

Significance
ESCC is a lethal cancer in urgent need of treatment and prevention strategies. It occurs with widely varying frequency in specific global human populations and likely has a significant dietary-deficiency association. In the Zn-deficient rat ESCC model, sequential delivery of antimiRs to downmodulate Zn-controlled expression of miR-31 and miR-21 led to reduced inflammation and upregulated apoptosis, suppressing ESCC. In parallel, ESCC-bearing ZD rats receiving Zn medication showed ~50% ESCC decrease by affecting the same miRs-31/21 controlled biological processes. Thus, Zn treatment reduced inflammation, promoted apoptosis, and reversed classic cancer cell metabolic phenotypes such as increased glycolysis and nucleoside intermediates. We conclude that Zn represents an ESCC preventive opportunity and, Zn and antimiRs-31/21 have therapeutic potential.
Keywords: zinc deficiency and esophageal squamous cell carcinoma, zinc deficiency-promoted esophageal cancer rat model, sequential antimiR-31 and antimiR-21 delivery in vivo, zinc therapy, metabolomics
Abstract
Esophageal squamous cell carcinoma (ESCC) is a deadly disease with few prevention or treatment options. ESCC development in humans and rodents is associated with Zn deficiency (ZD), inflammation, and overexpression of oncogenic microRNAs: miR-31 and miR-21. In a ZD-promoted ESCC rat model with upregulation of these miRs, systemic antimiR-31 suppresses the miR-31-EGLN3/STK40-NF-κB–controlled inflammatory pathway and ESCC. In this model, systemic delivery of Zn-regulated antimiR-31, followed by antimiR-21, restored expression of tumor-suppressor proteins targeted by these specific miRs: STK40/EGLN3 (miR-31), PDCD4 (miR-21), suppressing inflammation, promoting apoptosis, and inhibiting ESCC development. Moreover, ESCC-bearing Zn-deficient (ZD) rats receiving Zn medication showed a 47% decrease in ESCC incidence vs. Zn-untreated controls. Zn treatment eliminated ESCCs by affecting a spectrum of biological processes that included downregulation of expression of the two miRs and miR-31-controlled inflammatory pathway, stimulation of miR-21-PDCD4 axis apoptosis, and reversal of the ESCC metabolome: with decrease in putrescine, increase in glucose, accompanied by downregulation of metabolite enzymes ODC and HK2. Thus, Zn treatment or miR-31/21 silencing are effective therapeutic strategies for ESCC in this rodent model and should be examined in the human counterpart exhibiting the same biological processes.
Esophageal squamous cell carcinoma (ESCC) is a male predominant cancer subtype. It has a deadly prognosis due to late-stage diagnosis and lack of effective treatment options. Risk factors include alcohol consumption, cigarette smoking, exposure to environmental carcinogens such as N-nitrosomethylbenzylamine (NMBA) (1), and nutritional deficiencies. In high incidence populations in Africa, India, Iran, and China (2–5), and in individuals with chronic alcohol use (6), increased ESCC risk is commonly associated with nutritional Zn deficiency (ZD). Using esophageal tissue biopsy specimens in a prospective observational study, Abnet et al. (3) demonstrated that esophageal tissue Zn concentration is inversely associated with risk of incident ESCC, thereby providing evidence for ZD as a contributing factor in ESCC development in humans. In a recent study of genome sequences of ESCC cases from eight countries with widely differing incidences, neither mutational signatures defining mutations caused by specific exogenous or endogenous processes, nor driver gene mutations, were found to vary significantly across these cancers from high and low incidence populations (7, 8), though authors reported few associations with lifestyle, environmental or nutritional risk factors. An unidentified Zn nutritional deficiency such as noted by Abnet et al. (3) in high incidence countries, likely would not directly cause a specific mutational signature or driver gene alteration.
Zn deficiency is a public health problem (9, 10) that affects ~31% of global populations. ZD is prevalent in individuals who eat little Zn-rich meat and/or consume high-phytate–containing vegetarian diet. Zn is required for activity of >300 enzymes, for immune function, and for conformation of many transcription factors controlling cell proliferation and apoptosis signaling pathways (11, 12). ZD can adversely affect these processes and increase the risk of developing various diseases, including ESCC, in which ZD is implicated in the pathogenesis of ESCC in many populations (2–5).
Our systematically well-characterized ZD-promoted ESCC male rat model recapitulates aspects of human ESCC, including dietary ZD, ESCC-associated signatures in inflammation genes (13), microRNAs (miRNAs, miRs) (14), and metabolic changes (15). Rats on a ZD diet for ~5 wk develop hyperplastic esophagi with upregulation of proinflammation mediators (16) and oncogenic miR-31 (17). Prolonged ZD induces an expanded inflammatory program (13) and pronounced upregulation of miR-31 and miR-21 (14) that when combined with low NMBA doses produces high ESCC tumor burdens (13–15). Notably, oncomiRs miR-31 (18) and miR-21 (19) are overexpressed in human ESCCs.
miR expression levels are altered in all human cancers (20). Each miR can target multiple genes, to regulate a range of biological processes, including proliferation, differentiation, apoptosis, and inflammation. miRs can act as oncogenes or tumor suppressors and have emerged as therapeutic targets for cancer (21). Suppression of oncomiR activity can be accomplished by miRNA inhibitors or anti-miR oligomers such as locked nucleic acid (LNA)-mediated RNA oligonucleotides that exhibit high biostability, low toxicity, and adequate biodistribution in vivo (22). Using the ZD rat ESCC model with miR-31 as the top upregulated miR (14), we previously found that LNA-antimiR-31 reduced the miR-31-EGLN3/STK40-NF-κB controlled inflammatory process, suppressing ESCC development in the ZD rats (15). The STK40/EGLN3 pathway is a negative regulator of NF-κΒ mediated transcription (23, 24) and a direct miR-31 target (15, 17, 25, 26). Since miR-31 and miR-21 are prominently overexpressed in hyperplastic ZD esophagus (27) and ESCC-bearing ZD esophagus (14), we next wished to determine if successive delivery of two antimiRs in vivo, namely antimiR-31 followed by antimiR-21, would suppress ESCC development more effectively through their associated tumor suppressor pathways. In addition, since Zn itself has chemopreventive activity at both the dysplastic and neoplastic stages of ESCC development (13), and in regression of tongue squamous cell carcinomas (SCCs) in Zn-supplemented rats (28), we asked if Zn medication as a controller of these same miR signal pathways would eliminate established ESCCs in ZD rats.
Results
Sequential AntimiR-31 and AntimiR-21 Delivery Inhibits ESCC Development in ZD Rats.
To determine whether successive delivery of two antimiRs in vivo, i.e., antimiR-31 followed by antimiR-21, would suppress ESCC development more effectively than single antimiR-31 delivery (15), we performed a 20-wk anticancer study in the ZD-promoted ESCC rat model with cancer-associated inflammatory gene signature (13) and upregulation of the oncomiRs miR-31 and miR-21 (14). We used LNA–mediated-inhibitors, rno-miR-31a-5p and rno-miR-21, to inhibit esophageal expression of the corresponding miRs. The sequential antimiR delivery study was conducted simultaneously with the previous single antimiR-31 delivery study (15) and, thus, shared the same control rat groups described in the prior study (15): untreated ZD:CTRL(control), untreated Zn-sufficient (ZS):CTRL, and ZD:antimiR-31/5-wk that received 10 intravenous (i.v.) doses of antimiR-31 over the first 5 wk (15); and utilized the same antimiR and carcinogen treatment timing/schedule as the single antimiR-31 delivery study (15). As depicted in Fig. 1A (treatment details in Materials and Methods), along with the ZD control rat cohorts, 4-wk–old rat antimiR-31 and antimiR-21 cohort (n = 11) were fed ZD diet. Over the first 5-wk period, this cohort received 10 i.v. doses of antimiR-31 to inhibit ZD-induced esophageal proliferation (17). At week 5 the animals received intragastric NMBA doses once a week for 4 wk (15). Immediately after the first NMBA dose and the remaining 15 wk, the animals received 20 i.v. doses of antimiR-21. Thus, antimiR-31 and antimiR-21 cohort received a total of 30 i.v. doses of anti-miRs over the 20-wk study. The animals were observed daily and killed 15 wk post first NMBA dose or 48 h after the final antimiR-21 dose for tumor evaluation and esophageal tissue isolation (15).
Fig. 1.
