

Abstract
Weight loss from an overweight state is associated with a disproportionate decrease in whole-body energy expenditure that may contribute to the heightened risk for weight regain. Evidence suggests that this energetic mismatch originates from lean tissue. Although this phenomenon is well documented, the mechanisms have remained elusive. We hypothesized that increased mitochondrial energy efficiency in skeletal muscle is associated with reduced expenditure under weight loss. Wildtype (WT) male C57BL6/N mice were fed with high fat diet for 10 weeks, followed by a subset of mice that were maintained on the obesogenic diet (OB) or switched to standard chow to promote weight loss (WL) for additional 6 weeks. Mitochondrial energy efficiency was evaluated using high-resolution respirometry and fluorometry. Mass spectrometric analyses were employed to describe the mitochondrial proteome and lipidome. Weight loss promoted ~50% increase in the efficiency of oxidative phosphorylation (ATP produced per O2 consumed, or P/O) in skeletal muscle. However, weight loss did not appear to induce significant changes in mitochondrial proteome, nor any changes in respiratory supercomplex formation. Instead, it accelerated the remodeling of mitochondrial cardiolipin (CL) acyl-chains to increase tetralinoleoyl CL (TLCL) content, a species of lipids thought to be functionally critical for the respiratory enzymes. We further show that lowering TLCL by deleting the CL transacylase tafazzin was sufficient to reduce skeletal muscle P/O and protect mice from diet-induced weight gain. These findings implicate skeletal muscle mitochondrial efficiency as a novel mechanism by which weight loss reduces energy expenditure in obesity.
Keywords: energy efficiency, energy expenditure, mitochondria, oxidative phosphorylation, phospholipids, weight loss
Introduction
Obesity imposes tremendous health risks, but efforts to lose weight are often met with limited success [1, 2]. Many find weight loss achieved by weight loss regimens difficult to sustain. As observed in multiple studies including that of a reality television show [3], it has been postulated that weight loss from an overweight state is associated with an increase in energy efficiency, promoting lower metabolic rate [4−7]. The increase in energy efficiency can last several years, suggesting that these individuals remain their high propensity for weight regain for an extended period of time. These observations are found not only in human weight loss but also in model organisms [8, 9]. It is unclear what mechanisms contribute to reduced energy expenditure in the weight-loss state.
The alteration in systemic metabolic rate with weight loss likely represents a part of adaptive responses that are known to occur with changes in diet, exercise, or environment. Evidence suggests a non-leptin system that defends against weight gain [10]. Exercise training is known to increase the fuel economy of a subsequent single bout of exercise that in turn facilitates performance [11]. Exercise-induced increase in energy efficiency is due to an increase in mechanical efficiency for movement as well as an increase in energy efficiency of skeletal muscle mitochondria [12, 13]. Adaptation to low oxygen availability in high altitude is also known to alter skeletal muscle mitochondrial efficiency [14].
Leibel and others demonstrate that weight loss is associated with a decrease in activity-associated energy expenditure in addition to basal metabolic rate [15−18]. Skeletal muscle work efficiency appears to be the primary determinant of the disproportionate decline in 24-hour energy expenditure following weight loss, with additional declines in resting energy expenditure [18]. Skeletal muscle contributes to the majority of increased energy expenditure induced during physical activity. Previous studies have demonstrated that weight loss may alter skeletal muscle mitochondrial respiration [20−23], though it is unclear whether these changes reflect alteration in energy efficiency. In this study, we tested our hypothesis that weight loss must improve the efficiency of skeletal muscle mitochondrial respiration.
Results
Weight loss in obese mice reduces whole-body energy expenditure
Wildtype C57BL6/N mice were fed with a Western diet (Envigo, TD.88137) for 10 weeks, followed by a subset of mice that were maintained on the obesogenic diet (OB), while others were switched to standard chow to promote weight loss (WL) (Fig. 1a). Body weights in the WL group decreased between 10 to 12 weeks and were at steady state thereafter. We performed all terminal phenotyping at 16 weeks to exclude potential acute effects that changes in diet or body weight may have on metabolism. Another group of mice were fed standard chow diet (SCD) for 16 weeks as a lean (LN) reference control group. This strategy successfully and consistently produced groups of mice with divergent body weights and adiposity without altering lean mass (Fig. 1b–e).
Figure 1. Weight loss promotes a decrease in whole-body energy expenditure.
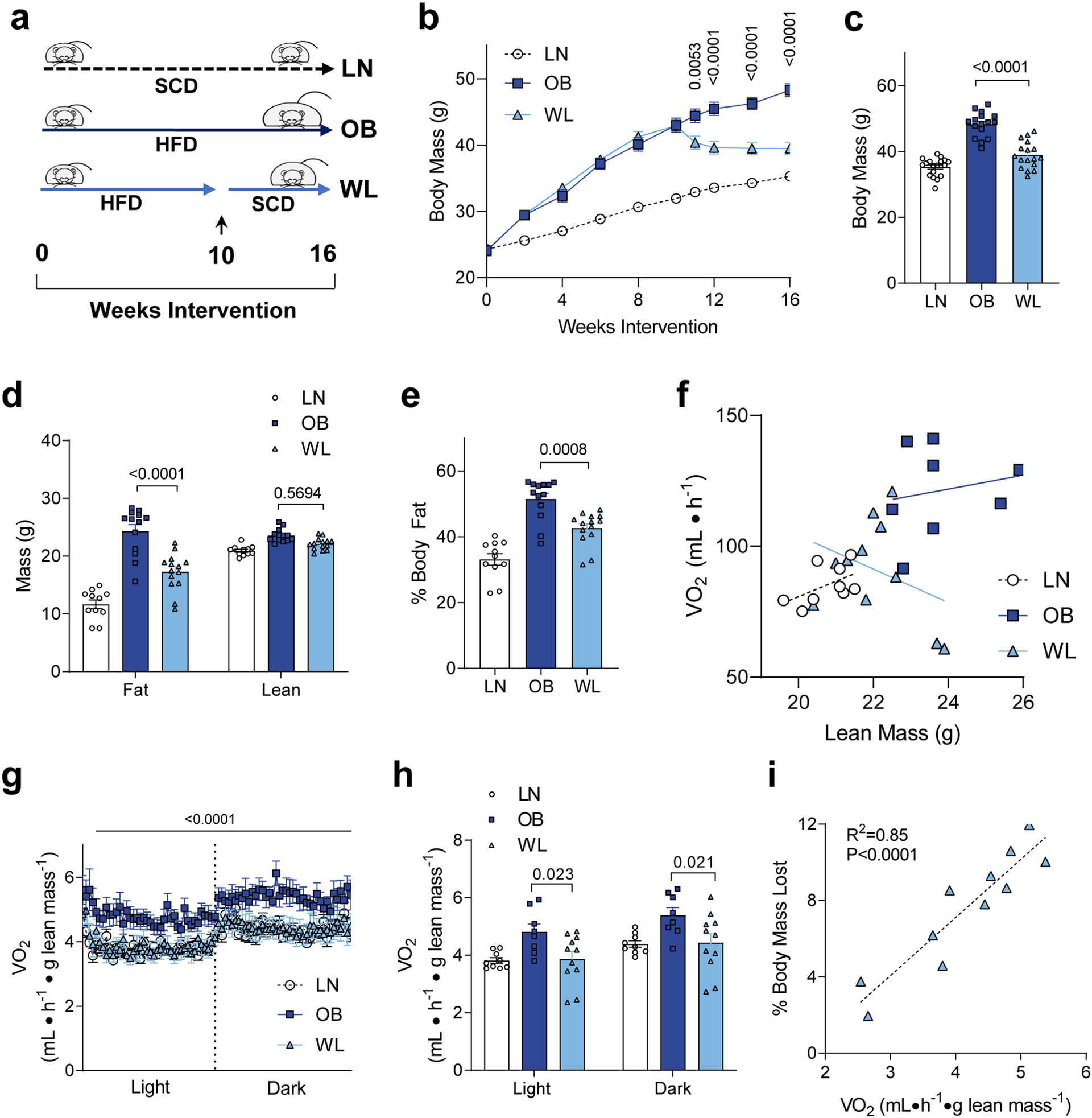
(a) Feeding timeline. LN: mice fed standard chow diet (SCD). OB: mice fed obesogenic high-fat diet (HFD). WL: mice with weight loss induced by switching from HFD to SCD at week 10. Ad lib fed for all mice. (b) Body mass of LN, OB and WL mice over the 16-week diet intervention. (c) Body mass just before terminal experiments. n =18 for LN, n =16 for OB, n =19 for WL for b and c. (d and e) Body composition measured just before terminal experiments. n =11 for LN, n =13 for OB, n =14 for WL for both d and e. (f) Relationship between lean mass and total VO2 (unnormalized) in LN, OB, and WL groups (n =9 for LN, n =8 for OB, n =11 for WL). (g and h) Whole-body oxygen consumption normalized to lean mass (n =9 for LN, n =8 for OB, n =11 for WL) at week 15. (i) Pearson correlation analyses of percent body weight loss to the oxygen consumption among the WL group (n =11). All data are represented as mean ± SEM. Two-way ANOVA with Sidak multiple comparisons (b, d, g, h) or one-way ANOVA with multiple comparisons (c, e). P-values indicate statistical significance between OB and WL groups.
