

Abstract
Objective
To characterize distinct comorbidities, outcomes, and treatment patterns in children with Down syndrome and pulmonary hypertension in a large, multicenter pediatric pulmonary hypertension registry.
Study design
We analyzed data from the Pediatric Pulmonary Hypertension Network (PPHNet) Registry, comparing demographic and clinical characteristics of children with Down syndrome and children without Down syndrome. We examined factors associated with pulmonary hypertension resolution and a composite outcome of pulmonary hypertension severity in the cohort with Down syndrome.
Results
Of 1475 pediatric patients with pulmonary hypertension, 158 (11%) had Down syndrome. The median age at diagnosis of pulmonary hypertension in patients with Down syndrome was 0.49 year (IQR, 0.21–1.77 years), similar to that in patients without Down syndrome. There was no difference in rates of cardiac catheterization and prescribed pulmonary hypertension medications in children with Down syndrome and those without Down syndrome. Comorbidities in Down syndrome included congenital heart disease (95%; repaired in 68%), sleep apnea (56%), prematurity (49%), recurrent respiratory exacerbations (35%), gastroesophageal reflux (38%), and aspiration (31%). Pulmonary hypertension resolved in 43% after 3 years, associated with a diagnosis of pulmonary hypertension at age <6 months (54% vs 29%; P = .002) and a pretricuspid shunt (65% vs 38%; P = .02). Five-year transplantation-free survival was 88% (95% CI, 80%–97%). Tracheostomy (hazard ratio [HR], 3.29; 95% CI, 1.61–6.69) and reflux medication use (HR, 2.08; 95% CI, 1.11–3.90) were independently associated with a composite outcome of severe pulmonary hypertension.
Conclusions
Despite high rates of cardiac and respiratory comorbidities that influence the severity of pulmonary hypertension, children with Down syndrome–associated pulmonary hypertension generally have a survival rate similar to that of children with non–Down syndrome–associated pulmonary hypertension. Resolution of pulmonary hypertension is common but reduced in children with complicated respiratory comorbidities.
Infants and children with Down syndrome, or trisomy 21, develop pulmonary hypertension at a reported prevalence as high as 28%,1 representing 5%–17% of cases recorded in international pediatric pulmonary hypertension registries.2–5 In addition to a genetic predisposition from trisomy 21, pulmonary hypertension in Down syndrome may result from congenital heart disease (CHD), developmental lung disease, and airway obstruction.
Pulmonary hypertension in infants with Down syndrome is often diagnosed shortly after birth, with persistent pulmonary hypertension of the newborn (PPHN) a common diagnosis.1,6 However, pulmonary hypertension can persist beyond the neonatal period and can be diagnosed later in childhood. One study identified a median age at diagnosis of pulmonary hypertension of 5 days, with 38% of those cases considered transient PPHN.1
CHD is a frequent cause of pulmonary hypertension in individuals with Down syndrome, categorized by the World Symposium on Pulmonary Hypertension (WSPH) as group 1 pulmonary hypertension, or pulmonary arterial hypertension.7 However, a wide spectrum of anatomic cardiac lesions occur in patients with Down syndrome, ranging from simple shunts to more complex lesions, most commonly atrioventricular septal defects (AVSDs).6,8–10 Pulmonary hypetension in patients with Down syndrome likely results from increased hemodynamic stress from left-to-right shunt lesions causing excess pulmonary blood flow, with elements of related or intrinsic endothelial dysfunction.11
In children with Down syndrome without CHD, pulmonary hypertension is most often associated with chronic lung disease, included in WSPH group 3. Autopsy studies have reported abnormal lung structure in children with Down syndrome.12–14 Increased expression of antiangiogenic factors encoded on chromosome 21, including collagen 18a1 (endostatin), β-amyloid peptide, and Down Syndrome Critical Region 1, may contribute to abnormal lung vascular growth and predispose to pulmonary vascular disease.15,16 Circulating biomarkers, including endothelin, endostatin, and galectin-3, are elevated in the blood of children with Down syndrome and pulmonary hypertension.17,18
With increased recognition of structural lung abnormalities in infants and children with Down syndrome, the Pediatric Task Force of the Sixth WSPH recognized Down syndrome as a “developmental lung disease” associated with pulmonary hypertension.19 These newer guidelines recommend that pulmonary hypertension in the absence of significant CHD should be categorized primarily as WSPH group 3 (pulmonary hypertension related to lung disease). Children with Down syndrome often have comorbidities, including upper airway obstruction, swallowing dysfunction, gastroesophageal reflux, and aspiration, which may lead to intermittent hypoxia, lung inflammation, and increased risk for pulmonary hypertension. Thus, it remains challenging to determine the influence of diverse intrinsic and extrinsic contributors to pulmonary hypertension in children with Down syndrome, especially in those with both CHD and lung disease.
