

Abstract
Powdery mildew resistance genes restrict infection attempts at different stages of pathogenesis. Here, a strong and rapid powdery mildew resistance phenotype was discovered from Vitis amurensis ‘PI 588631’ that rapidly stopped over 97% of Erysiphe necator conidia, before or immediately after emergence of a secondary hypha from appressoria. This resistance was effective across multiple years of vineyard evaluation on leaves, stems, rachises, and fruit and against a diverse array of E. necator laboratory isolates. Using core genome rhAmpSeq markers, resistance mapped to a single dominant locus (here named REN12) on chromosome 13 near 22.8–27.0 Mb, irrespective of tissue type, explaining up to 86.9% of the phenotypic variation observed on leaves. Shotgun sequencing of recombinant vines using skim-seq technology enabled the locus to be further resolved to a 780 kb region, from 25.15 to 25.93 Mb. RNASeq analysis indicated the allele-specific expression of four resistance genes (NLRs) from the resistant parent. REN12 is one of the strongest powdery mildew resistance loci in grapevine yet documented, and the rhAmpSeq sequences presented here can be directly used for marker-assisted selection or converted to other genotyping platforms. While no virulent isolates were identified among the genetically diverse isolates and wild populations of E. necator tested here, NLR loci like REN12 are often race-specific. Thus, stacking of multiple resistance genes and minimal use of fungicides should enhance the durability of resistance and could enable a 90% reduction in fungicides in low-rainfall climates where few other pathogens attack the foliage or fruit.
Introduction
Plants and pathogens have interacted over an evolutionary scale of time within the context of an arms race involving host resistance genes (nucleotide-binding site leucine-rich repeats, or NLRs) and pathogen effectors. Reasonably, researchers expect to find host resistance within geographic regions where co-evolution occurred. Based on historical records and population genetic analysis, eastern North America has been documented as a center of diversity for Erysiphe necator (Schwein.) Burr [1], which causes grapevine powdery mildew (GPM). As expected, most Vitis species native to eastern North America exhibit resistance to GPM, and this resistance typically maps to NLR loci, such as RUN1, REN2, REN3, REN11 [2–8]. Surprisingly, several Vitis species from Eurasia also express qualitative resistance that maps to NLR loci, such as REN1, REN4, REN6, REN7 [9–11].
A similar scenario exists for Plasmopara viticola, which causes grapevine downy mildew. Both pathogens have centers of diversity in the Eastern US, were introduced to Europe in the mid-1800s, and were subsequently distributed worldwide [12], and yet host resistance occurs in apparently non-adapted wild Vitis [13]. An excellent example is the Amur grapevine V. amurensis, from Northeastern Asia. Some accessions of V. amurensis have been used for breeding for resistance to downy mildew [14–17] and ripe rot [18], as well as for cold hardiness [19] and for the presence of compounds of medicinal interest [20]. While there are reports of GPM resistance from V. amurensis [13, 21], the genetic basis is unknown. This is in sharp contrast to the at least eight downy mildew resistance loci that have been mapped in V. amurensis: RPV8, 10, 12, and 22 to 26 [14–16, 22].
The obligate ascomycete E. necator infects each green tissue: leaf, stem, rachis, and fruit. This causes a decrease in yield, vine growth, winter hardiness, and fruit and wine quality [23, 24]. The disease cycle begins after a viable conidium or ascospore is deposited on host tissue, germinates, and forms a primary appressorium. The appressorium produces a penetration peg that pierces the host cuticle and epidermal cell wall, then advances to form a feeding structure called a haustorium. Haustoria do not disrupt the host cell membrane, but rather invaginate it; the resultant double-membrane structure acts as a reservoir for water and nutrients exported to the developing epiphytic hyphae [25]. On the host surface, hyphae elongate, branch and infect additional epidermal cells to form a colony. Conidiation upon this colony is dependent upon achieving a critical hyphal density that triggers production of conidiophores and sporulation [25–27].
The disease cycle is halted or slowed down if the host/pathogen interaction is incompatible (Cadle-Davidson 2007). This resistance mechanism is often characterized by the rate at which the pathogen successfully forms a secondary hypha as an indicator of haustorial formation (penetration success) and the rate by which programmed cell death (PCD) halts further colonization (colony success), or by reduced hyphal length or extended latent period [13, 28, 29]. Host resistance need not prevent infection entirely to have a disproportionate effect upon epidemic development, as reducing the density of hyphal colonization can substantially delay or prevent the production of secondary inoculum [27]. A few strong resistance genes have been shown to act quickly and qualitatively reduce the formation of secondary hyphae on leaves, among these most notably RUN1, REN4, and REN6 [10, 30–32]. Most of the time, GPM resistance on leaves is an excellent indicator of resistance on other tissues, though a counter example was recently found in that REN11 confers reproducibly strong resistance on all green tissues except stems [6].
Grape breeders routinely use DNA markers for parent selection, genetic mapping of traits, and introgression and selection of multiple resistance genes for durable resistance. However, breeders continue to seek a better marker platform that is cost-effective and can combine multiple resistance genes simultaneously with high accuracy [33]. A multiallelic, local haplotype-based marker platform was recently developed to target the core Vitis genome for improved marker transferability, using rhAmpseq chemistry [34]. While this marker panel can be routinely used for QTL analysis and marker-assisted selection, it currently genotypes only 2000 sites across the genome, limiting its applications for fine mapping. Because next-generation sequencing continues to have rapid cost decreases, simplified and cost-efficient library preparation, and improved imputation methods, whole genome shotgun sequencing like skim-seq has gained more attention for QTL fine-mapping [35].
Here, we discovered and characterized the genetics and phenotypes of a novel GPM resistance source from V. amurensis “PI 588631”. A combination of traditional and next-generation genotyping and phenotyping tools enabled us to make efficient progress to determine the significance of this resistance, identify candidate genes likely controlling resistance, and develop DNA markers and germplasm for grape genetic improvement.
Results
Resistance mechanism and race specificity
To characterize the mechanism and race specificity of resistance from the grapevine powdery mildew (GPM) resistance source V. amurensis “PI 588631”, a diversity panel of E. necator isolates was used for controlled inoculations observed at 48 hpi. Resistance was similarly strong, rapid, and effective against all isolates, with over 97% of conidia being stopped before a second hypha or a hyphal branch was formed (Figure S1 & S2). Resistance was characterized as a significant reduction in the frequency of a secondary hypha formed from the primary appressorium (P < 2 × 10−16), loosely indicative of penetration success rate (Figure 1). Further, the conidia that successfully formed a secondary hypha had a significant reduction in multiple or secondary hyphal branching (P < 2 × 10−16), indicative of colony success rate, frequently associated with host epidermal necrosis and typical of a programmed cell death (PCD) response (Figure 1).
Figure 1.