To assess the energy utilization in these animals, indirect calorimetry experiments were performed at week 15. Total unnormalized VO2 was substantially elevated in the OB group compared to WL or LN group (Supplementary Fig. S1a and b). Correlation analyses indicated that lean mass exhibited a substantially different relationship to total VO2 in WL group compared to OB group (Fig. 1f, similar relationship with total body mass shown in Supplementary Fig. S1c), suggesting that the lean mass differentially contributes to metabolic rate between OB and WL groups. Indeed, VO2 normalized to lean mass was ~20% lower in WL compared to OB group (Fig. 1g and h). Spontaneous movement did not explain the differences in VO2 (Supplementary Fig. S1d and e). There was no relationship between spontaneous movement and VO2 in any of the groups (Supplementary Fig. S1f), suggesting that physical activity does not significantly contribute to 24-hour energy expenditure [24]. In contrast, VCO2 was largely unaffected between the groups (Supplementary Fig. S1g–j). Respiratory exchange ratio (RER) was significantly reduced in OB group compared to WL or LN groups (Supplementary Fig. S1k and l), likely representing the lower carbohydrate composition of their diets. Strikingly, in WL mice, there was a very strong positive correlation between VO2 to % decrease in body mass (Fig. 1i), consistent with the notion that higher energy expenditure contributes to weight loss.
Weight loss in obese mice increases skeletal muscle oxidative phosphorylation (OXPHOS) efficiency
Thermogenesis in the brown adipose tissue substantially contributes to whole-body metabolic rate in mice [25]. Mitochondrial uncoupling driven by the uncoupling protein 1 (UCP1) resides at the inner mitochondrial membrane to dissipate the proton gradient, driving mitochondrial uncoupling and brown adipose thermogenesis. However, brown adipose tissues from LN, OB, and WL groups did not look different, and had similar mitochondrial density and UCP1 content (Supplementary Fig. S2a).
Previously, we found that the propensity for obesity may be affected by the energy efficiency of sarco/endoplasmic reticulum (SR) Ca2+-ATPase (SERCA) pump, a highly abundant ATPase that contributes to ~30% of skeletal muscle energy expenditure [26, 27]. However, SERCA energy efficiency or abundance in skeletal muscle was not different among the groups (Fig. 2a and b). In contrast, weight loss in obese mice induced a robust increase (~50%) in the efficiency of OXPHOS quantified by ATP produced (energy OUT) per O2 consumed (energy IN) or “P/O” in skeletal muscle (Fig. 2c). The increase in muscle P/O induced by weight loss was exclusively due to a decrease in O2 requirement (Supplementary Fig. S2b) without compromising the capacity for ATP synthesis (Supplementary Fig. S2c). The increase in muscle OXPHOS efficiency was a unique feature of weight loss in obese mice, as P/O was not different between LN and OB groups.
Figure 2. Weight loss improves OXPHOS efficiency in skeletal muscle.
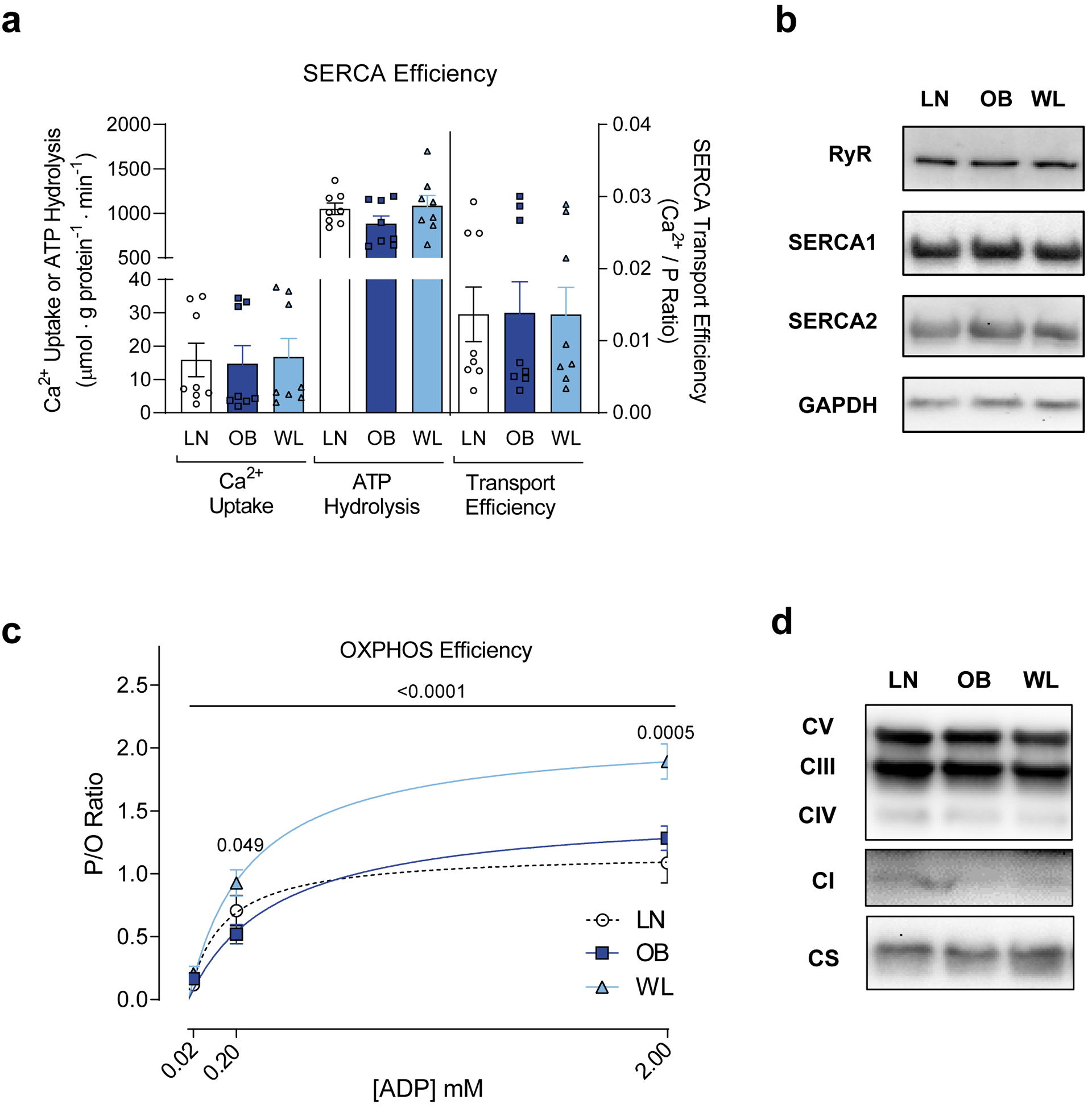
(a) Rates of Ca2+ uptake, SERCA ATP hydrolysis, and SERCA transport efficiency in skeletal muscle (n =8 for all groups). (b) Representative western blots of proteins involved in Ca2+ transport. (c) Skeletal muscle P/O ratio in fiber bundles isolated from gastrocnemius muscles (n =6 for LN, n =8 for OB, n =10 for WL). P-values indicate statistical significance between OB and WL groups. (d) Representative western blots of OXPHOS subunits and citrate synthase (CS) in isolated mitochondria. One-way ANOVA with multiple comparisons (a) or two-way ANOVA with Sidak multiple comparisons (c). P-values except in panel I indicate statistical significance between OB and WL groups. P-value in panel I shows statistical significance for correlation. Data are represented as mean ± SEM.
Weight loss in obese mice does not robustly alter skeletal muscle mitochondrial proteome
Next, we explored the molecular mechanisms by which weight loss improves skeletal muscle OXPHOS efficiency. Western blot analyses of OXPHOS subunits or citrate synthase did not reveal differences in these proteins in whole tissue lysate (Supplementary Fig. S2d), suggesting that these interventions did not alter skeletal muscle mitochondrial content. Western blotting also did not reveal differences in these OXPHOS subunits in isolated mitochondria (Fig. 2d, and Supplementary Fig. S2e). To more comprehensively understand how weight loss influences skeletal muscle OXPHOS enzymes, we analyzed the mitochondrial proteome using mass spectrometry [28]. However, weight loss did not induce changes in any of the OXPHOS subunits (Fig. 3a–e). In fact, mass spectrometry-based proteomic analyses did not detect a single mitochondrial protein whose abundance was statistically different between OB and WL group (Fig. 3f, comparisons with LN group in Supplementary Fig. S3a and b). The principal component analysis also showed the lack of divergence in mitochondrial proteome among the three groups (Fig. 3g). Clustering analyses revealed no pattern in the mitochondrial proteome (Supplementary Fig. S3c). We also quantified OXPHOS supercomplex assembly, which also did not reveal any differences (Fig. 3h).
Figure 3. Weight loss does not alter skeletal muscle mitochondrial proteome.
