

Abstract
B and T cells are integral parts of the immune system and are implicated in many diseases, e.g. autoimmunity. Towards understanding the biology of B and T cells and subsets thereof, their transcriptomes can be analyzed using single-cell RNA sequencing. In some studies, the V(D)J transcripts encoding the variable regions of the B- and T-cell antigen receptors have been removed before the analyses. However, a systematic analysis of the effects of including versus excluding these genes is currently lacking. We have investigated the effects of these transcripts on unsupervised clustering and down-stream analyses of single-cell RNA sequencing data from B and T cells. We found that exclusion of the B−/T-cell receptor genes prior to unsupervised clustering resulted in clusters that represented biologically meaningful subsets, such as subsets of memory B and memory T cells. Furthermore, pseudo-time and trajectory inference analyses of early B-lineage cells resulted in a developmental pathway from progenitor to immature B cells. In contrast, when the B−/T-cell receptor genes were not removed, with the PCs used for clustering consisting of up to 70% V-genes, this resulted in some clusters being defined exclusively by V-gene segments. These did not represent biologically meaningful subsets; for instance in the early B-lineage cells, these clusters contained cells representing all developmental stages. Thus, in studies of B and T cells, to derive biologically meaningful results, it is imperative to remove the gene sequences that encode B- and T-cell receptors.
Keywords: single-cell RNA sequencing, scRNA-seq, immunoglobulins, BCR-genes, TCR-genes, interference
Introduction
B cells express on their cell surfaces a B-cell antigen receptor (BCR). The fact that each B cell expresses a unique BCR—in its secreted form an antibody—ensures that we have immune protection against invading pathogens. The BCR is composed of antibody heavy and κ or λ light chains, each containing a variable region, which together determine the antigen specificity, and a constant region, whereby the heavy chain, e.g. IgM or IgG, is responsible for the effector function. However, the number of genes in the genome (2 × 104) does not correspond to the number of B cells in the human body. Instead, a sophisticated system is in place whereby immunoglobulin (Ig) V(D)J gene segments, which encode the respective variable regions, undergo recombination [1, 2]. In theory, this gives rise to more than 1011 specificities [3]. By analogy, T cells express on their cell surfaces a T-cell receptor (TCR), which is composed of α- and β- or γ- and δ-chains, in which the respective variable regions are generated by V(D)J recombination. The recombination process ensures diversity among the billions of B and T cells and their immune repertoire. Multiple studies have excluded BCR- and TCR-genes from the analyses of scRNA-seq data to prevent dimensionality reduction and clustering to be influenced by individual clones [4–8]. However, a systematic analysis of the impact of BCR- and TCR-genes in scRNA-seq data analysis is lacking. Here, we have investigated whether the BCR-genes and TCR-genes influence unsupervised clustering and down-stream analyses of scRNA-seq data obtained from peripheral blood B cells and T cells, as well as bone marrow (BM) early B-lineage cells, with implications for our understanding of the biology of these cell types.
Results
BCR-genes influence clustering of peripheral blood memory B cells
To determine more precisely, the effects of the BCR-genes on the clustering and interpretation of single-cell RNA sequencing (sc-RNAseq) data, we first analyzed sorted peripheral blood (PB) memory B cells (n = 4085). To exclude the BCR-genes, all the count data related to IGH and IGL genes, i.e. V(D)J and constant regions (Figure S1), were removed from the dataset. As clustering and PCA reduction are calculated from the highly variable genes (HVGs), an alternative to excluding all BCR-genes would be to exclude BCR transcripts from the list of HVGs. The exclusion of all BCR genes, as in our case, does not prevent the analyses of these genes, as the count data for BCR transcripts can be added back and be analyzed, as shown below. Using an Elbow plot, we set the number of principal components (PCs) to 35, to cover the elbow with reasonable margin, and using a Clustree plot the resolution was set to 0.25 (Figure 1A and B). Unsupervised clustering with these settings resulted in five distinct clusters (Figure 1C). The top-10 up-regulated differentially expressed genes (DEGs) per cluster are shown in a heat map (Figure 1D, Table S1). Plotting a few of the DEGs revealed high levels of expression of CD69 in cluster 0, COCH in cluster 1 and CRIP2 in cluster 2 (Figure 1E), as expected from the heat map. To determine whether the unsupervised clustering resulted in biologically relevant clusters, we utilized the VDJ-sequencing data from the same sample to assess BCR isotype expression. As the analyzed cells represented memory B cells, we would expect some of the cells to express IgM, representing unswitched memory B cells, and other cells to have undergone class-switch recombination, such that they expressed isotypes other than IgM and representing switched memory B cells. The VDJ data are more reliable, as they allow one to assess the expression of productively rearranged VDJ-genes, i.e. those that result in a protein. This contrasts with the transcriptomics data, which do not distinguish between productive and unproductive transcripts (albeit with a relatively similar expression pattern for this isotype), where the unproductive transcripts do not result in proteins (Figure 1F, Figure S2). Nevertheless, displaying cells that expressed IgM showed that most of these were found in clusters 0 and 4. Thus, the cells in these two clusters represented unswitched memory B cells, whereas clusters 1–3 represented switched memory B cells. TEX9 and TOX are two genes that are expressed mainly in switched memory B cells [9], which fits well with their observed expression in IgM-negative, switched cells (Figure 1G; data not shown). Moreover, we have shown that cell surface expression of the β1-integrin CD29 (ITGB) is higher in switched than in unswitched memory B cells [10], and is consistent with the expression pattern of ITGB1 in our single-cell analyses. Based on these results, we conclude that unsupervised clustering after excluding the BCR-genes gives rise to biologically relevant B-cell subsets that separate the cells into clusters of switched and unswitched memory B cells.
Figure 1.

Unsupervised clustering of peripheral blood memory B cells after removal of the BCR-genes, with results for five clusters. A-G, Analyses of single-cell RNA sequencing data from sorted PB memory B cells after the BCR-genes were removed. A, Elbow plot showing the standard deviation explained by each PC; the dashed line depicts the number of PCs used for the unsupervised clustering. B, Clustree plot showing the size and the number of clusters for different values of the Seurat::FindClusters resolution parameter. C, UMAP projection and unsupervised clustering of PB memory B cells. D, Heat map displaying the scaled relative expression of the top-10 significantly up-regulated genes for each cluster (Bonferroni-adjusted P-values <0.05 of the Seurat negative binomial generalized linear model). Gene list in Table S1. Asterisks indicate genes plotted in E, and the black horizontal lines indicate which DEGs are found in each cluster. E-G, UMAP projection of all the cells: E, colour scheme according to the scaled level of gene expression; F, cells expressing a productively re-arranged IgM are highlighted; and G, colour scheme according to the scaled level of gene expression.
To determine whether inclusion of the BCR-genes resulted in any major differences, we analyzed the same dataset using the same settings as described above (Figure 2A and B). Unsupervised clustering resulted in 12 clusters (Figure 2C), as compared to the five clusters that emerged when the BCR-genes were excluded. Furthermore, analysis of the top-10 up-regulated DEGs per cluster revealed a completely different pattern, with fewer than 10 DEGs per cluster (Figure 2D, Table S1). In fact, in most of the 12 clusters, it was possible to identify only 1, 2 or 3 DEGS, rather than 10 DEGs. For instance, in cluster 0, NFKBIA was the only gene that distinguished this cluster from the other clusters. However, plotting the expression of this gene in a UMAP projection did not generate a distinct pattern (Figure 2E). Moreover, in 8 of the 12 clusters, we identified mainly BCR-genes, including Ig heavy (IGH) and Ig κ- and λ-light (ΙGK/IGL) chain V-genes (Figure 2D). For instance, clusters 4, 5 and 6 contained fewer than 10 DEGs, with at least 1 out of the 1–3 genes being a BCR-gene, e.g. IGHV3-7, IGKV4-1, and IGLV2-14, indicating that some clusters were defined exclusively by a few BCR-genes. Plotting the expression of the afore-mentioned BCR-genes in a UMAP projection revealed that each of these was localized primarily to a single cluster, i.e. to cluster 4, 5 or 6 (Figure 2F). To investigate the basis for this, we identified the 50 genes that accounted for most of the variance in each PC, for PCs 1–50. For the dataset analyzed here, the first 35 PCs were used for the unsupervised clustering (Figure 2A). When we analyzed the variance observed for many of the PCs, we found that it was defined by having more than 50% BCR-genes (Figure 2G). In fact, the proportion of BCR-genes was ≥20% from PC4 to PC50, with the exception of PC10. Thereafter, we analyzed the expression patterns of the three DEGs identified after exclusion of the BCR-genes (Figure 1E). We found that they no longer exhibited a distinct expression pattern but instead had a pattern whereby some cells in almost every cluster expressed these genes, with this being most evident for CD69 and COCH (Figure 2H). In addition, IgM (from the VDJ-seq data), as well as TEX9 and ITGB1 characterized both positive and negative cells in almost every cluster (Figure 2I and J). Next, we wanted to determine whether the cells that expressed IGHV3-7, IGKV4-1, and IGLV2-14, which were localized to clusters 4, 5 and 6 (Figure 2F), still clustered together after excluding the BCR-genes. We could still analyze the expression pattern of these genes by adding back the count data for all the BCR-genes. If they represented a particular subset of cells defined by their BCR-genes, we would expect them to still cluster together. However, our analysis showed that the cells that expressed IGHV3-7, IGKV4-1, and IGLV2-14 were now distributed across the clusters (Figure 2K). Since B cells express a large repertoire of V-genes, irrespective of whether they are naïve B, memory B or plasma cells, this is the pattern that would be expected. Thus, when the BCR-genes are retained, they influence the unsupervised clustering, as it results in many clusters being defined exclusively by a few BCR-genes. In contrast, excluding the BCR-genes results in clusters that are defined by other genes, and allows separation of the cells into biologically relevant subsets, distinguishing, for instance, between switched and unswitched memory B cells.
Figure 2.
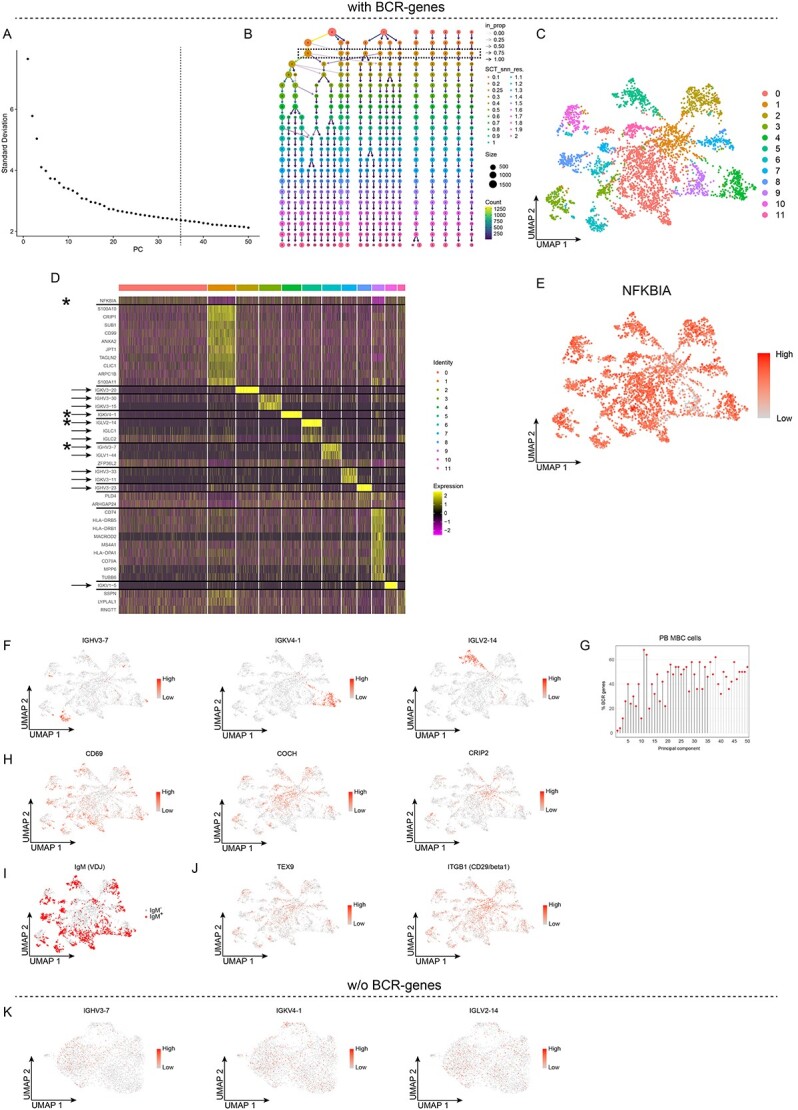
Unsupervised clustering of peripheral blood memory B cells retaining the BCR-genes, with results for 12 clusters. A-J, Analyses of single-cell RNA sequencing data for sorted PB memory B cells with the BCR-genes retained. A, Elbow plot showing the standard deviation explained by each PC; the dashed line depicts the number of PCs used for the unsupervised clustering. B, Clustree plot showing the size and the number of clusters for different values of the Seurat::FindClusters resolution parameter. C, UMAP projection and unsupervised clustering of PB memory B cells. D, Heat map displaying the scaled relative expression of the top-10 significantly up-regulated genes for each cluster (Bonferroni-adjusted P-values <0.05 of the Seurat negative binomial generalized linear model). Gene list in Table S1. The asterisks indicate genes plotted in E and F, the black horizontal lines indicate which DEGs are found in each cluster, and the arrows indicate the BCR-genes. E, F, H and J, UMAP projection of all the cells, coloured according to the scaled level of gene expression. G, Lollipop plot displaying the percentages of BCR-genes among the top-50 genes in the first 50 PCs; PCs used for unsupervised clustering are indicated by black stems. I, UMAP projection of all cells, with cells expressing a productively re-arranged IgM highlighted in red. K, UMAP projection of PB memory B cells clustered without BCR-genes, whereby the BCR-genes were then added back, and their scaled levels of gene expression are shown.
TCR-genes influence clustering of peripheral blood T cells
As T cells express a TCR with a variable region that consists of V(D)J-gene segments (Figure S1), we investigated whether the TCR-genes influence the unsupervised clustering of T cells. To this end, we analyzed a public scRNA-seq dataset obtained from PB CD8+ T cells (see Materials and methods section), applying the same workflow as above (Figure S3). Following removal of all the TCR-genes, unsupervised clustering of the cells (n = 13 309) resulted in 11 clusters (Figure 3A, Figure S3). Moreover, the top-10 up-regulated DEGs supported the notion that they represent different CD8+ T-cell subsets, such that for instance CD160 is prominently expressed in cluster 1, FGFBP2 in cluster 2 and KIR3DL1 in cluster 8 (Figure 3B, Table S2). Next, we performed unsupervised clustering after inclusion of the TCR genes, using the same parameters as above (Figure S3), which resulted in 29 clusters (Figure 3C, Figure S3). Analyzing the up-regulated DEGs for each cluster revealed that, to an even greater extent than was observed for the memory B cells, several of the clusters contained relatively few cells and many of the up-regulated DEGs were TCR-genes, including TCR α (TRA) and TCR β (TRB) chain V-genes (Figure 3D, Table S2). For instance, TRAV39 and TRBV9 pre-dominated in cluster 13, and the latter pre-dominated also in cluster 14, while TRBV20-1 pre-dominated in clusters 15 and 16 (Figure 3E). Analyzing the CD8+ T cells in a manner similar to that used for the memory B cells, we found that also here the variance observed for many of the PCs was explained by more than 50% TCR-genes, and for PC5 to PC50 (with the exceptions of PC6 and PC7) more than 20% were TCR-genes (Figure 3F). Thereafter, we added back the count data for the TCR-genes and analyzed their expression pattern in the cells clustered after exclusion of the TCR genes. This showed that the expression patterns of TRAV39, TRBV9 and TRBV20-1, previously assigned to clusters 13–16, were now distributed across most of the clusters (Figure 3G). To determine whether clustering in the absence of the TCR-genes was biologically relevant, we analyzed the expression patterns of TBX21 (Tbet) and GZMK (granzyme K). These represent two genes that are expressed by some but not all CD8+ memory T cells, and not by naïve CD8+ T cells [11, 12]. This showed that the levels of TBX21 were for instance, relatively high in cluster 2 but not in cluster 1 whereas GZMK was expressed in cluster 1 but not as strongly in cluster 2 (Figure 3H). The expression of these two genes was barely detectable in clusters 0, 6 and 7, whereas these clusters expressed CCR7, a gene with the highest expression levels in naïve CD8+ T cells. These results demonstrate that not only the genes that encode a BCR, but also those encoding a TCR influence the unsupervised clustering of lymphoid cells. The results also demonstrate that excluding the TCR-genes results in biologically relevant clusters, such as CD8+ memory T cells.
Figure 3.
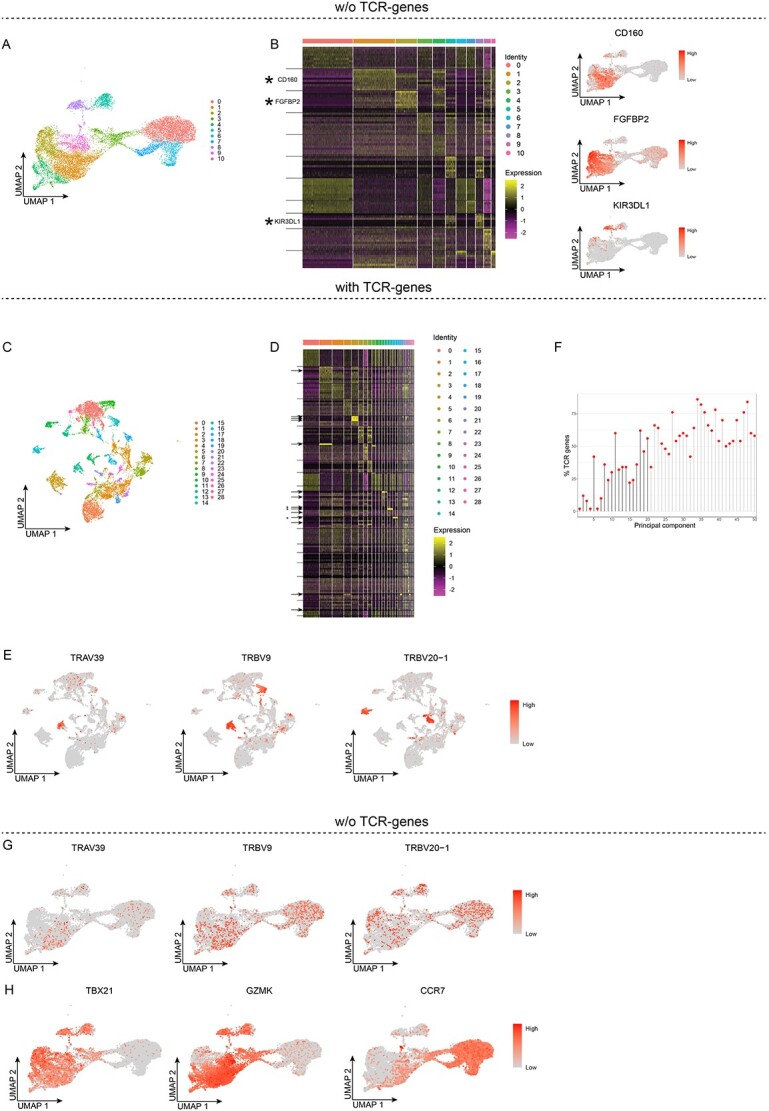
TCR-genes influence unsupervised clustering of peripheral blood CD8+ T cells. A and B, Unsupervised clustering without TCR-genes. A, UMAP projection and unsupervised clustering of PB CD8+ T cells. B, Heat map displaying the scaled relative expression levels of the top-10 significantly up-regulated genes in each cluster (Bonferroni-adjusted P-values <0.05 of the Seurat negative binomial generalized linear model). The black horizontal lines indicate which DEGs are found in each cluster. Gene list in Table S2. C-F, Unsupervised clustering with the TCR-genes retained. C, UMAP projection and unsupervised clustering. D, Heat map displaying the scaled relative expression levels of the top-10 significantly up-regulated genes in each cluster (Bonferroni-adjusted P-values <0.05 of the Seurat negative binomial generalized linear model). The asterisks indicate genes plotted in E, the arrows indicate the TCR-genes, and the black horizontal lines indicate which DEGs are found in each cluster. Gene list in Table S2. E, UMAP projection of all cells, coloured according to the scaled gene expression level. F, Lollipop plot displaying the percentages of TCR-genes among the top-50 genes in the first 50 PCs; PCs used for unsupervised clustering are indicated by black stems. G and H, UMAP projection of PB CD8+ T cells clustered without TCR-genes, whereby the TCR-genes were added back, and their scaled gene expression is shown.