Using data from the multicenter Pediatric Pulmonary Hypertension Network (PPHNet) patient registry of infants and children with pulmonary hypertension,20 we characterized the distinct comorbidities and outcomes in children with Down syndrome–related pulmonary hypertension with a focus on CHD and respiratory disease. In addition, we explored the clinical factors associated with both pulmonary hypertension resolution and adverse long-term outcomes in children with Down syndrome–associated pulmonary hypertension.
Methods
The PPHNet registry includes comprehensive clinical data from 8 centers in the US and Canada that enrolled patients between October 2014 and February 2018 and includes follow-up data submitted through May 18, 2020.20 The PPHNet registry was supported in part by a National Heart, Lung, and Blood Institute–funded U01 program (U01 HL12118; Data Fusion: A Sustainable, Open Source Registry Advancing Pediatric Pulmonary Vascular Disease Research; S.A. and K.M., principal investigators). The study protocol was approved by the Institutional Review Board of each participating center. Parents and/or guardians provided signed informed consent, and participants gave assent when appropriate, according to institutional guidelines.
Inclusion criteria for the PPHNet registry have been published previously and include a clinical diagnosis of pulmonary hypertension based on echocardiography or cardiac catheterization before age 18 years, defined according to established guidelines from the joint American Heart Association/American Thoracic Society guidelines for pediatric pulmonary hypertension.21 Demographic and clinical data were collected as described previously.20 Case report forms for clinical data, including cardiac hemodynamics, diagnostic imaging, laboratory information, and drug therapies, were completed at the time of enrollment and included some retrospective data from the time of pulmonary hypertension diagnosis and any clinical comorbidities, if applicable. We enrolled both incident and prevalent patients; therefore, some were enrolled at the time of diagnosis of pulmonary hypertension and some were enrolled after. Prospective clinical and outcomes data were collected up to the registry data freeze for this report in May 2020.20
The enrolling investigator at each site identified subjects with Down syndrome using data from the medical record. CHD-specific data collection was included for all subjects in the registry identified as having CHD, excluding patent foramen ovale. The CHD diagnoses provided in the registry were reviewed by 3 pediatric cardiologists, who categorized diagnoses by the primary lesion most likely affecting cardiac physiology, using the following categories: pretricuspid shunt, posttricuspid shunt, left-sided obstructive lesion (including pulmonary vein stenosis), or complex/mixed CHD. Patients without surgical or catheter-based repair data were categorized as unrepaired.
Pulmonary hypertension medications are defined as inhaled nitric oxide, calcium channel blockers, phosphodiesterase type V inhibitors, endothelin receptor antagonists, prostacyclins, and selexipag (prostacyclin receptor agonist). Resolution of pulmonary hypertension is defined as cessation of pulmonary hypertension medications. This definition was applied to WSPH groups 1 and 3, without Eisenmenger syndrome; other patients were classified as nonresolved. For patients with pulmonary hypertension–targeted medication stops and restarts, the latest date on record for cessation of pulmonary hypertension–targeted medications served as the event time, not the initial date of cessation. If a patient never had pulmonary hypertension medications listed on the registry medications form, the time to medication discontinuation equalled the years from diagnosis to consent. This timing accounts for the fact that a patient might have been on pulmonary hypertension medications between the time of diagnosis and enrollment, because no medication data were collected for this interval.
Statistical Analyses
Descriptive statistics include count and percentage for categorical variables and mean ± SD or median and IQR for continuous variables. Categorical characteristics for the Down syndrome and non-Down syndrome groups were compared using the Fisher exact test; continuous characteristics were compared using the t-test (mean) and Wilcoxon rank-sum test (median).
The subdistribution proportional hazards model of Fine and Gray22 was used to estimate time from the diagnosis of pulmonary hypertension to the earliest occurrence of a qualifying event in a competing risks framework (death vs lung transplant vs pulmonary hypertension resolution). The competing risks analysis was restricted to the incident case cohort, defined as cases with a diagnosis of pulmonary hypertension at or after inception of the registry in October 2014.
