

Abstract
The NGF metabolic pathway entails the proteins that mature pro-nerve growth factor (proNGF) to NGF and those that degrade NGF. Basal forebrain cholinergic neurons require NGF, are critical for cognition, and degenerate early in Alzheimer’s disease (AD). In AD, NGF metabolism is altered, but it is not known whether this is an early phenomenon, nor how it relates to AD pathology and symptomology. We acquired frontal cortex samples from individuals with Alzheimer’s dementia, Mild Cognitive Impairment (MCI), or no cognitive impairment with high (HA-NCI) and low (LA-NCI) brain Aβ (Religious Orders Study). Cortical proNGF protein, but not mRNA, was higher in AD, MCI, and HA-NCI, while mature NGF was lower. Plasminogen protein was higher in MCI and AD brain tissue, with similar mRNA, suggesting diminished activation of the proNGF convertase, plasmin. The plasminogen activator tPA was lower in HA-NCI while neuroserpin, the CNS tPA inhibitor, was higher in AD and MCI cortical samples. Matrix metalloproteinase 9 (MMP-9), which degrades NGF, was overactive in MCI and AD. Transcription of the MMP-9 inhibitor TIMP-1 was lower in HA-NCI. ProNGF levels correlated to plasminogen and neuroserpin while NGF correlated to MMP-9 activity. In NCI, proNGF correlated with cerebral Aβ and tau deposition and to cognitive performance. In summary, frontal cortex proNGF maturation is impaired in preclinical and clinical AD while mature NGF degradation is enhanced. These differences correlate with cognition and may provide a platform for novel biomarkers and therapeutic targets.
Introduction
Alzheimer’s disease (AD) pathomechanistic alterations commence 10 to 20 years before cognitive decline manifests (1, 2). As a likely irreversible loss of functional cells and synapses underlies disease symptoms (3, 4), there is a need for new therapeutics targeting key AD pathophysiological mechanisms at the earliest possible stage. As such, a comprehensive understanding of early AD pathophysiology will be essential for the development of disease-modifying therapies (5).
Acetylcholinesterase inhibitors offer transient relief from cognitive decline (6) by potentiating the activity of acetylcholine at cortical and hippocampal cholinergic synapses, thereby partially compensating for a loss of cholinergic tone (7–10). Indeed, basal forebrain cholinergic neurons (BFCNs) lose their phenotype and functionality early in the course of AD (10–12). In early to moderate AD, the extent of the cholinergic deficit correlates well to the cognitive impairment (13, 14) and the efficacy of AChEIs correlates well to their pro-cognitive effects (15). AChEIs improve cholinergic tone but do not prevent or slow the degeneration of the BFCN. A more sophisticated therapeutic capable of maintaining cholinergic function would likely provide greater relief for patients with Alzheimer’s dementia. However, the development of such a drug requires an understanding of the causes of cholinergic degeneration.
BFCN neurons, uniquely in the CNS, depend exclusively on the retrograde supply of nerve growth factor (NGF) for the maintenance of their cholinergic phenotype (16–18). In AD, however, NGF transcription is normal (19) and the NGF precursor, proNGF, which lacks the same trophic function of mature NGF (20–23), is elevated (24).
Insight into this apparent paradox was provided by an ex vivo study in the rat cortex demonstrating that proNGF, and not mature NGF, was secreted in an activity-dependent manner alongside a set of proteins (the NGF metabolic pathway) responsible for converting proNGF to mature NGF and subsequently degrading mature NGF (25). Briefly, proNGF is converted to NGF by plasmin, which is derived from the inactive zymogen, plasminogen, by tissue plasminogen activator (tPA). The activity of tPA is regulated by its central inhibitor, neuroserpin (26, 27). Mature NGF is degraded by matrix metallo-protease 9 (MMP9), which is inhibited by TIMP1 (28, 29). This pathway (Figure 1A) has been shown to regulate the phenotype of cortical cholinergic synapses (30) and basal forebrain cholinergic cell bodies (31).
Figure 1. Schematic representation of the NGF metabolic pathway and its compromise in the continuum of Alzheimer’s disease.
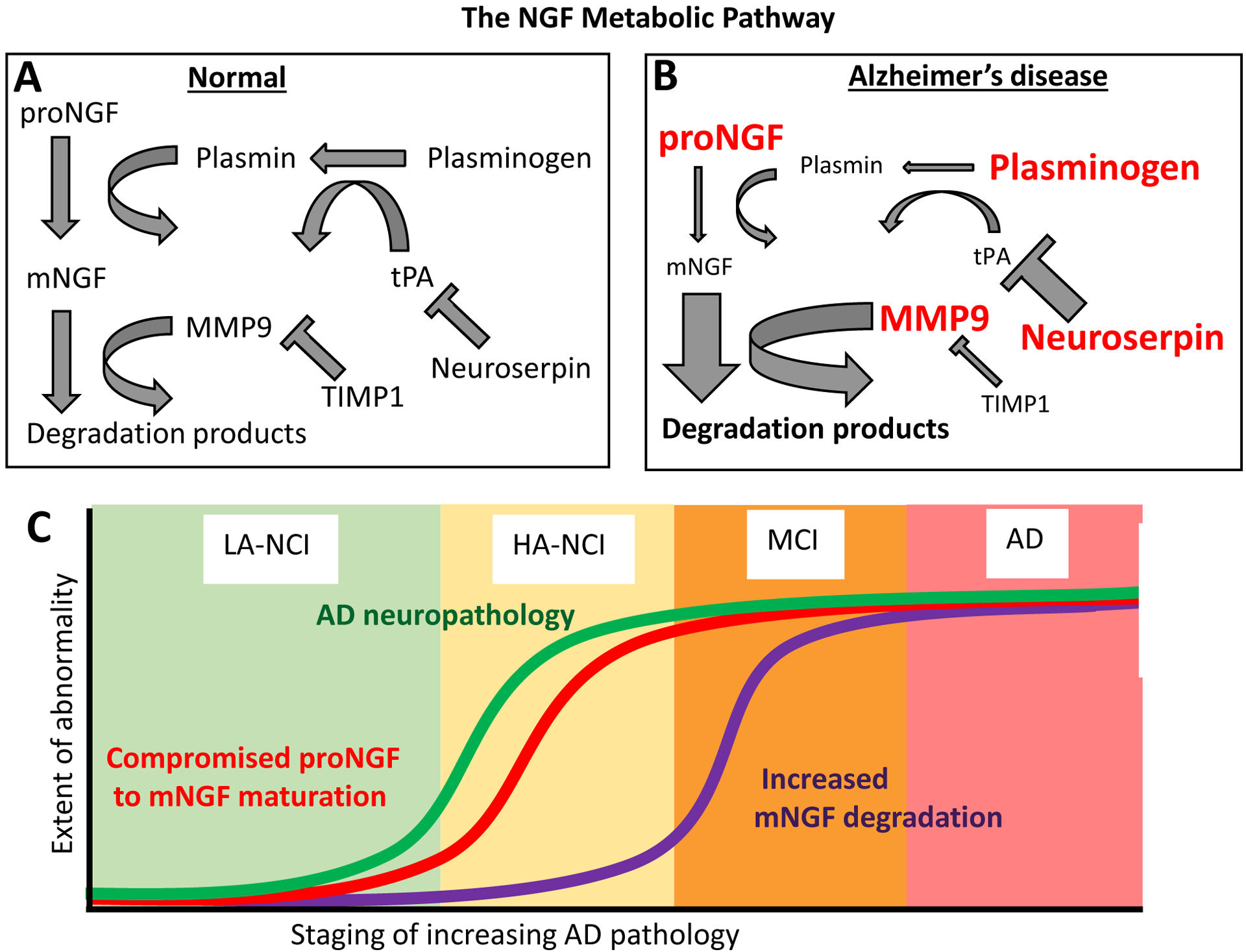
A: ProNGF secreted from BFCN target cells is converted extracellularly to mature NGF (mNGF) by plasmin, which is derived from plasminogen by tissue plasminogen activator (tPA). The latter is regulated by its endogenous inhibitor, neuroserpin. Mature NGF is degraded by matrix metallo-protease 9 (MMP9), which is regulated by its endogenous inhibitor TIMP1 (tissue inhibitor of metallo-proteinases 1). B: In Alzheimer’s disease, neuroserpin levels are increased. The resulting deficit in tPA activity leads to impaired maturation of plasminogen to plasmin, evidenced by accumulating plasminogen. ProNGF likewise accumulates as it fails to be converted to mature NGF, which is decreased. Simultaneously, MMP9 activity is increased while its inhibitor, TIMP1, is decreased. C: While deficits in proNGF maturation are evident alongside AD-like Aβ accumulation in individuals with no cognitive impairment (NCI), excessive NGF degradation is only evident in individuals diagnosed as MCI or AD.
The proNGF to mNGF conversion has been shown to be compromised in human brain material from individuals with AD as early as in Mild Cognitive Impairment (MCI) (32, 33) and tPA activity was shown to be reduced in the brains of AD patients (34). A transgenic rat modeling the human amyloid pathology recapitulates such NGF metabolic deficits (35) at stages with intracellular and extracellular amyloid deposition in the absence of tau, suggesting that NGF dysmetabolism could occur prior to the onset of dementia. However, the NGF metabolic pathway has not been fully assessed across the continuum of human AD pathology.