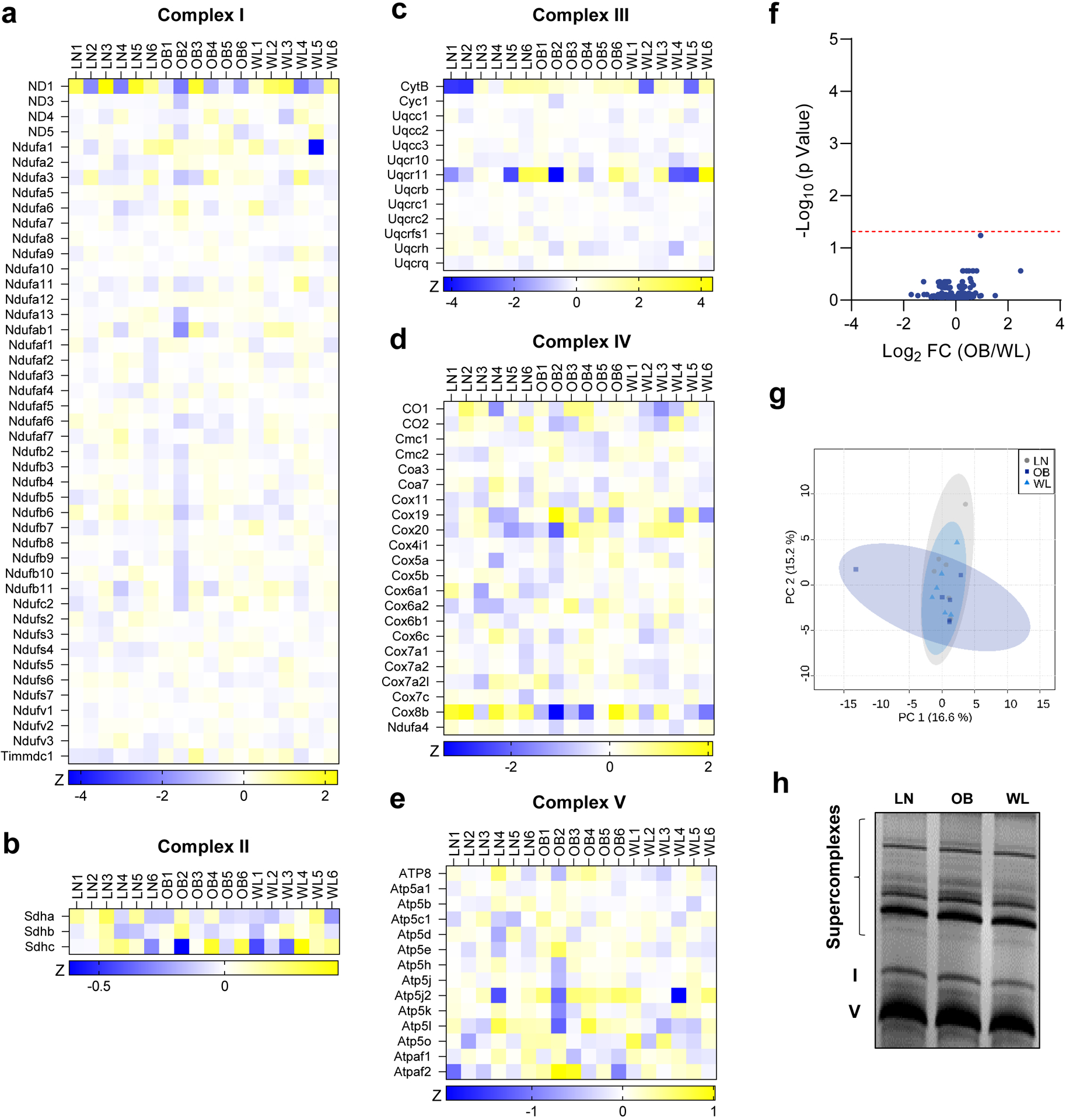
(a−e) Heatmap of abundance of OXPHOS subunits measured with mass spectrometry. (f) Volcano plot of differentially abundant mitochondria proteins between OB and WL groups (significance threshold shown with dotted red line). (g) Principal component analysis of mitochondrial proteomic data. (h) Abundance of respiratory supercomplex formation in isolated mitochondria.
Weight loss in obese mice alters skeletal muscle mitochondrial lipidome
OXPHOS enzymes are imbedded in the phospholipid bilayer of the inner mitochondrial membrane [29, 30]. Indeed, energy-transducing steps of OXPHOS occurs in (electron transfer) and through (proton transport) the lipid environment. Thus, we examined the mitochondrial lipidome in muscles from LN, OB, and WL mice (Fig. 4a). Unlike the mitochondrial proteome, there were some changes in the skeletal muscle mitochondrial lipidome induced by weight fluctuation (Fig. 4a and b). Importantly, mitochondria from OB and WL groups demonstrated lower lipid-to-protein ratio compared to LN group (Fig. 4c), suggesting that muscle mitochondrial membranes become more protein-rich with obesity and remain protein-rich in the weight-loss state. Such change would be predicted to have complex biophysical consequences on OXPHOS dynamics. Unfortunately, it also makes it difficult to interpret differences in mitochondrial lipid composition between LN and the other two groups. For this reason, we decided to focus on the differences in mitochondrial lipidome between the OB and WL groups (Supplementary Fig. S4a−g and Fig. 4d). We felt that this is a justifiable strategy given that P/O values were not different between LN and OB groups (Fig. 2c), suggesting that differences in mitochondrial membrane lipids between LN and OB is not sufficient to influence OXPHOS efficiency.
Figure 4. Weight loss accelerates skeletal muscle CL remodeling.
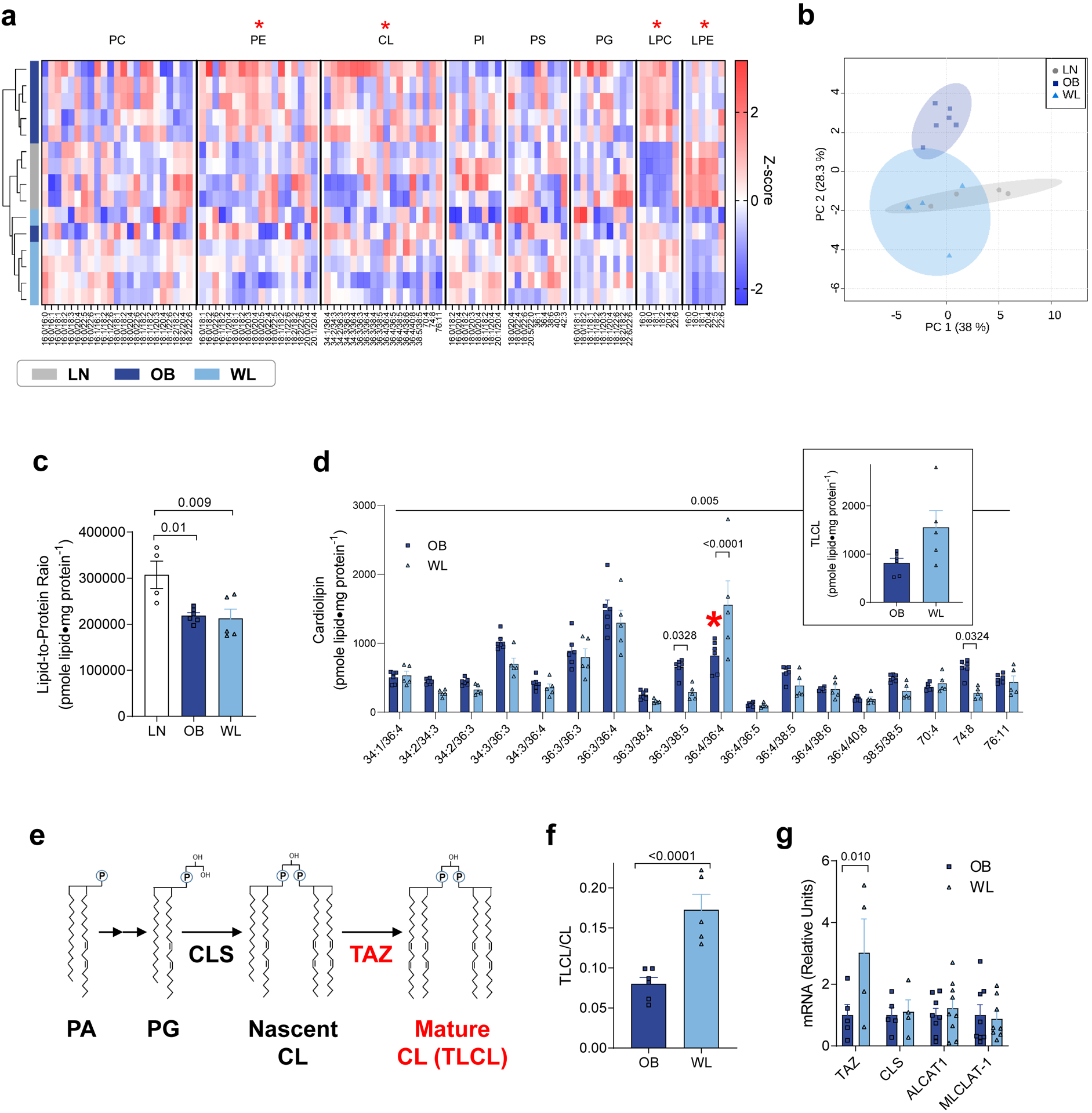
(a) Heatmap of relative abundance of mitochondrial lipids. X-axis represents individual lipid species classified according to lipid classes. Hierarchical clustering is shown on the left. Red asterisks indicate the main effect of weight loss (OB vs. WL). (b) Principal component analysis shows divergence in the mitochondrial lipidome. (c) Lipid-to-protein ratio. n =4 for LN, n =6 for OB, n =5 for WL for a−c. (d) Abundance of individual CL species in isolated mitochondria. TLCL is shown in red asterisk and separately as an insert (n =6 for OB, n =5 for WL). (e) Schematic of CL synthesis and remodeling that occurs in the inner mitochondrial membrane. (f) TLCL to total CL ratio (n =6 for OB, n =5 for WL). (g) TAZ, CLS, ALCAT1, and MLCLAT-1 mRNA levels in skeletal muscle (n =5 for OB, n =4 for WL). One-way ANOVA with multiple comparisons (c), two-way ANOVA with Sidak multiple comparisons (d, g) or unpaired t-test (f). All P-values except for those in panel c indicate statistical significance between OB and WL groups. P-values in panel c show comparisons between LN and OB and LN and WL. Data are represented as mean ± SEM.
