

Abstract
The nuclear factor κB (NF-κB) system is critical for various biological functions in numerous cell types, including the inflammatory response, cell proliferation, survival, and differentiation, and pathogenic responses. Each cell type is characterized by a subset of 15 NF-κB dimers whose activity is regulated in a stimulus-responsive manner. Numerous studies have produced different mathematical models that account for cell type–specific NF-κB activities. However, whereas the concentrations or abundances of NF-κB subunits may differ between cell types, the biochemical interactions that constitute the NF-κB signaling system do not. Here, we synthesized a consensus mathematical model of the NF-κB multidimer system, which could account for the cell type–specific repertoires of NF-κB dimers and their cell type–specific activation and crosstalk. Our review demonstrates that these distinct cell type–specific properties of NF-κB signaling can be explained largely as emergent effects of the cell type–specific expression of NF-κB monomers. The consensus systems model represents a knowledge base that may be used to gain insights into the control and function of NF-κB in diverse physiological and pathological scenarios and that describes a path for generating similar regulatory knowledge bases for other pleiotropic signaling systems.
Introduction
A key goal of systems biology is to represent molecular mechanistic knowledge that is informed by a plethora of experimental observations in mathematical models so that it can be used for predictive simulations. Indeed, constructing mathematical models involves summarizing accumulated knowledge into wiring diagrams and encoding these in equations whose constants are specified by biophysical, biochemical, or cell biological measurements. As such, models may function as knowledge bases of molecular mechanisms. Common model repositories [for example, Biomodels (1)] are fueled by the vision that such mathematically encoded knowledge bases will drive quantitative systems biology studies broadly. Indeed, models of metabolic networks in bacteria, yeast, or humans have enhanced research of microbial growth and metabolic control (2, 3). Yet, in the fields of signaling, gene expression, and regulatory biology, the adoption and application of models produced by one laboratory by other researchers has been disappointingly rare.
The reliability of a model as a knowledge base is evaluated by its ability to recapitulate experimental observations and make accurate predictions that can be experimentally confirmed. However, models are necessarily an abstraction of the true molecular regulatory network. One consequence is that an abstracted model built to explore a functional characteristic in one cell type may not capture the functional characteristics observed in another cell type. To capture diverse biological phenomena in different cell types, we suggest that the mathematical model must be sufficiently detailed that the genetically encoded biochemical mechanisms are represented explicitly, such that nongenetic, cell-type differences may be accounted for by altering merely expression or synthesis (not interaction or catalytic) rate constants based on readily accessible mRNA or protein abundance measurements.
The NF-κB transcription factor family plays a critical role in the regulation of inflammation, immunity, cell development, cell survival, and proliferation. The dynamic control of the ubiquitous NF-κB family member RelA has been the subject of numerous mathematical models, and these have been reviewed previously (4, 5). However, much NF-κB–associated biology and pathology is mediated by other NF-κB family members, such as RelB, cRel, p50, and p52, which, together with RelA, may potentially form 15 distinct dimers whose activities are controlled by two stimulus-responsive signaling pathways (6, 7). Several studies involving models of the multidimer NF-κB system have explored diverse biological scenarios, ranging from B cells (8–10) to dendritic cells (DCs) (11) and fibroblasts (11–13). It remains unclear whether the knowledge base of molecular mechanisms encoded by these models of the multidimer NF-κB system may by represented by a single consensus model that may then be applied to diverse physiological and pathological contexts.
Here, we reviewed the accumulated molecular mechanistic knowledge contained in published mathematical models of the NF-κB multidimer system to produce a consensus model. Our analysis and synthesis were guided by the following principles: (i) that biochemical interactions mediated by protein surfaces are encoded by the genome and hence are conserved between cell types, and (ii) that the abundances of NF-κB monomers are a function of the epigenome and hence are cell type–specific. We thereby showed that the NF-κB multi-dimer system consensus model is a knowledge base that may be used to explore NF-κB control and function in diverse physiological and pathological contexts.
Components and topology of the NF-κB signaling network in different cell types
Canonical and noncanonical NF-κB signaling
Two NF-κB activation pathways have been described: the canonical, NEMO-dependent pathway and the noncanonical, NF-κB-inducing kinase (NIK)–dependent pathway (14) (Fig. 1). Generally, pathogen and inflammatory signals activate the canonical pathway, whereas developmental signals activate the noncanonical pathway (15). The canonical activation of NF-κB in response to immune threats plays a critical role in acute and reversible immune and inflammatory responses, whereas noncanonical NF-κB activation by developmental signals may promote long-lasting or irreversible cell survival, cell differentiation, or organogenesis. The canonical NF-κB pathway can reach its peak response to inflammatory stimuli within 30 min, whereas the noncanonical pathway responding to pro-survival stimuli can remain active beyond 12 hours (15–17).
Fig. 1. Two signaling pathways are mediated by a single NF-κB network.
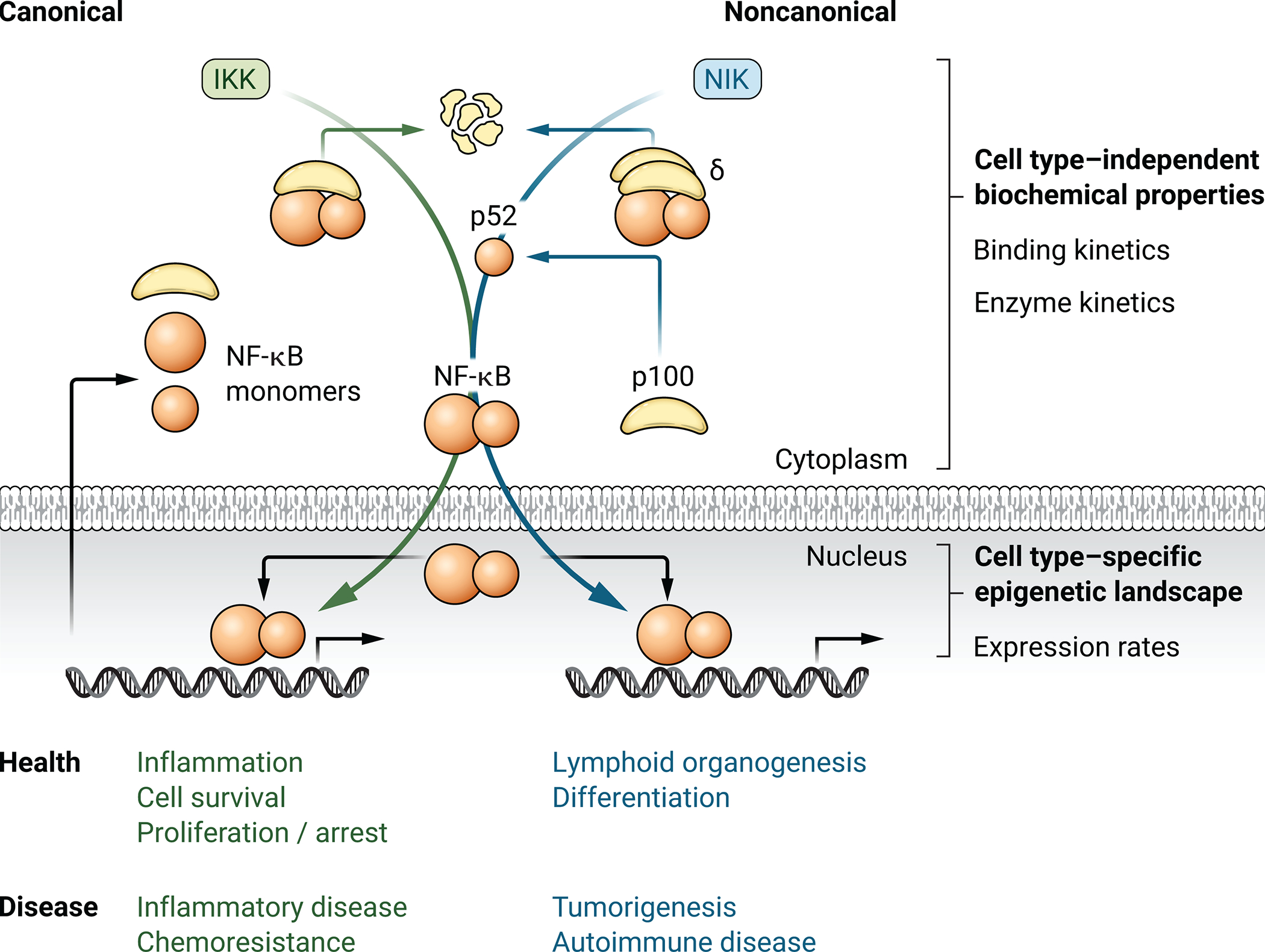
Schematic illustration of the canonical and noncanonical NF-κB activation pathways that regulate the indicated cellular functions. These functions are broadly characterized as transient and reversible immune responses and longer lasting immune development, respectively. Whereas the regulatory architecture of the signaling networks is conserved between cell types, cell types differ in which NF-κB dimers they generate and activate in response to a given stimulus. The cell type–specific dimer repertoire is, at least in part, a function of the cell type–specific abundances of NF-κB monomers. Dysregulation of the network in any of the many cells it operates in may result in diverse pathologies, including cancer, inflammatory disease, and autoimmune disease.
The molecular mechanism of canonical pathway activation by Toll-like receptors (TLRs), tumor necrosis factor receptors (TNFRs), B cell receptors (BCRs) and T cell receptors (TCRs), involves activation of the NF-κB essential modulator (NEMO)-IKK complex, which phosphorylates inhibitory NF-κB proteins (IκBs) and targets them for proteasomal degradation (18). Upon IκB degradation, the NF-κB dimers that were associated with them, such as RelA:p50, RelA:RelA, cRel:p50, RelA:cRel, and RelB:p50, are released and translocate to the nucleus to activate target gene transcription.
Activation of the noncanonical NF-κB pathway occurs after the ligation of receptors such as LTβR, CD40, BAFFR, RANK, TNFR2, or CD27. Upon receptor ligation, proteasomal degradation of the E3 ubiquitin ligase TRAF3 results in the accumulation of NIK (19), and hence activation of the IKK1-containing, NEMO-independent IKK complex. Once activated, IKK1 phosphorylates p100 at specific C-terminal serine residues, promoting ubiquitylation and proteasomal degradation of its C-terminal inhibitory domain (20). This enables two activities to result from noncanonical NF-κB activation. First, degradation of the C-terminal inhibitory domain enables the release of trapped NF-κB dimers, such as RelA:p50 and RelB:p50. Second, p100 processing leads to the generation of p52, enabling the generation and nuclear translocation of the RelB:p52 dimer (15).
