. The cells were also treated with control siRNA (sictrl) or siAR to knockdown endogenous AR. Western blot analysis demonstrated comparable expression levels of AR WT and K913R in rescuing the AR levels in the cells (SI Appendix, Fig. S16A). We first examined the E2F activity through the E2F-luciferase reporter assay. As shown in SI Appendix, Fig. S16B, AR knockdown decreased E2F activity while overexpression of AR WT but not K913R rescued this effect. Similar effects were also found for the expression levels of many TRAF4-up-regulated target genes, including UBE2C and CDC20, as shown in SI Appendix, Fig. S16C. Furthermore, we examined the effect of AR ubiquitination mutation on TRAF4-promoted androgen-independent cell growth and found that this K913R mutation also failed to restore androgen-independent cell growth after AR knockdown in contrast to AR WT (SI Appendix, Fig. S16D). These results further substantiate the importance of TRAF4-mediated AR ubiquitination in promoting E2F activity and androgen-independent PCa cell growth.
We next determined whether inhibition of the OR-cAMP-protein kinase A (PKA) signaling pathway could affect TRAF4-promoted androgen-independent growth. To this end, we compared the growth of control and TRAF4-overexpressing LNCaP cells in the absence or presence of a PKA inhibitor H89 under androgen-deprivation conditions (Fig. 7H). As expected, we observed increased androgen-independent cell growth in TRAF4 stable cells as compared to control cells in the vehicle control-treated groups. H89 treatment significantly decreased the cell proliferation in TRAF4 stable cells while it only had a modest effect on control cells. These results substantiate that TRAF4-mediated AR ubiquitination can promote androgen-independent cell growth through the OR-cAMP-PKA signaling pathway.
Altogether, we demonstrate that under an androgen deprivation condition overexpression of TRAF4 in PCa promotes K27-linked AR ubiquitination at its C-terminal tail, which increases its interaction with FOXA1 and triggers ubiquitinated AR binding to a distinct set of genomic loci to induce a different transcriptional program, including ORs and other CRPC-associated (such as mitotic genes UBE2C and CDC20). Upregulation of OR gene expression then increases intracellular cAMP levels and subsequently activates the E2F transcription factor. Ultimately, these pathway activations facilitate cell growth in the absence of androgen, leading to CRPC (Fig. 8).
Fig. 8.

Illustration of the role of TRAF4 in AR signaling and CRPC development. The androgen receptor plays an important role in both androgen-dependent and androgen-independent prostate cancer. During androgen deprivation condition, high TRAF4 expression promotes K27-linked AR ubiquitination at its C-terminal end K913 residue. Ubiquitinated AR has increased interaction with pioneer factor FOXA1. As a result, AR binds to a distinct set of genomic loci enriched with FOXA1 binding motif to drive different transcriptional programs including olfactory transduction pathway, as well as CRPC-associated genes such as UBE2C and CDC20. Upregulation of OR gene expression increases intracellular cAMP levels and E2F transcription factor activity, together with other CRPC-associated mitotic genes, ultimately promoting cell proliferation under androgen deprivation condition.
Discussion
AR remains active and alters its transcriptional program in CRPCs (2–6). Understanding the events triggering the switch of AR-regulated transcriptional profile under castration conditions is critical for more effectively treating CRPCs. Here we demonstrate that TRAF4-mediated atypical AR ubiquitination plays an important role in switching the AR genomic binding profiles and promoting CRPC development.