Mitochondrial phosphatidylethanolamine (PE) was significantly reduced (Fig. 4a and Supplementary Fig. S4b) in WL group compared to the OB group. Mitochondrial PE consists 25−40% of mitochondrial lipids and is primarily synthesized by the phosphatidylserine decarboxylase (PSD) that resides in the inner mitochondrial membrane [31]. To determine whether reduction of mitochondrial PE contributes to the increased muscle P/O, we performed lentivirus-mediated knockdown of PSD in murine C2C12 myotubes (Supplementary Fig. S5a). However, PSD knockdown reduced, not increased, mitochondrial P/O (Supplementary Fig. S5b). Thus, reduction in mitochondrial PE observed with weight loss is unlikely to explain the increased P/O.
Weight loss in obese mice also reduced mitochondrial cardiolipin (CL) in skeletal muscle (Fig. 4d). Mitochondrial CL consists 10%−20% of mitochondrial lipids and is known to bind with high affinity to OXPHOS enzymes and affect their functions [31]. Previous studies suggest that CL may have an impact on OXPHOS efficiency [33−35]. CL consists of two phosphatidic acid moieties linked by a central glycerol backbone. CL is almost exclusively localized in the inner mitochondrial membrane, and is synthesized by a series of enzymes localized in the inner mitochondrial membrane including CL synthase (CLS) and CL transacylation enzymes [32] (Fig. 4e). For reasons that are not completely understood, CL molecules generated by CLS with uneven acyl-chains are not fully functional and referred as “nascent CL”. These nascent CL molecules are then transacylated to become tetralinoleoyl-CL (TLCL or 18:2/18:2/18:2/18:2-CL), a reaction primarily driven by CL transacylase tafazzin (TAZ) [32, 34]. TLCL is thought to be fully functional and referred as “mature CL” (Fig. 4e). Thus, we examined the mitochondrial CL portfolio between muscles from OB and WL groups (Fig. 4d). Strikingly, even though many of the CL species were lower in WL compared to OB, TLCL was almost twice as highly abundant in WL compared to OB (Fig. 4d red asterisk and insert). Indeed, the ratio of TLCL to total CL was 2.2-fold greater in WL compared to OB (Fig. 4f). This increase in TLCL content was likely explained by a greater TAZ expression in the WL compared to OB without altering the expression of other CL-synthesizing enzymes (Fig. 4g).
Deficiency in CL remodeling is sufficient to reduce muscle OXPHOS efficiency
We investigated whether TLCL influences OXPHOS efficiency to alter the propensity for weight gain. For these experiments, we utilized mice with doxycycline-induced whole-body knockdown of TAZ (TAZKD mice) compared to doxycycline fed WT littermates (Fig. 5a). Previous studies have shown that TAZKD mice exhibit greater energy expenditure and are protected from diet-induced obesity [34, 36]. Our observations recapitulated these findings (Fig. 5b and c). We then examined skeletal muscle tissues from these mice to study the role of CL remodeling in OXPHOS efficiency. As expected, the intervention successfully reduced TAZ expression in skeletal muscle (Fig. 5d). We quantified the CL species from skeletal muscle mitochondria using mass spectrometry (Fig. 5e). Previous studies showed that virtually all CL species are reduced in TAZKD mice [34, 37], suggesting that nascent CL maybe targeted for degradation without the presence of mature CL. Nevertheless, TAZKD had a disproportionately greater effect to reduce TLCL (Fig. 5e, red asterisk) compared to other CL species, shown by a substantial reduction in TLCL to total CL ratio (Fig. 5f). The trace amount of TLCL present in these tissues likely arose from alternate CL transacylases such as MLCLAT-1 and ALCAT-1 that are not highly expressed in skeletal muscle. Importantly, we phenotyped skeletal muscle mitochondria from WT and TAZKD mice with high-resolution respirometry and fluorometry. Consistent with previous findings, TAZ deletion reduced the capacity for mitochondrial respiration and ATP production [34] (Fig. 5g). However, TAZ deletion had a disproportionately greater effect to reduce ATP production than O2 consumption, which consequently reduced P/O ratio (Fig. 5g). Together, these observations indicate that TAZ deletion is sufficient to reduce skeletal muscle OXPHOS efficiency. In turn, these findings suggest that accelerated CL remodeling in weight loss state may explain the greater OXPHOS efficiency in skeletal muscle.
Figure 5. TLCL deficiency reduces OXPHOS efficiency.
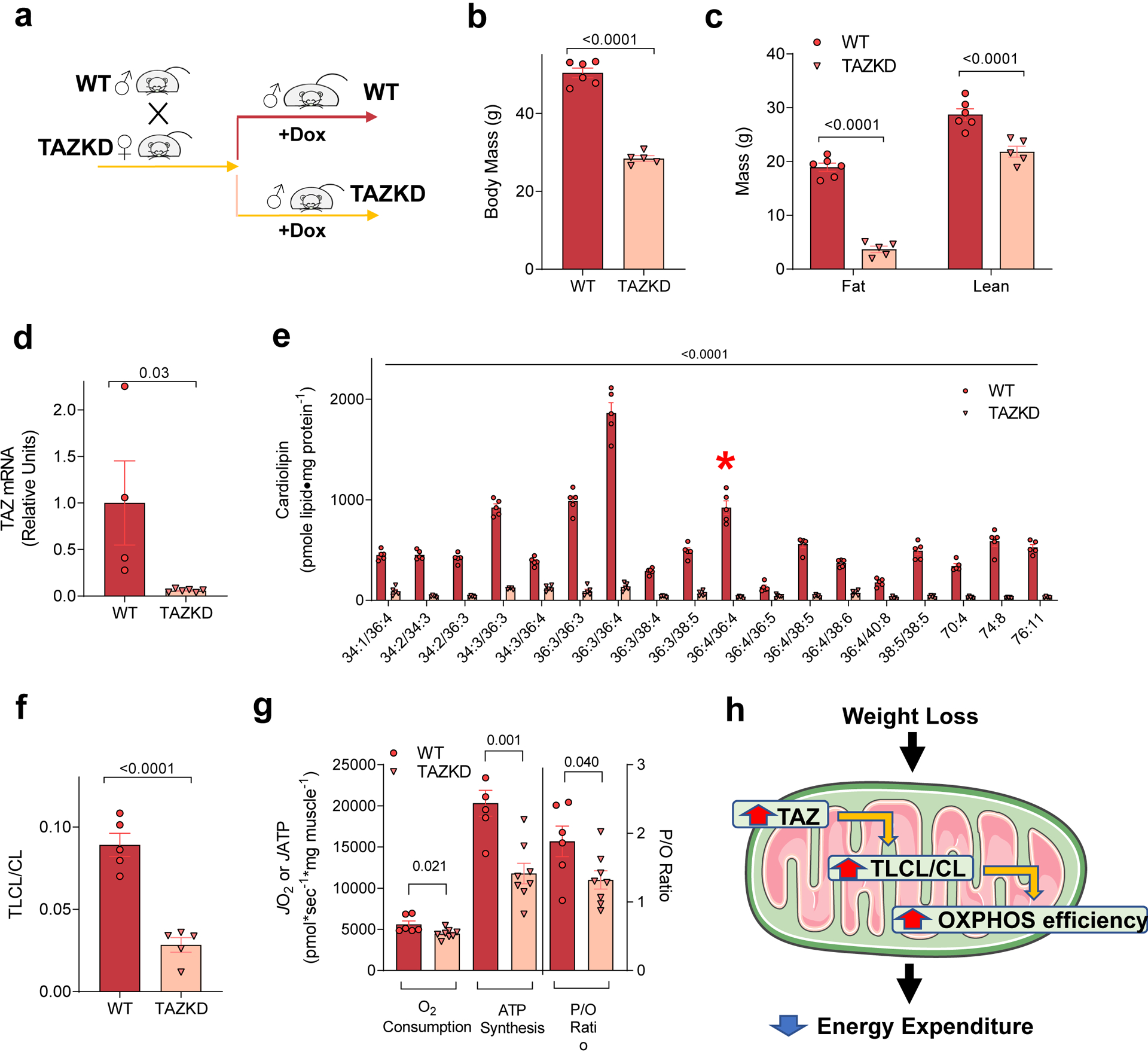
(a) Schematic of doxycycline intervention in wildtype (WT) and TAZKD littermates. (b) Body mass of WT and TAZKD mice (n =6 for WT, n =5 for TAZKD). (c) Body composition of WT and TAZKD mice (n =6 for WT, n =5 for TAZKD). (d) TAZ mRNA abundance in skeletal muscle from WT and TAZKD mice (n =4 for WT, n =6 for TAZKD). (e) Abundance of individual CL species in isolated mitochondria (n =5 for both groups). TLCL is shown in red asterisk. (f) TLCL to total CL ratio (n =5 for both groups). (g) Rates for O2 consumption, ATP production, and P/O ratio in skeletal muscles from WT and TAZKD mice with 200 μmol/L of ADP (n =6 for WT, n =8 for TAZKD). (h) Proposed mechanism for how weight loss improves OXPHOS efficiency. Two-way ANOVA with Sidak multiple comparisons (c, e) or unpaired t-test (b, d, f, g). All P-values indicate statistical significance between WT and TAZKD groups. Data are represented as mean ± SEM.