Cell type–specific NF-κB dimer repertoires
Although the canonical and noncanonical signaling pathways exist in many cells, they stimulate different sets of NF-κB dimers in each cell type. A number of previous studies have established the prevalence of specific NF-κB dimers in specific cell types, including resting dendritic cells (DCs), which containing a high abundance of RelB:p50 (11), whereas resting mouse embryonic fibroblasts (MEFs) primarily have RelA:p50 with RelA:RelA homodimers upon activation (21, 22), and both resting and activated B cells have cRel:p50 in addition to RelB:p52 as their predominant NF-κB dimers (23–25). Different cells in the B lineage exhibit differential abundances of NF-κB dimer, with RelA:p50 as a major dimer in pre-B cells, cRel:p50 as a major dimer in mature B cells, and RelB:p52 as a major dimer in plasma cells (8, 26). Upon entering the germinal center (GC), antigen-selected B cells are dependent on cRel and RelB:p52 to traffic from light zones to dark zones. The dynamic switch from cRel-containing to RelA-containing NF-κB dimers controls the switch from proliferative GC B cells to differentiated antibody-secreting B cells (8, 27). A RelA:cRel dimer may also play a role in macrophages, increasing the expression of genes encoding proinflammatory factors, such as TNF (28).
A single NF-κB signaling network
The biochemical interactions that constitute the NF-κB signaling network are conserved across cell types; however, this network is also capable of generating the cell type–specific repertoires of NF-κB dimers and their activation. The NF-κB network consists of three types of biochemical processes (29): (i) molecular mechanisms that control NF-κB dimer generation; (ii) the binding of IκB inhibitors which block NF-κB dimers from activating transcription; and (iii) enzyme-mediated phosphorylation and degradation of inhibitors to release NF-κB dimers so that they can translocate to the nucleus for DNA binding and transcriptional activation. Mathematical modeling of these processes using appropriate binding affinities and enzymatic kinetic rates has recapitulated myriad experimental results and provided insight into previously uncharacterized regulatory interactions and signal transduction in health and disease (5, 6, 8, 14, 30–32).
Review of these diverse experimental studies has produced a self-consistent knowledge base of the interaction network that enables NF-κB signaling in different cell types (fig. S1). This standard signaling network has been mathematically encoded, parameterized, and tested iteratively with experiments in multiple studies and multiple experimental systems. Thus, the interaction network, its network topology, and the biochemical interaction parameters represent a knowledge base that applies generally to mammalian cells. Many biochemical interactions and kinetic rate constants are expected to remain unchanged by cellular context. For example, one would not expect the affinity of IκB for a particular NF-κB dimer to vary between cell types. Where mammalian cell types differ is in their chromatin epigenome, which controls the expression of genes, including those that encode NF-κB monomers. Indeed, analysis of the relative amounts of NF-κB monomers in various cell types shows substantial differences in RelA, RelB, and cRel (fig. S2) (33). This cell type–specific difference in gene expression may propagate through a cell type–independent molecular interaction network to confer cell type–specific responses. We will now review whether cell type–specific cell expression of NF-κB constituents may thus account for the cell type–specific functionality of the NF-κB signaling network.
Predicting the cell type–specific and stimulus-specific activation of NF-κB dimers
Basal cell type–specific NF-κB and IκB repertoires
Combining published measurements of the expression of genes encoding NF-κB monomers in MEFs, B cells, T cells, and DCs (fig. S2) (33) with the standard NF-κB signaling network model, results in cell type–specific NF-κB signaling systems that produce cell type–specific NF-κB dimer repertoires (Fig. 2A; for a description of the modeling methodology, see data file S1). Analyzing the steady-state abundances of NF-κB dimers reached by each cell type–specific model recapitulates many of the cell type–specific NF-κB dimer abundances observed in experimental studies. Consistent with experimental observations, simulated MEFs and T cells almost exclusively contain RelA:p50, whereas in silico, naïve B cells contain both cRel:p50 and RelA:p50, with DCs containing predominantly RelB bound to either p50 or p52 (Fig. 2B) (8, 9, 11, 34). The cell type–specific dimer repertoire is maintained in both the inhibited (latent) NF-κB dimers bound by IκBs and the much smaller proportion of unbound NF-κB proteins under unstimulated, steady-state conditions (Fig. 2, B and C).
Fig. 2. Cell type–specific NF-κB repertoires generated by cell type–specific simulations of the NF-κB network model.
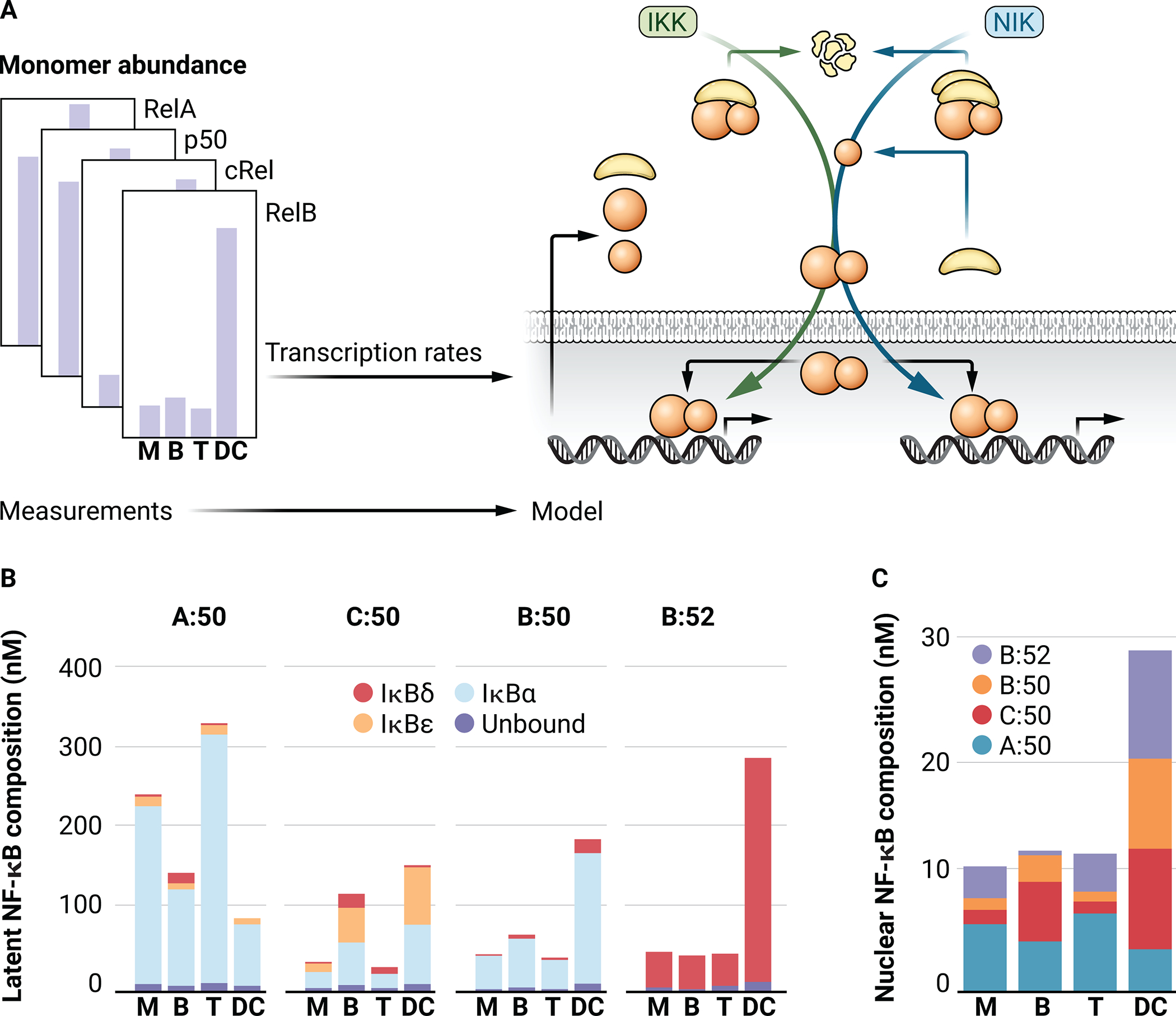
(A) Schematic of the process used to generate cell type–specific models through varying only the abundances of the indicated NF-κB monomers while conserving all reactions and kinetic parameters. M, MEF; B, B cell; T, T cell; DC, dendritic cell. (B) Bar graph showing the predicted abundances of RelA:p50 (A:50), cRel:p50 (C:50), RelB:p50 (B:50), and RelB:p52 (B:52) at steady state in cell type–specific simulations. Colors indicate the inhibitory (IκB) binding partners of each NF-κB dimer. (C) Bar graph showing the NF-κB-dimer composition of unbound NF-κB [dark blue in (B)].
The cell type–specific dimerization repertoire results in a cell type–specific repertoire of inhibitors, to which NF-κB dimers are bound under unstimulated conditions. For p50-containing dimers (RelA:p50, cRel:p50, and RelB:p50), IκBα is the primary inhibitor, which enables the rapid activation [based on its scaffolding function (35)], which is a hallmark of canonical NF-κB activation. IκBε is predicted to play a more important role in B cells and DCs, due to the increased abundance of cRel:p50 in these cells (Fig. 2B). Indeed, IκBε has a functionally important role in B cell proliferation (9). The noncanonical NF-κB dimer RelB:p52 is predicted to be at low abundance in all cell types except for DCs, which exhibit high amounts of Relb and nfkB2 mRNAs and increased NIK stability compared to other cell types (11). RelB:p52 is predicted to be predominantly inhibited by higher molecular-weight complexes of p100 (IκBδ) (Fig. 2, B and C). Cell type–specific NIK stability in DCs may be attributed to the altered abundances of the mediators of NIK degradation, such as TRAF3, which were not explicitly modelled here. Increasing the stability of NIK in MEFs, through reducing TRAF3 abundance, results in “DC-like” NF-κB signaling in MEFs (11). In silico modeling demonstrates substantial differences in NF-κB dimer abundance, IκB inhibitor repertoire, and basal NF-κB activity, as measured in multiple experimental studies, which may largely be an emergent behavior of the cell–type specific basal amounts of NF-κB monomers.