Cox proportional hazards regression with delayed entry to risk set methodology to accommodate left truncation23,24 was used to explore associations between patient comorbidity factors and time to a composite outcome of pulmonary hypertension severity, defined as the earliest occurrence of any of the following events: death, lung transplant, Potts shunt, continuous intravenous or subcutaneous prostacyclin at any time, hospitalization attributed to pulmonary hypertension occurring after the diagnosis visit, and Pediatric Panama or World Health Organization (WHO) functional class IV at any time.25 For 5 patients, the outcome was achieved at the time of diagnosis; an event time of 1 day was assigned for these cases. The multivariable model stepwise selection used as candidate predictors variables with a univariate P value <.20.
The distribution of transplant-free survival was estimated using Kaplan–Meier methodology with delayed entry into risk sets,23,24 defined by the time of enrollment in the registry relative to the time of pulmonary hypertension diagnosis, for the composite outcome of death/Potts shunt/lung transplant. All available follow-up was used, but graphs were truncated at 20 years. Follow-up duration was calculated as the time between pulmonary hypertension diagnosis and the latest physical examination date if none of the aforementioned events occurred; otherwise, follow-up ended at the time of the earliest event.
A P value <.05 was defined as statistically significant. All analyses were conducted using SAS version 9.4 (SAS Institute) and R version 4.03.26
Results
Of the 1475 children enrolled in the PPHNet Registry, 158 (10.7%) were identified as subjects with Down syndrome.20 There was a slight preponderance of males in the Down syndrome group (53.8%). The proportion of patients with pulmonary hypertension with Down syndrome differed by study site, ranging from 6% to 22%. Compared with the non-Down syndrome cohort, patients with Down syndrome were less likely to be White and more likely to be Asian (P < .001). There were 12 deaths and no lung transplants in the Down syndrome cohort, which was followed for a median of 2.9 years. There was no difference in the proportion of patients with death/transplant for the Down syndrome and non-Down syndrome cohorts (Table I).
Table I.
Demographics, clinical characteristics, and diagnostic evaluation of patients with pulmonary hypertension with Down syndrome compared with patients without Down syndrome in the PPHNet Registry
| Variables at pulmonary hypertension diagnosis | Down syndrome (N = 158) | Non-Down syndrome (N = 1317) | P value |
|---|---|---|---|
|
| |||
| Sex, n (%) | .56 | ||
| Male | 85 (53.8) | 673 (51.1) | |
| Female | 73 (46.2) | 644 (48.9) | |
| Age, y, median (IQR) | 0.49 (0.21–1.77) | 0.50 (0.08–3.69) | .80 |
| Race, n (%) | .003 | ||
| White | 87 (62.1%) | 812 (69.9) | |
| Black | 22 (15.7%) | 169 (14.5) | |
| Asian | 21 (15.0%) | 114 (9.8) | |
| American Indian or Alaska Native | 6 (4.3%) | 9 (0.8) | |
| Native Hawaiian or other Pacific Islander | 2 (1.4) | 8 (0.7) | |
| Multiple races | 2 (1.4) | 50 (4.3) | |
| WSPH Nice classification, n (%) | <.001 | ||
| Group 1 | 110 (69.6) | 492 (37.3) | |
| Group 1′ | 2 (1.3) | 9 (0.7) | |
| Group 1″ | 4 (2.5) | 46 (3.5) | |
| Group 2 | 2 (1.3) | 52 (3.9) | |
| Group 3 | 33 (20.9) | 687 (52.2) | |
| Group 4 | 0 | 8 (0.6) | |
| Group 5 | 7 (4.4) | 23 (1.7) | |
| Testing at diagnosis, n (%) | |||
| Echocardiography | .33 | ||
| No | 2 (1.3) | 9 (0.7) | |
| Yes | 151 (98.7) | 1282 (99.3) | |
| Cardiac catheterization | .86 | ||
| No | 77 (50.7) | 638 (49.9) | |
| Yes | 75 (49.3) | 641 (50.1) | |
| Electrocardiography | .048 | ||
| No | 44 (29.5) | 479 (38.0) | |
| Yes | 105 (70.5) | 783 (62.0) | |
| 6-minute walk test | .02 | ||
| No | 148 (98.0) | 1185 (93.2) | |
| Yes | 3 (2.0) | 86 (6.8) | |
| 6-minute walk test, aged >4 y | .03 | ||
| No | 155 (98.1) | 1235 (93.8) | |
| Yes | 3 (1.9) | 82 (6.2) | |
| Exercise test | .07 | ||
| No | 151 (100.0) | 1239 (97.6) | |
| Yes | 0 (0.0) | 30 (2.4) | |
| Cardiac magnetic resonance imaging | .43 | ||
| No | 145 (98.6) | 1224 (97.0) | |