Discussion
Weight loss reduces whole-body energy expenditure that likely promotes weight regain [4−7]. In the current study, we report that weight loss in overweight mice increases skeletal muscle OXPHOS efficiency concomitant to a decrease in whole-body energy expenditure. As one of the organs with large contributions to resting and non-resting energy expenditures, improved skeletal muscle energy efficiency would be predicted to explain a substantial component of a reduction in metabolic rate that occurs with weight loss [38−40]. These findings are consistent with previous reports that weight loss is associated with a decrease in activity-associated energy expenditure in addition to basal metabolic rate [15−18].
Skeletal muscle OXPHOS efficiency was increased by ~50% with weight loss. This is a striking increase in energy efficiency that would be predicted to lower muscle energy expenditure, requiring 50% more work to expend equivalent calories. Nevertheless, the increase in OXPHOS efficiency did not coincide with changes in abundance of OXPHOS subunits. Indeed, there was not a single mitochondrial protein whose abundance was detected to be significantly affected with weight loss. Instead, weight loss had a more substantial effect on the lipidomic landscape of skeletal muscle mitochondria. One of these changes was an increase in the concentration of mitochondrial TLCL.
We then demonstrated that genetically-induced deficiency in TLCL biosynthesis was sufficient to promote muscle OXPHOS inefficiency and protect mice from diet-induced obesity. These results are consistent with the notion that increased TLCL may contribute to increased OXPHOS efficiency and reduced energy expenditure with weight loss. There are a few caveats to these results pertaining to the use of the TAZKD mice. First, deletion of TAZ is not specific to muscle, so we cannot rule out the possibility that reduced muscle OXPHOS efficiency in these mice could occur indirectly through other tissues. We are currently in a process of developing mice with skeletal muscle-specific knockout of TAZ though these studies are beyond the scope of the current manuscript. Second, doxycycline is known to influence mitochondrial function [41], making this system not ideal for studying bioenergetics. Nevertheless, we treated WT littermates also with doxycycline to control to the best of our abilities. Third, TAZ knockdown lowered the content of all CL species including TLCL, even though TLCL was reduced disproportionately more compared to others. Last but not least, we did not test whether TAZ knockdown would make mice resistant to weight loss-induced reduction in energy expenditure and an increase in muscle OXPHOS efficiency. We plan on performing these experiments as well as the studies on propensity for weight regain in mice with muscle-specific knockout of TAZ.
For the current study, we initially considered placing TAZKD mice on the weight loss regimen as described in Fig. 1a. If performed correctly, such experiment would help provide a better insight into the role of TAZ or TLCL with improved OXPHOS efficiency with weight loss. However, we decided to wait to perform these experiments until tamoxifen-inducible skeletal muscle-specific TAZ mice are in hand. This is because performing this experiment in whole-body TAZKD mice presents the following difficulties. First, TAZ deletion is induced by doxycycline, which increases food intake. We reason that body weight changes for doxycycline fed WT and TAZKD mice will not mirror those performed in Fig. 1, making it difficult to interpret data. Second, due to the slow turnover rate of membrane lipids, it is predicted to take approximately 4 weeks for doxycycline treatment to alter mitochondrial CL milieu. This means that we would need to switch mice from being fed high fat diet (HFD) to doxycycline-containing HFD 4 weeks prior to switching to chow diet. We felt that this added additional layer of complexity that made the design less than ideal. Finally, local doxycycline injection to muscle might be used to circumvent the food intake issue, but doxycycline would need to be injected daily throughout the study as the TAZKD model utilizes a shTAZ-mediated silencing strategy instead of knockout. Tamoxifen-inducible, TAZ conditional knockout system would be able to circumvent these issues to better address the role of this pathway in regulating OXPHOS efficiency with weight loss.
Findings from this study should not be interpreted to mean that all interventions that promote weight loss increase skeletal muscle OXPHOS efficiency. First, the current study is limited to observations in overweight mice with diet-induced obesity. The results might not be applicable to weight loss in other states. Second, because OB and WL mice were fed diet with different compositions, we cannot rule out the possibility that some of the effects we have observed is not necessarily driven by the effects of weight loss per se. In the WL group, switching to standard chow promptly induced weight loss during the first 2 weeks, followed by a 4-week period of steady body mass. In contrast, OB group continued to gain weight between week 10 and 16. Third, we did not quantify energy efficiency during muscle contraction, and some evidence suggests disconnect between P/O and muscle contractile efficiency [42]. Last but not least, the current study was performed in room temperature. It would be important to study how these findings are recapitulated in thermoneutrality. It is important to interpret our findings in the context of these caveats.
Changes in mitochondrial energy efficiency may be a part of adaptive responses that occur in response to a variety of energetic insults. As discussed above, exercise training improves the fuel economy for a single bout of exercise, partly driven by an increase in skeletal muscle OXPHOS efficiency [13]. Similarly, skeletal muscle undergoes mitochondrial adaptations in response to low oxygen availability at high altitude [14, 43]. Whether they are driven by changes in energy supply, energy demand, or oxygen availability, alterations in mitochondrial energy efficiency likely represents a fundamental adaptation to counterbalance changes in energy dynamics.
In summary, weight loss in obese mice promotes an increase in skeletal muscle OXPHOS efficiency that likely contributes to reduced whole-body energy expenditure. Weight loss also coincided with increased mitochondrial TLCL, and deficiency of TLCL was sufficient to reduce muscle OXPHOS efficiency and make mice more resistant to weight gain. We interpret these findings to propose that weight loss accelerates CL remodeling in skeletal muscle to improve OXPHOS efficiency and lower whole-body energy expenditure (Fig. 5h). We speculate that such decrease in metabolic rate arose from a tremendous evolutionary pressure to conserve energy in states of energy deprivation. In our age, these adaptive responses likely strongly contribute to increased propensity for a rebound in adiposity.
Materials and methods
Animals and diet intervention
Male C57BL/6NCrl (Charles River: 027) mice were used for the weight loss study. At 10 weeks of age, mice were either maintained on SCD (Envigo 2920X) or HFD (42% calories from fat; Envigo: TD88137). After 10 weeks of HFD feeding, a subset of mice were switched back to SCD while the others continued HFD feeding for another 6 weeks. Heterozygous TAZKD mice were obtained from the Jackson Laboratory (Stock number 014648, B6.Cg- Gt(ROSA)26S or tm37(H1/tetO-RNAi:Tafazzin)Arte/ZkhuJ). TAZ knockdown was induced in utero by supplying 625 mg/kg doxycycline chow (Envigo, TD.09628) as previously described [44]. Briefly, female TAZKD mice were maintained on doxycycline chow (625 mg/kg) at least 5 days before being mated with male WT mice. The doxycycline diet was removed during the mating period, and following copulation, males were removed and the doxycycline diet was reintroduced for the duration of gestation. Following weaning, all offspring were maintained on the doxycycline diet for the remainder of the study. For all experiments, mice were provided access to food ad libitum, maintained on a 12-hour light/dark cycle, and fasted for ~4 hours prior to terminal experiments. For terminal experiments, mice were given intraperitoneal injection of 80 mg/kg ketamine and 10 mg/kg xylazine, after which tissues were harvested. All terminal experiments were performed between 10 am and 2 pm, with the order of dissections randomized to avoid bias. All animals were randomized and no animals were excluded from the analyses.
Metabolic cage and body composition
Whole mouse indirect calorimetry and body composition were measured as previously described [26]. Columbus Instruments Lab Monitoring Systems were used to measure VO2, VCO2, respiratory exchange ratio (RER; VCO2/VO2), and activity. Mice were housed individually and acclimated for at least 24 hours before data collections. Data from the final complete light/dark cycle were used for analysis. Bruker Minispec NMR (Bruker, Germany) was used to determine composition of fat and fat-free mass.