Cell type–specific inducible NF-κB responses
Different cell types exhibit the activation of different NF-κB dimers in response to the same stimuli. For example, RelA:p50 is the near-exclusive dimer in fibroblasts that is activated in response to inflammatory stimuli (22, 36), whereas RelB:p50 is strongly stimulated during DC activation (11). In response to stimuli, cell-type differences in the different NF-κB dimers that are activated may be influenced by several factors, including receptor abundances, kinase activity levels, and the abundances of components of the NF-κB signaling network before stimulation. However, if in a given cell a specific receptor signaling pathway is functional, the NF-κB dimers that are activated through the NF-κB signaling network depend on the cell type–specific basal NF-κB and IκB repertoires. We simulated canonical IKK activation and noncanonical NIK activation using the cell type–specific models of the NF-κB signaling network (Fig. 3A). In response to canonical IKK activation, the NF-κB response in MEFs primarily shows dynamic RelA:p50 nuclear activity, as expected (Fig. 3B). Virtual B cells show a transient peak of activation of all of the canonical NF-κB dimers, which is followed by the increased late-phase activation of RelA:p50 and cRel:p50. This is consistent with studies showing a role for cRel:p50 in controlling B cell proliferation, as well as for RelA:p50 in differentiation (8–10). In simulations of DCs, both RelB:p50 and RelB:p52 play more substantial roles, consistent with studies identifying an important role for RelB in DC activation (11). In response to noncanonical activation through NIK, all cell types respond similarly, with RelB:p52 being stimulated with similar dynamics (Fig. 3C). This indicates that the architecture of the NF-κB signaling network enables relative monomer abundance to tune cell type–specific canonical NF-κB responses, while maintaining reliable and conserved responses to noncanonical, developmental stimuli (Fig. 3, B and C).
Fig 3. NF-κB dimer activation in response to canonical and noncanonical stimuli in the cell type–specific computational models.
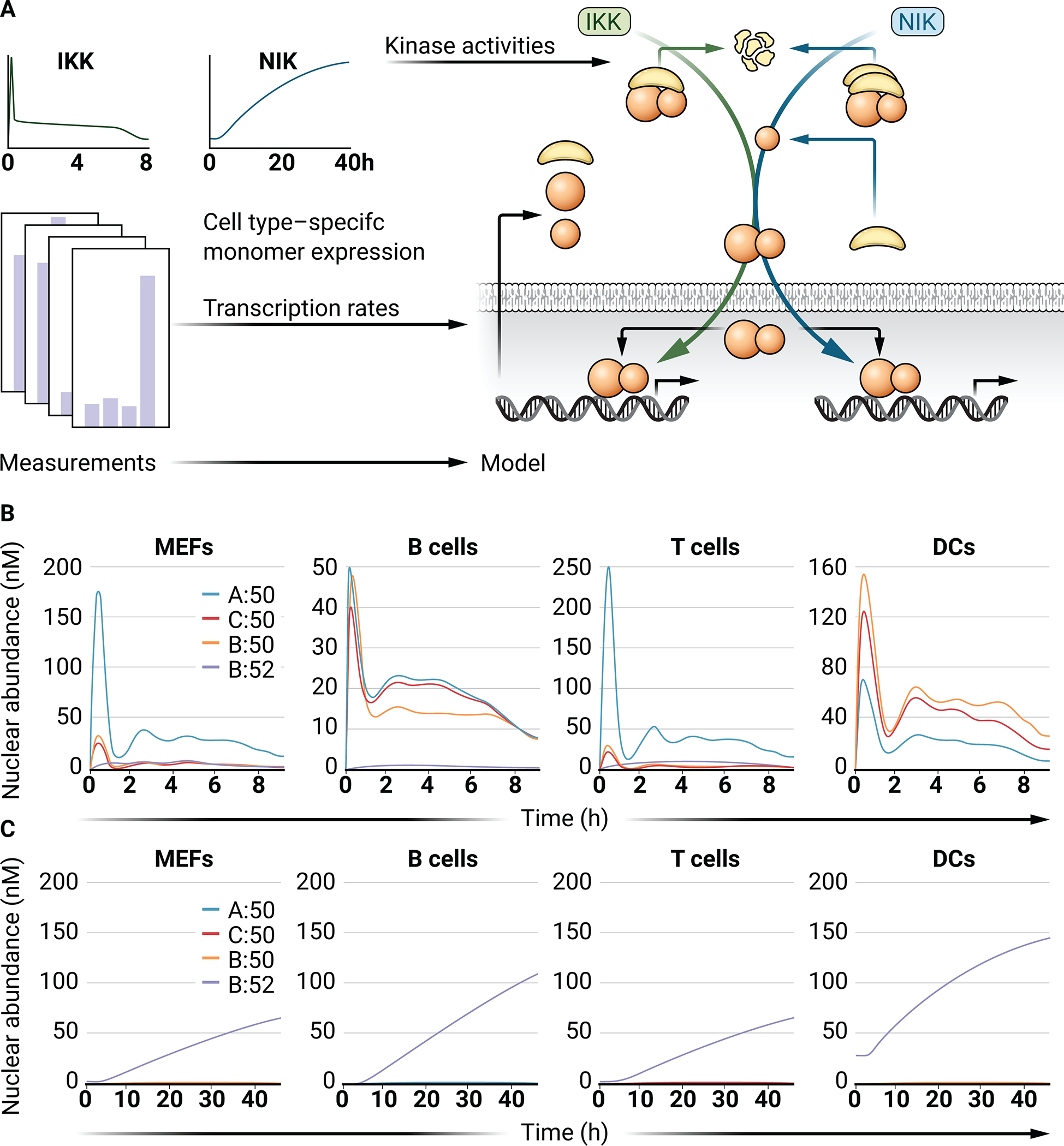
(A) Schematic showing the cell-specific NF-κB monomer abundances that were used to generate cell-specific models that all received the same canonical (IKK-dependent) or noncanonical (NIK-dependent) activity profiles representing activation of canonical and noncanonical pathways. (B) Line graphs of the abundances of the indicated NF-κB dimers in response to the canonical (IKK-dependent) stimuli indicated in (A) in each cell type–specific model. (C) Line graphs of the indicated NF-κB dimer abundances in response to the noncanonical (NIK-dependent) stimuli indicated in (A).
Canonical and noncanonical NF-κB crosstalk
Whereas the two NF-κB pathways are often described as separate, there are numerous interconnections between them. Studies have indicated that higher molecular weight complexes of p100 (IκBδ) and p105 (IκBγ), termed the “IκBsome” complex, are capable of inhibiting RelA:p50 activity (37). Thus, NIK-dependent p100 processing enables RelA:p50 activation after simulation of the noncanonical pathway (38). These studies suggest that chronic inflammatory signals should not give rise to increased RelA activity, because IκBδ controls excess RelA:p50. However, given an environment in which there are tonic developmental signals in addition to chronic inflammatory signals, enhanced and sustained RelA activity may result. Additionally, previous work established the role of canonical NF-κB activity in providing substrates for noncanonical NF-κB activation (6, 39). The synthesis of RelB and p100 is dependent on RelA activity, and reconstitution of RelB in RelA-deficient MEFs restores noncanonical NF-κB activation (6, 39). These studies have implicated p100 as an important mediator between the two activation pathways, with important functions in regulating basal and activated NF-κB. However, overexpression of p100 in RelA-deficient MEFs fails to restore noncanonical RelB activity (6). These studies suggest that whereas RelB activation is regulated by basal RelA activity, p100 synthesis induced by RelA activation is a critical determinant of activation of the noncanonical pathway (6). Despite this substantial potential for crosstalk, competition between the roles of NIK in p52 processing and p100 release limits the effect of canonical signaling on the noncanonical NF-κB pathway in inflammatory environments (40). Pathway insulation may also be mediated by other, receptor-proximal mechanisms (13).
Cell type–specificity of canonical and noncanonical NF-κB crosstalk
In principle, the mechanisms reviewed earlier together with cell type–specific NF-κB monomer expression may lead to distinct crosstalk between the two NF-κB signaling pathways in different cell types. Modeling that encapsulates this knowledge enables exploration of how developmental stimuli affect signaling through the canonical, inflammatory NF-κB pathway. This crosstalk is investigated in multiple cell types by simulating the response to IKK activity (maintained from Fig. 3A) in the context of different basal amounts of NIK (Fig. 4, A and B). Increasing NIK activity amplifies the dynamic response of canonical NF-κB dimers to IKK activation (Fig. 4C). The first peak of the canonical NF-κB dimer response is amplified by increasing NIK activity together with moderate amplification of late-phase activity, particularly for cRel:p50 and RelB:p50 in B cells (Fig. 4C). Simultaneous, noncanonical activation extends cRel responses in B cells (12). The activation of RelB:p52 is predicted to be largely independent of canonical pathway activity across multiple levels of basal NIK activity (Fig. 4C).
Fig. 4. Simulation of canonical (IKK-mediated) pathway activation at different basal levels of NIK activity.
