

Significance
The molecular circadian clock regulates daily rhythms in gene expression, cellular function, and animal behavior. Clock proteins can also regulate noncircadian functions that are highly cell-type specific. Regulation of proteostasis in different cell types in the brain to remove misfolded proteins is critical for healthy brain aging. However, the role of clock proteins in the control of degradation pathways has been poorly investigated in the brain. Here, we demonstrate that deletion of the core clock protein BMAL1 leads to increased astrocyte endolysosomal function and extracellular protein degradation. Our findings implicate BMAL1 in astrocyte proteostasis and brain health.
Keywords: astrocyte, circadian, Bmal1, lysosome, autophagy
Abstract
An emerging role for the circadian clock in autophagy and lysosome function has opened new avenues for exploration in the field of neurodegeneration. The daily rhythms of circadian clock proteins may coordinate gene expression programs involved not only in daily rhythms but in many cellular processes. In the brain, astrocytes are critical for sensing and responding to extracellular cues to support neurons. The core clock protein BMAL1 serves as the primary positive circadian transcriptional regulator and its depletion in astrocytes not only disrupts circadian function but also leads to a unique cell-autonomous activation phenotype. We report here that astrocyte-specific deletion of Bmal1 influences endolysosome function, autophagy, and protein degradation dynamics. In vitro, Bmal1-deficient astrocytes exhibit increased endocytosis, lysosome-dependent protein cleavage, and accumulation of LAMP1- and RAB7-positive organelles. In vivo, astrocyte-specific Bmal1 knockout (aKO) brains show accumulation of autophagosome-like structures within astrocytes by electron microscopy. Transcriptional analysis of isolated astrocytes from young and aged Bmal1 aKO mice indicates broad dysregulation of pathways involved in lysosome function which occur independently of TFEB activation. Since a clear link has been established between neurodegeneration and endolysosome dysfunction over the course of aging, this work implicates BMAL1 as a key regulator of these crucial astrocyte functions in health and disease.
A common feature of brain aging and neurodegenerative diseases is the aggregation of misfolded proteins: amyloid beta (Aβ) plaques and tau neurofibrillary tangles in Alzheimer’s Disease, alpha-synuclein Lewy bodies in Parkinson’s Disease, TDP-43 aggregates in Amytrophic Lateral Sclerosis, and others. In addition, patients with neurodegenerative conditions often have a combination of these different protein aggregates, implicating disrupted proteostasis as a potentially common disease mechanism. Thus, cellular mechanisms responsible for maintaining a healthy proteome are critical to preventing toxic aggregation and neurodegeneration. Activated glial cells interact with toxic protein aggregates in neurodegenerative diseases and play a role in their pathogenesis. Direct uptake and intracellular degradation by microglia and astrocytes as well as extracellular clearance by secreted proteolytic enzymes have been demonstrated for tau, Aβ, and alpha-synuclein (1, 2). For example, inducing lysosome biogenesis in astrocytes enhances phagocytosis and degradation of both Aβ and tau and prevents pathology in mouse models (3, 4). However, glia may also potentiate disease through the propagation of pathology and exacerbation of the inflammatory environment (2, 5). Cell–cell transmission of tau has been shown between astrocytes and neurons (6) as well as alpha-synuclein between astrocytes (7). Therefore, identifying mechanisms by which glia prevent or contribute to protein pathology and neurodegeneration has significant implications for combating neurodegenerative disease.
The circadian clock is composed of transcription–translation feedback loops that coordinate daily rhythms in gene expression, physiology, and behavior. The core clock protein BMAL1 (aka ARNTL) is critical for circadian transcription, and Bmal1 deletion abrogates circadian rhythms in cells and mice (8–10). Previous work in our lab has identified BMAL1 as a major regulator of astrocyte function. Deletion of Bmal1 in astrocytes induces a unique cell-autonomous activation state, alters astrocyte gene expression patterns, and impairs their support of neurons in culture (10, 11). This work suggests that various biological processes may be perturbed in the Bmal1-disrupted astrocyte. Accumulating evidence from peripheral cell types has begun to reveal mechanisms by which the core circadian clock might influence astrocyte protein degradation in the brain. For example, in addition to regulating endolysosomal and autophagy genes, the master lysosomal transcription factor, Transcription Factor EB (TFEB), drives expression of the circadian clock component REV-ERBα, which in turn inhibits TFEB’s own target genes and helps to drive rhythms in these processes (12). Similarly, the core clock has been bidirectionally linked to nutrient sensing through the autophagy and endocytosis regulator mTOR. BMAL1 represses mTOR activity (13), and in turn, mTOR activity leads to phosphorylation of BMAL1 by the downstream regulator S6K1 (14). Autophagy is under clock control and exhibits circadian rhythms in the brain and periphery. For example, chaperone-mediated autophagy exhibits BMAL1-dependent rhythms in the liver, heart, and kidney (15). These rhythms differ between these tissues and are likely to also differ from the brain and even between different neural cell types. Expression and processing of LC3, a marker of autophagy, exhibit circadian rhythms in the hippocampus, but this has not been explored in depth in other brain regions or in a cell-type-specific manner (16). Considering the multitude of genes regulated by the clock—with over 43% of protein-coding genes exhibiting rhythmic transcription in mice (17) and 80% of genes rhythmically expressed in one or more tissues in primates (18)—it is likely that many genes involved in proteostasis may be influenced by the circadian clock. The coordination of these transcriptional programs by the clock may translate short-term rhythms in protein degradation to longer-term effects on diseases of toxic aggregation.
The goal of the current study was to investigate the role of BMAL1 in regulating the endolysosomal system of astrocytes. We report here that deletion of Bmal1 in murine astrocytes in vitro and in vivo enhances endocytosis, lysosomal activity, and autophagy in a manner independent of TFEB. These findings suggest that BMAL1 is a key regulator of astrocyte proteostasis and may mediate astrocyte functions in health and disease.
Results
Bmal1 Deletion Leads to Broad Dysregulation of Endolysosomal Transcriptional Pathways.
We have previously developed and characterized mice with inducible Cre-driven astrocyte-specific excision of Bmal1 (Aldh1l1-CreERT2;Bmal1fl/fl, termed Bmal1 aKO) and have shown that these mice develop spontaneous astrocyte activation (10). While we have previously reported transcriptional changes in bulk cortex tissue from Bmal1 aKO mice, here, we investigated the astrocyte-specific transcriptional changes in Bmal1 aKO brains by performing RNA sequencing on isolated astrocytes from adult mice. Magnetic bead sorting allowed us to collect whole-brain astrocytes from young adult (~7 mo) and aged (~20 mo) Bmal1 aKO mice (and Cre- littermate controls), all of which were treated with tamoxifen at 2 months of age to induce Bmal1 deletion (SI Appendix, Fig. S1A). We used the EdgeR, Limma, and Voom with Quality Weights packages to normalize and filter data before applying linear modeling and the empirical Bayes method to test for differentially regulated genes (19). Cell-type specific markers confirmed enrichment of astrocyte genes and dilution of microglial genes in astrocyte samples, although some contamination from neurons and oligodendrocytes was present (SI Appendix, Fig. S1B). The markers for Bmal1-disrupted astrocytes that we previously reported (10, 11), such as Fabp7 and Cxcl5, were again altered as expected in the Bmal1 aKO astrocytes, demonstrating effective gene deletion and reliable cellular isolation and gene expression analysis (Fig. 1A). Differentially expressed gene lists with cutoffs of FDR < 0.15, unadjusted P < 0.01, and unadjusted P < 0.05 can be found in Dataset S1. Due to the wide diversity of dysregulated genes in Bmal1 aKO, we performed GO pathway analysis to evaluate pathway disruption in the absence of Bmal1. Gene set enrichment was performed with the Generally Applicable Gene-set Enrichment (GAGE) package using the mouse Go.db annotation package for gene sets. Analysis of the top 20 upregulated GO cellular component pathways indicated several terms associated with vesicle and lysosome function in both young and aged astrocytes, suggesting a persistent effect on lysosomal genes throughout aging (Fig. 1B).
Fig. 1.