Mitochondria and SR enrichment
Gastrocnemius muscles were used to isolate fractions enriched in mitochondria or SR as previously described [26, 34]. For mitochondria enrichment, muscles were minced in mitochondria isolation medium (300 mmol/L sucrose, 10 mmol/L HEPES, 1 mmol/L EGTA, and 1 mg/mL BSA) and subsequently homogenized using a Teflon-glass system. Homogenates were then centrifuged at 800 × g for 10 min, after which the supernatant was taken and centrifuged at 12,000 × g for 10 min. The resulting mitochondrial pellet was carefully resuspended in mitochondria isolation medium without BSA. For SR isolations, muscles were homogenized [300 mmol/L sucrose, 20 mmol/L HEPES pH 7.4, Halt protease (78430)] and underwent differential centrifugation (1,300 × g for 10 min, 20,000 × g for 20 min, 180,000 × g for 2 hours and 15 min) [45] to pellet an SR-enriched fraction, which was resuspended in SR isolation buffer.
High-resolution respirometry and fluorimetry
Respiration in permeabilized muscle fiber bundles was performed as previously described [31, 34]. Briefly, a small portion of freshly dissected red gastrocnemius muscle tissue was placed in buffer × [7.23 mmol/L K2EGTA, 2.77 mmol/L Ca K2EGTA, 20 mmol/L imidazole, 20 mmol/L taurine, 5.7 mmol/L ATP, 14.3 mmol/L phosphocreatine, 6.56 mmol/L MgCl2·6H2O, and 50 mmol/L K-MES (pH 7.1)]. Fiber bundles were separated and permeabilized for 30 minutes at 4°C with saponin (30 μg/mL), and immediately washed in buffer Z [105 mmol/L K-MES, 30 mmol/L KCl, 10 mmol/L K2HPO4, 5 mmol/L MgCl2·6H2O, 0.5 mg/mL BSA, and 1 mmol/L EGTA (pH 7.4)] for 15 min. After washing, high-resolution respiration rates were measured using an OROBOROS Oxygraph-2k. The muscle fibers were suspended in buffer Z with 20 mmol/L creatine monohydrate and 10 μmol/L blebbistatin to inhibit myosin adenosine triphosphatases during respiration measurements. Fiber bundles were added to the oxygraph chambers containing assay buffer (105 mmol/L MES potassium salt, 30 mmol/L KCl, 10 mmol/L K2HPO4, 5 mmol/L MgCl2, 0.5 mg/mL BSA). ATP production was measured fluorometrically using a Horiba Fluoromax-4 (Horiba Scientific), by enzymatically coupling ATP production to NADPH synthesis as previously described [46]. Respiration and ATP production were measured in the presence of 20, 200, or 2000 μmol/L ADP.
SR ATPase efficiency assay
SERCA efficiency assays were performed as previously described [26]. SR-fraction was quantified using BCA protein assay (Pierce, 23225) and 10 μg of SR protein was used in each replicate for SERCA-dependent Ca2+-uptake and ATPase activity assay. SERCA-dependent Ca2+ uptake and ATPase activity assays were performed in buffer containing 60 mmol/L HEPES, 200 mmol/L KCl, 15 mmol/L MgCl2, 10 mmol/L NaN3, 1 mmol/L EGTA, and 0.005% Triton-X at a pCa of 5.15 and with or without 15 μmol/L thapsigargin. Free Ca2+ was determined using Maxchelator Ca-Mg-ATP-EDTA Calculator using constants from the NIST database #46 v8 at 37°C, pH 7.3, and ionic constant of 0.25N [47]. ATP SERCA-dependent measurements were calculated by taking the difference of values without thapsigargin to values with thapsigargin [Total (without thapsigargin) – SERCA independent (with thapsigargin) = SERCA-dependent]. Ca2+ uptake assay buffer additionally contained 5 mmol/L of (COOK)2. ATPase activity assay buffer additionally contained 10 mmol/L PEP, 1.5 mmol/L NADH, 2.4–4 units of pyruvate kinase/3.6–5.6 units lactate dehydrogenases enzymes (Sigma, P0294).
Calcium uptake assays were performed as previously described [27]. Reactions were started by the addition of 4 mmol/L ATP and 45CaCl2 (Perkin Elmer, NEZ013001MC) to assay buffer with sample. After incubation for 15 minutes at 37°C with 300 RPM rotation, assay was quenched with the addition of 150 mmol/L KCl and 1 mmol/L LaCl3 and placed on ice. Samples were then filtered on to a 0.22 μm PES membrane filter (Millipore, GPWP02500), rinsed 3 × 5 mL PBS, and processed for scintillation counting. SERCA ATPase activity assay [48] was performed on a 96-well plate reader. The assay was initiated by the addition of 4 mmol/L ATP and the absorbance at 340 nm was recorded every 60 seconds for 30 minutes at 37°C. SERCA transport efficiency was determined by the ratio of SERCA-dependent Ca2+ uptake to SERCA-dependent ATPase hydrolysis.
Western blot
Western blots were performed as previously described [49]. Protein homogenates were analyzed for abundance of ryanodine receptor (RyR; Santa Cruz 13942), SERCA1 (Abcam 2818), SERCA2 (Abcam 3625), glyceraldehyde 3-phosphate dehydrogenase (GAPDH: Cell Signal 2118), mitochondrial complexes I-V (Abcam 110413), citrate synthase (Abcam 96600), and uncoupling protein-1 (UCP1: Alpha Diagnostic UCP11-A).
Sample preparation and nLC-MS/MS label free proteomic analysis
Mitochondria were purified and subjected to label free proteomic screening as previously described [50]. Isolated mitochondria were lysed in buffer D (8 mol/L urea in 40 mmol/L Tris, 30 mmol/L NaCl, 1 mmol/L CaCl2, 1 × cOmplete ULTRA mini EDTA-free protease inhibitor tablet; pH = 8.0), as described previously [28]. The samples were subjected to three freeze–thaw cycles, and sonication with a probe sonicator in three 5-second bursts (Q Sonica #CL-188; amplitude of 30). Samples were then centrifuged at 10,000 × g for 10 minutes at 4°C. Protein concentration was determined by BCA protein assay. Equal amounts of protein were reduced with 5 mmol/L DTT at 37°C for 30 min, and then alkylated with 15 mmol/L iodoacetamide at room temperature for 30 minutes in the dark. Unreacted iodoacetamide was quenched with DTT up to 15 mmol/L. Initial digestion was performed with Lys C (ThermoFisher Cat# 90,307; 1:100 w:w; 2 μg enzyme per 200 μg protein) for 4 hours at 37°C. Following dilution to 1.5 mol/L urea with 40 mmol/L Tris (pH = 8.0), 30 mmol/L NaCl, 1 mmol/L CaCl2, samples were digested overnight with trypsin (Promega; Cat# V5113; 50:1 w/w, protein:enzyme) at 37°C. Samples were acidified to 0.5% TFA and then centrifuged at 4000×g for 10 minutes at 4°C. Supernatant containing soluble peptides was desalted as described previously [28], and then eluate was frozen and lyophilized.
Final peptides were suspended in 0.1% formic acid, quantified (ThermoFisher Cat# 23,275), and then diluted to a final concentration of 0.25 μg/μL. Samples were subjected to nLC-MS/MS analysis using an UltiMate 3000 RSLCnano system (ThermoFisher) coupled to a Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer (ThermoFisher) via a nanoelectrospray ionization source. For each injection, 4 μL (1 μg) of sample was first trapped on an Acclaim PepMap 100 20 mm × 0.075 mm trapping column (ThermoFisher Cat# 164,535; 5 μL/min at 98/2 v/v water/acetonitrile with 0.1% formic acid). Analytical separation was then performed over a 95 minutes gradient (flow rate of 250 nL/min) of 4%–25% acetonitrile using a 2 μm EASY-Spray PepMap RSLC C18 75 μm × 250 mm column (ThermoFisher Cat# ES802A) with a column temperature of 45°C. MS1 was performed at 70,000 resolution, with an AGC target of 3 × 106 ions and a maximum injection time (IT) of 100 ms. MS2 spectra were collected by data-dependent acquisition (DDA) of the top 15 most abundant precursor ions with a charge greater than 1 per MS1 scan, with dynamic exclusion enabled for 20 s. Precursor ions isolation window was 1.5 m/z and normalized collision energy was 27. MS2 scans were performed at 17,500 resolution, maximum IT of 50 ms, and AGC target of 1 × 105 ions.
Proteome Discoverer 2.2 (PDv2.2) was used for raw data analysis, with default search parameters including oxidation (15.995 Da on M) as a variable modification and carbamidomethyl (57.021 Da on C) as a fixed modification. Data were searched against the Uniprot Mus musculus reference proteome (Proteome ID: UP 000000589), as well as the mouse Mito Carta 2.0 database [51]. PSMs were filtered to a 1% FDR and grouped to unique peptides while maintaining a 1% FDR at the peptide level. Peptides were grouped to proteins using the rules of strict parsimony and proteins were filtered to 1% FDR. Peptide quantification was done using the MS1 precursor intensity. Imputation was performed via low abundance resampling. Using only high confidence master proteins, mitochondrial enrichment factor (MEF) was determined by comparing mitochondrial protein abundance (i.e., proteins identified to be mitochondrial by cross-reference with the MitoCarta 2.0 database) to total protein abundance. A total of 644 mitochondrial proteins were detected out of 1158 proteins in the MitoCarta. CL synthesis enzymes were not detected by the proteomic analyses likely due to their low abundance.