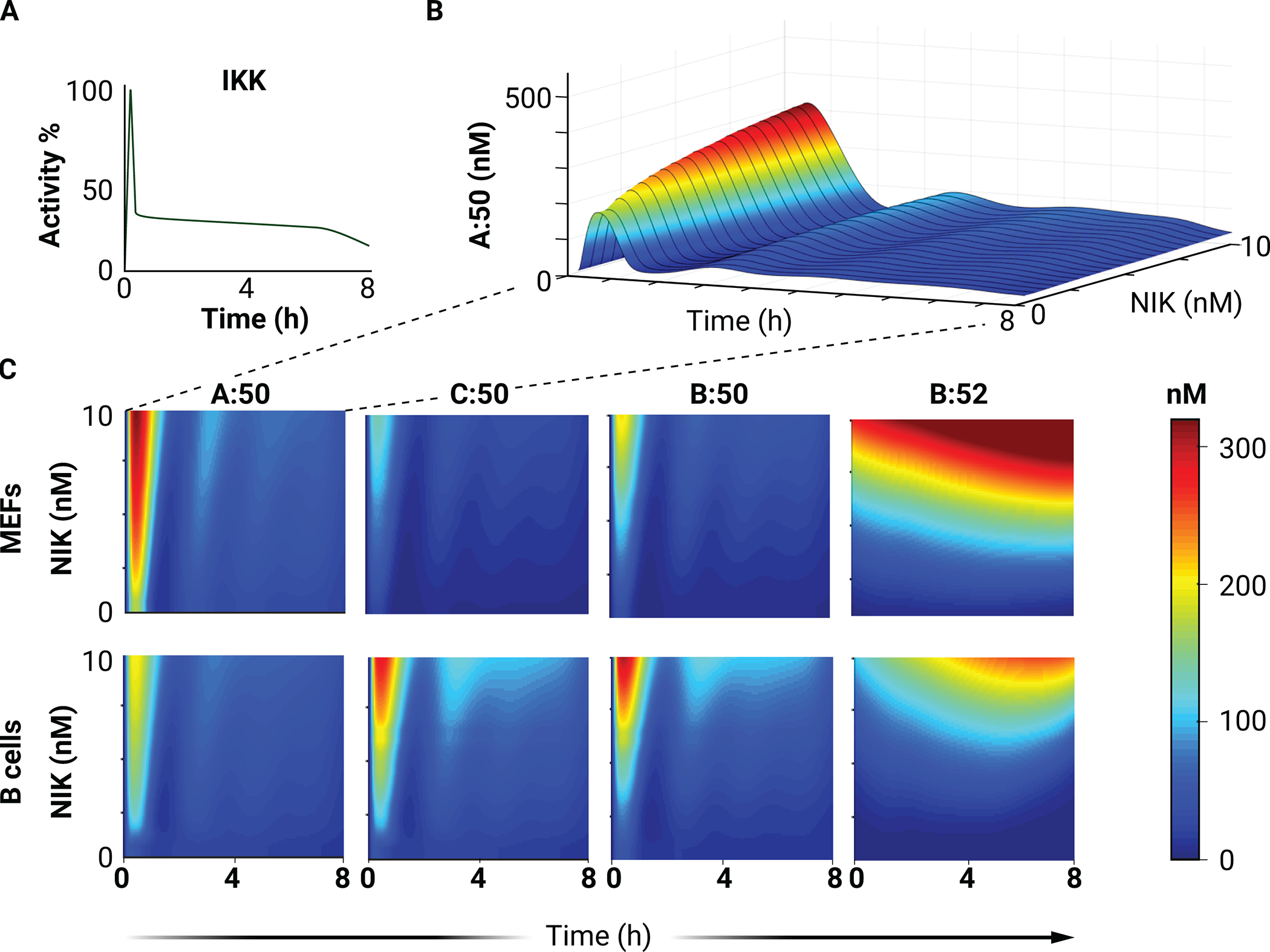
(A) The physiological IKK activity curve used as input for all heatmaps. (B) Surface plot of RelA:p50 (A:50) abundance (A) in simulations with a physiological IKK activity curve and the indicated NIK abundances. (C) Heatmaps of the nuclear concentrations of the indicated NF-κB dimers in simulations with a physiological IKK activity curve and the indicated NIK abundances.
Conversely, simulations with a consistent physiological NIK activation profile (consistent with Fig. 3A) at varying levels of basal IKK activation (Fig. 5A) enable investigation of the crosstalk from inflammatory status on the response of a cell to developmental, noncanonical stimuli. As expected, the predominant dimer stimulated by NIK activation is RelB:p52, and this activation is largely unaffected by canonical pathway activity in both B cells and MEFs (Fig. 5, B and C). An increase in cRel:p50 abundance in response to NIK activation through BAFF is amplified at later times (6, 12, 24, and 40 hours) by the addition of IKK-activating IgM (12), which is recapitulated in our simulations (Fig. 5B). Whereas p52 is thought to primarily bind to RelB, simulations in MEFs with high NEMO activity and increased NIK stability predict the formation of RelA:p52 dimer at amounts comparable to those of RelA:p50 (fig. S3). This is consistent with experiments performed in MEFs, intestinal epithelial cells, and macrophages, showing concurrent activation of canonical and noncanonical signaling (41–43). Indeed, in both MEFs and B cells, the model predicts that the canonical dimers RelA:p50 and cRel:p50 can be moderately activated by noncanonical stimuli in environments with high basal inflammation (high tonic IKK activity) (Fig. 5C; compare the response to increasing NIK activity in the context of 10 vs. 1% IKK activity). Aberrant stimulation of cRel activity is oncogenic; after all, it was named for being the cellular homolog of the oncogene v-Rel, which is encoded by the avian reticuloendotheliosis virus. The predicted activation of cRel in response to noncanonical (NIK-mediated) pathway activation may lead to oncogenic activation of cRel in response to noncanonical signals, such as CD40 receptor-ligand binding on the surface of GC B cells.
Fig. 5. Simulation of noncanonical (NIK-mediated) pathway activation at different basal levels of IKK activity.
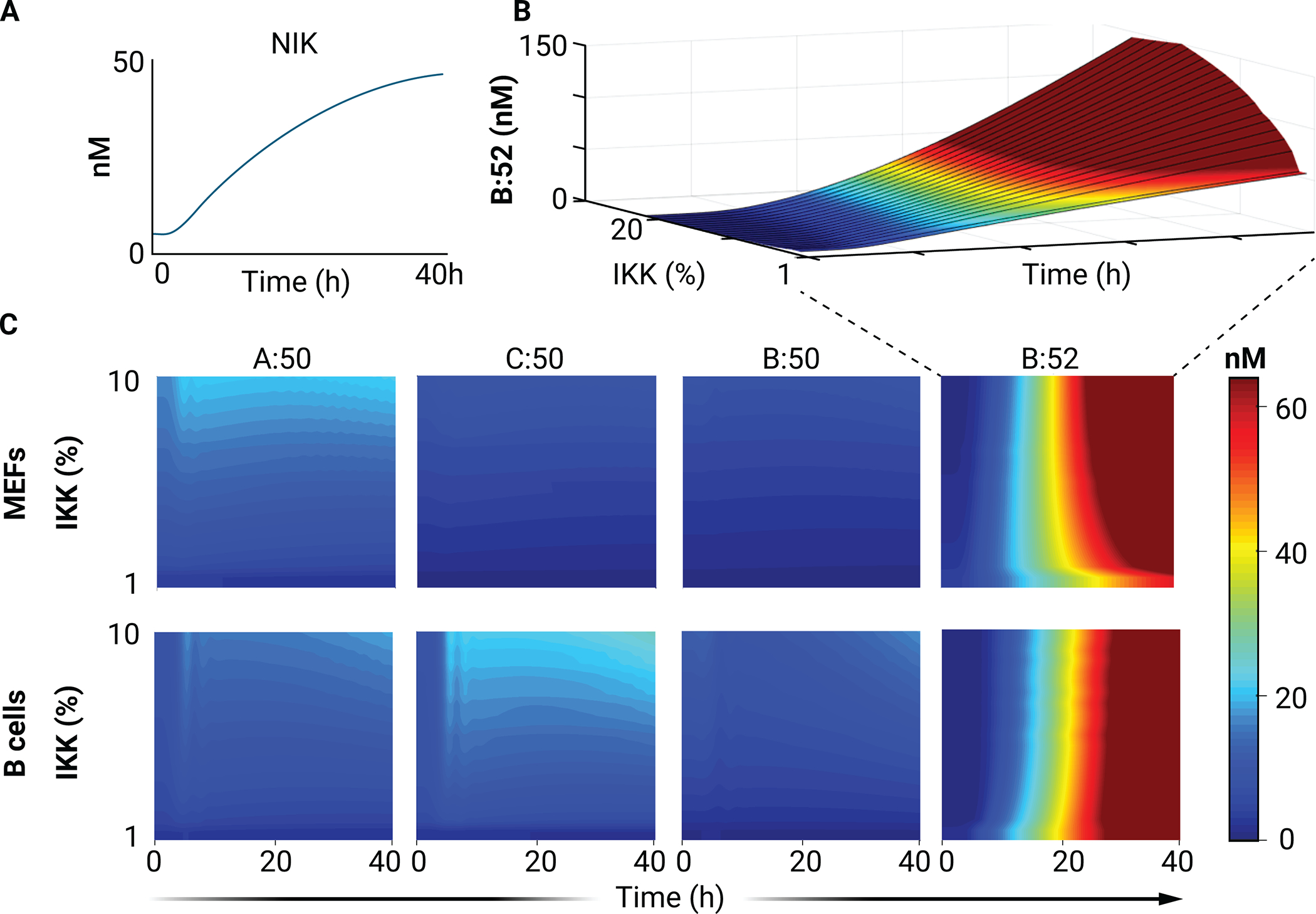
(A) The physiological NIK activity curve used as input for all subsequent heatmaps. (B) Surface plot of RelB:p52 (B:52) abundance in simulations with the physiological NIK activity curve and the indicated basal IKK abundances. (C) Heatmaps of the nuclear concentrations of the indicated NF-κB dimers in simulations with a physiological NIK activity curve with the indicated basal IKK abundances.
Predicting the regulatory consequences of cancer-associated NF-κB mutations
Mutations that affect NF-κB signaling system components are associated with various diseases, including multiple types of cancer, atherosclerosis, lupus, and Hodgkin’s disease (44, 45). Particularly in lymphoid malignancies, it is well established that NF-κB dysregulation is involved (46, 47); however, it is not always clear which of the NF-κB dimers is affected or dysregulated (48, 49).
Hodgkin’s B cell lymphoma
Hodgkin’s disease is one of the most frequent lymphomas and is caused by Hodgkin/Reed-Sternberg (H/RS) cells that are derived from B lymphocytes. Many H/RS cell lines show multiple coexisting mutations, and strong NF-κB activity results from many of these mutations, playing a causal role in the pathogenesis of these cells (50). Some 10 to 20% of classical Hodgkin’s disease cases have a mutation in nfkbia, which encodes IκBα, a primary inhibitor of NF-κB in the canonical pathway (51). Adapting the mathematical B cell model with an IκBα deletion enables examination of the signaling effect of this mutation within the context of the knowledge base of NF-κB signaling. IκBα knockout simulations predict increased amounts of all canonical dimers under basal conditions, which is consistent with canonical pathway activation being consistently observed in H/RS patient samples and cell lines (51). Furthermore, the model predicts a marked increase in nuclear RelB:p50 NF-κB abundance, recapitulating results showing that nuclear RelB:p50 is ubiquitously detected in Hodgkin’s lymphoma (HL) cell lines (Fig. 6A) (52). In addition to basal increases in canonical NF-κB dimer activity in IκBα knockout cells, the model also predicts that the activation of dimers is considerably affected, with RelA:p50 and cRel:p50 being largely nonresponsive to canonical pathway stimulation (Fig. 6A). HL cell lines also show increased nuclear RelB:p52 abundance, which is not predicted by simulations (52). Some of these discrepancies can be attributed to the EBV-positivity of some of the lines tested (for example, L591 cells) because EBV infection mimics CD40 activation through the viral protein LMP1, which activates noncanonical signaling (52, 53). However, even EBV-negative HL cell lines and almost all patient samples show constitutive increases in NIK abundance, and many of these cases can be attributed to the loss of TRAF2 or TRAF3 (negative regulators of NIK) or copy number gains of the gene that encodes NIK (MAP3K14) (54, 55). Although these NIK-activating mechanisms are not considered in our simulations (Fig. 6), the model provides a tool with which to disentangle the predicted effects of the recurrent loss of IκBα from the co-occurring mutations found in many HL cells. These simulations (Fig. 6A) show that the IκBα mutation alone can result in a substantial shift toward RelB-containing NF-κB dimers, even without concurrent NIK activation.