| Yes | 2 (1.4) | 38 (3.0) | |
| Chest radiography | .56 | ||
| No | 42 (28.6) | 333 (26.4) | |
| Yes | 105 (71.4) | 929 (73.6) | |
| Chest CT | .003 | ||
| No | 132 (89.8) | 1009 (79.7) | |
| Yes | 15 (10.2) | 257 (20.3) | |
| Pulmonary function tests | .001 | ||
| No | 152 (100.0) | 1204 (95.0) | |
| Yes | 0 (0.0) | 64 (5.0) | |
| Lung biopsy | .17 | ||
| No | 150 (99.3) | 1235 (97.3) | |
| Yes | 1 (0.7) | 34 (2.7) | |
The distribution of the primary WSPH classification of pulmonary hypertension differed between the Down syndrome and non-Down syndrome cohorts (P < .001). The Down syndrome cohort was more commonly group 1 (pulmonary arterial hypertension) compared with the non-Down syndrome cohort (69.6% vs 37.3%), and group 3 pulmonary hypertension (related to lung disease) was more common in the non-Down syndrome group (52.2% vs 20.9%) (Table I). Of the 110 patients with Down syndrome categorized as having group 1 pulmonary hypertension (pulmonary arterial hypertension), 109 were further classified: 103 (94.5%) with pulmonary arterial hypertension associated with CHD, 5 (4.6%) with idiopathic pulmonary arterial hypertension, and 1 (0.9%) with pulmonary arterial hypertension induced by drugs and toxins. Relatively few patients were classified as group 10 (pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis), group 1″ (PPHN), and group 2 pulmonary hypertension (related to left heart disease), and prevalence was similar in children with Down syndrome and those without Down syndrome. No patients were classified as having chronic thromboembolic pulmonary hypertension (group 4). Group 5 (multifactorial) pulmonary hypertension was uncommon across the registry (Table I).
For patients with Down syndrome, the median age at pulmonary hypertension diagnosis was 0.49 year (IQR, 0.21–1.77 years), similar to that of patients without Down syndrome. Nearly all subjects in the registry underwent echocardiography as part of their pulmonary hypertension diagnosis; however, there were some significant differences in the diagnostic testing performed in patients with Down syndrome at the time of pulmonary hypertension diagnosis compared with those without Down syndrome: the former were more likely to undergo electrocardiography (70.5% vs 62.0%; P = .048) and less likely to undergo chest computed tomography (CT) scan (10.2% vs 20.3%; P = .003), pulmonary function testing (0 vs 5.0%; P = .001), and the 6-minute walk test (2.0% vs 6.8%; P = .02), even for patients aged >4 years (1.9% vs 6.2%; P = .03) (Table I).
Approximately one-half of the subjects in the PPHNet registry underwent cardiac catheterization at the time of pulmonary hypertension diagnosis. To control for differences in WSPH classification between the cohorts of patients with Down syndrome vs those without Down syndrome, we compared hemodynamics restricted to patients with group 1 pulmonary hypertension (Table II). Patients with Down syndrome underwent cardiac catheterization at a younger median age than those without Down syndrome (1.3 [IQR 0.5, 5.5] years vs 3.7 [IQR, 0.7–8.9 years]; P = .004). Overall, hemodynamic measurements revealed less severe disease in subjects with Down syndrome, with lower mean right ventricular systolic pressure (61.7 ± 21.0 mmHg vs 74.2 ± 25.6 mmHg; P = .01), mean pulmonary arterial pressure (40.6 ± 15.6 mmHg vs 47.6 ± 21.0 mmHg; P = .002), and median pulmonary vascular resistance indexed to body surface area (PVRi) (7.2 [IQR, 4.4–10.2] WU*m2 vs 9.2 [IQR, 5.0–16.9] WU*m2; P = .03). Despite significantly lower systemic arterial systolic and diastolic pressures, patients with Down syndrome had a lower mean ratio of systolic pulmonary arterial pressure to systemic arterial pressure (0.71 ± 0.25 vs 0.80 ± 0.32; P = .02); however, the ratio of PVRi to systemic vascular resistance (SVRi) did not differ between the 2 groups. The median net left-to-right shunt (Qp:Qs) was higher in the Down syndrome cohort (1.2 [IQR, 1.0–1.6] vs 1.0 [IQR, 1.0–1.2]; P = .02), which is consistent with a high rate of CHD associated with Down syndrome. The cardiac index did not differ between subjects with Down syndrome and those without Down syndrome (Table II).