In this study, we investigated the protein and transcript levels of key players in NGF metabolism in the brains of individuals with Alzheimer’s dementia, MCI, and cognitively normal individuals with (and without) Alzheimer’s dementia-like levels of Aβ pathology. We also investigated the relationship between NGF metabolic markers and AD neuropathology at preclinical stages. Finally, we assessed the associations of NGF metabolic pathway proteins to cognitive scores in cognitively normal individuals. We demonstrate that proNGF maturation is reduced in preclinical and clinical AD while mature NGF degradation is enhanced. These changes correlate with preclinical cognition and AD neuropathology, and may provide a platform for novel biomarkers and therapeutics.
Methods
Demographic characteristics of the study sample cohort
The Religious Orders Study (ROS) is a cohort of American clergy that undergo yearly cognitive evaluations, annual clinical assessments, and post-mortem brain donation (36). Cognitive examinations include 21 tests of episodic memory, semantic memory, working memory, perceptual orientation, and processing speed which can be summarized as a Global Cognitive Score (GCS). Post-mortem data follow a uniform structured evaluation and provide measures of Aβ load (percent area of 6E10 Aβ immunoreactivity in eight regions: the CA1 and subiculum, the angular gyrus, the entorhinal cortex, the superior frontal cortex, the dorsolateral prefrontal cortex, the inferior temporal cortex, the anterior cingulate cortex and the calcarine cortex) and tangle pathology (provided by stereological assessment of AT8 immunoreactivity across the entorhinal cortex, CA1, superior frontal cortex, mid frontal cortex, inferior temporal cortex, angular gyrus, cingulate gyrus, and calcarine cortex) (37). Staging according to Braak, Reagan, and CERAD classifications were accomplished based on the nature and quantity of neuritic plaques and neurofibrillary tangles visualized with modified Bielschowsky silver staining.
We obtained ninety-eight frontal cortex samples comprising 59 individuals with no cognitive impairment (NCI), 19 individuals diagnosed with MCI, and 20 individuals with clinically diagnosed Alzheimer’s dementia. Diagnoses of Alzheimer’s dementia were made using the clinical criteria for AD recommended by the National Institute of Neurologic and Communicative Disorders and Stroke/AD and Related Disorders Association (NINCDS/ADRDA) (38). Patients were diagnosed with MCI if determined by a blinded neuropsychologist to have a cognitive impairment while deemed by a clinician to not meet criteria for dementia. The demographic characteristics of the study groups are illustrated in Table 1.
Table 1:
Demographic and neuropathological characteristics of ROS brain donors included in this study.
| LA-NCI | HA-NCI | MCI | AD | p value (ANOVA) | |
|---|---|---|---|---|---|
| n = | 43 | 16 | 19 | 20 | |
| Percent female | 53% | 48% | 58% | 55% | 0.82 |
| Age (years) | 78.2 ± 7.5 | 84.5 ± 7.9 | 83.9 ± 7.6 | 82.7 ± 8.2 | 0.01 |
| Post-mortem interval (hr) | 10.4 ± 8.3 | 8.5 ± 10.0 | 8.9 ± 5.6 | 5.7 ± 3.7 | 0.16 |
| Years of education | 17.8 ± 3.9 | 18.3 ± 2.8 | 18.2 ± 4.0 | 18.1 ± 3.9 | 0.73 |
| Cerebral amyloid-β percent area | 0.4 ± 0.6 | 2.2 ± 2.1 | 2.8 ± 2.4 | 4.3 ± 3.5 | <0.0001 |
| NFT density/mm2 | 1.6 ± 1.8 | 3.6 ± 4.8 | 5.1 ± 6.7 | 13.7 ± 19.1 | <0.0001 |
| CERAD | 3.3 ± 1.1 | 2.3 ± 0.6 | 2.8 ± 1.1 | 1.9 ± 0.8 | <0.0001 |
| REAGAN | 2.9 ± 0.5 | 2.3 ± 0.5 | 2.4 ± 0.7 | 2.2 ± 0.8 | <0.0001 |
| BRAAK | 2.1 ± 1.2 | 3.7 ± 1.1 | 3.5 ± 0.8 | 3.6 ± 1.1 | <0.0001 |
| APOE ε4 allele frequency | 0.12 | 0.20 | 0.18 | 0.17 | 0.31 |
| Global Cognitive Score (z score) | 0.18 ± 0.33 | −0.04 ± 0.28 | −0.22 ± 0.30 | −2.44 ± 1.11 | <0.0001 |
Data is presented as mean ± standard deviation. LA-NCI: low amyloid-no cognitive impairment, HA-NCI: high amyloid-no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, NFT: neurofibrillary tangles. The p value column gives the result of a one-way ANOVA comparing the means of all four pathological group.
Classification of preclinical Aβ pathology
To determine the relationship between Aß-amyloidosis and NGF dysmetabolism in AD-asymptomatic Aß-positive individuals, we established a cut-off point equivalent to two standard errors below the mean Aβ scores of the MCI group which divided NCI into low-Aβ expressing (LA-NCI; n = 43) and high-Aβ expressing (HA-NCI; n = 16) groups, with the latter representing an AD/MCI-like Aβ.
Analysis of the NGF metabolic pathway by qPCR, Western Blotting, ELISA, and gelatin zymography
Protein and transcript levels of NGF metabolic pathway proteins were assays by qPCR, Western Blotting, and ELISA using established techniques (25, 33), as detailed in the Supplementary Methods. Western blotting parameters are outlined in Table 2, while qPCR primers are listed in Table 3. For NGF, Western Blotting was preceded by a chloroform/methanol protein extraction. MMP9 and proMMP9 activity was assayed using gelatin zymography, following (32, 39).
Table 2:
Western blotting parameters
| Antibody (target) | Clonality | Source | Concentration | SDS-PAGE Gel % | Molecular weight |
|---|---|---|---|---|---|
| Ab9795 (NGF) | Polyclonal | Abcam (rabbit) | 1:1000 | 12% | 13 kDa |
| ANT-005 (proNGF) | Polyclonal | Alomone labs (rabbit) | 1: 2500 | 10% | 27 kDa; 41 kDa |
| Ab154560 (plasminogen) | Polyclonal | Abcam (rabbit) | 1: 5000 | 8% | 88 kDa |
| Anti-neuroserpin | Polyclonal | Dr. Daniel Lawrence, U. Michigan (rabbit) | 1:10 000 | 10% | 47 kDa |
| Ab52915 (MMP3) | Monoclonal | Abcam (rabbit) | 1:10 000 | 10% | 54 kDa |
| MAB374 (GAPDH) | Monoclonal | Millipore (mouse) | 1:10 000 | 8–12% | 37 kDa |
Table 3:
Primers for mRNA analysis of NGF metabolic pathway markers
| Target gene | Forward primer (5’-3’) | Reverse primer (5’-3’) |
|---|---|---|
| NGF | CATCATCCCATCCCATCTTC | GTCTGTGGCGGTGGTCTTAT |
| PLASMINOGEN | GCCCCATAGACACAGCATTT | TAGCACCAGGGACCACCTAC |
| NEUROSERPIN | GTAGCCGTGGCCAACTACAT | CCCTTGGGGATACCAAATCT |
| TPA | GACGTGGGAGTACTGTGATGTG | CCCTCCTTTGATGCGAAACTGA |
| MMP-9 | GCCATTCACGTCGTCCTTAT | TTGACAGCGACAAGAAGTGG |
| TIMP-1 | TGACATCCGGTTCGTCTACA | TGCAGTTTTCCAGCAATGAG |
| HPRT | TTGCTTTCCTTGGTCAGGCA | ATCCAACACTTCGTGGGGTC |
| ACTIN | ACAGCCTGGATAGCAACG | CACCAACTGGGACGACAT |
| GAPDH | CCCCACTTGATTTTGGAGGGA | AGGGCTGCTTTTAACTCTGGT |
Statistical analysis
Statistics were performed using GraphPad Prism 5 (GRAPHPAD Software, San Diego, California, USA). One-way ANOVAs with Bonferroni post-hoc tests or Kruskal-Wallis tests with Dunn’s post-hoc corrections were used to compare group means, depending on the adherence of each sample to a normal distribution as assessed by a D’Agostino-Pearson Omnibus Normality Test. Correlations were examined in R using multiple linear regression, with age and sex included as cofactors and partial r2 and p values reported for the species of interest. All values were normalized to the mean of the LA-NCI group and represented as a fold-change from that mean. Differences in demographic characteristics were examined using linear regression or chi-square tests as appropriate (see Table 1). The effects of age and sex on each protein or transcript were examined using linear regression and unpaired Student’s t-tests (see Supplementary Table 1).
Results
Demographic characteristics of the study cohort (see Table 1)
No differences in sex proportions, years of education, post-mortem interval, or APOE ε4 allele frequency were observed between groups. The low- Aβ NCI group was younger than the high- Aβ NCI group. As expected, the HA-NCI, MCI, and AD groups had higher Aβ deposition than the LA-NCI group; this area was greater in AD compared to HA-NCI and MCI, though there was no difference between the latter groups. The AD groups exhibited more cortical tangles than the LA-NCI and HA-NCI; no other differences were observed. Average CERAD scores were higher in HA-NCI, MCI, and AD compared to LA-NCI. Average Reagan diagnoses (low, intermediate, and high likelihood of AD, coded 1–4), were likewise higher in HA-NCI and AD compared to the LA-NCI group; no other differences were observed. Similarly, average Braak stage was higher in HA-NCI, MCI , and AD versus LA-NCI. Global Cognitive Scores were lower in AD compared to each of LA-NCI, HA-NCI, and MCI; no other differences were observed.
Effects of age and sex
Weak correlations were observed between MMP9 activity and age and proNGF 41 kDa protein and age. All other associations were not significant and no effects of sex were observed (see Supplementary Table 1).
Protein and mRNA expression of NGF across the AD continuum
We investigated both mature NGF and proNGF protein levels, as well as the associated NGF transcript, to assess the likely availability of trophic support to BFCN neurons in the continuum of AD pathology. We assessed both 27 kDa and 41 kDa proNGF variants, with the former representing unprocessed proNGF and the latter representing the secreted form (25).
While we observed no difference in the expression of the NGF transcript between the different clinical groups (Figure 2A; Table 3), the mature NGF protein was lower in frontal cortex homogenates from MCI and AD cases compared to LA-NCI, with a trend towards a reduction in the HA-NCI group (Figure 2B; Table 3). Both 27 kDa and 41 kDa proNGF protein immunoreactivity in individuals classified as HA-NCI, MCI, or AD as compared to those classified as LA-NCI were higher (Figure 2C–D; Table 3).
Figure 2. Normal NGF synthesis in the continuum of AD pathology is accompanied by an increase of proNGF and decrease of mNGF, beginning at preclinical stages, as well as abnormal expression of proteins participating in proNGF maturation.
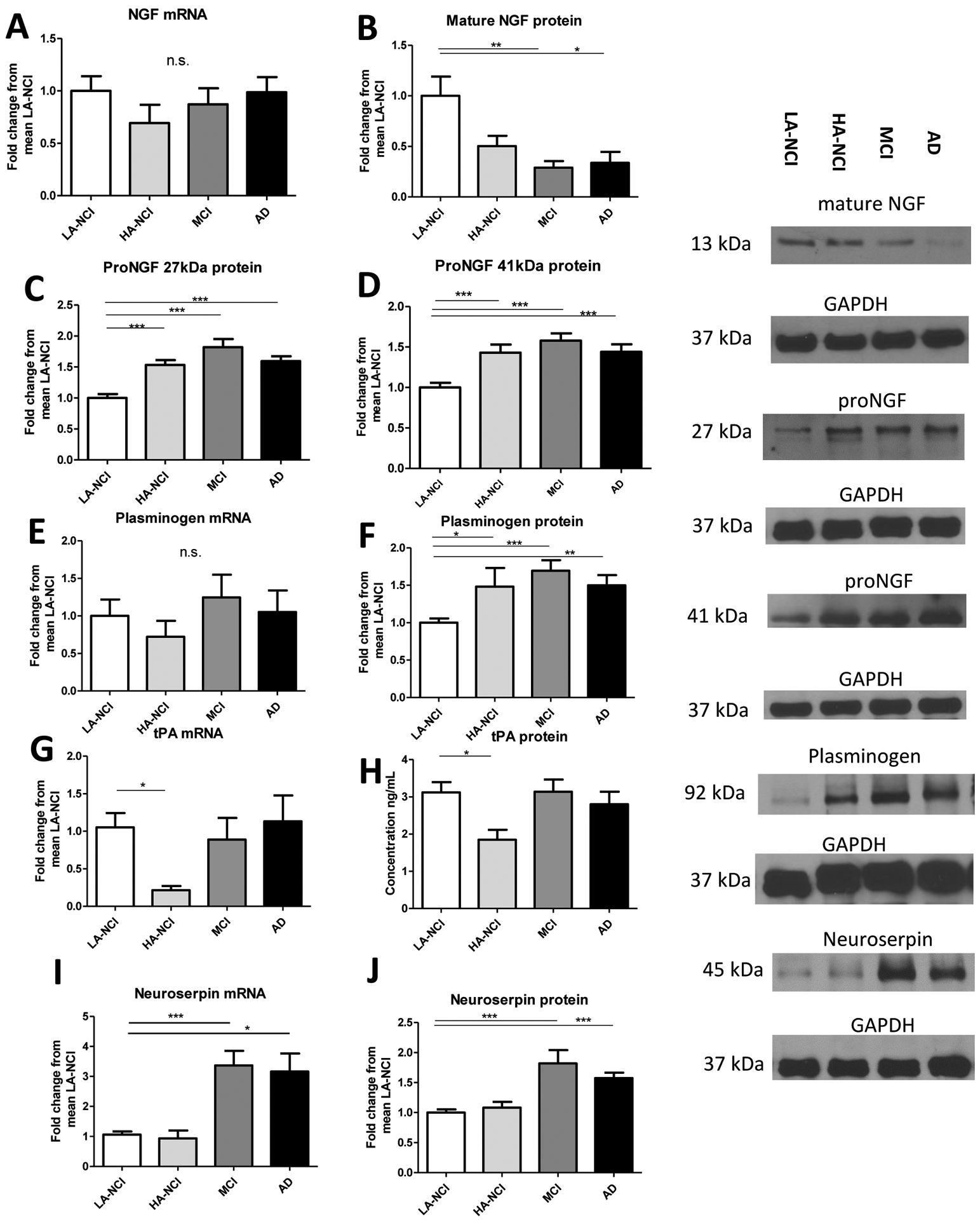
A: no differences in NGF mRNA were observed by qPCR. B: decreased mNGF immunoreactivity at 13 kDa in MCI/AD brains. C: increased proNGF immunoreactivity at 27 kDa in HA-NCI/MCI/AD brains. D: increased proNGF immunoreactivity at 41 kDa in HA-NCI/MCI/AD brains. E: frontal cortex plasminogen mRNA does not differ between LA-NCI, HA-NCI, MCI, and AD. F: Plasminogen protein is elevated in frontal cortex homogenates from HA-NCI, MCI, and AD individuals versus those from LA-NCI individuals. G: tPA mRNA in frontal cortex homogenates is decreased in HA-NCI and unchanged in MCI and AD versus LA-NCI. H: tPA protein is likewise solely decreased in HA-NCI vs. LA-NCI. I: neuroserpin mRNA is increased in frontal cortex homogenates in AD and MCI versus LA-NCI. J: levels of neuroserpin protein are higher in frontal cortex homogenates from individuals diagnosed with MCI/AD. Representative Western blots are shown for NGF at 13 kDa, proNGF at 27 and 41 kDa, plasminogen at 92 kDa, and neuroserpin at 45 kDa with 37 kDa GAPDH as the reference protein. Groups were LA-NCI; n = 43, HA-NCI; n = 16, MCI; n = 20, or AD; n = 19. All comparisons performed with a one-way ANOVA and Bonferroni post-hoc tests or a Kruskal-Wallace test and Dunn’s post-hoc tests. All bars indicate mean + SEM.
Investigation of the plasmin activating system across the AD continuum
To determine the impact of AD pathology on the conversion of proNGF to mature NGF and possible underlying mechanisms, we investigated the protein and transcript levels of plasminogen, tPA, and neuroserpin.
Although plasminogen mRNA levels were comparable between groups (Figure 2E; Table 3), plasminogen protein expression was higher in frontal cortex homogenates from HA-NCI, AD, and MCI individuals (Figure 2F; Table 3). The expression of tissue plasminogen activator (tPA) was lower in HA-NCI compared to LA-NCI, but not in MCI or AD, both as a transcript (Figure 2G) and as protein (Figure 2H; Table 3). However, the mRNA levels of the tPA inhibitor, neuroserpin, were higher in MCI and AD (Figure 2I; Table 3), which was mirrored by the expression of neuroserpin protein (Figure 2J; Table 3).
Protein and mRNA expression of MMP9 and TIMP1 across the AD continuum
AD and MCI individuals showed increased mRNA levels of MMP9, a prominent NGF-degrading protease (25), though there was no difference between low and high Aβ NCI individuals (Figure 3A; Table 3). Frontal cortex homogenates from such cases also had higher MMP9 proteolytic activity as measured by gelatin zymography at MCI and AD clinical stages (Figure 3B; Table 3). At the same stages, the activity of proMMP9 was correspondingly increased (Figure 3C; Table 3). Consistently with the above, transcript levels of the MMP9 inhibitor TIMP1 were lower in MCI and AD frontal cortex but not in HA-NCI (Figure 3D; Table 3).
Figure 3. Increased levels and protease activity of MMP9, a mature-NGF degrading protease, and diminished expression of its endogenous inhibitor, TIMP1, at AD clinical stages.