Fig. 6. NF-κB dimer repertoire in simulations of IκBα loss and nfkb2 truncation cancers.
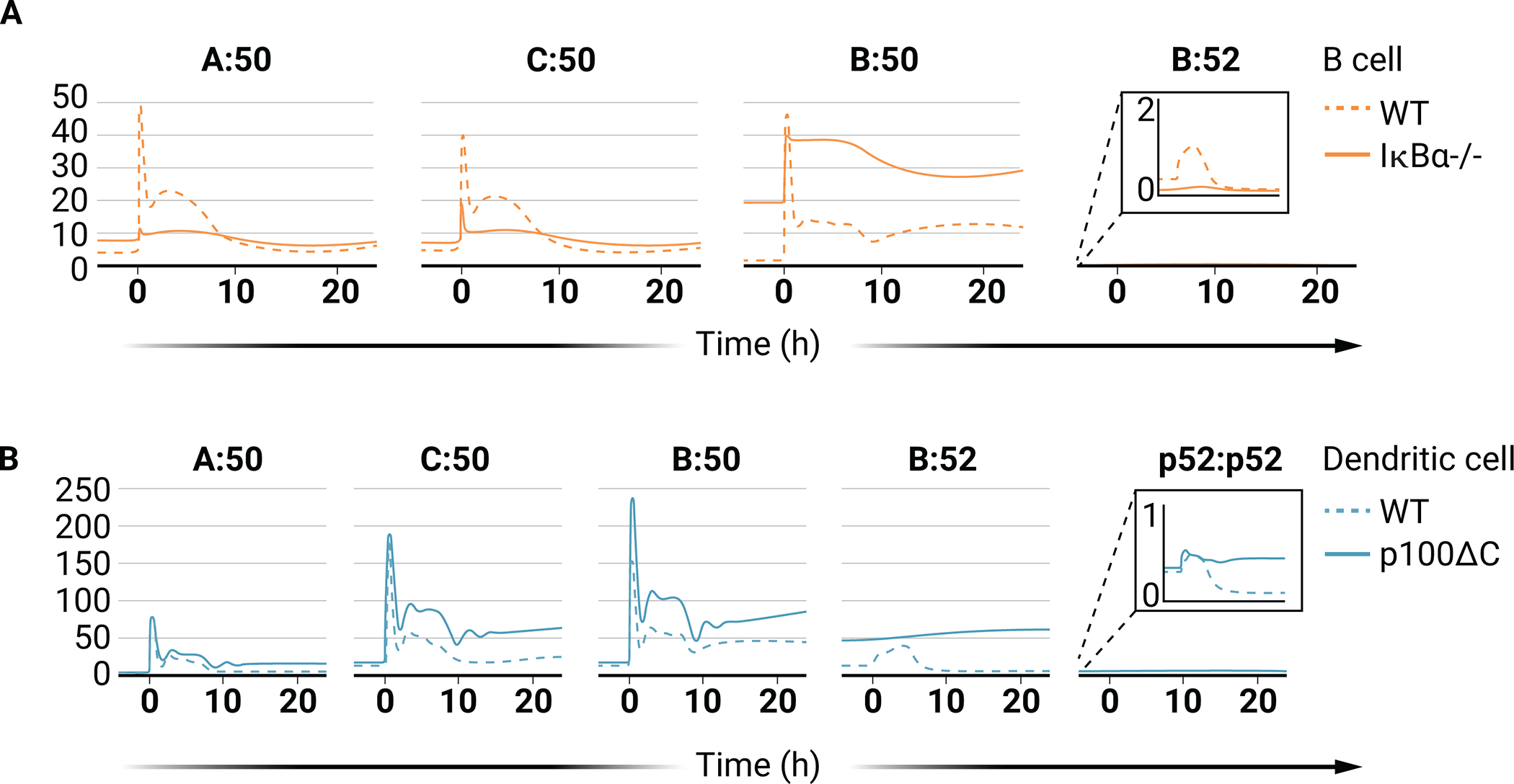
(A) Line graphs of the abundances of the indicated NF-κB dimers in simulations of WT B cells (dashed lines) and IκBα-deficient B cells (solid lines). (B) Line graphs of the abundances of the indicated NF-κB dimers in simulations of WT DCs (dashed lines) and DCs expressing truncated nfkb2 (p100ΔC, solid lines). For (A) and (B), the IKK input curve used is from Fig. 4A and the abundance of NIK is kept constant.
NFkB2-truncation cancers
Various lymphomas and leukemias, such as B cell chronic lymphocytic leukemia, multiple myeloma, and adult T-cell leukemia/lymphoma, have mutations in the nfkb2 gene, which encodes the protein p100 (56–58). Total loss of p100 in both knockout cell lines and multiple myeloma cells prolongs the NF-κB response to stimuli, providing a pro-survival response (30, 59). Mutations in the gene encoding p100 often result in the deletion of its C-terminal, inhibitory ankyrin repeats (60), causing constitutive generation of a p52-like protein that can bind to Rel monomers and translocate to the nucleus (20, 61, 62). Whereas constitutive processing of p100 results in oncogenesis, the mechanisms remain unclear (63). Constitutive generation of p52 due to rearrangements in nfkb2 results in both the increased abundance of p52 and loss of the higher molecular weight inhibitory complexes formed by p100 (IκBδ) (62). Both mechanisms may result in increased and oncogenic NF-κB activity. We can disentangle these two effects by comparing the effect of p100 truncation mutations (resulting in increased amounts of p52 and reduced amounts of IκBδ) with nfkb2 deletion (resulting in loss of both p52 and IκBδ). Nfkb2 knockout mice do not form tumors, suggesting that the generation of increased amounts of p52 resulting from C-terminal mutations is responsible for oncogenesis (64). Because p52 transgenic mice expressing wild-type p100 do not develop tumors (65), these studies suggest that both the loss of IκBδ function and the gain of transcriptional p52 function contribute to the oncogenesis of cells that contain rearrangements in nfkb2. Reports have suggested that increased nuclear RelA:p52 activity results from these genetic events, whereas other reports have suggested the formation of a p52 homodimer that is capable of binding to κB sites in the DNA (62, 66).
The identities of the specific NF-κB dimers that are activated in these tumors because of nfkb2 rearrangement are still unclear. The mathematical model enables exploration of the signaling effect of the tumor-associated nfkb2 mutations in the context of the conserved NF-κB signaling network. The nfkb2 rearrangement is simulated by eliminating IκBδ activity and by removing the NIK dependence of p100 processing to p52, so that the processing is purely constitutive. These simulations predict minimal difference in basal canonical dimers in unstimulated DCs harboring nfkb2 truncation mutants (Fig. 6B). The model also predicts that RelB:p52 is the primary NF-κB dimer that is increased in abundance in unstimulated DCs that have the nfkb2 mutation (Fig. 6B). The absence of IκBδ is predicted to only mildly affect basal amounts of canonical dimers due to compensation by other members of the IκB family. However, in response to a canonical, IKK-activating stimulus, the increased inhibition of NF-κB dimers by IKK-responsive IκBs results in a hyper-responsive induction of the activities of RelA:p50, cRel:p50, and, to a lesser extent, RelB:p50 (Fig. 6B). Encoding available biochemical knowledge into the mathematical model demonstrates that the nfkb2 truncation mutants can result in increases in RelB:p52 nuclear abundance. Simulations of Nfkb2 truncation mutants also predict that this mutation may sensitize the canonical pathway to activation, indicating another crosstalk mechanism by which mutations affecting the noncanonical pathway may lead to oncogenic signaling in the canonical NF-κB pathway.
MYD88-mutated diffuse large B cell lymphoma and Waldenström Macroglobulinemia
In diffuse large B cell lymphoma (DLBCL), nuclear translocation of one or more of the five NF-κB monomers (cRel, RelA, RelB, p50, and p52) has been identified, with each subunit being activated with varying frequency in patient samples (67–72). RelB is activated in about 60% of patient samples, whereas RelA is almost always active (72). Gain-of-function mutations in MYD88 are found in many of these cancers (73–77). The gain-of-function mutation of MYD88 in DLBCLs results in high constitutive NF-κB activity (78). MYD88L265P also occurs in ~90% of Waldenström Macroglobulinemia (WM) cases. MYD88L265P results in activation of the canonical NF-κB pathway and RelA-dependent gene expression (79). However, introducing MYD88L265P into splenic murine B cells does not increase RelA activation (80). Therefore, it is unclear to what extent inducible NF-κB inhibitors may overcome the activating effect of MYD88L265P and which of the specific NF-κB dimers are activated by this mutation.
The scope of the consensus NF-κB system model does not include receptor-proximal interactions that converge on IKK. To explore the effect of MYD88 mutations, we leveraged a previously constructed mathematical model that represents a knowledge base of TLR-proximal signaling mechanisms (Fig. 7A) (81). MYD88L265P results in spontaneous activation of downstream signaling (82), and analogous model simulations show increased TRAF6 activity, as was seen experimentally (Fig. 7A) (82). By taking the predicted increase in IKK activity with MYD88L265P as an input to the NF-κB signaling networks, the change in the basal and inducible NF-κB dimer repertoire is predicted. This increased IKK activity is combined with the different extents of NIK activity that may be the result of the tumor microenvironment, such as interactions with T cells or secreted cytokines (Fig. 7B).
Fig. 7. NF-κB dimer repertoire in simulations of DLBCL.