Table II.
Baseline hemodynamic data from diagnostic cardiac catheterization of patients with WSPH group 1 pulmonary hypertension with Down syndrome vs those without Down syndrome
| Measurements | Group 1 Down syndrome (n = 65) | Group 1 non-Down syndrome (n = 391) | P value |
|---|---|---|---|
|
| |||
| Age at catheterization (years) | |||
| No. of patients | 65 | 391 | |
| Median (IQR) | 1.3 (0.5–5.5) | 3.7 (0.7–8.9) | .004 |
| Time since diagnosis, y, mean ± SD | 0.11 ± 0.5 | 0.07 ± 1.8 | .70 |
| RAP, mmHg | |||
| No. of patients | 59 | 347 | |
| Mean ± SD | 7.2 ± 2.3 | 7.1 ± 3.4 | .74 |
| RV systolic pressure, mmHg | |||
| No. of patients | 32 | 196 | |
| Mean ± SD | 61.7 ± 21.0 | 74.2 ± 25.6 | .01 |
| RV end-diastolic pressure, mmHg | |||
| No. of patients | 33 | 185 | |
| Mean ± SD | 9.2 ± 3.5 | 9.6 ± 4.3 | .62 |
| PA systolic pressure, mmHg | |||
| No. of patients | 61 | 360 | |
| Mean ± SD | 60.0 ± 19.9 | 69.7 ± 27.8 | .001 |
| PA diastolic pressure, mmHg | |||
| No. of patients | 61 | 362 | |
| Mean ± SD | 27.0 ± 12.1 | 32.1 ± 17.8 | .005 |
| PA mean pressure, mmHg | |||
| No. of patients | 63 | 374 | |
| Mean ± SD | 40.6 ± 15.6 | 47.6 ± 21.0 | .002 |
| PCWP, mmHg | |||
| No. of patients | 51 | 308 | |
| Mean ± SD | 9.4 ± 2.6 | 9.3 ± 3.7 | .79 |
| LV end-diastolic pressure, mmHg | |||
| No. of patients | 29 | 103 | |
| Mean ± SD | 10.4 ± 3.7 | 9.5 ± 4.1 | .27 |
| Systemic arterial systolic pressure, mmHg | |||
| No. of patients | 49 | 285 | |
| Mean ± SD | 77.0 ± 13.8 | 84.7 ± 18.5 | .001 |
| Systemic arterial diastolic pressure, mmHg | |||
| No. of patients | 49 | 285 | |
| Mean ± SD | 42.6 ± 8.8 | 48.9 ± 13.1 | <.001 |
| PA mean pressure: systemic arterial mean pressure | |||
| No. of patients | 49 | 282 | |
| Mean ± SD | 0.71 ± 0.25 | 0.8 ± 0.32 | .02 |
| PVRi (WU*m2) | |||
| No. of patients | 50 | 338 | |
| Median (IQR) | 7.2 (4.4–10.2) | 9.2 (5.0–16.9) | .03 |
| SVRi (WU*m2) | |||
| Count | 45 | 297 | |
| Median (IQR) | 15.8 (11.6–20.8) | 16.5 (12.0–21.7) | .38 |
| PVRi:SVRi | |||
| No. of patients | 45 | 291 | |
| Median (IQR) | 0.5 (0.3–0.8) | 0.6 (0.4–0.9) | .17 |
| Cardiac Index, L/min/m2* | |||
| No. of patients | 48 | 319 | |
| Median (IQR) | 3.1 (2.7, 3.9) | 3.3 (2.6, 4.2) | .63 |
| Qp/Qs | |||
| No. of patients | 47 | 249 | |
| Median (IQR) | 1.2 (1.0,1.6) | 1.0 (1.0,1.2) | .02 |
LV, left ventricular; PA, pulmonary arterial; PCWP, pulmonary capillary wedge pressure; Qp/Qs, ratio of total pulmonary blood flow to total systemic blood flow; RV, right ventricular; SVRi, systemic vascular resistance.
Cardiac Index based on thermodilution, Fick if thermodilution value unavailable, or indexed Qs if Fick value unavailable.