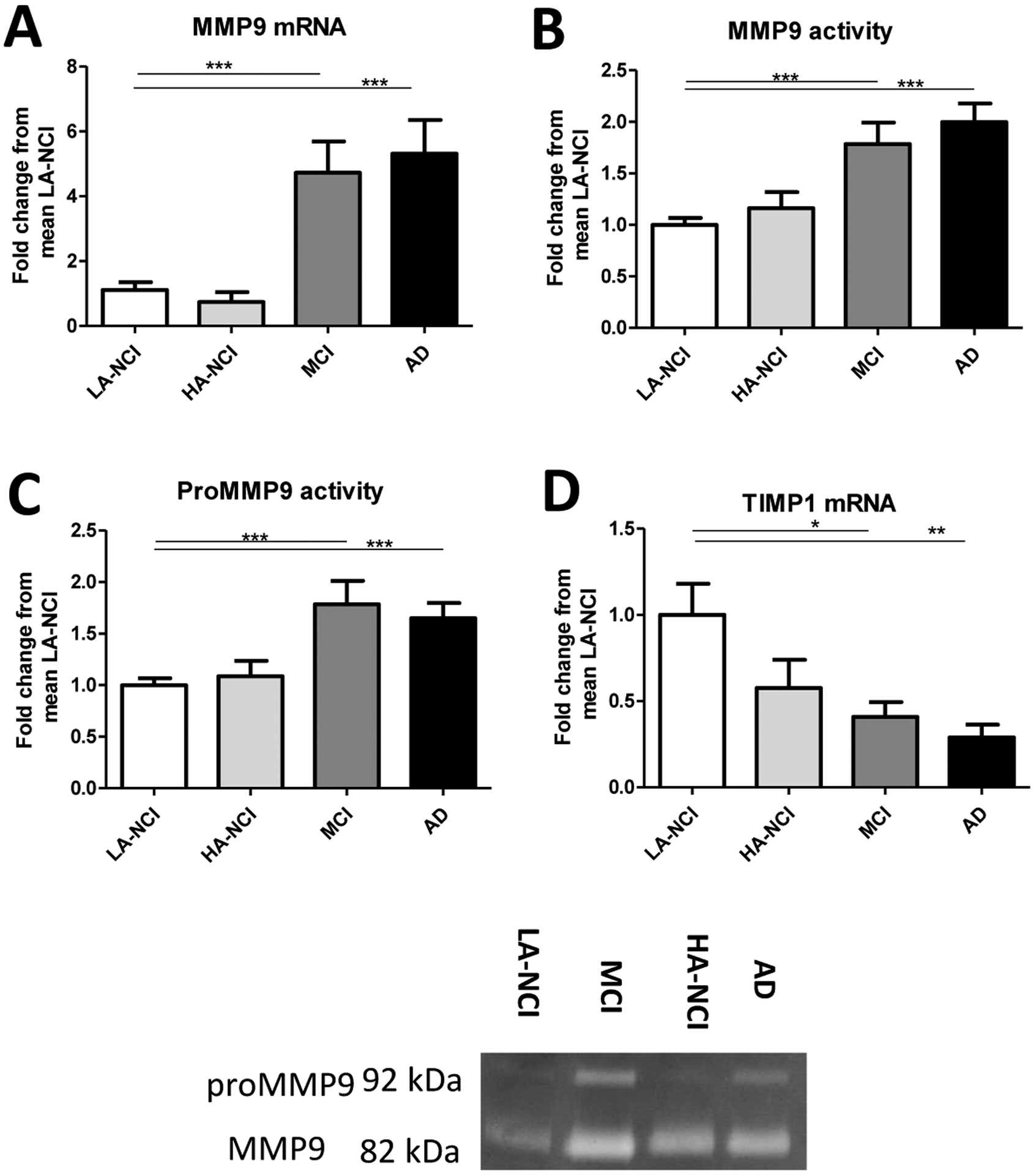
A: increased expression of MMP9 mRNA in MCI and AD was observed by qPCR. B: Gelatin zymography revealed increased MMP9 activity in MCI and AD individuals. C: proMMP9 activity was increased in MCI and AD. Representative zymograms are shown for MMP9 at 82 kDa and proMMP9 at 92 kDa. D: Using qPCR, we observed less TIMP1 mRNA in individuals with MCI or AD. Refer to Figure 1 legend for cases, groups, and statistics.
Associations between the NGF metabolic pathway proteins and proNGF
NGF dysmetabolism would predict higher proNGF levels with higher plasminogen and neuroserpin, reflecting a diminished proNGF conversion to mature NGF, causing an accumulation of unmatured proNGF. Indeed, levels of the 27 kDa proNGF protein were positively correlated to levels of plasminogen (Supplementary Figure 1A; Supplementary Table 2) and to neuroserpin (Supplementary Figure 1B; Supplementary Table 2) in the whole sample, as were levels of 41 kDa proNGF (Supplementary Figure 1C; Supplementary Table 2). Levels of neuroserpin, the endogenous tPA inhibitor, also correlated to levels of plasminogen (Supplementary Figure 1E; Supplementary Table 2). TIMP1 mRNA correlated negatively to MMP9 activity (Figure 1F; Supplementary Table 2). As predicted, levels of mature NGF (but not proNGF) correlated to MMP9 activity (Figure 1G; Supplementary Table 2).
Associations between NGF pathway markers and neuropathological measures in individuals with no cognitive impairment (NCI)
To investigate the relationship between preclinical AD pathology and NGF metabolism, we correlated quantitative measures of Aβ and tau pathology to proNGF protein levels in individuals with NCI.
The 27kDa variant of proNGF positively correlated to cerebral Aβ deposition (Figure 4A; Table 4). This form of proNGF also correlated to a score of cortical tangle pathology (Figure 4B; Table 4). The 41 kDa variant of proNGF also correlated to cortical Aβ deposition (Figure 4C; Table 4) and tangle burden (Figure 4D; Table 4). Furthermore, proNGF 41 kDa protein levels were differentially expressed according to Reagan diagnosis in NCI individuals, with brains from individuals classified as Reagan 3 or 4 expressing more proNGF compared to that of those classified as 2, and 4 having higher proNGF than 3 (Figure 4E; Table 4). When NCI cases were separated according to the Braak index of tauopathy, higher proNGF 41 kDa protein was found in individuals classified as Braak stage 4–5 than those classified as Braak 0–1 (Figure 4F; Table 4). No correlations to or differential expression of other NGF pathway markers by these variables were observed.
Figure 4. Association between elevations of frontal cortex proNGF protein levels and hallmarks of Alzheimer’s neuropathology and to cognitive scores in individuals with no cognitive impairment (n = 59).
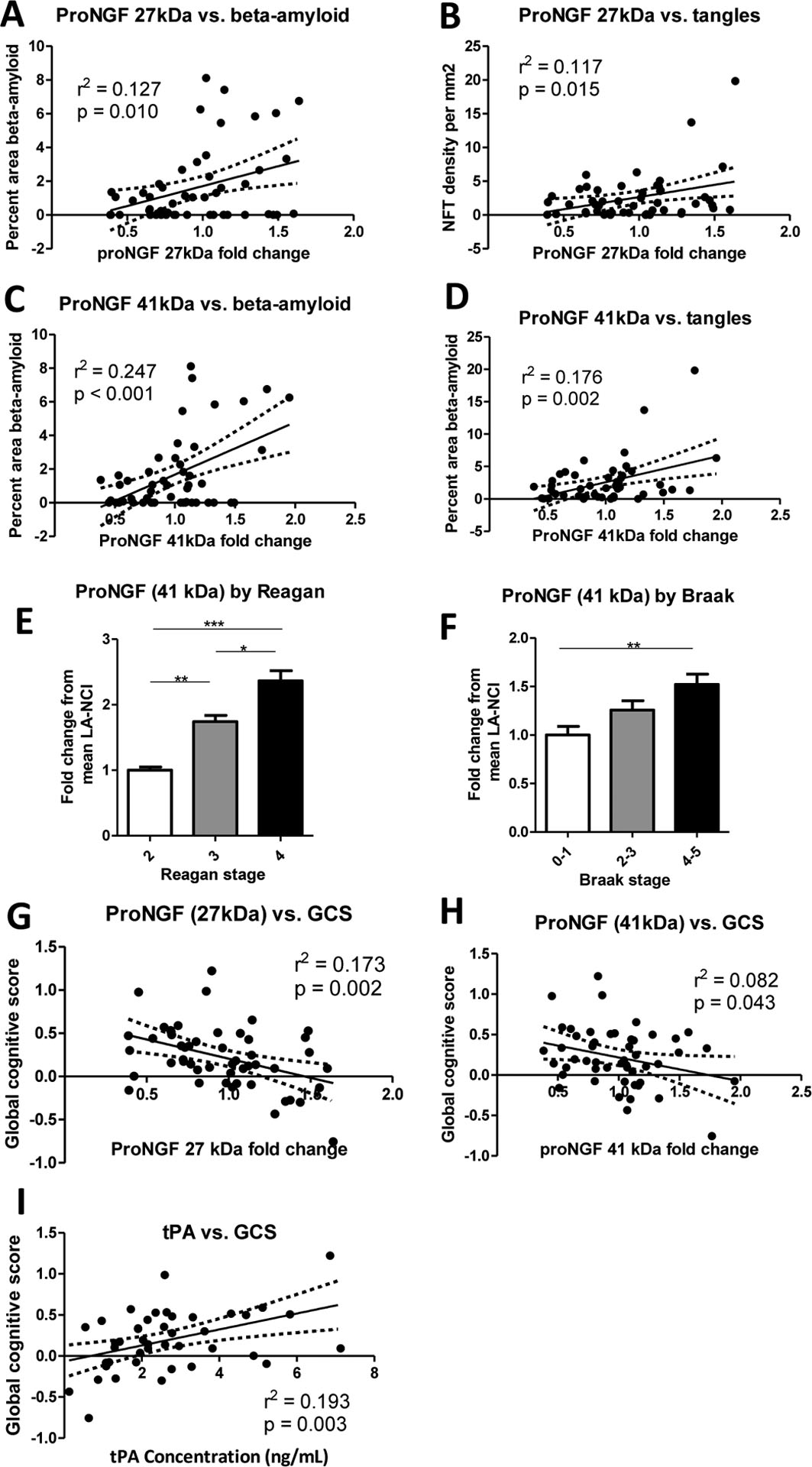
A: The 27 kDa variant of proNGF correlates to amyloid deposition in aged individuals without cognitive impairment. B: The 27 kDa variant of proNGF correlates to an index of tangle pathology. C: ProNGF immunoreactivity at 41 kDa also correlates to amyloid deposition. ProNGF 41 kDa protein correlates to tangle pathology. E: ProNGF 41 kDa protein is differentially regulated by Reagan score, with higher levels in individuals classified as Reagan 2 (n = 20) and Reagan 3 (n = 31) than in Reagan 4 (n = 8), as well as in Reagan 2 compared to Reagan 3. F: The 41 kDa isoform of proNGF was also differentially expressed by Braak stage, with higher levels in brains classified as Braak 4–5 (n = 17) compared to those classified as Braak 0–1 (n = 27), with the intermediate Braak 2–3 brains (n = 15) not significantly different from either. G: ProNGF 27 kDa protein correlates negatively to Global Cognitive Score z-scores. H: ProNGF immunoreactivity at 41 kDa correlates to Global Cognitive Score z-scores. I: tPA protein concentrations measured by ELISA correlate to Global Cognitive Score z-scores. All comparisons performed with a one-way ANOVA and Bonferroni post-hoc tests or a Kruskal-Wallace test and Dunn’s post-hoc test. Bars indicate mean + SEM. All relationships assessed by multiple linear regression with age and sex as covariates, with 95% confidence intervals and partial r2 and p values displayed.
Table 4:
Comparisons of NGF metabolic pathway proteins in frontal cortex between individuals classified LA-NCI (n = 43), HA-NCI (n = 16), MCI (n = 20), and AD (n = 19).
| Species | LA-NCI vs HA-NCI | LA-NCI vs. MCI | LA-NCI vs AD | Overall |
|---|---|---|---|---|
| NGF mRNA | n.s. | n.s. | n.s. | df=3,95; H = 2.4; p > 0.05 |
| NGF protein | rs = 8.842, p = 0.06 | rs = 15.73, p < 0.01 | rs = 17.14, p < 0.01 | df = 2,52; H = 14.38; p < 0.01 |
| proNGF 27 kDa | t=4.095, p < 0.001 | t = 7.058, p < 0.001 | t = 4.984 p < 0.001 | df = 3,95; F = 19.96; p < 0.0001 |
| proNGF 41 kDa | t = 3.795, p < 0.001 | t = 5.399, p < 0.001 | t = 4.035, p < 0.001 | df = 3,95; F = 12.68; p < 0.0001 |
| Plasminogen mRNA | n.s | n.s | n.s | df = 3,95; H = 0.87; p > 0.05 |
| Plasminogen protein | t = 2.605, p < 0.05 | t = 4.035, p < 0.001 | t = 3.070 p < 0.01 | df = 3,95; F = 26.80; p < 0.0001 |
| tPA mRNA | rs = 22.37, p < 0.05 | n.s | n.s | df = 3,95; H = 7.652; p = 0.05 |
| tPA protein | rs = 23.46, p < 0.05 | n.s | n.s | df = 3,95; H = 8.754, p < 0.05 |
| Neuroserpin mRNA | n.s | rs = 30.91, p < 0.001 | rs = 22.26, p < 0.05 | df = 3,95; H = 29.23; p < 0.0001 |
| Neuroserpin protein | n.s | rs = −34.41, p < 0.001 | rs = −33.39 p < 0.001 | df = 3,95; H = 30.18; p < 0.0001 |
| MMP9 mRNA | n.s | rs = 26.67, p < 0.001 | rs = 28.63, p < 0.001 | df = 3,95; H = 25.44; p < 0.0001 |
| MMP9 activity | n.s | rs = 27.55, p < 0.001 | rs = 33.91 p < 0.001 | df = 3,95; H = 29.33; p < 0.0001 |
| proMMP9 activity | n.s | rs = 27.55, p < 0.001 | t = 33.91 p < 0.001 | df = 3,95; H = 29.33; p < 0.0001 |
| TIMP1 mRNA | n.s | rs = 18.90, p < 0.05 | rs = 26.40, p < 0.01 | df = 3,95; H = 15.03; p < 0.01 |
Results of ANOVA/Kruskal-Wallis comparison and Bonferroni/Dunn’s post-hoc tests are reported.
Associations between NGF pathway markers and pre-mortem cognitive scores in NCI
To understand the consequences of NGF dysmetabolism prior to the onset of symptomatic AD, we correlated protein levels of NGF pathway markers in individuals with NCI to cognitive scores, given by the overall z-scores of the last Global Cognitive Score (CGS) assessment prior to death. GCS z-scores correlated negatively to the 27 kDa band immunoreactive for proNGF (Figure 4G; Table 4) and to the 41 kDa proNGF variant (Figure 4H; Table 4). We also observed a positive correlation between tPA protein levels and GCS z-scores (Figure 4I; Table 4) in patients without overt cognitive impairment. No other association with changes in cognitive scores were observed in the NCI group.
Discussion
In this report we provide a thorough analysis of the NGF metabolic pathway across the continuum of AD pathology, including preclinical and clinical stages, in the frontal cortices of individuals with AD, MCI and AD-asymptomatic individuals with AD-like Aβ deposition. We demonstrate higher levels of proNGF and plasminogen protein, in the absence of transcriptional differences in associated genes, suggesting impairments to the plasminogen activating system throughout HA-NCI, MCI, and AD. We confirmed this by demonstrating higher neuroserpin in MCI and AD and lower tPA in NCI individuals with higher brain Aβ deposition, which suggest decreased plasmin maturation resulting in impaired proNGF maturation to mNGF. Simultaneously, higher MMP9 occurs concurrently with lower TIMP1, resulting in lower NGF as evidenced by the reduced levels of mature NGF in MCI and AD (see Figure 1B). Our results suggest that the conversion of proNGF is impaired alongside Aβ pathology in preclinical AD, while excessive NGF degradation is a feature of clinically established AD and MCI (see Figure 1C).
We validate the NGF metabolic pathway by demonstrating correlations between proNGF or mature NGF levels and the levels of relevant maturing or degrading enzymes, supporting its role as a critical determinant of trophic support to BFCN. Most importantly, we illustrate the association of proNGF and tPA with cognitive function in AD-asymptomatic cognitively normal individuals. These results support the hypothesis that the AD-related atrophy of NGF-dependent BFCN is a consequence of the withdrawal of their trophic support, a concept demonstrated in vivo by the immunoneutralization of endogenous NGF or the blocking of TrkA receptors (40), as well as pharmacologically by the inhibition of plasmin activation and therefore the conversion of proNGF to mNGF (30, 31). As such, alterations to the NGF metabolic pathway could serve as a platform for developing novel pro-cognitive cholinergic therapeutics or biomarkers of cognitive decline.
A link between amyloid-β pathology and NGF metabolism
Herewith we found consistent associations between levels of Aβ pathology and the various dysregulated markers of NGF metabolism, in particular proNGF, at preclinical disease stages. These results accord with our previous findings in individuals with Down syndrome, a condition characterized by lifelong amyloidosis, (41, 42), in which we described a similar dysregulation of NGF metabolism in frontal cortex which correlated to Aβ pathology (39). We have also previously shown that injected soluble Aβ oligomers per se can induce rapid proNGF accumulation and increased MMP9 activation in the brains of naïve rats (33) and that both rats and mice transgenic for human mutated APP display a lifelong deregulation of NGF metabolism in the absence of tau pathology (33, 35). While we did observe certain correlations between tauopathy burden and markers of NGF dysmetabolism, these were less strong and consistent than those with Aβ. These studies therefore support NGF metabolic dysfunction as following primarily, though not necessarily exclusively, from Aβ accumulation in AD.
While this study did not investigate the direct mechanism by which Aβ disrupts NGF metabolism, previous results have demonstrated that the application of an anti-inflammatory is sufficient to resolve the NGF dysmetabolism induced by the injection of Aβ oligomers into the hippocampi of naïve rats (33). Indeed, many proteins involved in the NGF metabolic pathway, such as the plasmin activating system and MMP9, have roles in pro-inflammatory pathways including cytokine activation, glial remodelling, and phagocytosis (43–45). Aβ is known to induce strong and inflammatory reactions that become chronic in the context of progressive amyloid pathology (46, 47) and growing evidence suggests that early CNS inflammation plays a critical role in the pathogenesis of AD (48). NGF dysmetabolism and cholinergic degeneration may therefore be a consequence of this prolonged inflammatory activation provoked by Aβ (49, 50). Intriguingly, the cholinergic system has an established anti-inflammatory role that might permit it to attenuate the degenerative effects of chronic neuroinflammation in AD (51).
A revisited model explaining cholinergic degeneration in Alzheimer’s disease
Several explanations for the preferential vulnerability of BFCN in AD have been proffered, including reductions in TrkA levels in the context of normal p75ntr expression (52), vulnerability to tauopathy (53), and impaired axonal transport (54, 55). While further research is warranted, a dysfunctional NGF metabolism could lie at the root of the cholinergic pathology. The expression of the NTRK1 (TrkA) gene is ultimately dependent on NGF signalling through its high affinity receptor: TrkA (56, 57), as is the cholinergic gene locus containing ChAT and VAChT (58–63) and genes critical for axonal integrity and transport (64, 65). Therefore, the decline of TrkA levels in AD and the consequent predomination of p75/sortilin-mediated signalling could result from the compromise of NGF metabolism and the subsequent reduction of mature NGF available to the basal forebrain. Furthermore, the NGF metabolic pathway has been demonstrated to be responsible for the maintenance of the synaptic (30) and somato-dendritic (31) cholinergic phenotype of BFCNs in vivo.
The NGF metabolic pathway and biomarker studies
We have previously shown that proteins involved in NGF metabolism, measured in biofluids, may serve as biomarkers of AD pathology and AD-associated cognitive decline. Self-to-self increases in proNGF levels measured in the plasma of AD-asymptomatic individuals with Down syndrome were effective predictors of cognitive decline across 2 years of follow-up (66). Retrospective analysis of a cohort of plasma from the general population showed significant associations between various markers of NGF metabolism such as MMP9 and the risk of dementia onset (67). Furthermore, CSF from patients with MCI and AD showed altered levels of metallo-proteases and the plasminogen activating system, both critical elements of the NGF metabolic pathway (68). These results are in line with reports that CSF proNGF is increased in AD and that this increase correlates to disease staging and cognitive impairments (69). This study indicates that such changes occur in step with similar stepwise changes in the brain (see Figure 1B–C), with Aβ as the most likely mediator, beginning at the earliest stages of the disease. As new, reliable, and cost effective biomarkers of incipient Alzheimer’s pathology are a critical unmet need for the treatment of AD (70), it will be important to validate proNGF and related markers in other cohorts in this capacity. If positive, it will also be essential to develop reliable ELISA assays or PET tracers capable of distinguishing proNGF from mature NGF.
Relevance of NGF metabolism for cognition and therapeutics
The forebrain cholinergic system plays a vital role in cognition by inducing specific information-processing states in target tissues (71). Previous studies have heavily implicated the cholinergic system in the cognitive deficit in early-to-moderate Alzheimer’s dementia (72).
Our results support a role for cholinergic dysfunction in early cognitive decline in AD, demonstrating that correlations between proNGF processing and reduced performance on cognitive tests can be observed even in ostensibly healthy, cognitively unimpaired patients.
While ACHEIs are only transiently efficacious, it is remarkable that cholinergic therapy can still exert pro-cognitive effects at stages with devastating brain damage already established. A more sophisticated therapy aimed at re-establishing a sustained cholinergic tone, perhaps by enhancing proNGF maturation of reducing mNGF degradation, might achieve a far more significant pro-cognitive effect. Even a symptomatic treatment extending good cognition for five years would translate to a 41% reduction in the global incidence of Alzheimer’s dementia (73).
Emerging evidence suggests that cholinergic drugs might have some disease modifying properties, albeit to a restricted extent, as we have discussed in a recent paper by the “Cholinergic System Working Group” (74). To this effect, individuals with suspected prodromal Alzheimer’s dementia taking donepezil show reduced rates of hippocampal, cortical, and BFCN atrophy (75, 76). Conversely, aged individuals taking anti-cholinergic drugs have a higher incidence of Alzheimer’s dementia and greater rates of hippocampal atrophy (77–80). Furthermore, the degeneration of the BFCN precedes and predicts the degeneration of its target tissues in the entorhinal and cerebral cortices (81, 82). It is possible that these effects are mediated by M1 AChR signalling in its capacity to reduce amyloidogenic processing of APP (83, 84). Indeed, a combined M1 AChR/σ1 agonist has shown the ability to decrease amyloid pathology, attenuate CNS inflammation and reverse cognitive deficits in a rat model of AD-like amyloid pathology (85). As such, therapeutics capable of sustaining a fully functional cholinergic system further through the continuum of AD pathology and would therefore constitute a disease-modifying, though not curative, therapy. Such a therapy could perhaps target NGF dysmetabolism, as here demonstrated across the continuum of AD with relevance to cognitive outcomes.
Supplementary Material
Table 5:
Associations between NGF metabolic pathway proteins and AD neuropathology, comparisons of NGF metabolic pathway proteins between Reagan and Braak classifications, and associations of NGF metabolic pathway markers with preclinical cognition in individuals classified as NCI (n = 59).
| Protein | Correlate | Statistics (partial r2 and p values for correlates in an age/sex corrected multiple regression model) |
|---|---|---|
| 27 kDa proNGF | Aβ deposition | r2 = 0.127; p = 0.01 |
| 27 kDa proNGF | tangles | r2 = 0.117; p < 0.05 |
| 41 kDa proNGF | Aβ deposition | r2 = 0.247, p < 0.001 |
| 41 kDa proNGF | tangles | r2 = 0.176, p < 0.01 |
| 41 kDa proNGF | Reagan stage | df = 2, 56; F = 14.40; p = 0.007; Reagan 2 vs Reagan 3, t = 2.825 p < 0.051; Reagan 2 vs Reagan 4, t = 4.965 p < 0.001; Reagan 3 vs Reagan 4, t = 3.753 p < 0.05 |
| 41 kDa proNGF | Braak stage | df = 2, 56; F = 14.40; p = 0.007; Reagan 2 vs Reagan 3, t = 2.825 p < 0.051; Reagan 2 vs Reagan 4, t = 4.965 p < 0.001; Reagan 3 vs Reagan 4, t = 3.753 p < 0.05 |
| 27 kDa proNGF | Global cognitive z-scores | r2 = 0.173, p < 0.01 |
| 41 kDa proNGF | Global cognitive z-scores | r2 = 0.082, p < 0.05 |
| tPA protein | Global cognitive z-scores | r2 = 0.193, p < 0.01 |
Acknowledgements
The authors express gratitude to the participants in the ROS study as well as to the team at the RUSH Medical Center for performing the cognitive testing and neuropathological analyses. The authors thank Dr. Daniel Lawrence from the Michigan Center for Integrative Research in Critical Care, USA for graciously providing the anti-neuroserpin antibody used in this study. The authors are also grateful for the revisions and suggestions provided by Drs. Ezio Giacobini, Harald Hampel, and Giancarlo Pepeu on this manuscript.
ACC acknowledges financial support from the Canadian Institutes of Health Research (CIHR) and the Alzheimer Society of Canada. He holds the McGill University Charles E. Frosst/Merck Chair in Pharmacology and is a member of the Canadian Consortium of Neurodegeneration in Aging. ACC wishes to thank Merck Canada for their unrestricted support. DAB was supported by grants P30AG10161 and R01AG15819 from the NIA. RP was the recipient of a Student Fellowship from the McGill Integrated Program in Neuroscience and CIHR Doctoral Award. MFI acknowledges support from a Bourse Postdoctorale from the Fonds de Recherche du Quebec Santé (FRQS). The funding bodies had no role in the design of the study or in the collection, analysis, and interpretation of data or in writing the manuscript.
Footnotes
Conflict of interest
The authors have no conflict of interest to report.
References
- 1.Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. The Lancet Neurology. 2013;12(2):207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7(3):280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gauthier S, Albert M, Fox N, Goedert M, Kivipelto M, Mestre-Ferrandiz J, et al. Why has therapy development for dementia failed in the last two decades? Alzheimer’s & Dementia. 2016;12(1):60–4. [DOI] [PubMed] [Google Scholar]
- 4.McDade E, Bateman RJ. Stop Alzheimer’s before it starts. Nature. 2017;547(7662):153. [DOI] [PubMed] [Google Scholar]
- 5.Epelbaum S, Genthon R, Cavedo E, Habert MO, Lamari F, Gagliardi G, et al. Preclinical Alzheimer’s disease: A systematic review of the cohorts underlying the concept. Alzheimer’s & Dementia. 2017. [DOI] [PubMed] [Google Scholar]
- 6.Lanctôt KL, Herrmann N, Yau KK, Khan LR, Liu BA, LouLou MM, et al. Efficacy and safety of cholinesterase inhibitors in Alzheimer’s disease: a meta-analysis. Canadian Medical Association Journal. 2003;169(6):557–64. [PMC free article] [PubMed] [Google Scholar]
- 7.Perry E, Tomlinson B, Blessed G, Perry R, Cross A, Crow T. Noradrenergic and cholinergic systems in senile dementia of Alzheimer type. The Lancet. 1981;318(8238):149. [DOI] [PubMed] [Google Scholar]
- 8.Davies P, Maloney A. Selective loss of central cholinergic neurons in Alzheimer’s disease. The Lancet. 1976;308(8000):1403. [DOI] [PubMed] [Google Scholar]
- 9.Bowen DM, Smith CB, White P, Davison AN. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain: a journal of neurology. 1976;99(3):459–96. [DOI] [PubMed] [Google Scholar]
- 10.Coyle JT, Price DL, Delong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science. 1983;219(4589):1184–90. [DOI] [PubMed] [Google Scholar]
- 11.Pearson R, Sofroniew M, Cuello A, Powell T, Eckenstein F, Esiri M, et al. Persistence of cholinergic neurons in the basal nucleus in a brain with senile dementia of the Alzheimer’s type demonstrated by immunohistochemical staining for choline acetyltransferase. Brain research. 1983;289(1):375–9. [DOI] [PubMed] [Google Scholar]
- 12.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215(4537):1237–9. [DOI] [PubMed] [Google Scholar]
- 13.Jacobs RW, Butcher LL. Pathology of the basal forebrain in Alzheimer’s disease and other dementias. The biological substrates of Alzheimer’s disease: Academic Press; New York; 1986. p. 87–100. [Google Scholar]
- 14.DeKosky DST, Harbaugh RE, Schmitt FA, Bakay RA, Chui HC, Knopman DS, et al. Cortical biopsy in Alzheimer’s disease: diagnostic accuracy and neurochemical, neuropathological, and cognitive correlations. Annals of neurology. 1992;32(5):625–32. [DOI] [PubMed] [Google Scholar]
- 15.Giacobini E, Spiegel R, Enz A, Veroff A, Cutler N. Inhibition of acetyl-and butyryl-cholinesterase in the cerebrospinal fluid of patients with Alzheimer’s disease by rivastigmine: correlation with cognitive benefit. Journal of Neural Transmission. 2002;109(7):1053–65. [DOI] [PubMed] [Google Scholar]
- 16.Hefti F Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1986;6(8):2155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuello AC. Trophic responses of forebrain cholinergic neurons. Progress in brain research. 1993;98:265-. [DOI] [PubMed] [Google Scholar]
- 18.Gage FH, Armstrong DM, Williams LR, Varon S. Morphological response of axotomized septal neurons to nerve growth factor. Journal of Comparative Neurology. 1988;269(1):147–55. [DOI] [PubMed] [Google Scholar]
- 19.Goedert M, Fine A, Hunt S, Ullrich A. Nerve growth factor mRNA in peripheral and central rat tissues and in the human central nervous system: lesion effects in the rat brain and levels in Alzheimer’s disease. Molecular Brain Research. 1986;1(1):85–92. [DOI] [PubMed] [Google Scholar]
- 20.Fahnestock M, Yu G, Michalski B, Mathew S, Colquhoun A, Ross GM, et al. The nerve growth factor precursor proNGF exhibits neurotrophic activity but is less active than mature nerve growth factor. Journal of neurochemistry. 2004;89(3):581–92. [DOI] [PubMed] [Google Scholar]
- 21.Ioannou MS, Fahnestock M. ProNGF, but Not NGF, Switches from Neurotrophic to Apoptotic Activity in Response to Reductions in TrkA Receptor Levels. International journal of molecular sciences. 2017;18(3):599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pedraza CE, Podlesniy P, Vidal N, Arévalo JC, Lee R, Hempstead B, et al. Pro-NGF isolated from the human brain affected by Alzheimer’s disease induces neuronal apoptosis mediated by p75NTR. The American journal of pathology. 2005;166(2):533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427(6977):843–8. [DOI] [PubMed] [Google Scholar]
- 24.Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Molecular and Cellular Neuroscience. 2001;18(2):210–20. [DOI] [PubMed] [Google Scholar]
- 25.Bruno MA, Cuello AC. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proceedings of the National Academy of Sciences. 2006;103(17):6735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osterwalder T, Contartese J, Stoeckli E, Kuhn T, Sonderegger P. Neuroserpin, an axonally secreted serine protease inhibitor. The EMBO journal. 1996;15(12):2944–53. [PMC free article] [PubMed] [Google Scholar]
- 27.Krueger SR, Ghisu G-P, Cinelli P, Gschwend TP, Osterwalder T, Wolfer DP, et al. Expression of neuroserpin, an inhibitor of tissue plasminogen activator, in the developing and adult nervous system of the mouse. The Journal of neuroscience. 1997;17(23):8984–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cawston TE, Galloway WA, Mercer E, Murphy G, Reynolds JJ. Purification of rabbit bone inhibitor of collagenase. Biochemical Journal. 1981;195(1):159–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Docherty AJ, Lyons A, Smith BJ, Wright EM, Stephens PE, Harris TJ, et al. Sequence of human tissue inhibitor of metalloproteinases and its identity to erythroid-potentiating activity. Nature. 1985;318(6041):66. [DOI] [PubMed] [Google Scholar]
- 30.Allard S, Leon WC, Pakavathkumar P, Bruno MA, Ribeiro-da-Silva A, Cuello AC. Impact of the NGF maturation and degradation pathway on the cortical cholinergic system phenotype. The Journal of Neuroscience. 2012;32(6):2002–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allard S, Jacobs ML, Do Carmo S, Cuello AC. Compromise of cortical proNGF maturation causes selective retrograde atrophy in cholinergic nucleus basalis neurons. Neurobiology of aging. 2018;67:10–20. [DOI] [PubMed] [Google Scholar]
- 32.Bruno MA, Mufson EJ, Wuu J, Cuello AC. Increased Matrix Metalloproteinase-9 Activity in Mild Cognitive Impairment. Journal of neuropathology and experimental neurology. 2009;68(12):1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruno MA, Leon WC, Fragoso G, Mushynski WE, Almazan G, Cuello AC. Amyloid β-induced nerve growth factor dysmetabolism in Alzheimer disease. Journal of Neuropathology & Experimental Neurology. 2009;68(8):857–69. [DOI] [PubMed] [Google Scholar]
- 34.Fabbro S, Seeds NW. Plasminogen activator activity is inhibited while neuroserpin is up‐regulated in the Alzheimer disease brain. Journal of neurochemistry. 2009;109(2):303–15. [DOI] [PubMed] [Google Scholar]
- 35.Iulita MF, Bistue Millon MB, Pentz R, Aguilar LF, Do Carmo S, Allard S, et al. Differential deregulation of NGF and BDNF neurotrophins in a transgenic rat model of Alzheimer’s disease. Neurobiol Dis. 2017;108:307–23. [DOI] [PubMed] [Google Scholar]
- 36.A Bennett D A Schneider J, Arvanitakis Z, S Wilson R. Overview and findings from the religious orders study. Current Alzheimer Research. 2012;9(6):628–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett D, Schneider J, Arvanitakis Z, Kelly J, Aggarwal N, Shah R, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–44. [DOI] [PubMed] [Google Scholar]
- 38.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS‐ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939-. [DOI] [PubMed] [Google Scholar]
- 39.Iulita MF, Do Carmo S, Ower AK, Fortress AM, Aguilar LF, Hanna M, et al. Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain. 2014:awt372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Debeir T, Saragovi HU, Cuello AC. A nerve growth factor mimetic TrkA antagonist causes withdrawal of cortical cholinergic boutons in the adult rat. Proceedings of the National Academy of Sciences. 1999;96(7):4067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Head E, Lott IT. Down syndrome and beta-amyloid deposition. Current opinion in neurology. 2004;17(2):95–100. [DOI] [PubMed] [Google Scholar]
- 42.Lott IT, Head E. Dementia in Down syndrome: unique insights for Alzheimer disease research. Nature Reviews Neurology. 2019:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nature Reviews Immunology. 2004;4(8):617. [DOI] [PubMed] [Google Scholar]
- 44.Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nature Reviews Immunology. 2013;13(9):649. [DOI] [PubMed] [Google Scholar]
- 45.Nissinen L, Kähäri V-M. Matrix metalloproteinases in inflammation. Biochimica et Biophysica Acta (BBA)-General Subjects. 2014;1840(8):2571–80. [DOI] [PubMed] [Google Scholar]
- 46.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. The Lancet Neurology. 2015;14(4):388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogers J Principles for central nervous system inflammation research: A call for a consortium approach. Alzheimer’s & Dementia. 2018;14(11):1553–9. [DOI] [PubMed] [Google Scholar]
- 48.Cuello AC. Early and late CNS inflammation in Alzheimer’s disease: two extremes of a continuum? Trends in pharmacological sciences. 2017. [DOI] [PubMed] [Google Scholar]
- 49.Iulita MF, Caraci F, Cuello AC. A Link Between Nerve Growth Factor Metabolic Deregulation and Amyloid-beta-Driven Inflammation in Down Syndrome. CNS & neurological disorders drug targets. 2016;15(4):434–47. [DOI] [PubMed] [Google Scholar]
- 50.Iulita MF, Cuello AC. Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends in pharmacological sciences. 2014;35(7):338–48. [DOI] [PubMed] [Google Scholar]
- 51.Rosas‐Ballina M, Tracey K. Cholinergic control of inflammation. Journal of internal medicine. 2009;265(6):663–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Counts SE, Mufson EJ. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. Journal of Neuropathology & Experimental Neurology. 2005;64(4):263–72. [DOI] [PubMed] [Google Scholar]
- 53.Mesulam M, Shaw P, Mash D, Weintraub S. Cholinergic nucleus basalis tauopathy emerges early in the aging‐MCI‐AD continuum. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 2004;55(6):815–28. [DOI] [PubMed] [Google Scholar]
- 54.Cooper JD, Salehi A, Delcroix J-D, Howe CL, Belichenko PV, Chua-Couzens J, et al. Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proceedings of the National Academy of Sciences. 2001;98(18):10439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salehi A, Delcroix J-D, Belichenko PV, Zhan K, Wu C, Valletta JS, et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51(1):29–42. [DOI] [PubMed] [Google Scholar]
- 56.Venero J, Knüsel B, Beck K, Hefti F. Expression of neurotrophin and trk receptor genes in adult rats with fimbria transections: effect of intraventricular nerve growth factor and brain-derived neurotrophic factor administration. Neuroscience. 1994;59(4):797–815. [DOI] [PubMed] [Google Scholar]
- 57.Figueiredo B, Skup M, Bedard A, Tetzlaff W, Cuello A. Differential expression of p140trk, p75NGFR and growth-associated phosphoprotein-43 genes in nucleus basalis magnocellularis, thalamus and adjacent cortex following neocortical infarction and nerve growth factor treatment. Neuroscience. 1995;68(1):29–45. [DOI] [PubMed] [Google Scholar]
- 58.Gnahn H, Hefti F, Heumann R, Schwab M, Thoenen H. NGF-mediated increase of choline acetyltransferase (ChAT) in the neonatal rat forebrain: evidence for a physiological role of NGF in the brain? Developmental Brain Research. 1983;9(1):45–52. [DOI] [PubMed] [Google Scholar]
- 59.Stephens P, Cuello A, Sofroniew M, Pearson R, Tagari P. Effect of unilateral decortication on choline acetyltransferase activity in the nucleus basalis and other areas of the rat brain. Journal of neurochemistry. 1985;45(4):1021–6. [DOI] [PubMed] [Google Scholar]
- 60.Hartikka J, Hefti F. Development of septal cholinergic neurons in culture: plating density and glial cells modulate effects of NGF on survival, fiber growth, and expression of transmitter-specific enzymes. Journal of Neuroscience. 1988;8(8):2967–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pongrac JL, Rylett RJ. NGF-induction of the expression of ChAT mRNA in PC12 cells and primary cultures of embryonic rat basal forebrain. Molecular brain research. 1998;62(1):25–34. [DOI] [PubMed] [Google Scholar]
- 62.Berse B, Lopez-Coviella I, Blusztajn JK. Activation of TrkA by nerve growth factor upregulates expression of the cholinergic gene locus but attenuates the response to ciliary neurotrophic growth factor. Biochemical Journal. 1999;342(2):301–8. [PMC free article] [PubMed] [Google Scholar]
- 63.Madziar B, Lopez‐Coviella I, Zemelko V, Berse B. Regulation of cholinergic gene expression by nerve growth factor depends on the phosphatidylinositol‐3′‐kinase pathway. Journal of neurochemistry. 2005;92(4):767–79. [DOI] [PubMed] [Google Scholar]
- 64.Villarin JM, McCurdy EP, Martínez JC, Hengst U. Local synthesis of dynein cofactors matches retrograde transport to acutely changing demands. Nature communications. 2016;7:13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoon BC, Jung H, Dwivedy A, O’Hare CM, Zivraj KH, Holt CE. Local translation of extranuclear lamin B promotes axon maintenance. Cell. 2012;148(4):752–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iulita MF, Ower A, Barone C, Pentz R, Gubert P, Romano C, et al. An inflammatory and trophic disconnect biomarker profile revealed in Down syndrome plasma: Relation to cognitive decline and longitudinal evaluation. Alzheimer’s & Dementia. 2016. [DOI] [PubMed] [Google Scholar]
- 67.Iulita MF, Ganesh A, Pentz R, Flores Aguilar L, Gubert P, Ducatenzeiler A, et al. Identification and Preliminary Validation of a Plasma Profile Associated with Cognitive Decline in Dementia and At-Risk Individuals: A Retrospective Cohort Analysis. Journal of Alzheimer’s Disease. 2019;67(1):327–41. [DOI] [PubMed] [Google Scholar]
- 68.Hanzel CE, Iulita MF, Eyjolfsdottir H, Hjorth E, Schultzberg M, Eriksdotter M, et al. Analysis of matrix metallo-proteases and the plasminogen system in mild cognitive impairment and Alzheimer’s disease cerebrospinal fluid. Journal of Alzheimer’s disease. 2014;40(3):667–78. [DOI] [PubMed] [Google Scholar]
- 69.E Counts S, He B, G Prout J, Michalski B, Farotti L, Fahnestock M, et al. Cerebrospinal fluid proNGF: a putative biomarker for early Alzheimer’s disease. Current Alzheimer Research. 2016;13(7):800–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blennow K, Zetterberg H. Biomarkers for Alzheimer’s disease: current status and prospects for the future. Journal of internal medicine. 2018;284(6):643–63. [DOI] [PubMed] [Google Scholar]
- 71.Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annual review of psychology. 1997;48(1):649–84. [DOI] [PubMed] [Google Scholar]
- 72.Mesulam M The cholinergic lesion of Alzheimer’s disease: pivotal factor or side show? Learning & Memory. 2004;11(1):43–9. [DOI] [PubMed] [Google Scholar]
- 73.Zissimopoulos J, Crimmins E, Clair PS, editors. The value of delaying Alzheimer’s disease onset. Forum for Health Economics and Policy; 2015: De Gruyter. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hampel H, Mesulam M-M, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 2018;141(7):1917–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cavedo E, Dubois B, Colliot O, Lista S, Croisile B, Tisserand GL, et al. Reduced regional cortical thickness rate of change in donepezil-treated subjects with suspected prodromal Alzheimer’s disease. Journal of Clinical Psychiatry. 2016. [DOI] [PubMed] [Google Scholar]
- 76.Dubois B, Chupin M, Hampel H, Lista S, Cavedo E, Croisile B, et al. Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1041–9. [DOI] [PubMed] [Google Scholar]
- 77.Fox C, Richardson K, Maidment ID, Savva GM, Matthews FE, Smithard D, et al. Anticholinergic medication use and cognitive impairment in the older population: the medical research council cognitive function and ageing study. Journal of the American Geriatrics Society. 2011;59(8):1477–83. [DOI] [PubMed] [Google Scholar]
- 78.Risacher SL, McDonald BC, Tallman EF, West JD, Farlow MR, Unverzagt FW, et al. Association between anticholinergic medication use and cognition, brain metabolism, and brain atrophy in cognitively normal older adults. JAMA neurology. 2016;73(6):721–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Campbell NL, Lane KA, Gao S, Boustani MA, Unverzagt F. Anticholinergics influence transition from normal cognition to mild cognitive impairment in older adults in primary care. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy. 2018;38(5):511–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Richardson K, Fox C, Maidment I, Steel N, Loke YK, Arthur A, et al. Anticholinergic drugs and risk of dementia: case-control study. bmj. 2018;361:k1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schmitz TW, Mur M, Aghourian M, Bedard MA, Spreng RN, Alzheimer’s Disease Neuroimaging I. Longitudinal Alzheimer’s Degeneration Reflects the Spatial Topography of Cholinergic Basal Forebrain Projections. Cell Rep. 2018;24(1):38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schmitz TW, Nathan Spreng R, Alzheimer’s Disease Neuroimaging I. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat Commun. 2016;7:13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258(5080):304–7. [DOI] [PubMed] [Google Scholar]
- 84.Fisher A Cholinergic modulation of amyloid precursor protein processing with emphasis on M1 muscarinic receptor: perspectives and challenges in treatment of Alzheimer’s disease. Journal of neurochemistry. 2012;120:22–33. [DOI] [PubMed] [Google Scholar]
- 85.Hall H, Iulita MF, Gubert P, Flores Aguilar L, Ducatenzeiler A, Fisher A, et al. AF710B, an M1/sigma-1 receptor agonist with long-lasting disease-modifying properties in a transgenic rat model of Alzheimer’s disease. Alzheimers Dement. 2018;14(6):811–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.