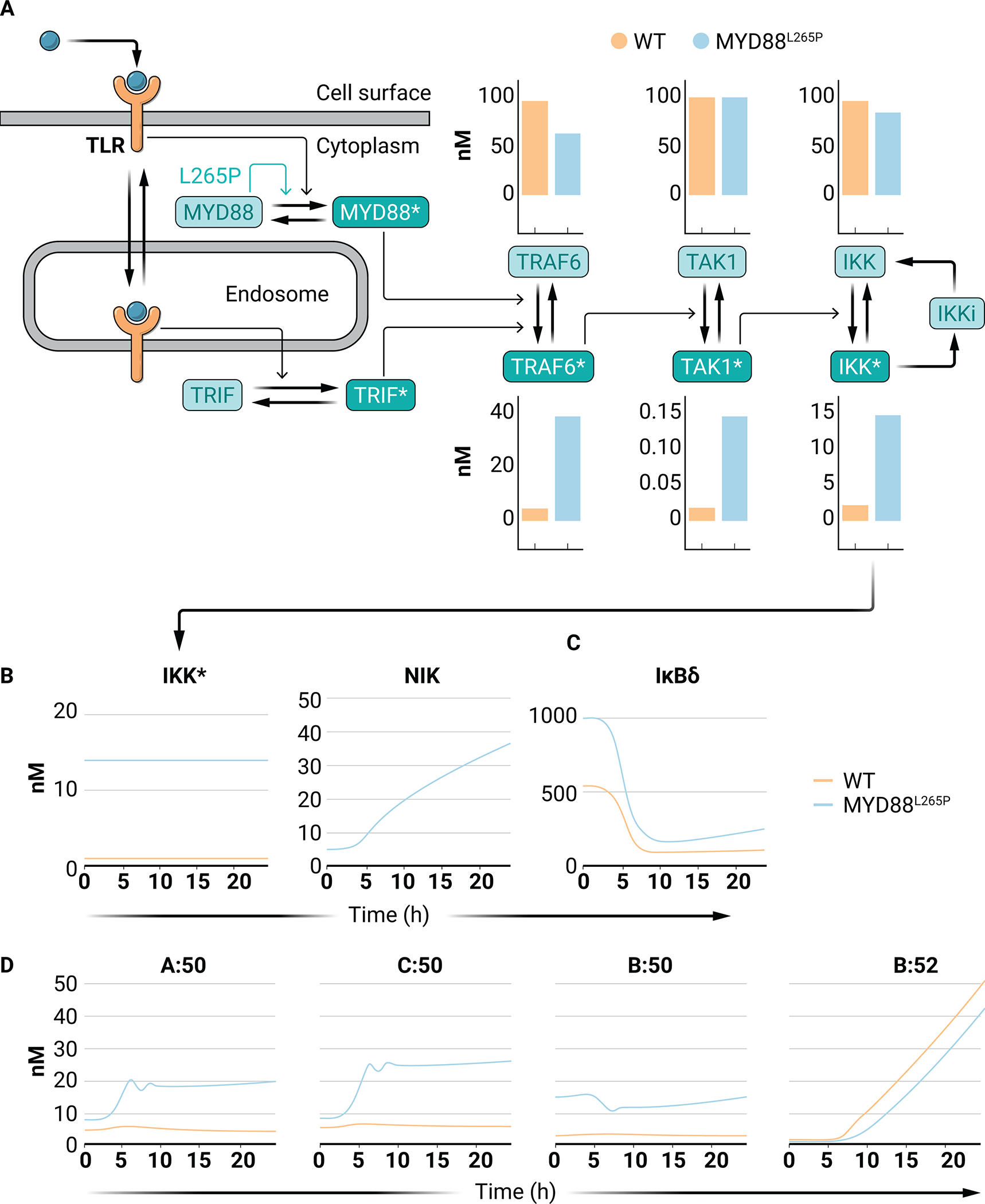
(A) Schematic of the model of Cheng et al. (2015) that was used to simulate signaling from a TLR to active IKK (IKK*). The effect of MYD88L265P is indicated by the green arrow (see data file S1). Constitutive receptor shuttling, independent of ligand binding, is omitted from the diagram. Bar graphs indicate the abundances of the indicated molecular species in WT (orange) and MYD88L265P (blue) simulations. (B) Line graphs of IKK (left) and NIK (right) activity curves for WT B cells (orange) and MYD88L265P B cells (blue). (C) Line graphs of IκBδ responses in WT B cells (orange) and MYD88L265P B cells (blue) in response to the indicated kinase activities in (B). (D) Line graphs of the abundances of the indicated NF-κB dimers in simulations of WT B cells (orange) and MYD88L265P B cells (blue) in response to the indicated kinase activities in (B).
Simulations of WT vs. MYD88L265P B cells predict a large increase in the abundances of complexes of p100 (IκBδ) in unstimulated MYD88L265P B cells (Fig. 7C). Indeed, p100-mediated signaling has been reported in ABC-DLBCL patient samples, particularly in tumors harboring MYD88L265P mutations (83, 84). Increases in RelA:p50 and cRel:p50 abundance are also predicted under unstimulated conditions but they remain largely inactive because they are bound by inhibitory IκBδ (Fig. 7D). In response to increasing NIK activity, this inhibitory IκBδ is degraded (Fig. 7C) resulting in substantial induction of nuclear RelA:p50 and cRel:p50 activities in MYD88L265P B cells (Fig. 7D). In contrast, RelB:p50 is predicted to be increased in MYD88L265P B cells under basal conditions, but the abundance of RelB:p50 in MYD88L265P B cells is not substantially increased by NIK activity. This is a result of the NIK-mediated induction of p52 activity and hence RelB:p52 dimer formation, thereby limiting the amount of RelB available for RelB:p50 formation. Furthermore, due to the abundance of IκBδ in MYD88L265P mutant simulations, RelB:p52 is slightly reduced due to “substrate complex competition” (40). Together, these simulations predict that Myd88L265P may unlock crosstalk from noncanonical pathway activation to canonical cRel- and RelA-containing NF-κB dimers. The result is that when Myd88L265P is present in B cells, developmental signals such as those from CD40 may lead to the oncogenic activation of cRel. Incorporating knowledge of cancer-associated signaling mutations into the regulatory knowledge encoded in a mathematical model illustrates how microenvironmental signals may have unexpected, nonlinear effects in tumor cells.
Conclusions
A key goal of systems biology is to represent our understanding of a biological regulatory system in the language of mathematics, whose precision and quantitative claims drive iterative experimental studies and thus research progress. Such mathematical models may then function as knowledge bases that can be leveraged for studying diverse biological phenomena. The best characterized molecular network may be the one responsible for the metabolism of sugars and carbohydrates (2, 3). This knowledge base has been exploited to drive research in numerous prokaryotic and eukaryotic organisms and in the analysis of numerous human cell types and disease contexts. Here, we showed that the same goals may be achieved with regulatory or signaling network models. The NF-κB signaling network model may be applied to different cell types, considering different stimulation contexts, which govern pathway crosstalk, and in how disease-causing mutations affect stimulus-response behavior. The mathematical model enables the use of cell type–specific gene expression data alone to generate insights about cell type–specific signaling behavior.
The present knowledge base is restricted to the NF-κB signaling systems themselves; however, a similar approach may enable extension of the model to receptor-associated signaling modules that activate this pathway (15, 81, 85) or downstream gene regulatory modules (81, 86, 87) Other efforts in the field have focused on single-cell macrophage responses to proinflammatory stimuli, requiring models to account for the observed cell-to-cell heterogeneity (81, 88). In all cases, the goal of representing all observations by a single model whose expression parameters are cell type–specifically adjusted and potentially distributed to account for cell-to-cell heterogeneity is in principle achievable.
Given the increasing availability of single-cell RNA-seq (scRNA-seq) data and descriptions of cell differentiation trajectories within a Waddington landscape, mathematical models that are reliable knowledge bases of quantitative interaction networks may be used to extrapolate from scRNA-seq datasets to predict the trajectory of how the functional responses of an individual cell to stimuli alter during development. Further studies focused on diseased tissues could extrapolate functional characteristics of cells based merely on gene expression data. Therefore, leveraging the knowledge base of a reliable mathematical model may enable unprecedented insight for research about diverse physiological and pathological questions. Whereas broad NF-κB inhibition often results in on-target toxicity, efforts to develop therapeutic compounds that target specific NF-κB dimers in specific pathological settings may also benefit from iterative systems biology studies with the described consensus NF-κB multidimer system model.
Supplementary Material
Acknowledgments:
The authors thank S. Basak, C. Pepper, U. Klein, E, Jayawant, and H. Huang for helpful discussions and critical reading of the manuscript.
Funding:
S.M. is funded by a Leukaemia UK John Goldman Fellowship (2020/JGF/003) and a UKRI Future Leaders Fellowship (MR/T041889/1). A.H. acknowledges the NIH for funding the described work in R01AI127864, R01AI132835, R01AI132731, and R01AI127867.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Model simulations were undertaken in MATLAB 2020a using Ode15s. All of the code to run the models is provided on Github (https://github.com/SiFTW/reviewModel). The code is also available at Zenodo (https://doi.org/10.5281/zenodo.xxxxxxx).