CHD in Down Syndrome
In the registry, CHD was common in patients with Down syndrome, with 149 (94%) having 1 or more CHD lesions across a spectrum of severity. Of these, 81 patients (54%) had multiple CHD lesions. When stratified by primary type of CHD, the majority of patients with Down syndrome with CHD had a posttricuspid shunt (76.5%), including 36% with an AVSD, 36% with a ventricular septal defect (VSD), and 28% with a patent ductus arteriosus. Isolated pretricuspid shunts were relatively uncommon (13%), consisting exclusively of ostium primum or secundum atrial septal defects (ASDs). ASDs were repaired in more than one-half of affected patients (55%). ASDs were commonly associated with other CHD lesions, with 53 of the 129 patients with other forms of CHD (41%) also having an ASD (Table III; available at www.jpeds.com).
Table III.
Primary CHD type and repair status in patients with Down syndrome and pulmonary hypertension
| Primary CHD | Down syndrome and CHD, n (%) | Repaired, n (%) |
|---|---|---|
|
| ||
| Total | 149 | 102 (68.5) |
| Pretricuspid shunt | 20 (13.4) | 11 (55) |
| ASD, ostium primum | 5 (25.0) | 4 (80.0) |
| ASD, ostium secundum | 15 (75.0) | 7 (46.7) |
| Post-tricuspid shunt | 114 (76.5) | 79 (69.2) |
| AVSD | 41 (36.0) | 32 (78.0) |
| VSD | 41 (36.0) | 29 (70.7) |
| Patent ductus arteriosus | 32 (28.1) | 18 (56.3) |
| Left-sided obstructive lesion | 3 (2.0) | 2 (66.7) |
| Coarctation of the aorta | 2 (66.7) | 1 (50.0) |
| Pulmonary vein stenosis | 1 (33.3) | 1 (100) |
| Complex/mixed CHD | 12 (8.1) | 10 (83.3) |
| VSD, coarctation of the aorta | 3 (25.0) | 1 (33.3) |
| AVSD, coarctation of the aorta | 2 (16.7) | 2 (100) |
| VSD, pulmonary vein stenosis | 1 (8.3) | 1 (100) |
| Tetralogy of Fallot | 1 (8.3) | 1 (100) |
| Tetralogy of Fallot and AVSD | 2 (16.7) | 2 (100) |
| Unbalanced AVSD, single ventricle physiology | 1 (8.3) | 1 (100) |
| Pulmonary atresia and Ebstein anomaly | 1 (8.3) | 1 (100) |
| Pulmonary atresia, AVSD, pulmonary vein stenosis | 1 (8.3) | 1 (100) |
Percentages in italics use the leading defect as the denominator, not the total cohort. Nine of 158 patients without CHD are not shown.
The majority of the patients with a posttricuspid shunt as their primary lesion underwent surgical repair (69.2%). Left-sided obstructive heart disease was rare, with few cases of aortic coarctation and pulmonary vein stenosis in isolation (2%) or as part of more complex CHD (4%). Twelve patients (8.1%) were identified as having complex or mixed CHD, most of whom (83.3%) underwent repair (Table III).
Respiratory and Other Comorbidities in Down Syndrome
Respiratory comorbidities are highly prevalent in patients with Down syndrome, with apnea the most common, reported in 56% of the patients with Down syndrome in the registry. Additional respiratory diagnoses included recurrent respiratory exacerbations (35%), tracheomalacia (14%), chronic lung disease (4%), tracheal stenosis (4%), and other upper airway lesions (2%). One-quarter of the patients underwent tonsillectomy and/or adenoidectomy, and 12% underwent an additional ear, nose, and throat procedure (not specified). In addition, aspiration was reported in 31% of patients with Down syndrome, with 14% undergoing Nissen fundoplication and 5% having nasojejunal or jejunal feeding tube placement. Gastroesophageal reflux medications were reported in 38%. The median gestational age was 37.0 weeks (IQR, 35.0–38.7 weeks; n = 138), with nearly one-half (49%) being born prematurely. Hypothyroidism was noted in 20 patients (12.7%) (Table IV; available at www.jpeds.com).
Table IV.