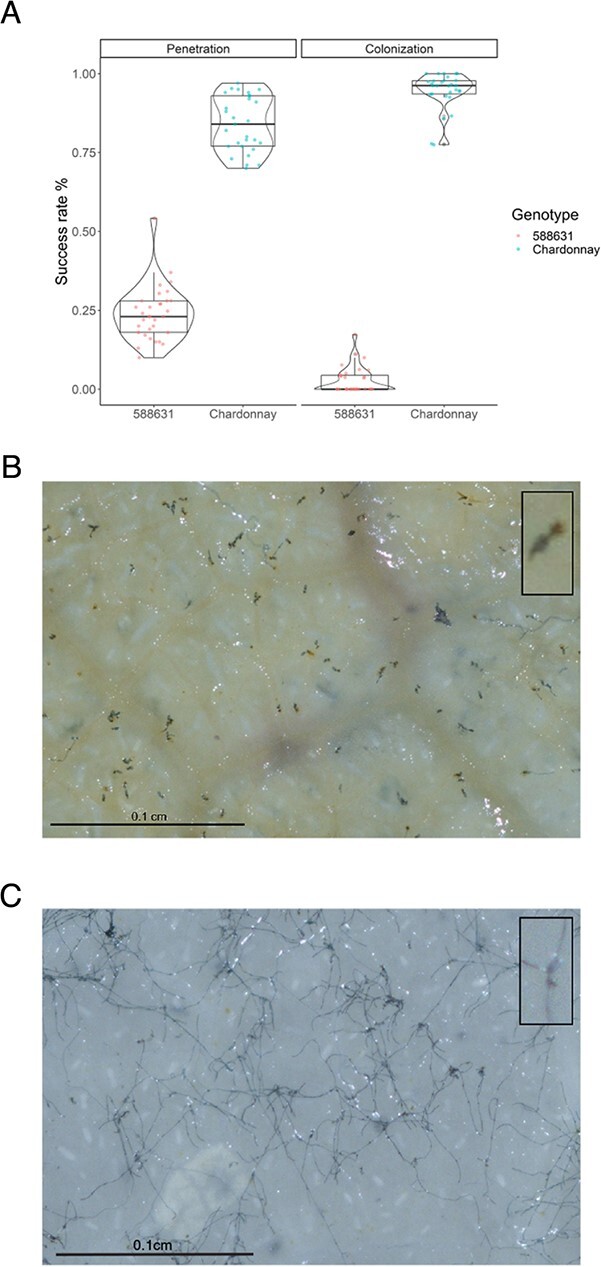
Vitis amurensis “PI 588631” exhibits a rapid and strong resistance response associated with host necrosis at attempted powdery mildew penetration sites. A) Overall distribution of penetration and colony success rate between the powdery mildew resistant accession V. amurensis “PI 588631” and susceptible control Vitis vinifera ‘Chardonnay’. Penetration was measured as the percentage of germinated conidia forming at least one secondary hypha, and colony success rate as the percentage of penetrating conidia forming multiple or branched hypha. Each dot within the plots represents a mean value of a sample, and bars within the boxplot represent inter-quartile ranges. B) Micrograph of of Erysiphe necator isolate MUSC5 inoculated onto V. amurensis “PI 588631”(left) in 2019 showing the representative host necrosis (brown areas in the corner inset picture) associated with attempted penetration, and a comparison of C) susceptible control ‘Chardonnay’ with successful penetration (inset) and subsequent hyphal growth. Images with additional isolates are shown in Figure S2.
Segregation of the phenotypes
Evaluation in the field
Grapevine powdery mildew resistance was evaluated in the vineyard after natural infection of the Vitis amurensis “PI 588631” × V. vinifera ‘Valley Pearl’ F1 individuals. Disease severity increased on all tissue types within each year on individual vines and newly symptomatic vines as the planting and inoculum became established (Figure 2). In 2016, GPM was found only on leaves, with 98.4% of vines being asymptomatic. By October 2017, approximately 25% of vines had GPM on leaves, stems and fruit. By September 2018, GPM was established on 50% of vines for these three tissues, suggesting 1:1 segregation, and on 13.4% of fruit (Figure 2).
Figure 2.
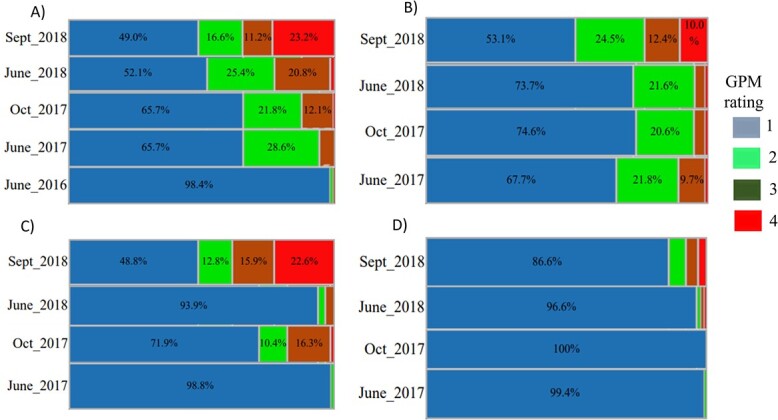
Grapevine powdery mildew (GPM) resistance segregation in the Vitis amurensis “PI 588631” × Vitis vinifera ‘Valley Pearl’ F1 family at the Parlier, CA vineyard on: A) leaf, B) stem, C) rachis, and D) fruit. Disease severity was evaluated for three years for leaf and two years for stem, rachis and fruit. Symptoms were classified from 0 to 4 (no disease to >50% severity). GPM severity increased over time irrespective of the tissue evaluated.
Computer vision analysis of the controlled inoculations
Vineyard phenotypes of the parents and F1 family were validated using controlled inoculation of leaf disks with E. necator isolate MUSC4, monitored by automated live imaging and neural network analysis of hyphal growth over time at 5, 7, 9 and 10 dpi. Phenotypic segregation of the controlled inoculation showed a bimodal distribution irrespective of timepoint (Figure 3, Figure S4). There was a strong correlation between lab and 2018 field phenotypes (r = 0.68–0.76), with lower correlation (r = 0.43–0.50) between lab and 2017 field phenotypes and no significant correlation with 2016 field phenotypes (Table S3).
Figure 3.
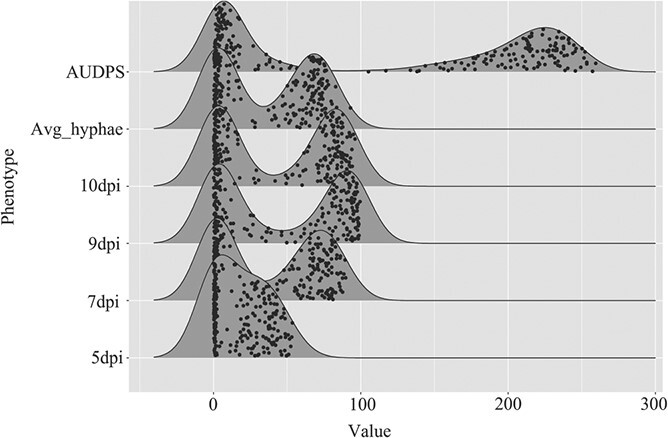
Powdery mildew growth over time on leaf disks using controlled inoculations. Images were taken at 5-, 7-, 9- and 10-days post-inoculation (dpi) using the PMbot microscopy robot. Disease severity was quantified for each leaf disk using a convolutional neural network based on GoogLeNet. The parameter Avg_hyphae was obtained by averaging hyphal growth over 5, 7, 9 and 10 dpi. AUDPS is the area under disease progress stairs, the sum of these same timepoints.