References and Notes
- 1.Malik-Sheriff RS, Glont M, Nguyen TV, Tiwari K, Roberts MG, Xavier A, Vu MT, Men J, Maire M, Kananathan S, BioModels—15 years of sharing computational models in life science. Nucleic acids research 48, D407–D415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teusink B, Passarge J, Reijenga CA, Esgalhado E, Van der Weijden CC, Schepper M, Walsh MC, Bakker BM, Van Dam K, Westerhoff HV, Can yeast glycolysis be understood in terms of in vitro kinetics of the constituent enzymes? Testing biochemistry. European Journal of Biochemistry 267, 5313–5329 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Thiele I, Swainston N, Fleming RM, Hoppe A, Sahoo S, Aurich MK, Haraldsdottir H, Mo ML, Rolfsson O, Stobbe MD, A community-driven global reconstruction of human metabolism. Nature biotechnology 31, 419–425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheong R, Hoffmann A, Levchenko A, Understanding NF-kappaB signaling via mathematical modeling. Mol Syst Biol 4, 192 (2008) 10.1038/msb.2008.30). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basak S, Behar M, Hoffmann A, Lessons from mathematically modeling the NF-kappaB pathway. Immunol Rev 246, 221–238 (2012); published online EpubMar ( 10.1111/j.1600-065X.2011.01092.x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basak S, Shih VF-S, Hoffmann A, Generation and activation of multiple dimeric transcription factors within the NF-κB signaling system. Molecular and cellular biology 28, 3139–3150 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann A, Natoli G, Ghosh G, Transcriptional regulation via the NF-κ B signaling module. Oncogene 25, 6706–6716 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Roy K, Mitchell S, Liu Y, Ohta S, Y.-s. Lin, M. O. Metzig, S. L. Nutt, A. Hoffmann, A Regulatory Circuit Controlling the Dynamics of NFκB cRel Transitions B Cells from Proliferation to Plasma Cell Differentiation. Immunity, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alves BN, Tsui R, Almaden J, Shokhirev MN, Davis-Turak J, Fujimoto J, Birnbaum H, Ponomarenko J, Hoffmann A, IkappaBepsilon is a key regulator of B cell expansion by providing negative feedback on cRel and RelA in a stimulus-specific manner. Journal of immunology (Baltimore, Md. : 1950) 192, 3121–3132 (2014); published online EpubApr 1 ( 10.4049/jimmunol.1302351). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shokhirev MN, Almaden J, Davis‐Turak J, Birnbaum HA, Russell TM, Vargas JA, Hoffmann A, A multi‐scale approach reveals that NF‐κB cRel enforces a B‐cell decision to divide. Molecular systems biology 11, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shih VF, Davis-Turak J, Macal M, Huang JQ, Ponomarenko J, Kearns JD, Yu T, Fagerlund R, Asagiri M, Zuniga EI, Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-κB pathways. Nature immunology 13, 1162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almaden JV, Tsui R, Liu YC, Birnbaum H, Shokhirev MN, Ngo KA, Davis-Turak JC, Otero D, Basak S, Rickert RC, A Pathway Switch Directs BAFF Signaling to Distinct NFκB Transcription Factors in Maturing and Proliferating B Cells. Cell reports 9, 2098–2111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mukherjee T, Chatterjee B, Dhar A, Bais SS, Chawla M, Roy P, George A, Bal V, Rath S, Basak S, A TNF-p100 pathway subverts noncanonical NF-kappaB signaling in inflamed secondary lymphoid organs. EMBO J 36, 3501–3516 (2017); published online EpubDec 1 ( 10.15252/embj.201796919). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell S, Vargas J, Hoffmann A, Signaling via the NFκB system. WIREs: Systems Biology and Medicine 8, 227–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shih VF-S, Tsui R, Caldwell A, Hoffmann A, A single NFκB system for both canonical and non-canonical signaling. Cell research 21, 86 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pomerantz JL, Baltimore D, Two pathways to NF-kappaB. Mol Cell 10, 693–695 (2002); published online EpubOct ( 10.1016/s1097-2765(02)00697-4). [DOI] [PubMed] [Google Scholar]
- 17.Oeckinghaus A, Ghosh S, The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1, a000034 (2009); published online EpubOct ( 10.1101/cshperspect.a000034). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwai K, Diverse ubiquitin signaling in NF-κB activation. Trends in cell biology 22, 355–364 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng P-H, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M, Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nature immunology 9, 1364 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao G, Harhaj EW, Sun S-C, NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Molecular cell 7, 401–409 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann A, Leung TH, Baltimore D, Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J 22, 5530–5539 (2003); published online EpubOct 15 ( 10.1093/emboj/cdg534). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsui R, Kearns JD, Lynch C, Vu D, Ngo KA, Basak S, Ghosh G, Hoffmann A, IkappaBbeta enhances the generation of the low-affinity NFkappaB/RelA homodimer. Nat Commun 6, 7068 (2015); published online EpubMay 7 ( 10.1038/ncomms8068). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerondakis S, Banerjee A, Grigoriadis G, Vasanthakumar A, Gugasyan R, Sidwell T, Grumont RJ, NF‐κB subunit specificity in hemopoiesis. Immunological reviews 246, 272–285 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Gerondakis S, Grumont R, Rourke I, Grossmann M, The regulation and roles of Rel/NF-κB transcription factors during lymphocyte activation. Current opinion in immunology 10, 353–359 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Grumont RJ, Gerondakis S, The subunit composition of NF-kappa B complexes changes during B-cell development. Cell growth & differentiation: the molecular biology journal of the American Association for Cancer Research 5, 1321–1331 (1994). [PubMed] [Google Scholar]
- 26.Liou H-C, Sha WC, Scott ML, Baltimore D, Sequential induction of NF-kappa B/Rel family proteins during B-cell terminal differentiation. Molecular and cellular biology 14, 5349–5359 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, Klein U, Germinal center B cell maintenance and differentiation are controlled by distinct NF-κB transcription factor subunits. J Exp Med 211, 2103–2118 (2014); published online EpubSep 22 ( 10.1084/jem.20132613). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao P, Hayden MS, Long M, Scott ML, West AP, Zhang D, Oeckinghaus A, Lynch C, Hoffmann A, Baltimore D, IκBβ acts to inhibit and activate gene expression during the inflammatory response. Nature 466, 1115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Dea E, Hoffmann A, The regulatory logic of the NF-κB signaling system. Cold Spring Harbor perspectives in biology 2, a000216 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy P, Mukherjee T, Chatterjee B, Vijayaragavan B, Banoth B, Basak S, Non-canonical NFkappaB mutations reinforce pro-survival TNF response in multiple myeloma through an autoregulatory RelB:p50 NFkappaB pathway. Oncogene 36, 1417–1429 (2017); published online EpubMar ( 10.1038/onc.2016.309). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chawla M, Mukherjee T, Deka A, Chatterjee B, Sarkar UA, Singh AK, Kedia S, Lum J, Dhillon MK, Banoth B, Biswas SK, Ahuja V, Basak S, An epithelial Nfkb2 pathway exacerbates intestinal inflammation by supplementing latent RelA dimers to the canonical NF-kappaB module. Proc Natl Acad Sci U S A 118, (2021); published online EpubJun 22 ( 10.1073/pnas.2024828118). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dhar A, Chawla M, Chattopadhyay S, Oswal N, Umar D, Gupta S, Bal V, Rath S, George A, Arimbasseri GA, Basak S, Role of NF-kappaB2-p100 in regulatory T cell homeostasis and activation. Sci Rep 9, 13867 (2019); published online EpubSep 25 ( 10.1038/s41598-019-50454-z). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsui RW, Cell Context-Dependent Control of NF-kappaB Dimers. (University of California, San Diego, 2014). [Google Scholar]
- 34.Wang J, Basagoudanavar SH, Wang X, Hopewell E, Albrecht R, García-Sastre A, Balachandran S, Beg AA, NF-κB RelA subunit is crucial for early IFN-β expression and resistance to RNA virus replication. The Journal of Immunology 185, 1720–1729 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schrofelbauer B, Polley S, Behar M, Ghosh G, Hoffmann A, NEMO ensures signaling specificity of the pleiotropic IKKbeta by directing its kinase activity toward IkappaBalpha. Mol Cell 47, 111–121 (2012); published online EpubJul 13 ( 10.1016/j.molcel.2012.04.020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gapuzan ME, Schmah O, Pollock AD, Hoffmann A, Gilmore TD, Immortalized fibroblasts from NF-kappaB RelA knockout mice show phenotypic heterogeneity and maintain increased sensitivity to tumor necrosis factor alpha after transformation by v-Ras. Oncogene 24, 6574–6583 (2005); published online EpubSep 29 ( 10.1038/sj.onc.1208809). [DOI] [PubMed] [Google Scholar]
- 37.Savinova OV, Hoffmann A, Ghosh G, The Nfkb1 and Nfkb2 proteins p105 and p100 function as the core of high-molecular-weight heterogeneous complexes. Molecular cell 34, 591–602 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basak S, Kim H, Kearns JD, Tergaonkar V, O’Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A, A fourth IkappaB protein within the NF-kappaB signaling module. Cell 128, 369–381 (2007); published online EpubJan 26 ( 10.1016/j.cell.2006.12.033). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Müller JR, Siebenlist U, Lymphotoxin β receptor induces sequential activation of distinct NF-κB factors via separate signaling pathways. Journal of Biological Chemistry 278, 12006–12012 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Mitchell S, Hoffmann A, Substrate complex competition is a regulatory motif that allows NFκB RelA to license but not amplify NFκB RelB. Proceedings of the National Academy of Sciences 116, 10592–10597 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banoth B, Chatterjee B, Vijayaragavan B, Prasad M, Roy P, Basak S, Stimulus-selective crosstalk via the NF-κB signaling system reinforces innate immune response to alleviate gut infection. Elife 4, e05648 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chatterjee B, Banoth B, Mukherjee T, Taye N, Vijayaragavan B, Chattopadhyay S, Gomes J, Basak S, Late-phase synthesis of IκBα insulates the TLR4-activated canonical NF-κB pathway from noncanonical NF-κB signaling in macrophages. Science signaling 9, ra120–ra120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fusco AJ, Savinova OV, Talwar R, Kearns JD, Hoffmann A, Ghosh G, Stabilization of RelB requires multidomain interactions with p100/p52. J Biol Chem 283, 12324–12332 (2008); published online EpubMay 2 ( 10.1074/jbc.M707898200). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taniguchi K, Karin M, NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 18, 309–324 (2018); published online EpubMay ( 10.1038/nri.2017.142). [DOI] [PubMed] [Google Scholar]
- 45.Zhang Q, Lenardo MJ, Baltimore D, 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 168, 37–57 (2017); published online EpubJan 12 ( 10.1016/j.cell.2016.12.012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaffer AL 3rd, Young RM, Staudt LM, Pathogenesis of human B cell lymphomas. Annu Rev Immunol 30, 565–610 (2012) 10.1146/annurev-immunol-020711-075027). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pasqualucci L, Dalla-Favera R, Genetics of diffuse large B-cell lymphoma. Blood 131, 2307–2319 (2018); published online EpubMay 24 ( 10.1182/blood-2017-11-764332). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kennedy R, Klein U, Aberrant activation of NF-κB signalling in aggressive lymphoid malignancies. Cells 7, 189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitchell S, What Will B Will B: Identifying Molecular Determinants of Diverse B-Cell Fate Decisions Through Systems Biology. Frontiers in Cell and Developmental Biology 8, 1649 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuppers R, Engert A, Hansmann ML, Hodgkin lymphoma. J Clin Invest 122, 3439–3447 (2012); published online EpubOct ( 10.1172/jci61245). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weniger MA, Küppers R, in Seminars in cancer biology. (Elsevier, 2016), vol. 39, pp. 32–39. [DOI] [PubMed] [Google Scholar]
- 52.de Oliveira KA, Kaergel E, Heinig M, Fontaine J-F, Patone G, Muro EM, Mathas S, Hummel M, Andrade-Navarro MA, Hübner N, A roadmap of constitutive NF-κB activity in Hodgkin lymphoma: Dominant roles of p50 and p52 revealed by genome-wide analyses. Genome medicine 8, 28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rastelli J, Hömig-Hölzel C, Seagal J, Müller W, Hermann AC, Rajewsky K, Zimber-Strobl U, LMP1 signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood 111, 1448–1455 (2008); published online EpubFeb 1 ( 10.1182/blood-2007-10-117655). [DOI] [PubMed] [Google Scholar]
- 54.Otto C, Giefing M, Massow A, Vater I, Gesk S, Schlesner M, Richter J, Klapper W, Hansmann M-L, Siebert R, Küppers R, Genetic lesions of the TRAF3 and MAP3K14 genes in classical Hodgkin lymphoma. British Journal of Haematology 157, 702–708 (2012) 10.1111/j.1365-2141.2012.09113.x). [DOI] [PubMed] [Google Scholar]
- 55.Ranuncolo SM, Pittaluga S, Evbuomwan MO, Jaffe ES, Lewis BA, Hodgkin lymphoma requires stabilized NIK and constitutive RelB expression for survival. Blood 120, 3756–3763 (2012); published online EpubNov 1 ( 10.1182/blood-2012-01-405951). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rayet B, Gelinas C, Aberrant rel/nfkb genes and activity in human cancer. Oncogene 18, 6938–6947 (1999). [DOI] [PubMed] [Google Scholar]
- 57.Sun S-C, Xiao G, Deregulation of NF-κB and its upstream kinases in cancer. Cancer and Metastasis Reviews 22, 405–422 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Xiao G, Fu J, NF-κB and cancer: a paradigm of Yin-Yang. American journal of cancer research 1, 192 (2011). [PMC free article] [PubMed] [Google Scholar]
- 59.Chatterjee B, Roy P, Sarkar UA, Zhao M, Ratra Y, Singh A, Chawla M, De S, Gomes J, Sen R, Basak S, Immune Differentiation Regulator p100 Tunes NF-kappaB Responses to TNF. Front Immunol 10, 997 (2019) 10.3389/fimmu.2019.00997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiao G, Rabson AB, Young W, Qing G, Qu Z, Alternative pathways of NF-κB activation: a double-edged sword in health and disease. Cytokine & growth factor reviews 17, 281–293 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Qing G, Qu Z, Xiao G, Regulation of NF-κB2 p100 processing by its cis-acting domain. Journal of Biological Chemistry 280, 18–27 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Qing G, Qu Z, Xiao G, Endoproteolytic processing of C-terminally truncated NF-κB2 precursors at κB-containing promoters. Proceedings of the National Academy of Sciences 104, 5324–5329 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ciana P, Neri A, Cappellini C, Cavallo F, Pomati M, Chang C-C, Maiolo AT, Lombardi L, Constitutive expression of lymphoma-associated NFKB-2/Lyt-10 proteins is tumorigenic in murine fibroblasts. Oncogene 14, 1805–1810 (1997). [DOI] [PubMed] [Google Scholar]
- 64.Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, Grinberg A, Tran T, Scharton-Kersten T, Anver M, Mice deficient in nuclear factor (NF)-κB/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. The Journal of experimental medicine 187, 147–159 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Z, Zhang B, Yang L, Ding J, Ding H-F, Constitutive production of NF-κB2 p52 is not tumorigenic but predisposes mice to inflammatory autoimmune disease by repressing Bim expression. Journal of Biological Chemistry 283, 10698–10706 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim K-E, Gu C, Thakur S, Vieira E, Lin JC, Rabson AB, Transcriptional regulatory effects of lymphoma-associated NFKB2/lyt10 protooncogenes. Oncogene 19, 1334–1345 (2000). [DOI] [PubMed] [Google Scholar]
- 67.Shaffer AL, Rosenwald A, Staudt LM, Lymphoid malignancies: the dark side of B-cell differentiation. Nat Rev Immunol 2, 920–932 (2002); published online EpubDec ( 10.1038/nri953). [DOI] [PubMed] [Google Scholar]
- 68.Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, Bhagat G, Chadburn A, Dalla-Favera R, Pasqualucci L, Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 459, 717–721 (2009); published online EpubJun 4 ( 10.1038/nature07968). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis RE, Brown KD, Siebenlist U, Staudt LM, Constitutive nuclear factor κB activity is required for survival of activated B cell–like diffuse large B cell lymphoma cells. Journal of Experimental Medicine 194, 1861–1874 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. New England Journal of Medicine 346, 1937–1947 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Zhang M, Xu-Monette ZY, Li L, Manyam GC, Visco C, Tzankov A, Wang J, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Richards KL, Hsi ED, Choi WW, Han van Krieken J, Huh J, Ponzoni M, Ferreri AJ, Møller MB, Parsons BM, Winter JN, Piris MA, Medeiros LJ, Pham LV, Young KH, RelA NF-κB subunit activation as a therapeutic target in diffuse large B-cell lymphoma. Aging (Albany NY) 8, 3321–3340 (2016); published online EpubDec 8 ( 10.18632/aging.101121). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eluard B, Nuan-Aliman S, Faumont N, Collares D, Bordereaux D, Montagne A, Martins I, Cagnard N, Caly M, Taoui O, The alternative RelB NF-κB subunit is a novel critical player in diffuse large B-cell lymphoma. Blood, (2021). [DOI] [PubMed] [Google Scholar]
- 73.Lee J-H, Jeong H, Choi J-W, Oh H, Kim Y-S, Clinicopathologic significance of MYD88 L265P mutation in diffuse large B-cell lymphoma: a meta-analysis. Scientific reports 7, 1785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jeelall YS, Horikawa K, Oncogenic MYD88 mutation drives Toll pathway to lymphoma. Immunology and cell biology 89, 659 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Staudt LM, Oncogenically active MYD88 mutations in human lymphoma. Nature 470, 115–119 (2011); published online EpubFeb 3 ( 10.1038/nature09671). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MG, Li AJ, Ziepert M, Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nature medicine 24, 679 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, Wang JQ, Schmitz R, Morin RD, Tang J, Jiang A, Bagaev A, Plotnikova O, Kotlov N, Johnson CA, Wilson WH, Scott DW, Staudt LM, A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 37, 551–568.e514 (2020); published online EpubApr 13 ( 10.1016/j.ccell.2020.03.015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, Arcaini L, Pinkus GS, Rodig SJ, Sohani AR, Harris NL, Laramie JM, Skifter DA, Lincoln SE, Hunter ZR, MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 367, 826–833 (2012); published online EpubAug 30 ( 10.1056/NEJMoa1200710). [DOI] [PubMed] [Google Scholar]
- 79.Ouk C, Roland L, Gachard N, Poulain S, Oblet C, Rizzo D, Saintamand A, Lemasson Q, Carrion C, Thomas M, Continuous MYD88 Activation Is Associated With Expansion and Then Transformation of IgM Differentiating Plasma Cells. Frontiers in immunology 12, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang JQ, Jeelall YS, Beutler B, Horikawa K, Goodnow CC, Consequences of the recurrent MYD88L265P somatic mutation for B cell tolerance. Journal of Experimental Medicine 211, 413–426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheng Z, Taylor B, Ourthiague DR, Hoffmann A, Distinct single-cell signaling characteristics are conferred by the MyD88 and TRIF pathways during TLR4 activation. Science signaling 8, ra69–ra69 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ansell S, Hodge L, Secreto F, Manske M, Braggio E, Price-Troska T, Ziesmer S, Li Y, Johnson S, Hart S, Activation of TAK1 by MYD88 L265P drives malignant B-cell Growth in non-Hodgkin lymphoma. Blood cancer journal 4, e183–e183 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guo X, Koff J, Moffitt A, Cinar M, Ramachandiran S, Chen Z, Switchenko J, Mosunjac M, Neill S, Mann K, Molecular impact of selective NFKB1 and NFKB2 signaling on DLBCL phenotype. Oncogene 36, 4224–4232 (2017). [DOI] [PubMed] [Google Scholar]
- 84.Ramachandiran S, Adon A, Guo X, Wang Y, Wang H, Chen Z, Kowalski J, Sunay UR, Young AN, Brown T, Mar JC, Du Y, Fu H, Mann KP, Natkunam Y, Boise LH, Saavedra HI, Lossos IS, Bernal-Mizrachi L, Chromosome instability in diffuse large B cell lymphomas is suppressed by activation of the noncanonical NF-κB pathway. Int J Cancer 136, 2341–2351 (2015); published online EpubMay 15 ( 10.1002/ijc.29301). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Adelaja A, Taylor B, Sheu KM, Liu Y, Luecke S, Hoffmann A, Six distinct NFκB signaling codons convey discrete information to distinguish stimuli and enable appropriate macrophage responses. Immunity 54, 916–930. e917 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ngo KA, Kishimoto K, Davis-Turak J, Pimplaskar A, Cheng Z, Spreafico R, Chen EY, Tam A, Ghosh G, Mitchell S, Hoffmann A, Dissecting the Regulatory Strategies of NF-κB RelA Target Genes in the Inflammatory Response Reveals Differential Transactivation Logics. Cell Rep 30, 2758–2775.e2756 (2020); published online EpubFeb 25 ( 10.1016/j.celrep.2020.01.108). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sen S, Cheng Z, Sheu KM, Chen YH, Hoffmann A, Gene Regulatory Strategies that Decode the Duration of NFκB Dynamics Contribute to LPS- versus TNF-Specific Gene Expression. Cell Syst 10, 169–182.e165 (2020); published online EpubFeb 26 ( 10.1016/j.cels.2019.12.004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Junkin M, Kaestli AJ, Cheng Z, Jordi C, Albayrak C, Hoffmann A, Tay S, High-Content Quantification of Single-Cell Immune Dynamics. Cell Rep 15, 411–422 (2016); published online EpubApr 12 ( 10.1016/j.celrep.2016.03.033). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Model simulations were undertaken in MATLAB 2020a using Ode15s. All of the code to run the models is provided on Github (https://github.com/SiFTW/reviewModel). The code is also available at Zenodo (https://doi.org/10.5281/zenodo.xxxxxxx).