Respiratory and other comorbidities and pulmonary hypertension resolution in patients with Down syndrome and pulmonary hypertension
| Pulmonary hypertension resolution (off medication) |
|||
|---|---|---|---|
| Comorbidities | Group N | Resolution, n (%) | P value |
|
| |||
| Primary WSPH group | .01 | ||
| Group 1/1′/1″ | 116 | 48 (41.4) | |
| Group 2 | 2 | 0 (0.0) | |
| Group 3 | 33 | 18 (54.5) | |
| Group 5 | 7 | 0 (0.0) | .002 |
| Age at pulmonary hypertension diagnosis | |||
| ≤6 mo | 82 | 44 (53.7) | |
| >6 mo | 76 | 22 (28.9) | |
| Recurrent respiratory exacerbations | .13 | ||
| No | 81 | 40 (40.6) | |
| Yes | 43 | 15 (25.9) | |
| Apnea* | .87 | ||
| No | 69 | 28 (28.9) | |
| Yes | 88 | 38 (43.2) | |
| Tracheomalacia | .61 | ||
| No | 118 | 52 (44.1) | |
| Yes | 19 | 10 (52.6) | |
| Upper airway lesions | |||
| No | 130 | 61 (46.9) | .50 |
| Yes | 2 | 0 (0.0) | |
| Tracheal stenosis | |||
| No | 132 | 59 (44.7) | .66 |
| Yes | 5 | 3 (60.0) | |
| Tonsillectomy and/or adenoidectomy | .45 | ||
| No | 111 | 46 (41.4) | |
| Yes | 37 | 18 (48.6) | |
| Tracheostomy | .10 | ||
| No | 134 | 61 (45.5) | |
| Yes | 14 | 3 (21.4) | |
| Other ENT procedure | .45 | ||
| No | 129 | 58 (45.0) | |
| Yes | 18 | 6 (33.0) | |
| Chronic lung disease* | .70 | ||
| No | 151 | 64 (42.4) | |
| Yes | 7 | 2 (28.60) | |
| Aspiration | .71 | ||
| No | 90 | 42 (46.7) | |
| Yes | 40 | 17 (42.5) | |
| Nissen | 1.00 | ||
| No | 119 | 54 (45.4) | |
| Yes | 19 | 8 (42.1) | |
| NJ/J tube | 1.00 | ||
| No | 126 | 56 (44.4) | |
| Yes | 6 | 3 (50.0) | |
| Reflux medications | .37 | ||
| No | 81 | 38 (46.9) | |
| Yes | 50 | 19 (38.0) | |
| Premature | 1.00 | ||
| No | 70 | 32 (45.7) | |
| Yes | 68 | 32 (47.1) | |
| Hypothyroidism* | .15 | ||
| No | 138 | 61 (44.2) | |
| Yes | 20 | 5 (25.0) | |
| CHD | .31 | ||
| No | 9 | 2 (22.2) | |
| Yes | 149 | 64 (43.0) | |
| CHD repair status | .87 | ||
| No | 57 | 23 (40.4) | |
| Yes | 101 | 43 (42.7) | |
| Posttricuspid shunt | .60 | ||
| No | 48 | 22 (45.8) | |
| Yes | 110 | 44 (40) | |
| Pretricuspid shunt | .02 | ||
| No | 135 | 51 (37.8) | |
| Yes | 23 | 15 (65.2) | |
| Complex CHD | .36 | ||
| No | 146 | 63 (43.2) | |
| Yes | 12 | 3 (25.0) | |
| Left-sided obstructive lesion | 1.00 | ||
| No | 156 | 65(41.2) | |
| Yes | 2 | 1 (50.0) | |
ENT, ear, nose, and throat; NJ/J, nasojejunal/jejunostomy.
Data obtained from write-in of diagnosis, not specifically queried in the case report form of the registry.
Outcomes
There were 12 deaths and no lung transplants or Potts shunts in the cohort with Down syndrome. Estimated 3-and 5-year survival rates were 90% (95% CI, 82%–98%) and 88% (95% CI, 80%–97%), respectively (Figure, A).
Figure.
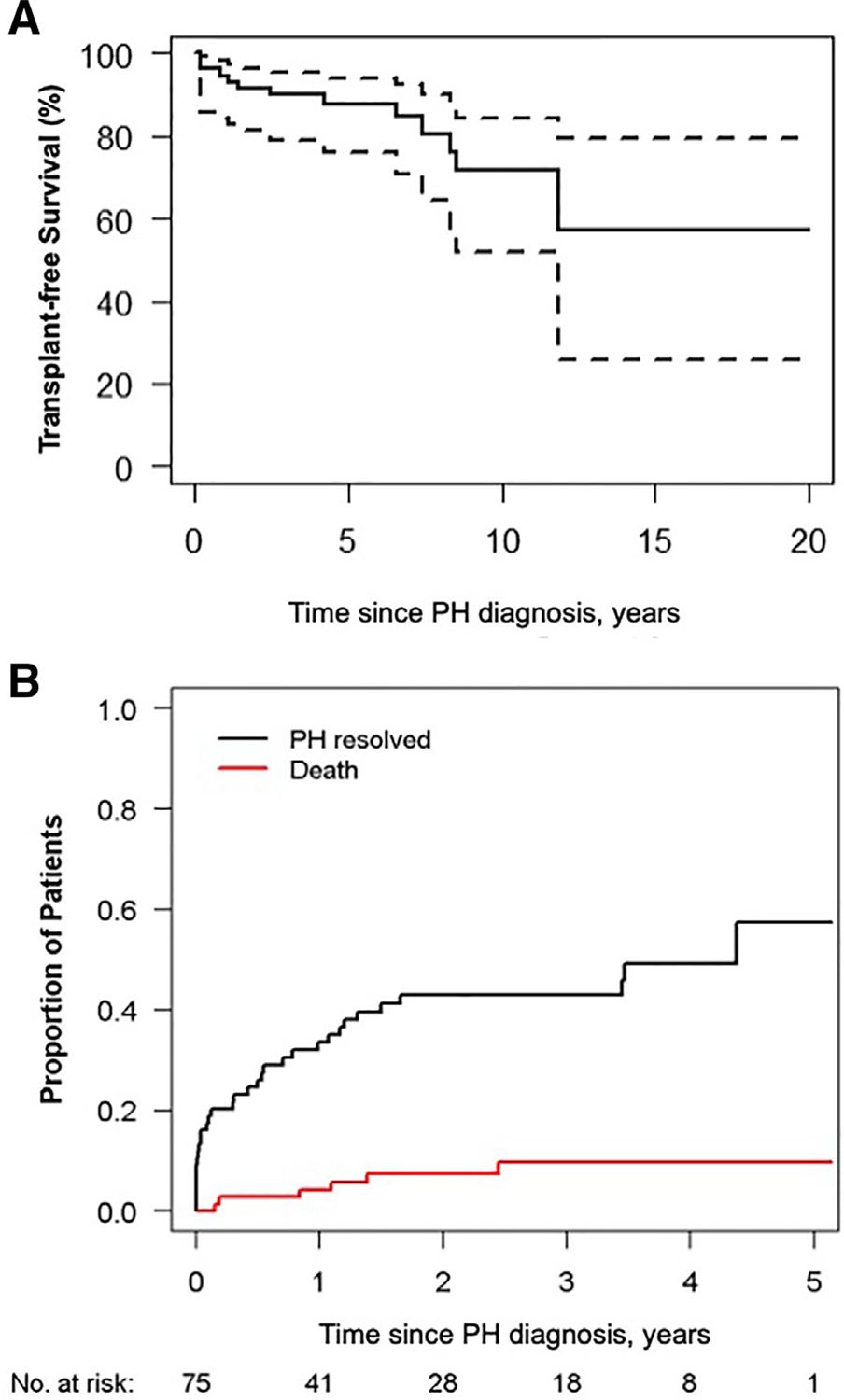
Outcomes in pediatric patients with Down syndrome and pulmonary hypertension. A, Overall survival of children with Down syndrome in the PPHNNet registry (n = 158). No lung transplantations were performed in this cohort. All available follow-up data were used, but the graph is truncated at 20 years. Dashed lines denote 95% pointwise CI bands. B, Display of competing risks estimates including death, lung transplantation, alive with pulmonary hypertension, and alive off pulmonary hypertension medications for the PPHNet incident cohort of patients with Down syndrome (n = 75). Estimated mortality is 4% at 1 year postdiagnosis and 10% at 3 years postdiagnosis. Estimated pulmonary hypertension resolution rate (defined as cessation of pulmonary hypertension medications; see Methods) is 34% at 1 year postdiagnosis and 43% at 3 years postdiagnosis.
Figure B, displays competing risk estimates including death, lung transplant, alive with pulmonary hypertension, and pulmonary hypertension resolution for the Down syndrome incident cohort (enrolled in the Registry at time of pulmonary hypertension diagnosis, n = 75). Estimated mortality is 4% at 1 year, and 10% at 3 years post-pulmonary hypertension diagnosis. