

Abstract
Severe hemorrhage associated with trauma, surgery, and congenital or drug-induced coagulopathies can be life-threatening and requires rapid hemostatic management via topical, intracavitary, or intravenous routes. For injuries that are not easily accessible externally, intravenous hemostatic approaches are needed. The clinical gold standard for this is transfusion of blood products, but due to donor dependence, specialized storage requirements, high risk of contamination, and short shelf life, blood product use faces significant challenges. Consequently, recent research efforts are being focused on designing biosynthetic intravenous hemostats, using intravenous nanoparticles and polymer systems. Here we report on the design and evaluation of thrombin-loaded injury-site-targeted lipid nanoparticles (t-TLNPs) that can specifically localize at an injury site via platelet-mimetic anchorage to the von Willebrand factor (vWF) and collagen and directly release thrombin via diffusion and phospholipase-triggered particle destabilization, which can locally augment fibrin generation from fibrinogen for hemostatic action. We evaluated t-TLNPs in vitro in human blood and plasma, where hemostatic defects were created by platelet depletion and anticoagulation. Spectrophotometric studies of fibrin generation, rotational thromboelastometry (ROTEM)-based studies of clot viscoelasticity, and BioFlux-based real-time imaging of fibrin generation under simulated vascular flow conditions confirmed that t-TLNPs can restore fibrin in hemostatic dysfunction settings. Finally, the in vivo feasibility of t-TLNPs was tested by prophylactic administration in a tail-clip model and emergency administration in a liver-laceration model in mice with induced hemostatic defects. Treatment with t-TLNPs was able to significantly reduce bleeding in both models. Our studies demonstrate an intravenous nanomedicine approach for injury-site-targeted direct delivery of thrombin to augment hemostasis.
Keywords: Hemostasis, Platelets, Thrombin, Fibrin, Nanoparticle, Targeted delivery
Graphical Abstract
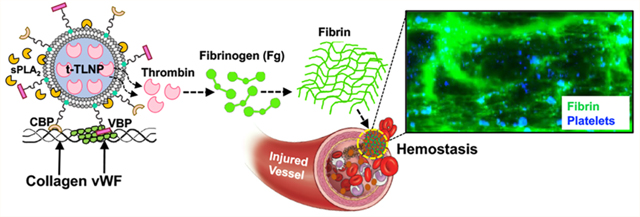
Noncompressible uncontrolled hemorrhage remains a major cause of mortality from traumatic injuries.1–4 Additionally, patients with congenital, disease-associated, and drug-induced hemostatic dysfunctions may often be at risk of excessive bleeding.5–8 Therefore, technologies that enable the rapid stoppage of bleeding (hemostasis) are clinically significant in stabilizing hemorrhaging patients and potentially saving lives. To this end, externally accessible injuries can be effectively managed by topical or externally administered hemostatic agents, e.g. tourniquets, pressure dressings, foams, QuikClot, self-expanding sponges, tissue-adhesive glues, etc., many of which are already in clinical and field use.9 Several additional technologies are in preclinical development, e.g. hemostatic-drug-loaded self-propelled calcium carbonate (CaCO3) microparticles, self-assembling hemostatic peptide nanofibers, polyethylenimine (PEI)-based charged hemostatic nanoparticles, multimaterial composite hemostatic gels, etc.10–16 However, for noncompressible hemorrhages and systemic bleeding dysfunctions (e.g., polytrauma, internal bleeding, platelet and coagulation factor defects, etc.), such technologies are unsuitable and intravenous hemostasis approaches are required.9,17,18
The clinical gold standard for such an approach is the transfusion of whole blood (WB) or blood components (e.g., controlled ratios of platelets, RBCs, and plasma), as evidenced by several robust clinical studies (e.g., PROPPR, PROMMTT, and PAMPer).19–22 However, such blood products have limited availability due to their donor dependence, and their portability and shelf life is limited due to contamination risks.23–26 Extensive preclinical and clinical research efforts are currently focused on improving the logistics of blood product availability and use, both within hospitals and prehospital (e.g., civilian accidents, battlefield injuries, etc.), including blood processing by pathogen reduction technologies (PRT), cold-stored WB, low titer group O whole blood (LTOWB), cold-stored platelets (CSP), freeze-dried platelets (e.g., Thrombosome by Cell-Phire), and freeze-dried plasma (e.g., FDP by Teleflex).27–34 While these approaches may improve blood transfusion logistics, these products are still donor dependent and continue to face challenges regarding portability, storage, and timely availability. Therefore, a parallel research focus has emerged in designing nanomaterial-based intravenously administrable technologies (nanoparticles, polymers, etc.) that can provide specific functional attributes of hemostasis while allowing donor-independent manufacturing, scale-up, and on-demand availability for bleeding management. Prominent recent examples of these are (i) “synthetic platelet” (SynthoPlate) nanoparticles that recapitulate platelets’ hemostatically relevant binding interactions with von Willebrand factor (vWF), collagen, and active platelet integrin GPIIb-IIIa, (ii) platelet-like particles (PLPs) that bind fibrin to recapitulate platelets’ hemostatically important biomechanical property of clot contraction, (iii) fibrinogen-function-mimicking nanoparticles that improve the aggregation of active platelets, (iv) tissue factor targeted injectable peptide amphiphiles, (v) a PolySTAT polymer that recapitulates factor FXIIIa function of fibrin stabilization and (vi) a HAPPI polymer that can accumulate at sites of high vWF and collagen exposure.35–45
In this framework, here we report on the investigation of intravenously administered, injury-site-targeted, enzyme-responsive direct delivery of thrombin using platelet-inspired nanoparticles to site-specifically augment hemostasis. Our design inspiration stems from platelets’ critical mechanisms of hemostasis by (i) rapidly adhering and aggregating at the injury site to form a plug (primary hemostasis) and (ii) serving as a coagulation amplifier via presenting an anionic phospholipid such as phosphatidylserine (PS) on the activated platelet surface to render coagulation factor assemblies forming tenase and prothrombinase complexes leading to thrombin (FIIa) burst, which then locally converts fibrinogen (Fg) to fibrin for hemostatic action (Figure 1A).46–48 Without platelets, only a modest amount of thrombin is created by the tissue factor (TF)-FVIIa pathway, which is responsible for initiation of the hemostatic process but not sufficient to rapidly amplify fibrin formation for a stable clot.49,50 This is possibly why clinical studies have indicated major survival benefits of early platelet transfusion in trauma.22 However, there are tremendous challenges for platelet usage, due to the limited donor availability, portability, special storage requirements, high risks of bacterial contamination, and very short shelf life (∼5 days at room temperature).25 Platelet cooling and freeze-drying may allow a slightly increased shelf life while partially conserving hemostatic functions, but due to their dependence on donors they cannot be the standalone solutions to address the “platelet need”. This essentially is the translational premise of our research design. On the basis of the platelet mechanisms shown in Figure 1A, we hypothesized that a lipid nanoparticle (LNP) system that can specifically accumulate at the injury site and directly deliver thrombin can site-specifically augment fibrin independent of native platelet and coagulation status or therapeutic availability of blood products. Thrombin delivery to augment hemostasis is clinically well accepted, as exemplified by products such as Tisseel, Surgiflo, Floseal, etc., where thrombin is delivered via a syringe device directly at the wound site to form fibrin in situ. Research has also been dedicated toward loading thrombin into hemostatic bandages/dressings, as well as topically administering thrombin-loaded particles to mitigate bleeding.51,52 However, these systems are only suitable for external administration scenarios and cannot be used intravenously. Interestingly, a recent research explored the possibility of encapsulating thrombin-loaded liposomes inside actual platelets with the idea of the thrombin being released (analogous to platelet granule secretion) upon platelet activation.53 However, this was only demonstrated in vitro because there are logistical challenges regarding collecting platelets, loading drug-carrying nanoparticles in them with reproducible quality, ensuring their consistent release kinetics upon platelet activation in vivo, and optimizing this complex strategy for consistent function. Our approach efficiently addresses these challenges by (i) loading a consistent amount of thrombin in LNPs, (ii) directly targeting LNPs to the injury site via specific binding to vWF and collagen, and (iii) releasing the thrombin with reproducible kinetics triggered by the injury-site-specific enzyme secretory phospholipase A2 (sPLA2) for site-localized fibrin production (Figure 1B). Here we report the evaluation of these injury-site-targeted thrombin-loaded LNPs (t-TLNPs) in vitro in human blood and plasma, where hemostatic defects were created by platelet depletion and anticoagulant treatment. Spectrophotometric studies of fibrin generation, rotational thromboelastometry (ROTEM) based studies of clot characteristics, and BioFlux microfluidics based real-time imaging of fibrin generation under simulated vascular flow confirmed the ability of t-TLNPs to restore fibrin in hemostatic dysfunction settings. Finally, the in vivo feasibility of t-TLNPs was tested in a mouse tail-bleed model, where a combination of antibody-induced thrombocytopenia plus heparin-induced anticoagulation was used to render significant hemostatic defects and treatment with t-TLNPs was able to effectively reduce bleeding. The t-TLNPs were also evaluated in a traumatic liver injury bleeding model in anticoagulated (with heparin) mice and here treatment with t-TLNPs post-injury significantly reduced bleeding while maintaining systemic safety.
Figure 1.

Platelet-mediated hemostatic mechanism and platelet-inspired t-TLNP design. (A) Platelets rapidly adhere at a vascular injury site by binding to von Willebrand factor (vWF, via platelet surface GPIbα) and collagen (via platelet surface GPIa/IIa and GPVI) exposed at the site and present high amounts of an anionic phospholipid such as phosphatidylserine (PS) on the activated platelet procoagulant membrane surface to enable the assembly of coagulation factors to form tenase (FVIIa + FIXa + FX) and prothrombinase (FXa + FVa + FII) complexes, ultimately leading to the amplified generation of thrombin (thrombin burst); the thrombin locally converts fibrinogen (Fg) to fibrin that gets cross-linked by FXIIIa for hemostatic clot formation. (B) t-TLNPs can undergo platelet-mimetic adhesion at the vascular injury site by anchoring to vWF via vWF-binding peptide (VBP) and collagen via collagen-binding peptide (CBP) and release thrombin at the site via diffusion as well as injury site secreted phospholipase A2 (sPLA2) triggered particle destabilization; this thrombin can locally convert fibrinogen (Fg) to fibrin for hemostatic action.
RESULTS
Targeted Thrombin-Loaded Lipid Nanoparticle (t-TLNP) Manufacture and Characterization.
For targeting to an injury site via platelet-inspired mechanisms, the liposomal LNPs were surface-decorated with vWF-binding peptides (VBPs) and collagen-binding peptides (CBPs). The VBP sequence TRYLRIHPQSWVHQI was chosen from a peptide library reported by Nogami et al. that mimicd FVIII C2 domain sequences with binding specificity to the D′-D3 domain of vWF.54 The CBP sequence [GPO]7 was chosen on the basis of reports of high helicogenic affinity of this sequence to fibrillar collagen, and such sequences have been used for molecular imaging of collagen fibers.55,56 Both VBP and CBP sequences were cysteinylated to enable thiol-mediated conjugation to maleimide-terminated lipid molecules for subsequent incorporation into liposomal LNPs. Figure 2A depicts the chemical structures of lipid–peptide conjugation reactions for t-TLNP manufacturing, and Figure 2B depicts the molecular components used in t-TLNP assembly. In current studies the total “DSPE-PEG-VBP + DSPE-PEG-CBP” composition in t-TLNPs was kept at 2.5 mol % of total lipid per batch of particle manufacture, with a 1:1 ratio (1.25 mol % of each).
Figure 2.

Manufacture and characterization of t-TLNPs. (A) Bioconjugation schematics of reacting cysteine-terminated peptides to maleimide-terminated DSPE-PEG2K utilizing thiol–maleimide chemistry to synthesize DSPE-PEG2K-peptide molecules. (B) Molecular components of t-TLNP manufacture. (C) Dynamic light scattering (DLS) analysis of five representative t-TLNP batches showing nanoparticle size reproducibility. (D) Cryo-transmission electron microscopy (Cryo-TEM) images of t-TLNPs (scale bar: 100 nm) showing a particle diameter of ∼175 nm. (E) Representative images from BioFlux experiments where Rhodamine B labeled control (undecorated) vs targeted nanoparticles (“VBP + CBP”-decorated) were flowed at 25 dyn/cm2 over “vWF + collagen”-coated channels and targeted nanoparticles were also flowed over albumin-coated channels, showing substantially high adhesion of targeted nanoparticles to “vWF + collagen”-coated surface but not of control particles to the “vWF + collagen”-coated surface or targeted nanoparticles to the albumin-coated surface. (F) Spectrometric analysis of three representative t-TLNP batches showing a mean thrombin loading of 114.3 ±14.2 nM. (G) Thrombin release analysis showing that t-TLNPs can slowly release low amounts of thrombin by diffusion, whereas exposure to sPLA2 significantly enhances thrombin release. *p≤ 0.05, **p≤ 0.01.
Figure S1 in the Supporting Information shows the maleimide–thiol chemistry and representative mass spectroscopy characterization data for the DSPE–PEG–peptide conjugates. Figure 2C shows dynamic light scattering (DLS) data of size distribution over five representative batches of “VBP + CBP”-decorated LNPs, and Figure S2 in the Supporting Information shows a representative DLS analysis data for such an LNP size. Figure 2D showsa representative cryo-transmission electron microscopy (Cryo-TEM) image of “VBP + CBP”-decorated LNPs. As is evident from these analyses, the nanoparticles could be manufactured reproducibly with a consistent particle diameter of approximately 175 nm. The nanoparticles thus manufactured were evaluated for their specific adhesion capability on the injury-site-relevant “vWF + collagen” surface under a hemodynamically relevant flow environment using a BioFlux microfluidic setup. For these studies, Rhodamine B (RhB, red fluorescence) labeled control nanoparticles (undecorated liposomes) and targeted nanoparticles were suspended in saline and flowed over “vWF + collagen”-coated microfluidic channels at 25 dyn/cm2 shear stress. Additionally, the targeted nanoparticles in saline were also flowed over a “negative control” surface, namely albumin-coated microchannels, since VBP and CBP are not expected to have any adhesion specificity to albumin. The experimental setup is shown in Figure S3A in the Supporting Information. As shown in representative images in Figure 2E, control nanoparticles were unable to bind to the “vWF + collagen”-coated surface while the “VBP + CBP”-decorated nanoparticles showed a high degree of adhesion on this surface. Additionally, these targeted nanoparticles did not show any adhesion to the albumin-coated surface. Representative Movies M1, M2, and M3 in the Supporting Information show minimal binding of control (undecorated) nanoparticles on the “vWF + collagen”-coated surface, substantial binding of targeted nanoparticles on the “WF + collagen”-coated surface, and minimal binding of the targeted nanoparticles on the albumin-coated surface, respectively, in the BioFlux setup. The results in Figure 2G as well as in these movies clearly indicate that the t-TLNPs have specific binding ability to vWF and collagen via VBP- and CBP-mediated interactions, respectively.
Additional studies were done to assess whether the thiol–maleimide reaction mediated conjugation of VBP and CBP on t-TLNPs remains stable in plasma. This is because, although the thiol–maleimide mediated bioconjugation is an established approach in the pharmaceutical field, it has been reported that in vivo some instability may occur in such bioconjugated molecules due to retro-Michael reactions with serum thiols.57,58 For these studies, control nanoparticles and “VBP + CBP”-decorated nanoparticles were incubated in human platelet-free plasma (PFP) for 60 min, and then these particles in the plasma suspension were flowed over the “vWF + collagen”-coated surface in the BioFlux channels. Figure S3B in the Supporting Information shows representative images from these studies, indicating that the control nanoparticles were unable to bind to the “vWF + collagen”-coated surface irrespective of plasma incubation, while the targeted nanoparticles retained their ability to bind to such surface even after incubation with plasma. Movies M4 and M5 in the Supporting Information show binding of plasma-incubated control nanoparticles and targeted nanoparticles to the “vWF + collagen”-coated channel surface. These results indicate that the thiol–maleimide reaction mediated VBP and CBP decorations on the targeted nanoparticle surface remain mostly stable in plasma and retain their specific binding ability to vWF and collagen, respectively.
Figure 2F shows thrombin-loading data for three representative batches of t-TLNPs, demonstrating that thrombin could be loaded at an average concentration of 114.3 ±14.2 nM. Figure 2G shows the thrombin release profile from t-TLNPs in PBS at 37 °C, in the absence versus the presence of sPLA2 over the course of 2 h. The diffusive release of thrombin from the t-TLNPs was slow (only ∼20% released over 2 h, magenta line), while in the presence of sPLA2 the release was significantly enhanced (∼60% released over 2 h, green line). The release rate of thrombin from t-TLNPs was also increased in the presence of sPLA2. These data strongly suggest that sPLA2 can accelerate the release of thrombin from t-TLNPs and that this property can be advantageous for enhanced thrombin release from injury-site-anchored t-TLNPs, since activated platelets and macrophages upregulate sPLA2 secretion at a vascular injury site.59,60 Additionally, we studied whether shear forces relevant to the circulation environment could destabilize the t-TLNPs, resulting in increased payload release, because this can be a potential systemic prothrombotic risk from off-target thrombin release in vivo. For this, we loaded carboxyfluorescein (CF) as a model payload in the targeted LNPs. Of note, CF loaded at a high concentration in the particle core remains self-quenched, giving a low fluorescence signal, but as CF is released it gets diluted and the fluorescence signal is enhanced, providing a way to use fluorescence spectrometry to measure payload release. For comparison studies, first the CF-loaded targeted LNPs were added to saline in a well plate and CF fluorescence was monitored for diffusive release, release triggered by sPLA2 added to a well, and exhaustive release due to complete destabilization of the particles by incubating with 1/1 methanol/chloroform mixture. Subsequently, the CF-loaded targeted LNPs were incubated on “vWF + collagen”-coated microchannels, the resultant bound CF-loaded LNPs were exposed to flow of saline at low to high shear (5–50 dyn/cm2) for 20 min in the BioFlux system, and the effluent from such experiments in the outlet well of the BioFlux setup was analyzed for “released” CF fluorescence. As shown in the data in Figure S4 in the Supporting Information, in comparison to sPLA2-triggered release and chloroform/methanol-induced exhaustive release, exposure to shear caused very minimal CF release, indicating that the particles are quite stable even under high shear (50 dyn/ cm2). On consideration that physiological shear forces in venous and arterial circulation are usually in the range of 5–30 dyn/cm2, it can be rationalized that thrombin loaded in such LNPs will have minimal release under physiological shear in circulation and will not pose a high systemic thrombotic risk. Once the particles bind to the target injury site, the thrombin release is expected to occur by diffusion and sPLA2-triggered destabilization, and if higher shear at the injury site induces more thrombin release, that could be further beneficial to site-specific hemostatic augmentation.
Evaluation of Biosafety Characteristics of Peptide-Decorated Nanoparticles.
The “VBP + CBP”-decorated nanoparticles were evaluated for biosafety characteristics using four complementary assays. In the first assay, it was evaluated whether these nanoparticles activate healthy endothelial cells, because this would indicate potential safety concerns toward healthy vasculature in vivo. For these experiments, human pulmonary microvascular endothelial cells (HPMEC) were cultured in custom-made microfluidic channels coated with 0.2 mg/mL of fibronectin for 1 h at 37 °C and maintained with 5% CO2 at 37 °C under 100 μL/min media flow for 48–72 h until a confluent monolayer was formed. As a “positive control” for EC activation, this EC monolayer was exposed to TNF-α (20 μM) and immunostaining of vWF expression on these stimulated ECs was used as an activation marker.61 In analogous experiments, similar EC monolayers were exposed to incubation of “VBP + CBP”-decorated particles or control undecorated particles (4.54 × 1012 particles/mL for 2 h at 37 °C), and the vWF expression was similarly imaged. As shown in Figure 3A, in comparison to TNF-α treatment, neither the peptide-decorated particles nor the control particles were found to activate ECs (minimal vWF staining). This indicates that the nanoparticles do not interact with and activate healthy ECs. In another assay, the VBP and CBP peptides as well as “VBP + CBP”-decorated nanoparticles were evaluated using platelet lumi-aggregometry to assess whether they activate and aggregate resting platelets, because this would indicate the risk of systemic thrombosis. As a positive control, adenosine diphosphate (ADP) was used as a platelet agonist which causes substantial platelet activation and aggregation.62 The results from these studies are shown in Figure 3B, with raw aggregometry profiles being shown in Figure S5A,B in the Supporting Information. As is evident from these data, neither the free VBP and CBP peptides nor the “VBP + CBP”-decorated nanoparticles (labeled as targeted LNPs or t-LNPs) were found to cause any platelet activation and aggregation in PRP (aggregation percent similar to that of PRP without ADP). In a third assay, the “VBP + CBP”-decorated nanoparticles (t-LNPs) were evaluated via ELISA for their ability to activate complement C3 to C3a, as this would indicate a systemic complement-mediated immunological risk.63 As is shown is Figure 3C, the C3a/C3 ratio in plasma remained similar to the baseline (saline), on incubation with control nanoparticles or t-LNPs, indicating that in vivo these particles would have minimal complement activation risks. In a fourth assay, the control particles as well as t-LNPs were evaluated for their ability to activate neutrophils, because this would indicate an unwanted stimulation of the innate immune defense mechanism. Neutrophils, when they are stimulated, exhibit a specific mechanism of deconvoluting their DNA via histone citrullination and extruding the DNA as neutrophil extracellular traps (NETs) which can be stained by Sytox Green.64 Therefore, for these studies, the calcium ionophore A23187 was used as a positive control, as it is well-known to induce NET formation.65 As shown in representative images (DAPI, blue nuclei of neutrophils; Sytox Green, NETs) and quantitative data shown in Figure S5C in the Supporting Information, in comparison to A23187 neither control nanoparticles nor t-LNPs were found to stimulate neutrophils, indicating potential safety toward the innate immune system. Altogether, results from these four complementary bioassays strongly suggest that the “VBP + CBP”-decorated nanoparticles are expected to have an inherently safe profile in circulation.
Figure 3.

Evaluation of biosafety characteristics of peptide-decorated nanoparticles. (A) Monolayers of healthy human pulmonary microvascular endothelial cells (HPMEC, nuclei stained with blue DAPI) were exposed to TNF-α (a known endothelial activator), media only, control (undecorated) nanoparticles or targeted (“VBP + CBP”-decorated) nanoparticles and vWF expression on endothelium was stained (green vWF antibody) as a marker for endothelial activation. In comparison to TNF-α-induced stimulation (high vWF staining), neither control particles nor targeted particles showed endothelial activation (low vWF staining, similar to that of the “media only” group). (B) Platelet lumi-aggregometry studies with human platelet-rich plasma (PRP) showed that addition of ADP (platelet agonist) induced significant platelet aggregation but addition of the targeted nanoparticles (t-LNPs) did not induce such aggregation (aggregation percent similar to that of the “no ADP” group); (C) ELISA-based complement C3 activation assay studies using human plasma incubated with control nanoparticles or targeted nanoparticles (t-LNPs) indicated that neither control nor targeted nanoparticles activate C3 (C3a/C3 ratio similar to the baseline of saline-incubated plasma).
t-TLNPs Restore Fibrin Generation Capability in Anticoagulant-Treated and Platelet-Depleted Human Plasma.
Since our in vitro characterization studies demonstrated that t-TLNPs can release thrombin diffusively as well as at enhanced levels triggered by sPLA2, we investigated whether such thrombin release from t-TLNPs can restore fibrin generation in human plasma in settings of a clinically relevant “hemostatic defect”. On the basis of the hemostatic mechanism of thrombin generation depicted in Figure 1, either inhibition of coagulation factor (e.g., FXa) or depletion of platelets as a whole (hence reducing the availability of the PS-rich procoagulant platelet surface) is expected to reduce overall thrombin production and thereby reduce the corresponding fibrin generation. Therefore, these are the two characteristic “hemostatic defects” that were induced in human plasma (Figure 4A) and t-TLNPs were evaluated for their ability to rescue/restore fibrin generation via direct release of thrombin. The fibrin generation kinetics in plasma was studied using spectrophotometric measurements (absorbance at 405 nm) of the optical density (OD) change in plasma due to fibrin generation and polymerization and thus monitoring the overall coagulation potential (OCP), onset of fibrin generation (OFG), and maximum hemostatic potential (MHP) parameters.66 The raw data showing fibrin generation curves and changes in these parameters due to anticoagulant effect and platelet depletion effect are shown in Figure S6 and S7 in the Supporting Information. Additionally, in vitro data in Figure S8 in the Supporting Information show that, in such anticoagulated or platelet-depleted plasma, directly adding thrombin (positive control) is able to restore fibrin generation, but such a direct administration of thrombin cannot be used intravenously in vivo due to systemic coagulation risks. For the various defects and t-TLNP treatment effects we present the parametric results in Figure 4B,C as percent (%) deviations from the healthy plasma baseline, since the goal was to evaluate whether t-TLNP treatment could restore the OCP, OFG, and MHP parameters in defective plasma to be closer to the healthy plasma baseline values. Treatment with empty (unloaded) nanoparticles (UNPs) was used as a control. As shown in Figure 4B1–B3, treatment of human plasma with Apixaban (an FXa inhibitor) induced a substantial debilitation (negative percent deviation for OCP and MHP, positive percent deviation for OFG) of fibrin generation/polymerization (a black curve for healthy plasma vs a blue curve for Apixaban-treated plasma are shown in representative OD curves and corresponding values in Figure S6 in the Supporting Information). Treatment with t-TLNPs, but without sPLA2 addition, resulted in partial restoration of OCP and MHP parameters toward healthy (i.e., Apixaban-free) plasma baselines but not as much of an improvement in OFG, indicating some recovery in fibrin generation/polymerization over the 60 min assay period due to diffusive release of thrombin over this time. When the Apixaban-treated plasma was treated with “t-TLNP + sPLA2” to simulate “enzyme-triggered thrombin release”, the OCP, OFG, and MHP parameters were all significantly restored toward the “healthy plasma” baseline. Treatment with UNPs was unable to achieve such a restoration of fibrin generation/polymerization parameters. Representative raw OD data in Figure S6A in the Supporting Information clearly show the improvement of fibrin generation when anticoagulated plasma (deep blue curve) is treated with t-TLNP (cyan curve), which have shifted toward the black curve (healthy plasma) and purple curve (t-TLNP + sPLA2 treatment) performing better than the black curve (healthy plasma). This demonstrates rescue of fibrin generation in comparison to the deep blue curve (Apixaban-treated plasma) and red curve (UNP treatment). Overall, these studies indicate that t-TLNPs can potentially restore fibrin generation kinetics as well as amount, in scenarios where coagulation factor dysfunction (simulated by FXa inhibition here) induces a hemostatic defect.
Figure 4.

Evaluation of t-TLNPs in restoring fibrin generation in anticoagulated and platelet-depleted human plasma. (A) Schematic of the experimental design where human whole blood (WB) was centrifuged to obtain platelet-rich plasma (PRP) and the PRP was further centrifuged to obtain either platelet-poor plasma (PPP) or platelet-free plasma (PFP); The PPP was treated with anticoagulant Apixaban (FXa inhibitor). PFP and Apixaban-treated PPP were both subjected to spectrophotometric monitoring of fibrin generation (measuring optical density of formed/polymerized fibrin over time at 405 nm) and the onset of fibrin generation (OFG), maximum optical density (also called maximum hemostatic potential or MHP) and area under the curve (also called overall coagulation potential or OCP) was recorded. (B1–B3) Effect of adding t-TLNP vs UNP in Apixaban-treated PPP, demonstrating that thrombin released by t-TLNP can restore OCP, OFG, and MHP parameters closer to the normal plasma baseline (increased OCP, reduced OFG, increased MHP) and this effect is enhanced when sPLA2 is added to accelerate thrombin release. (C1–C3) Effect of adding t-TLNP vs UNP in PFP, demonstrating that thrombin released by t-TLNP can restore OCP, OFG, and MHP parameters closer to the normal plasma baseline and this effect is enhanced when sPLA2 is added to accelerate thrombin release. * p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
Figure 4C1–C3 (and corresponding raw OD data and quantitative comparison in Figure S7 in the Supporting Information) show the detrimental effect of extreme platelet depletion (going from platelet-rich plasma or PRP to platelet-free plasma or PFP) on fibrin OCP, OFG, and MHP parameters and to what extent t-TLNPs are able to restore them. In comparison to the PRP baseline, PFP showed substantial debilitation (negative percent deviation for OCP and MHP parameters, positive percent deviation for OFG parameter) of fibrin (black curve for PRP vs deep blue curve for PFP in Figure S7 in the Supporting Information). Treatment of PFP with t-TLNPs significantly restored OFG closer to the PRP baseline and led to significant recovery in OCP and MHP toward PRP levels (raw OD data as the cyan curve in Figure S7A in the Supporting Information). Adding sPLA2 to t-TLNP treatment in PFP led to a further improvement in OFG, OCP, and MHP parameters (raw OD data as the purple curve in Figure S7A in the Supporting Information). UNPs were unable to recover any such parameters in PFP (raw OD data as the red curve in Figure S7A in the Supporting Information). Additional in vitro studies were carried out to assess whether t-TLNPs can have a dose response effect on fibrin generation in Apixaban-treated (i.e., anticoagulated) or platelet-depleted plasma. For these studies, the same fibrin generation assay in anticoagulated plasma or PFP was carried out with various doses of t-TLNPs that correspond to various doses of encapsulated thrombin (0.25–5 nM). Representative results from these studies are shown in Figure S9 in the Supporting Information, which indicate that indeed the modulation of t-TLNP dose (and hence encapsulated thrombin dose) can allow for modulation of fibrin restoration response, in both anticoagulated plasma and platelet-depleted plasma. Overall, these studies indicate, that even in an extreme platelet-depleted scenario (e.g., severe hemorrhage, severe thrombocytopenia, etc.), t-TLNPs can directly deliver thrombin to potentially rescue fibrin kinetics and restore hemostasis.
t-TLNPs Enhance Viscoelastometric Characteristics of Clots in Anticoagulant-Treated and Platelet-Depleted Human Whole Blood.
On the basis of the finding that t-TLNPs can restore fibrin generation and polymerization in anticoagulant-treated as well as platelet-depleted human plasma, we studied whether this capability of t-TLNPs can enhance the viscoelastometric properties of clots in human whole blood with similar anticoagulation-induced as well as platelet depletioninduced defects. For these studies the rotational thromboelastometry (ROTEM) methodology was used (Figure 5A), since ROTEM-based diagnostics have become clinically significant in the assessment of hemostatic defects in trauma and surgery.67 To induce an anticoagulation effect, human whole blood was directly pretreated with Apixaban (FXa inhibitor). To create platelet-depleted (thrombocytopenic, TC) blood, the whole blood was first fractionated into its components (RBC, leukocytes, platelets, plasma) and then reconstituted with a low (<20000/μL) platelet count. The resultant blood samples were subjected to ROTEM analysis in NATEM mode (CaCl2-induced clotting), and the clot formation time (CFT), clot formation rate (alpha angle), and early amplitude at 10 min (A10) were recorded. The rationale was that debilitation in thrombin generation due to an anticoagulant effect as well as a platelet depletion effect is expected to reduce the early kinetics of clot formation and also possibly affect the firmness (amplitude) of the growing clot. Treatment of such anticoagulated or platelet-depleted blood samples with t-TLNPs or “t-TLNPs + sPLA2” was studied to see whether this restores the clot kinetics and amplitude. Treatment with unloaded (empty) nanoparticles (UNPs) was used as a negative control. As before for fibrin generation studies in plasma, for the ROTEM studies on the various defects and t-TLNP treatment effects on whole blood we present the ROTEM parametric results in Figure 5B,C as percent (%) deviations from the healthy whole blood (WB) baseline, since the goal was to evaluate how close to the healthy WB baseline values the parameters could be restored upon t-TLNP treatment. As is evident from Figure 4B1–B3 (corresponding raw TEM-ograms and quantitative comparison are shown in Figure S10 in the Supporting Information) Apixaban-treated WB showed significantly prolonged CFT, reduced alpha angle, and reduced A10 in comparison to healthy WB, indicating slower clot formation and clot growth, as well as weaker clot quality.
Figure 5.

Evaluation of t-TLNPs in restoring clot viscoelastic parameters as measured by rotational thromboelastometry (ROTEM). (A) Schematic of the experimental design where human whole blood (WB) was directly treated with the anticoagulant Apixaban (FXa inhibitor) or was fractionated into blood components (RBC, platelets, leukocytes, plasma) and then reconstituted with a reduced number of platelets to create thrombocytopenic whole blood (TC Blood). Anticoagulated blood and thrombocytopenic blood were analyzed in ROTEM in NATEM mode (CaCl2-induced blood clotting resisting pin rotation), and the clot formation time (CFT), clot formation rate (also called alpha angle), and early clot amplitude at 10 min (also called A10) were monitored. (B1–B3) Effect of adding t-TLNP vs UNP in Apixaban-treated WB, demonstrating that thrombin released by t-TLNP can significantly restore CFT, alpha angle, and A10 parameters closer to the normal WB baseline (reduced CFT, increased alpha angle, increased A10) and this effect is enhanced when sPLA2 is added to accelerate thrombin release. (C1–C3) Effect of adding t-TLNP vs UNP in thrombocytopenic blood (TC Blood) demonstrating that thrombin released by t-TLNP can partially restore CFT, alpha angle, and A10 parameters closer to the normal WB baseline, but no statistical significance was observed in this improvement without or with sPLA2. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
Treatment of this anticoagulated blood with t-TLNPs but without sPLA2 significantly improved these parameters, reducing CFT and increasing alpha angle as well as A10 values toward WB baseline levels. These parameter improvements were further amplified when t-TLNPs and sPLA2 were added together, suggesting that diffusive as well as sPAL2-triggered release of thrombin from t-TLNPs could directly generate fibrin from fibrinogen to restore clot kinetics and clot robustness, even when the native prothrombinase activity is inhibited in the blood (e.g., by FXa inhibition here). UNP treatment did not indicate any such improvement and in fact worsened the parameters further, possibly due to a dilution effect. Figure 5C1–C3 (and corresponding raw data in Figure S11 in the Supporting Information) show the detrimental effects of the aforementioned ROTEM parameters in thrombocytopenic blood (TC blood). The mean CFT of TC blood was higher, and the mean alpha angle and A10 of TC blood were lower than those of healthy WB, suggesting an impairment in clot kinetics and robustness. Treatment of the TC blood with t-TLNPs as well as “t-TLNPs + sPLA2” was found to partially improve the CFT, alpha angle and A10 characteristics, but not at statistically significant levels in comparison to TC blood parameters. Treatment with UNPs did not show any such improvement trend. These results indicate that severe depletion of platelets (reducing the count from the normal ∼200000/μL to <20000/ μL) creates more severe debilitation of clot characteristics in comparison to coagulation factor inhibition, and while the t-TLNPs are highly efficient in restoring clot viscoelastometric characteristics in a “coagulation inhibition” setting, in a “severe platelet depletion” setting the t-TLNPs can only partially restore these characteristics.
t-TLNPs Enhance Fibrin Generation under Simulated Vascular Flow Environment Imaged in Real-Time Using BioFlux Microfluidic Setup.
The BioFlux microfluidic setup (Fluxion Biosciences, California, USA) allows a simulation of blood or plasma flow at physiological and pathological shear rates over bioactive-molecule-coated microchannels, using a customized flow controller system, and the channels can be imaged in real time using a fluorescence microscope to assess specific cellular and molecular processes (Figure 6A). This system was previously described in the assessment of “VBP + CBP”-decorated (i.e., targeted) vs control particle adhesion under a 25 dyn/cm2 shear stress flow on “vWF + collagen”-coated microchannels. Since t-TLNPs showed enhanced binding on the “vWF + collagen”-coated surface and also showed the capability of restoring/improving fibrin generation as well as ROTEM-based clot characteristics in anticoagulant-treated and platelet-depleted plasma and blood samples (results from Figures 4 and 5), we sought to study whether these capabilities of t-TLNPs enable enhanced fibrin generation/ restoration in hemostatically defective plasma under a simulated vascular flow environment on “VWF + collagen”-coated microchannels in the BioFlux setup. For these studies, the hemostatic defect was induced by a combination of “Apixaban anticoagulation + platelet depletion” in plasma, to render a drastic debilitation in endogenous thrombin generation capability of the plasma, which in turn would render a drastic reduction in fibrin. The treatment comparison was done between “t-TLNP + sPLA2” vs “UNP + sPLA2” to simulate the injury-site-relevant enzyme-triggered payload release.
Figure 6.

Evaluation of t-TLNPs in restoring fibrin generation under a simulated vascular flow environment in human plasma containing the combined hemostatic defect of platelet depletion plus anticoagulation. (A) Schematic of the BioFlux microfluidic setup and experimental design where human plasma containing fluorescently labeled platelets and fibrinogen (by Calcein and AlexaFluor647, respectively) were flowed over “vWF + collagen”-coated microchannel and fibrin formation was imaged in real time. (B) Representative fluorescence images of fibrin formation over time (0–12 min) in the microchannel with flows of platelet-rich plasma (PRP), platelet-depleted plus anticoagulated (Defective) plasma, Defective plasma treated with “t-TLNPs + sPLA2” and Defective plasma treated with “UNPs + sPLA2” showing that the combined defect of platelet depletion plus anticoagulation in plasma drastically reduces fibrin formation in comparison to that in PRP and treatment with “t-TLNPs + sPLA2” is able to restore fibrin generation even when platelet numbers were low. Treatment with “UNP + sPLA2” was unable to restore fibrin. (C) Representative dual-fluorescence images of the full microchannel surface at the experiment end point (12 min) showing a substantial number of blue platelets enmeshed in green fibrin in the channel containing PRP flow. In comparison, the channel with Defective plasma showed sparse platelets and minimal fibrin and Defective plasma treated with “t-TLNPs + sPLA2” showed fibrin recovery even though the platelets were sparse, while treatment with “UNPs + sPLA2” showed no such recovery. (D) Surface-averaged fluorescence intensity quantification of fibrin corroborating that treatment of Defective plasma with “t-TLNP + sPLA2” restores fibrin generation comparably to that of PRP. (E) D-dimer ELISA based quantification of digested fibrin from the microchannels further confirming that treatment of Defective plasma with “t-TLNP + sPLA2” restores the formation of cross-linked fibrin at concentrations comparable to those of PRP. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
The platelets in the plasma were prestained with calcein, and the plasma was also spiked with AlexaFluor-647-labeled fluorescent fibrinogen, such that the platelet accumulation and fibrin formation on the “vWF + collagen”-coated microchannel surface can be imaged under a fluorescence microscope. The flow experiments were maintained for 12 min, and the end point surface-averaged fibrin fluorescence intensity was analyzed. Following this the plasma flow was stopped, the channels were washed with a flow of saline, and the fibrin that formed within the channels was digested with plasmin to be quantified by a D-Dimer assay. Figure 6B shows representative images of fibrin fluorescence (pseudocolored green) in the channels over 0–12 min for healthy platelet-rich plasma (PRP), “Apixaban-treated + platelet-depleted” plasma (Defect plasma), and treatment effect of this “Defect plasma” with t-TLNPs vs UNPs in the presence of sPLA2. Representative movies of these four conditions are shown in Movie M6 in the Supporting Information (PRP), Movie M7 in the Supporting Information (Defect plasma), Movie M8 in the Supporting Information (Defect Plasma + t-TLNPs + sPLA2), and Movie M9 in the Supporting Information(Defect Plasma + UNPs + sPLA2), while the representative end point dual-fluorescence (platelets, blue; fibrin, green) images for each of the conditions at the completion of the experiment are shown in Figure 6C. As is evident from these movies and images, healthy PRP resulted in significant fibrin generation and the end point image showed substantial platelet accumulation within the fibrin mesh. In contrast, “anticoagulation + platelet depletion” resulted in drastic debilitation of fibrin formation in the channel and the end point image showed the accumulation of a small number of platelets and almost no fibrin. When “t-TLNPs + sPLA2” was added in this drastic defect condition, the fibrin fluorescence was restored over time and the end point image showed significant fibrin formation even though the platelet presence was low. Adding “UNPs + sPLA2” was unable to have any such fibrin restoration effect, and the images looked similar to those obtained for the defective plasma itself. Figure 6D shows surface-averaged fluorescence intensity based “net fibrin” quantification data at the experiment end point for each condition tested, and these results emphasized the capability of the “t-TLNPs + sPLA2” treatment group to significantly restore/ improve fibrin formation in the defective plasma. Figure 6E shows the D-Dimer ELISA based quantification of digested fibrin from the various experiment channel conditions. The D-Dimer concentration is a surrogate measurement for cross-linked fibrin concentration, and these data further corroborated that treatment of defective plasma with “t-TLNPs + sPLA2” was able to restore fibrin amounts comparable to those of PRP. The D-Dimer concentration for healthy platelet-rich plasma (PRP) was 120000 ± 15900 pg/mL, while that for “Defect plasma” was 16500 ± 7100 pg/mL, indicating a drastic reduction of fibrin generation when endogenous platelet-mediated and FXa-mediated mechanisms of thrombin amplification were reduced. These analyses also indicated that small amounts of fibrin might have formed in the “Defect plasma” channels but were too dispersed and not discernible by fluorescence imaging. In contrast, the D-Dimer concentration for defective plasma channels treated with “t-TLNPs + sPLA2” was 161000 ± 17800 pg/mL, while that for defective plasma channels treated with “UNPs + sPLA2” was 45000 ± 23300 pg/mL. This further confirms that sPLA2-triggered direct release of thrombin from “vWF + collagen” surface-adhered t-TLNPs was able to significantly restore fibrin formation from fibrinogen in “hemostatic defect” settings where endogenous abilities for thrombin generation (and hence fibrin formation) are debilitated due to combined anticoagulation and platelet depletion.
Prophylactic Dose of t-TLNPs Improve Hemostatic Efficacy in Tail-Clip Bleeding Model in Mice Suffering from Combined Effect of Platelet Depletion and Anticoagulation.
The in vivo feasibility of t-TLNPs was tested in mice using a tail-clip model, where the significant fibrin detriment caused by the combination of platelet depletion and anticoagulation, evident in the in vitro BioFlux studies, was induced in vivo (Figure 7A). Systemic administration of anti-CD42b (anti-GPIb) antibody can cause transient platelet clearance in mice, leading to a temporary thrombocytopenic state and impaired hemostatic ability.68 The tail-clip bleeding model in mice is also a standardized model to study the efficacy of hemostatic agents, where the tail is cut at 1–3 mm from the tip and the time for bleeding to stop as well as the total blood loss in that time is measured.69 Usually, bleeding from a clipped tail in thrombocytopenic (TC) mice is substantially prolonged in comparison to normal mice but ultimately stops (2–5 min in normal mice vs 15–20 min in thrombocytopenic mice; representative data are shown in Figure S12 in the Supporting Information). Since our BioFlux studies indicated that combining anticoagulation with platelet depletion drastically reduces fibrin formation, we hypothesized that this “combination effect” would lead to a hemostatic defect in mice more severe than that caused by thrombocytopenia alone and possibly lead to uncontrolled hemorrhage (bleeding does not stop). Before the experiments in mouse tail-clip model were conducted, studies were done to assess the circulation lifetime and biodistribution of t-TLNPs in mice. For this, Rhodamine B (RhB) labeled t-TLNPs were injected retro-orbitally in mice (125 μL injection volume per mouse at 1.14 × 1012 particles/ mL) and at various time points over 24 h (1, 6, 12, and 24 h) the blood was harvested from the inferior vena cava (IVC), the mouse was sacrificed, and the clearance organs (heart, lung, kidney, spleen, liver) were also harvested and homogenized. The RhB signal in harvested blood as well as the harvested clearance organ homogenate were measured via fluorescence spectrometry and quantified to particle concentration by utilizing a calibration curve of RhB-labeled t-TLNP particle concentrations against the corresponding fluorescence intensity. This enabled the quantification of percent (%) injected dose in blood and clearance organs over the 24 h period. As shown in Figure S13 in the Supporting Information, the circulation lifetime of the particles in mice was about 24 h, with low accumulation in the clearance organs over time. Of note, the t-TLNP-injected mice did not show any sign of systemic distress, further indicating that the particles are potentially safe in vivo. This allowed subsequent prophylactic administration in the mouse tail-clip model. When mice were administered with anti-CD42b antibody (platelet-depleting antibody) plus low-molecular-weight heparin (LMWH Enoxaparin, anticoagulant), the combined coagulopathic effect led to drastic impairment of hemostasis such that bleeding from clipped tail did not stop at all even beyond 15 min. Therefore, we rationalized that bleeding for 15 min in these “thrombocytopenic + coagulopathic” mice could be considered as the experiment end point to euthanize the mice and the treatment effect of t-TLNPs vs UNPs in restoring hemostatic capability in such mice could be expressed as “percent (%) of 15 min” for which bleeding occurred. Blood loss from the tail clip at 15 min (or earlier if hemostasis occurred) was measured by a spectrophotometric assessment of hemoglobin. Figure 7B shows the bleeding time (as a percent of 15 min), and Figure 7C shows blood loss results from these studies.
Figure 7.

Evaluation of prophylactic administration of t-TLNPs in restoring hemostatic efficacy in the tail-clip model in mice with significant bleeding due to the combined effect of platelet depletion and anticoagulation. (A) Schematic of the experimental design where mice were first made thrombocytopenic (TC Mouse) by anti-CD42b dose induced platelet clearance and then further dosed with anticoagulant (Enoxaparin) to induce combine a hemostatic defect (“Defect” mouse). t-TLNP or UNP treatment was administered in the “Defect” mice via an intravenous (retroorbital) route and allowed to circulate for 15 min, and then a tail-clip injury was performed to measure the bleeding time and blood loss. (B) Bleeding time data as a percent of 15 min time period showing that normal mice stopped bleeding in 3.11 ± 0.44 min while the combined effect of thrombocytopenia and anticoagulation in “Defect” mice resulted in continuous bleeding for 15 min (and beyond). Treatment of t-TLNPs in defect mice significantly restores the hemostatic capability, with the mice stopping bleeding in 6.18 ± 3.19 min, while treatment with UNPs has no such effect (mice continue bleeding for 15 min and beyond). (C) Blood loss analysis via a spectrophotometric measurement of hemoglobin in shed blood indicating that the combined effect of thrombocytopenia and anticoagulation in “Defect” mice results in significantly increased blood loss over the 15 min time period in comparison to normal mice. Treatment of “Defect” mice with t-TLNPs significantly reduces blood loss. In contrast, treatment with UNPs did not reduce blood loss but rather exacerbated it, possibly due to a dilution effect. *≤ 0.05, **p≤ 0.01, ***p ≤ 0.001.
As isevident from Figure 7B, normal mice stopped bleeding in 3.11 ± 0.44 min, whereas mice administered with anti-CD42b plus Enoxaparin (labeled as “Defect”) continued bleeding at 15 min (and beyond, thus designated as 100% bleeding at 15 min). Treatment with t-TLNPs was able to significantly reduce this bleeding, restoring bleeding cessation at 6.18 ± 3.19 min, whereas treatment with UNPs had no effect (bleeding continued at 15 min and beyond). The bleeding time data for the 15 min time window is shown as a Kaplan–Meier curve in Figure S14 in the Supporting Information, where the ability of t-TLNPs to significantly reduce bleeding time (the purple line shifting left toward the blue line) is evident. The corresponding blood loss data at the 15 min time point (Figure 7C) showed that normal mice had a blood loss of 18.06 ± 4.13 μL, whereas mice with a severe hemostatic defect had a significantly increased blood loss of 77 ± 10.41 μL (monitoring stopped at 15 min). Treatment of such “defect” mice with t-TLNPs reduced blood loss to 16.61 ± 7.82 μL, which was comparable to that in normal mice. Interestingly, the treatment of “defect” mice with UNPs further worsened blood loss (177.5 ± 29.47 μL), and this is possibly due to the dilution effect of the administered UNP injection volume which exacerbates coagulopathy without providing any hemostatic benefit. Altogether, these data indicate that in a setting of drastic coagulopathy and hemorrhage where the endogenous thrombin generation (hence fibrin formation) capability is considerably impaired, treatment with t-TLNPs can potentially restore hemostatic capability and reduce bleeding.
Emergency Dose of t-TLNPs Improve Hemostatic Efficacy in Liver Laceration Bleeding Model in Mice Suffering from Effect of Anticoagulation.
On consideration of the fact that a technology such as t-TLNPs could be potentially used not only for prophylactic management of bleeding risks but also for emergency management of acute hemorrhage (e.g., in trauma), studies were performed in a traumatic liver laceration injury model in mice where the particles were administered intravenously postinjury.70 In this model, 30 min prior to injury, mice were dosed intravenously with 1 U/g of unfractionated heparin (UFH) to induce a coagulation defect. At the 30 min time point, a midline laparotomy incision was made, preweighed gauze absorption triangles were inserted inside the abdominal cavity of the mouse without touching the liver, and then the right middle lobe of the liver was lacerated and resected, resulting in heavy bleeding in the abdominal cavity. Treatment (sham saline, control UNP, or t-TLNP) was administered via the femoral vein 1 min postinjury (125 μL of injection volume). The abdominal cavity was then stapled close, and the mean arterial pressure (MAP) of the mice was monitored for 20 min. At this 20 min time point the cavity was reopened to retrieve absorption triangles, and the difference between the preinjury and postinjury weight of the absorption triangles was recorded to assess blood loss in grams. Figure 8A shows the schematic of this experimental setup, with a representative anatomic image of the liver injury site. Following retrieval of the absorption triangles, the abdominal cavity was stapled close again and the mice were kept under observation for 3 h from the time of liver injury. At the 3 h time point the mice were sacrificed, and their clearance organs (uninjured part of liver, heart, lung, kidney, spleen) were harvested for histological staining and imaging with Carstair’s stain to assess the off-target clotting risk.71 Figure 8B shows the blood loss data from these experiments (n = 10 per group). As is evident from the data, heparin treatment resulted in a significant increase in blood loss from the liver injury (normal vs defect) and treatment with UNPs was unable to reduce blood loss in the “defect” mice, whereas treatment with t-TLNPs significantly reduced blood loss in the “defect” mice. Figure S15 in the Supporting Information shows the MAP data in “defect” mice from these studies, while Figure S16 in the Supporting Information shows representative histology images (Carstair’s staining) of the harvested organs postsacrifice. The MAP data show that administration of the UNPs or t-TLNPs did not cause any drastic fluctuation of arterial pressure in comparison to sham (saline treatment), which is indicative of the systemic safety of the particles. The histology data of the organs did not show any sign of clotting in clearance organs, since a fibrin stain (usually bright red in Carstair’s stain) was not visible in any of the organs for UNP-treated or t-TLNP-treated mice (histological staining similar to that for saline-treated sham mice). Altogether these results suggest that postinjury emergency administration of t-TLNPs can render targeted hemostatic efficacy while maintaining systemic safety, in an acute traumatic hemorrhage model.
Figure 8.

Evaluation of emergency administration of t-TLNPs in restoring hemostatic efficacy in liver laceration bleeding model in anticoagulated mice. (A) Schematic and representative anatomic picture of liver laceration model in mice where treatment (sham saline, UNP, or t-TLNP) was administered post-injury and blood loss from the injured liver was measured by preweighed gauze. (B) Blood loss data (in grams, g) from mouse liver injury model studies showing a significant increase in bleeding from an injured liver in defect (heparinized) mice in comparison to normal (nonheparinized) mice. Treatment with UNPs was unable to reduce blood loss, but treatment with t-TLNPs was able to significantly reduce blood loss. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
DISCUSSION
Severe hemorrhage associated with trauma, surgery, congenital or drug-induced coagulopathies, etc. can be life-threatening and necessitates rapid hemostatic management via external/topical, intracavitary, and intravenous routes to reduce mortality risks. While bleeding from accessible and external injuries can be efficiently mitigated by tourniquets, bandages, pressure dressings, foams, and glues, options to manage internal and diffuse bleeding are limited to the transfusion of blood products (whole blood, blood components, concentrated are recombinant coagulation factors, etc.). The major hemostatic players in blood are platelets and coagulation factors, which through a complex concert of interactions and reactions localized at the injury site generate thrombin, which then converts fibrinogen to fibrin for hemostatic action. Therefore, the transfusion of platelets and coagulation factors (as plasma, prothrombin complex concentrate, cryoprecipitate, or individual recombinant factors) has become clinically significant in hemorrhage management. However, such products are donor dependent, are highly expensive, and often have a short shelf life due to contamination risks, which effect on-demand availability. Pathogen reduction technologies (PRTs) as well as reduced temperature processing and storage (cooling, freeze-drying, cryopreservation, etc.) are currently being studied for these products to improve their availability, portability, and transfusion logistics. In parallel, significant research interest has emerged regarding the design of donor-independent biosynthetic intravenous hemostatic technologies using distinct biomaterials and nanotechnology approaches.35–45
To this end, one approach that has not been extensively explored is the direct delivery of thrombin to a vascular injury site using an intravenous route. It is well-recognized that thrombin-induced conversion of fibrinogen to fibrin is critical for hemostatic clot formation and stability, and this has led to several technologies (e.g., Tisseel and Floseal by Baxter, Surgiflo by Ethicon, etc.) that deliver thrombin topically to externally accessible wounds (e.g., localized use in surgery) to accelerate hemostasis. However, thrombin cannot be directly delivered via an intravenous route in systemic circulation because this will cause fibrin generation in circulation, leading to systemic clotting risks. In fact, circulating thrombin is short-lived and is promptly neutralized by circulating inhibitors such as antithrombin III (AT III), whereas at the vascular injury site the procoagulant platelet-mediated “thrombin burst” results in locally amplified uninhibited thrombin concentrations that result in fibrin clot formation for hemostasis. Therefore, a targeted delivery strategy that can encapsulate thrombin to minimize its exposure in circulation while enabling active localization and release at the vascular injury site, can become a promising bioinspired approach for hemostatic applications. Interest in this strategy is evident from recent studies in loading thrombin in iron oxide nanoparticles, in liposomes which are then loaded within actual platelets, in chitosan nanoparticles, etc. These approaches have been predominantly evaluated in vitro for their ability to generate fibrin in normal plasma and in vivo in some cases for topical/external applications. The thrombin-loaded chitosan nanoparticles have been tested in vivo in an intravenous delivery route, but only in a cancer model to induce tumor vasculature infarction.72 Also, none of the above particle designs has any active targeting capability to the vascular injury site and none of them has been tested in drastic coagulopathic and hemorrhagic settings regarding the recovery of hemostatic fibrin. Therefore, we sought to address these technological and functional gaps with our t-TLNP design and evaluation.
The t-TLNP system actively targets the vascular injury site via peptide-mediated anchorage to vWF and collagen, which mimics the injury site-specific binding mechanisms of our natural hemostatic cells, the platelets. Our microfluidic studies with fluorescently labeled “VBP + CBP”-decorated particles flowed over a “vWF + collagen”-coated surface vs albumin-coated surface confirmed this specific anchorage ability. Additionally, our biosafety assays confirmed that the “VBP + CBP”-decorated particles do not have unwanted activation of healthy endothelial cells, resting platelets, neutrophils, and complement C3, suggesting that the particles would be physiologically safe in vivo. Our studies also showed that the diffusive release of the particle-encapsulated payload was very low, and the shear-induced release of the payload was also very low, in comparison to the significantly higher release of the payload upon particle destabilization by the enzyme sPLA2 (predominantly due to hydrolysis of sn-2 ester bond of phospholipids in the liposome membrane lipids). Such phospholipase enzymes have been reported to be upregulated at vascular injury sites due to production from activated platelets, and therefore we rationalize that injury-site-anchored t-TLNPs can be amenable to sPLA2-triggered enhanced release of encapsulated thrombin.59,60 For our in vitro and in vivo studies the t-TLNPs were used to render a thrombin concentration of 1–2.5 nM (please see Methods for dosage details), which is at the lower end of what is needed to render fibrin formation.73 In circulation there are thrombin inhibitors such as antithrombin and α-macroglobulin that can neutralize thrombin rapidly in circulation, but thrombin is protected from such inhibition at the injury site when it associates with fibrin.74 Therefore, a low fraction of the 1–2.5 nM of thrombin that may leak out from the t-TLNPs by diffusion is expected to be quickly neutralized in circulation, but when the particle-encapsulated thrombin is released at higher amounts due to “diffusion + sPLA2-triggered particle degradation” at the injury site from site-localized t-TLNPs, this thrombin will be protected from inhibition by the fibrin forming at the site and will continue to augment hemostasis.
Our in vitro studies utilizing a spectrophotometric assessment of fibrin generation in human plasma and a ROTEM-based assessment of clot kinetics and viscoelastic properties in human blood indicated that thrombin released from t-TLNPs can indeed restore fibrin generation and the corresponding clot characteristics when the endogenous thrombin (and hence fibrin) generation capability is drastically affected due to anticoagulation or platelet depletion. Therefore, we combined these two detrimental effects (anticoagulation + platelet depletion) to render a drastic coagulopathy condition in human plasma, and our BioFlux-based in vitro microfluidic studies indicated that, even under such severe hemostatic impairment condition, t-TLNPs can restore fibrin generation and polymerization comparable to those of the positive control of healthy PRP. Subsequently, we created this severe hemostatic impairment in vivo in mice systemically administered with a combination of antiplatelet antibody and anticoagulant agent, and these mice showed incessant bleeding from a tail-clip injury for over 15 min. Intravenous treatment with t-TLNPs prophylactically administered in these mice was able to render improved hemostasis and significantly reduced blood loss. Additional in vivo studies using a liver laceration acute hemorrhage model in heparinized mice showed that postinjury intravenous administration of t-TLNPs could significantly reduce blood loss. In hemodynamic observations (e.g., of MAP) as well as posteuthanasia histology analyses of clearance organs, no indication of off-target clotting was found. Therefore, considering the fact that our studies showed a t-TLNP circulation lifetime of ∼24 h in mice, one can envision the translational advancement of such a technology in both prophylactic and emergency settings. Altogether, our in vitro and in vivo studies provide exciting evidence for t-TLNPs as an effective platelet-inspired intravenous hemostatic nanomedicine.
Our current studies have a few potential limitations that need to be addressed in continued and future phases of our studies. First, for all of our studies we have used only a single dose or concentration of t-TLNPs; future studies will be directed at a detailed evaluation of the pharmacology and toxicology profiles of these nanoparticles at escalating and recurrent doses, such that the therapeutic window and maximum tolerated dose can be determined. In association with this, we will also need to determine dose-dependent systemic thrombotic risks, if any. Our in vitro studies showed that fibrin kinetics and output levels could be modulated in a dose-dependent manner with t-TLNPs, and this provides a rationale to expand such dose modulation studies in vivo in the future to assess safety and efficacy. Second, an evaluation of escalating doses and recurrent doses will need to include a detailed immunogenicity assessment (complement activation, cytokine profile, etc.), especially in larger animal models. Third, the t-TLNP system will need to be evaluated in emergency applications in more complex trauma models of hemorrhagic shock, where such technologies could have the highest life-saving impact. Trauma-induced coagulopathy (TIC) presents highly complex scenarios where a combination of platelet depletion and dysfunction, coagulation factor depletion, and maladaptive inflammatory responses result in significant hemostatic impairment.75–79 Future studies will need to evaluate whether in such complex hemorrhagic settings t-TLNPs can bypass all of the coagulatory dysfunctions and directly deliver sufficient thrombin at the bleeding injury site for rapid hemostasis and hemodynamic stabilization. Additionally, future studies can explore the potential of combining t-LNPs (a fibrin-generating system) with other hemostatic agents (e.g., tranexamic acid or TXA, a plasmin-inhibiting fibrin-stabilizing drug) to enable complementary hemostatic benefit.80 Such approaches for t-TLNPs can also be envisioned with regard to integration with other biomaterial-based fibrin-stabilizing hemostats (e.g., PLPs, PolySTAT, etc.) for combinatorial effects.
CONCLUSION
Drawing inspiration from the natural platelet’s hemostatic mechanisms of injury-site-specific adhesion via binding to vWF and collagen and amplification of thrombin via membrane-surface-catalyzed procoagulant activity, we have designed a nanomedicine system, termed a targeted thrombin-loaded lipid nanoparticle (t-TLNP), that can undergo peptide-ligand-mediated adhesion to vWF and collagen and can release thrombin at its site of adhesion. The t-TLNP system demonstrated a promising ability to restore fibrin generation and clot characteristics in human plasma and blood when the natural hemostatic abilities were impaired by anticoagulation and platelet depletion. This ability of t-TLNPs was also demonstrated with real-time imaging under a simulated vascular flow environment in a microfluidic setup. The in vivo safety and hemostatic efficacy of t-TLNPs was established by their ability to significantly reduce bleeding in a tail-clip injury model in coagulopathic mice with prophylactic (preinjury) administration, as well as in a liver-laceration acute hemorrhagic injury model in coagulopathic mice with emergency (postinjury) administration. In conclusion, our studies demonstrate the significant promise of t-TLNPs as an intravenous nanomedicine that can directly deliver thrombin in a vascular-injury-site-targeted fashion, to enable site-localized fibrin generation for hemorrhage control.
METHODS
Study Design.
The purpose of the study was to test the hypothesis that nanoparticles which mimic injury-site-targeted specific adhesion mechanisms of platelets via binding to vWF and collagen, and mimic the procoagulant thrombin amplification function of platelets by the site-localized direct delivery of thrombin, can locally generate fibrin from fibrinogen to augment hemostasis even when the native platelets are depleted and endogenous coagulation reactions are inhibited. Such an intravenous nanomedicine system enabling targeted direct delivery of thrombin can provide significant benefit in treating bleeding complications stemming from congenital, drug-related, disease-associated, and trauma-induced platelet and coagulation dysfunctions. To achieve platelet-mimetic vWF binding and collagen binding, small-molecular-weight peptides having vWF and collagen adhesion specificities were synthesized and decorated on a liposomal nanoparticle surface. Liposomes were used as a model nanoparticle platform here because of their established clinical history and their capability to enable combinatorial peptide decorations via lipid–peptide self-assembly. The resultant “VBP + CBP”-decorated liposomes were loaded with thrombin. DSPC was used as one of the lipid components of these liposomal systems, since DSPC is amenable to degradation by injury-site-specific enzyme sPLA2, resulting in the destabilization of the injury-site-anchored liposomes for enhanced release of thrombin that can then generate fibrin locally at a rapid rate for hemostatic action. These targeted thrombin-loaded lipid nanoparticles (t-TLNPs) were characterized for their size using dynamic light scattering (DLS) and cryo-transmission electron microscopy (Cryo-TEM). The thrombin loading and release kinetics from t-TLNPs were characterized by spectrophotometric assays using an appropriate thrombin ELISA. The ability of t-TLNPs to undergo platelet-mimetic adhesion on an injury-site-relevant “vWF + collagen”-coated surface under flow was confirmed using BioFlux microfluidics imaged with inverted fluorescence microscopy. The physiologically relevant safety parameters of t-TLNPs were assessed by evaluating the t-TLNP incubation effect on healthy endothelial monolayer activation, resting platelet activation/aggregation, neutrophil activation/NET-osis, and plasma complement factor C3 activation. The ability of t-TLNPs (without and with sPLA2 trigger) to rescue fibrin generation in anticoagulant-treated and platelet-depleted human plasma was established using an optical density (OD) based method to record fibrin kinetics. In these studies, normal plasma (no platelet depletion or anticoagulation-associated hemostatic defect) was used as a positive control. The effect of this t-TLNP-mediated fibrin rescue in restoring clot kinetics and robustness in anticoagulant-treated and platelet-depleted human blood was established by rotational thromboelsatometry (ROTEM). In these studies, normal whole blood (no platelet depletion or anticoagulation-associated hemostatic defect) was used as a positive control. Additionally, the ability of t-TLNPs to rescue fibrin in plasma with a severe hemostatic defect due to combined anticoagulation and platelet depletion was established using real-time imaging under simulated vascular flow conditions using BioFlux microfluidics imaged with inverted fluorescence microscopy. All human plasma and human blood studies described above were performed with human blood drawn from healthy donors using the Case Western Institutional Review Board (IRB) approved protocol. Finally, the hemostatic efficacy of t-TLNPs was evaluated in vivo using prophylactic administration in a tail-clip ableeding model in mice bearing a severe hemostatic defect due to combined anticoagulation and platelet depletion (n = 3 per group), as well as emergency administration in a liver-laceration acute bleeding model in mice bearing a severe hemostatic defect due to anticoagulation (n = 10 per group). The tail-clip injury model in mouse was used under protocols approved by the Case Western Reserve University Institutional Animal Care and Use Committee (IACUC), and since the “anticoagulation + platelet depletion” induced combined hemostatic defect leads to uncontrolled incessant bleeding from the tail-clip injury for 15 min and beyond, the experiment end point was considered to be at 15 min, when mice were humanely euthanized. The liver-laceration injury model in mouse was used under protocols approved by University of Pittsburgh IACUC, and here the blood loss was measured at the 20 min time point following injury and the mice were observed for an additional time (up to 3 h following injury) followed by euthanasia, organ harvesting, and histology.
Materials.
Human alpha thrombin and plasmin were obtained from Haematologic Technologies (Essex Junction, VT, USA). 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)1000] (DSPE-mPEG1000), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)2000] maleimide (DSPE-PEG2K-Mal), and cholesterol were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Rhodamine B dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DHPE-RhB) was purchased from Invitrogen (Carlsbad, CA, USA). The peptides C(GPO)7 (CBP) and CTRYL-RIHPQSWVHQI (VBP) were purchased from Bachem (Torrance, CA, USA). Sterile saline (0.9% NaCl) was purchased from Baxter (Deerfield, IL, USA). Cellulose dialysis tubing (MWCO 100 K), calcium chloride, chloroform, methanol, dimethyl sulfoxide, apixaban, 4% paraformaldehyde, BeadBug homogenizing tubes, AlexaFluor 647 conjugated fibrinogen, calcein, collagen (equine Type I), von Willebrand factor and D-Simer Human ELISA Kits were purchased from Fisher Scientific (Pittsburgh, PA, USA). Cholesterol, calcium ionophore A23187, fibrinogen, phospholipase A2 (bovine pancreas), and enoxaparin sodium were purchased from MilliPore Sigma (Burlington, MA, USA). Human Thrombin ELISA Kits were purchased from Abcam (Cambridge, MA, USA). The Complement C3 and Complement C3a des Arg ELISA kit was purchased from Promega Corporation (Madison, WI, USA). Econo-Column Chromatography columns were purchased from Bio-Rad (Hercules, CA, USA). G-100 Sephadex beads were purchased from GE Healthcare (Chicago, IL, USA). For ROTEM studies, all reagents were purchased from Werfen USA (Bedford, MA, USA). For platelet lumi-aggregometry studies, cuvets, stir bars, and adenosine diphosphate (ADP) were purchased from Bio/Data (Horsham, PA, USA). For BioFlux studies, the flow controller, tubings, and microfluidic plates were purchased from Fluxion Biosciences (Alameda, CA, USA). Sytox Green and glass slides were purchased from Themo Fisher Scientific (Waltham, MA). VECTASHIELD Antifade Mounting containing DAPI was purchased from Vector Laboratories (Newark, CA). The EasySep direct human neutrophil isolation kit was purchased form STEMCELL Technologies (Vancouver, Canada). For in vivo studies with mice, C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). Mouse platelet depleting anti-CD42b antibodies were obtained from Emfret Analytics (Eibelstadt, Germany). Ketamine and xylazine were obtained from Patterson Veterinary (Greeley, CO, USA).
Manufacture and Characterization of t-TLNPs.
The cysteinylated VBP and CBP peptides were conjugated to DSPE-PEG2000-Mal via a maleimide–thiol reaction to obtain DSPE-PEG2K-VBP and DSPE-PEG2K-CBP. These conjugates were combined at 1.25 mol % each with DSPC (41.5 mol %), cholesterol (40 mol %), DSPE-mPEG1000 (15 mol %). and DHPE-RhB (1 mol %) in 1/1 chloroform/methanol, and the solvent mixture was rotary-evaporated under vacuum. The resultant thin lipid film was rehydrated with a solution of 540 nM human alpha thrombin in Tris buffered saline (TBS), sonicated for 30 min, and then subsequently extruded five times through a 200 nm pore size polycarbonate filter to yield thrombin-loaded “VBP + CBP”-decorated liposomal vesicles (t-TLNPs). These t-TLNPs were passed through a column packed with TBS-swelled G-100 Sephadex beads, to remove unencapsulated thrombin, and the isolated t-TLNPs were characterized by DLS and Cryo-TEM to the measure size distribution. The “VBP + CBP”-decorated nanoparticles and control (undecorated) nanoparticles were evaluated for their specific adhesion capabilities utilizing a BioFlux 200 microfluidic system (Fluxion Biosciences) and observed under inverted fluorescence microscope imaging. This system allows simulation of a physiologically relevant vascular flow environment. Here, the microfluidic channels were incubated with either 0.3 wt % bovine serum albumin (BSA) in water or 40 μg/mL equine type 1 fibrillar collagen and 10 μg/mL vWF in 20 μM acetic acid for 1.5 h. Unbound albumin and collagen were removed with a saline rinse. Next, control nanoparticles (undecorated) or targeted nanoparticles (“VBP + CBP”-decorated) were flowed over the collagen- or albumin-coated channels under a shear stress of 25 dyn/cm2 for 10 min. Additionally, for some experiments control and targeted nanoparticles were first incubated in platelet-free plasma (PFP) for 60 min and then flowed over the coated channels in PFP instead of saline. Fluorescence microscopy imaging of particle Rhodamine B label was used to quantify the extent of particle adhesion to the coated surfaces. Thrombin release kinetics was measured by sealing the column-separated t-TLNPs in a MWCO 100 K dialysis bag, placing the bag in a TBS reservoir, and removing aliquots from the reservoir at predefined time points over the course of 6 h (fresh TBS was added back to the reservoir appropriately after each aliquot removal). In separate studies, similarly prepared t-TLNPs were mixed with 25 μg/mL sPLA2, sealed in dialysis tubing, and subjected to a similar analysis. Thrombin concentrations from the collected aliquots were evaluated using a human thrombin ELISA and plotted over time to determine the release profile. The total thrombin loading was determined by adding a 1/1 methanol/chloroform mixture to t-TLNPs to induce complete particle dissolution and exhaustive release of thrombin and determining this exhaustively released thrombin concentration with ELISA. In addition to t-TLNPs, control untargeted empty nanoparticles (UNPs) were manufactured using the same methods described above, but with the lipid film rehydration performed with buffer only (i.e., no thrombin). Additionally, to characterize the particle stability under shear, experiments were performed with targeted nanoparticles loaded with carboxyfluorescein (CF) instead of thrombin. Here, the targeted particles were first flowed over “vWF + collagen”-coated microchannel surfaces to allow them to bind and become immobilized and then subjected to a flow of fresh saline at a range of shear stresses (5–50 dyn/cm2) for 30 min. The effluent was collected and transferred to a black 96-well plate in a plate reader, and the fluorescence intensity of CF (excitation 485 nm, emission 528 nm) was measured in the effluent. These data were converted to CF concentration using a standard calibration curve generated using serial dilutions of CF. Shear-induced CF release was compared to diffusive, sPLA2-triggered, and exhaustive (induced by 1/1 methanol/chloroform) release of CF.
Evaluation of Peptide-Decorated Nanoparticles with Healthy Endothelial Cells, Resting Platelets, Resting Neutrophils, and Complement C3.
For endothelial incubation studies, microfluidic channels were fabricated by assembling three layers, including a glass slide as the bottom layer, a double-sided adhesive (DSA) film as the middle layer, and poly(methyl methacrylate) (PMMA) as the top layer. The microchannels were incubated with 0.2 mg/mL fibronectin for 1 h at 37 °C. Human pulmonary microvascular endothelial cells (HPMECs, from Lonza, Basel, Switzerland) were seeded onto the microchannels and cultured with 5% CO2 at 37 °C under a 100 μL/min flow of the medium for 48–72 h until a confluent monolayer was formed over the fibronectin-coated surface. Cultured confluent HPMEC monolayers were washed with fresh culture medium and then incubated with TNF-α at 20 μM or control nanoparticles (UNP) or targeted nanoparticles (t-LNP) particles at 4.54 × 1012 particles/mL for 2 h at 37 °C. The HPMECs were then rinsed again with fresh culture medium and fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature. Fixed HPMECs were rinsed twice with PBS and blocked with 2% BSA for 1 h at room temperature. After washing with PBS, HPMECs were incubated with DAPI and sheep polyclonal antihuman vWF antibody (Abcam) conjugated with fluorescein isothiocyanate (FITC, 1/100 v/v dilution) for 1 h at room temperature in the dark. Images were then acquired across the microchannel at 10× using a fluorescence microscope. For platelet lumi-aggregometry, human blood was centrifuged (150g, 15 min) to obtain platelet-rich plasma (PRP), 400 μL of PRP was added to cuvets with ADP or t-LNP, and platelet aggregation was monitored on a BioData platelet aggregometer. For neutrophil studies, neutrophils were isolated from human blood using immunomagnetic separation. Isolated neutrophils (1 × 106 cells/mL) were plated on fibrinogen-coated glass slides and allowed to adhere for 30 min. Control particles or t-LNPs were then added on the slides and incubated with the neutrophils for 1h. Following this, the neutrophils were gently washed with saline. Similarly isolated neutrophils treated with 25 μM A23187 (calcium ionophore) were used as a positive control. The slides were then fixed with 4% PFA for 5 min, washed with saline, stained with 167 nM Sytox Green for 15 min, washed again with saline, mounted with a VectaShield mounting solution, secured with a coverslip, and imaged using a Leica HyVolution SP8 confocal microscope. The fluorescence intensity of the Sytox Green signal was quantified using ImageJ software, as a marker of neutrophil activation and NET-osis. A Complement C3 activation assay was carried out by incubating platelet-rich plasma with saline or control (undecorated) nanoparticles or targeted (“VBP + CBP”-decorated) nanoparticles (t-LNPs) and analyzing using ELISA kits for C3 and C3a des Arg.
Fibrin Generation Assay with t-TLNPs in Human Plasma with Induced Hemostatic Defects.
The optical density (OD) based fibrin generation assay (FGA) was adapted from methods described by Curnow et al.66 In this assay, coagulation in plasma is initiated by CaCl2 and the temporal OD change of the plasma due to formation and polymerization of fibrin (from plasma fibrinogen) is monitored by measuring the absorbance at 405 nm. Citrated human whole blood (WB) was centrifuged at 150g for 15 min at room temperature to obtain platelet-rich plasma (PRP), which was further centrifuged at 2000g for 20 min to obtain platelet-poor plasma (PPP, platelet count <50000/ μL) and at 13000g for 5 min to obtain platelet-free plasma (PFP, platelet count <5000/μL). An assay-specific coagulation buffer consisting of CaCl2 (35 mM) and s trace amount of thrombin (1 U/ ml) in TBS (66 mM Tris and 130 mM NaCl, pH 7) was prepared. In a 96-well plate, 60 μL of PPP was combined with 40 μL of buffer. Using a plate reader, absorbance values at 405 nm were recorded every 1 min for 1 h to construct the OD-based fibrin generation curves. The coagulation defect in PPP was induced by preincubating PPP with Apixaban (FXa inhibitor) at a concentration of 120 nM for 5 min before commencing the assay. We rationalized that, since FXa is a major component of the prothrombinase complex, inhibition of FXa would reduce thrombin generation and hence reduce fibrin formation. Fibrin generation curves in Apixaban-treated PPP were compared to those of PPP alone. In separate experiments, a variation of this fibrin generation assay was performed directly with PFP without Apixaban treatment. Treatments of t-TLNP, “t-TLNP + sPLA2”, or UNP (concentration of 5.675 × 1011 particles/mL) were added to “PPP + Apixaban” or PFP, and the resultant fibrin generations were compared to those in PPP and in PRP. This t-TLNP dose concentration was to achieve a 2.5 nM “exogenously delivered” thrombin concentration for each test condition in the assay. In both variations of the fibrin generation assay, the parameters of overall coagulation potential (OCP), onset of fibrin generation (OFG), and maximum hemostatic potential (MHP) were monitored. OCP is the total area under the OD curve and represents the total amount of fibrin formed during the experiment duration. OFG is the time at which fibrin generation begins, and MHP is the maximum absorbance value of the curve that reflects the maximum steady-state level of fibrin formed. For studies involving treatment of “PPP + Apixaban” with t-TLNP, “t-TLNP + sPLA2” or UNP, data were presented as percent (%) deviations from PPP plasma baseline values of OCP, OFG, and MHP. For studies involving treatment of PFP with t-TLNP, “t-TLNP + sPLA2” or UNP, data were presented as percent (%) deviations from PRP baseline values of OCP, OFG, and MHP.
ROTEM Studies with t-TLNPs in Human Blood with Induced Hemostatic Defects.
Rotational thromboelastometry (ROTEM) is a clinically established method for real-time monitoring of whole blood clotting kinetics and clot mechanical properties. In this method, 340 μL of “blood + reagents” is held in a cup and a pin suspended on a ball-bearing mechanism is lowered into the cup to rotationally oscillate through 4°75′ every 6 s with a constant force. As the blood clot forms and grows in strength, it impedes the rotation of the pin and this mechanical impedance is detected optically using a charge-coupled-device image sensor system.67 The CaCl2-induced blood clotting modality (termed NATEM in ROTEM) allows real-time monitoring of this process as “endogenous clotting capability”, and any defect in the clotting mechanism (e.g., coagulation factor deficiency, platelet deficiency, etc.) results in a delay of clot formation, clot growth rate, and reduction in mechanical impedance. Therefore, this method enabled the investigation of the effect of t-TLNPs, “t-TLNPs + sPLA2”, and UNPs on CaCl2-induced blood clotting in human whole blood (WB) with either a coagulation defect or a platelet depletion (thrombocytopenic, TC) defect. Since the defects in thrombin generation and hence fibrin formation drastically affect the initial phases of clot growth, we monitored the clot formation time (CFT), clot growth rate (alpha angle), and clot amplitude at 10 min (A10) parameters in the ROTEM profile. Human WB was obtained from healthy donors with consent using an IRB-approved protocol. The coagulation defect was induced by preincubating WB with Apixaban (FXa inhibitor) at a concentration of 120 nM for 5 min before commencing the assay. The platelet-depleted TC Blood was made by (1) first centrifuging WB to isolating RBCs, PRP, and PFP, (2) then diluting PRP with PFP to form TC plasma containing <20000/μL of platelets, and (3) finally reconstituting the RBCs with TC plasma. The two defects (FXa inhibition and platelet depletion) were studied independently. Similarly to the fibrin generation assay, the nanoparticles (t-TLNPs and UNPs) were added at concentration of 5.67 × 1011 particles/mL to the blood samples to achieve a 2.5 nM “exogenously delivered” thrombin concentration in each ROTEM cup. For the “t-TLNP + sPLA2” group the sPLA2 was added with the t-TLNPs at a concentration of 25 μg/mL, and this mixture was added to the blood sample to commence the ROTEM assay. For studies involving the treatment of “WB + Apixaban” and TC Blood with t-TLNP, “t-TLNP + sPLA2”, or UNP, data were presented as percent (%) deviations from WB baseline values of CFT, alpha angle, and A10 parameters on the ROTEM NATEM modality.
BioFlux Microfluidic Studies of t-TLNP Effect on Fibrin Generation under Flow in Coagulopathic Plasma.
The BioFlux 200 microfluidic system observed under inverted fluorescence imaging was used to simulate a physiologically relevant vascular flow environment to study injury-site-targeted fibrin formation by t-TLNPs in coagulopathic plasma. To simulate an injury-site-relevant subendothelial matrix surface presenting vWF and collagen, microchannels in a 24-well BioFlux plate were coated by incubating with a solution of type I equine collagen (40 μg/mL) and vWF (10 μg/mL) in 20 μM acetic acid for 1.5 h and subsequently washing off with saline. Highly coagulopathic plasma was made by combining the Apixaban-induced anticoagulation effect with the platelet depletion effect by first creating TC plasma from PRP and then incubating it with 120 nM Apixaban. We rationalized that, since in our FGA studies with human plasma as well as ROTEM studies with human blood, both Apixaban-induced FXa inhibition and platelet depletion individually led to a drastic detriment in fibrin formation that could be rescued by t-TLNPs, therefore combining these two scenarios (platelet defect + coagulation defect) would enable evaluation of the “fibrin rescue” ability of t-TLNPs in a more severe coagulopathy setting. For each channel, 500 μL of plasma (PRP or defective) with calcein-stained platelets and AlexaFluor-647 stained fibrinogen was introduced into the inlet well and then flowed over the coated channel surface at a shear stress of 25 dyn/cm2 for 12 min, and “platelet accumulation + fibrin formation” was imaged under a Zeiss AxioObserver inverted fluorescence microscope with images being automatically captured every 15 s. The effect of “t-TLNPs + sPLA2” vs “UNPs + sPLA2” on fibrin formation in the defective plasma was studied by adding the nanoparticles (5.67 × 1011 particles/mL concentration) with sPLA2 (25 μg/mL) in the flow volume. After completion of the time-lapse experiments, the channels were washed with a saline flow and end point images taken. The surface-averaged fibrin fluorescence intensity (SAFI) was determined in these end-point images to be presented as “net fibrin generation” fluorescence data. Clots were then digested/lysed by flowing plasmin over them, and the lysis product was analyzed by D-Dimer ELISA to quantify the amount of cross-linked fibrin that had formed in each channel. Each condition (PRP vs defect plasma vs defect plasma with t-TLNP treatment vs defect plasma with UNP treatment) was studied for both SAFI and D-Dimer analyses in triplicate.
Evaluation of Circulation Lifetime and Organ Biodistribution Studies of t-TLNPs in Mice.
Mice were anesthetized (using 2% isoflurane gas) and retro-orbitally injected with 125 μL of Rhodamine B-labeled nanoparticles (NPs) at a concentration of 1.14 × 1012 particles/mL and allowed to recover. After 1, 6, 12, or 24 h, the mice were anesthetized and underwent a midline laparotomy, and blood was collected from the inferior vena cava (IVC). Livers, lungs, kidneys, spleens, and hearts were excised and placed in preweighed homogenizing tubes. The samples were then freeze-dried and their dry weights recorded. The dry organs were homogenized with a BeadBug Microtube Homogenizer. The blood and homogenized organs were then mixed vigorously with 1/1 methanol/chloroform to disassemble the Rhodamine B labeled lipids of nanoparticles in the samples. The samples were then centrifuged (20 min, 12000g) to separate the organ tissue from the supernatant containing Rhodamine B labeled lipids. The supernatant was analyzed using a Biotek Synergy H1 Plate Reader (λex = 560 nm, λem = 580 nm) to assess Rhodamine B fluorescence. The biodistribution of the nanoparticles was determined by calculating the percentage of injected dose per organ utilizing an appropriate calibration curve that correlated the RhB fluorescence intensity with the particle concentration.
Evaluation of t-TLNPs in Tail-Clip Bleeding Model in Severely Coagulopathic Mouse.
All experiments were carried out in accordance with protocols approved by the CWRU IACUC. Wild-type C57/BL6 mice (average weight 20 g) were injected intraperitoneally with anti-CD42b (anti-GPIbα) antibody at a dose of 0.5 μg/kg. The normal platelet count is mice is approximately 1500/nL and upon a platelet-depleting anti-CD42b antibody dose, thrombocytopenia TC (<500 platelets/nL) was confirmed 18–24 h later, by drawing blood retro-orbitally and measuring the platelet count using a Heska HemaTrue system. After the confirmation of TC mice, these mice were further dosed with low-molecular-weight heparin (LMWH or Enoxaparin at 12.3 mg/kg) to induce an additional anticoagulant effect. After 1 h, a tail-clip was done on normal (no TC and anticoagulation) and defect (TC + anticoagulation) mice by transecting 3 mm from the tail tip with a surgical scalpel and the clipped tail was immediately immersed in warm (37 °C) saline. Bleeding was monitored over time, and if bleeding did not stop beyond 15 min, then the 15 min time point was considered as the experiment end point. In separate experiments, t-TLNPs or UNPs (stock concentration of 1.14 × 1012 particles/mL) were administered retro-orbitally in “defect mice” 15 min before tail-clip and bleeding was similarly monitored by immersing the injured tail in warm saline. In all mice experimental groups, the bleeding time (time for bleeding to stop) was recorded as a percentage (%) of 15 min. At the 15 min time point, the collected blood was analyzed for hemoglobin by the sodium lauryl sulfate method using UV spectrometry measurements at 550 nm, and these data were used to calculate blood loss. The t-TLNP particle administration volume was calculated to achieve an initial encapsulated circulating concentration of 1 nM thrombin or an exogenous thrombin dose of 0.031 mg/kg per animal.
Evaluation of t-TLNPs in Traumatic Liver Injury Model in Severely Coagulopathic Mouse.
C57BL/6J mice, 8–9 weeks old, were anesthetized using 70 mg/kg of sodium pentobarbital via an intraperitoneal injection. The femoral artery and vein were cannulated for hemodynamic monitoring and administration of intravenous treatment, respectively. Thirty (30) minutes prior to injury, mice were treated intravenously with 1 U/g of unfractionated heparin (UFH), followed by a flush of normal saline of equal volume to ensure complete infusion, through a catheter placed in the femoral vein. Then, mice were subjected to a previously validated model of uncontrolled hemorrhage that utilizes liver laceration.70 Briefly, following a midline laparotomy incision, preweighed absorption triangles were inserted inside the abdominal cavity against the abdominal wall without touching the liver, then the right middle lobe of the liver was lacerated. The resected liver weight was recorded in g immediately after. Treatment (sham saline or UNP or t-TLNP) was administered via a femoral vein catheter, 1 min after the liver laceration. The abdominal cavity was stapled close. Twenty (20) min following the injury and postinjury treatment, the abdomen was reopened and the absorption triangles were retrieved and weighed. Blood loss was calculated as the difference between the pre- and postliver laceration weights of the absorption triangles and recorded in grams (g). The abdominal cavity was then closed, and the mice were observed for 3 h from the time of liver laceration. At the 3 h time point, the mice were sacrificed, and their organs were harvested for histological staining and imaging.
Statistical Analysis.
All data from the in vitro fibrin generation, ROTEM, and BioFlux microfluidic assays, as well as blood loss data from the in vivo tail-clip and liver-laceration injury studies, were analyzed using one-way ANOVA with Tukey’s multiple comparisons test. For tail-clip studies, the bleeding time was assessed with a Log-rank (Mantel–Cox) test and Gehan–Breslow–Wilcoxon test. In all analyses, significance was considered to be p < 0.05. For all data shown, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grants R01 HL121212, R01 HL141080 to A.S.G., R01 HL098217 to M.N., and R01 HL141080 and R35 GM119526 to M.D.N. The work at Case Western made use of biomedical engineering research facilities built with funding from National Center for Research Resources Grant Number C06 RR1246301 (PI: Kenneth Kutina). The Leica confocal microscope was acquired via an NIH Shared Instrumentation Grant S10-OD024996. The authors acknowledge the assistance of the Case Western Reserve University School of Medicine Light Microscopy Core Facility for confocal microscopy studies. The authors also acknowledge the assistance of Dr. Patricia A. Loughran at The Department of Surgery and the Biospecimen Core at University of Pittsburgh for assistance with organ harvesting from mice in liver laceration injury studies and paraffin embedding for histological staining. Additionally, the authors acknowledge the assistance of the Histology Core at the Cleveland Clinic Foundation Lerner Research Institute for assistance in Carstair’s staining and imaging.
Footnotes
The authors declare the following competing financial interest(s): A.S.G. is an inventor and co-founder of Haima Therapeutics LLC, a biotechnology start-up company focused on the research and development of bioinspired hemostatic technologies. M.D.N. is a scientific advisory board member of Haima Therapeutics LLC. A.S.G. is a co-inventor on patents US 9107845, US 9636383, US 10426820, US 10434149 that are licensed to Haima.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.2c05306.
Bioconjugation reaction scheme and representative mass spectroscopy characterization data for DSPE-PEG-VBP and DSPE-PEG-CBP, representative dynamic light scattering (DLS) data for t-TLNP size characterization, Schematic of BioFlux microfluidic setup for nanoparticle adhesion studies and representative fluorescence images of Rhodamine B labeled (red fluorescent) control nanoparticles (no peptide decoration) vs targeted nanoparticles (“VBP + CBP”-decorated) binding to “vWF + collagen”-coated surface pre- and postincubation in human platelet-free plasma, carboxyfluorescein (CF) concentration vs fluorescence calibration curve and CF release levels from t-TLNPs via diffusion vs sPLA2-triggered particle degradation vs chloroform/methanol-induced exhaustive destabilization vs exposure to low to high shear stress, representative aggregometry profile of human platelet-rich plasma (PRP) at baseline (no ADP) vs with agonist (ADP) or free VBP or free CBP, representative aggregometry profile of human platelet-rich plasma (PRP) at the baseline (no ADP) vs with agonist (ADP) or t-LNP, and representative fluorescent images of neutrophils (DAPI, blue nuclei; Sytox Green, extracellularized DNA NETs) at the baseline (w/o stimulation) in comparison to neutrophils incubated with control nanoparticles (no peptide decoration), targeted nanoparticles (“VBP + CBP”-decorated), or calcium ionophore A23187 and quantitative data of Sytox Green fluorescence comparing these groups, representative optical density (OD) data for fibrin generation in plasma, plasma anticoagulated with Apixaban, and anticoagulated plasma treated with t-TLNPs, “t-TLNPs + sPLA2”, and UNPs, representative OD data for fibrin generation in platelet-rich plasma (PRP), platelet-free plasma (PFP), and PFP treated with t-TLNPs, “t-TLNPs + sPLA2”, and UNPs, representative OD data for fibrin generation in plasma, plasma anticoagulated with Apixaban, and this anticoagulated plasma treated with directly added thrombin and representative OD data for fibrin generation in platelet-rich plasma (PRP), platelet-free plasma (PFP), and this PFP treated with directly added thrombin, representative OD data for fibrin generation in plasma, plasma anticoagulated with Apixaban, and this anticoagulated plasma treated with various doses of t-TLNPs (hence various concentrations of encapsulated thrombin) and representative optical density (OD) data for fibrin generation in platelet-rich plasma (PRP), platelet-free plasma (PFP), and this PFP treated with various doses of t-TLNPs (hence various concentrations of encapsulated thrombin), representative ROTEM data showing debilitation of CFT, alpha angle, and A10 parameters when WB is anticoagulated with Apixaban and the corresponding effects of treatment with t-TLNPs, “t-TLNPs + sPLA2”, and UNPs, representative ROTEM data showing debilitation of CFT, alpha angle and A10 parameters when platelets are depleted from WB to make TC Blood and corresponding effects of treatment with t-TLNPs, “t-TLNPs + sPLA2”, and UNPs, effect of anti-CD42b antibody dose on platelet count in mice showing significant thrombocytopenia (TC) and bleeding time analysis in tail-clip injury model in TC mice in comparison to wild type (WT) normal mice, circulation lifetime characterization of t-TLNPs in mice and organ biodistribution studies of t-TLNPs in mice over a 24 h period, tail-clip injury bleeding time data in WT mice vs coagulopathic mice (thrombocytopenia plus anticoagulation) and effect of t-LNP treatment vs UNP treatment on bleeding time in these coagulopathic mice represented in a Kaplan–Meyer format, mean arterial pressure (MAP) characterization of mice subjected to liver laceration injury and observed for over 20 min period postinjury before retrieval of absorbent triangles, and representative histology images (Carstair’s staining) of uninjured liver section and clearance organs harvested from mice subjected to liver injury model, postinjury treatment with saline or UNP or t-TLNP, and euthanasia at 3 h time point postinjury (PDF)
Binding of control (undecorated) nanoparticles on “vWF + collagen”-coated channel surface in BioFlux microfluidic setup prior to incubation in platelet-free plasma (MP4)
Binding of targeted (“VBP + CBP”-decorated) nanoparticles on “vWF + collagen”-coated channel surface in BioFlux microfluidic setup prior to incubation in platelet-free plasma (MP4)
Binding of targeted (“VBP + CBP”-decorated) nanoparticles on albumin-coated channel surface in BioFlux microfluidic setup (MP4)
Binding of control (undecorated) nanoparticles on “vWF + collagen”-coated channel surface in BioFlux microfluidic setup after incubation in platelet-free plasma (MP4)
Binding of targeted (“VBP + CBP”-decorated) nanoparticles on “vWF + collagen”-coated channel surface in BioFlux microfluidic setup prior after incubation in platelet-free plasma (MP4)
Fibrin generation in platelet-rich plasma (PRP) under flow (blue, platelets; green, fibrin) on “vWF + collagen”-coated channel surface in BioFlux microfluidics (MP4)
Fibrin generation in “anticoagulated + platelet-depleted” plasma (Defect plasma) under flow (blue, platelets; green, fibrin) on “vWF + collagen”-coated channel surface in BioFlux microfluidics (MP4)
Fibrin generation in Defect plasma treated with “t-TLNP + sPLA2” under flow (blue, platelets; green, fibrin) on “vWF + collagen”-coated channel surface in BioFlux microfluidics (MP4)
Fibrin generation in Defect plasma treated with “UNP + sPLA2” under flow (blue, platelets; green, fibrin) on “vWF + collagen”-coated channel surface in BioFlux microfluidics (MP4)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsnano.2c05306
Contributor Information
Aditya Girish, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Ketan Jolly, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Nijmeh Alsaadi, Department of Surgery, University of Pittsburgh, Pittsburgh, Pennsylvania 15123, United States.
Maria de la Fuente, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Arielle Recchione, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Ran An, Department of Mechanical and Aerospace Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Dante Disharoon, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Zachary Secunda, Department of Surgery, University of Pittsburgh, Pittsburgh, Pennsylvania 15123, United States.
Shruti Raghunathan, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Norman F Luc, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Cian Desai, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Elizabeth Knauss, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Xu Han, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Keren Hu, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Hanyang Wang, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Ujjal Didar Singh Sekhon, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Nathan Rohner, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Umut A Gurkan, Department of Mechanical and Aerospace Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Marvin Nieman, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Matthew D. Neal, Department of Surgery, University of Pittsburgh, Pittsburgh, Pennsylvania 15123, United States
Anirban Sen Gupta, Department of Biomedical Engineering and Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio 44106, United States.
REFERENCES
- (1).Bulger EM; Snyder D; Schoelles K; Gotschall C; Dawson D; Lang E; Sanddal ND; Butler FK; Fallat M; Taillac P; White L; Salomone JP; Seifarth W; Betzner MJ; Johannigman J; McSwain N. An evidence-based prehospital guideline for external hemorrhage control: American College of Surgeons Committee on Trauma. Prehospital Emergency Care 2014, 18, 163–173. [DOI] [PubMed] [Google Scholar]
- (2).Eastridge BJ; Holcomb JB; Shackelford S. Outcomes of traumatic hemorrhagic shock and epidemiology of preventable death from injury. Transfusion 2019, 59, 1423–1428. [DOI] [PubMed] [Google Scholar]
- (3).Buehner MF; Eastridge BJ; Aden JK; DuBose JJ; Blackbourne LH; Cestro RF Combat casualties and severe shock: Risk factors for death at Role 3 military facilities. MILITARY MEDICINE 2017, 182 (9), e1922. [DOI] [PubMed] [Google Scholar]
- (4).Spinella PC; Cap AP Prehospital hemostatic resuscitation to achieve zero preventable deaths after traumatic injury. Curr. Opin. Hematol 2017, 24, 529–535. [DOI] [PubMed] [Google Scholar]
- (5).Klok FA; Kooiman J; Huisman MV; Konstantinides S; Lankeit M. Predicting anticoagulant-related bleeding in patients with venous thromboembolism: A clinically oriented review. Eur. Respir. J 2015, 45, 201–210. [DOI] [PubMed] [Google Scholar]
- (6).Hunt BJ Bleeding and coagulopathies in critical care. N. Engl. J. Med 2014, 370, 847–859. [DOI] [PubMed] [Google Scholar]
- (7).Deshpande NV; Admane P; Mardikar HM Bleeding on dual antiplatelet therapy: real-life challenges. Eur. Heart. J. Suppl 2018, 20, B1–B9. [Google Scholar]
- (8).Piran S; Schulman S. Treatment of bleeding complications in patients on anticoagulant therapy. Blood 2019, 133, 425–435. [DOI] [PubMed] [Google Scholar]
- (9).Hickman DA; Pawlowski CL; Sekhon UDS; Marks J; Sen Gupta A. Biomaterials and advanced technologies for hemostatic management of bleeding. Adv. Mater 2018, 30, 1700859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Baylis JR; Yeon JH; Thomson MH; Kazerooni A; Wang X; John AE St; Lim EB; Chien D; Lee A; Zhang JQ; Piret JM; Machan LS; Burke TF; White NJ; Kastrup CJ Self-propelling particles that transport cargo through flowing blood and halt hemorrhage. Science Adv. 2015, 1, e1500379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hsu BB; Conway W; Tschabrunn CM; Mehta M; Perez-Cuevas MB; Zhang S; Hammond PT Clotting mimicry from robust hemostatic bandages based on self-assembling peptides. ACS Nano 2015, 9, 9394–9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cheng J; Feng S; Han S; Zhang X; Chen Y; Zhou X; Wang R; Li X; Hu H; Zhang J. Facile assembly of cost-effective and locally applicable or injectable nanohemostats for hemorrhage control. ACS Nano 2016, 10, 9957–9973. [DOI] [PubMed] [Google Scholar]
- (13).Sakoda M; Kaneko M; Ohta S; Qi P; Ichimura S; Yatomi Y; Ito T. Injectable hemostat composed of a polyphosphate-conjugated hyaluronan hydrogel. Biomacromolecules 2018, 19, 3280–3290. [DOI] [PubMed] [Google Scholar]
- (14).Li M; Zhang Z; Liang Y; He J; Guo B. Multifunctional tissue-adhesive cryogel wound dressing for rapid nonpressing surface hemorrhage and wound repair. ACS Appl. Mater. Interfaces 2020, 12, 35856–35872. [DOI] [PubMed] [Google Scholar]
- (15).Kim K; Ryu JH; Koh M-Y; Yun SP; Kim S; Park JP; Jung C-W; Lee MS; Seo H-I; Kim JH; Lee H. Coagulopathy-independent bioinspired hemostatic materials: A full research story from preclinical models to a human clinical trial. Science Adv. 2021, 7, eabc9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Guo Y; Wang Y; Zhao X; Li X; Wang Q; Zhong W; Mequanint K; Zhan R; Xing M; Luo G. Snake extract-laden hemostatic bioadhesive gel crosslinked by visible light. Science Adv. 2021, 7, eabf9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lashof-Sullivan M; Shoffstall A; Lavik EB Intravenous hemostats: Challenges in translation to patients. Nanoscale 2013, 5, 10719–10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chan LW; White NJ; Pun SH Synthetic strategies for engineering intravenous hemostats. Bioconjugate Chem. 2015, 26, 1224–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cohen MJ; Kutcher M; Redick B; Nelson M; Call M; Knudson MM; Schreiber MA; Bulger EM; Muskat P; Alarcon LH; Myers JG; Rahbar MH; Brasel KJ; Phelan HA; del Junco DJ; Fox EE; Wade CE; Holcomb JB; Cotton BA; Matijevic N. Clinical and mechanistic drivers of acute traumatic coagulopathy. J. Trauma Acute Care Surg 2013, 75, S40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Holcomb JB; Tilley BC; Baraniuk S; Fox EE; Wade CE; Podbielski JM; del Junco DJ; Brasel KJ; Bulger EM; Callcut RA; Cohen MJ; Cotton BA; Fabian TC; Inaba K; Kerby JD; Muskat P; O’Keeffe T; Rizoli S; Robinson BRH; Scalea TM; Schreiber MA; Stein DM; Weinberg JA; Callum JL; Hess JR; Matijevic N; Miller CN; Pittet J-F; Hoyt DB; Pearson GD; Leroux B; van Belle G. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015, 313, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sperry JL; Guyette FX; Brown JB; Yazer MH; Triluzi DJ; Early-Young BJ; Adams PW; Daley BJ; Miller RS; Harbrecht BG; Claridge JA; Phelan HA; Witham WR; Putnam AT; Duane TM; Alarcon LH; Callaway CW; Zuckerbraun BS; Neal MD; Rosengart MR; Forsythe RM; Billiar TR; Yealy DM; Peitzman AB; Zenati MS Prehospital Plasma during Air Medical Transport in Trauma Patients at Risk for Hemorrhagic Shock. N Engl J. Med 2018, 379, 315–326. [DOI] [PubMed] [Google Scholar]
- (22).Cardenas JC; Zhang X; Fox EE; Cotton BA; Hess JR; Schreiber MA; Wade CE; Holcomb JB Platelet Transfusions Improve Hemostasis and Survival in a substudy of the Prospective, Randomized PROPPR Trial. Blood Adv. 2018, 2, 1696–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Spinella PC; Dunne j.; GBeilman GJ; O’Connell RJ; Borgman MA; Cap AP; Rentas F. Constant challenges and evolution of US military transfusion medicine and blood operations in combat. Transfusion 2012, 52, 1146–1153. [DOI] [PubMed] [Google Scholar]
- (24).Lambert MP; Sullivan SK; Fuentes R; French DL; Poncz M. Challenges and promises for the development of donor-independent platelet transfusions. Blood 2013, 121, 3319–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Humbrecht C; Kientz D; Gachet C. Platelet transfusion: Current challenges. Transfus. Clin. Biol 2018, 25, 151–164. [DOI] [PubMed] [Google Scholar]
- (26).Shaffer L. Making and storing blood to save lives. Proc. Nat. Acad. Sci 2020, 117, 7542–7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Picker SM Current methods for the reduction of blood-borne pathogens: a comprehensive literature review. Blood Transfusion 2013, 11, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Leeper CM; Yazer M; Cladis F; Saladino R; Triluzi DJ; Gaines B. Cold-stored whole blood platelet function is preserved in injured children with hemorrhagic shock. J. Trauma Acute Care Surg 2019, 87, 49–53. [DOI] [PubMed] [Google Scholar]
- (29).Strandenes G; Berseus O; Cap AP; Hervig T; Reade M; Prat N; Sailliol A; Gonzales R; Simn CD; Ness P; Doughty HA; Spinella PC; Kristoffersen EK Low titer Group O whole blood in emergency situations. SHOCK 2014, 41, 70–75. [DOI] [PubMed] [Google Scholar]
- (30).McGinity AC; Zhu CS; Greebon L; Xenakis E; Waltman E; Epley E; Cobb D; Jonas R; Nicholson SE; Eastridge BJ; Stewart RM; Jenkins DH Prehospital low-titer cold-stored whole blood: Philosophy for ubiquitous utilization of O-positive product for emergency use in hemorrhage due to injury. J. Trauma Acute Care Surg 2018, 84, S115–S119. [DOI] [PubMed] [Google Scholar]
- (31).Dumont LJ; Slichter SJ; Reade MC Cryopreserved platelets: Frozen in a logjam? Transfusion 2014, 54, 1907–1910. [DOI] [PubMed] [Google Scholar]
- (32).Reddoch-Cardenas KM; Bynum JA; Meledeo MA; Nair PM; Wu X; Darlington DN; Ramasubramanian AK; Cap AP Cold-stored platelets: A product with function optimized for hemorrhage control. Transfus. Apher. Sci 2019, 58, 16–22. [DOI] [PubMed] [Google Scholar]
- (33).Bynum JA; Meledeo MA; Peltier GC; McIntosh CS; Taylor AS; Montgomery RK; Reddoch-Kardenas KM; Getz TM; Fitzpatrick MJ; Cap AP Evaluation of a lyophilized platelet-derived hemostatic product. Transfusion 2019, 59, 1490–1498. [DOI] [PubMed] [Google Scholar]
- (34).Feuerstein SJ; Skovmand K; Møller AM; Wildgaard K. Freeze-dried plasma in major hemorrhage: A systematic review. Vox Sanguinis 2020, 115, 263–274. [DOI] [PubMed] [Google Scholar]
- (35).Shukla M; Sekhon UDS; Betapudi V; Li W; Hickman DA; Pawlowski CL; Dyer MR; Neal MD; McCrae KR; Sen Gupta A. In vitro characterization of SynthoPlate (synthetic platelet) technology and its in vivo evaluation in severely thrombocytopenic mice. J. Thromb. Haemost 2017, 15, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dyer MR; Hickman D; Luc N; Haldeman S; Loughran P; Pawlowski C; Sen Gupta A; Neal MD Intravenous administration of synthetic platelets (SynthoPlate) in a mouse liver injury model of uncontrolled hemorrhage improves hemostasis. J. Trauma Acute Care Surg 2018, 84, 917–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hickman DA; Pawlowski CL; Shevitz A; Luc NF; Kim A; Girish A; Marks J; Ganjoo S; Huang S; Niedoba E; Sekhon UDS; Sun M; Dyer M; Neal MD; Kashyap VS; Sen Gupta A. Intravenous synthetic platelet (SynthoPlate) nanoconstructs reduce bleeding and improve ‘golden hour’ survival in a porcine model of traumatic arterial hemorrhage. Sci. Rep 2018, 8, 3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Brown AC; Stabenfeldt SE; Ahn B; Hannan RT; Dhada KS; Herman ES; Stefanelli V; Guzetta N; Alexeev A; Lam WA; Lyon LA; Barker TH Ultrasoft microgels displaying emergent platelet-like behaviors. Nat. Mater 2014, 13, 1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bertram JP; Williams CA; Robinson R; Segal SS; Flynn NT; Lavik EB Intravenous hemostat: Nanotechnology to halt bleeding. Science Trans. Med 2009, 1, 11ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lashof-Sullivan MM; Shoffstall E; Atkins KT; Keanie N; Bir C; VandeVord P; Lavik EB Intravenously administered nanoparticles increase survival following blast trauma. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 10293–10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gkikas M; Peponis T; Mesar T; Hong C; Avery RK; Roussakis E; Yoo H-J; Parakh A; Patino M; Sahani DV; Watkins MT; Oklu R; Evans CL; Albadawi H; Velhamos G; Olsen BD Systemically administered hemostatic nanoparticles for identification and treatment of internal bleeding. ACS Biomater. Sci. Eng 2019, 5, 2563–2576. [DOI] [PubMed] [Google Scholar]
- (42).Morgan CE; Dombrowski AW; Rubert Perez CM; Bahnson ESM; Tsihlis ND; Jiang W; Vercammen JM; Prakash VS; Pritts TA; Stupp SI; Kibbe MR Tissue-factor targeted peptide amphiphile nanofibers as an injectable therapy to control hemorrhage. ACS Nano 2016, 10, 899–909. [DOI] [PubMed] [Google Scholar]
- (43).Klein MK; Kassam HA; Lee RH; Bergmeier W; Peters EB; Gillis DC; Dandurand BR; Rouan JR; Karver MR; Struble MD; Clemons TD; Palmer LC; Gavitt B; Pritts TA; Tsihlis ND; Stupp SI; Kibbe MR Development of optimized tissue-factor-targeted peptide amphiphile nanofibers to slow non-compressible torso hemorrhage. ACS Nano 2020, 14, 6649–6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chan LW; Wang X; Wei H; Pozzo LD; White NJ; Pun SH A synthetic fibrin cross-linking polymer for modulating clot properties and inducing hemostasis. Sci. Trans. Med 2015, 7, 277ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gao Y; Sarode A; Kokoroskos N; Ukidve A; Zhao Z; Guo S; Flaumenhaft R; Sen Gupta A; Saillant N; Mitragotri S. A polymer-based systemic hemostatic agent. Science Adv. 2020, 6, eaba0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hoffman M; Monroe DM III A cell-based model of hemostasis. Thromb Haemost 2001, 85, 958–965. [PubMed] [Google Scholar]
- (47).Wolberg AS; Campbell RA Thrombin generation, fibrin clot formation and hemostasis. Transfus. Apher. Sci 2008, 38, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Versteeg HH; Heemskerk JWM; Levi M; Reitsma PH New fundamentals in hemostasis. Physiol. Rev 2013, 93, 327–358. [DOI] [PubMed] [Google Scholar]
- (49).Walker CPR; Royston D. Thrombin generation and its inhibition: a review of the scientific basis and mechanism of action of anticoagulant therapies. Br. J. Anaesth 2002, 88, 848–863. [DOI] [PubMed] [Google Scholar]
- (50).Mackman N. The role of Tissue Factor and Factor VIIa in hemostasis. Anesth. Analg 2009, 108, 1447–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Shefa AA; Taz M; Lee SY; Lee B-T Enhancement of hemostatic property of plant derived oxidized nitrocellulose-silk fibroin based scaffolds by thrombin loading. Carbohydr. Polym 2019, 208, 168–179. [DOI] [PubMed] [Google Scholar]
- (52).Baylis JR; St. John, A. E.; Wang, X.; Lim, E. B.; Statz, M. L.; Chien, D.; Simonson, E.; Stern, S. A.; Liggins, R. T.; White, N. J.; Kastrup, C. J. Self-propelled dressings containing thrombin and tranexamic acid improve short-term survival in a swine model of lethal junctional hemorrhage. SHOCK 2016, 46, 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Chan V; Sarkari M; Sunderland R; John AE St; White NJ; Kastrup CJ Platelets loaded with liposome-encapsulated thrombin have increased coagulability. J. Thromb. Haemost 2018, 16, 1226–1235. [DOI] [PubMed] [Google Scholar]
- (54).Nogami K; Shima M; Giddings JC; Takeyama M; Tanaka I; Yoshioka A. Relationship between the binding sites of von Willebrand Factor, phospholipid, and huma Factor VIII C2 inhibitoralloantibodies within the Factor VIII C2 domain. Int. J. Hematol 2007, 85, 317–322. [DOI] [PubMed] [Google Scholar]
- (55).Nuyttens BP; Thijs T; Deckmyn H; Broos K. Platelet adhesion to collagen. Thromb. Res 2011, 127, S26–S29. [DOI] [PubMed] [Google Scholar]
- (56).Mo X; An Y; Yun C-S; Yu SM Nanoparticle-assisted visualization of binding interactions between collagen mimetic peptide and collagen fibers. Angew. Chem., Int. Ed 2006, 45, 2267–2270. [DOI] [PubMed] [Google Scholar]
- (57).Ross PL; Wolfe JL Physical and chemical stability of antibody drug conjugates: current status. J. Pharm. Sci 2016, 105, 391–397. [DOI] [PubMed] [Google Scholar]
- (58).Turell L; Radi R; Alvarez B. The thiol pool in human plasma: The central contribution of albumin to redox processes. Free. Radic. Biol. Med 2013, 65, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Emadi S; Mirshahi M; Elalamy I; Nicolas C; Vargarftig BB; Hatmi M. Cellular source of human platelet secretory phospholipase A2. Br. J. Hamaetol 1998, 100, 365–373. [DOI] [PubMed] [Google Scholar]
- (60).Blache D; Gautier T; Tietge UJF; Lagrost L. Activated platelets contribute to oxidized low-density lipoproteins and dysfunctional high-density lipoproteins through a phospholipase A2-dependent mechanism. FASEB J. 2012, 26, 927–937. [DOI] [PubMed] [Google Scholar]
- (61).Bernardo A; Ball C; Nolasco L; Moake JF; Dong J. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand Factor multimers under flow. Blood 2004, 104, 100–106. [DOI] [PubMed] [Google Scholar]
- (62).Koltai K; Kesmarky G; Feher G; Tibold A; Toth K. Platelet aggregometry testing: Molecular mechanisms, techniques and clinical implications. Int. J. Mol. Sci 2017, 18, 1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).La-Beck NM; Islam MR; Markiewski MM Nanoparticle-induced complement activation: Implications for cancer nanomedicine. Front. Immun 2021, 11, 603039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Gupta S; Chan DW; Zaal KJ; Kaplan MJ A high throughput real-time imaging technique to quantify NETosis and distinguish mechanisms of cell death in human neutrophils. J. Immunol 2018, 200, 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kenny EF; Herzig A; Krüger, R.; Muth, A.; Mondal, S.; Thompson, P. R.; Brinkmann, V.; von Bernuth, H.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife 2017, 6, e24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Curnow JL; Morel-Kopp M-C; Roddie C; Aboud M; Ward CM Reduced fibrinolysis and increased fibrin generation can be detected in hypercoagulable patients using the overall hemostatic potential assay. J. Thromb. Haemost 2007, 5, 528–534. [DOI] [PubMed] [Google Scholar]
- (67).Tanaka KA; Bolliger D; Vadlamudi R; Nimmo A. Rotational thromboelastometry (ROTEM)-based coagulation management in cardiac surgery and major trauma. J. Cardiothorac. Vasc. Anesth 2012, 26, 1083–1093. [DOI] [PubMed] [Google Scholar]
- (68).Nieswandt B; Bergmeier W; Rackebrandt K; Gessner JE; Zirngibl H. Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood 2000, 96, 2520–2527. [PubMed] [Google Scholar]
- (69).Greene TK; Schiviz A; Hoellriegl W; Poncz M; Muchitsch E-M On behalf of the animal model subcommittee of the scientific and standardization committee of the ISTH. J. Thromb. Haemost 2010, 8, 2820–2822. [DOI] [PubMed] [Google Scholar]
- (70).Dyer M; Haldeman S; Gutierrez A; Kohut L; Sen Gupta A; Neal MD Uncontrolled hemorrhagic shock modeled via liver laceration in mice with real time hemodynamic monitoring. J. Vis. Exp 2017, 123, 55554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Gu Y; Bai Y; Wu J; Hu L; Gao B. Establishment and characterization of an experimental model of coronary thrombotic microembolism in rats. Am. J. Pathol 2010, 177, 1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Li S; Zhang Y; Ho S-H; Li B; Wang M; Deng X; Yang N; Liu G; Lu Z; Xu J; Shi Q; Han J-Y; Zhang L; Wu Y; Zhao Y; Nie G. Combination of tumor-infarction therapy and chemotherapy via the co-delivery of doxorubicin and thrombin encapsulated in tumour-targeted nanoparticles. Nature Biomed. Eng 2020, 4, 732–742. [DOI] [PubMed] [Google Scholar]
- (73).Wolberg AS; Campbell RA Thrombin generation, fibrin clot formation and hemostasis. Transfus. Apher. Sci 2008, 38, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Hulshof A-M; Hemker HC; Spronk HMH; Henskens YMC; ten Cate H. Thrombin-Fibrin(ogen) interactions, host defense and risk of thrombosis. Int. J. Mol. Sci 2021, 22, 2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Chang R; Cardenas JC; Wade CE; Holcomb JB Advances in the understanding of trauma-induced coagulopathy. Blood 2016, 128, 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Simmons JW; Powell MF Acute traumatic coagulopathy: pathophysiology and resuscitation. Br. J. Anaesth 2016, 117, iii31–iii43. [DOI] [PubMed] [Google Scholar]
- (77).Kornblith LZ; Moore HB; Cohen MJ Trauma-induced coagulopathy: The past, present, and future. J. Thromb. Haemost 2019, 17, 852–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Peralta R; Al Thani H; Rizoli S. Coagulopathy in the surgical patient: trauma-induced and drug-induced coagulopathies. Curr. Opin. Crit. Care 2019, 25, 668–674. [DOI] [PubMed] [Google Scholar]
- (79).Moore EE; Moore HB; Kornblith LZ; Neal MD; Hoffman M; Mutch NJ; Schochl H; Hunt BJ; Sauaia A. Trauma-induced coagulopathy. Nature Rev. Disease Primers 2021, 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Ockerman A; Vanassche T; Garip M; Vandenbriele C; Engelen MM; Martens J; Politis C; Jacobs R; Verhamme P. Tranexamic acid for the prevention and treatment of bleeding in surgery, trauma and bleeding disorders: a narrative review. Thrombosis J. 2021, 19, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.