TRAF4 is frequently overexpressed in advanced PCas (14). We demonstrate that its overexpression converts androgen-sensitive PCa cells into castration-resistant cells both in vitro and in vivo (Fig. 1). Importantly, the E3 ubiquitin ligase activity of TRAF4 is required for this function. Ubiquitination is a reversible posttranslational modification. Different ubiquitin linkages (K6, K11, K27, K29, K33, K48, and K63) formed at target proteins could produce distinct structural conformations and serve as an ubiquitin code for interacting with different readers, leading to different outcomes of ubiquitinated proteins (56). We report here that TRAF4 promotes AR ubiquitination primarily through an atypical K27-linkage. Unlike K11 and K48-linked ubiquitination, which mainly function as protein degradation signals (57), K27 linkage frequently has been associated with protein recruitment (58–60) where the ubiquitin chain serves as an interaction platform to allow protein binding. We found that TRAF4-mediated AR ubiquitination occurs at the C-terminal tail. The C-terminal end of AR plays a role in the ligand-binding function and the consequent transcriptional activity (61). We found that ubiquitination at this end does not promote AR turnover. Instead, it dramatically increases the association of AR with pioneer factor FOXA1. FOXA1 physically interacts with the DBD/hinge region of AR (41). It is possible that the ubiquitin chain at the C-terminal end serves as a scaffold to stabilize the interaction between AR and FOXA1.
FOXA1 reprograms AR cistrome in prostate tumors (38). It is highly expressed (62–65) and frequently mutated in metastatic CRPCs (62). Recent comprehensive mutation analyses of FOXA1 suggest that these are activating mutations and establish FOXA1 as a principle oncogene in AR-driven PCas (62, 66). Elevated FOXA1 levels have been shown to expand AR binding to the regions that normally are not occupied by AR to promote PCa cell proliferation under an androgen-depleted state (65). It appears to be essential for AR to switch its transcriptional program in androgen-independent LNCaP-Abl cells to regulate CRPC-associated genes, such as UBE2C and CDC20, which are not regulated by AR in androgen-sensitive LNCaP cells (4). Our finding that TRAF4-mediated AR ubiquitination increases its interaction with FOXA1 could be another layer of posttranslational modification to alter the AR-regulated transcriptional programs and promote CRPC, especially in a TRAF4 overexpressed condition. With increased affinity, it is likely that FOXA1 directs ubiquitinated AR binding to a number of new genomic loci, therefore up-regulating pathways beneficiary for cell proliferation under androgen-deprived conditions.
Our pathway analysis of top genomic binding sites occupied by AR upon TRAF4 overexpression surprisingly reveals a role for the olfactory transduction and its downstream cAMP signaling pathway as the most-enriched pathway bound by AR. ORs are expressed in a number of human tissues outside of nasal epithelium. Overexpression of ORs has been linked to tumor growth, invasion, metastasis, inflammation, and apoptosis. We found that TRAF4 promotes AR binding to a set of OR gene enhancers and up-regulates their expression levels in PCa cells, including OR51E1 and OR51E2, which are previously known as prostate-specific G-protein-coupled receptors and biomarkers of PCa progression (46, 47, 54, 67). OR51E2 was also found to promote neuroendocrine transdifferentiation of PCa cells (55). OR activation agents are not limited to odor molecules. A recent metabolite screen of OR51E2 agonists identified 19-hydroxyandrostenedione, a product of the aromatase reaction, together with several other metabolites as endogenous ligands (55). Thus, upregulation of these OR genes by TRAF4 and ubiquitinated AR likely results in amplified OR signaling activated by these endogenous ligands. Consistently, we found that TRAF4 overexpressing cells have higher levels of intracellular cAMP and are highly responsive to OR agonist stimulation in contrast to control cells. The finding that cAMP effector PKA inhibitor treatment abolishes TRAF4-promoted androgen-independent growth further substantiates the important role of OR signaling in this process.
The cAMP pathway may stimulate or inhibit cell proliferation depending on the cell types (68, 69). cAMP can promote PCa cell proliferation and differentiation (70–73). Low concentration of cAMP stimulates cell growth while high concentration of cAMP promotes cell differentiation (69). We found that potent cAMP stimulator, forskolin treatment in LNCaP cells inhibits E2F activity. However, OR agonist treatment stimulates E2F activity in TRAF4 overexpressing cells (Fig. 7G). The potential mechanism may be involved in cAMP-mediated extracellular signal-regulated kinase (ERK) stimulation (69) and subsequent ERK-activated cyclin D1 upregulation as well as cyclin D1-CDK4/6 complex assembly followed by Rb phosphorylation, and ultimately E2F activation (74). This implies that upregulation of cAMP signaling at the receptor expression level through ubiquitinated AR could mildly enhance the cAMP level to promote PCa cell survival and growth under androgen deprivation conditions. In line with this, a previous report indicates that low nanomolar concentrations of a neuropeptide, acting through its G stimulatory protein-coupled receptor and the cAMP/PKA pathway, induced androgen-sensitive prostate cell growth in the absence of androgen, and this action is dependent on functional AR (75).
Although multiple mechanisms are involved in castration-resistant PCa, the E2F pathway has emerged as a central pathway in this process (5, 20, 21, 29). Disruption of RB/E2F axis also was observed in the transition to CRPCs (31). Consistently, a cell cycle proliferation gene signature predicts poor outcome of patients with PCa (76). Our AR ChIP seq analysis and xenograft tumor RNA-seq studies also emphasize the importance of E2F transcriptional activity. Inhibition of CDK4/6, the cyclin-dependent kinase inducing Rb phosphorylation and E2F hyperactivation, abolishes TRAF4-promoted androgen-independent growth. The inhibitor palbociclib is now in phase II clinical trial for treating metastatic CRPC (NCT02905318). The markers predicting patient responses to the drug are not yet clear. Our study here implies that CRPC patients with high TRAF4 expression have a hyperactivated E2F pathway and likely are more responsive to palbociclib treatment.
Materials and Methods
Castration Surgery and CRPC Tumor Study.
All animal experiments were performed in accordance with the Institutional Animal Care and Use Committees (IACUCs) at Baylor College of Medicine. For in vivo LNCaP studies, 5-to 6-wk-old male SCID mice (The Jackson Laboratory, 5 mice each group) were used for experimental castration-resistant PCa study. Ten million TRAF4-overexpressing or control LNCaP cells were injected into NOD/SCID mice subcutaneously. When tumors reach approximately 1 cm in diameter, mice were anesthetized using isoflourane and surgically castrated using the standard surgical technique. The tumor growth was monitored every week through tumor volume measurement. For 22Rv1 xenograft studies, SCID mice (6 mice each group) were castrated; the testis and associated tissues were surgically removed 1 wk before injection. Matrigel matrix was mixed with pTRIZ-shTRAF4 or control vector stably infected 22Rv1 cells before injection. One million 22Rv1 cells were injected subcutaneously. Mice began receiving 2 mg/mL Doxycycline, 10% sucrose in drinking water for inducible expression of shTRAF4 in the xenografts. Tumor growth was monitored at the indicated time after the injection.
E2F Luciferase Reporter assay.
Cells were cotransfected with 500 ng pE2F-TA-Luc plasmid, 100 ng pRL-Renilla (encoding the Renilla luciferase gene for standardization), and empty expression vector or increasing amount of TRAF4 expression vectors. Cells were harvested 48 h after transfection, and firefly luciferase activity was measured on a Synergy LX multimode reader (BioTek, USA) using a Dual Luciferase Reporter Assay (Promega) and according to the manufacturer’s protocol. Firefly luciferase experimental reporter was normalized to Renilla luciferase.
ChIP-Seq Analysis.
The ChIP-Seq yielded 25 to 28 million 75 single end reads. Data were mapped to the human genome build hg19 using bwa v 0.7.12 (77). Peaks were called using MACS 2.1.0 with default settings (78). Venn diagrams of peaks were generated using bedtools (79). ChIP-Seq normalized signal plots were generated using HOMER (80). We used publicly available FOXA1 ChIP-Seq datasets, and HOXB13 datasets (38, 81–83). The relative intensity of each AR-binding site is calculated via the computeMatrix function in Galaxy Deeptool with the RPKM normalized bigwig files and bed files from the peak calling as inputs by calculating the area under the curve. bamCompare function in Galaxy Deeptool was used to compute the log2 ratio of AR-binding intensity in TARF4-overexpression and control conditions, and the heatmap was generated by plotHeatmap function accordingly. Cis-regulatory element annotation system function in Galaxy/Cistrome was applied to calculate the enrichment of the binding sites in the promoter, exon, intron, untranslated region (UTR) and other genomic regions against the mappable human genome using the binomial model. Motif analysis was performed with the SeqPos motif tool (version 0.590) embedded in Galaxy Cistrome.
GO Analysis.
DAVID (Version 6.8) (84) and GREAT (Version 3.0.0) (85) were used to perform gene ontology analysis. Briefly, gene names were first converted to DAVID-recognizable IDs using Gene Accession Conversion Tool. The updated gene list was then subject to GO analysis using all human genes as background and with functional annotation chart function. KEGG_PATHWAY, were used as GO categories for all GO analyses.
cAMP Assay.
cAMP levels were determined by the cAMP enzyme immunoassay kit following the manufacturer's instructions (Cayman Chemical Co., Ann Arbor, MI, USA, Item no. 581001). Intracellular cAMP was determined in LNCaP cells with and without TRAF4 overexpression using the acid extraction method. Briefly, cells were plated on a 6-well plate overnight and exposed to different concentrations of b-ionone for 15 min. After treatment, the medium was removed and 300 μL 0.1 M HCl was added. These plates were then incubated at room temperature for 20 min. Cells were directly scraped off, transferred into a fresh tube, and centrifuged. Fifty microliters of this extract supernatant was measured in the cAMP enzyme-linked immunoassay (ELISA).
Statistics.
Unless otherwise indicated, all results represent mean ± SEM, and statistical comparisons between different groups were performed using the 2-tailed Student’s t test or 1-way ANOVA with multiple comparisons corrections. For all statistical analyses, differences of P < 0.05 were considered statistically significant, and experiments were repeated in triplicate at least two times. GraphPad Prism software version 4.0/7.0 (GraphPad Software) was used for data analysis.
Study Approval.
All animal experiments were approved by the IACUCs at Baylor College of Medicine.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Acknowledgments
This work was supported by grants from DAMDW81XWH-15-1-0536 and W81XWH-21-1-0404 to P.Y., NIH-NICHD grants (HD8818, HD07857) to B.W.O.; Pilot Project and Subsidy Program under NIEHS Award Number P30ES030285 to H.M.; NCI Cancer Center Support Grant P30CA125123 (BCM Monoclonal Antibody/recombinant Protein Expression Core Facility) and the Cancer Prevention & Research Institute of Texas Proteomics & Metabolomics Core Facility Support Award RP170005, and NIEHS CG-CPEH grant 1P30ES030285 to C.C.
Author contributions
R.S., B.W.O., and P.Y. designed research; R.S., T.S., L.E.V.L., S.N., H.S., S.D., L.Q., and D.K. performed research; H.M., B.Z., F.Y., and C.C. analyzed data; and R.S. and P.Y. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
The RNA seq data and the ChIP seq data have been deposited in the GeneExpression Omnibus database (accession nos. GSE225509 (86), GSE154029 (87), and GSE154001 (88), respectively).
Supporting Information
References
- 1.Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Shen M. M., Abate-Shen C., Molecular genetics of prostate cancer: New prospects for old challenges. Genes. Dev. 24, 1967–2000 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li B., Lu W., Chen Z., Regulation of androgen receptor by E3 Ubiquitin ligases: For more or less. Recept. Clin. Investig. 1, 10.14800%2Frci.122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Q., et al. , Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138, 245–256 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma N. L., et al. , The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 23, 35–47 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Decker K. F., et al. , Persistent androgen receptor-mediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Res. 40, 10765–10779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faus H., Haendler B., Post-translational modifications of steroid receptors. Biomed. Pharmacother. 60, 520–528 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Mahajan N. P., et al. , Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 104, 8438–8443 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu K., et al. , Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell 15, 270–282 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haglund K., Dikic I., Ubiquitylation and cell signaling. EMBO J. 24, 3353–3359 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qi J., et al. , The E3 ubiquitin ligase Siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity. Cancer Cell 23, 332–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo X., Cao J., Zhang C., Huang H., Liu J., TRAF4 promotes the malignant progression of high-grade serous ovarian cancer by activating YAP pathway. Biochem. Biophys. Res. Commun. 627, 68–75 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Ruan X., et al. , The research progress in physiological and pathological functions of TRAF4. Front. Oncol. 12, 842072 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh R., et al. , TRAF4-mediated ubiquitination of NGF receptor TrkA regulates prostate cancer metastasis. J. Clin. Invest. 128, 3129–3143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varambally S., et al. , Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 8, 393–406 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Shah R. B., et al. , Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 64, 9209–9216 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Chandran U. R., et al. , Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 7, 64 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grasso C. S., et al. , The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor B. S., et al. , Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Handle F., et al. , Drivers of AR indifferent anti-androgen resistance in prostate cancer cells. Sci. Rep. 9, 13786 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramos-Montoya A., et al. , HES6 drives a critical AR transcriptional programme to induce castration-resistant prostate cancer through activation of an E2F1-mediated cell cycle network. EMBO Mol. Med. 6, 651–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni M., et al. , Amplitude modulation of androgen signaling by c-MYC. Genes. Dev. 27, 734–748 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao L., et al. , Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS One 8, e63563 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leung J. Y., Ehmann G. L., Giangrande P. H., Nevins J. R., A role for Myc in facilitating transcription activation by E2F1. Oncogene 27, 4172–4179 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Leone G., DeGregori J., Sears R., Jakoi L., Nevins J. R., Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 387, 422–426 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Oswald F., Lovec H., Moroy T., Lipp M., E2F-dependent regulation of human MYC: Trans-activation by cyclins D1 and A overrides tumour suppressor protein functions. Oncogene 9, 2029–2036 (1994). [PubMed] [Google Scholar]
- 27.Roussel M. F., Davis J. N., Cleveland J. L., Ghysdael J., Hiebert S. W., Dual control of myc expression through a single DNA binding site targeted by ets family proteins and E2F–1. Oncogene 9, 405–415 (1994). [PubMed] [Google Scholar]
- 28.Thalmeier K., Synovzik H., Mertz R., Winnacker E. L., Lipp M., Nuclear factor E2F mediates basic transcription and trans-activation by E1a of the human MYC promoter. Genes. Dev. 3, 527–536 (1989). [DOI] [PubMed] [Google Scholar]
- 29.Kim Y. R., et al. , HOXB13 promotes androgen independent growth of LNCaP prostate cancer cells by the activation of E2F signaling. Mol. Cancer 9, 124 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis J. N., et al. , Elevated E2F1 inhibits transcription of the androgen receptor in metastatic hormone-resistant prostate cancer. Cancer Res. 66, 11897–11906 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Sharma A., et al. , The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Invest. 120, 4478–4492 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scher H. I., Buchanan G., Gerald W., Butler L. M., Tilley W. D., Targeting the androgen receptor: Improving outcomes for castration-resistant prostate cancer. Endocr. Relat. Cancer 11, 459–476 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Chen C. D., et al. , Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 10, 33–39 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Jernberg E., Bergh A., Wikstrom P., Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 6, R146–R161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L., et al. , TRAF4 promotes TGF-beta receptor signaling and drives breast cancer metastasis. Mol. Cell 51, 559–572 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Li W., et al. , TRAF4 is a critical molecule for Akt activation in lung cancer. Cancer Res. 73, 6938–6950 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu W., Singh R., Wang Z., O’Malley B. W., Yi P., The E3 ligase TRAF4 promotes IGF signaling by mediating atypical ubiquitination of IRS-1. J. Biol. Chem. 296, 100739 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pomerantz M. M., et al. , The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 47, 1346–1351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Norris J. D., et al. , The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol. Cell 36, 405–416 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sahu B., et al. , Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. Embo J. 30, 3962–3976 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao N., et al. , The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 17, 1484–1507 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Firestein S., How the olfactory system makes sense of scents. Nature 413, 211–218 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Massberg D., et al. , Monoterpene (-)-citronellal affects hepatocarcinoma cell signaling via an olfactory receptor. Arch. Biochem. Biophys. 566, 100–109 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Gelis L., et al. , Functional characterization of the odorant receptor 51E2 in human melanocytes. J. Biol. Chem. 291, 17772–17786 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schweda F., Friis U., Wagner C., Skott O., Kurtz A., Renin release. Physiology (Bethesda) 22, 310–319 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Weng J., et al. , PSGR2, a novel G-protein coupled receptor, is overexpressed in human prostate cancer. Int. J. Cancer 118, 1471–1480 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Xu L. L., et al. , PSGR, a novel prostate-specific gene with homology to a G protein-coupled receptor, is overexpressed in prostate cancer. Cancer Res. 60, 6568–6572 (2000). [PubMed] [Google Scholar]
- 48.Xia C., Ma W., Wang F., Hua S., Liu M., Identification of a prostate-specific G-protein coupled receptor in prostate cancer. Oncogene 20, 5903–5907 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Chen Z., Zhao H., Fu N., Chen L., The diversified function and potential therapy of ectopic olfactory receptors in non-olfactory tissues. J. Cell Physiol. 233, 2104–2115 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Masjedi S., Zwiebel L. J., Giorgio T. D., Olfactory receptor gene abundance in invasive breast carcinoma. Sci. Rep. 9, 13736 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.An S. S., Liggett S. B., Taste and smell GPCRs in the lung: Evidence for a previously unrecognized widespread chemosensory system. Cell Signal 41, 82–88 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez M., et al. , Prostate-specific G-protein-coupled receptor collaborates with loss of PTEN to promote prostate cancer progression. Oncogene 35, 1153–1162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanz G., et al. , Promotion of cancer cell invasiveness and metastasis emergence caused by olfactory receptor stimulation. PLoS One 9, e85110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez M., Siwko S., Liu M., Prostate-specific G-protein coupled receptor, an emerging biomarker regulating inflammation and prostate cancer invasion. Curr. Mol. Med. 16, 526–532 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Abaffy T., et al. , A testosterone metabolite 19-Hydroxyandrostenedione induces neuroendocrine trans-differentiation of prostate cancer cells via an ectopic olfactory receptor. Front. Oncol. 8, 162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Komander D., Rape M., The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Ravid T., Hochstrasser M., Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 9, 679–690 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Huizen M., Kikkert M., The role of atypical ubiquitin chains in the regulation of the antiviral innate immune response. Front. Cell Dev. Biol. 7, 392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin Q., et al. , K27-linked ubiquitination of BRAF by ITCH engages cytokine response to maintain MEK-ERK signaling. Nat. Commun. 10, 1870 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gatti M., et al. , RNF168 promotes noncanonical K27 ubiquitination to signal DNA damage. Cell Rep. 10, 226–238 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Tahiri B., Auzou G., Nicolas J. C., Sultan C., Lupo B., Participation of critical residues from the extreme C-terminal end of the human androgen receptor in the ligand binding function. Biochemistry 40, 8431–8437 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Parolia A., et al. , Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 571, 413–418 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jain R. K., Mehta R. J., Nakshatri H., Idrees M. T., Badve S. S., High-level expression of forkhead-box protein A1 in metastatic prostate cancer. Histopathology 58, 766–772 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Gerhardt J., et al. , FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am. J. Pathol. 180, 848–861 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Robinson J. L., et al. , Elevated levels of FOXA1 facilitate androgen receptor chromatin binding resulting in a CRPC-like phenotype. Oncogene 33, 5666–5674 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adams E. J., et al. , FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature 571, 408–412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang J., et al. , The prostate-specific G-protein coupled receptors PSGR and PSGR2 are prostate cancer biomarkers that are complementary to alpha-methylacyl-CoA racemase. Prostate 66, 847–857 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Gottesman M. M., Fleischmann R. D., The role of cAMP in regulating tumour cell growth. Cancer Surv. 5, 291–308 (1986). [PubMed] [Google Scholar]
- 69.Stork P. J., Schmitt J. M., Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 12, 258–266 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Merkle D., Hoffmann R., Roles of cAMP and cAMP-dependent protein kinase in the progression of prostate cancer: Cross-talk with the androgen receptor. Cell Signal 23, 507–515 (2011). [DOI] [PubMed] [Google Scholar]
- 71.Shah G. V., et al. , Calcitonin stimulates growth of human prostate cancer cells through receptor-mediated increase in cyclic adenosine 3’,5’-monophosphates and cytoplasmic Ca2+ transients. Endocrinology 134, 596–602 (1994). [DOI] [PubMed] [Google Scholar]
- 72.Chen T., Cho R. W., Stork P. J., Weber M. J., Elevation of cyclic adenosine 3’,5’-monophosphate potentiates activation of mitogen-activated protein kinase by growth factors in LNCaP prostate cancer cells. Cancer Res. 59, 213–218 (1999). [PubMed] [Google Scholar]
- 73.Deeble P. D., Murphy D. J., Parsons S. J., Cox M. E., Interleukin-6- and cyclic AMP-mediated signaling potentiates neuroendocrine differentiation of LNCaP prostate tumor cells. Mol. Cell Biol. 21, 8471–8482 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang W., Liu H. T., MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12, 9–18 (2002). [DOI] [PubMed] [Google Scholar]
- 75.Xie Y., Wolff D. W., Lin M. F., Tu Y., Vasoactive intestinal peptide transactivates the androgen receptor through a protein kinase A-dependent extracellular signal-regulated kinase pathway in prostate cancer LNCaP cells. Mol. Pharmacol. 72, 73–85 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Cuzick J., et al. , Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 12, 245–255 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Y., et al. , Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heinz S., et al. , Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Malinen M., Niskanen E. A., Kaikkonen M. U., Palvimo J. J., Crosstalk between androgen and pro-inflammatory signaling remodels androgen receptor and NF-kappaB cistrome to reprogram the prostate cancer cell transcriptome. Nucleic Acids Res. 45, 619–630 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.He B., et al. , GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proc. Natl. Acad. Sci. U.S.A. 111, 18261–18266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sunkel B., et al. , Integrative analysis identifies targetable CREB1/FoxA1 transcriptional co-regulation as a predictor of prostate cancer recurrence. Nucleic Acids Res. 44, 4105–4122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.W. Huang da, B. T. Sherman, R. A. Lempicki, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 85.McLean C. Y., et al. , GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meng H., Total RNA-Seq in control and TRAF4-overexpressing LNCaP cells. NCBI Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE225509. Deposited 17 February 2023.
- 87.Meng H., Total RNA-Seq in TRAF4-overexpressing castration-resistant xenograft tumors and the control castration-responsive tumors. NCBI Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154029. Deposited 8 July 2020.
- 88.Yi P., Genome-wide AR binding under androgen deprivation condition upon TRAF4 overexpression. NCBI Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154001. Deposited 8 July 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Data Availability Statement
The RNA seq data and the ChIP seq data have been deposited in the GeneExpression Omnibus database (accession nos. GSE225509 (86), GSE154029 (87), and GSE154001 (88), respectively).