Genetic analysis
rhAmpSeq-based genetic map and collinearity
Of the 2000 rhAmpSeq core genome markers tested, 874 markers were polymorphic across 248 F1 progeny. Overall, there was a high degree of collinearity between the parental maps (Figure 4). In the resistant parent, V. amurensis “PI 588631”, 862 markers were mapped with an inter-marker distance of 1.37 cM spanning 1121 cM across the genome (Table S4, Figure S5). Chromosomes (Chr) 8, 12, 13, 16 and 18 had inter-marker distances of more than 10 cM, often adjacent to long physical distances with no recombination, especially on ‘Valley Pearl’ Chr 12, 13, 16 and 18 (Figure 4).
Figure 4.

Collinearity between physical (Mb) and genetic map (cM) for the 19 different chromosomes in the Vitis amurensis “PI 588631” × Vitis vinifera ‘Valley Pearl’ F1 family. Overall, there was good synteny between the maps with some chromosomal rearrangements in susceptible parent ‘Valley Pearl’.
Reproducible major QTL across different tissue types
A total of 24 disease severity phenotypes were used to identify QTL. A significant QTL was consistently detected on Chr 13 near 22.8–27.0 Mb, irrespective of tissue types, with various degrees of strength across observations (Figure 5, Figure S6 & S7). This locus was assigned the name REN12 (Resistance to E. necator-12), and it explained variability in disease severity more significantly on leaf tissue, followed by stem, rachis, and fruit with maximum LOD values of 101.5, 34.1, 32.2, and 4.1 respectively. Phenotypes from June 2016 did not detect any QTL, and no other QTL were detected apart from REN12 (Table 1). The highest proportion of the phenotypic variance explained (86.9%) was obtained using AUDPS (area under disease progress stairs, which sums disease severity across timepoints) of controlled inoculation, automated imaging, and convolutional neural network analysis. The REN12 locus explained little of the variation in fruit severity, likely due to susceptible vines escaping infection, as reflected by the presence of disease affecting only 0.6 to 14.5% of vines (Figure 2). On all tissues, the observed increase in the number of symptomatic vines from 2016 to 2018 corresponded with increased significance and phenotypic variance explained by REN12, suggesting that susceptible vines escaped infection early in vineyard establishment.
Figure 5.

Genetic location of REN12 powdery mildew resistance in the Vitis amurensis “PI 588631” × Vitis vinifera ‘Valley Pearl’ mapping family. The horizontal red dashed line in the upper panel represents a logarithm of odds (LOD) significance threshold of 4.1. In the lower panel, interval mapping shows REN12 was reproducible across different tissue types with the leaf being most significant. The red dotted box indicates the most probable boundary that spans 4.2 Mb.
Table 1. Summary of QTL for powdery mildew severity detected across various tissue types in the Vitis amurensis “PI 588631” × Vitis vinifera ‘Valley Pearl’ mapping family.
| Tissue type | Location | Phenotype | LOD threshold | LOD max | LOD max marker | Confidence interval (Mb) [LOD max − 1] | Variance explained (%) |
|---|---|---|---|---|---|---|---|
| Leaf | Field | June_2016 | - | - | - | - | - |
| Leaf | Field | June_2017 | 4.3 | 19.1 | 13_25013004 | 22.8–27.0 | 29.9 |
| Field | Oct_2017 | 4.2 | 18.9 | 13_25145371 | 22.4–27.0 | 29.7 | |
| Field | June_2018 | 4.2 | 42.0 | 13_25145371 | 22.8–27.0 | 55.9 | |
| Field | Sept_2018 | 4.1 | 53.8 | 13_25280719 | 22.8–27.0 | 64.2 | |
| Lab | 5dpi | 4.1 | 53.5 | 13_25145371 | 22.8–27.0 | 63.7 | |
| Lab | 7dpi | 4.1 | 88.2 | 13_25280719 | 22.8–27.0 | 81.3 | |
| Lab | 9dpi | 4.1 | 82.3 | 13_25145371 | 22.8–27.0 | 80.8 | |
| Lab | 10dpi | 4.1 | 87.7 | 13_25280719 | 22.8–27.0 | 80.8 | |
| Lab | Average | 4.2 | 84.8 | 13_25145371 | 22.8–27.0 | 84.8 | |
| Lab | AUDPS | 4.1 | 101.5 | 13_25145371 | 22.8–27.0 | 86.9 | |
| Stem | Field | June_2017 | 4.2 | 4.1 | 13_25145371 | 0.4–28.9 | 7.3 |
| Field | Oct_2017 | 4.2 | 5.4 | 13_26962710 | 22.4–28.9 | 9.6 | |
| Field | June_2018 | 4.2 | 9.9 | 13_25145371 | 19.7–27.7 | 17.6 | |
| Field | Sept_2018 | 4.1 | 32.2 | 13_25280719 | 22.8–27.7 | 45.9 | |
| Rachis | Field | June_2017 | - | - | - | - | - |
| Field | Oct_2017 | 4.0 | 10.4 | 13_25013004 | 22.4–27.7 | 29.8 | |
| Field | June_2018 | - | - | - | - | - | |
| Field | Sept_2018 | 4.2 | 34.1 | 13_25280719 | 22.8–27.0 | 61.6 | |
| Fruit | Field | June_2017 | - | - | - | - | - |
| Field | Oct_2017 | - | - | - | - | - | |
| Field | June_2018 | - | - | - | - | - | |
| Field | Sept_2018 | 4.1 | 4.1 | 13_26962710 | 7.0–28.9 | 10.9 |
Fine mapping
To estimate the QTL boundary of REN12 more accurately, we analyzed the position of the recombination events on the resistant donor parent V. amurensis “PI 588631” using both rhAmpSeq and skim-seq technologies (Figure 6). The rhAmpSeq markers delimited a region spanning from 22.7 Mb to 27.0 Mb on Chr 13. The desirable alleles in this haploblock (Table S3) are available for marker-assisted selection. When increasing the marker density using skim-seq technology, the most probable region lies from 25.15 Mb to 25.93 Mb. Considering the phenotype of 540_121 was moderate between resistance and susceptibility, the QTL might extend to 26.3 Mb. On the reference genome PN40024 12X.v2 [17], a total of 92 genes were annotated in the VCost.v3 annotation from 25.00 Mb to 26.5 Mb. After building a reference-guided transcriptome assembly from the RNA-seq of inoculated and mock-inoculated resistant vines, we found 51 newly annotated genes in this region. Among these 143 genes, we annotated 12 resistance-related genes and 13 NLRs (including complete, pseudogene, and partial NLRs) (Figure 7, Table S5). In this region, 52 genes were expressed, and five of those are significantly differentially expressed among inoculated and non-inoculated samples (Benjamini-Hochberg adjusted P-value <0.01, Wald test by Deseq2). When considering the allele-specific expression, there are 11 genes including four NLRs that were significantly differentially expressed (P < 0.01, Wald test by Deseq2). Recently, genome sequence for a different accession of V. amurensis was published, a variety called ‘Shanputao’ [36]. A genome alignment of the REN12 candidate region between PN40024 and V. amurensis ‘Shanputao’ revealed structural variations in this region and approximately double the number of NLRs (including complete, pseudogene, and partial) in the V. amurensis ‘Shanputao’ locus (Figure S8). Genome-wide, 320 differentially expressed genes (DEGs) were identified after inoculation of resistant grapevines (Figure S9). These DEGs are enriched in genes involved in response to cold and chitin, kinase, and transcription factors (Figure S10 & S11).
