

Significance
The marine picocyanobacteria Prochlorococcus and Synechococcus are generally thought to be free-living planktonic cells. We show that some lineages can attach to, and use, particulate chitin, changing our view of their ecology. We also show that this trait was acquired around the time that arthropods, the main source of oceanic chitin, underwent a major ecological expansion. We propose the “chitin raft hypothesis”: Benthic ancestors of picocyanobacteria colonized the ocean alongside arthropods by attaching to free-floating exoskeleton particles. Co-expansion of both groups into the open ocean helped seed the rise of modern marine ecosystems. Loss of chitin utilization and transition to an exclusively planktonic lifestyle later occurred within a major subgroup of Prochlorococcus. It now numerically dominates the extant ocean.
Keywords: marine picocyanobacteria, chitin, evolution, lifestyle
Abstract
Marine picocyanobacteria Prochlorococcus and Synechococcus, the most abundant photosynthetic cells in the oceans, are generally thought to have a primarily single-celled and free-living lifestyle. However, while studying the ability of picocyanobacteria to supplement photosynthetic carbon fixation with the use of exogenous organic carbon, we found the widespread occurrence of genes for breaking down chitin, an abundant source of organic carbon that exists primarily as particles. We show that cells that encode a chitin degradation pathway display chitin degradation activity, attach to chitin particles, and show enhanced growth under low light conditions when exposed to chitosan, a partially deacetylated soluble form of chitin. Marine chitin is largely derived from arthropods, which underwent major diversifications 520 to 535 Mya, close to when marine picocyanobacteria are inferred to have appeared in the ocean. Phylogenetic analyses confirm that the chitin utilization trait was acquired at the root of marine picocyanobacteria. Together this leads us to postulate that attachment to chitin particles allowed benthic cyanobacteria to emulate their mat-based lifestyle in the water column, initiating their expansion into the open ocean, seeding the rise of modern marine ecosystems. Subsequently, transitioning to a constitutive planktonic life without chitin associations led to cellular and genomic streamlining along a major early branch within Prochlorococcus. Our work highlights how the emergence of associations between organisms from different trophic levels, and their coevolution, creates opportunities for colonizing new environments. In this view, the rise of ecological complexity and the expansion of the biosphere are deeply intertwined processes.
Prochlorococcus and its sister group marine Synechococcus (together “marine picocyanobacteria” or “marine SynPro”) constitute a highly abundant group of oceanic phytoplankton that perform about ~25% of oceanic CO2-fixation (1–3). In addition to their role as primary producers, it is increasingly clear that marine picocyanobacteria also use organic carbon as a supplemental carbon and energy source (i.e., mixotrophy) (4–6), especially in the light-limited deep euphotic zone (7, 8). While studying genes involved in mixotrophy in this group (9), we noticed that some strains have chitinase genes, indicating the potential for using chitin, one of the most abundant sources of organic carbon in the ocean (10, 11). However, chitin in the marine environment typically exists as particles, primarily derived from arthropod exoskeletons (10, 11), which are broken down by microbial consortia (12). Marine picocyanobacteria are typically considered to live an exclusively single-celled planktonic lifestyle (13, 14), but are also sometimes found within particulate organic matter aggregates (15) that sink out of the euphotic zone (16). Together this suggests that chitin utilization might be a functional trait in marine picocyanobacteria.
Results and Discussion
To explore this possibility, we began by examining the distribution of genes involved in chitin utilization in Prochlorococcus and Synechococcus genomes (Fig. 1). This comparison is complicated by the significant overlap in gene content between pathways for chitin utilization and peptidoglycan recycling (Fig. 1). Peptidoglycan, which is chemically similar to chitin, forms a single cell-size polymeric structure (the sacculus) that is a component of the bacterial cell wall, and recycling of peptidoglycan fragments inevitably generated during cell growth and division is widespread in bacteria (17). We therefore searched 702 partial and complete genomes for genes required for both chitin utilization and peptidoglycan recycling. Except for the ChiA/ChiA-like chitinases, genes from these pathways occur at a frequency similar to, or higher than, the average genome completeness of ~75% (Materials and Methods) in Synechococcus and deeply branching “low-light-adapted IV” (LLIV) Prochlorococcus, suggesting they are universal or nearly universal in these clades (Fig. 1 and Dataset S1). Indeed, the full suite of non-ChiA genes occurs in all 24 complete Synechococcus genomes and all 20 complete LLIV Prochlorococcus genomes in our dataset (Fig. 1 and Dataset S1), consistent with their universality in these clades.
Fig. 1.
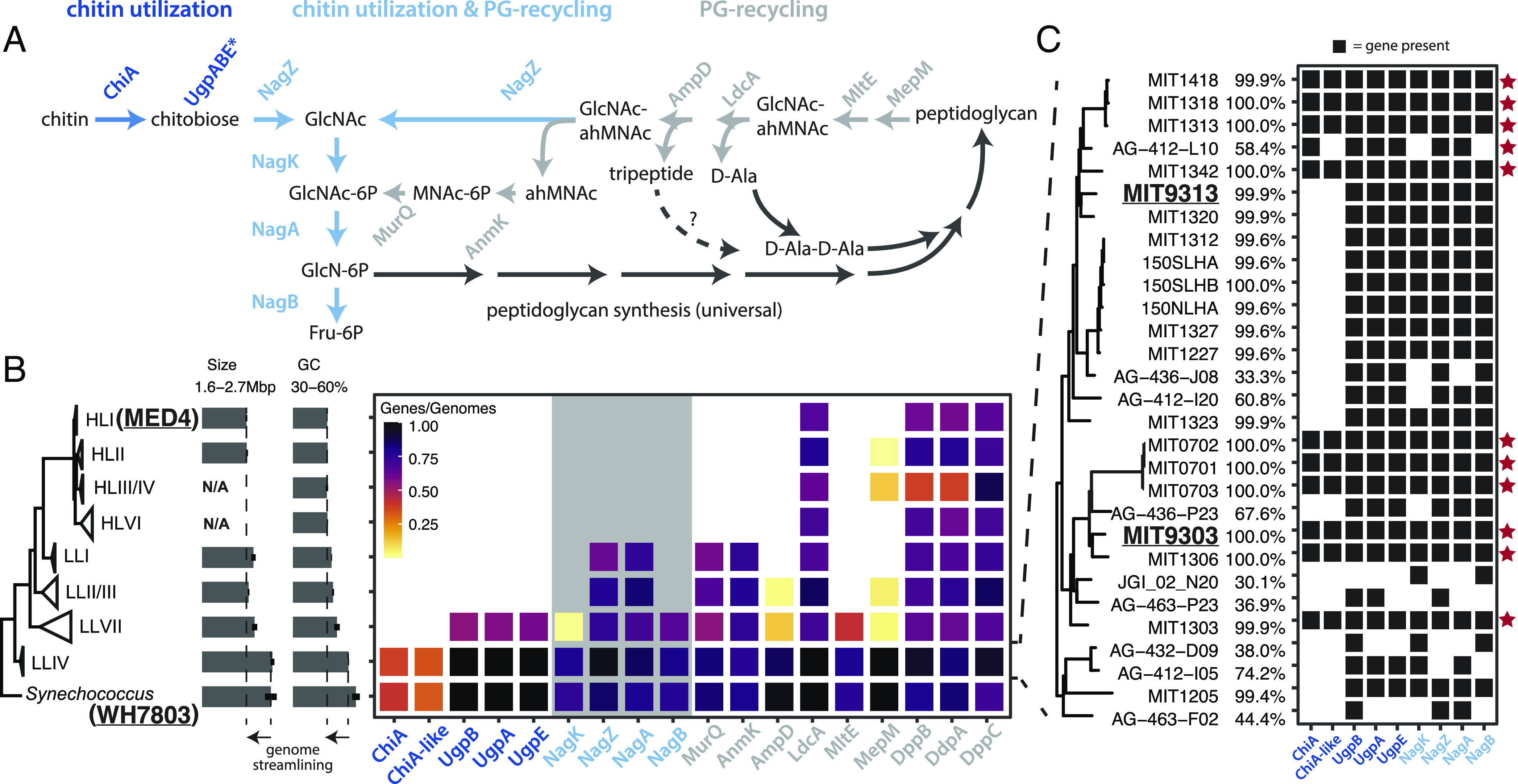
Distribution of chitin utilization genes in marine picocyanobacteria (A) Pathway for chitin degradation (dark blue) and its reactions that overlap (blue) with peptidoglycan metabolic recycling (gray). ChiA is annotated as a putative chitinase enzyme, while ChiA-like is a homolog of the substrate-binding domain of ChiA and annotated as a putative chitin-binding domain protein. The putative chitobiose transporter UgpBAE is highlighted with an asterisk (*). (B) Average frequency of occurrence of chitin utilization and peptidoglycan recycling genes estimated from both partial and complete genome sequences available in Synechococcus and the major clades of Prochlorococcus shown in the tree on the left. Average completeness of genomes in our sample is ~75% (Materials and Methods). The clade membership of the strains used in the experiments are highlighted in bold and underlined. The gray background area in the gene frequency frame highlights genes shared between the chitin utilization and peptidoglycan pathways (dark blue genes in Fig. 1A). Average size and guanine-cytosine (GC) content of genomes are reported for each clade to illustrate the genome streamlining associated with the loss of the chitin utilization pathway (C) Detailed genomic profiles of members of the LLIV clade of Prochlorococcus, which reveals putative primary chitin degraders that possess chitinase (red star) and putative secondary chitin degraders that lack chitinase. The percentage genome completeness is reported for each clade. Abbreviations: GlcNAc—N-acetyl-glucosamine, GlcNAc-6P—N-acetyl-glucosamine 6-phosphate, GlcN-6P—glucosamine 6-phosphate, F6P—fructose 6-phosphate, ahMNAc—anhydro-N-acetyl-beta-muramate, MNAc-6P—N-acetyl-muramate 6 phosphate.
The lone exception to the apparent universality of chitin utilization genes within Synechococcus and LLIV Prochlorococcus is the ChiA/ChiA-like chitinases that perform the first hydrolysis step in the pathway. These genes occur at about half the frequency of downstream genes in the pathway, i.e., in ~40% of genomes in our full dataset and ~50% of complete genomes (Dataset S1). It is known from other systems that chitin utilization is a complex ecological process (18, 19) with hydrolysis of chitin polymers occurring in the extracellular milieu, making chitin fragments available to all cells in the community. Consequently, chitin breakdown involves niche partitioning into groups that specialize in performing the initial hydrolysis steps and others that specialize in using small chitin oligosaccharides (20). Our results suggest a similar niche partitioning into “primary” and “secondary” degraders may occur among chitin-using members of the marine picocyanobacteria.
In contrast to the widespread occurrence of chitin utilization and peptidoglycan recycling genes in Synechococcus and LLIV Prochlorococcus, these genes are almost universally absent in “high-light-adapted” Prochlorococcus (Fig. 1 and Dataset S1). All LL clades of Prochlorococcus possess peptidoglycan recycling genes, but chitin utilization genes are nearly exclusive to the deeply branching LLIV clade (Fig. 1). Subunits of the putative chitobiose transporter UgpBAE also occur at high frequency within low-light-adapted VII (LLVII) Prochlorococcus (Fig. 1 and Dataset S1), but the specificity of these transporters has not been determined, and they are not diagnostic of chitin utilization. [Transporters are often thought to be multifunctional in oligotrophic bacteria such as Prochlorococcus (21, 22)]. ChiA and NagK, without which chitin utilization cannot proceed, are nearly absent in LLVII Prochlorococcus genomes in our sample (Fig. 1 and Dataset S1). These observations suggest that chitin utilization and peptidoglycan recycling were both present in the last common ancestor of marine picocyanobacteria, but were sequentially lost during the diversification of extant Prochlorococcus groups.
To determine whether the putative chitin users (Fig. 1) actually metabolize chitin, we first used enzyme assays to examine whether the cells display chitinase activity. Both Synechococcus and Prochlorococcus strains that are putative primary degraders of chitin (i.e., possessing the full complement of chitin utilization genes) displayed chitinase activity in cell-free spent media, and the activity disappeared upon boiling the media (Fig. 2 and SI Appendix, Fig. S1)—consistent with the production of extracellular chitinase enzymes that are denatured upon heating. In agreement with the genomic predictions, a Prochlorococcus strain (MIT9313, Fig. 1) that is a putative secondary chitin degrader did not display extracellular chitinase activity in spent media (Fig. 2 and SI Appendix, Fig. S1).
Fig. 2.
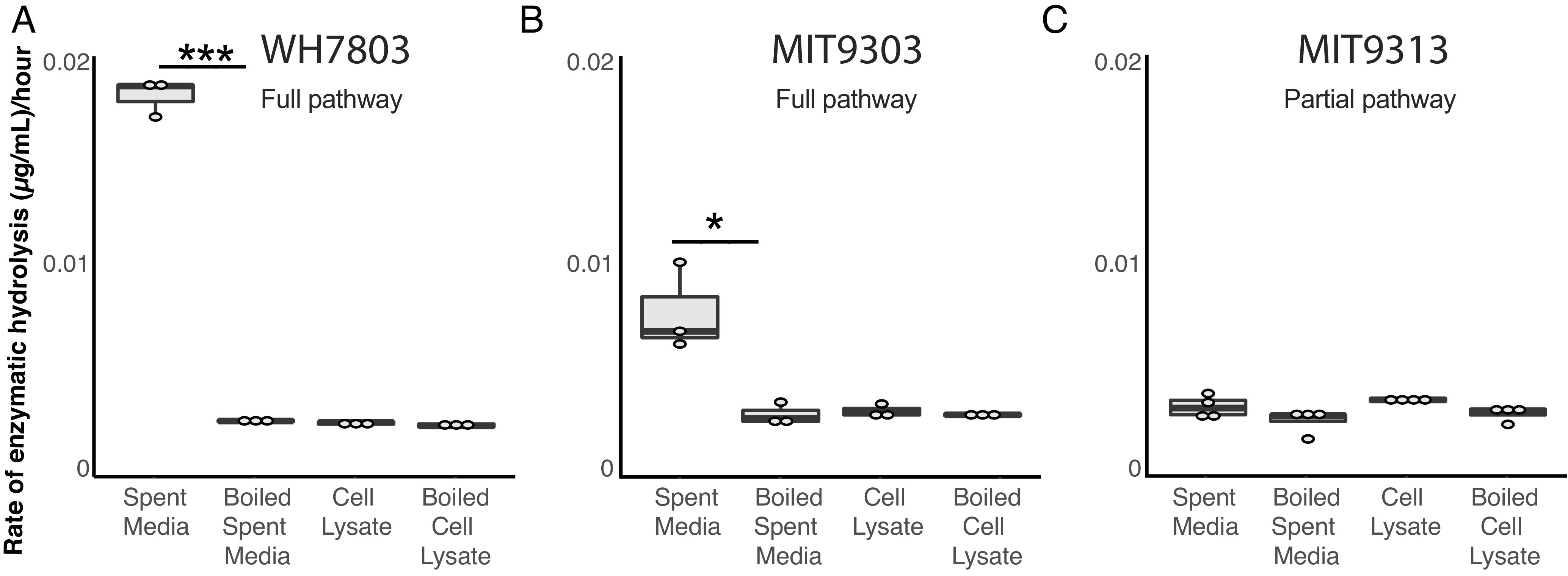
Chitinase activity in Prochlorococcus and Synechococcus. Endochitinase activity in Synechococcus WH7803 (A), Prochlorococcus MIT9303 (B), and Prochlorococcus MIT9313 (C) measured in spent media and cell lysates. Strains with the complete chitin degradation pathway display such activity in the spent media. Activity is lost after boiling. *P < 0.05, ***P < 0.001. Exochitinase activity is shown in SI Appendix, Fig. S1.
The next question we addressed was whether or not Synechococcus and Prochlorococcus cells with the chitin utilization pathway colonize chitin surfaces, by adding hydrogel chitin beads to cultures. The Prochlorococcus strains used were from the two subgroups we identified as putative primary (MIT9303) and secondary (MIT9313) degraders (Fig. 1), as well as cells lacking the complete pathway for chitin degradation (MED4). The Synechococcus strain used was WH7803, a primary chitin degrader with a complete set of chitin utilization genes. We found that both Synechococcus and Prochlorococcus cells with chitin utilization genes attach to chitin particles, while Prochlorococcus lacking chitin degradation genes do not (Fig. 3). Primary degraders attach both to the surface of particles and accumulate in “pockets” within the beads (Fig. 3 A, C, D, and F), while secondary degraders only accumulate in pockets (Fig. 3 A, B, and F), potentially reflecting the inferred niche partitioning between them. Finally, we found that Prochlorococcus attaches only to chitin particles and not to agarose, but that Synechococcus attaches to both (Fig. 3A and SI Appendix, Fig. S2), suggesting that Synechococcus has broader surface attachment abilities than Prochlorococcus.
Fig. 3.
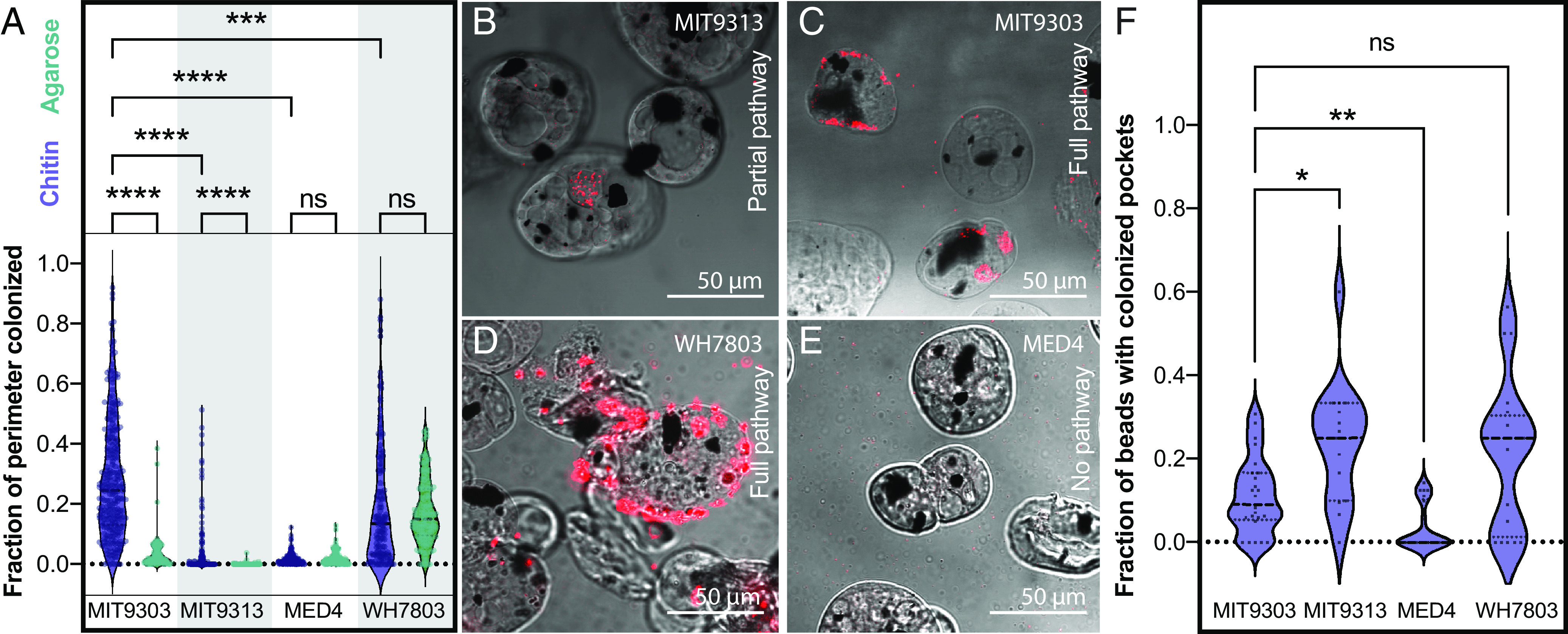
Colonization of artificial chitin particles by Prochlorococcus and Synechococcus. (A) Fraction of particle perimeter colonized by a strain of Synechococcus (WH7803) and three strains of Prochlorococcus (MED4, MIT 9313, and MIT 9303) exposed to agarose (green) and chitin (purple) particles. Synechococcus WH7803 and Prochlorococcus MIT9303 are putative primary chitin degraders, Prochlorococcus MIT9313 is a putative secondary chitin degrader (i.e., it lacks chitinase), and Prochlorococcus MED4 lacks all chitin degradation genes. (B–E). Confocal sections of chitin particles quantified in A. Chitin particles are shown in bright field mode; the dark spots are magnetic beads embedded in the particles. Synechococcus and Prochlorococcus are detected by their red autofluorescence. Cells were sometimes observed contained in pockets within the particles (as shown in B and C, for example). Quantification of this phenomenon is shown in F. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns = not significant.
As a material resource, chitin potentially provides energy, carbon, and/or nitrogen. Prochlorococcus niche partitioning provides relevant clues on what it derives from chitin, as chitin utilization is only retained within the LLIV clade (Fig. 1), which dominates at the bottom of the euphotic zone (23). At these depths, light goes to extinction and Prochlorococcus is thought to derive a significant fraction of its carbon through mixotrophy rather than photosynthesis (7, 8). This suggests chitin could provide Prochlorococcus a supplemental carbon and energy source that is beneficial under low light conditions. To test this hypothesis, we grew Prochlorococcus cells representing both the primary and secondary degrader genotypes (Fig. 1) at several light levels, with and without the addition of chitosan, a form of chitin that is solubilized through partial deacetylation. Differences due to chitosan additions emerged only at the lowest light level (Fig. 4), where chitosan-amended cultures achieved both a higher growth rate and higher final density than unamended cultures. A growth boost due to chitin utilization only under light-limitation is consistent with results in other cyanobacteria showing that wild-type cells able to recycle peptidoglycan fragments generated during the normal course of growth have higher growth rates than mutants without this ability, but only at low light levels (24). We also examined whether Prochlorococcus can use chitosan as a source of nitrogen by adding it to the growth media of nitrogen-starved cells, but relief from nitrogen stress was not apparent (SI Appendix, Figs. S3 and S4). Together, this suggests that in its role as a resource, chitin primarily acts as a supplemental carbon source to light-limited Prochlorococcus cells deep in the water column.
Fig. 4.
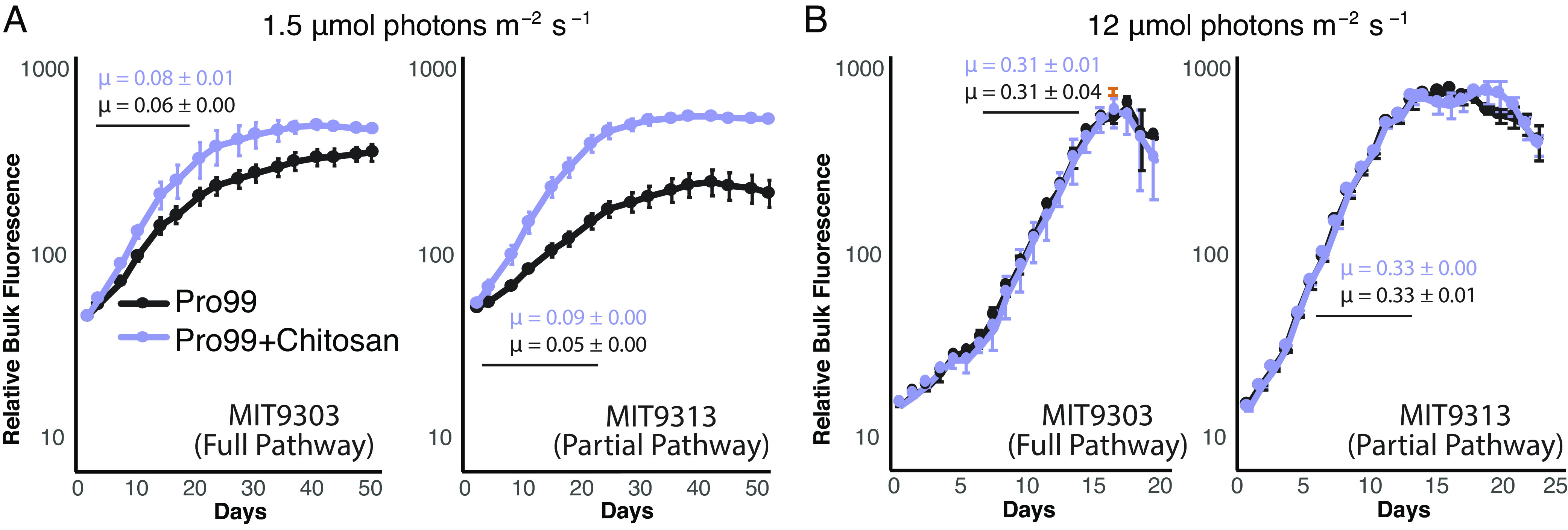
Effect of chitosan addition on growth of two Prochlorococcus strains as a function of light intensity. MIT9303 is a primary chitinase degrader and MIT9313 is a secondary degrader, missing the chitinase genes. Cultures were grown in Pro99 media with (purple) and without (black) added chitosan, at two different light intensities: 1.5 μmol photons m−2 s−1 (A) and 12 μmol photons m−2 s−1 (B), the former being growth-limiting for these strains as reflected in the difference in growth rates, μ, at the different light levels. Growth was monitored by bulk chlorophyll fluorescence and cell counts were measured using flow cytometry in cultures growing at 1.5 μmol photons m−2 s−1 (SI Appendix, Fig. S5). Error bars show SD between the three biological replicates. The average growth rate and associated SD (μ, in units day−1) were calculated in exponential phase (marked with a black line) and is shown for each curve.
To investigate how cells respond to chitin addition at the molecular level, we used qPCR to measure the expression of genes in the chitin degradation pathway in Prochlorococcus cultures grown with and without chitosan. All genes were expressed under both conditions (Dataset S2), including the chitinase and chitobiose transporter genes that are specific to chitin degradation (Fig. 5A). This suggests cells constitutively express this pathway, perhaps so they are poised to use chitin when it appears in the environment. Furthermore, while changes in expression due to chitosan exposure were not statistically significant for most genes, we observed a clear trend across the full suite of genes. That is, shortly after chitosan addition, expression of chitinase genes was higher relative to unamended cultures, while expression of downstream genes was lower (Fig. 5A). At later time points, the expression of chitinase was lower and downstream genes higher in amended relative to unamended cultures (Fig. 5A). These patterns suggest that Prochlorococcus cells increase the activity of chitin degradation enzymes in the order in which pathway intermediates become available, which is broadly similar to how chitin degradation pathways operate in other chitinolytic bacteria (25).
Fig. 5.
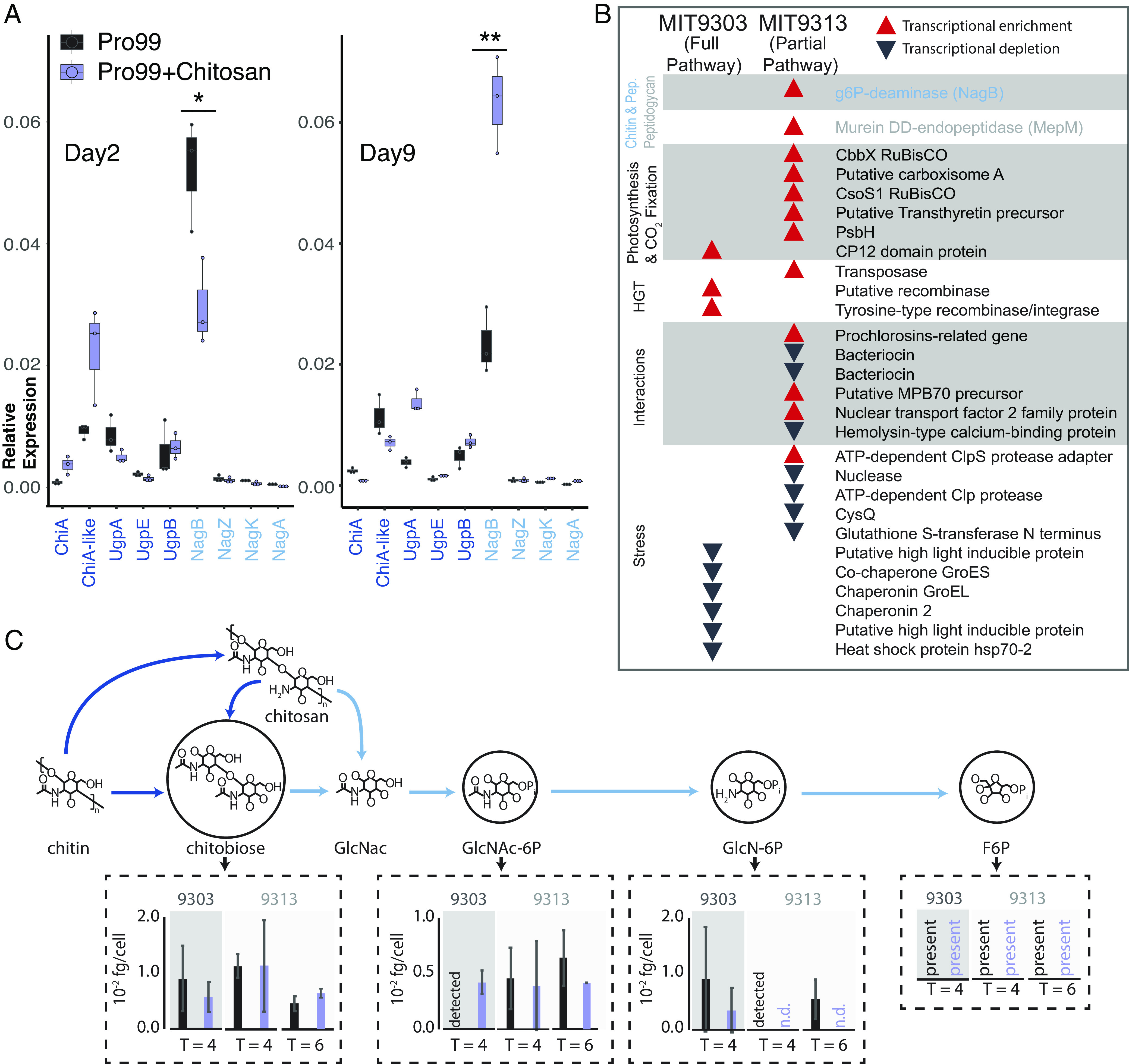
Gene expression and metabolomic analysis in Prochlorococcus in response to the addition of chitosan. (A) Expression (measured by qPCR) of all the chitin-related genes in MIT9303 (primary degrader) in relation to the housekeeping gene, rnpB, gene in natural seawater-based Pro99 medium in presence and absence of chitosan at two time points over the growth curve. Cells were in early exponential growth on day 2 and mid-exponential on day 9. *P < 0.05, **P < 0.01. (B) Qualitative representation of the relative expression of genes (measured using RNA-Seq) in the two Prochlorococcus strains 24 h after addition of chitosan (SI Appendix, Fig. S6). Red upward pointing arrows represent statistically significant transcript enrichment in presence of chitosan while black downward pointing arrows represent statistically significant transcript depletion. Genes are grouped by functional categories, which are shown in shaded/unshaded sections. (C) Concentrations of intermediates of chitin degradation on days 4 and 6 after chitosan additions (purple) compared to unamended controls (black). Error bars show the SDs of three biological replicates for MIT9303 and MIT9313 on day 4, and of two replicates for MIT9313 on day 6. Molecular abbreviations and color scheme is the same as in Fig. 1. Metabolites are labeled as “detected” in samples where they were observed in only a single replicate, and as “n.d.” (not detected) in samples where they were not observed in any replicates. Fructose-6-P was present in most samples, but its levels could not be quantified due to interference by the organic carbon matrix.
To augment the qPCR results and gain a systems-level understanding of how the availability of chitin shapes Prochlorococcus, we performed RNA sequencing (RNA-Seq) and metabolomic analyses of cultures exposed to chitosan. While expression of many genes changed in response to the addition of chitosan, relatively few of those changes were significant, and they disappeared after 48 h (Fig. 5B and SI Appendix, Fig. S6). As with our analysis of the expression of individual genes by qPCR, this suggests a mild and transient response to chitosan additions. We further observed transcripts of all genes of the chitin utilization pathway and accumulation of all its metabolic intermediates under all conditions (Fig. 5C and Datasets S2 and S3), consistent with the idea that cells constitutively express this pathway and not only when chitin appears in the environment, as also suggested by qPCR data (Fig. 5A).
While the chitin utilization pathway is always active, Prochlorococcus metabolism also responds to chitosan additions: in one strain, MIT9313 (secondary degrader), the expression of two genes involved in peptidoglycan recycling (NagB, MepM), one of which is also involved in chitin utilization (NagB), significantly increased (Fig. 5B), consistent with the noted molecular, physiological and ecological links between pathways (Fig. 1). Similarly, concentrations of chitin degradation intermediates also changed upon addition of chitosan, although most changes were not significant (Fig. 5C). Additional changes occurred in downstream metabolic processes. Transcripts of several genes involved in photosynthesis and carbon fixation were enriched in the presence of chitosan in MIT9313, a secondary degrader of chitin, which could have indirectly contributed to the difference in cell number observed in low light levels (Fig. 4A). Transcripts for CP12, which inhibits carbon fixation (26), were enriched in the presence of chitosan in MIT9303, a primary degrader of chitin (Figs. 1 and 3B). Finally, the concentration of some intermediates of core carbohydrate metabolism changed in the presence of chitosan (SI Appendix, Fig. S7), although changes were again largely not significant. While changes in gene transcripts and metabolites are minimal under the conditions we tested, above observations are consistent with chitosan exposure modifies the processing of carbon through core metabolic pathways.
Changes in a number of other functional categories in cells exposed to chitosan can be grouped together in terms of their inferred ecological function. Transcripts of various genes involved in horizontal gene transfer (HGT) (e.g., transposases and recombinases), and microbial interactions (e.g., antibiotics, lanthipeptides, secreted proteins) were enriched, while transcripts of various genes involved in mitigating stress (e.g., chaperonins, high light-inducible proteins, proteases, and genes related to glutathione) were depleted (Fig. 5B and SI Appendix, Fig. S6). Since life in biofilms increases microbial interactions (27), promotes HGT (28), helps buffer against external stresses (27), and increases likelihood of the light-limited conditions under which peptidoglycan recycling becomes beneficial (24), we interpret these collective changes as cells preparing to attach to chitin particles and switch to a surface-attached lifestyle.
To gain insight into the Earth-historical context of the acquisition of the chitin utilization trait in Prochlorococcus and Synechococcus, we examined when this acquisition happened relative to the divergence of marine picocyanobacteria from other cyanobacteria. Picocyanobacterial sequences encoding NagZ, NagA, and NagB—each of which are involved in both chitin utilization and peptidoglycan recycling (Fig. 1)—are nested within a broader diversity of cyanobacterial proteins in phylogenetic analyses (Fig. 6 and SI Appendix, Fig. S8), indicating ancestral vertical inheritance of this part of the pathway. In contrast, picocyanobacterial sequences for ChiA/ChiA-like and NagK proteins group within noncyanobacterial diversity, indicating HGT to picocyanobacteria after their divergence from other cyanobacteria (Fig. 6A and SI Appendix, Fig. S8). NagK is present in deeply branching picocyanobacterial groups (i.e., including Cyanobium and related Synechococcus that bridge the fresh-salt water divide), but chitinase genes are exclusive to marine SynPro (Fig. 6A and SI Appendix, Fig. S8). NagK fortifies peptidoglycan recycling by completing a secondary metabolic pathway involved in metabolizing GlcNAc, but is obligately required for chitin utilization (Figs. 1 and 6B). These observations indicate that peptidoglycan recycling is ancestral to marine SynPro, and provided a preadaptation to chitin utilization. Finally, picocyanobacterial sequences for subunits of the putative chitobiose transporter UgpBAE are also nested within a broader diversity of cyanobacterial proteins (SI Appendix, Fig. S8), suggesting it may have been recruited to participate in the chitin utilization trait from another function in ancestral cyanobacteria. This is also consistent with its broader distribution in Prochlococcus than some other genes involved in chitin utilization (Fig. 1). Collected observations thus indicate that chitinase activity and chitin utilization is a derived trait in marine SynPro, acquired via HGT of chitinase genes in the stem lineage preceding the diversification of extant crown-group marine SynPro (Fig. 6B).
Fig. 6.

Phylogenies of ChiA and NagK genes in marine picocyanobacteria. (A) Red arrows indicate inferred HGTs into SynPro ancestor (stem) lineages. Branch lengths are indicated by included scale bars (average substitutions/site). High-level taxonomic identities are indicated by color coding, with specific represented groups labeled for terminal and collapsed groups. In each case, trees are rooted at the branch closest to the midpoint that preserves the monophyly of Synechococcus groups including SynPro. Branch supports (approximate likelihood ratio test/bootstrap value) are included for bipartitions with less than 90% support for either metric. Black circles at the internal node represent branches with both supports >90%. Phylogenies of NagZ, NagA, NagB, UgpA, and UgpE are found in SI Appendix, Fig. S8. (B) Pathways for chitin utilization and peptidoglycan metabolic recycling. Genes exclusive to chitin utilization are shown in dark blue, and genes shared between the two pathways are shown in blue. Genes highlighted with dashed circles predate evolution of marine picocyanobacteria, while genes highlighted with solid circles were acquired along the stem leading to their crown group.
To better understand the context of the transfer of chitinase genes to the ancestors of marine SynPro we examined their phylogenetic relationships to similar sequences found within other bacterial groups. Picocyanobacterial chitinases contain two major chitin-binding domains that show homology to different chitinase sequence variants found within other bacterial genomes. Gene sequence alignments suggest that the marine SynPro variant is likely the product of a fusion of two genes that were both acquired from Planctomycetes (SI Appendix, Text 1). However, the sampling of Planctomycete diversity is too sparse to determine if the gene fusion event occurred before or after the transfers into ancestral marine SynPro (see SI Appendix, Text 1 and https://doi.org/10.5061/dryad.gxd2547pp for detailed discussion). Planctomycete genomes and metagenomes containing chitinases are found in both fresh and marine environments, preventing us from determining if the gene transfers to picocyanobacteria occurred in freshwater environments prior to colonization of the ocean, or within the marine planktonic environment. Recipients of chitinase genes from Planctomycetes include both marine and freshwater bacteria (SI Appendix, Text 1), suggesting the lack of a strong congruence between ecological and phylogenetic signals.
Palaeobiological evidence provides further insight into the early evolution of chitin utilization in marine picocyanobacteria. First, molecular clocks constrained by horizontally transferred genes that link the timing of cyanobacterial evolution to that of other bacterial phyla indicate that the stem lineage leading to crown group SynPro existed during the interval of ~570 to 420 Ma (29) (Fig. 7A). Second, both fossils and fossil-calibrated molecular clocks indicate that marine arthropods also underwent a major ecological expansion between ~535 to 400 Ma (30–32). Finally, while the body fossil record from open ocean environments is limited because of a high likelihood of degradation before preservation, the second half of this period (~480 to 400 Ma), nearer to the calculated timing of crown group SynPro (Fig. 7A), is thought to have seen an increase in pelagic planktonic arthropods (33) and the emergence of more complex trophic structures in the open ocean (33–36). These dates are consistent with the hypothesis that the early evolution of marine picocyanobacteria and arthropods was intertwined, and played a role in establishing modern marine ecosystems.
Fig. 7.
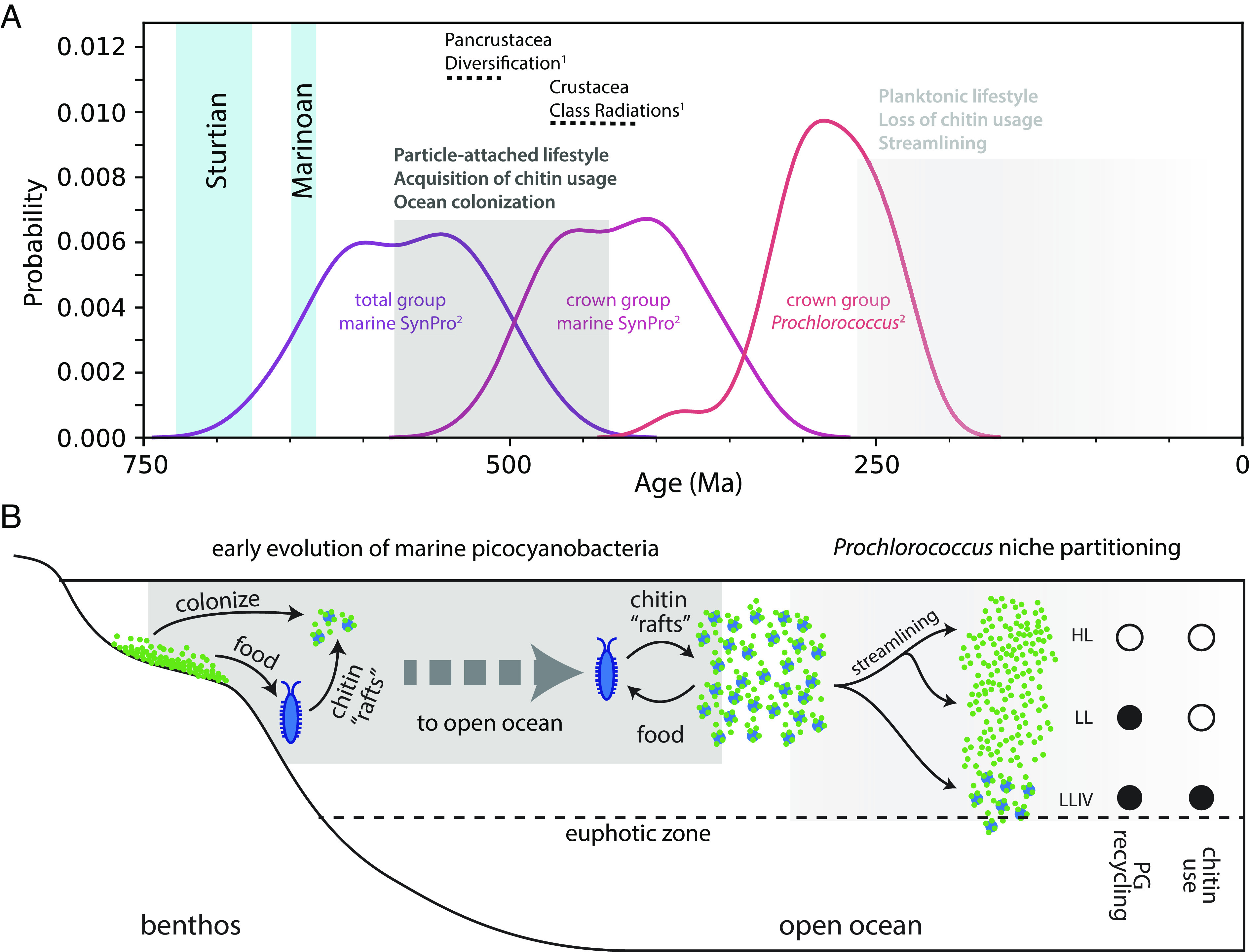
Hypothesized timing and ecological context of the rise of marine picocyanobacteria. (A) Summary of divergence time estimates for the evolution of 1) arthropods (30) and 2) marine picocyanobacteria (29), which we postulate are linked via chitin utilization metabolism. “Total group marine SynPro” refers to the point of divergence of marine SynPro from other nonmarine picocyanobacteria, which constitutes the older-bound on the acquisition of chitinase genes. “Crown-group marine SynPro” refers to the last common ancestor of extant marine SynPro lineages, which constitutes the younger bound on the acquisition of chitinase genes. Vertical blue bars represent the snowball Earth intervals preceding the rise of marine picocyanobacteria. Gray boxes in B indicate the timeline in which events illustrated in C occurred. (B) The “chitin-raft hypothesis” for the rise of marine picocyanobacteria as outlined in the main text. Chitinaceous waste of marine arthropods provided a substrate in the pelagic environment that benthic cyanobacteria could colonize while preserving their surface-attached lifestyle. The chitin utilization trait was later lost in most Prochlorococcus lineages, but preserved in the LLIV clade of Prochlorococcus, giving cells access to a supplemental carbon source under the low light conditions at the bottom of the euphotic zone.
Given chitin utilization by marine picocyanobacteria and their apparent contemporaneous evolution with arthropods, one might expect existence of direct associations with arthropods in the extant biosphere. However, while Synechococcus has been found to be abundant in copepod guts (37), we found no reports of direct associations with the exoskeletons of living copepods or other arthropods (37, 38). It is possible that chitin utilization by picocyanobacteria has become primarily restricted to detrital particulate matter, derived from exoskeletal molts and dead bodies. Alternatively, direct arthropod-picocyanobacterial associations may yet be discovered now that there is added motivation to look for them. Indeed, picocyanobacterial symbionts have been discovered in association with an increasingly diverse set of algae in recent years, including dinoflagellates, foraminifera, radiolarians, and tintinnids (15, 39–43).
The emergence of chitin attachment and utilization during the rise of marine picocyanobacteria helps address a conundrum regarding the evolution of lifestyle in this group. In most environments other than lakes and oceans, microbes predominantly live as aggregates attached to surfaces, which provides protection from external stresses and allows efficient recycling of nutrients (27). The earliest marine cyanobacteria likely also predominantly lived in mats in the benthic environment (13, 44–46). In contrast, extant marine picocyanobacteria are generally thought to live a single-celled planktonic life (13, 14), despite existing in a saline, ultraviolet radiation (UV) rich, and nutrient-poor environment, which are all factors associated with inducing aggregation of cells and attachment to surfaces (27, 47–49). This raises the question: what was the sequence of innovations that allowed cyanobacteria to make the evolutionary transition from life in benthic mats to life as individual planktonic cells in the open ocean?
To resolve this conundrum, we propose the “chitin raft hypothesis,” in which picocyanobacteria and arthropods colonized the open ocean in tandem (Fig. 7B). Accumulated detrital chitin and other organic material derived from the expansion of arthropods could have provided “rafts” that allowed picocyanobacteria to maintain a surface-attached lifestyle while expanding into the ocean. By promoting collective nutrient recycling and stress mitigation, life in chitin particle-attached communities would have provided refugia from the harsh environment of the open ocean, allowing cells over millions of years of evolution to acquire necessary adaptations for an eventual transition to a single-celled planktonic life. Indeed, evolution of marine picocyanobacteria involved acquisition of genes for synthesis of unique compatible solutes for mitigating osmotic stress in saline environments (50, 51), various genes involved in mitigating light/UV stress or repairing light damage (52, 53), a number of changes to the membrane, genome, and proteome that are thought to lower cellular nutrient requirements (54–56), and a general remodeling of metabolism that is thought to enhance cellular nutrient affinity (57). Each of these innovations acts to mitigate against stresses that in other systems and/or other contexts are linked to triggering aggregation of cells and attachment to surfaces (27, 47–49).
This sequence of innovations culminated with a period of dramatic genomic and proteomic streamlining (54, 56) (Fig. 1) and accumulation of nonsynonymous nucleotide substitutions (58) along the branch separating the LLIV clade and all other Prochlorococcus, indicating the occurrence of a population bottleneck and a shift in the selective landscape. This transition also involved a shift in other Prochlorococcus lineages toward a smaller and rounder cell (59, 60) and the loss of both chitin utilization genes (Fig. 1) and the ability to attach to chitin particles (e.g., MED4 in Fig. 3). Together these observations lead us to conclude that the divergence between LLIV and other Prochlorococcus involved a transition from a facultatively particle-attached to a constitutively planktonic lifestyle. Metabolic reconstructions indicate that streamlining was part of a drive toward greater cellular energy flux with as a by-product increased organic carbon exudation, in turn driving co-evolution with co-occurring heterotrophs (57). Added context from our findings here suggests that various adaptations over the course of Prochlorococcus evolution collectively mediated an ecological transition from physical-chemical interactions at microscopic scales on particles to primarily chemical interactions at ecosystem scales in the water column.
Materials and Methods
Culture Conditions for Growth Curves.
Prochlorococcus or Synechococcus cells were grown under constant light flux at 24 °C in natural seawater-based Pro99 medium containing 0.2-μm-filtered Sargasso Sea water, amended with Pro99 nutrients (N, P, and trace metals) prepared as previously described (61). The low light experiment in Prochlorococcus MIT9303 and MIT9313 was performed at a light level of 1.5 μmol quanta m−2 s−1. The other experiments in Prochlorococcus or Synechococcus were performed at 12 or 15 μmol quanta m−2 s−1 as specified in each legend. The final concentrations of NH4+were 800 μM in Pro99, 250 μM in Pro5, 150 μM in Pro3, and 50 μM in Pro1. In these experiments, half of the samples were amended with high molecular weight chitosan (Millipore Sigma) to a final concentration of 56 μg/mL. All the growth curves were performed in triplicates.
Identification of Genes Involved in Chitin Degradation and Peptidoglycan Recycling.
To identify genes potentially involved in chitin-degradation, we searched for homologs to known chitin degradation genes (12) in Prochlorococcus strains MIT9313 and MIT1318 using blastp (62) v2.12.0+ with default settings. Similarly, for peptidoglycan recycling, we searched for homologs to known peptidoglycan recycling genes (17). We found homologues for most genes of the peptidoglycan recycling pathway employed by Escherichia coli (17) in Prochlorococcus, most of which had previous functional annotations consistent with our findings. We next assigned each of these sequences to National Center for Biotechnology Information (NCBI) clusters of orthologous groups (COGs) (63) based on reciprocal best hits (Dataset S1_chitin-cogs)
We then expanded these annotations to our entire collection: 623 Prochlorococcus (64) and 79 Synechococcus genomes (65) (Dataset S1_pro-syn-genomes). Of note, the majority of these genomes were derived from single-cell sequencing projects and often are not complete. The estimated average completeness for genomes in this set is 75%. We first clustered all proteins using mmseqs (66) (--cov-mode 5 -c 0.5), then annotated the cluster representatives with ggnog-mapper (67) to obtain NCBI COG labels for each cluster, and, finally, crossmatched the clusters with the COG-labels assigned to our seed sequence set. We manually checked and if necessary refined each matched protein cluster for consistency and correct functional assignment. For the final list of annotated genes see (Dataset S1 pro-chitin-genes).
Quantitative PCR Analysis.
Prochlorococcus MIT9303 cells grown at 11 μmol photons m−2 s−1 were collected from the samples by centrifugation in triplicates at each time point and each condition. RNA samples were extracted with a standard acidic Phenol:Chloroform protocol and measured with Nanodrop (Thermo Scientific). RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) with random primers was used to obtain complementary DNA (cDNA). Quantitative PCR reactions were performed in a CFX96 thermocycler (Bio-Rad) using the primers listed in SI Appendix, Table S1. The expression of rnpB gene was used to normalize the results.
Chitinase Assay.
Prochlorococcus MIT9303 and MIT9313 and Synechococcus WH7803 cultures were grown at 15 μmol quanta m−2 s−1 in Pro99 media amended with high molecular weight chitosan (Millipore Sigma) to a final concentration of 56 μg/mL in triplicates. A volume of 50 mL of culture in mid-exponential was then centrifuged to separate the cells fraction and the spent media. The pellet was flash frozen, resuspended in sterile MilliQ water to a volume of 2 mL, sonicated, and filtered through a Spin-X centrifuge tube filter (Costar). The supernatant was filtered through a 0.45-μm filter and concentrated using 30 kDa Amicon® Ultra-15 Centrifugal Filter Units (Millipore) to a volume of 1.5 mL. Half volume of each sample has been boiled at 90 °C for 45 min to serve as controls. Protease inhibitor (Roche) was added to all samples. Each sample was then divided into three aliquots. Each aliquot was tested with one of the three substrates contained in the Chitinase kit (Sigma): 4-methylumbelliferyl N,N′-diacetyl-β-D-chitobioside (substrate suitable for exochitinase activity detection or chitobiosidase activity), 4-methylumbelliferyl N-acetyl-β-D-glucosaminide (substrate suitable for exochitinase activity detection or β-N-acetylglucosaminidase activity), and 4-methylumbelliferyl β-D-N,N′,N′′-triacetylchitotriose (substrate suitable for endochitinase activity detection).
The aliquots were then mixed with each of these substrates, incubated in darkness and the fluorescence of the 4-methyl-umbelliferone released by the chitinase activity in the sample was measured every 24 h on a plate reader set at excitation 360 nm and emission at 450 nm.
Beads and Microscopy.
Chitin magnetic beads (NEB Cat#E8036L), or Agarose magnetic beads (Pierce™ Glutathione Magnetic Agarose Beads Thermo Scientific™ Cat#78602) were washed in Pro99 media and size selected using cell strainers. Chitin beads smaller than 40 µm or larger than 100 µm have been discarded during the washes with Pro99. The media enriched with beads has then been inoculated with the Prochlorococcus or Synechococcus strains of interest, in triplicates in light level of 30 μmol quanta m−2 s−1 (for MED4 and WH7803) and 11 μmol quanta m−2 s−1 (for MIT9303 and MIT9313). Growth was monitored using bulk culture fluorescence measured with a 10AU fluorometer (Turner Designs) and samples were collected in late exponential, at a cell density of 108. A volume of 1 mL of each sample was poured on a 35-mm Petri dish with 20-mm glass bottom microwell dish (Mat Tek). To avoid excessive fluorescent signal from floating cyanobacteria, the samples were washed five times with fresh media. Floating culture was removed by placing the mini dish on a magnet (to keep the magnetic beads on the bottom) and then substituted with fresh Pro99 media before placing the sample on the scope. Images were taken with a LSM780 Zeiss, capturing the beads on the bright field and the Prochlorococcus by its autofluorescence with laser 561.
Analysis of the Colonized Perimeter and Cells Colonizing Pockets.
To quantify the fraction of cells colonizing the perimeter of chitin or agarose magnetic beads, we wrote a custom analysis script using MATLAB (version R2020a, Mathworks. Code available at https://github.com/jaschwartzman/prochlorococcus). The script segmented the brightfield image of magnetic beads to create a binary mask for the bead perimeter. To account for the fact that chitin magnetic beads are semi-transparent, the mask was created using the gradient of the image, which defines the shadow around the perimeter of the beads. This area was filled and eroded to define the bead center. The center area was subtracted from the total area to define the perimeter zone of the beads. Any cells found in this zone are assumed to be attached to the bead surface. To define cell area, we segmented the accompanying image of cell autofluorescence. Briefly, cells were segmented by defining a linear threshold based on the magnitude of the gradient of the autofluorescence image. This threshold was defined for each day of experimental acquisition, to account for variation in acquisition parameters. For each bead, the perimeter area, and the subset of the perimeter area colonized by cells were quantified. The fraction of the perimeter area colonized was defined by dividing the total perimeter area by the cell area. An assumption of this quantification is that cells from different taxa occupy approximately the same two-dimensional area.
The chitin magnetic beads were found to contain pockets, in which we sometimes observed cells. The irregularities and pockets internal to the chitin hydrogel were visible due to the weak autofluorescence of chitin detected in the 561-nm channel and were also identifiable in brightfield images. In the latter scenario, the greater depth of field in the brightfield image creates the possibility that cells are sitting on a surface above a hollow feature. Due to the diverse morphologies of pockets, we were unable to define a consistent set of characteristics to segment these structures using a script. Accordingly, we quantified the number of particles in which we observed cells colonizing internal pockets manually, by counting instances of cells colonizing either a zone with less autofluorescence within a particle and verifying the presence of a pocket in the brightfield image.
RNA-Seq and Metabolites Analysis.
Prochlorococcus MIT9303 and MIT9313 cells were grown under constant light flux at 11 μmol photons m−2 s−1 at 24 °C in artificial AMP1 medium (61). Cultures were transferred to AMP1 from Pro99 and kept in AMP1 for at least three transfers before performing the experiment to avoid any potential contributions to observed pathway dynamics by background levels of uncharacterized organic polymers in natural seawater. Half of the samples were supplemented with chitosan (Millipore Sigma) to a final concentration of 56 μg/mL. The growth curves were monitored using bulk culture fluorescence measured with a 10AU fluorometer (Turner Designs) and the cell count was recorded using Guava® easyCyte™ High-throughput Flow Cytometer. Samples were collected on day 1 and day 3 after inoculation for RNA extraction and day 4 and day 6 for the metabolomics analysis.
For the RNA-Seq Prochlorococcus cells were collected from the samples by centrifugation in triplicates at each time point and each condition. RNA samples were extracted with a standard acidic Phenol:Chloroform protocol, rRNA was depleted with NEBNext® rRNA Depletion Kit (Bacteria); libraries were prepared with KAPA HyperPrep Kit (Roche) and the multiplexed samples were run on an Illumina HiSeq with a 75nt NextSeq sequencing program.
RNA-Seq Analysis.
The raw Illumina reads were trimmed of adapters using bbduk v38.16 (68) (ktrim = r, k = 23, mink = 11, hdist = 1). Low-quality regions were removed from the adapter-trimmed sequences with bbduk v38.16 (68) (qtrim = rl, trimq = 6). The trimmed RNA-Seq reads from Prochlorococcus MIT9303 and MIT9313 cultures were aligned to the MIT9303 and MIT9313 reference genomes, respectively (available from https://github.com/thackl/pro-tycheposons/) with the Burrows–Wheeler Aligner (BWA) v0.7.16a-r1181 (69), using the BWA-backtrack algorithm. The number of reads that aligned to each annotated open reading frame (ORF) in the “sense” orientation was determined using the HTSeq package v0.11.2 (70) (default parameters, “nonunique all”). Counts of reads that aligned to each ORF (excluding rRNAs and tRNAs) were compiled across replicates. MIT9303 and MIT9313 reads were analyzed separately using the DESeq2 R package v1.24.0 (71) to determine differentially expressed genes. The standard DESeq2 functions and workflow were implemented to normalize samples by library sequencing depth and estimate gene dispersion. Differential expression tests were performed on comparisons of cultures with chitosan vs. control cultures 1 and 3 d after chitosan addition. Significance was determined with the Wald test, using a negative binomial generalized linear model, and P values were corrected for multiple testing with the Benjamini–Hochberg procedure. Following the guidance of the DESeq2 authors (71), genes were considered to be significantly differentially expressed between a given pair of treatments if the adjusted P value was <0.1. Gene expression results were visualized with ggplot2 (72).
Metabolite Extractions.
For the metabolomics analysis, Prochlorococcus cells were filtered under gentle vacuum (to prevent cell lysis) in a glass filtration tower onto 0.1 µM Omnipore filters (Millipore). Filters were then folded and placed into a cryovial, and frozen at −80 °C until processed. We extracted intracellular metabolites using a method described in ref. 73 and modified as described in ref. 74. Briefly, we extracted each filter with ice-cold extraction solvent (40:40:20 acetonitrile: methanol: water with 0.1 M formic acid), neutralized the combined extracts with 6M ammonium hydroxide, and dried them in a vacufuge until near dryness. Prior to liquid chromatography-mass spectrometry (LC/MS) analysis, we reconstituted all extracts in 245 μL of solvent (30 μL Milli-Q pure water, 70 μL methanol, and 145 μL acetonitrile) with isotopically labeled injection standards (50 pg/μL each; d2 biotin, d6 succinic acid, d4 cholic acid, d7 indole-3-acetic acid, and 13C1-phenylalanine). We generated a pooled sample for quality control (QC) by combining 25 μL aliquots from each sample.
LC/MS Analysis of Metabolites.
The organic matter extracts were analyzed using targeted LC/MS. A mix of metabolite standards (Dataset S3) was made in 80:20 acetonitrile:Milli-Q water at 10 µg/mL. Matrix-matched calibration solutions were prepared by diluting his mix in the pooled sample with methanol and water added to match the composition of the samples. The concentrations prepared included 0.25, 0.5, 2.5, 5, 12.5, 25, 50, 125, 250, and 500 ng/mL. Separate hydrophilic interaction liquid chromatography (HILIC) methods were developed to separate metabolites in negative and positive ionization modes. For negative mode, a 2.1 × 100 mm, 2.7 µm Infinity Lab Poroshell HILIC-Z, P column (Agilent Technologies) was used. A mobile phase at pH 9 was prepared for gradient elution with 10 mM ammonium acetate in water (A) and 10 mM ammonium acetate in 90% acetonitrile (B) and pH adjusted with ammonium hydroxide. The additive medronic acid (5 mM methylenediphosphonic acid) was added to A and B at final 5 µM concentration to improve peak shape (75). A sample volume of 5 µL was injected onto the column held at 30 °C, and eluted at a flow rate of 0.3 mL/min with the following gradient: hold at 90% B from 0 to 1.7 min, 90 to 60% from 1.7 to 10 min, hold at 60% B from 10.2 to 12.7 min, return to 90% B at 13.5 min, and equilibrate with 90% B till 20 min. For positive ion mode analysis, a 2.1 × 100 mm, 1.7 µm Acquity ethylene bridged hybrid Amide column (Waters Corporation) was used. A mobile phase at pH 10 was prepared and composed of 0.1% ammonium hydroxide in Milli-Q water (A) and 0.1% ammonium hydroxide in 90% acetonitrile. A 5 µL sample volume was injected onto the column held at 40 °C, and eluted at 0.3 mL/min with the same gradient as above. The autosampler was washed between injections to prevent carryover with two successive wash solutions, 50:50 acetonitrile/water followed by 95:5 acetonitrile/water.
To quantify metabolites, we used liquid chromatography (Accela Open Autosampler and Accela 1250 Pump, Thermo Scientific) coupled to a heated electrospray ionization source and a triple quadrupole mass spectrometer (TSQ Vantage, Thermo Scientific) operated under selected reaction monitoring (SRM) mode. Optimal SRM parameters (s-lens, collision energy, fragment ions) for each target compound were determined with authentic standards purchased from Sigma. The complete list of metabolites is provided in SI Appendix, Table S2. We monitored two SRM transitions per compound for quantification and confirmation and generated external calibration curves based on peak area for each compound. We converted raw data files from proprietary Thermo (.RAW) format to mzML using the msConvert tool (76) prior to processing with El-MAVEN (77). Samples were analyzed in random order within each batch, and the pooled QC sample was analyzed every six injections.
Phylogenetic Analysis.
Sequences were collected from the Genbank database (78) for the following chitin degradation pathway proteins: The N-terminal region of ChiA/ChiA-like, the C-terminal region of ChiA/ChiA-like, UgpA, UgpE, NagZ, NagK, NagA, and NagB. Orthologs found within Prochlorococcus MIT1303 were used as protein search queries using BLASTP (79), with the top 500 or 250 hits recovered in each case. For BLAST, blastp 2.11.0 program was used with default parameters (BLOSUM62 matrix, Word size 6, Expect value 0.05, Hitlist size 500, Gapcosts 11,1. 500 hits were collected for the initial ChiA/ChiA-like BLASTP search and in an additional follow up search, the top 250 Blast hits were retrieved for three independent regions (N-terminal, linker region containing CBDII domains, C-terminal region containing GH18 domain). 250 Blast hits were retrieved for NAG_Kinase, NagA, NagB, NagZ, UgpA, UgpE respectively. Each set of sequences were then aligned in MAFFT (80) with the automatic algorithm selection option. Aligned sequences were then used for phylogenetic reconstruction using IQTree (81) with automatic best-fitting model selection. All sequence alignment and phylogenetic data files are available in SI data (Dataset S1 and SI Appendix, Text 1 files), with alignment and tree filenames in each case describing the algorithms and parameters used for these reconstructions. Several BLAST hits for the ChiA and ChiA-like genes in SynPro overlapped, with some protein sequences containing multiple domains homologous to different chitinase orthologs in other bacteria. A phylogenomic analysis of our alignment data showed that the SynPro variant was likely the result of a fusion between genes from Planctomycetes, before or after HGT into SynPro (see SI Appendix, Text 1 and SI Appendix, Text 1 files for a detailed analysis of the protein fusion history).
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (CSV)
Dataset S05 (CSV)
Acknowledgments
We thank members of the Chisholm lab for thoughtful discussions, and Nikolai Radzinski for insightful discussions and useful references regarding microbial aggregation. This research was supported by grants from the Simons Foundation (Award ID 509034SCFY20 to R.B. and S.W.C., Award ID 509034FY20 to E.B.K., and SCOPE Award ID 329108 to M.J. Follows; Life Sciences Award IDs 337262, 647135, and 736564 to S.W.C.; SCOPE Award ID 329108 and 721246 to S.W.C.). G.C. was also supported by the European Molecular Biology Organization (EMBO) Long-Term Fellowship (ALTF 904-2018) and by the Human Frontier Science Program (LT000069/2019-L). This paper is a contribution from the Simons Collaboration on Ocean Processes and Ecology (SCOPE).
Author contributions
G.C., R.B., G.P.F., T.H., and A.Y. designed research; G.C., R.B., G.P.F., T.H., J.S., X.L., K.L., M.C.K.S., E.T., G.S., A.M., J.G.P., K.G.C., J.R.W., and E.B.K. performed research; G.C., R.B., G.P.F., J.S., E.T., J.G.P., and O.X.C. analyzed data; G.C., R.B., and G.P.F., interpreted data; G.C., X.L., A.M., and K.C. performed culturing, molecular biology and imaging experiments; R.B., T.H., and E.T. performed bioinformatic analyses; R.B. developed macroevolutionary model; G.F. and J.G.P. performed phylogenetic analyses; K.L., M.C.K.S., G.S., and E.B.K. performed metabolite analyses; and G.C., R.B., G.P.F., and S.W.C. wrote the paper with input from all co-authors.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Giovanna Capovilla, Email: giocapo@mit.edu.
Rogier Braakman, Email: braakman@mit.edu.
Sallie W. Chisholm, Email: chisholm@mit.edu.
Data, Materials, and Software Availability
All other data are included in the manuscript and/or supporting information. All 886 raw data for the main text and figures are available for free download at European Nucleotide Archive (ENA) #PRJEB53957 (RNA-Seq data); Metabo-Lights #MTBLS5229 (metabolites data); and Dryad (https://doi.org/10.5061/dryad.gxd2547pp) (82) (Phylogenomic data, including SI Appendix, Supplementary Text 1 file).
Supporting Information
References
- 1.Waterbury J. B., Watson S. W., Guillard R. R. L., Brand L. E., Widespread occurrence of a unicellular, marine, planktonic, cyanobacterium. Nature 277, 293–294 (1979). [Google Scholar]
- 2.Chisholm S. W., et al. , A novel free-living prochlorophyte abundant in the oceanic euphotic zone. Nature 334, 340 (1988). [Google Scholar]
- 3.Flombaum P., et al. , Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. U.S.A. 110, 9824–9829 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gómez-Baena G., et al. , Glucose uptake and its effect on gene expression in prochlorococcus. PLoS One 3, e3416 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muñoz-Marín M. C., et al. , Mixotrophy in marine picocyanobacteria: Use of organic compounds by Prochlorococcus and Synechococcus. ISME J. 14, 1065–1073 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muñoz-Marín M. del C., et al. , Prochlorococcus can use the Pro1404 transporter to take up glucose at nanomolar concentrations in the Atlantic Ocean. Proc. Natl. Acad. Sci. U.S.A. 110, 8597–8602 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Z., et al. , Significant organic carbon acquisition by Prochlorococcus in the oceans. bioRxiv (2022.01.14.476346) (2022).
- 8.Casey J. R., et al. , Basin-scale biogeography of marine phytoplankton reflects cellular-scale optimization of metabolism and physiology. Sci. Adv. 8, eabl4930 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yelton A. P., et al. , Global genetic capacity for mixotrophy in marine picocyanobacteria. ISME J. 10, 2946–2957 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cauchie H.-M., Chitin production by arthropods in the hydrosphere. Hydrobiologia 470, 63–95 (2002). [Google Scholar]
- 11.Tang K. W., Turk V., Grossart H. P., Linkage between crustacean zooplankton and aquatic bacteria. Aquat. Microb. Ecol. 61, 261 (2010). [Google Scholar]
- 12.Beier S., Bertilsson S., Bacterial chitin degradation-mechanisms and ecophysiological strategies. Front. Microbiol. 4, 149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sánchez-Baracaldo P., Origin of marine planktonic cyanobacteria. Sci. Rep. 5, 17418 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aguilo-Ferretjans M. del M., et al. , Pili allow dominant marine cyanobacteria to avoid sinking and evade predation. Nat. Commun. 12, 1–10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pierella Karlusich J. J., et al. , A robust approach to estimate relative phytoplankton cell abundances from metagenomes. Mol. Ecol. Resour. 23, 16–40 (2022), 10.1111/1755-0998.13592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lomas M. W., Moran S. B., Evidence for aggregation and export of cyanobacteria and nano-eukaryotes from the Sargasso Sea euphotic zone. Biogeosciences 8, 203–216 (2011). [Google Scholar]
- 17.Mayer C., et al. , Bacteria’s different ways to recycle their own cell wall. Int. J. Med. Microbiol. 309, 151326 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Noell S. E., Giovannoni S. J., SAR11 bacteria have a high affinity and multifunctional glycine betaine transporter. Environ. Microbiol. 21, 2559–2575 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Noell S. E., et al. , SAR11 cells rely on enzyme multifunctionality to transport and metabolize a range of polyamine compounds. bioRxiv (2021.05.13.444117) (2021). [DOI] [PMC free article] [PubMed]
- 20.Ebrahimi A., Schwartzman J., Cordero O. X., Cooperation and spatial self-organization determine rate and efficiency of particulate organic matter degradation in marine bacteria. Proc. Natl. Acad. Sci. U.S.A. 116, 23309–23316 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Datta M. S., Sliwerska E., Gore J., Polz M. F., Cordero O. X., Microbial interactions lead to rapid micro-scale successions on model marine particles. Nat. Commun. 7, 11965 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enke T. N., et al. , Modular assembly of polysaccharide-degrading marine microbial communities. Curr. Biol. 29, 1528–1535.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Malmstrom R. R., et al. , Temporal dynamics of Prochlorococcus ecotypes in the Atlantic and Pacific oceans. ISME J. 4, 1252–1264 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Haibo Jiang, Kong Renqiu Xu., Xudong, The N-Acetylmuramic acid 6-phosphate etherase gene promotes growth and cell differentiation of cyanobacteria under light-limiting conditions. J. Bacteriol. 192, 2239–2245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saito Y., Okano T., Gaill F., Chanzy H., Putaux J.-L., Structural data on the intra-crystalline swelling of β-chitin. Int. J. Biol. Macromol. 28, 81–88 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Thompson L. R., et al. , Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci. U.S.A. 108, E757–E764 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flemming H.-C., Wuertz S., Bacteria and archaea on Earth and their abundance in biofilms. Nat. Rev. Microbiol. 17, 247–260 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Abe K., Nomura N., Suzuki S., Biofilms: Hot spots of horizontal gene transfer (HGT) in aquatic environments, with a focus on a new HGT mechanism. FEMS Microbiol. Ecol. 96 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fournier G. P., et al. , The Archean origin of oxygenic photosynthesis and extant cyanobacterial lineages. Proc. Biol. Sci. 288, 20210675 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rota-Stabelli O., Daley A. C., Pisani D., Molecular timetrees reveal a cambrian colonization of land and a new scenario for ecdysozoan evolution. Curr. Biol. 23, 392–398 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Harvey T. H. P., Vélez M. I., Butterfield N. J., Exceptionally preserved crustaceans from western Canada reveal a cryptic Cambrian radiation. Proc. Natl. Acad. Sci. U.S.A. 109, 1589–1594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams M., Vandenbroucke T. R. A., Perrier V., Siveter D. J., Servais T., A link in the chain of the Cambrian zooplankton: Bradoriid arthropods invade the water column. Geol. Mag. 152, 923–934 (2015). [Google Scholar]
- 33.Perrier V., Williams M., Siveter D. J., The fossil record and palaeoenvironmental significance of marine arthropod zooplankton. Earth-Sci. Rev. 146, 146–162 (2015). [Google Scholar]
- 34.Servais T., et al. , The onset of the “Ordovician Plankton Revolution” in the late Cambrian. Palaeogeogr. Palaeoclimatol. Palaeoecol. 458, 12–28 (2016). [Google Scholar]
- 35.Kröger B., Servais T., Zhang Y., The origin and initial rise of pelagic cephalopods in the Ordovician. PLoS One 4, e7262 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blieck A., The Andre Dumont medallist lecture: From adaptive radiations to biotic crises in palaeozoic vertebrates: A geobiological approach/Des radiations adaptatives aux crises biologiques des vertebres paleozoiques: une approche geobiologique. Geol. Belgica 14, 203+ (2011). [Google Scholar]
- 37.Shoemaker K. M., Duhamel S., Moisander P. H., Copepods promote bacterial community changes in surrounding seawater through farming and nutrient enrichment. Environ. Microbiol. 21, 3737–3750 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Datta M. S., et al. , Inter-individual variability in copepod microbiomes reveals bacterial networks linked to host physiology. ISME J. 12, 2103–2113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foster R. A., Collier J. L., Carpenter E. J., Reverse transcription pcr amplification of cyanobacterial symbiont 16s rRNA sequences from single non-photosynthetic eukaryotic marine planktonic host cells1. J. Phycol. 42, 243–250 (2006). [Google Scholar]
- 40.Yuasa T., Horiguchi T., Mayama S., Matsuoka A., Takahashi O., Ultrastructural and molecular characterization of cyanobacterial symbionts in Dictyocoryne profunda (polycystine radiolaria). Symbiosis 57, 51–55 (2012). [Google Scholar]
- 41.Bird C., et al. , Cyanobacterial endobionts within a major marine planktonic calcifier (Globigerina bulloides, Foraminifera) revealed by 16S rRNA metabarcoding. Biogeosciences 14, 901–920 (2017). [Google Scholar]
- 42.Nakayama T., et al. , Single-cell genomics unveiled a cryptic cyanobacterial lineage with a worldwide distribution hidden by a dinoflagellate host. Proc. Natl. Acad. Sci. U.S.A. 116, 15973–15978 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim M., Choi D. H., Park M. G., Cyanobiont genetic diversity and host specificity of cyanobiont-bearing dinoflagellate Ornithocercus in temperate coastal waters. Sci. Rep. 11, 1–13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bosak T., Liang B., Sim M. S., Petroff A. P., Morphological record of oxygenic photosynthesis in conical stromatolites. Proc. Natl. Acad. Sci. U.S.A. 106, 10939–10943 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blank C. E., Sánchez-Baracaldo P., Timing of morphological and ecological innovations in the cyanobacteria–a key to understanding the rise in atmospheric oxygen. Geobiology 8, 1–23 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Lalonde S. V., Konhauser K. O., Benthic perspective on Earth’s oldest evidence for oxygenic photosynthesis. Proc. Natl. Acad. Sci. U.S.A. 112, 995–1000 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flemming H.-C., et al. , Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 14, 563–575 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Allen R., Rittmann B. E., Curtiss R. 3rd, Axenic biofilm formation and aggregation by Synechocystis sp. Strain PCC 6803 are induced by changes in nutrient concentration and require cell surface structures. Appl. Environ. Microbiol. 85, e02192-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Callieri C., Lami A., Bertoni R., Microcolony formation by single-cell Synechococcus strains as a fast response to UV radiation. Appl. Environ. Microbiol. 77, 7533–7540 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klähn S., Steglich C., Hess W. R., Hagemann M., Glucosylglycerate: A secondary compatible solute common to marine cyanobacteria from nitrogen-poor environments. Environ. Microbiol. 12, 83–94 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Blank C. E., Phylogenetic distribution of compatible solute synthesis genes support a freshwater origin for cyanobacteria. J. Phycol. 49, 880–895 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Kettler G. C., et al. , Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet. 3, e231 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Partensky F., Garczarek L., Prochlorococcus: Advantages and limits of minimalism. Ann. Rev. Mar. Sci. 2, 305–331 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Dufresne A., Garczarek L., Partensky F., Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol. 6, R14 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Mooy B. A. S., Rocap G., Fredricks H. F., Evans C. T., Devol A. H., Sulfolipids dramatically decrease phosphorus demand by picocyanobacteria in oligotrophic marine environments. Proc. Natl. Acad. Sci. U.S.A. 103, 8607–8612 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grzymski J. J., Dussaq A. M., The significance of nitrogen cost minimization in proteomes of marine microorganisms. ISME J. 6, 71–80 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braakman R., Follows M. J., Chisholm S. W., Metabolic evolution and the self-organization of ecosystems. Proc. Natl. Acad. Sci. U.S.A. 114, E3091–E3100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo H., Huang Y., Stepanauskas R., Tang J., Excess of non-conservative amino acid changes in marine bacterioplankton lineages with reduced genomes. Nat. Microbiol. 2, 1–9 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Ting C. S., Hsieh C., Sundararaman S., Mannella C., Marko M., Cryo-electron tomography reveals the comparative three-dimensional architecture of Prochlorococcus, a globally important marine cyanobacterium. J. Bacteriol. 189, 4485–4493 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cermak N., et al. , High-throughput measurement of single-cell growth rates using serial microfluidic mass sensor arrays. Nat. Biotechnol. 34, 1052–1059 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moore L. R., et al. , Culturing the marine cyanobacterium Prochlorococcus. Limnol. Oceanogr. Methods 5, 353–362 (2007). [Google Scholar]
- 62.Camacho C., et al. , BLAST+: Architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galperin M. Y., Makarova K. S., Wolf Y. I., Koonin E. V., Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hackl T., et al. , Novel integrative elements and genomic plasticity in ocean ecosystems. Cell 186, 47–62.e16 (2023). [DOI] [PubMed] [Google Scholar]
- 65.Hogle S. L., et al. , Siderophores as an iron refrefsource for picocyanobacteria in deep chlorophyll maximum layers of the oligotrophic ocean. ISME J. 16, 1636–1646 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steinegger M., Söding J., Clustering huge protein sequence sets in linear time. Nat. Commun. 9, 2542 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huerta-Cepas J., et al. , eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bushnell B., BBTools software package. URL http://sourceforge.net/projects/bbmap (2014).
- 69.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anders S., Pyl P. T., Huber W., HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wickham H., ggplot2. WIREs Comp. Stat 3, 180–185 (2011). [Google Scholar]
- 73.Rabinowitz J. D., Kimball E., Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal. Chem. 79, 6167–6173 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Kido Soule M. C., Longnecker K., Johnson W. M., Kujawinski E. B., Environmental metabolomics: Analytical strategies. Mar. Chem. 177, 374–387 (2015). [Google Scholar]
- 75.Hsiao J. J., Potter O. G., Chu T.-W., Yin H., Improved LC/MS methods for the analysis of metal-sensitive analytes using medronic acid as a mobile phase additive. Anal. Chem. 90, 9457–9464 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Chambers M. C., et al. , A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Agrawal S., et al. , El-MAVEN: A fast, robust, and user-friendly mass spectrometry data processing engine for metabolomics. Methods Mol. Biol. 1978, 301–321 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Benson D. A., et al. , GenBank. Nucleic Acids Res. 46, D41–D47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 80.Katoh K., Kuma K.-I., Toh H., Miyata T., MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nguyen L.-T., Schmidt H. A., von Haeseler A., Minh B. Q., IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Capovilla G., et al. , Phylogenomic analysis of chitinase. Dryad. https://datadryad.org/stash/dataset/doi:10.5061/dryad.gxd2547pp. Deposited 7 August 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (CSV)
Dataset S05 (CSV)
Data Availability Statement
All other data are included in the manuscript and/or supporting information. All 886 raw data for the main text and figures are available for free download at European Nucleotide Archive (ENA) #PRJEB53957 (RNA-Seq data); Metabo-Lights #MTBLS5229 (metabolites data); and Dryad (https://doi.org/10.5061/dryad.gxd2547pp) (82) (Phylogenomic data, including SI Appendix, Supplementary Text 1 file).