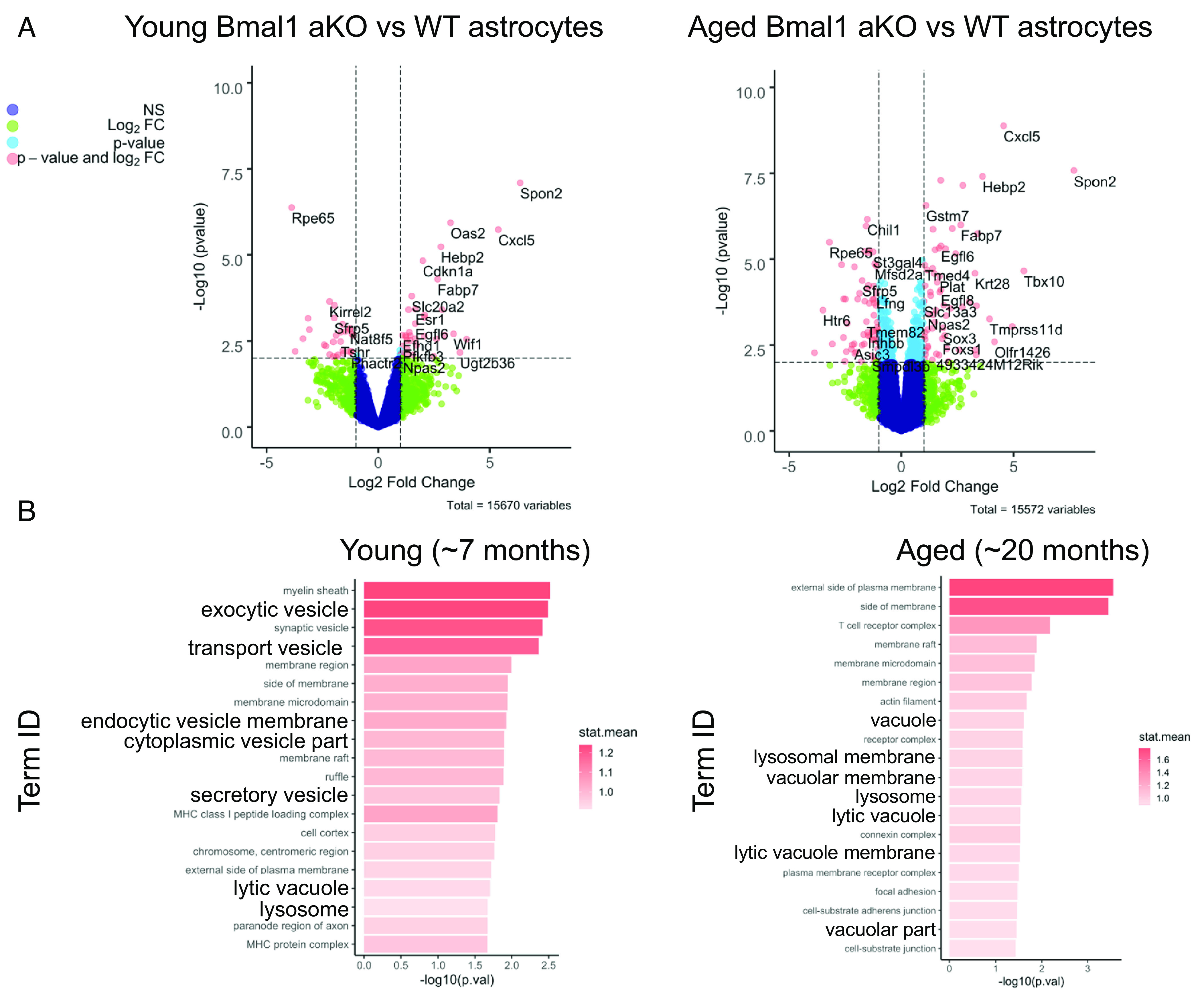
Bmal1 deletion in astrocytes induces upregulation of endolysosomal pathways. RNAseq analysis comparing isolated astrocytes from control (Cre−) and Bmal1 aKO brains. Young mice (n = 3 control, n = 5 Bmal1 aKO) were harvested at 7 mo of age and aged mice (n = 4 per group) were harvested at 20 mo of age. (A) Volcano plots showing differential gene expression comparing young (Left) or aged (Right) astrocytes from control and Bmal1 aKO brains (Log2 fold change cutoff = 1.0, −Log10pvalue cutoff = 2.0, corresponds to P value of 0.01). (B) Top 20 upregulated GO cellular component pathways obtained from GAGE pathway analysis. Pathways involved in the endolysosome system are in bold.
We analyzed the 10 most differentially expressed genes within the “lysosome” GO pathway but did not find any large-magnitude changes in specific lysosomal genes such as lysosomal proteases or membrane proteins. We found both up- and down-regulated genes, although individually these were not significantly different by a cutoff of FDR < 0.15 except for Litaf, Naaa, and Marcks in the aged astrocytes (SI Appendix, Fig. S2A). In both young and aged Bmal1 aKO mice, Akr1b8 was down-regulated and Litaf was up-regulated. Interestingly, Litaf autosomal dominant mutations cause the demyelinating Charcot-Marie-Tooth disease type 1C and have been linked to endolysosomal dysfunction in patient fibroblasts (20). Litaf has also been implicated in the induction of autophagy (21). We conclude that Bmal1 deletion induces modest fold changes in a number of lysosome-related genes, suggesting a broad transcriptional response as opposed to strong, direct regulation of a few key genes. These changes could represent direct regulation by BMAL1 but may more likely represent a compensatory response to the shift in overall astrocyte health as a result of Bmal1 deletion. Thus, we focused our attention on the functional consequences of Bmal1 deletion rather than the transcriptional mechanism by which it regulates lysosomal genes.
Astrocytes Lacking Bmal1 Exhibit Elevated Levels of Lysosome Markers.
In order to understand the role of BMAL1 in astrocyte lysosome function, we next investigated how BMAL1 affects lysosome abundance and activity. We have previously reported (10) and shown here (SI Appendix, Fig. S3A) that siRNA-mediated knockdown of Bmal1 in primary postnatal mouse astrocytes (siBmal1) recapitulates many of the changes to clock gene expression seen in vivo and, therefore, used this system for functional assays. We observed a striking increase in Lysotracker signal in live siBmal1 astrocytes (Fig. 2 A and B), suggesting a shift to greater lysosomal abundance. Due to the role of BMAL1 in generating rhythms as part of the circadian clock, we investigated whether this observation is due to a shift in lysosomal rhythms as a result of deleting Bmal1 in astrocytes. We synchronized the circadian clocks of astrocytes by media change as previously described (22) and collected cells at 6-h intervals across their circadian cycle. We observed that Lysotracker signal did not appear rhythmic in either siScramble (control) or siBmal1 astrocytes (Fig. 1C). In addition, increased Lysotracker signal is consistently observed across the circadian cycle in siBmal1 astrocytes. We confirmed that media change did result in rhythms in clock gene expression (Bmal1 and Nr1d1) in astrocytes collected for RNA during these same experiments (SI Appendix, Fig. S4A). Thus, deletion of Bmal1 appears to increase lysosomal abundance in a noncircadian manner in rhythmic astrocytes.
Fig. 2.
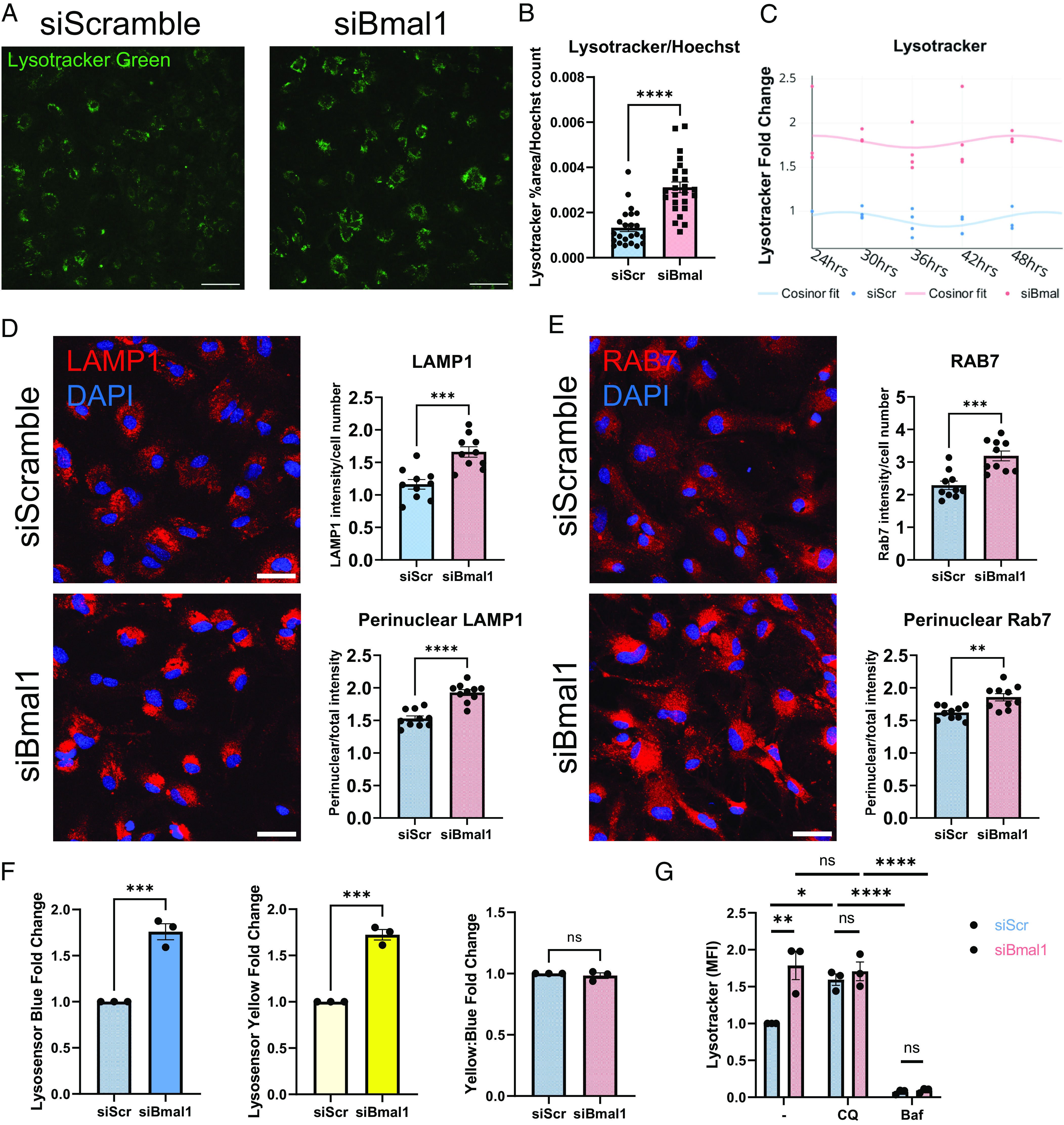
Astrocytes lacking Bmal1 exhibit elevated levels of lysosome markers. (A) Lysotracker Green in Bmal1 knockdown and scramble siRNA-treated astrocytes. (Scale bars, 100 µm.) (B) Lysotracker %area above threshold per image, normalized to Hoechst cell count per well. N = 23 to 25 wells per condition, representative of three independent experiments. **** = P < 0.0001 by t test. (C) Flow cytometry of Lysotracker in astrocytes collected at listed time points after synchronization by media change. N = 3 to 4 pups over two independent experiments (some overlapping points not visible), MFI values normalized to each pup’s siScramble 24-h value. (D and E) Confocal imaging and quantification for LAMP1 and RAB7 in siBmal1 and siScramble-treated astrocytes. N = 10 wells per condition, representative of three independent experiments. (Scale bars, 50 µm.) * = P < 0.05, ** = P < 0.005, *** =P < 0.0005, **** =P < 0.0001 by t test. (F) Flow cytometry of Lysosensor Yellow/Blue DND-160 in siBmal1 and siScramble-treated astrocytes. N = 3 independent experiments, MFI values normalized per experiment to siScramble controls. *** =P < 0.0005 by t test. (G) Flow cytometry of Lysotracker in siScramble and siBmal1 astrocytes treated for 3 h with either serum-free medium, 20 µM chloroquine (CQ), or 50 nM Bafilomycin (BAF). N = 3 independent experiments, MFI values normalized per experiment to serum-free media-treated siScramble controls. * =P < 0.05, **** =P < 0.0001 by two-way ANOVA with Sidak’s multiple comparisons test.
As a pH-sensitive dye, Lysotracker accumulates and fluoresces in any acidic cellular environment, including lysosomes and late endosomes. We hypothesized that an increase in this signal must be due to either an increase in abundance or acidification of acidic organelles. We therefore stained for the lysosome membrane protein LAMP1 and the late endosome marker RAB7 and observed increases in both LAMP1 and RAB7 intensity in siBmal1 compared to siScramble astrocytes (Fig. 2 D and E). The localization of LAMP1 and RAB7 also became more condensed in a perinuclear distribution in siBmal1 astrocytes. As endolysosomal and autophagic organelles move from the cell periphery toward the nucleus in order to fuse with lysosomes and degrade cargo (23), these data suggest increased retrograde transport of these organelles and fusion with lysosomes in siBmal1 astrocytes.
Next, we examined the effect of Bmal1 knockdown on the acidification of astrocyte lysosomes using Lysosensor Yellow/Blue, a dual fluorescent ratiometric indicator of acidic and neutral pH within lysosomes (24). After applying Lysosensor to astrocytes, we used flow cytometry to measure the fluorescence of acidic (yellow) and neutral (blue) lysosomes. Similar to our Lysotracker measurements, siBmal1 astrocytes showed an increase in both the acidic and neutral lysosome signal by Lysosensor, but the pH ratio remained unchanged (Fig. 2F). We also explored the effect of the lysosome inhibitors chloroquine and bafilomycin in this system. At high concentrations, these drugs both block lysosomal acidification: Chloroquine directly enters the lysosome and acts as a base (25, 26), and bafilomycin A1 directly inhibits the V-ATPase to block lysosomal acidification (27) (both also block autophagosome fusion with lysosomes which will be addressed later). However, at a lower concentration, chloroquine can actually induce lysosomal biogenesis through activation of TFEB family transcription factors (28). Both chloroquine and bafilomycin also block the fusion of lysosomes with other organelles and degradation of their components, which can lead to the accumulation of these organelles and their substrates. We found that siScramble astrocytes exhibit increased Lysotracker signal with chloroquine treatment, indicating either increased lysosomal biogenesis or accumulation of alkalized organelles (due to inhibited fusion with autophagosome). Treatment with bafilomycin completely eliminated Lysotracker signal, indicating deacidification (Fig. 2G). Bafilomycin also eliminated Lysotracker signal in siBmal1 astrocytes; however, chloroquine treatment did not significantly affect Lysotracker in siBmal1 astrocytes. This observation in the chloroquine-treated astrocytes suggests that either lysosomal biogenesis in siBmal1 astrocytes is already at its upper limit (and that choloroquine increases lysosomal biogenesis in siScramble cells to reach a similar limit) or that endolysosomal fusion is already significantly blocked in siBmal1 cells such that chloroquine cannot increase this. Due to these complex possibilities, we chose bafilomycin as the lysosomal inhibitor treatment for the remainder of our experiments.
Bmal1 Deletion in Astrocytes Boosts Extracellular Protein Uptake and Degradation.
While our Lysotracker, LAMP1, RAB7, and Lysosensor experiments suggest increased lysosomal abundance in siBmal1 astrocytes, it remained unclear if this leads to increased functional protein uptake and degradation. To address this, we next turned to assays of protein uptake and degradation to confirm enhanced lysosome function rather than accumulation of dysfunctional lysosomes in siBmal1 astrocytes. We treated siRNA-transfected astrocytes with Alexa Fluor 647-labeled bovine serum albumin (BSA-647) for 3 h and quantified the fluorescence by flow cytometry and imaging as a measure of endocytosis. We found that siBmal1 astrocytes take up more BSA-647 than siScramble cells using both flow cytometry and imaging endpoints (Fig. 3 A–D). This uptake was reduced with bafilomycin (Fig. 3D) indicating that disrupted lysosomal function likely provides feedback to mechanisms that regulate endocytosis. We also observed a stronger decrease in BSA-647 uptake in siBmal1 than siScramble cells when treating with the dynamin inhibitor Dynasore which broadly prevents endocytosis (Fig. 3E). Dynasore treatment abrogated the effect of Bmal1 knockdown on astrocyte uptake of BSA, indicating a strong dependence on dynamin-dependent endocytosis with loss of Bmal1 (Fig. 3E).
Fig. 3.
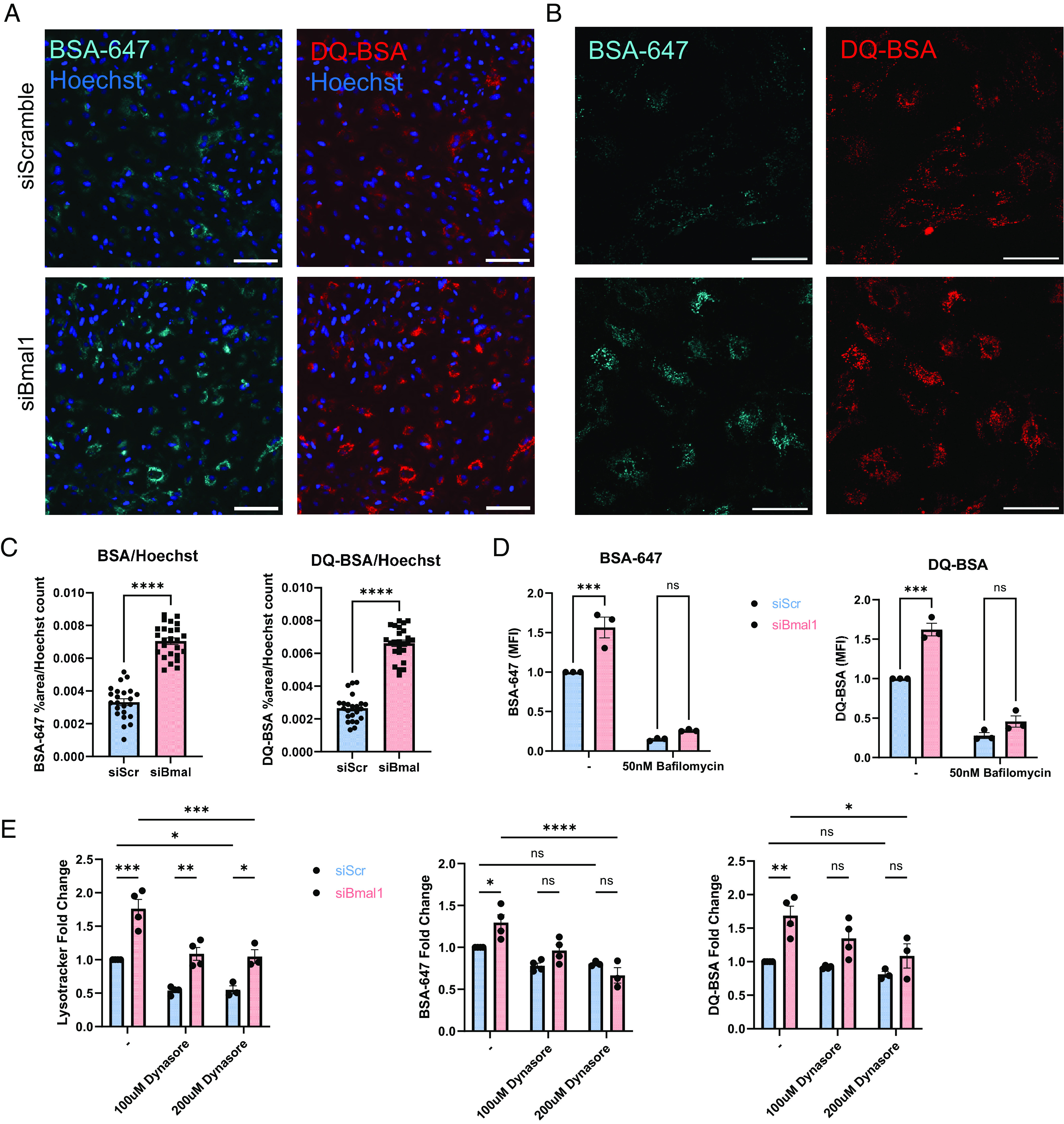
Bmal1 deletion in astrocytes boosts extracellular protein uptake and degradation. (A) 10X epifluorescence live cell imaging of siScramble and siBmal1 astrocytes treated with 1 µg/mL BSA-647 and DQ-BSA for 3 h. (Scale bars, 100 µm.) (B) Example 40X confocal live cell imaging of siScramble and siBmal1 astrocytes treated with 1 µg/mL BSA-647 and DQ-BSA for 3 h. (Scale bars, 50 µm.) (C) Quantification from epifluorescence imaging of BSA-647 and DQ-BSA uptake in live siScramble and siBmal1 astrocytes, %area above threshold normalized to Hoechst cell count per field of view. N = 23 to 25 wells per condition, representative of two independent experiments. (D) Flow cytometry quantification of BSA-647 and DQ-BSA uptake in live siScramble and siBmal1 astrocytes treated with serum-free medium or 50nM Bafilomycin for 3 h. N = 3 independent experiments, MFI normalized per experiment to serum-free media-treated siScramble controls. (E) Flow cytometry quantification of Lysotracker, BSA-647, and DQ-BSA in siScramble and siBmal1 astrocytes treated with either serum-free medium, 100 µM Dynasore, or 200 µM Dynasore for 3 h during BSA incubation. N = 3 to 4 independent experiments, MFI normalized per experiment to serum-free media-treated siScramble controls. (D and E) * =P < 0.05, ** =P < 0.005, *** =P < 0.0005, **** =P < 0.0001 by two-way ANOVA with Sidak multiple comparisons test.
To measure lysosome-dependent protein degradation, we used dye-quenched bovine serum albumin (DQ-BSA), a self-quenching BODIPY dye that fluoresces only upon cleavage in the lysosome and release of the isolated fluorophores (29). Similar to our results with BSA-647, DQ-BSA also shows greater signal in siBmal1 astrocytes by both flow cytometry and imaging, indicating elevated protein degradation capacity in these cells (Fig. 3 A–D). Proteolysis of DQ-BSA was inhibited as expected by bafilomycin and Dynasore treatments which interfere with lysosome and endosome function (Fig. 3 D and E). However, at the 100µM concentration, Dynasore normalized BSA endocytosis and DQ-BSA cleavage to control levels. This suggests that the increased abundance of acidic organelles is the primary effect of Bmal1 knockdown that may feedback onto endocytosis mechanisms to induce greater protein uptake and degradation. Alternatively, Bmal1 depletion may exert effects on lysosome abundance and protein endocytosis via distinct mechanisms. Taken together, these results indicate that both uptake and degradation of proteins through the endolysosomal system are elevated with depletion of Bmal1.
Bmal1 Deletion via Tamoxifen-Induced Cre Recombination Elevates Endolysosomal Function.
In order to confirm that the changes to endolysosomal function we observed are the result of Bmal1 depletion, as opposed to an off-target effect of our siRNAs, we performed the same flow cytometry uptake and degradation experiments as described above with an alternate method of deleting Bmal1. We generated astrocyte cultures from CAG-CreERT2;Bmal1fl/fl and Cre-; Bmal1fl/fl control pups and then treated them in vitro with 4-hydroxy-tamoxifen to induce Bmal1 deletion in Cre+ cells. After 4 to 5 d of tamoxifen exposure, we confirmed efficient deletion of Bmal1 and reduction of its downstream target Nr1d1 by qPCR (SI Appendix, Fig. S5B). We also observed a similar increase in lysotracker signal, BSA-647 uptake, and DQ-BSA cleavage (SI Appendix, Fig. S5A) as in our siRNA-treated astrocytes (Fig. 3 A–D). Thus, we have shown two methods of depleting Bmal1 that both result in increased endolysosomal function.
Bmal1 Deletion in Microglia Mildly Increases Protein Uptake but Not Lysosomal Function.
In order to ascertain whether BMAL1 influences endolysosomal function in microglia in the same manner as in astrocytes, we performed the same siRNA knockdown experiments in primary murine microglia using BSA-647, DQ-BSA, and Lysotracker (SI Appendix, Fig. S6A). Contrary to Bmal1 knockdown in astrocytes, siBmal1 microglia showed no increase in Lysotracker signal and responded the same to chloroquine or bafilomycin treatment as siScramble microglia. BSA-647 uptake was mildly increased in siBmal1 microglia, although at a 1.13-fold increase, this effect was smaller than that in astrocytes. We also confirmed efficient deletion of Bmal1 and reduction of its downstream target Nr1d1 by qPCR (SI Appendix, Fig. S6B). This observation matches previous work showing that Bmal1 deletion in macrophages enhances phagocytic activity (30). We have also reported increased uptake of amyloid-beta in primary microglia with knockdown of Nr1d1/Nr1d2 (Rev-erbα/β), two clock genes which interact with BMAL1 (31). However, we observed no increase in DQ-BSA cleavage in siBmal1 microglia, suggesting that the influence of BMAL1 on lysosomal abundance and function is specific to astrocytes.
Bmal1 Deletion in Astrocytes Does Not Influence TFEB Activation.
Due to the lysosomal pathways identified in our GO analysis and our measures of increased lysosomal markers and endolysosomal function, we were intrigued by the possibility that BMAL1 may control expression of the critical endolysosomal regulator TFEB or its targets. It is already known that another clock protein, REV-ERBα, shares an inverse relationship with TFEB family transcription factors in the induction of autophagy (12). Therefore, we assessed transcriptional differences in TFEB and its family members and targets. Surprisingly, our RNAseq data indicated no differential regulation of these genes in young or aged astrocytes (SI Appendix, Fig. S7A). We also examined TFEB and its family member TFE3 by immunofluorescence in siRNA-treated astrocytes to ascertain whether they became activated and translocated to the nucleus differently with Bmal1 knockdown. When kept in normal growth medium, we observed no difference in the nuclear translocation of TFEB or TFE3 between siScramble and siBmal1 astrocytes (SI Appendix, Fig. S7B). However, with 3 h of serum-free medium treatment, siBmal1 astrocytes showed more nuclear signal for TFEB. In addition, we used Torin1 treatment to inhibit mTOR and activate TFEB and TFE3 as a positive control. Surprisingly, siBmal1 astrocytes showed greater nuclear translocation of TFEB and TFE3 in response to Torin1 treatment than siScramble astrocytes. This suggests that Bmal1 deletion may disrupt some feedback mechanism between mTOR and TFEB or that Bmal1-deficient astrocytes may be more sensitive to mTOR inhibition due to some metabolic dysfunction. However, Bmal1 knockdown at baseline does not change TFEB or TFE3 activation. Thus, we concluded that BMAL1 may regulate the endolysosomal system through a separate mechanism than TFEB as a master regulator of these genes. We believe that this likely involves BMAL1 targeting many separate genes in these pathways as a transcription factor or causing some larger metabolic change that leads to lysosomal compensation, rather than targeting a single master regulator such as TFEB.
Bmal1 Deletion in Astrocytes Induces Autophagy without Increasing p62 Degradation.
We next considered the possibility that autophagy may be affected by Bmal1 deletion since Lysotracker has been shown to track autophagosomes as well as lysosomes and other acidic cellular compartments (32–34). In order to examine autophagy flux, we performed LC3 western blots on siBmal1 astrocytes to gauge the conjugation of LC3 to phosphatidylethanolamine in the formation of autophagosomes. We found that the LC3 II/I ratio is greater in siBmal1 astrocytes, indicating an increase in the formation of autophagosomes (Fig. 4A). In samples treated with the lysosomal inhibitor bafilomycin, we saw an expected accumulation of LC3 in both control and Bmal1 knockdown astrocytes, indicating that lysosomal function in the fusion and turnover of autophagosomes is intact in these cells. We also assessed the degradation of the autophagy substrate p62/SQSTM1. p62 serves as a cargo-binding protein to target proteins to autophagosomes for degradation and is itself degraded upon lysosomal fusion (34). In our western blots, we saw no change in p62 levels in Bmal1 knockdown astrocytes and normal levels of p62 accumulation in cells treated with bafilomycin (Fig. 4B). This surprising finding suggests that without Bmal1 expression, astrocytes boost autophagy, without a basal increase in degradation of the intracellular autophagy chaperone p62, suggesting a possible selectivity for certain substrates.
Fig. 4.
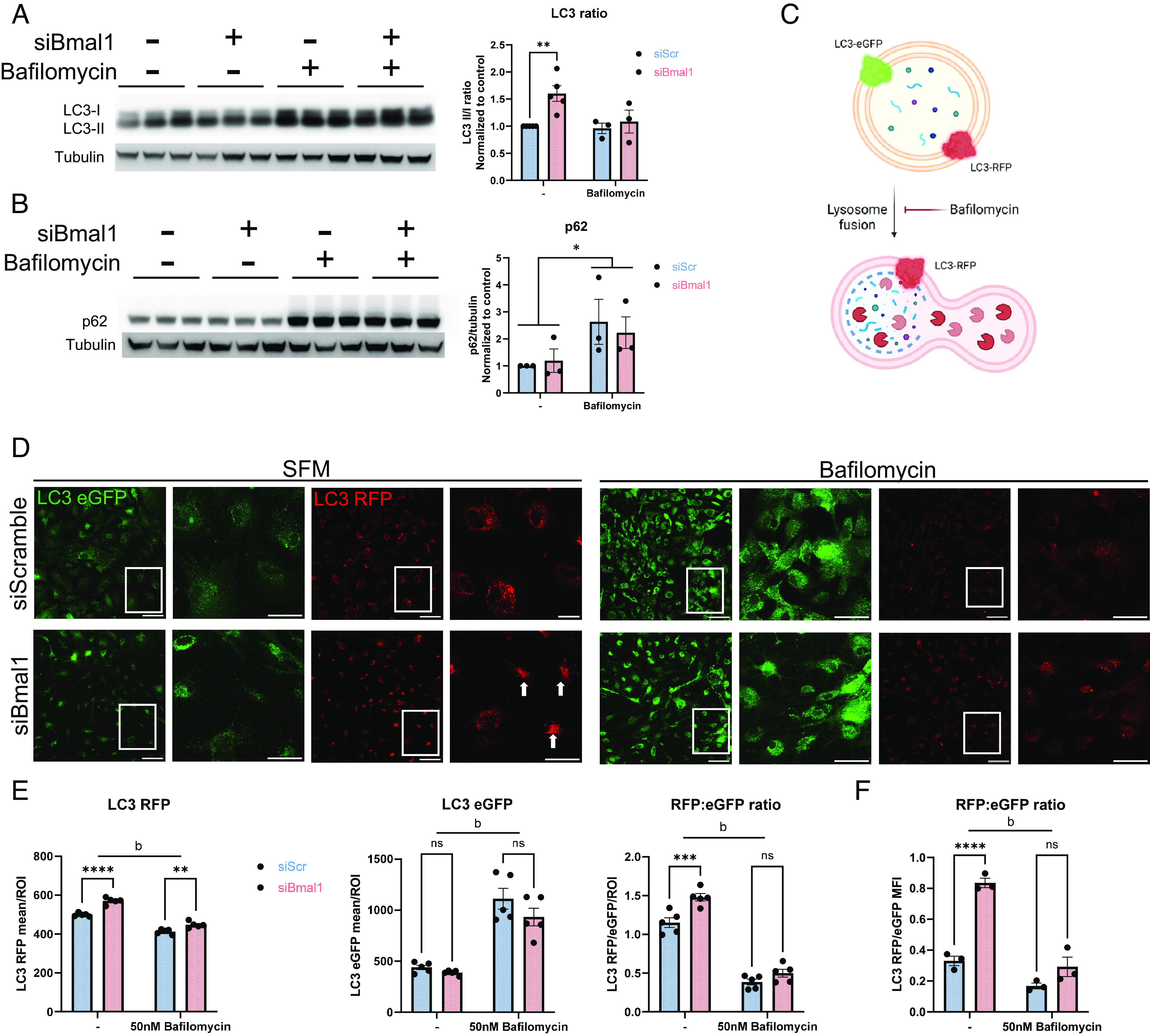
Bmal1 deletion in astrocytes induces autophagy without increasing degradation. (A and B) Western blot of siScramble and siBmal1 astrocytes treated with either vehicle (DMSO) or 50nM Bafilomycin in serum-free media for 6 h. (A) LC3 blot image and quantification. N = 3 to 5 independent experiments, density quantifications normalized per experiment to vehicle-treated siScramble controls. β-tubulin is shown as a loading control. ** = P < 0.005 compared to control by two-way ANOVA with Dunnett’s multiple comparison test. (B) p62 blot image and quantification. N = 3 independent experiments, density quantifications normalized per experiment to vehicle-treated siScramble controls. β-tubulin is shown as a loading control. * =P < 0.05 main effect of bafilomycin compared to untreated groups by two-way ANOVA. (C) Schematic of LC3 eGFP/RFP reporter signal in autophagosomes. Created with Biorender.com (D) Fixed cell confocal imaging of LC3 eGFP/RFP primary astrocytes transfected with siScramble or siBmal1 and treated with serum-free medium alone or with 50 nM Bafilomycin for 3 h. For each treatment, left images are 20× confocal images, scale bars, 100 µm and right images are insets, scale bars, 50 µm. (E) Quantification of confocal imaging from D, plotted as LC3 RFP or eGFP mean intensity per LC3 RFP thresholded ROI, or as the LC3 RFP intensity: eGFP intensity ratio per image. (F) LC3 RFP:eGFP ratio from flow cytometry experiments of primary astrocytes treated as in D and E. N = 3 independent experiments, MFI normalized per experiment to serum-free media-treated siScramble controls. (E and F) * =P < 0.05, ** =P < 0.005, *** =P < 0.0005, **** =P < 0.0001, b = bafilomycin main effect of P < 0.0001 by two-way ANOVA with Sidak multiple comparison test.
In order to further investigate autophagy activity post-autophagosome formation, we used CAG-RFP-EGFP-LC3 transgenic mice in order to generate LC3 reporter astrocytes (Fig. 4C). These mice express both RFP and eGFP linked to LC3 in all cells. The pH-sensitive eGFP signal is quenched in the acidic environment of the lysosome, while the LC3-RFP signal is pH-insensitive (35). Thus, red LC3-RFP signal indicates the overall abundance of autophagosomes, while green eGFP signal represents only autophagosomes that have yet to acidify. Using confocal microscopy, we observed that, within LC3-RFP-positive regions-of-interest (RFP+ ROIs), the LC3-RFP signal was brighter within siBmal1 astrocytes, indicating more total autophagosomes (acidified and neutral) (Fig. 4 D and E). Importantly, the LC3-eGFP intensity within RFP+ ROIs was not different from siScramble cells, indicating that the increase in RFP-LC3 signal is due to an increase in acidified autophagosomes. Qualitatively, we also observed higher perinuclear clustering of autophagosomes in siBmal1 astrocytes (Fig. 4D, arrows), similar to our observations with lysosomes and late endosomes in Fig. 2 D and E. As expected, bafilomycin treatment prevented acidification and raised the eGFP signal, therefore lowering the RFP:GFP ratio. Similar to our western blot data, these results indicate that with Bmal1 deletion, astrocytes have a higher pool of autophagosomes, but suggest that many of these are fused RFP+eGFP- autolysosomes.
Finally, to investigate autophagy in vivo, we prepared hippocampal sections for transmission electron microscopy from adult 7-mo-old Bmal1 aKO mice and Cre- littermate controls treated with tamoxifen at 2 mo of age. Considering our observations of endosomes, lysosomes, and autophagosomes in cultured astrocytes, we decided to quantify these three structures within hippocampal astrocytes identified within these sections. Hippocampal astrocytes were identified as containing a round or oval nucleus, electrolucent cytoplasm, and intermediate filament bundles (36, 37), which was more pronounced in Bmal1 knockout astrocytes as they express more GFAP. Early-late endosomes were identified as pleiomorphic to spherical vacuolar organelles with singular membranes, while lysosomes were identified as roughly spherical, electron-dense organelles (38). Autophagosomes were identified as multilamellar structures with distinguishable electron-lucent space between membranes (34). Surprisingly, in vivo the number of endosomes and lysosomes did not significantly differ from controls (Fig. 5, although endosomes were trending toward greater abundance in Bmal1 aKO, P = 0.07). However, the number of multilamellar structures, which we have considered to be autophagosomes, was significantly greater in astrocytes in the brain of Bmal1 aKO mice, in agreement with our in vitro data that suggests the total pool of autophagosomes is higher in these cells. These data also suggest that increased autophagosome content may partially explain the increase in Lysotracker signal in siBmal1 astrocytes, as acidic autophagosomes can also take up Lysotracker (34).
Fig. 5.
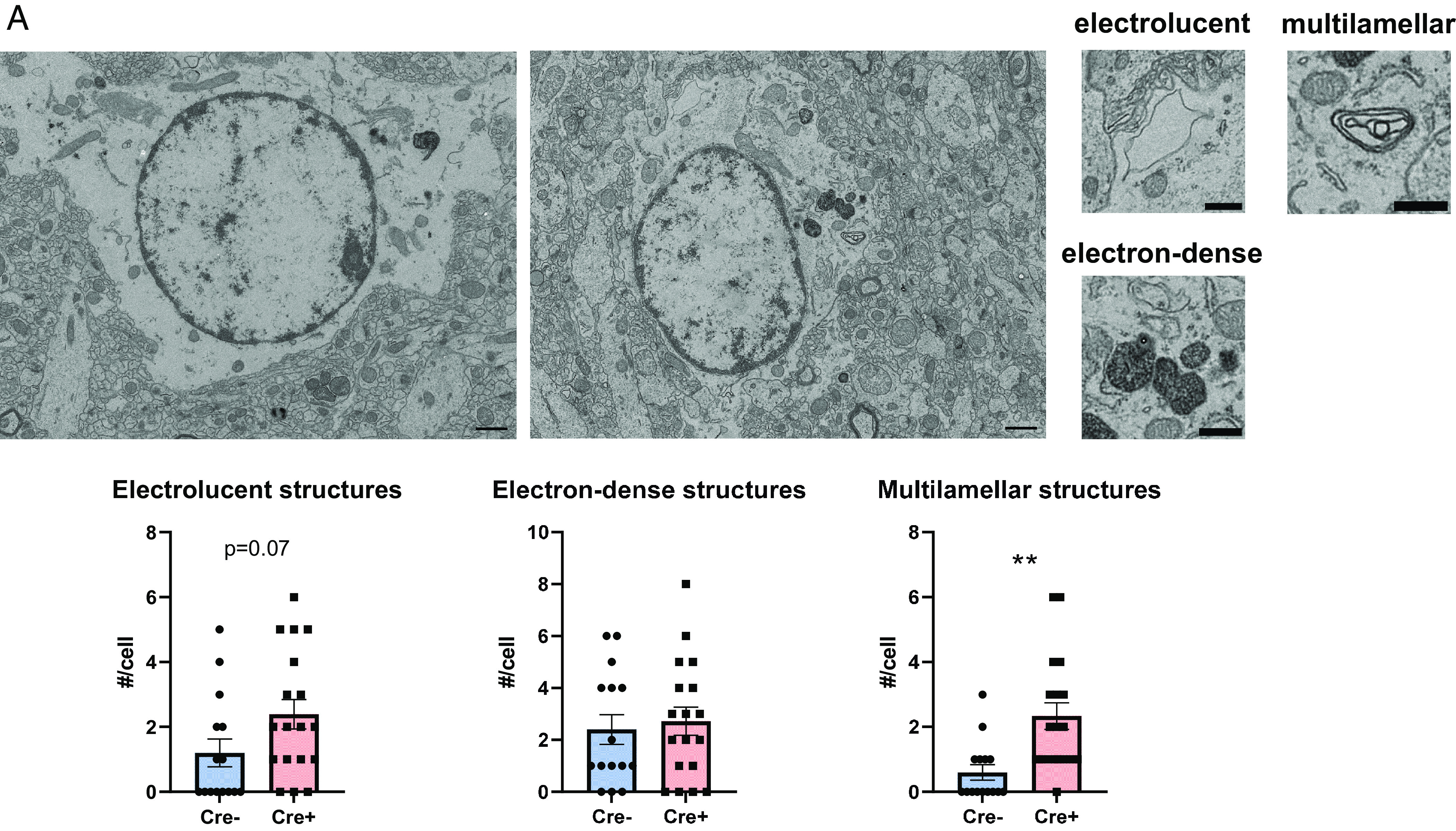
Bmal1 deletion leads to autophagosome accumulation in astrocytes in vivo. (A) Transmission electron microscopy images of astrocytes from Cre- control (Left) and Bmal1 aKO (Right) hippocampus (Scale bars, 1 µm). Example images of electrolucent (endosomes), electron-dense (lysosomes), and multilamellar (autophagosomes) structures (Scale bars, 0.5 µm). Plots show counts of each organelle per cell. N = 15 to 19 cells from three to four mice per group. ** =P < 0.005 by t test.
These data lead us to propose a model in which Bmal1 knockout/knockdown in astrocytes increases lysosomal degradation of endocytosed extracellular proteins (Fig. 3). Bmal1 depletion also induces autophagosome formation, but despite a larger pool of functional autophagosomes, these astrocytes do not efficiently improve their degradation of intracellular autophagy substrate p62 under basal conditions (Fig. 4B). However, we cannot rule out the possibility that for other chaperones or for nonselective autophagy, the larger pool of autophagosomes and their degradation dynamics may alter degradation and recycling of cellular components. Alternatively, the LC3 processing and increase in LC3 RFP+ organelles may be taking part in LC3-associated endocytosis (LANDO) (39) and degradation of extracellular rather than intracellular proteins.
Discussion
We have shown here that Bmal1 knockout/knockdown in astrocytes reprograms endolysosomal transcriptional pathways and induces endocytosis, lysosomal degradation, and autophagy activity within these cells. While the exact mechanism by which BMAL1 influences these astrocyte functions is unclear, we suggest that Bmal1 deletion influences many genes associated with the endolysosomal system. These findings help support the evidence that circadian clock proteins play a role in neurodegenerative diseases by implicating BMAL1 in the control of proteostasis in astrocytes.
The possibility that genes downstream of BMAL1 may act to temporally regulate proteostasis opens up therapeutic opportunities for targeting these processes in disease. Considering the variety of genes that BMAL1 regulates, we do not believe it likely that an individual gene causes these effects. This idea is exemplified by our finding that TFEB-target genes are not significantly dysregulated in Bmal1 knockdown astrocytes. An intriguing possibility is that mTOR could be the major regulator of endolysosome and autophagy function in astrocytes downstream of BMAL1. Here, we have shown that inhibition of mTOR with Torin1 treatment induces higher activation of TFEB and TFE3 in siBmal1 astrocytes than control astrocytes. mTOR activity exhibits circadian rhythms (40–42) which are influenced by BMAL1 and PER2 (13, 43). However, it has been shown that mTOR activity increases in several tissues in Bmal1 global knockout mice (13), which would decrease rather than increase autophagy as we have reported here. Thus, we suggest that BMAL1 may directly regulate many genes involved in these processes as shown in our pathway analysis indicating lysosomal dysregulation. However, BMAL1 may also indirectly regulate other processes that interact with the endolysosomal system and autophagy. For example, BMAL1 regulates phagocytosis in macrophages through the activation of RhoA and reorganization of the actin cytoskeleton (30). Similarly, in astrocytes, it has also been shown that BMAL1 regulates the actin cytoskeleton through the level of cortactin and Rho-GTPase activity (44). Thus, it is possible that our observations of endolysosome and autophagy activity relate to the cytoskeletal changes in the context of BMAL1 deletion. In addition, we have previously shown that deletion of Bmal1 induces oxidative stress in the brain (9), which has also been shown in several other systems (45–50). It is well established that oxidative stress induces autophagy in order to clear toxic proteins and damaged organelles such as mitochondria that lead to ROS generation (51). While it remains unclear what the primary event is that transforms the intracellular biology of astrocytes with Bmal1 deletion, we speculate that broad dysregulation of processes like oxidative metabolism or cytoskeletal organization contribute to the changes that we have described here.
It has previously been shown that Bmal1 knockdown in cultured microglia increases their phagocytosis of microbeads (52). This finding has also been recapitulated in vivo in Bmal1 microglia-specific knockout mice which exhibit increased microglial phagocytosis of synapses in response to a high-fat diet (53). Here, we have shown increased endocytosis in Bmal1 knockdown astrocytes and microglia. These data invite speculation that BMAL1 may influence various forms of endocytosis across many cell types both in the brain and periphery and that it will be important to untangle the effects of BMAL1 and other clock genes on cellular metabolism and endocytosis. If manipulating clock genes shifts the cell’s metabolism from a “fed” to “starved” state, it will be important to understand how that shift affects other cellular functions, especially those responsible for supporting neurons and synapses in the face of stress.
The ability of astrocytes to take up and degrade proteins is critical not just for their own health but also for the neurons they support. This is exemplified by a model of multiple sulfatase deficiency in which astrocyte lysosome dysfunction results in degeneration of cortical neurons (54). In addition, in neurodegenerative models of Aβ and tau accumulation, enhancing astrocyte lysosome biogenesis through TFEB activation can help to clear pathological protein aggregates (3, 4). However, when compared to microglia, astrocytes may be less efficient at degrading protein aggregates and may resecrete them and propagate pathologies, particularly in the case of alpha-synuclein (7, 55, 56). Thus, it will be crucial to understand the benefits and potential risks of manipulating uptake and degradation of proteins in astrocytes in disease models in order to boost the clearance of toxic proteins without exacerbating neuroinflammation or pathology spread. We previously found that astrocyte-specific Bmal1 deletion did not alter amyloid beta plaque deposition in mouse APP/PS1 models (11). However, this could suggest that increased uptake of Aβ aggregates by astrocytes was counteracted by some other pro-amyloidogenic function of Bmal1 KO astrocytes, such as increased inflammatory gene expression. Further study of astrocyte Bmal1 in other models of protein aggregation is needed to fully understand the pathogenic and therapeutic implications of manipulating BMAL1 in astrocytes.
BMAL1 is required for circadian function in cells and mice but can also regulate the expression of some nonrhythmic transcripts and cellular processes. Previous studies have demonstrated BMAL1-dependent rhythms in certain aspects of endolysosomal function, particularly chaperone-mediated autophagy. Chaperone-mediated autophagy exhibits BMAL1-dependent rhythms in the liver, heart, and kidney and remodels the lysosomal proteome at different times of day (15). In our studies, it is unknown if the effects of BMAL1 deletion on endolysosomal function are due to altered circadian rhythmicity or dysregulation of nonrhythmic transcripts. It should be noted that astrocyte-specific deletion of Bmal1 leads to only very mild changes in whole-animal circadian rhythms, which can only be detected under constant darkness conditions (our mice were all kept under 12 h:12 h light:dark) (57, 58). Thus, the effects are presumably not due to alterations in behavioral rhythms in the mice. It remains possible that disruption of the cellular transcriptional rhythms within astrocytes, as occurs with astrocytic Bmal1 deletion (10), could cause the endolysosomal changes observed. However, considering that lysosomal abundance as measured by Lysotracker does not show a rhythm in astrocytes and that Lysotracker signal is increased across all timepoints in siBmal1 astrocytes (Fig. 2C), it seems more likely that Bmal1 deletion exerts effects through nonrhythmic pathways. Regardless, because the abundance of BMAL1 in cells varies in a circadian manner, it is reasonable to predict that perturbations that alter circadian expression of BMAL1, particularly over longer periods of time, could potentially influence endolysosomal activity in astrocytes. Moreover, pharmacological manipulation of BMAL1 expression/function, whether directly or through other clock proteins (such as REV-ERBα-targeted drugs), could also impact astrocyte protein degradation pathways, creating a novel therapeutic opportunity for neurodegenerative diseases.
In conclusion, we have shown that Bmal1 deletion in astrocytes increases protein endocytosis as well as autophagosome content and activity, increasing extracellular protein degradation. This effect of BMAL1 seems to occur through transcriptional regulation of numerous endolysomal genes. Our results demonstrate that the unique astrocyte activation state conferred by Bmal1 deletion encompasses increased capacity for protein uptake and degradation, suggesting that further study of the role of astrocyte BMAL1 in protein aggregation disorders of the brain could have promising therapeutic implications.
Materials and Methods
Mice.
All mouse experiments were conducted in accordance with protocols approved by the Washington University Institutional Animal Care and Use Committee (IACUC), which is accredited by AAALAC. Aldh1L1-CreERT2 and Bmal1fl/fl mice were obtained from The Jackson Laboratory (Bar Harbor, ME; stock #031008 and 007668, respectively) and were bred at Washington University. C57BL/6-Tg(CAG-RFP/EGFP/Map1lc3b)1Hill/J (Jackson Laboratory stock # 027139) were a gift from Jeffrey Haspel at Washington University in St. Louis. All cohorts of mice were mixed sex and consisted of Cre+ and Cre− littermates from several breeding cages. All mice were maintained on a C57Bl\6 background and housed under 12-h light/12-h dark conditions. All Aldh1L1-CreERT2 (Cre− and Cre+) were given tamoxifen (Sigma, dissolved in corn oil, 2 mg/mouse/day for 5 d) by oral gavage at 2 mo of age to induce Bmal1 astrocyte-specific deletion. For tamoxifen-induced Bmal1 knockout in vitro, CAG-CreERT2 mice were obtained from The Jackson Laboratory and crossed to Bmal1fl/fl mice in house to generate CAG-CreERT2, Bmal1fl/fl pups from which astrocytes were cultured as described below. All mice were group housed with food and water available ad libitum.
Adult Astrocyte Isolation.
Adult astrocytes were isolated from 7-mo-old or 20-mo-old WT and Bmal1 aKO mice. Mice were anesthetized with an i.p. injection of Fatal Plus pentobarbital and then perfused with ice-cold DPBS. Mice were then decapitated, and the brain was dissected into a petri dish of digestion buffer (Collagenase D, TLCK trypsin inhibitor, DNAse I, Hepes pH 7.4, and HBSS). The brain stem and ventral white matter as well as the olfactory bulb were removed, and the remaining rest of the brain was minced with a clean razor blade. Each sample was placed on ice until all samples were ready for digestion. The brain samples were then incubated at 37 °C for 25 min with mixing.
Following digestion, cell suspensions were generated by pushing the digested samples through a 70-µm cell strainer with a syringe plunger, adding ice-cold dPBS to rinse cells off the strainer. Cells were centrifuged at 300g and then incubated with debris removal solution (Miltenyi MACS). Cells were centrifuged at 3,000 g, and debris was removed from the top phases of the gradient formed. Cells were then incubated with red blood cell removal solution (Miltenyi MACS), centrifuged at 300 g, and resuspended in MACS buffer (dPBS, 0.5% BSA, 2mM EDTA).
Astrocytes were then magnetically labeled and separated from the rest of the cells. Each sample was first resuspended in 80 µL of MACS buffer with 10 µL of FcR Blocking reagent (Miltenyi MACS). After blocking, 10uL of Anti-ACSA-2 microbeads were added and incubated for 15 min at 4 °C. Cells were then washed and resuspended in 500uL of MACS buffer. MS columns (Miltenyi) were placed onto an OCTO MACS Separator (Miltenyi), and each sample was loaded into a column. Cells were washed with MACS buffer and flow-through was collected for each sample. Columns were then removed and placed onto collection tubes, and then, 1 mL of MACS buffer was quickly plunged through each column to collect labeled astrocytes. Astrocyte samples were then centrifuged, lysed in Trizol, and stored at −80 °C until RNA isolation.
RNA Isolation and qPCR.
RNA was isolated from Trizol samples as described previously (11). Briefly, chloroform was added to samples to separate RNA from other contaminants and then extracted using a PureLink RNA Mini Kit (Life Technologies). For qPCR analysis, RNA concentration was determined (Thermo Nanodrop 8000) and then converted to cDNA with a reverse transcription kit (Applied Biosystems).
Taqman probes (Applied Biosystems) were used to amplify sequences on a StepOnePLus Real-Time PCR thermocycler (Applied Biosystems). For Fluidigm micro-fluidic qPCR, cDNA was sent with Taqman probes to the Washington University Genome Technology Access Center for analysis on a Fluidigm BioMark HD system. Data were analyzed using the delta-delta Ct method with Gapdh as a housekeeping gene.
RNA Sequencing.
Bulk tissue RNA sequencing and analysis were performed by the Genome Technology Access Center at Washington University using their standard methods, which are described in SI Appendix. Differential expression analysis was performed to analyze for differences between conditions, and the results were plotted using the Enhanced Volcano package with a −log10 P value cutoff of 2.0 (corresponds to a P value cutoff of 0.01) and Log2 fold change cutoff of 1.0. Gene set enrichment was performed with the Generally Applicable Gene-set Enrichment (GAGE) version 2.36.0 package using the mouse Go.db version 3.10.0 annotation package for gene sets. For the GO lysosome pathway, the gene list was obtained by filtering by the GO lysosome term ID (GO:0005764) in the Ensembl BioMart web tool (http://useast.ensembl.org/biomart/martview/e5264c3c8048cf6ddc7bb63edff294c6). This gene list was then merged with differential expression data and filtered to produce pathway-specific heatmaps of top-most differentially expressed genes. All of the RNAseq data are publicly available through the NIH GEO website under accession number GSE227932 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227932)
Primary Astrocyte Culture.
Primary astrocyte cortical cultures were generated as previously described (10). For wild-type astrocyte experiments with siRNA, CD1 pregnant dams were ordered from Charles River. CAG-CreERT2, Bmal1fl/flpups were generated and genotyped in house. Briefly, brains were harvested in a dissection hood from P2-P3 pups and placed into cold DMEM in a petri dish. The meninges were peeled off the surface of the brain and hippocampi and subcortical structures were removed. Cortices were digested in 0.05% trypsin at 37 °C before being dissociated with a P1000 pipet. Cells were then diluted and grown in DMEM + 10% fetal bovine serum + 1% penicillin-streptomycin. Cells were plated on T75 flasks coated in 100nM poly-d-lysine (Corning). After about a week, cells became confluent and flasks were shaken at 225 rpm for 2 h to remove nonadherent cells such as microglia and debris before being split into PDL-coated plates for experiments.
siRNA Transfections.
First, 48 to 72 h after replating, astrocytes were transfected with siRNA using lipofectamine RNAiMAX (Life Technologies) in Optimem (LifeTechnologies) according to the manufacturer’s instructions. siRNAs targeting mouse Bmal1 or control nontargeting (designed to target no known mouse gene) were obtained from Horizon. ON-TARGETplus Mouse SmartPool siRNA was used, which contains five separate siRNAs targeting the gene of interest which are pooled together (Bmal1: product L-040483-01-0020, non-targeting: D-001810-01-05). siRNAs were then added at a 50 nM working concentration. Twenty-four hours after transfection, media were replaced with an astrocyte growth medium described above. All cell imaging and flow cytometry experiments were performed at days 3 to 4 post transfection.
Cell Treatments.
Dynasore (Sigma) was dissolved in DMSO and stored in aliquots at −20 °C. Stocks were diluted to 100 to 200 µM in a serum-free medium (DMEM + 1% penicillin-streptomycin) before being added to cells. Chloroquine (Sigma) was dissolved in sterile water and stored in aliquots at −20 °C. Stocks were diluted to 20 or 40 µM in a serum-free medium before being added to cells. Bafilomycin A1 (Sigma) was bought ready to use, stored at −20 °C, and diluted to 50 nM in a serum-free medium before being added to cells. Torin1 (Tocris Biosience) was dissolved in DMSO and stored in aliquots at −20 °C. Stocks were diluted to 200 nM in a serum-free medium before being added to cells. On the day of the experiment, cells were treated with the above inhibitors either alone for western blot or at the same time as BSA for uptake assays. For BSA uptake assays, BSA-647 (Thermo Fisher) and DQ red BSA (Thermo Fisher) were each dissolved to 1 μg/mL in a serum-free medium and added to cells for 3 h. In Lysotracker Green (Thermo Fisher, 200 nM) and Lysosensor Yellow/Blue (Thermo Fisher, 10µM) experiments, each dye was incubated with cells in a serum-free medium for 30 min at the end of the experiment. For cell synchronization in rhythm assays, astrocytes were synchronized via media change and rhythms were analyzed with the Nitecap circadian analysis web application as described in the SI Appendix methods (59).
Flow Cytometry.
Cells were then trypsinized with TryplE, collected in astrocyte growth medium, and centrifuged at 1,000 g for 5 min. Live cells were resuspended in flow buffer (dPBS, 1% FBS, 1mM EDTA) and kept on ice briefly before flow cytometry. Approximately 2,000 to 3,000 live, single cells were counted for each sample on a BD X20 or LSR Fortessa FACS analyzer. Data were analyzed in Flowjo software v10.8.1. After performing compensation with single-stained controls, cells were gated for live and single cells, and median fluorescence intensity was recorded. Data were plotted in Graphpad Prism, and significance was determined with either Student’s t test or two-way ANOVA with Sidak’s multiple comparisons tests.
Immunocytochemistry.
Live cells were first washed with dPBS and then fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. Following fixation, cells were washed in PBS and either stored at 4°C or stained immediately. Cells were first blocked in PBS with 3% goat serum and 0.4% Triton X-100 for 25 min at room temperature. Primary antibodies (LAMP1, 1:1,000, DSHB; RAB7, 1:1,000, Cell Signaling Technology; TFEB, 1:500, Bethyl Laboratories; TFE3, 1:500, Sigma-Aldrich) were diluted in PBS with 1% serum and 0.4% Triton X-100 and incubated with cells overnight at 4 °C. After washing cells in PBS, secondary antibodies diluted in PBS with 0.25% Triton X-100 were incubated on cells for 1 h at room temperature. Cells were then washed and stored in PBS at 4 °C until the day of imaging.
For imaging analysis, fixed cells were imaged on a Nikon A1R confocal microscope. Live cells for BSA assays were washed in warm HBSS, given imaging buffer (HBSS, 1% FBS, 25mM HEPES), and imaged on a BZX-800 Keyence microscope (10× epifluorescence images) or a Zeiss LSM 880 confocal microscope (40× confocal images) to visualize puncta. For all image analysis, z-stack images were loaded into Fiji v 1.53s and max projected. The particle analysis tool was used to draw regions of interest around the DAPI/Hoechst channel to count the number of nuclei per image. Intensity measurements or thresholded percent area measurements were normalized to the number of nuclei per image. For nuclear analysis of TFEB/TFE3, the ROI drawn on the DAPI channel was applied to measure the average intensity of TFEB/TFE3 in the nucleus. For perinuclear analysis, the combined selection of DAPI nuclei was extended to draw a 7-µm shell around each nucleus, and then intensity measurements were obtained from this 7-µm extended “perinuclear region.” For LC3 RFP/eGFP image analysis, the particle analysis tool was used to draw a region of interest including only the LC3 RFP particles above threshold. The LC3 eGFP and RFP intensity within this region was then recorded. This RFP particle region of interest was used to measure only the LC3 eGFP that was also RFP+, thus giving a measure of the “yellow” neutral autophagosome signal.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Acknowledgments
We thank the Washington University Department of Pathology and Immunology’s Flow Cytometry Core for their training and assistance with flow cytometry. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant# UL1TR002345 from the National Center for Research Resources (NCRR), a component of the NIH, and NIH Roadmap for Medical Research. We acknowledge the assistance of Dr. Sanja Sviben and Dr. James Fitzpatrick at the Washington University Center for Cellular Imaging (WUCCI) in electron microscopy studies, which is supported by Washington University School of Medicine, The Children’s Discovery Institute of University and St. Louis Children’s Hospital (CDI-CORE-2015-505 and CDI-CORE-2019-813) and the Foundation for Barnes-Jewish Hospital (3770). TEM images were acquired using an AMT Imaging Nanosprint15-MkII sCMOS camera, which was purchased with support from the Office of Research Infrastructure Programs (ORIP), a part of the NIH Office of the Director under grant OD032186. We thank the laboratory of Dr. Abhinav Diwan for technical assistance and advice. Finally, we thank Julie Dimitry for mouse and reagent assistance, as well as the rest of the Musiek lab for insightful discussions.
Author contributions
C.A.M. and E.S.M. designed research; C.A.M., A.J.P., and M.W.K. performed research; C.A.M. and E.S.M. analyzed data; and C.A.M., A.J.P., and E.S.M. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
RNAseq data have been deposited in GEO: young isolated astrocytes [GSE227633 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227633)] (60), old isolated astrocytes [GSE227932 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227932)] (61).
Supporting Information
References
- 1.Peng C., Trojanowski J. Q., Lee V. M. Y., Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 16, 199–212 (2020), 10.1038/s41582-020-0333-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Q., Haney M. S., The role of glia in protein aggregation. Neurobiol. Dis. 143, 105015 (2020), 10.1016/j.nbd.2020.105015. [DOI] [PubMed] [Google Scholar]
- 3.Xiao Q., et al. , enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 34, 9607–9620 (2014), 10.1523/JNEUROSCI.3788-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martini-Stoica H., et al. , TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 215, 2355–2377 (2018), 10.1084/jem.20172158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang P., Ye Y., Astrocytes in neurodegenerative diseases: A perspective from Tauopathy and α-Synucleinopathy. Life 11, 938 (2021), 10.3390/life11090938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maté de Gérando A., et al. , Neuronal tau species transfer to astrocytes and induce their loss according to tau aggregation state. Brain 144, 1167–1182 (2021), 10.1093/brain/awab011. [DOI] [PubMed] [Google Scholar]
- 7.Rostami J., et al. , Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J. Neurosci. 37, 11835–11853 (2017), 10.1523/JNEUROSCI.0983-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bunger M. K., et al. , Mop3 Is an essential component of the master circadian pacemaker in mammals. Cell 103, 1009–1017 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Musiek E. S., et al. , Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Invest. 123, 5389–5400 (2013), 10.1172/JCI70317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lananna B. V., et al. , Cell-autonomous regulation of astrocyte activation by the circadian clock protein BMAL1. Cell Rep. 25, 1–9.e5 (2018), 10.1016/j.celrep.2018.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKee C. A., et al. , Astrocytes deficient in circadian clock gene Bmal1 show enhanced activation responses to amyloid-beta pathology without changing plaque burden. Sci. Rep. 12, 1796 (2022), 10.1038/s41598-022-05862-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pastore N., et al. , Nutrient-sensitive transcription factors TFEB and TFE3 couple autophagy and metabolism to the peripheral clock. EMBO J. 38, e101347 (2019), 10.15252/embj.2018101347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khapre R. V., et al. , BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging 6, 48–57 (2014), 10.18632/aging.100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lipton J. O., et al. , The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell 161, 1138–1151 (2015), 10.1016/j.cell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juste Y. R., et al. , Reciprocal regulation of chaperone-mediated autophagy and the circadian clock. Nat. Cell Biol. 23, 1255–1270 (2021), 10.1038/s41556-021-00800-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Y., et al. , Circadian rhythm of autophagy proteins in hippocampus is blunted by sleep fragmentation. Chronobiol. Int. 33, 553–560 (2016), 10.3109/07420528.2015.1137581. [DOI] [PubMed] [Google Scholar]
- 17.Zhang R., Lahens N. F., Ballance H. I., Hughes M. E., Hogenesch J. B., A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. U.S.A. 111, 16219–16224 (2014), 10.1073/pnas.1408886111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mure L. S., et al. , Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 359, eaao0318 (2018), 10.1126/science.aao0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Law C. W., et al. , RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res. 5, ISCB Comm J-1408 (2018), 10.12688/f1000research.9005.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgar J. R., et al. , A dysfunctional endolysosomal pathway common to two sub-types of demyelinating Charcot–Marie–Tooth disease. Acta Neuropathol. Commun. 8, 165 (2020), 10.1186/s40478-020-01043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertolo C., et al. , LITAF, a BCL6 target gene, regulates autophagy in mature B-cell lymphomas. Br. J. Haematol. 162, 621–630 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prolo L. M., Takahashi J. S., Herzog E. D., Circadian rhythm generation and entrainment in astrocytes. J. Neurosci. 25, 404–408 (2005), 10.1523/JNEUROSCI.4133-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pu J., Guardia C. M., Keren-Kaplan T., Bonifacino J. S., Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339 (2016), 10.1242/jcs.196287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diwu Z., Chen C. S., Zhang C., Klaubert D. H., Haugland R. P., A novel acidotropic pH indicator and its potential application in labeling acidic organelles of live cells. Chem. Biol. 6, 411–418 (1999), 10.1016/S1074-5521(99)80059-3. [DOI] [PubMed] [Google Scholar]
- 25.Poole B., Ohkuma S., Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J. Cell Biol. 90, 665–669 (1981), 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wibo M., Poole B., Protein degradation in cultured cells: II. The uptake of chloroquine by rat fibroblasts and the inhibition of cellular protein degradation and Cathepsin B1. J. Cell Biol. 63, 430–440 (1974), 10.1083/jcb.63.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang R., et al. , Molecular basis of V-ATPase inhibition by bafilomycin A1. Nat. Commun. 12, 1782 (2021), 10.1038/s41467-021-22111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu S., Sung T., Lin N., Abraham R. T., Jessen B. A., Lysosomal adaptation: How cells respond to lysosomotropic compounds. PLoS ONE. 12, e0173771 (2017), 10.1371/journal.pone.0173771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vázquez C. L., Colombo M. I., “Chapter 6 Assays toassess autophagy induction and fusion of autophagic vacuoles with a degradative compartment, using monodansylcadaverine (MDC) and DQ- ‐BSA” in Methods in Enzymology. Autophagy in Mammalian Systems, Part B. (Academic Press, 2009), pp. 85–95, 10.1016/S0076-6879(08)03606-9, Vol. 452. [DOI] [PubMed] [Google Scholar]
- 30.Kitchen G. B., et al. , The clock gene Bmal1 inhibits macrophage motility, phagocytosis, and impairs defense against pneumonia. Proc. Natl. Acad. Sci. U.S.A. 117, 1543–1551. 10.1073/pnas.1915932117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J., et al. , Inhibition of REV-ERBs stimulates microglial amyloid-beta clearance and reduces amyloid plaque deposition in the 5XFAD mouse model of Alzheimer’s disease. Aging Cell. 19, e13078 (2020), 10.1111/acel.13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chikte S., Panchal N., Warnes G., Use of LysoTracker dyes: A flow cytometric study of autophagy. Cytometry Part A. 85, 169–178 (2014), 10.1002/cyto.a.22312. [DOI] [PubMed] [Google Scholar]
- 33.González-Polo R. A., et al. , The apoptosis/autophagy paradox: Autophagic vacuolization before apoptotic death. J. Cell Sci. 118, 3091–3102 (2005), 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 34.Klionsky D. J., et al. , Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 17, 1–382 (2021), 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L., Wang Z. V., Hill J. A., Lin F., New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J. Am. Soc. Nephrol. 25, 305–315 (2014), 10.1681/ASN.2013040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de la Rosa C., Cano J., Reinoso-suárez F., An electron microscopic study of astroglia and oligodendroglia in the lateral geniculate nucleus of aged rats. Mech. Ageing Dev. 29, 267–281 (1985), 10.1016/0047-6374(85)90067-3. [DOI] [PubMed] [Google Scholar]
- 37.Maxwell D. S., Kruger L., The fine structure of astrocytes in the cerebral cortex and their response to focal injury produced by heavy ionizing particles. J. Cell Biol. 25, 141–157 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klumperman J., Raposo G., The complex ultrastructure of the endolysosomal system. Cold Spring Harb. Perspect. Biol. 6, a016857 (2014), 10.1101/cshperspect.a016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heckmann B. L., et al. , LC3-associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell 178, 536–551.e14 (2019), 10.1016/j.cell.2019.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cornu M., et al. , Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc. Natl. Acad. Sci. U.S.A. 111, 11592–11599 (2014), 10.1073/pnas.1412047111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jouffe C., et al. , The circadian clock coordinates ribosome biogenesis. PLOS Biol. 11, e1001455 (2013), 10.1371/journal.pbio.1001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robles M. S., Humphrey S. J., Mann M., Phosphorylation is a central mechanism for circadian control of metabolism and physiology. Cell Metab. 25, 118–127 (2017), 10.1016/j.cmet.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Wu R., et al. , The circadian protein period2 suppresses mTORC1 activity via recruiting Tsc1 to mTORC1 complex. Cell Metab. 29, 653–667.e6 (2019), 10.1016/j.cmet.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 44.Ali A. A. H., et al. , Bmal1-deficiency affects glial synaptic coverage of the hippocampal mossy fiber synapse and the actin cytoskeleton in astrocytes. Glia 68, 947–962 (2020), 10.1002/glia.23754. [DOI] [PubMed] [Google Scholar]
- 45.Alexander R. K., et al. , Bmal1 integrates mitochondrial metabolism and macrophage activation. eLife 9, e54090 (2020), 10.7554/eLife.54090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Early J. O., et al. , Circadian clock protein BMAL1 regulates IL-1β in macrophages via NRF2. Proc. Natl. Acad. Sci. U.S.A. 115, E8460–E8468 (2018), 10.1073/pnas.1800431115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacobi D., et al. , Hepatic Bmal1 regulates rhythmic mitochondrial dynamics and promotes metabolic fitness. Cell Metab. 22, 709–720 (2015), 10.1016/j.cmet.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee J., et al. , Bmal1 and β-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced β-cell failure in mice. Mol. Cell Biol. 33, 2327–2338 (2013), 10.1128/MCB.01421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chhunchha B., Kubo E., Singh D. P., Clock protein Bmal1 and Nrf2 cooperatively control aging or oxidative response and redox homeostasis by regulating rhythmic expression of Prdx6. Cells 9, 1861 (2020), 10.3390/cells9081861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ali A. A. H., et al. , Deficiency of the clock gene Bmal1 affects neural progenitor cell migration. Brain Struct. Funct. 224, 373–386 (2019), 10.1007/s00429-018-1775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee J., Giordano S., Zhang J., Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 441, 523–540 (2011), 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X. L., et al. , Deficiency of the circadian clock gene Bmal1 reduces microglial immunometabolism. Front. Immunol. 11, 586399 (2020), 10.3389/fimmu.2020.586399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X. L., et al. , Microglia-specific knock-down of Bmal1 improves memory and protects mice from high fat diet-induced obesity. Mol. Psychiatry 26, 6336–6349 (2021), 10.1038/s41380-021-01169-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Malta C., Fryer J. D., Settembre C., Ballabio A., Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorder. Proc. Natl. Acad. Sci. U.S.A. 109, E2334–E2342 (2012), 10.1073/pnas.1209577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rostami J., et al. , Crosstalk between astrocytes and microglia results in increased degradation of α-synuclein and amyloid-β aggregates. J. Neuroinflam. 18, 124 (2021), 10.1186/s12974-021-02158-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valdinocci D., Radford R. A. W., Siow S. M., Chung R. S., Pountney D. L., Potential modes of intercellular α-synuclein transmission. Int. J. Mol. Sci. 18, 469 (2017), 10.3390/ijms18020469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tso C. F., et al. , Astrocytes regulate daily rhythms in the suprachiasmatic nucleus and behavior. Curr. Biol. 27, 1055–1061 (2017), 10.1016/j.cub.2017.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barca-Mayo O., et al. , Astrocyte deletion of Bmal1 alters daily locomotor activity and cognitive functions via GABA signalling. Nat. Commun. 8, 14336 (2017), 10.1038/ncomms1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brooks T. G., et al. , Nitecap: An exploratory circadian analysis web application. J. Biol. Rhythms 37, 43–52 (2022), 10.1177/07487304211054408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McKee C. A., Musiek E. S., Isolated astrocytes from young BMAL1aKO and wildtype mice. GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227633. Accessed 17 March 2023.
- 61.McKee C. A., Musiek E. S., Isolated astrocytes from aged BMAL1aKO and wildtype mice. GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227932. Accessed 22 March 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Data Availability Statement
RNAseq data have been deposited in GEO: young isolated astrocytes [GSE227633 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227633)] (60), old isolated astrocytes [GSE227932 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227932)] (61).