Figure 6.

The recombinations inferred from rhAmpSeq and skim-seq analysis define the boundary of the REN12 resistance QTL. (A) Recombinations were estimated from 31 rhAmpseq markers (rows) for 72 vines (columns). The red dotted box indicates the most probable boundary that spans 4.2 Mb. (B) The most probable boundary of REN12 is reduced to a 0.78 Mb region based on 1607 skim-seq variants from 31 vines. Each column represents an individual vine, and rows represent a phenotype or a rhAmpSeq marker or skim-seq variant ordered by their physical position.
Figure 7.
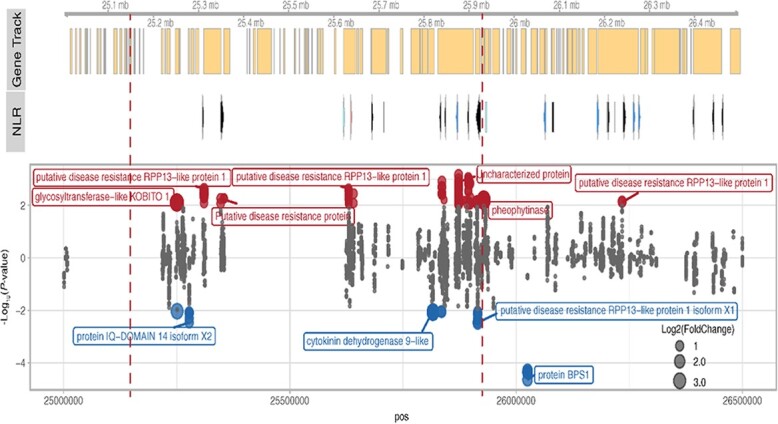
Gene annotation and allele-specific expression at the REN12 resistance locus. Gene Track: gene models combined from COST.v3 annotation and reference guided transcriptome annotation. NLR: Predicted resistance genes (NLRs) in this region. The color of the arrow or box indicates different status of the gene. Black, complete NLR; blue, complete but pseudogene; light blue, partial pseudogene; rose, partial NLRs. Lower Panel: Differentially expressed genes between the inoculated and mock experiment at resistant allele only. The y-axis is an adjusted −log10 P value, and the negative values indicate down regulated after inoculation. The sizes of the dots illustrate the log2 fold change of the gene. The color of the dots indicates the up- (red) or down- (blue) regulation of the gene.
Discussion
Breeders constantly search for beneficial alleles for use in crop improvement. Because the European grapevine V. vinifera evolved in the absence of selection pressure from grapevine powdery mildew (GPM), nearly all cultivars are highly susceptible. Thus, grape breeders look for desirable alleles among Vitis species with a significant coevolutionary history with E. necator to introgress into high quality V. vinifera genetic backgrounds. Here, we report a novel resistance locus REN12, conferring a strong and rapid resistance to GPM inherited from the wild Amur grapevine V. amurensis “PI 588631”, a species with no historical record of an evolutionary association with the causal agent of GPM. Several accessions of V. amurensis have been used to study resistance against grapevine downy mildew caused by P. viticola [14–16, 22], but this is the first study to report its ability to strongly resist GPM growth.
For more than a decade we have used genetically diverse isolates of E. necator collected across the Eastern U.S. center of diversity [1, 37] to characterize the relative strength, speed, and race specificity of GPM resistance alleles. In this study, against all isolates tested, REN12 prevented at least 97% of conidia from forming two short hypha or a hyphal branch by 48 hpi. This is as strong and rapid as any resistance we have observed in these bioassays. To our knowledge, there are only three resistance alleles for which we or others have not found race specificity following natural infection or when challenged with these diverse isolates: REN12, REN5, and REN4 [34, 38]. Thus, not only is REN12 among the strongest resistance loci we have characterized (similar to RUN1, REN4, and REN6), but it also appears to confer resistance to diverse isolates of the pathogen. Still, given that REN12, REN5, and REN4 map to NLR loci, we anticipate they will eventually be shown to confer race-specific resistance.
The V. amurensis “PI 588631” × V. vinifera ‘Valley Pearl’ mapping family was generated in 2014 and planted to a Parlier, CA research vineyard in 2015. In this first year, 98.4% of vines had no visible GPM, and as expected, no QTL were detected. As GPM increased over years and within growing seasons, QTL associated with all vineyard phenotypes became more significant, suggesting that escaped infection on susceptible vines was affecting the results. An alternative hypothesis that additional resistance loci may have been effective early in the study and diminished due to race specificity is not supported since no other loci were detected in any QTL analysis here.
The rhAmpSeq core genome strategy for genetic map construction has proven to be a simple and efficient method for construction of genetic maps and marker-assisted selection. By publishing the desirable local haplotype amplicon sequences, rhAmpSeq QTL studies enable breeders to use the markers directly or convert them to other genotyping platforms. However, when converting to other platforms, the caveat is that individual polymorphisms in the amplicon do not track ancestral differences as accurately as the full haplotype of multiple polymorphisms.
Because the informative rhAmpSeq core genome markers in this family were separated by 550 kb on average, they lacked the resolution for fine mapping and identification of candidate genes. In this case, the rhAmpSeq markers delimited a region spanning from 22.7 Mb to 27.0 Mb on Chr 13. To overcome this, we used the rhAmpSeq markers to identify vines with recombinations at or near REN12, then used skim-seq as a cost-effective tool for shotgun genome sequencing of the recombinants. This narrowed the region from 4.3 Mb to 780 kb. The challenge then is identifying candidate genes on the resistant haplotype in the absence of genome sequence for this resistance source. Although a reference genome exists for V. amurensis ‘Shanputao’ [36], the fact that so many resistance QTL have been found in diverse accessions of V. amurensis and are not shared among diverse accessions suggests that a genome specific to each study should be assembled for each accession of this species. In lieu of this, we used allele-specific RNASeq analysis to identify four NLRs that were significantly differentially expressed from the resistant haplotype, which are the most likely candidate genes. The functional testing of expressed NLRs genes to be identified in the resistant haplotype of V. amurensis “588 631” genome is a logical next step.
The REN12 locus could become widely used in traditional grape breeding and is publicly available from the USDA cold-hardy grapevine repository in Geneva, NY. While this locus could reduce powdery mildew fungicide applications by up to 90% in commercial plantings [39], there are a limited number of strong resistance alleles in Vitis, which should be protected for future generations. To preserve what is likely an at-risk but highly useful asset for disease suppression, we recommend the stacking of resistance loci and that their deployment be combined with other suppressive tactics, including minimal fungicide programs, cultural practices to reduce disease pressure, and any other means (e.g. UV applications) to minimize the selection of virulent isolates.
Material and methods
Plant material
The mapping family consisted of F1 hybrids derived by crossing a female resistant parent V. amurensis Rupr. “PI 588631” with a pollen donor Vitis vinifera “Valley Pearl”. The pollen parent “Valley Pearl” is an early to midseason, white seedless table grape (V. vinifera) obtained by crossing table cross selections A60–42 and C77–79 at the ARS San Joaquin Valley Agricultural Sciences Center in Parlier, CA [40]. The resistant parent V. amurensis “PI 588631” is a dioecious female grown at the USDA-ARS repository in Geneva, NY. A cross between these parents were made in 2014. These hybrids were planted at the ARS San Joaquin Valley Agricultural Sciences Center (latitude 36°81′N: longitude 119°72′W) with a row spacing of 3.66 m and 0.91 m between plants and were spur pruned on a single T-trellis. Vines were drip-irrigated and were grown in fine sandy loam soil. The 248 F1 hybrids obtained by crossing these two parents were used to study GPM resistance. No fungicides were applied for GPM evaluation [11].
Phenotyping
Powdery mildew assessment in Parlier, CA
Vineyard rating following natural infections was performed from the year 2016 to 2018 as previously described [41]. Briefly, GPM severity was rated subjectively by one expert on leaves, stems, rachis, and berries with the following scale: 1 = no observed infections; 2 = very few small colonies; 3 ≤ 50% of the area affected; and 4 ≥ 50% of the area affected. GPM susceptible vines ‘Ruby Seedless’ and ‘Thompson Seedless’ were interplanted within the family and were used as susceptible controls. Vines were evaluated between June and October each year, only when susceptible check vines showed >50% foliar disease severity.
Validation in the lab using PMbot
To validate phenotypes from the field, a controlled experiment was conducted for foliar resistance at [42] phenotyping center in Geneva, NY as previously described [41, 43]. Briefly, one leaf from each of four replicated shoots was sampled at the 3rd node from each vine from the vineyard at Parlier, CA, and was shipped overnight at 4°C for next-day processing. From each leaf, two subsampled leaf disks were created using a 1-cm cork borer, and one leaf disk from each vine plus susceptible and resistant checks were arrayed on 1% water agar media in a 32 × 26 × 2 cm Pyrex® tray (adaxial surface up), with 8 replicate trays.
Erysiphe necator isolate MUSC4, an isolate collected from Vitis rotundifolia and fully virulent on RUN1 vines, was used to study phenotype segregation. MUSC4 was propagated and maintained using surface-sterilized detached leaves of disease-free ‘Chardonnay’ or ‘Thompson Seedless’ vines grown in greenhouses at Cornell AgriTech, Geneva NY. The leaf disks were spray inoculated using a Preval paint sprayer (Coal City, IL, USA) to apply 5 × 104 conidia per ml suspended in distilled water containing 0.001% Tween-20 until visible droplets were seen on the disks. The trays were allowed to dry, then covered to maintain high humidity. To provide a stable condition for GPM growth, inoculated trays were incubated in a growth chamber maintained at 23°C with a 12-hour photoperiod (45 μmol m−2 s−1 of photosynthetically active radiation, PAR) irradiance until and between imaging.
Automated microscopy data acquisition
The inoculated trays were imaged at 5-, 7-, 9-, and 10-days post-inoculation (dpi) using an automated system for image capture called PMbot, as described in Bierman et al. [43]. Briefly, the system consists of a Nikon model D850 46 MP digital SLR camera with 60 MM F/2.8 D Micro Autofocus lens, an automated robotic Z-axis positioner for focusing, and oblique LED illumination of sample trays sitting on an automated X-Y robotic stage for movement between samples. To capture an image, a sample tray is positioned against the corner guide rails of the stage platform, and the sample layout is loaded into the software written in MATLAB® 2018B. The system captures multiple images (typically 3–10) for each leaf disk, depending upon the uniformity of the surface. These sub-images are stacked into a single image using Helicon Focus 6 software [44].
For this experiment, images were analyzed using trained convolutional neural networks based on a modified GoogLeNet from the MATLAB® Deep Learning Toolbox, version 18.1.0. During the process, a single image is classified into sub-images of 224 × 224 pixels each, which are assessed as the binary of either presence or absence of hypha. The incidence of detection for the whole leaf disk gives a disease severity percentage for that particular leaf disk.
Isolate-specific quantification of penetration and colony success rates
To study the mechanism of resistance, four fully expanded leaves from the 3rd node were collected from both V. amurensis “PI 588631” and V. vinifera ‘Chardonnay’ vines and were processed as described above. Each leaf disk was challenged with six different E. necator isolates (NY19, NY90, RoACS, NY1–137, MUSC4, and MUSC5) as described in Table S3. These isolates were propagated and maintained using V. vinifera ‘Chardonnay’ or ‘Thompson Seedless’ vines grown in greenhouses at Cornell AgriTech, Geneva NY. To quantify penetration and colony success rates, leaf disks were cleared 48 hours after inoculation in 3:1 (vol/vol) ethanol:acetic acid until the tissue was completely bleached, then were stained with 0.5% Chlorazol Black E aqueous solution for 16 hours. After staining, disks were rinsed briefly in distilled water and placed onto an agar imaging tray along with a drop of 1:1 (vol/vol), glycerol:water solution for image collection. In 2019, a prototype imaging robot, PMbot, was used as detailed above [43]. In 2021, the commercially available Blackbird version of this imaging robot was used [45].
One hundred germinated conidia per leaf disk were counted within each image for the presence of: i) primary hypha leading to an appressorium, ii) a single unbranched secondary hypha at least twice as long as the conidium, or iii) multiple or branching secondary hyphae. The penetration success rate was quantified as the proportion of germinated conidia that formed secondary hypha or hyphae (ratio of category ii + iii to i + ii + iii), and the colony success rate was quantified as the percentage of penetrating conidia that formed multiple or branching secondary hyphae (ratio of category iii to ii + iii).
Statistical analyses
Phenotypic correlation among lab and different field tissue phenotypes (leaf, stem, rachis and fruit) were calculated using the chat. Correlation function in the R package “Performance Analytics” (Peterson et al. 2018), and were plotted using the “ggplot2” package [46] in R software version 3.6.3. The broad-sense heritability was estimated using linear mixed-effects models implemented in the R package “lme4” (Bates et al. 2008) as the proportion of the variance due to genotype relative to the total variance (genotype and environment) using a method detailed in Reshef et al. [47]. Normality of the phenotypes was visualized using histogram and QQ plots and was further quantitatively tested using Shapiro–Wilk test. Phenotypes that deviated from normality were Box-Cox transformed using the “MASS” package in R software version 3.6.3 (Ripley et al. 2013). Both non-parametric and normal QTL models were conducted using a four-way cross format in the “R/qtl” package [48].
rhAmpSeq genetic map construction
DNA was extracted by Intertek AgriTech (Alnarp, Sweden) and used without normalization target core genome haplotypes using rhAmpSeq chemistry as previously described in Zou et al. [34]. The rhAmpSeq markers were amplified with half-reactions of the rhAmpSeq Library Kit (IDTDNA, Redwood City, CA, USA), indexed, pooled, cleaned, and Illumina sequenced (2 × 150 bp read length) as previously detailed for local haplotype analysis and quality filtering [6, 34]. Four possible haplotypes (for diploid bi-parental family) were converted to a variant call format (VCF) file, with a pseudo-A, -C, −G, or -T allele (https://github.com/avinashkarn/analyze_amplicon/blob/master/haplotype_to_VCF.pl; [34]). NCBI BioProject PRJNA281110 contains these and other VitisGen genotype data.
Before constructing a genetic map, quality control analysis was performed using Multidimensional scaling (MDS) in TASSEL software and Mendelian error detection using the Mendelian plugin in BCFtool34 [49]. This eliminated seedlings that arose from self-hybridization, off-target pollination, or mislabeling. A genetic map was constructed using Lep-MAP3 v.0.2 (LM3) software, which follows the LM3 modules of parent calling, filtering distorted markers, separating chromosomes into 19 different linkage groups, and ordering markers within the linkage groups [50]. This map was loaded into “R/qtl” package [48] to construct a four-way cross-map, where: 1 1 = AC = 1; 1 2 = AD = 2; 2 1 = BC = 3; and 2 2 = BD = 4. Lastly, the synteny of the maps was checked by plotting genetic position against their physical coordinates from the V. vinifera PN40024 version 12X.v2 [51].
QTL analysis
Quantitative trait locus (QTL) mapping was performed using sex-averaged 4-way cross genetic mapping in “R/qtl” with default parameters for interval mapping. For this, the scanone function and Haley-Knot (HK) regression (Haley and Knott 1992) method was used. Genotype probabilities were calculated using calc.genoprob with step = 0 (probabilities were calculated only at the marker locations) and assumed genotyping error rate of 1.0 × 10−4. If the phenotype did not meet the assumptions for a normal model, a non-parametric model was used. Logarithm of the odds (LOD) significance thresholds were determined by 1000 permutation tests at an alpha of 0.1. Only the QTLs that exceeded the threshold LOD value were considered significant. Each QTL support interval was determined by calculating the 1.8 - LOD support intervals using lodint. The percentage of variance explained in the context of a full additive model was calculated using fitqtl. To determine alleles that were associated with resistance, an effect plot was constructed using effectplot.
RNA sampling and processing
Total RNA was extracted from five resistant F1 individuals at 24 hours post-inoculation (hpi) and non-inoculated controls to identify expressed candidate genes at the resistance locus and genes involved in an early resistance response rapidly induced by inoculation. Three leaf disk replicates of each genotype were collected in 1.5 ml PCR tubes, flash frozen, and stored at −80°C. The Spectrum Plant Total RNA Kit [52] was used with 2% PVP40 added to the extraction buffer, otherwise following the manufacturer’s protocol. After extraction, RNA samples were subjected to library preparation and sequencing at Novogene’s Sacramento, CA facility. After quality checks were passed, RNASeq libraries were prepared and sequenced using the Eukaryotic mRNA-seq, 6G raw data WOBI-Package (20 M 150 bp paired reads; 40 M raw) (Table S1). Poly-T oligo-attached magnetic beads were used to capture messenger RNA. First strand cDNA was synthesized using random hexamer primers. After second strand cDNA synthesis, end repair, A-tailing, adapter ligation, size selection, amplification, and purification, the library was sequenced.
After sequencing, adapters, poly-A tails, and low-quality regions were computationally removed by fastp (v.0.20.0, with parameters −5 -3 -W 4 -M 20 -l 15 -x -n 5), reads were mapped to the reference genome PN40024 12X.V2 [51] with HISAT2 (v2.2.1) allowing 5% mismatch; 86% of the reads were mapped uniquely. Because the family is a cross between V. amurensis and V. vinifera, a genome-guided transcriptome assembly using stringtie (v.2.1.6) was then performed to improve the gene annotation. Transcripts that overlapped 90% with annotated transposable elements (TE, based on VCost.v3) or contained a pfam domain that belonged to TE were marked as TE-related. Additionally, a de novo transcriptome assembly was conducted using Trinity (v2.10.0). The derived transcripts were further qualified using Salmon (v1.4.0). Genes with significantly different expression level between the inoculated and non-inoculated treatments were determined with DESeq2 (v.1.28.0). The newly assembled transcripts from stringtie and Trinity were then translated into protein with TransDecoder (v.5.5.0). Function and gene ontology annotations were predicted using BLAST2GO (v1.5.1, database: April, 2021) with the Uniref90 database. Genes belonging to the NLR family were identified with NLR-annotator [53–55] in both PN40024 12X.V2 [51] and V. amurensis ‘Shanputao’ [36].
Skim-seq and analysis
The two parents and 31 progeny vines that had a potential recombination near REN12 based on rhAmpSeq genotyping were sequenced using skim-seq technology using DNA previously isolated. Barcoded libraries were constructed using the Illumina Nextera DNA Library Preparation Kit (Illumina Inc., San Diego, CA, USA) with 1/3 volume reactions and sequenced with the HiSeq X sequencer (Illumina Inc., San Diego, CA, USA) at Novogene’s Sacramento, CA sequencing center. The reads were mapped to the PN40024 12X.V2 genome [51] using BWA MEM (v 0.7.17) with default parameters. Because the sequencing depth ranged from 1× to 8× (Table S2) for each descendent, pooled variance calling was first performed with all the reads in this family using freebayes (v1.3.5, —use-best-n-alleles 4 —pooled-continuous —min-mapping-quality 30) to capture the high-quality genetic variances. Complex alleles were decomposed using vcfallelicprimitives in vcflib (v1.0.1). Downstream analysis used SNPs supported by ten or more reads and at least 3 bp away from any indels. The high-quality variances were then called for each sample using freebayes (v1.3.5, —hwe-priors-off —min-mapping-quality 30). To further eliminate false positives, only SNPs in linkage with other SNPs were kept, using the criteria that the largest r2 between the SNPs in 100 kb should be larger than 0.8, calculated using plink (v.1.9).
In order to infer that recombination happened on the resistant donor (V. amurensis “PI 588631”), variants were kept heterozygous in V. amurensis “PI 588631” but homozygous in Valley Pearl. The two haplotypes in the V. amurensis “PI 588631” were denoted as A and a. Because the recombination rate is low in grapes with less than one recombination per chromosome per meiosis on average [34], the transition from haplotype A to haplotype a and from haplotype a to haplotype A is very low. As the probability for the state change to happen, 1/total number of variants was used and 1–1/total number of variants as the probability for remaining in the same status. The emission matrix represents how likely each genotype is to be observed given the haplotype. The most probable path of the haplotypes was estimated using the Viterbi algorithm. The script is available at https://bitbucket.org/cornell_bioinformatics/amplicon.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, BioProject PRJNA281110.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Data
Supplementary data is available at Horticulture Research online.
Supplementary Material
Contributor Information
Surya Sapkota, School of Integrative Plant Science, Cornell AgriTech, Cornell University, Geneva, NY, 14456, USA.
Cheng Zou, BRC Bioinformatics Facility, Institute of Biotechnology, Cornell University, Ithaca, NY, 14853, USA.
Craig Ledbetter, United States Department of Agriculture (USDA)-Agricultural Research Service (ARS), Crop Diseases, Pests and Genetics Research Unit, San Joaquin Valley Agricultural Sciences Center, Parlier, CA, 93648, USA.
Anna Underhill, USDA-ARS, Grape Genetics Research Unit, Geneva, NY, 14456, USA.
Qi Sun, BRC Bioinformatics Facility, Institute of Biotechnology, Cornell University, Ithaca, NY, 14853, USA.
David Gadoury, School of Integrative Plant Science, Cornell AgriTech, Cornell University, Geneva, NY, 14456, USA.
Lance Cadle-Davidson, School of Integrative Plant Science, Cornell AgriTech, Cornell University, Geneva, NY, 14456, USA; USDA-ARS, Grape Genetics Research Unit, Geneva, NY, 14456, USA.
References
- 1. Brewer MT, Milgroom MG. Phylogeography and population structure of the grape powdery mildew fungus, Erysiphe necator, from diverse Vitis species. BMC Evol Biol. 2010;10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akkurt M, Welter L, Maul Eet al. Development of SCAR markers linked to powdery mildew (Uncinula necator) resistance in grapevine (Vitis vinifera L. and Vitis sp.). Mol Breed. 2007;19:103–11. [Google Scholar]
- 3. Barker CL, Donald T, Pauquet Jet al. Genetic and physical mapping of the grapevine powdery mildew resistance gene, Run1, using a bacterial artificial chromosome library. Theor Appl Genet. 2005;111:370–7. [DOI] [PubMed] [Google Scholar]
- 4. Dalbo MA, Ye GN, Weeden NFet al. Marker-assisted selection for powdery mildew resistance in grapes. J Am Soc Hortic Sci. 2001;126:83–9. [Google Scholar]
- 5. Heerden CJ, Burger P, Vermeulen Aet al. Detection of downy and powdery mildew resistance QTL in a “regent” x “RedGlobe” population. Euphytica. 2014;200:281–95. [Google Scholar]
- 6. Karn A, Zou C, Brooks Set al. Discovery of the REN11 locus from Vitis aestivalis for stable resistance to grapevine powdery mildew in a family segregating for several unstable and tissue-specific quantitative resistance loci. Front Plant Sci. 2021;12:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Welter LJ, Gokturk-Baydar N, Akkurt Met al. Genetic mapping and localization of quantitative trait loci affecting fungal disease resistance and leaf morphology in grapevine (Vitis vinifera L). Mol Breed. 2007;20:359–74. [Google Scholar]
- 8. Zendler D, Schneider P, Toepfer Ret al. Fine mapping of Ren3 reveals two loci mediating hypersensitive response against Erysiphe necator in grapevine. Euphytica. 2017;213:68. [Google Scholar]
- 9. Hoffmann S, Di Gaspero G, Kovacs Let al. Resistance to Erysiphe necator in the grapevine “Kishmish vatkana” is controlled by a single locus through restriction of hyphal growth. Theor Appl Genet. 2008;116:427–38. [DOI] [PubMed] [Google Scholar]
- 10. Pap D, Riaz S, Dry IBet al. Identification of two novel powdery mildew resistance loci, Ren6 and Ren7, from the wild Chinese grape species Vitis piasezkii. BMC Plant Biol. 2016;16:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramming DW, Gabler F, Smilanick JLet al. Identification of race-specific resistance in north American Vitis spp. limiting Erysiphe necator hyphal growth. Phytopathology. 2012;102:83–93. [DOI] [PubMed] [Google Scholar]
- 12. Rouxel M, Mestre P, Baudoin Aet al. Geographic distribution of cryptic species of Plasmopara viticola causing downy mildew on wild and cultivated grape in eastern North America. Phytopathology. 2014;104:692–701. [DOI] [PubMed] [Google Scholar]
- 13. Cadle-Davidson L. Variation within and between Vitis spp. for foliar resistance to the downy mildew pathogen Plasmopara viticola. Plant Dis. 2008;92:1577–84. [DOI] [PubMed] [Google Scholar]
- 14. Blasi P, Blanc S, Wiedemann-Merdinoglu Set al. Construction of a reference linkage map of Vitis amurensis and genetic mapping of Rpv8, a locus conferring resistance to grapevine downy mildew. Theor Appl Genet. 2011;123:43–53. [DOI] [PubMed] [Google Scholar]
- 15. Fu P, Wu W, Lai Get al. Identifying Plasmopara viticola resistance loci in grapevine (Vitis amurensis) via genotyping-by-sequencing-based QTL mapping. Plant Physiol Biochem. 2020;154:75–84. [DOI] [PubMed] [Google Scholar]
- 16. Venuti S, Copetti D, Foria Set al. Historical introgression of the downy mildew resistance gene Rpv12 from the Asian species Vitis amurensis into grapevine varieties. PLoS One. 2013;8:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu J, Zhang Y, Zhang Het al. Whole genome wide expression profiles of Vitis amurensis grape responding to downy mildew by using Solexa sequencing technology. BMC Plant Biol. 2010;10:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fu P, Tian Q, Lai Get al. Cgr1, a ripe rot resistance QTL in Vitis amurensis “Shuang Hong” grapevine. Hortic Res. 2019;6:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Q, Zhang J, Wang Yet al. Breeding for cold-resistant, seedless grapes from Chinese wild Vitis amurensis using embryo rescue. N Z J Crop Hortic Sci. 2016;44:136–51. [Google Scholar]
- 20. Wang H, Yan A, Sun Let al. Novel stable QTLs identification for berry quality traits based on high-density genetic linkage map construction in table grape. 2020;20:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wan Y, He P, Wang Y. Inheritance of downy mildew resistance in two interspecific crosses between Chinese wild grapes and European grapes. Vitis. 2007;46:156–7. [Google Scholar]
- 22. Schwander F, Eibach R, Fechter Iet al. Rpv10: a new locus from the Asian Vitis gene pool for pyramiding downy mildew resistance loci in grapevine. Theor Appl Genet. 2012;124:163–76. [DOI] [PubMed] [Google Scholar]
- 23. Bulit J, Lafon R. Powdery mildew of the vine. In: Spencer DM, ed. In: “The Powdery Mildews”. Academic Press: New York, 1978,525–48. [Google Scholar]
- 24. Gadoury DM, Seem RC, Pearson RCet al. Effects of powdery mildew on vine growth, yield, and quality of Concord grapes. Plant Dis. 2001;85:137–40. [DOI] [PubMed] [Google Scholar]
- 25. Gadoury DM, Cadle-Davidson L, Wilco WFet al. Grapevine powdery mildew (Erysiphe necator): a fascinating system for the study of the biology, ecology, and epidemiology of an obligate biotroph. Mol Plant Pathol. 2011;13:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adam-Blondon A-F. Grapevine Genome Update and Beyond. In: Reisch BI, Londo J, eds. X International Conference on Grapevine Breeding and Genetics. Int Soc Horticultural Science: Leuven: 1, 2014,311–8. [Google Scholar]
- 27. Gadoury DM, Wakefield LM, Cadle-Davidson Let al. Effects of prior vegetative growth, inoculum density, light, and mating on conidiation of Erysiphe necator. Phytopathology. 2012;102:65–72. [DOI] [PubMed] [Google Scholar]
- 28. Barba P, Cadle-Davidson L, Galarneau Eet al. Vitis rupestris B38 confers isolate-specific quantitative resistance to penetration by Erysiphe necator. Phytopathology. 2015;105:1097–103. [DOI] [PubMed] [Google Scholar]
- 29. Possamai T, Wiedemann-Merdinoglu S, Merdinoglu Det al. Construction of a high-density genetic map and detection of a major QTL of resistance to powdery mildew (Erysiphe necator Sch.) in Caucasian grapes (Vitis vinifera L.). BMC Plant Biol. 2021;21:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dry IB, Feechan A, Anderson Cet al. Molecular strategies to enhance the genetic resistance of grapevines to powdery mildew. Aust J Grape Wine Res. 2010;16:94–105. [Google Scholar]
- 31. Feechan A, Kabbara S, Dry IB. Mechanisms of powdery mildew resistance in the Vitaceae family. Mol Plant Pathol. 2011;12:263–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramming DW, Gabler F, Smilanick Jet al. A single dominant locus, Ren4, confers rapid non-race-specific resistance to grapevine powdery mildew. Phytopathology. 2011;101:502–8. [DOI] [PubMed] [Google Scholar]
- 33. Cantu D, Walker MA. The Grape Genome. Cham, Switzerland: Springer; 2019. [Google Scholar]
- 34. Zou C, Karn A, Reisch Bet al. Haplotyping the Vitis collinear core genome with rhAmpSeq improves marker transferability in a diverse genus. Nat Commun. 2020;11:413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Malmberg MM, Barbulescu DM, Drayton MCet al. Evaluation and recommendations for routine genotyping using skim whole genome re-sequencing in canola. Front Plant Sci. 2018;9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang Y, Xin H, Fan Pet al. The genome of Shanputao (Vitis amurensis) provides a new insight into cold tolerance of grapevine. Plant J. 2021;105:1495–506. [DOI] [PubMed] [Google Scholar]
- 37. Frenkel O, Portillo I, Brewer MTet al. Development of microsatellite markers from the transcriptome of Erysiphe necator for analyzing population structure in North America and Europe. Plant Pathol. 2012;61:106–19. [Google Scholar]
- 38. Blanc S, Wiedemann-Merdinoglu S, Dumas Vet al. A reference genetic map of Muscadinia rotundifolia and identification of Ren5, a new major locus for resistance to grapevine powdery mildew. Theor Appl Genet. 2012;125:1663–75. [DOI] [PubMed] [Google Scholar]
- 39. Sambucci O, Alston JM, Fuller KBet al. The pecuniary and non-pecuniary costs of powdery mildew and the potential value of resistant varieties in California grapes. Am J Enol Vitic. 2019;70:177–87. [Google Scholar]
- 40. Ledbetter CA. ‘Valley pearl’ table grape. HortScience. 2016, 2016;51:772–4. [Google Scholar]
- 41. Cadle-Davidson L, Gadoury D, Fresnedo-Ramirez Jet al. Lessons from a phenotyping center revealed by the genome-guided mapping of powdery mildew resistance loci. Phytopathology. 2016;106:1159–69. [DOI] [PubMed] [Google Scholar]
- 42. VitisGen2 , https://www.vitisgen2.org/
- 43. Bierman A, LaPlumm T, Cadle-Davidson Let al. A high-throughput phenotyping system using machine vision to quantify severity of grapevine powdery mildew. Plant Phenomics. 2019;2019:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Helicon Focus 6, Rendering Methods . 2017, https://www .heliconsof.com/focus/help/english/HeliconFocus.html#HF METHODS
- 45. Printersys , https://printersys.com/
- 46. Wickham, H. (2016). Ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York. https://ggplot2.tidyverse.org. [Google Scholar]
- 47. Reshef N, Karn A, Manns DCet al. Stable QTL for malate levels in ripe fruit and their transferability across Vitis species. 2020;9:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Broman KW, Wu H, Sen Śet al. R/QTL: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–90. [DOI] [PubMed] [Google Scholar]
- 49. Bradbury PJ, Zhang Z, Kroon DEet al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5. [DOI] [PubMed] [Google Scholar]
- 50. Rastas P. Lep-MAP3: robust linkage mapping even for low-coverage whole genome sequencing data. Bioinformatics. 2017;33:3726–32. [DOI] [PubMed] [Google Scholar]
- 51. Canaguier A, Grimplet J, Di Gaspero Get al. A new version of the grapevine reference genome assembly (12X.v2) and of its annotation (VCost.v3). Genom Data. 2017;14:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sigma , https://www.sigmaaldrich.com/US/en
- 53. Bankar KG, Todur VN, Shukla RNet al. Ameliorated de novo transcriptome assembly using Illumina paired-end sequence data with trinity assembler. Genomics data. 2015;5:352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Patro R, Duggal G, Love MIet al. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Steuernagel B, Witek K, Krattinger SGet al. The NLR-annotator tool enables annotation of the intracellular immune receptor repertoire. Plant Physiol. 2020;183:468–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, BioProject PRJNA281110.