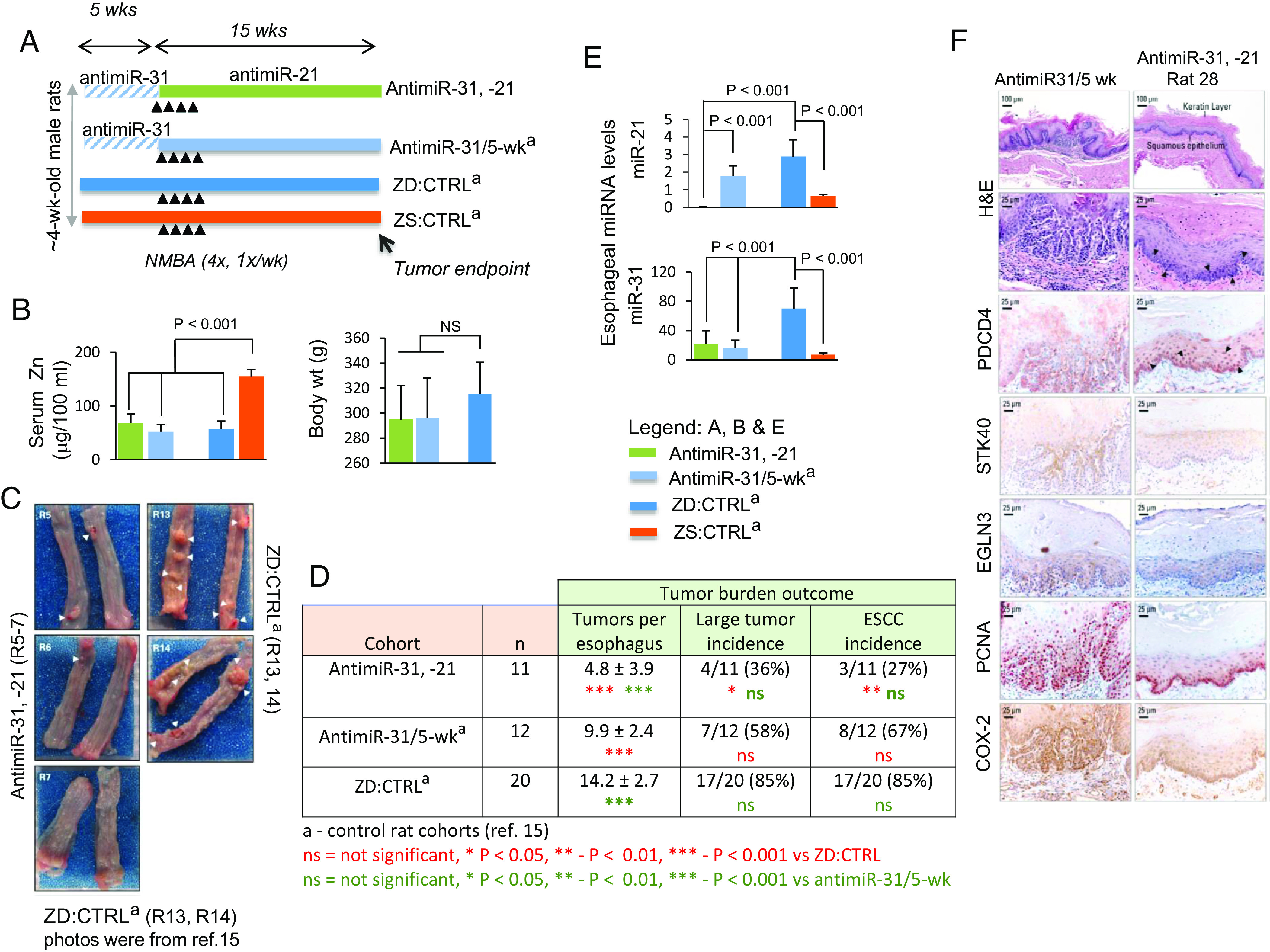
Sequential systemic antimiR-31 delivery followed by antimiR-21 suppresses esophageal carcinogenesis. (A) Study design: this sequential antimiR-31 and antimiR-21 research was done simultaneously with the previous single antimiR-31 delivery study (15) and thus shared the same control rat cohorts as described (15). Over the first 5 wk period, antimiR-31 and antimiR-21 rats (n = 11) were given 10 i.v. doses of antimiR-31. At week 5, the animals received intragastric NMBA doses once a week for 4 wk. Immediately after the first NMBA dose and over 15 wk, antimiR-31 and antimiR-21 rats received their 20 i.v. doses of antimiR-21. The animals were killed 15 wk after first NMBA dose or 48 h after the final antimiR-21 dose. (B) Body weights and serum Zn levels. (C) Macroscopic view of whole esophagus. Representative photos of antimiR-31 and antimiR-21 (R5-7) with isolated/small tumor (arrowheads) vs. representative photos obtained from ZD:CTRLa (R13, R14) (15), showing multiple/large sessile esophageal tumors. (D) Tumor multiplicity (number of tumors/esophagus, mean ± SD; Tukey-HSD (honestly significant difference) post hoc unpaired t test, n = 11 to 20 rats/cohort) (antimiR-31, -21 vs. ZD:CTRL, P < 0.001; antimiR-31 and antimiR-21 vs. antimiR-31/5-wk (P < 0.001); antimiR-31/5wk vs. ZD:CTRL, P < 0.001). Large tumor incidence (size >2 mm), antimiR-31, -21 vs. ZD:CTRL [36 vs. 85%, P < 0.05] and ESCC incidence (%), antimiR-31 and antimiR-21 vs. ZD:CTRL [27 vs. 85%, P = 0.004 (two-tailed Fisher’s Exact test; n = 10 to 20 rats/cohort). (E) qPCR analysis of esophageal miR-31 and miR-21, levels in antimiR-31 and antimiR-21 and control rat cohorts (rat snoRNA as normalizer, Tukey-HSD post hoc unpaired t test. Error bars represent SD; n = 8 rats per group). (F) Sequential delivery of antimiR-31 (5-wk) followed by antimiR-21 (15-wk) induces apoptosis in esophagus antimiR-31 and antimiR-21 (rat 28). Representative photos of antimiR-31/5-wk and antimiR-31 and antimiR-21 (rat 28) showing H&E staining, IHC staining for STK40, EGLN3, & COX-2 (brown, 3,3′-diaminobenzidine tetrahydrochloride, DAB) and PDCD4, & PCNA (red, 3-amino-9-ethylcarbazole substrate-chromogen, AEC). Arrowheads in H&E-stained section (rat 28) indicate the presence of apoptotic cells in esophageal epithelium; arrowheads in near serial PDCD4-stained sections (rat 28) showing abundant/intense nuclear overexpression of PDCD4 (arrowheads), as compared with lack of apoptotic cells (H&E-stained section) and absence of PDCD4 nuclear expression in esophagus of an antimiR-31/5-wk rat.
At tumor endpoint, all three ZD cohorts displayed similar body weights independent of antimiR dosing, indicating the LNA-mediated antimiR delivery was well-tolerated (Fig. 1B). Serum Zn level in all three ZD cohorts (~55 μg/100 mL) was significantly lower than in ZS rats (Fig. 1B) (14). Regardless of antimiR treatments, ZD cohorts had 100% overall esophageal tumor incidence. Macroscopically (Fig. 1C), antimiR-31 and antimiR-21 rats displayed isolated/small tumors in esophagi compared with the omnipresent multiple large/sessile tumors (>2 mm) in ZD:CTRL esophagus (15). Delivery of two antimiRs led to a large decrease in tumor multiplicity vs. ZD:CTRL (4.8 vs. 14, P < 0.001) and vs. antimiR-31/5-wk (4.8 vs. 9.9, P < 0.001) and in large tumors (>2 mm) vs. ZD:CTRL (36 vs. 85%, P < 0.05) (Fig. 1D). Histological examination of hematoxylin & eosin (H&E)-stained sections revealed that, despite sustained ZD promotional action toward ESCC progression, sequential antimiR-31 and antimiR-21 delivery reduced ESCC incidence from 85 to 27%, a decrease of 58% (P = 0.004). As found previously (15), while short-term antimiR-31 delivery (5 wk) led to a reduction in tumor multiplicity vs. ZD:CTRL rats (9.9 vs. 14, P < 0.001) but no effect on ESCC incidence, long-term delivery of antimiR-31 (20 wk) reduced ESCC incidence from 85 to 45%, a drop of 40%. These results suggest that delivery of two antimiRs for 20 wk (antimiR-31 followed by antimiR-21) is more effective in restraining ESCC development than a single antimiR-31 delivery through suppressing two miR-controlled tumor pathways.
Sequential Systemic AntimiR-31 and AntimiR-21 Delivery Knocks Down miR-21 and Promotes miR-21-PDCD4 Axis Apoptosis.
Several studies have shown that oncogenic miR-21 has antiapoptotic properties and exerts these effects by targeting its tumor suppressor targets PDCD4 (29, 30). miR-21 knockdown has been shown to lead to caspase activation and apoptotic cell death in glioblastoma cells (31). Here, sequential systemic antimiR-31 and antimiR-21 delivery resulted in >99% reduction of miR-21 vs. ZD:CTRL or antimiR-31/5wk (P < 0.001; Fig. 1 E, Upper). miR-31 levels in antimiR-31 and antimiR-21 and antimiR-31/5-wk esophagus assessed 15 wk after antimiR-31 dosing were ~70% lower than ZD:CTRL tissues (Fig. 1 E, Lower), consistent with the long-lasting effect of antimiR (15, 32). Thus, despite continued dietary ZD to upregulate miR-21 and miR-31 (P < 0.001, ZD:CTRL vs. ZS:CTRL; Fig. 1E), sequential delivery of two antimiRs effectively knocked down their corresponding miRs in ESCC development.
We hypothesized that a >99% miR-21 knockdown in the esophagus by systemic delivery of antimiR-21 would initiate apoptosis through upregulation of its tumor suppressor target PDCD4, an inducer of apoptosis (33). To test this, we used a) light microscopy in H&E-stained sections to identify morphological changes that occur during apoptosis in esophageal epithelia cells (34, 35) from antimiR-31 and antimiR-21 rats killed at 48 h after delivery of the final antimiR-21 dose and, b) immunohistochemistry (IHC) to analyze expression of PCDC4 protein. We found the antimiR-31 and antimiR-21 esophageal epithelium (H&E-stained sections) did display an apoptotic phenotype with numerous suprabasal and basal cells undergoing apoptosis, characterized by shrinkage of the cell/nucleus, condensation of cytoplasm/nuclear chromatin, and fragmented nuclei as compared with a highly proliferative antimiR-31/5-wk tumorous epithelium with sporadic/isolated apoptotic cells (Fig. 1F and SI Appendix, Fig. S1, H&E-stained sections). Moreover, 11 of 11 (100%) antimiR-31 and antimiR-21 rats had esophageal epithelia showing degrees of intense waves of apoptosis triggered by miR-21 knockdown, with representative esophagus from three individual antimiR-31 and antimiR-21 rats shown in Fig. 1 (rat 28) and SI Appendix, Fig. S1 (rat 23, 29). These data show that a massive wave of apoptotic activity was triggered in antimiR-31 and antimiR-21 esophagus following >99% knockdown of miR-21 by antimiR-21.
PDCD4, one of the most frequently downregulated proteins in human ESCC (36), is a well-characterized, multifunctional tumor suppressor that inhibits cell proliferation, invasion/metastasis and is also an inducer of apoptosis (33). Zhang et al. (37) reported that as cells undergo apoptosis, PDCD4-protein is accumulated in the nuclei. PDCD4 upregulation activates the pro-apoptotic member of the Bcl2 protein family, BAX, followed by the release of cytochrome C from mitochondria and activation of caspases 8, 9, and 3, bringing about induction of apoptosis in human hepatocellular carcinoma cells (33, 37). Strikingly, PDCD4-IHC analysis in near serial sections showed strong nuclear expression of PDCD4 in the cells undergoing apoptosis (H&E-stained section) in antimiR-31 and antimiR-21 rat esophagus vs. absence of PDCD4 nuclear expression in the esophagus of ESCC-bearing antimiR-31/5-wk rat lacking apoptotic cells (H&E-stained section) (PDCD4-IHC, Fig. 1F and SI Appendix, Fig. S1). In addition, PDCD4 nuclear overexpression in antimiR-31 and antimiR-21 esophagus was accompanied by strong cytoplasmic immunostaining of BAX protein (SI Appendix, Fig. S1, antimiR-31 and antimiR-21 rat 23 and 29). These results demonstrate that during ESCC development a >99% knockdown of miR-21 by antimiR-21 powerfully upregulated its direct tumor suppressor target PDCD4, triggering massive waves of apoptosis in esophageal epithelium in the same cells that overexpressed nuclear PDCD4. Additionally, IHC analysis showed moderate cytoplasmic staining for the direct tumor suppressor targets STK40/EGLN3 of miR-31 in antimiR-31 and antimiR-21 esophagus, indicating that these tumor suppressor targets were moderately upregulated by a 5-wk delivery of antimiR-31 (Fig. 1F). These data therefore show that sequential delivery of antimiR-31(5 wk) followed by antimiR-21 (15 wk) initiated waves of apoptosis, leading to a nonproliferative (PCNA [proliferating cell nuclear antigen], IHC) and less inflammatory esophagus (COX-2, IHC) compared with the proliferative and inflammatory ESCC bearing antimiR-31/5-wk esophagus (Fig. 1F).
To summarize, decreased tumor multiplicity/size and ESCC incidence; reduced tumor cell proliferation and inflammation, and notably amplified/intensified apoptosis were observed in NMBA-induced ZD rats upon sequential delivery of antimiR-31 (prior to NMBA administration) followed by delivery of antimiR-21 oligonucleotides (during onset and progression of ESCC development). Our findings suggest that sequential delivery of miR-31 plus miR-21 holds promise as a therapeutic approach.
Zn Medication Eliminates Established ESCCs in ZD Rats.
We previously showed that replenishing Zn in ZD rats and supplementing Zn in ZS rats inhibit the development of ESCC (13) and tongue SCC (28). To determine whether Zn has anticancer activity in established ESCC, we performed a 33-day Zn treatment study in 31 ESCC-bearing ZD male rats with 85% ESCC incidence (designated Zn-untreated) that were generated in a 20-wk ZD-promoted, NMBA-induced ESCC experiment. These ESCC-bearing ZD male rats demonstrated oncogenic miR-31 and antimiR-21 upregulation (14, 15), an ESCC-associated metabolomic profile and inflammatory gene signature (15). As shown in Fig. 2A, at 20 wk, 31 ESCC-bearing Zn-untreated rats were randomized into two groups, viz Zn-untreated (n = 10) and Zn-treated (n = 21). While Zn-untreated rats continued on ZD diet and deionized water, Zn-treated rats were given deionized water supplemented with 25 ppm Zn as Zn gluconate (28) and paired-fed a ZS diet to Zn-untreated rats. This Zn dose translates to human supplementation of ~100 mg Zn/day/70 kg individual, a dose comparable to those used for Zn supplementation in commercial preparations. This study was concluded after 33 d of Zn medication at which point the animals were euthanized for evaluation of the effectiveness of Zn medication in ESCC elimination.
Fig. 2.
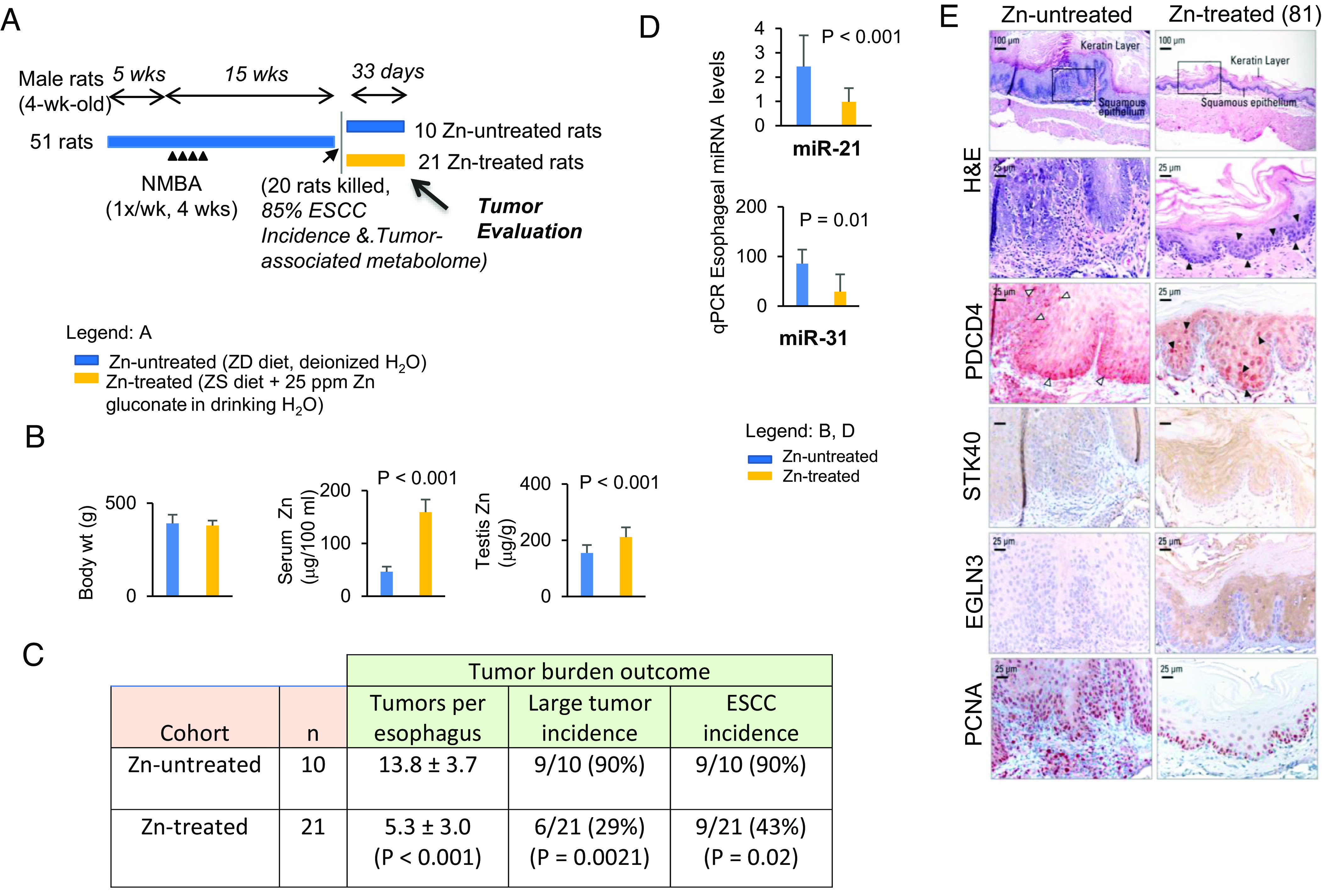
Zn medication eliminates established ESCCs in ZD rats. (A) Study design. (B) Body weights, serum Zn, and testis Zn levels. (C) Tumor multiplicity (number of tumors/esophagus, mean ± SD) (two-tailed Welch t test, n = 8 to 20 rats/cohort); large tumor (size >2 mm) and ESCC incidence (%) (two-tailed Fisher’s exact test; n = 8 to 20 rats/cohort). (D) qPCR analysis of esophageal miR-31 and miR-21 in Zn-treated and Zn-untreated rat esophagus (rat snoRNA as normalizer, two-tailed Welch t test. Error bars represent SD; n = 8 rats per group). (E) Zn treatment induces upregulation of tumor suppressor target PDCD4 (miR-21) and STK40/EGLN3 (miR-31). Black arrowheads in H&E-stained section indicate the presence of apoptotic cells in esophageal epithelium; black arrowheads in near serial PDCD4-stained sections showing abundant/intense nuclear overexpression of PDCD4 in Zn-treated esophagus, compared with sporadic occurrence of apoptotic cells (H&E section) and PDCD4 nuclear expression in isolated/sporadic cells of a Zn-untreated rat (white arrowheads). Representative photos of Zn-treated (n = 21) vs. Zn-untreated (n = 10) esophagus showing H&E staining, IHC staining for STK40, & EGLN3 (brown, 3,3′-diaminobenzidine tetrahydrochloride, DAB) and PDCD4, & PCNA (red, 3-amino-9-ethylcarbazole substrate-chromogen, AEC).
At intervention endpoint, Zn-untreated and pair-fed Zn-treated rats had similar body weights. The level of serum Zn in Zn-treated rats rose significantly from a ZD level of 46 to 159 µg/100 mL, and testis Zn rose from a ZD level of 155 to 211 µg/g (Fig. 2B). Zn treatment resulted in a significant decline in tumor multiplicity (5.3 vs. 14, P < 0.0001) and decline in large tumor (>2 mm) incidence (29% vs. 90%, P = 0.002) (Fig. 2C). Histological examination revealed that Zn therapy reduced ESCC incidence from 90 to 43%, a drop of 47% (P = 0.02). Histologically, Zn-treated rats demonstrated a thinned esophageal epithelium without the characteristic hyperkeratosis of the ESCC-bearing Zn-untreated rat esophagus (H&E-stained sections, Figs. 2E and 3A). Also, Zn-treatment led to a thinned epithelium with PCNA-positive nuclei largely restricted to the basal cell layers (PCNA-IHC, Zn-treated rat 81, 82, and 89 vs. Zn-untreated rat; Figs. 2E and 3B). By contrast, Zn-untreated rats displayed hyperplastic esophageal epithelium with abundant PCNA-positive S-phase nuclei in basal/suprabasal cell layers and tumor areas, evidence of uncontrolled cell proliferation. Thus, Zn treatment effectively eliminates esophageal tumors, and the Zn-treated esophagus assumes an overall non-proliferative phenotype. More prolonged Zn nutritional supplementation would have eliminated ESCCs.
Fig. 3.
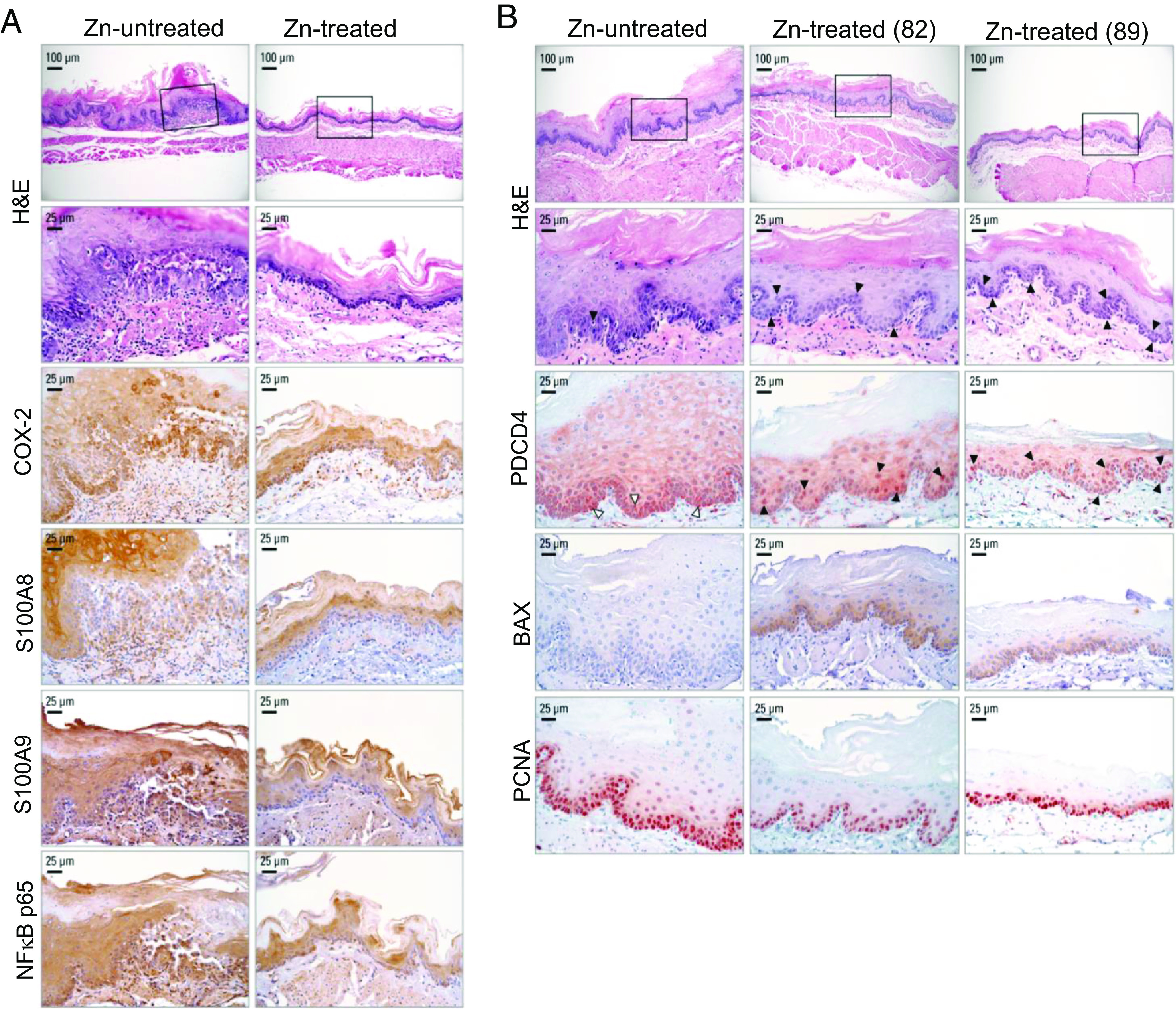
Zn treatment in ESCC-bearing ZD esophagus abolishes inflammation and promotes apoptosis. (A) Zn treatment abolished NF-κB p65-controlled inflammation network. Representative photos of Zn-untreated rats showing strong/abundant IHC staining for all four inflammation markers COX-2, S100A8, S100A9, and NF-κB p65 (brown, 3,3′-diaminobenzidine tetrahydrochloride, DAB) in ESCC bearing hyperplastic esophagus, compared with moderate/diffuse IHC staining for the same inflammation markers in non-proliferative Zn-treated esophagus. (B) Zn treatment triggers apoptosis in Zn-treated esophagus. Black arrowheads in H&E-stained section indicate the presence of apoptotic cells in esophageal epithelium; black arrowheads in near serial PDCD4-IHC stained sections showing abundant/intense nuclear overexpression of PDCD4 in Zn-treated esophagus compared with sporadic occurrence of apoptotic cells (H&E section) and PDCD4 nuclear expression in isolated/sporadic cells of a Zn-untreated rat (white arrowheads). Two esophageal samples from rats 82 and 89 were presented. Representative photos of Zn-treated vs. Zn-untreated esophagus showing H&E staining, IHC staining for PDCD4 (red, 3-amino-9-ethylcarbazole substrate chromogen, AEC), BAX (brown, 3,3′-diaminobenzidine tetrahydrochloride, DAB), and PCNA (red, AEC).
Zn Treatment Reduces miR-31-Controlled Inflammatory Pathways and Promotes miR-21-PDCD4 Axis Apoptosis.
qPCR analysis showed that after 33 d of Zn medication miR-21 and miR-31 levels were reduced by 63% and 66%, respectively (Fig. 2D, P < 0.01), events accompanied by upregulation of their corresponding tumor suppressor target protein, viz PDCD4 (miR-21), STK40/EGLN3 (miR-31), as revealed by IHC. Thus, Zn-treated esophageal epithelia cells showed strong to moderate immunostaining for PDCD4 and STK40/EGLN3 protein vs. weak to absent immunostaining for the same proteins in ESCC-bearing Zn-untreated counterpart (Fig. 2E). Thus, by knocking down critical ESCC-associated miR-31 and miR-21, Zn restored the protein expression of their respective tumor suppressor targets.
Notably, IHC analysis in near serial sections revealed strong nuclear expression of PDCD4 in numerous esophageal epithelia apoptotic cells in Zn-treated esophagi [PDCD4-IHC and H&E-stained sections—Fig. 2E (rat 81) and 3B (rat 82, 89)] vs. absence of PDCD4 nuclear expression in the esophagus of ESCC-bearing Zn-untreated esophagus that showed only sporadic occurrence of apoptotic cells (PDCD4-IHC and H&E-stained sections, Zn-untreated rats—Figs. 2E and 3B). In addition, consistent with previous findings (15), miR-31 overexpression in ESCC-bearing Zn-untreated esophagus (Fig. 2D) was associated with downregulation of EGLN3 and STK40 proteins (Fig. 2E) and accompanying upregulation of the entire the miR-31-EGLN3/STK40-NF-κB controlled COX-2/S100A8/S100A9 inflammation pathway as evidenced by strong/abundant IHC staining of the four inflammation markers (Fig. 3A). Expectedly (15), knockdown of miR-31 in Zn-treated esophagus upregulated EGLN3 and STK40 protein expression (EGLN3 and STK40, IHC analysis; Fig. 2E) and suppressed the same NF-κB p65-controlled COX-2/S100A8/S100A9 inflammation network (IHC analysis, Fig. 3A). Thus, by knocking down critical ESCC-associated miR-21 and miR-31, Zn restored the protein expression of their respective tumor suppressor targets, PDCD4 and EGLN3/STK40, thereby promoting apoptosis and suppressing inflammation.
We first reported (35) in 2001 that Zn replenishment of ZD rats exposed to a single NMBA dose rapidly induces apoptosis in esophageal epithelial cells and reduces the development of esophageal papillomas. Here in ESCC-bearing ZD rats, Zn-treatment knocked down miR-21, an anti-apoptosis miR in the esophagus, restoring the nuclear expression of its tumor suppressor target PDCD4, activated BAX protein expression, and triggering apoptosis in esophageal epithelial cells. Notably, 21 of 21 (100%) Zn-treated rat esophageal epithelia exhibited an apoptotic phenotype with numerous suprabasal and basal cells undergoing apoptosis in accordance with the morphological criteria of apoptosis (33) (H&E, Figs. 2E and 3B, representative esophagus from three Zn-treated rats, 81, 82, and 89). By contrast, 10 of 10 (100%) Zn-untreated ESCC-bearing rats showed only a few sporadic/isolated apoptotic cells in the proliferative esophageal epithelia (H&E, Figs. 2E and 3B, representative esophagus from two Zn-untreated rats).
As in the apoptotic antimiR-31 and antimiR-21 rat esophagus (Fig. 1F and SI Appendix, Fig. S1), Zn-treated rat esophagus displayed strong nuclear expression for PDCD4 in the numerous esophageal epithelial cells undergoing apoptosis (PDCD4, IHC; Figs. 2E and 3B), an event accompanied by strong cytoplasmic immunostaining for BAX protein (BAX-IHC, Fig. 3B). By contrast, Zn-untreated rat esophagus had only sporadic occurrence of apoptotic cells with PDCD4 nuclear expression (PDCD4, IHC; Figs. 2E and 3B) and absence of BAX expression (BAX-IHC, Fig. 3B). These in vivo data provide strong evidence of the mechanism by which Zn treatment triggers apoptosis and eliminates ESCC cells.
Zn Nutritional Supplementation Reverses Cancer-Associated Metabolic Reprogramming.
The ESCC-bearing ZD rats used for the current Zn-therapy study showed human ESCC-associated metabolic changes (15), with decreased levels of glycolytic intermediates and increased levels of anabolic/biosynthetic intermediates, changes pointing to a classic cancer cell remodeling of energy and nucleoside metabolism (38). We determined if Zn intervention, that reduced ESCC incidence from 90 to 43% (Fig. 2C), would induce metabolic changes to reverse the ESCC-associated metabolome (15). Using gas chromatography time-of-flight mass spectrometry GC-TOF MS (15), we performed untargeted metabolomic profiling on esophageal mucosa of Zn-treated vs. Zn-untreated rats (n = 10 rats per cohort). Thirty-eight significantly altered metabolites (24 downregulated, 14 upregulated, P < 0.05) were identified in the Zn-treated vs. Zn-untreated esophagus (Fig. 4A and SI Appendix, Table S1). Of the 24 downregulated metabolites, 15 (63%) were involved in anabolic/biosynthetic pathways, including amino acid/pyrimidine/purine metabolism and polyamine biosynthesis. Putrescine (intermediate in polyamine biosynthesis), shown to be up 6.4-fold in ESCC-bearing ZD esophagus (15), was downregulated −3.96-fold in Zn-treated esophagus (Fig. 4B and SI Appendix, Table S1). Ornithine decarboxylase (ODC) is the rate-liming enzyme in the polyamine biosynthetic pathway to form putrescine, which is converted into spermidine and spermine. Polyamines are indispensable for cell proliferation/tumor growth, and depletion of polyamines inhibits tumor growth (39). Using matrix-assisted laser desorption ionization mass spectrometry imaging, Sun et al. (40) reported that the polyamine-biosynthetic pathway was significantly altered in human ESCC, and IHC analysis of the metabolic enzyme ODC was higher in ESCC tissue than in paired normal tissue. Our IHC analysis showed strong/abundant cytoplasmic staining for ODC in cancerous Zn-untreated esophagus vs. weak/diffuse ODC staining in non-proliferative Zn-treated esophagus (Fig. 4C). Thus, Zn-treated esophagus had a significantly reduced immunoreactive score of ODC compared to Zn-untreated esophagus (Fig. 4D).
Fig. 4.

Zn treatment reverses ESCC metabolic phenotype in ZD rats. (A) Metabolomic profiling by GC-TOF MS on Zn-treated vs. Zn-untreated rat esophagus. A metabolomic network among the structurally identified metabolites was constructed (using the KEGG databases and PubChem compound identification [CID]) showing 14 up- and 24 downregulated metabolites. Each labeled node represents an identified and significantly altered metabolite (P < 0.05, t test; n = 10 rats per group). Blue and red nodes are significantly decreased and increased metabolites. The size of the nodes represents the size of the fold-change (Zn-treated vs. untreated). Blue lines are edges connected due to Tanimoto similarity; red lines are edges connected through KEGG reaction pairs. (B) Box and whisker plot (with median denoted by the line through the box and whiskers representing values within 1.5 IQR of the first and third quartile) showing distribution of putrescine and glucose metabolic activities and significantly reduced putrescine (P = 0.02) and increased glucose levels (P = 0.02) in Zn-treated vs. Zn-untreated esophagus. (C) IHC analysis of metabolite enzymes ODC (rate-limiting enzyme that converts ornithine to putrescine) expression and HK2 (rate-limiting enzyme and first obligatory step of glucose metabolism) expression in Zn-treated vs. Zn-untreated rat esophagus Representative esophageal photo showing moderate to strong immunostaining for ODC and HK2 in Zn-untreated esophagus as compared to weak and diffuse staining for the same protein in Zn-treated esophagus. HK2 and ODC expression (brown, 3,3′-diaminobenzidine tetrahydrochloride, DAB, counterstain, Harris modified hematoxylin). (D) Box and whisker plot showing distribution of ODC and HK2 immunoreactive score and significantly reduced ODC and reduced HK2 protein expression (P < 0.001) in Zn-treated vs. Zn-untreated esophagus.
A common characteristic of cancer cell metabolism is a high rate of aerobic glycolysis (the Warburg effect) to support the biosynthetic requirement of uncontrolled proliferation (41). This phenomenon has been shown in a variety of human cancers, including ESCC (42) and in our ESCC-bearing ZD rat esophagus (15). Of the 14 metabolites that were significantly upregulated in the Zn-treated esophagus, five (37%) were carbohydrates, including glucose, which is upregulated 3.4-fold (Fig. 4B and SI Appendix, Table S1), pointing to a decreased uptake of glucose and a reversal of the Warburg effect after Zn treatment. Critical to cancer aerobic glycolysis is the metabolic enzyme hexokinase 2 (HK2) that catalyzes the first committed step in glucose metabolism where glucose is phosphorylated to form glucose-6-phosphate. HK2 overexpression accounts for the high glycolytic rate in cancer cells (43). Additionally, HK2 is overexpressed in human cancers (44), including ESCC (45). Our IHC analysis of HK2 expression showed strong/abundant cytoplasmic staining for the metabolic enzyme HK2 in cancerous Zn-untreated esophagus vs. weak/diffuse HK2 staining in nonproliferative Zn-treated esophagus (Fig. 4C), producing a significantly reduced immunoreactive score for HK2 in ZD-treated vs. Zn-untreated esophagus (Fig. 4D).
To understand the inter-relationship of the metabolites that were significantly altered in response to Zn intervention, we performed chemical similarity enrichments analysis (ChemRICH) (46) and found that Zn-treated esophagus with reduced tumor burden was associated with increased hexoses levels (glucose, fructose, mannose) and decreased levels of biosynthetic compounds, including sugar acids (glyceric acid, saccharic acid), hydroxy acids (phenylpyruvate, 3-phenyllactic acid) (SI Appendix, Fig. S2 and Table S2A), and biogenic polyamines (putrescine) (SI Appendix, Table S2A). Thus, Zn therapy resulted in a reversal of the classic cancer cell metabolic phenotype of increased glycolysis and increased biosynthetic intermediates.
Discussion
Dietary ZD is associated with an increased risk of developing ESCC in humans (3), and overexpression of miR-31 (18) and miR-21 (19) is implicated in the pathogenesis of ESCC. Our rat model that recapitulates features of human ESCC, including ZD, miR-31/-21 overexpression, inflammation, and a cancer metabolome (3, 18, 19, 40, 47, 48), provides an opportunity to unravel the molecular mechanisms of oncogenic Zn-regulated miR-31/-21 in ESCC. In previous studies, we had focused on mechanisms through which Zn regulates miR-31 to promote ESCC (15, 17). In a ZD-promoted rat ESCC model with miR-31 controlled inflammation pathway upregulation, systemic antimiR-31 reduced miR-31 level and the miR-31-EGLN3/STK40-NF-κΒ controlled inflammatory process, suppressing ESCC development, and miR-31 gene knockout abrogated development of ESCC (15).
In the current study, we have now determined the effectiveness of successive delivery in vivo of two antimiRs, viz antimiR-31 followed by antimiR-21, to suppress ESCC development in a high ESCC burden ZD rat model (13–15). Importantly, antimiR-21 (15-wk) delivery in vivo elicited a >99% knockdown of miR-21 that effectively upregulated its direct tumor suppressor target PDCD4, thus triggering massive waves of apoptosis in the same esophageal epithelial cells that overexpressed nuclear PDCD4. Concurrently, a short-term delivery (5 wk) of antimiR-31 (assessed 15 wk later) led to only moderate cytoplasmic staining for the direct tumor suppressor targets STK40/EGLN3 of miR-31 in antimiR-31 and antimiR-21 esophagus, indicating that the miR-31 tumor suppressor targets were not robustly upregulated, though as noted in the introduction and the above preceding paragraph, long-term antimiR31 had shown very robust upregulation of the entire miR-31 inflammation signal pathway. Since a short-term antimiR-31 delivery had no effect on suppressing ESCC development (Fig. 1F), the resulting data showing that antimiR-21 delivery strongly upregulated the miR-21-PDCD4 axis apoptosis, argues that the observed ESCC suppression from 85 to 27% is mostly driven by antimiR-21 delivery, a new finding in this model echoing a previous report by Fassan et al. (36) in human ESCC tissues that loss of immunohistochemical expression of PDCD4 protein in the nucleus, and thus loss of PDCD4 induced apoptosis, is associated with unfavorable cancer outcome.
Studies in rodents and humans have shown that Zn has a cytotoxic effect on cancer cells without any harmful effects on normal cells; thus, Zn therapy for cancer has received renewed interest (49). Here, we demonstrate for the first time that in ZD rats with established ESCC, Zn treatment successfully eliminates ESCC by exerting broad biological effects. Notably, Zn reduces Zn-regulated miR-31 and miR-21 levels and restores expression of tumor-suppressor proteins targeted by these specific miRs, thus strongly suppressing the miR-31-EGLN3/STK40-NF-κB–controlled inflammatory pathway (Fig. 3A), as previously described with prolonged delivery antimiR-31 (20 wk), as well as vigorously stimulating the miR-21-PDCD4-Bax axis apoptosis (Figs. 2E and 3B), a very important hallmark of tumor suppression in many cancers, including human ESCCs. The functional link among oncogenic miR-31, EGLN3/STK40 downregulation, and inflammation was previously documented in human ESCC tissues by in situ hybridization and IHC (15). We did not study combined Zn medication with anti-miRs treatment in our rat model because it would have been prohibitively expensive, due to cost of the anti-miR oligos.
Zn treatment also leads to reversal of dysregulated tumor associated metabolic pathways, including the biosynthetic polyamine pathway with decreased putrescine levels and aerobic glycolytic pathway with an increase in glucose levels, driven by downregulation of respective metabolic enzymes ODC and HK2 (Fig. 4), also hallmarks of many cancers, though we have not yet we have not been able to link miR-31 or miR-21 dysregulation to metabolome dysregulation through mRNA profiling. Our prior work, by integration of metabolomics, transcriptomics, and miRNA expression profiling did reveal a miR-143-HK2-glucose network that underlies ZD-associated esophageal preneoplasia (45).
A limitation of this study is the absence thus far of well-designed human clinical trials showing efficacy of Zn supplementation in preventing cancer development or reducing cancer burdens in ESCCs, oral cancers, or skin cancers where animal models (13, 28, 50) have suggested efficacy. It seems this would be especially useful for the skin cancers predictably developed in immunosuppressed transplant patients. One cannot help but wonder if the simplicity involved in such a study, where addition of Zn supplementation (commercially easily and inexpensively available) as an additional arm to a planned clinical trial, with few side effects, could be unattractive because the resulting treatment, if eventually approved by regulatory agencies, would not generate large profits. Because this ESCC study also suggests that apoptotic and inflammation pathway targets should be pursued for treatment of this common, almost universally lethal cancer, we believe this model study may inspire research toward development of such future clinical trials.
In a phase 1 pilot study to determine whether the chemopreventive activity of Zn in rodents could be extended to humans with Barrett’s esophagus (BE), Valenzano et al. (51) placed patients with a prior BE diagnosis on oral Zn gluconate (26.4 mg, twice a day, for 14 d) or a placebo. Esophageal mucosal BE biopsies found Zn-induced mRNA changes for multiple transcripts, including downregulation of transcripts encoding proinflammatory proteins, upregulation of anti-inflammatory mediators, and downregulation of transcripts mediating epithelial-to-mesenchymal transition. MiRNA arrays showed significant upregulation of miRs with tumor suppressor activity. When these data are considered collectively, a cancer chemopreventive action by Zn in Barrett’s metaplasia may be possible. The BE results and our ZD ESCC model studies argue for a prospective clinical trial for this safe, easily administered, inexpensive micronutrient that could demonstrate human chemopreventive action by Zn, given that ESCC patients of various ethnicities exhibit decreased plasma/serum Zn level (52–54), indicative of ZD in ESCC patients. Taken together, these results could have significant relevance for human health.
Materials and Methods
Rats, Diets, and Carcinogen.
Weanling male Sprague-Dawley rats (55 ± 5 g) were from Taconic Laboratory. Custom-formulated ZD and ZS diets (Harlan Teklad) were identical except for the amount of Zn, which was 3 to 4 ppm for ZD and ~60 ppm for ZS diet. NMBA was from Midwest Research Institute. Animal protocols were approved by the Thomas Jefferson University Animal Care and Use Committee.
In Vivo LNA™ miRNA Inhibitors.
In vivo grade, custom miRCURY™ LNA-enhanced inhibitor of: rno-miR-31a-5p, rno-miR-21-5p, and Negative Control A were purchased from Exiqon (15). These inhibitor probes were dissolved in sterile PBS at a concentration of 5.2 mg/mL, aliquoted, and stored at −80 °C.
Anticancer Studies: Sequential Delivery of AntimiR-31 Followed by AntimiR-21 in ZD Rat ESCC Model.
As previously described (15), 4-wk-old male Sprague–Dawley rats were a ZD diet ad libitum. The ZD animals received PBS-formulated LNA-antimiR oligonucleotides, forming cohort antimiR-31 and antimiR-21 (n = 10). Over a period of the first 5 wk, this cohort received via tail vein a loading i.v. dose of antimiR-31 (20 mg kg−1), followed by nine maintenance i.v. doses (6 mg kg−1, twice weekly). Starting at week 5, the animals were given four intragastric doses of NMBA (2 mg kg−1, once/week for 4 wk). During and following NMBA administration (15 wk), antimiR-31 and antimiR-21 rats received 20 additional i.v. antimiR-21 doses (6 mg kg−1, twice/wk for 5 wk and then once/week for the remaining 10 wk). The animals were killed 48 h after the final dose of antimiR or 15 wk after the first NMBA dose. Blood was obtained from the retro-orbital venous plexus for serum preparation. Whole esophagus was excised, longitudinally slit open and photographed. Tumors >0.5 mm in diameter were mapped. Esophagi were cut into two equal portions. Esophageal epithelium was isolated, snap-frozen in liquid nitrogen, and stored at –80 °C. The remaining portion was fixed in 10% buffered formalin.
Zn Treatment of ESCC in ZD Rats with Pre-Existing ESCC.
The present study utilized 31 ESCC-bearing ZD male rats that were produced from a previous dietary ZD-promoted rat ESCC study (15) as follows. Weanling male rats on a custom formulated ZD and ZS diets were given four intragastric doses of NMBA at week 5 (2 mg/kg body weight, once per week for 4 wk). At 20 wk, a group of ZD rats and ZS rats were killed for tumor evaluation and 31 ZD rats were reserved for the present study. Expectedly, the overall esophageal tumor incidence was 100% in ZD rats vs. 25% in ZS counterpart; ESCC incidence was 85% in ZD rats, while ZS rats had no cancer. Thus, we expect an ~80% ESCC incidence in the 31 ZD rats.
Zn Medication.
Zn medication was administered in the form of Zn gluconate (a widely used dietary supplement) in deionized drinking water. Thus, 31 ZD animals were randomly divided into 2 groups, Zn-untreated (n = 10) and Zn-treated (n =21) that were administered deionized water supplemented with 0 and 25 ppm Zn, respectively. Also, Zn-untreated rats were continued on a ZD diet and Zn-treated rats were switched to ZS diet. Based on a daily water intake of ~22 mL, we estimated that the daily Zn intake by a 390 g animal (average body weight at study termination) from the 25 ppm Zn-supplemented water was 0.55 mg. This Zn dose translates to a human supplementation of 101 mg Zn/day for a 70 kg individual, a dose comparable to those used for Zn supplement in commercial preparations. The rats were monitored daily for signs of ill health and weighed weekly. After 5 wk of Zn intervention, the animals were killed for tumor incidence analysis as described above.
RNA Isolation.
Total RNA was extracted from the pulverized esophageal mucosal samples using RNA extraction Kit (Norgen Biotek #25700). The integrity of RNA was analyzed by Agilent 2100 Bioanalyzer and RNA integrity number was ≥8 for all samples.
TaqMan miRNA Assay.
Reverse transcription (Applied Biosystems) of miRNAs was performed. The qRT-PCR was performed using Taqman miRNA assays, mmu-miR-31 (ID 000185); hsa-miR-21 (ID 000397); endogenous controls snoRNA (ID 001718) and U87 (ID 001712). As an overall quality control, CT values above 35 were excluded from analysis.
IHC.
IHC was performed using primary antibodies for PCNA (clone PC-10, Ab-1, Thermo Scientific), COX-2 (#12282, Cell Signaling), S100A8 (T-1032, BMA, Augst, Switzerland), S100A9 (NB110-89726, Novus Biologicals), NF-κΒ p65 (ab7970, Abcam), STK40 (orb101780, Biorbyt), EGLN3 (orb443107, Biorbyt), PDCD4 (LS-B1388, Lifespan Biosciences, Seattle, WA), BAX (#14796, rabbit mAb, Cell Signaling), HK2 (NBP1-51643, Novus Biologicals), and ODC1 (NBP2-32887, Novus Biologicals). Protein was localized by incubation with 3-amino-9-ethyl-carbazole substrate-chromogen (Dako) or 3,3′-diaminobenzidine tetrahydrochloride (Sigma-Aldrich).
Immunoreactive scores were calculated by multiplying the percentage of positive cells by the grade of staining intensity. The percentage of positive cells was evaluated as follows: 0 = 0 to 5%, 1 = 6 to 25%, 2 = 26 to 50%, 3 = 51 to 75%, and 4 = 76 to 100%. The intensity of immunostaining was graded as follows: 0 = none, 1 = weak, 2 = moderate, and 3 = intense (35).
Apoptosis Analysis.
Apoptosis was assessed by morphologic characterization of cells in sections H&E–stained slides. The morphology of apoptotic cells depends on their stage in the process. Apoptotic morphologies include 1) diffuse cytoplasmic staining with only minimal nuclear condensation, 2) distinct apoptotic bodies resulting from nuclear disintegration, or 3) dense-staining nuclei with normal nuclear structure. All three forms are considered to be equivalent (34).
Metabolomic Profiling by GC-TOF MS.
Frozen rat esophageal mucosa were shipped to the NIH West Coast Metabolomics Center (University of California, Davis). Esophagus tissue was extracted, derivatized, and processed as described (15). For primary metabolites analysis by GC-TOF MS, the cold injection/automatic liner exchange (CIS-ALEX GC-TOF MS, Leco Pegasus IV) was employed using chromatographic and mass spectrometric parameters (15, 55).
ChemRICH.
ChemRICH analysis was performed on metabolomics data as described (46).
Metabolome Network Visualization.
A biochemical and chemical similarity network was calculated for all measured metabolites with Kyoto Encyclopedia of Genes and Genomes (KEGG) and PubChem CIDs, as previously described (15, 55).
Microscopy.
IHC analyses were performed by light microscopy using an Olympus BX51 microscope and photographs taken with a Spot RT3 camera and Spot software v. 5.2.
Zn Measurement.
Serum Zn content was determined using Atomic Absorption Spectrometer Analyst 400 (PerkinElmer).
Statistical Analysis.
ANOVA and Tukey post hoc t tests (or Kruskal–Wallis and pairwise Wilcoxon rans sum test were used for groups with non-normally distributed data) were used to determine whether the differences among three or more groups. For two groups, standard t tests were used to detect differences among two groups. Tumor and ESCC incidence data among the groups was evaluated using an overall Chi-squared test. Individual differences in incidence data were then assessed using the Fisher’s exact test. The Mann–Whitney U test was used to detect significant compounds in metabolomics. All statistical tests were two-sided and were considered significant at P < 0.05. Statistical analysis was performed by R (http://www.R-project.org).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by grants from the NIH (R01CA118560 to L.Y.F., R35CA197706 to C.M.C., and 5P30CA056036 to K.J.S., Kimmel Cancer Center Shared Resources, Thomas Jefferson University); the NIH West Coast Metabolomics Center (U24 DK097154 to O.F.). We thank Professor Stephan Peiper and the Department of Pathology, Anatomy, and Cell Biology, Thomas Jefferson University, for funds to purchase the custom miRCURY™ LNA-enhanced inhibitor of rno-miR-31a-5p, rno-miR-21-5p from Exiqon. We also thank Timothy Flanagan of Medical Media Services, Thomas Jefferson University, for assistance with figure preparation and Joseph Altemus of Office of Animal Resources, Thomas Jefferson University, for assistance with intravenous tail vein injections in the rat.
Author contributions
L.Y.F., K.H., and C.M.C. designed research; L.Y.F., R.J., and O.F. performed research; L.Y.F., K.H., K.J.S., C.R.B., O.F., J.L.F., and C.M.C. analyzed data; and L.Y.F., K.H., O.F., J.L.F., and C.M.C. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: G.A.C., The University of Texas MD Anderson Cancer Center; and R.D.-F., Columbia University Irving Medical Center.
Contributor Information
Louise Y. Fong, Email: louisefong@me.com.
Carlo M. Croce, Email: carlo.croce@osumc.edu.
Data, Materials, and Software Availability
Metabolomics profiling data were deposited in Metabolomics Workbench (56). All other data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Magee P. N., The experimental basis for the role of nitroso compounds in human cancer. Cancer Surv. 8, 207–239 (1989). [PubMed] [Google Scholar]
- 2.McCormack V. A., et al. , Informing etiologic research priorities for squamous cell esophageal cancer in Africa: A review of setting-specific exposures to known and putative risk factors. Int. J. Cancer 140, 259–271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abnet C. C., et al. , Zinc concentration in esophageal biopsy specimens measured by X-ray fluorescence and esophageal cancer risk. J. Natl. Cancer Inst. 97, 301–306 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Dar N. A., et al. , Association between copper excess, zinc deficiency, and TP53 mutations in esophageal squamous cell carcinoma from Kashmir Valley, India–a high risk area. Nutr. Cancer 60, 585–591 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Hashemian M., et al. , Dietary intake of minerals and risk of esophageal squamous cell carcinoma: Results from the Golestan Cohort Study. Am. J. Clin. Nutr. 102, 102–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McClain C. J., Su L. C., Zinc deficiency in the alcoholic: A review. Alcohol. Clin. Exp. Res. 7, 5–10 (1983). [DOI] [PubMed] [Google Scholar]
- 7.Moody S., et al. , Mutational signatures in esophageal squamous cell carcinoma from eight countries with varying incidence. Nat. Genet. 53, 1553–1563 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Giannakis M., Peters U., Esophageal cancer mutational signatures around the world. Nat. Genet. 53, 1522–1523 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Maret W., Zinc and human disease. Met Ions Life Sci. 13, 389–414 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Caulfield L. E., Black R. E., “Zinc deficiency” in Comparative Quantification of Health Risk, Ezzati M., Lopez A. D., Rodgers A., Murray C. J., Eds. (World Health Organization, Geneva, Switzerland, 2004), vol. 1, pp. 257–280. [Google Scholar]
- 11.Vallee B. L., Falchuk K. H., The biochemical basis of zinc physiology. Physiol. Rev. 73, 79–118 (1993). [DOI] [PubMed] [Google Scholar]
- 12.Rink L., Haase H., Zinc homeostasis and immunity. Trends Immunol. 28, 1–4 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Taccioli C., et al. , Dietary zinc deficiency fuels esophageal cancer development by inducing a distinct inflammatory signature. Oncogene 31, 4550–4558 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fong L. Y., et al. , MicroRNA dysregulation and esophageal cancer development depend on the extent of zinc dietary deficiency. Oncotarget 7, 10723–10738 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fong L. Y., et al. , Abrogation of esophageal carcinoma development in miR-31 knockout rats. Proc. Natl. Acad. Sci. U.S.A. 117, 6075–6085 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taccioli C., et al. , Zinc replenishment reverses overexpression of the proinflammatory mediator S100A8 and esophageal preneoplasia in the rat. Gastroenterology 136, 953–966 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taccioli C., et al. , Repression of esophageal neoplasia and inflammatory signaling by Anti-miR-31 delivery in vivo. J. Natl. Cancer Inst. 107, djv220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang T., et al. , The oncogenetic role of microRNA-31 as a potential biomarker in oesophageal squamous cell carcinoma. Clin. Sci. (Lond) 121, 437–447 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Mathe E. A., et al. , MicroRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: Associations with survival. Clin. Cancer Res. 15, 6192–6200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calin G. A., Croce C. M., MicroRNA signatures in human cancers. Nat. Rev. Cancer 6, 857–866 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Croce C. M., Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet 10, 704–714 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fluiter K., et al. , In vivo tumor growth inhibition and biodistribution studies of locked nucleic acid (LNA) antisense oligonucleotides. Nucleic Acids Res. 31, 953–962 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang J., et al. , Identification of a novel serine/threonine kinase that inhibits TNF-induced NF-kappaB activation and p53-induced transcription. Biochem. Biophys. Res. Commun. 309, 774–778 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Xue J., et al. , Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKbeta independent of hydroxylase activity. Gastroenterology 138, 606–615 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Creighton C. J., et al. , Molecular profiling uncovers a p53-associated role for microRNA-31 in inhibiting the proliferation of serous ovarian carcinomas and other cancers. Cancer Res. 70, 1906–1915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu N., et al. , MicroRNA-31 is overexpressed in psoriasis and modulates inflammatory cytokine and chemokine production in keratinocytes via targeting serine/threonine kinase 40. J. Immunol. 190, 678–688 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Alder H., et al. , Dysregulation of miR-31 and miR-21 induced by zinc deficiency promotes esophageal cancer. Carcinogenesis 33, 1736–1744 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fong L. Y., et al. , Zinc supplementation suppresses 4-nitroquinoline 1-oxide-induced rat oral carcinogenesis. Carcinogenesis 32, 554–560 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frankel L. B., et al. , Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 283, 1026–1033 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Asangani I. A., et al. , MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 27, 2128–2136 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Chan J. A., Krichevsky A. M., Kosik K. S., MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 65, 6029–6033 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Elmen J., et al. , LNA-mediated microRNA silencing in non-human primates. Nature 452, 896–899 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Matsuhashi S., Manirujjaman M., Hamajima H., Ozaki I., Control mechanisms of the tumor suppressor PDCD4: Expression and functions. Int. J. Mol. Sci. 20, 2304 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kerr J. F., Winterford C. M., Harmon B. V., Apoptosis. Its significance in cancer and cancer therapy. Cancer 73, 2013–2026 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Fong L. Y., Nguyen V. T., Farber J. L., Esophageal cancer prevention in zinc-deficient rats: Rapid induction of apoptosis by replenishing zinc. J. Natl. Cancer Inst. 93, 1525–1533 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Fassan M., et al. , Programmed cell death 4 protein in esophageal cancer. Oncol. Rep. 24, 135–139 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Zhang H., et al. , Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene 25, 6101–6112 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Ward P. S., Thompson C. B., Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pegg A. E., Polyamine metabolism and its importance in neoplastic growth and a target for chemotherapy. Cancer Res. 48, 759–774 (1988). [PubMed] [Google Scholar]
- 40.Sun C., et al. , Spatially resolved metabolomics to discover tumor-associated metabolic alterations. Proc. Natl. Acad. Sci. U.S.A. 116, 52–57 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Warburg O., On the origin of cancer cells. Science 123, 309–314 (1956). [DOI] [PubMed] [Google Scholar]
- 42.Zhu X., et al. , Metabolic perturbation and potential markers in patients with esophageal cancer. Gastroenterol. Res. Pract. 2017, 5469597 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathupala S. P., Ko Y. H., Pedersen P. L., Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25, 4777–4786 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ciscato F., Ferrone L., Masgras I., Laquatra C., Rasola A., Hexokinase 2 in cancer: A prima donna playing multiple characters. Int. J. Mol. Sci. 22, 4716 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fong L. Y., et al. , Integration of metabolomics, transcriptomics, and microRNA expression profiling reveals a miR-143-HK2-glucose network underlying zinc-deficiency-associated esophageal neoplasia. Oncotarget 8, 81910–81925 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barupal D. K., Fiehn O., Chemical similarity enrichment analysis (ChemRICH) as alternative to biochemical pathway mapping for metabolomic datasets. Sci. Rep. 7, 14567 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mandard A. M., Hainaut P., Hollstein M., Genetic steps in the development of squamous cell carcinoma of the esophagus. Mutat. Res. 462, 335–342 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Tokunaga M., et al. , Metabolome analysis of esophageal cancer tissues using capillary electrophoresis-time-of-flight mass spectrometry. Int. J. Oncol. 52, 1947–1958 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Gelbard A., Zinc in cancer therapy revisited. Isr. Med. Assoc. J. 24, 258–262 (2022). [PubMed] [Google Scholar]
- 50.Sun J., et al. , Reduction in squamous cell carcinomas in mouse skin by dietary zinc supplementation. Cancer Med. 5, 2032–2042 (2016), 10.1002/cam4.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valenzano M. C., et al. , Zinc gluconate induces potentially cancer chemopreventive activity in Barrett’s Esophagus: A phase 1 pilot study. Dig. Dis. Sci. 66, 1195–1211 (2020), 10.1007/s10620-020-06319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mellow M. H., et al. , Plasma zinc and vitamin A in human squamous carcinoma of the esophagus. Cancer 51, 1615–1620 (1983). [DOI] [PubMed] [Google Scholar]
- 53.Hashemi S. M., et al. , The relationship between serum selenium and zinc with gastroesophageal cancers in the southeast of Iran. Indian J. Med. Paediatr. Oncol. 38, 169–172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin H. J., Chan W. C., Fong Y. Y., Newberne P. M., Zin levels in serum, hair and tumors from patients with esophageal cancer. Nutrition Rep. Int. (USA) 15, 635–643 (1977). [Google Scholar]
- 55.Fong L. Y., et al. , Human-like hyperplastic prostate with low ZIP1 induced solely by Zn deficiency in rats. Proc. Natl. Acad. Sci. U.S.A. 115, E11091–E11100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fong L. Y., et al. , ST002585. Metabolomics Workbench. http://dev.metabolomicsworkbench.org:22222/data/DRCCMetadata.php?Mode=Study&StudyID=ST002585. Deposited 26 April 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Metabolomics profiling data were deposited in Metabolomics Workbench (56). All other data are included in the article and/or SI Appendix.