Mitochondrial lipid mass spectrometry
Lipids were extracted from mitochondria enriched fractions as previously described [31]. A mixture of cold methyl-tert-butyl ether, methanol, and internal standards (SPLASH Mix Avanti Polar Lipids 330707 and Cardiolipin Mix I Avanti Polar Lipids LM6003) was added to 125 μg of mitochondria protein. The samples were vortexed and then sonicated for 1 minutes. The samples were then incubated on ice for 15 minutes and vortexed every 5 minutes. Phase separation was induced by adding 300 μL of H2O and centrifuging at 15,000 × g, and the organic phase was then dried using a SpeedVac. The dried lipids were reconstituted in a 9:1 methanol:toluene mixture. Untargeted mass spectrometry was performed (Agilent 6530 UHPLC-QTOF mass spectrometer) and analyzed in negative (lyso-PC, lyso-PE, PC, PE, PS, PI, and PG) or positive (CL) modes. Lipid content reported was normalized to mitochondrial protein content. Quality control was performed for each sample by first analyzing MS spectrum for reproducibility and excluding samples that did not achieve less than 30% of coefficient variation.
Native PAGE
Mitochondria supercomplex analysis was performed as previously described [31]. Isolated mitochondria (100 μg) suspended in MIM were pelleted at 12,000 × g for 15 minutes and subsequently solubilized in 20 μL sample buffer (4% digitonin, 1x native PAGE sample buffer) for 20 minutes on ice, and then centrifuged at 20,000 × g for 30 minutes at 4°C. 15 μL of the supernatant (75 μg) was collected and placed into a new tube and mixed with 2 μL G-250 sample buffer additive. The samples and standards were then loaded onto a native PAGE 3% to 12% Bis-Tris Gel (BN1001BOX, Thermo Fisher Scientific), and electrophoresis was performed at 150 V for 3 hours on ice. The gel was then placed in de-stain solution (20% methanol and 10% acetic acid) then shaken on an orbital shaker for 10 minutes at room temperature. De-stain solution was discarded and fresh de-stain solution was added before shaking for 60 minutes at room temperature. After incubation, the gel was placed in de-staining solution and incubated overnight at 4°C on an orbital shaker.
Cell culture
C2C12 myoblasts were grown [high glucose DMEM + 10% fetal bovine serum (FBS) + 100 μg/mL of penicillin/streptomycin] and differentiated into myotubes [low glucose DMEM (1 g/L glucose, L-glutamine, 110 mg/L sodium pyruvate) + 2% horse serum + 100 μg/mL of penicillin/streptomycin]. HEK 293T cells were maintained in high glucose DMEM + 10% FBS + 100 μg/mL of penicillin/streptomycin. Lentivirus-mediated knockdown of PSD was performed as previously described [31]. Vectors were sourced from Sigma (St. Louis, MO) for shRNA for mouse PISD (shPSD: TRCN0000115415), and Addgene (Cambridge, MA) for psPAX2 (ID #12260), pMD2.G (ID #12259), and scrambled shRNA plasmid (SC: ID #1864).
Quantitative PCR
Samples were homogenized in TRIzol reagent (Life Technologies) to extract total RNA. One microgram RNA was reverse-transcribed using an IScript cDNA synthesis kit (Bio-Rad). Reverse transcription PCR (RT-PCR) was performed with the Viia 7 Real-Time PCR System (Life Technologies) using SYBR Green reagent (Life Technologies). All data were normalized to ribosomal L32 gene expression and were normalized to the mean of the control group. Primers were based on sequences in public databases.
Statistics
Statistical analysis was performed using GraphPad Prism 9 software. One-way ANOVA with multiple comparisons, two-way ANOVA with Sidak multiple comparisons, Pearson correlation analyses, or unpaired t-test were performed for group comparisons. Where multiple comparisons were made, P-values between OB and WL groups or WT and TAZKD groups are shown in the figures (see legends). Simple linear regressions were performed for correlation analysis. All data are represented as mean ± SEM and statistical significance was set at P ≤ 0.05.
Supplementary Material
Acknowledgements
This research is supported by NIH DK107397, DK127979, GM144613, AG074535, AG067186 (to K.F.), AG065993 (to A.C.), DK091317 (to M.J.L.), Department of Defense W81XWH-19-1-0213 (to K.H.F-W), American Heart Association 18PRE33960491 (to A.R.P.V.), 19PRE34380991 (to J.M.J.), and 915674 (P.S.), Larry H. & Gail Miller Family Foundation (to P.J.F.). University of Utah Metabolomics Core Facility is supported by S10 OD016232, S10 OD021505, and U54 DK110858.
Footnotes
Conflict of interest
The authors declare that no conflict of interest exists.
Ethics approval
All procedures were approved by the University of Utah Institutional Animal Care and Use Committee.
Supplementary data
Supplementary material is available at Life Metabolism online.
Data availability
All study data are included in the article and/or the supplementary data.
References
- 1.Weinsier RL, Nelson KM, Hensrud DD, Darnell BE, Hunter GR, and Schutz Y (1995) Metabolic predictors of obesity. Contribution of resting energy expenditure, thermic effect of food, and fuel utilization to four-year weight gain of post-obese and never-obese women. J Clin Invest 95, 980–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall KD, and Kahan S (2018) Maintenance of Lost Weight and Long-Term Management of Obesity. Med Clin North Am 102, 183–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fothergill E, Guo J, Howard L, Kerns JC, Knuth ND, Brychta R, Chen KY, Skarulis MC, Walter M, Walter PJ, and Hall KD (2016) Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity (Silver Spring) 24, 1612–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Apfelbaum M, Bostsarron J, and Lacatis D (1971) Effect of caloric restriction and excessive caloric intake on energy expenditure. Am J Clin Nutr 24, 1405–1409 [DOI] [PubMed] [Google Scholar]
- 5.Bray GA (1969) Effect of caloric restriction on energy expenditure in obese patients. Lancet 2, 397–398 [DOI] [PubMed] [Google Scholar]
- 6.Bessard T, Schutz Y, and Jequier E (1983) Energy expenditure and postprandial thermogenesis in obese women before and after weight loss. Am J Clin Nutr 38, 680–693 [DOI] [PubMed] [Google Scholar]
- 7.Astrup A, Gotzsche PC, van de Werken K, Ranneries C, Toubro S, Raben A, and Buemann B (1999) Meta-analysis of resting metabolic rate in formerly obese subjects. Am J Clin Nutr 69, 1117–1122 [DOI] [PubMed] [Google Scholar]
- 8.Ravussin Y, Gutman R, Diano S, Shanabrough M, Borok E, Sarman B, Lehmann A, LeDuc CA, Rosenbaum M, Horvath TL, and Leibel RL (2011) Effects of chronic weight perturbation on energy homeostasis and brain structure in mice. Am J Physiol Regul Integr Comp Physiol 300, R1352–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levin BE, and Keesey RE (1998) Defense of differing body weight set points in diet-induced obese and resistant rats. Am J Physiol 274, R412–419 [DOI] [PubMed] [Google Scholar]
- 10.Ravussin Y, Edwin E, Gallop M, Xu L, Bartolome A, Kraakman MJ, LeDuc CA, and Ferrante AW Jr. (2018) Evidence for a Non-leptin System that Defends against Weight Gain in Overfeeding. Cell Metab 28, 289–299 e285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joyner MJ, and Coyle EF (2008) Endurance exercise performance: the physiology of champions. J Physiol 586, 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whipp BJ, and Wasserman K (1969) Efficiency of muscular work. J Appl Physiol 26, 644–648 [DOI] [PubMed] [Google Scholar]
- 13.Zoladz JA, Koziel A, Woyda-Ploszczyca A, Celichowski J, and Jarmuszkiewicz W (2016) Endurance training increases the efficiency of rat skeletal muscle mitochondria. Pflugers Arch 468, 1709–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murray AJ, and Horscroft JA (2016) Mitochondrial function at extreme high altitude. J Physiol 594, 1137–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leibel RL, Rosenbaum M, and Hirsch J (1995) Changes in energy expenditure resulting from altered body weight. N Engl J Med 332, 621–628 [DOI] [PubMed] [Google Scholar]
- 16.Levine JA, Eberhardt NL, and Jensen MD (1999) Role of nonexercise activity thermogenesis in resistance to fat gain in humans. Science 283, 212–214 [DOI] [PubMed] [Google Scholar]
- 17.Rosenbaum M, Vandenborne K, Goldsmith R, Simoneau JA, Heymsfield S, Joanisse DR, Hirsch J, Murphy E, Matthews D, Segal KR, and Leibel RL (2003) Effects of experimental weight perturbation on skeletal muscle work efficiency in human subjects. Am J Physiol Regul Integr Comp Physiol 285, R183–192 [DOI] [PubMed] [Google Scholar]