BCR-genes influence down-stream analyses
To understand the developmental pathway and cell fate, clustering analyses are often followed by down-stream analyses such as pseudo-time and trajectory inference. Since the presence versus absence of BCR-genes and TCR-genes had such a strong effect on the clustering of B and T cells, respectively, we hypothesized that these types of down-stream analyses would also be affected, and not just the gene expression patterns. As earlier work based on cell surface markers and the recombination status of the BCR (Ig) genes uncovered a developmental pathway from progenitor to precursor to immature B cells [13], we focused on BM early B-lineage cells. Accordingly, these cells would be optimal for down-stream pseudo-time and trajectory inference analyses, in contrast to subsets of PB memory B and CD8+ T cells, where these pathways are not as well-known and the subject of investigation. Thus, we analyzed BM early B-lineage cells (progenitor, precursor and immature B cells). After analyzing the cells (n = 5016 cells) using an Elbow plot and Clustree plot (Figure S4), regressing cell cycle genes and removing the BCR-genes, we performed unsupervised clustering, which yielded 8 clusters (Figure 4A). These were all well-defined, as evidenced by a heat map showing the top-10 DEGs per cluster, exemplified by the expression of IGF2.1 mainly in cluster 0, HMHB1 in cluster 2 and LY9 in cluster 5 (Figure 4B, Table S3). In contrast, when the BCR-genes were retained, the unsupervised clustering resulted in 11 clusters, with some of these being defined by only a couple of genes, including BCR-genes (Figure 4C and D). For instance, clusters 6, 7 and 10 showed only one or two up-regulated DEGs each, of which at least one was a BCR V-gene (IGHV4-34, IGHV3-23 or IGHV3-33, respectively). Plotting their expression patterns showed that most of the cells that expressed these genes were localized to clusters 6, 7 and 10, respectively (Figure 4E). As was observed in the other datasets, some of the PCs in this dataset contained around 40% BCR-genes, and at least 10% were identified in PC12–PC50 (Figure 4F). The slightly smaller proportion of BCR-genes in the PCs in this dataset is likely due to the presence of cells that do not express BCR-genes, as they have not undergone VDJ (progenitor B cells) or VJ (precursor B cells) recombination of the IGH and IGL loci, respectively. Nevertheless, adding back the count data for the BCR genes on the cells clustered in their absence, demonstrated that the expression of IGHV4-34, IGHV3-23, and IGHV3-33 was spread throughout all the clusters, except for clusters 2 and 6, which did not express any of these three genes (Figure 4G).
Figure 4.

BCR-genes influence unsupervised clustering of bone marrow early B-lineage cells. A and B, Unsupervised clustering without BCR-genes. A, UMAP projection and unsupervised clustering of BM early B-lineage cells. B, Heat map displaying the scaled relative expression levels of the top-10 significantly up-regulated genes in each cluster (Bonferroni-adjusted P-values <0.05 of the Seurat negative binomial generalized linear model). Gene list in Table S3. The black horizontal lines indicate which DEGs are found in each cluster. C-F, Unsupervised clustering with BCR-genes retained. C, UMAP projection and unsupervised clustering. D, Heat map displaying the scaled relative expression levels of the top-10 significantly up-regulated genes in each cluster (Bonferroni-adjusted P-values <0.05 of the Seurat negative binomial generalized linear model). The asterisks indicate genes plotted in E, the arrows indicate the BCR-genes, and the black horizontal lines indicate which DEGs are found in each cluster. Gene list in Table S3. E, UMAP projection of all the cells, coloured according to scaled gene expression level. F, Lollipop plot displaying the percentages of BCR TCR-genes among the top-50 genes in the first 50 PCs; PCs used for unsupervised clustering are indicated by black stems. G, UMAP projection of BM early B-lineage cells clustered without BCR-genes, whereby the BCR-genes were added back, and their scaled gene expression is shown.
Previous work has shown that progenitor B cells express CD34, and that progenitor and precursor B cells consist of both resting and cycling (large) cells, whereas immature B cells are resting cells (Figure 5A) [13]. Consistent with the above observation that clusters 2 and 6 did not express the analyzed IGHV genes (Figure 4G), these cells expressed CD34 and, thus, represent progenitor B cells (Figure 5B). Analyzing the expression of MKI67 (Ki67), which is linked to proliferation, showed that it was expressed in clusters 6, 3 and 4 (Figure 5C). Moreover, the gene for CXCR5, a chemokine receptor that is indicative of cells migrating to the spleen, was expressed in cluster 5. Taken together, this suggested that clusters 2 and 6 represented resting and cycling progenitor cells, respectively, and that clusters 3 and 4 represented cycling precursor B cells, whereas the remaining cells would be resting precursor B and immature B cells, with the most-mature of the immature B cells being assigned to cluster 5. To analyze these early B-lineage cells for a developmental pathway, we used Monocle 3. Inference of the pseudo-time values and developmental trajectory, starting with the resting progenitor B cells, revealed that all the clusters were part of the developmental trajectory, following a pathway from resting to cycling progenitor B cells, to cycling precursor B cells via resting precursor B to immature B cells, and ending in cluster 5 (Figure 5D), which are the cells expressing CXCR5 (Figure 5C). Thus, a pathway from progenitor via precursor to immature B cells would be consistent with the current literature.
Figure 5.

BCR-genes influence pseudo-temporal ordering of bone marrow early B-lineage cells. A, Cartoon depicting the developmental stages of BM early B-lineage cells, with the expression of CD34, MKI67 and CXCR5 indicated. B-D, UMAP projection of unsupervised clustering without BCR-genes. E-G, UMAP projections of unsupervised clustering with BCR-genes retained. UMAP projection coloured according to scaled gene expression of CD34 (B and E), MKI67 and CXCR5 (C and F). D and G, UMAP projection coloured according to maturation level inferred from the pseudo-temporal analysis, with super-imposed developmental trajectory inferred from Monocle 3. Arrow shows the developmental origin (CD34-expressing cells). H, UMAP projection of unsupervised clustering with BCR-genes retained, with clusters 6, 7, and 10 highlighted with ellipses. I, UMAP projection of unsupervised clustering without BCR-genes, with the cells from clusters 6, 7, and 10 in Figure 5H highlighted.
Next, we performed the same analyses but with retention of the BCR-genes. The expression pattern of CD34 was similar to that described above, detecting two clusters (Figure 5E). Consistent with this, one of these two clusters expressed MKI67, suggesting that this represented the cycling progenitor B cells. MKI67 was also expressed in clusters 8 and 3, as well as in some of the cells in clusters 6 and 7 (Figure 5F). When inferring the pseudo-time values and developmental trajectory, starting with the resting progenitor B cells, we found that some of the cells/clusters were close to the developmental trajectory, while others were not, as if they were not part of the developmental pathway (Figure 5G). Among the latter were clusters 6, 7 and 10, which were defined by expression of the IGHV-genes (Figure 4E). Moreover, the developmental pathway seemingly had three end-points, of which two were defined by the expression of IGHV3-23 and IGHV3-33, i.e. clusters 7 and 10, respectively. The cells in the third end-point did express CXCR5 but did not represent the most mature of the immature B cells according to the pseudo-time analysis. Next, we focused on the cells in clusters 6, 7 and 10 that were found among the most-mature cells according to the pseudo-time values (Figure 5G) and projected these onto the clusters obtained in the absence of BCR genes. This showed that these cells were now spread throughout all the clusters having undergone VDJ-recombination, i.e. except for the progenitor B cells, and were thus found at all stages in the developmental pathway (Figure 5H and I). Thus, the BCR-genes interfere also with down-stream analyses of scRNA-seq data, such as analyses of developmental pathways and cell fate.
Discussion
Taken together, these results demonstrate that BCR-genes and TCR-genes interfere with scRNA-seq analyses of B and T cells, and this has implications for subsequent analyses. This is a crucial issue, as the initial clustering often forms the basis for down-stream analyses, for instance, comparison of DEGs in different clusters to identify cell subsets, cell fate and trajectories, and gene-set enrichment for pathway analyses. The validity of the clustering also affects the identification of disease-specific characteristics, treatment responses, and diagnostic and prognostic biomarkers based on scRNA-seq analyses.
A broad immune repertoire is essential for a healthy immune system, with BCRs and TCRs established according to the pairing of heavy and light chains, each of which uses unique combinations of V(D)J gene segments. Although the V(D)J-recombination process is to some extent random, it is evident from the VH-usage patterns observed in naïve B cells (and T cells) that usage is not proportional to the number of VH-genes in the genome [14, 15]. For instance, the Ig genes VH1-69 and VH4-34 are used in around 5%, respectively, while other VH-genes are used in smaller or even larger proportions. Nevertheless, there are some stereotyped receptors that use particular Ig V-genes, although these are mainly found in B-cell tumours and certain autoimmune diseases, e.g. mixed cryoglobulinemia and chronic B-lymphocytic leukaemia where VH1-69 and VH4-34 are frequently used, and in systemic lupus erythematosus where the usage of VH4-34 is particularly high. In terms of T cells there exist, for instance, MAIT cells that are defined by a semi-invariant TCR, and another example is the iNKT cells that use TRAV24 recombined with JA18, and often in combination with a certain TRBV11. In most of these cases, we would expect the cells to show similar transcriptomics profiles, which means that they would be assigned to the same cluster when analyzed in the absence of TCR/BCR gene transcripts.
The importance of excluding the BCR-/TCR-genes before the analysis of scRNA-seq datasets is perhaps best illustrated by our analyses of the BM early B-lineage cells. These represent the cells that after maturation form the pool of naïve B cells, so it is essential that they express as broad as possible a repertoire of BCRs. Our data show that when analyzing the early B-lineage cells in the presence of BCR transcripts they form clusters that express particular IGHV, IGKV and/or IGLV transcripts (e.g. IGHV4-34, IGHV3-23, and IGHV3-33). These clusters consist of relatively mature cells, according to the pseudo-time analysis, and in the trajectory inference analysis, they are found either outside of the developmental pathway or forming separate branches. However, when analyzed in the absence of BCR-transcripts, these same cells are found in all developmental stages, consistent with the formation of a large repertoire of BCRs, and biologically relevant.
Our results show that it is mostly the V-genes that dominate the clustering when retaining the BCR-/TCR-genes, although constant region genes from both BCR- and TCR-loci were found in several PCs and among the top-10 DEGs. We also found that if we excluded only the IGHV-gene transcripts that pre-dominated in the clustering, other BCR transcripts became dominant (data not shown). As a consequence, we chose to exclude all the BCR-/TCR-genes. In many instances, sc-RNAseq analyses are based on 2000-3000 HVGs. As the BCR-genes could correspond to as much as 5% of the HVGs, the BCR- (and TCR) genes were excluded before calculating the number of HVGs.
Conclusion
In order to answer with confidence scientific questions based on analyses of scRNA-seq data, the BCR-/TCR-genes need to be excluded from the clustering analyses.
Materials and methods
Sample collection
Human peripheral blood memory B cells
Frozen peripheral blood mononuclear cells (PBMCs) from a patient with rheumatoid arthritis were thawed as for the BM. CD20+ memory B cell subpopulations were sorted as one population containing CD27+IgD+, CD27+IgD− and CD27−IgD− cells to high purity with a Sony SH800. Dead cells were excluded by staining with a Fixable Viability Dye eFluor™ 506 (Invitrogen).
Human peripheral blood CD8+ T cells
Described at: https://www.10xgenomics.com/resources/datasets/cd-8-plus-t-cells-of-healthy-donor-1-1-standard-3-0-2.
Human early B-lineage bone marrow cells
Frozen bone marrow (BM) cells from two healthy donors were thawed and resuspended in RPMI-1640 10% FCS (ThermoFisher). CD24hi CD38hi early B-lineage cells were sorted to high purity with a Sony SH800. Dead cells were excluded by staining with a Fixable Viability Dye eFluor™ 506 (Invitrogen).