- 18.Goldsmith R, Joanisse DR, Gallagher D, Pavlovich K, Shamoon E, Leibel RL, and Rosenbaum M (2010) Effects of experimental weight perturbation on skeletal muscle work efficiency, fuel utilization, and biochemistry in human subjects. Am J Physiol Regul Integr Comp Physiol 298, R79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baldwin KM, Joanisse DR, Haddad F, Goldsmith RL, Gallagher D, Pavlovich KH, Shamoon EL, Leibel RL, and Rosenbaum M (2011) Effects of weight loss and leptin on skeletal muscle in human subjects. Am J Physiol Regul Integr Comp Physiol 301, R1259–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coen PM, Menshikova EV, Distefano G, Zheng D, Tanner CJ, Standley RA, Helbling NL, Dubis GS, Ritov VB, Xie H, Desimone ME, Smith SR, Stefanovic-Racic M, Toledo FG, Houmard JA, and Goodpaster BH (2015) Exercise and Weight Loss Improve Muscle Mitochondrial Respiration, Lipid Partitioning, and Insulin Sensitivity After Gastric Bypass Surgery. Diabetes 64, 3737–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabol R, Svendsen PF, Skovbro M, Boushel R, Haugaard SB, Schjerling P, Schrauwen P, Hesselink MK, Nilas L, Madsbad S, and Dela F (2009) Reduced skeletal muscle mitochondrial respiration and improved glucose metabolism in nondiabetic obese women during a very low calorie dietary intervention leading to rapid weight loss. Metabolism 58, 1145–1152 [DOI] [PubMed] [Google Scholar]
- 22.Menshikova EV, Ritov VB, Dube JJ, Amati F, Stefanovic-Racic M, Toledo FGS, Coen PM, and Goodpaster BH (2017) Calorie Restriction-induced Weight Loss and Exercise Have Differential Effects on Skeletal Muscle Mitochondria Despite Similar Effects on Insulin Sensitivity. J Gerontol A Biol Sci Med Sci 73, 81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toledo FG, Menshikova EV, Azuma K, Radikova Z, Kelley CA, Ritov VB, and Kelley DE (2008) Mitochondrial capacity in skeletal muscle is not stimulated by weight loss despite increases in insulin action and decreases in intramyocellular lipid content. Diabetes 57, 987–994 [DOI] [PubMed] [Google Scholar]
- 24.Virtue S, Even P, and Vidal-Puig A (2012) Below thermoneutrality, changes in activity do not drive changes in total daily energy expenditure between groups of mice. Cell Metab 16, 665–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chouchani ET, Kazak L, and Spiegelman BM (2019) New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab 29, 27–37 [DOI] [PubMed] [Google Scholar]
- 26.Verkerke ARP, Ferrara PJ, Lin C-T, Johnson JM, Ryan TE, Maschek JA, Eshima H, Paran CW, Laing BT, Siripoksup P, Tippetts TS, Wentzler EJ, Huang H, Spangenburg EE, Brault JJ, Villanueva CJ, Summers SA, Holland WL, Cox JE, Vance DE, Neufer PD, and Funai K (2019) Phospholipid methylation regulates muscle metabolic rate through Ca2+ transport efficiency. Nature Metabolism 1, 876–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paran CW, Zou K, Ferrara PJ, Song H, Turk J, and Funai K (2015) Lipogenesis mitigates dysregulated sarcoplasmic reticulum calcium uptake in muscular dystrophy. Biochim Biophys Acta 1851, 1530–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McLaughlin KL, Kew KA, McClung JM, and Fisher-Wellman KH (2020) Subcellular proteomics combined with bioenergetic phenotyping reveals protein biomarkers of respiratory insufficiency in the setting of proofreading-deficient mitochondrial polymerase. Sci Rep 10, 3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heden TD, Neufer PD, and Funai K (2016) Looking Beyond Structure: Membrane Phospholipids of Skeletal Muscle Mitochondria. Trends Endocrinol Metab 27, 553–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Funai K, Summers SA, and Rutter J (2020) Reign in the membrane: How common lipids govern mitochondrial function. Curr Opin Cell Biol 63, 162–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heden TD, Johnson JM, Ferrara PJ, Eshima H, Verkerke ARP, Wentzler EJ, Siripoksup P, Narowski TM, Coleman CB, Lin CT, Ryan TE, Reidy PT, de Castro Bras LE, Karner CM, Burant CF, Maschek JA, Cox JE, Mashek DG, Kardon G, Boudina S, Zeczycki TN, Rutter J, Shaikh SR, Vance JE, Drummond MJ, Neufer PD, and Funai K (2019) Mitochondrial PE potentiates respiratory enzymes to amplify skeletal muscle aerobic capacity. Science advances 5, eaax8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pennington ER, Funai K, Brown DA, and Shaikh SR (2019) The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim Biophys Acta Mol Cell Biol Lipids 1864, 1039–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang M, Mileykovskaya E, and Dowhan W (2002) Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 277, 43553–43556 [DOI] [PubMed] [Google Scholar]
- 34.Johnson JM, Ferrara PJ, Verkerke ARP, Coleman CB, Wentzler EJ, Neufer PD, Kew KA, de Castro Bras LE, and Funai K (2018) Targeted overexpression of catalase to mitochondria does not prevent cardioskeletal myopathy in Barth syndrome. Journal of molecular and cellular cardiology 121, 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prola A, Blondelle J, Vandestienne A, Piquereau J, Denis RGP, Guyot S, Chauvin H, Mourier A, Maurer M, Henry C, Khadhraoui N, Gallerne C, Molinie T, Courtin G, Guillaud L, Gressette M, Solgadi A, Dumont F, Castel J, Ternacle J, Demarquoy J, Malgoyre A, Koulmann N, Derumeaux G, Giraud MF, Joubert F, Veksler V, Luquet S, Relaix F, Tiret L, and Pilot-Storck F (2021) Cardiolipin content controls mitochondrial coupling and energetic efficiency in muscle. Science advances 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cole LK, Mejia EM, Vandel M, Sparagna GC, Claypool SM, Dyck-Chan L, Klein J, and Hatch GM (2016) Impaired Cardiolipin Biosynthesis Prevents Hepatic Steatosis and Diet-Induced Obesity. Diabetes 65, 3289–3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soustek MS, Falk DJ, Mah CS, Toth MJ, Schlame M, Lewin AS, and Byrne BJ (2011) Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum Gene Ther 22, 865–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersen P, and Saltin B (1985) Maximal perfusion of skeletal muscle in man. J Physiol 366, 233–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Funai K, and Semenkovich CF (2011) Skeletal muscle lipid flux: running water carries no poison. Am J Physiol Endocrinol Metab 301, E245–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zurlo F, Larson K, Bogardus C, and Ravussin E (1990) Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Invest 86, 1423–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B, Houtkooper RH, and Auwerx J (2015) Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep 10, 1681–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mogensen M, Bagger M, Pedersen PK, Fernstrom M, and Sahlin K (2006) Cycling efficiency in humans is related to low UCP3 content and to type I fibres but not to mitochondrial efficiency. J Physiol 571, 669–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chicco AJ, Le CH, Gnaiger E, Dreyer HC, Muyskens JB, D’Alessandro A, Nemkov T, Hocker AD, Prenni JE, Wolfe LM, Sindt NM, Lovering AT, Subudhi AW, and Roach RC (2018) Adaptive remodeling of skeletal muscle energy metabolism in high-altitude hypoxia: Lessons from AltitudeOmics. J Biol Chem 293, 6659–6671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acehan D, Vaz F, Houtkooper RH, James J, Moore V, Tokunaga C, Kulik W, Wansapura J, Toth MJ, Strauss A, and Khuchua Z (2011) Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem 286, 899–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Funai K, Song H, Yin L, Lodhi IJ, Wei X, Yoshino J, Coleman T, and Semenkovich CF (2013) Muscle lipogenesis balances insulin sensitivity and strength through calcium signaling. J Clin Invest 123, 1229–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lark DS, Torres MJ, Lin CT, Ryan TE, Anderson EJ, and Neufer PD (2016) Direct real-time quantification of mitochondrial oxidative phosphorylation efficiency in permeabilized skeletal muscle myofibers. Am J Physiol Cell Physiol 311, C239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bers DM, Patton CW, and Nuccitelli R (2010) A practical guide to the preparation of Ca(2+) buffers. Methods Cell Biol 99, 1–26 [DOI] [PubMed] [Google Scholar]
- 48.Simonides WS, and van Hardeveld C (1990) An assay for sarcoplasmic reticulum Ca2(+)-ATPase activity in muscle homogenates. Anal Biochem 191, 321–331 [DOI] [PubMed] [Google Scholar]
- 49.Heden TD, Ryan TE, Ferrara PJ, Hickner RC, Brophy PM, Neufer PD, McClung JM, and Funai K (2017) Greater Oxidative Capacity in Primary Myotubes from Endurance-trained Women. Med Sci Sports Exerc 49, 2151–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McLaughlin KL, Hagen JT, Coalson HS, Nelson MAM, Kew KA, Wooten AR, and Fisher-Wellman KH (2020) Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci Rep 10, 17599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calvo SE, Clauser KR, and Mootha VK (2016) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data are included in the article and/or the supplementary data.