Single-cell RNA sequencing
Sorted cellular suspensions were loaded into the Chromium apparatus (10x Genomics Inc., Pleasanton, CA, USA) to generate single-cell GEMs (Gel Bead-In EMulsions). The single-cell GEMs were then processed following the Chromium Single Cell V(D)J Reagent Kits Protocol (10x Genomics). Sequencing was performed using NextSeq (Illumina).
Analysis of single-cell RNA sequencing data
Early B-lineage BM cells and peripheral blood memory B cells
Raw base-calling files from the sequencing data were de-multiplexed, trimmed, filtered, and aligned to the GRCh38 genome assembly using the software suite Cell Ranger (v. 3.1.0; 10x Genomics). Gene expression count matrices were imported into the R package Seurat [16] (v. 4.0.4) using R (v. 4.0.4), where further quality control was performed. Genes found in fewer than 3 cells, and cells with fewer than 200 expressed genes were excluded. BCR-genes were removed from the count data using regular expression commands with the following patterns: ‘IG[HKL]V’, ‘IG[KL]J’, ‘IG[KL]C’, ‘IGH[ADEGM]’. Counts were log-normalized, scaled and centred. The 2000 most-variable features were calculated with variance-stabilizing transformation and used for the principal component (PC) analysis. The 10 first PCs (decided by Seurat::ElbowPlot) were used to construct an approximate nearest-neighbour graph, and clustering was performed with Seurat::FindClusters with the resolution set to 0.8 decided by Clustree [17]. Dimensionality reduction was performed with uniform manifold approximation and projection [18] (UMAP). A cluster with a high proportion of mitochondrial transcripts was identified and removed before the down-stream analyses. The single-cell transform wrapper [19] in Seurat (v. 0.3.2) was implemented using the first 20 PCs, and with the resolution parameter set to 0.8, decided by Clustree, as well as regression of ‘G2M.Score’ and ‘S.Score’ obtained from the Seurat::CellCycleScoring function. Ig-genes were added back by importing the count data again as a new assay to the Seurat object, without the filtering step to remove the Ig-genes. The two BM datasets were integrated using the SelectIntegrationFeatures, PrepSCTIntegration, FindIntegrationAnchors and IntegrateData functions from the Seurat package. Due to the larger number of cells, we detected two clusters with a high proportion of mitochondrial transcripts, which were removed from the downstream analyses. To identify genes that were differentially expressed between the clusters, we used a negative binomial generalized linear model with the Seurat Seurat::Findallmarkers function. The resulting list was filtered by selecting the lowest adjusted P-value (Bonferroni-corrected P-value <0.05), so as to identify the top 10 statistically significantly up-regulated genes for each cluster (although, for some clusters fewer than 10 genes were identified). Monocle 3 [20–22] (v. 0.2.3.0) was used for calculating pseudo-time values and inferring the developmental trajectory. The Seurat object was imported using the SeuratWrappers::as.cell_data_set (v. 0.3.0) programme and further analyzed using default settings with the root node set to the cluster that contained cells expressing CD34, as these were the earliest B-lineage cells in the dataset. For the analyses with the BCR-genes retained, we performed the same steps as described above.
Peripheral blood CD8+ T cells
Count data matrices were retrieved from the 10x Genomics website (https://www.10xgenomics.com/resources/datasets/cd-8-plus-t-cells-of-healthy-donor-1-1-standard-3-0-2). The analyses were performed in a manner similar to that used for the BM samples. However, cells for which >5% of the total transcripts were of mitochondrial origin were excluded. Seurat::FindClusters was run with the resolution set to 0.4. TCR-genes were removed from the count data using a regular expression command with the following pattern:’^TR[ABDG][VJC]’. A cluster containing cells positive for CD3E, CD19 and CD14 was discarded.
Key Points
The analyses of scRNA-seq datasets derived from various sources of B cells show that the genes that encode B-cell antigen receptors interfere with the process of unsupervised clustering, as well as the down-stream analyses of these cells.
The analyses of a publicly available T-cell scRNA-seq dataset show that the genes that encode T-cell receptors interfere with the unsupervised clustering of these cells.
This interference is likely due to the high frequencies of B- and T-cell receptor genes among the genes that account for most of the variance in each of the PCs used for the clustering.
The effects of the B-cell and T-cell receptor genes are abrogated upon their exclusion before clustering is undertaken.
Data availability
All scRNA-seq data from the PB and BM B cells are available at Array Express. Identifier: E-MTAB-12346. The PB CD8+ T cell dataset is available from 10x Genomics website.
Code availability
All custom codes used for data processing and computational analyses are available under project BCR_TCR_Interference at https://github.com/MartenssonLab/.
Author contributions
T.S. and I-L.M. designed the study. T.S., K.G., A.C. and I.G. contributed to the procurement and processing of samples and acquired the data. T.S. and K.G. analyzed the single-cell RNA sequencing data. I.G. and I-L.M. carried out or contributed essential reagents and materials for the experiments. K.G., A.C., A.T. and I.G. contributed substantially to the discussions. T.S. and I-L.M. wrote the manuscript with contributions from all the authors.
Supplementary Material
Acknowledgements
We thank Dr Vincent Collins for critically reading and editing the manuscript.
Author Biographies
Timothy Sundell is a PhD student in the Department of Rheumatology and Inflammation Research, University of Gothenburg, Sweden. He works on the analysis of single-cell RNA sequencing data.
Kristoffer Grimstad is a PhD student in the Department of Rheumatology and Inflammation Research, University of Gothenburg, and School of Bioscience, University of Skövde, Sweden. He works on the analysis of single-cell RNA sequencing data.
Alessandro Camponeschi is a postdoctoral researcher in the Department of Rheumatology and Inflammation Research, University of Gothenburg, Sweden. His work spans cellular and molecular biology in the field of B cells in health and disease, e.g. autoimmunity and cancer.
Andreas Tilevik is an associate professor at the School of Bioscience, University of Skövde, Sweden. His research focuses on the use of mathematic and statistical models for the understanding of the immune system.
Inger Gjertsson is a professor in the Department of Rheumatology and Inflammation Research, University of Gothenburg, Sweden. Her research focus is B cells and the contributions of cartilage-specific autoantibodies to cartilage destruction in rheumatoid arthritis.
Inga-Lill Mårtensson is a professor and Head of the Department of Rheumatology and Inflammation Research, University of Gothenburg, Sweden. Her research focus is B cells in health and disease, e.g. autoimmunity and cancer.
Contributor Information
Timothy Sundell, Department of Rheumatology and Inflammation Research, Institute of Medicine, The Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Kristoffer Grimstad, Department of Rheumatology and Inflammation Research, Institute of Medicine, The Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden; School of Bioscience, University of Skövde, Skövde, Sweden.
Alessandro Camponeschi, Department of Rheumatology and Inflammation Research, Institute of Medicine, The Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Andreas Tilevik, School of Bioscience, University of Skövde, Skövde, Sweden.
Inger Gjertsson, Department of Rheumatology and Inflammation Research, Institute of Medicine, The Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Inga-Lill Mårtensson, Department of Rheumatology and Inflammation Research, Institute of Medicine, The Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Funding
This work was supported by the Swedish Research Council, grants 2018-03128 and 2021-01150 (ILM) and 2016-01576 (IG); the Swedish Cancer Foundation, grant 19 0464 (ILM); the Swedish Childhood Cancer Fund, grants PR2018-0170 and PR2020-0147 (ILM), and TJ2019-0098 (AC); Assar Gabrielsson’s Foundation, FB21-104 (AC); Patient Association for Rheumatic Diseases, R-94129 (ILM) and R-940945 (IG); ALF (agreement between the Government of Sweden and the County Councils), ALFGBG-719631 (IG); Adlerbertska stiftelsen (TS); and the IngaBritt och Arne Lundbergs Foundation (ILM, IG).
References
- 1. Tonegawa S. Somatic generation of antibody diversity. Nature 1983;302:575–81. [DOI] [PubMed] [Google Scholar]
- 2. Rajewsky K. Clonal selection and learning in the antibody system. Nature 1996;381:751–8. [DOI] [PubMed] [Google Scholar]
- 3. Schroeder HW, Jr. Similarity and divergence in the development and expression of the mouse and human antibody repertoires. Dev Comp Immunol 2006;30:119–35. [DOI] [PubMed] [Google Scholar]
- 4. Stewart A, Ng JC, Wallis G, et al. Single-cell transcriptomic analyses define distinct peripheral B cell subsets and discrete development pathways. Front Immunol 2021;12:602539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mathew NR, Jayanthan JK, Smirnov IV, et al. Single-cell BCR and transcriptome analysis after influenza infection reveals spatiotemporal dynamics of antigen-specific B cells. Cell Rep 2021;35:109286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Siu JHY, Pitcher MJ, Tull TJ, et al. Two subsets of human marginal zone B cells resolved by global analysis of lymphoid tissues and blood. Sci Immunol 2022;7:eabm9060. [DOI] [PubMed] [Google Scholar]
- 7. Andreatta M, Tjitropranoto A, Sherman Z, et al. A CD4(+) T cell reference map delineates subtype-specific adaptation during acute and chronic viral infections. Elife 2022;11:e76339. 10.7554/eLife.76339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. King HW, Orban N, Riches JC, et al. Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci Immunol 2021;6:eabe6291. 10.1126/sciimmunol.abe6291. [DOI] [PubMed] [Google Scholar]
- 9. Moroney JB, Vasudev A, Pertsemlidis A, et al. Integrative transcriptome and chromatin landscape analysis reveals distinct epigenetic regulations in human memory B cells. Nat Commun 2020;11:5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Camponeschi A, Gerasimcik N, Wang Y, et al. Dissecting integrin expression and function on memory B cells in mice and humans in autoimmunity. Front Immunol 2019;10:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Galletti G, De Simone G, Mazza EMC, et al. Two subsets of stem-like CD8(+) memory T cell progenitors with distinct fate commitments in humans. Nat Immunol 2020;21:1552–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martin MD, Badovinac VP. Defining memory CD8 T cell. Front Immunol 2018;9:2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghia P, ten Boekel E, Sanz E, et al. Ordering of human bone marrow B lymphocyte precursors by single-cell polymerase chain reaction analyses of the rearrangement status of the immunoglobulin H and L chain gene loci. J Exp Med 1996;184:2217–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henry Dunand CJ, Wilson PC. Restricted, canonical, stereotyped and convergent immunoglobulin responses. Philos Trans R Soc Lond B Biol Sci 2015;370:20140238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Freeman JD, Warren RL, Webb JR, et al. Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res 2009;19:1817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zappia L, Oshlack A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience 2018;7:giy083. 10.1093/gigascience/giy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Becht E, McInnes L, Healy J, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 2019;37:38–44. [DOI] [PubMed] [Google Scholar]
- 19. Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 2019;20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qiu X, Mao Q, Tang Y, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 2017;14:979–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cao J, Spielmann M, Qiu X, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019;566:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All scRNA-seq data from the PB and BM B cells are available at Array Express. Identifier: E-MTAB-12346. The PB CD8+ T cell dataset is available from 10x Genomics website.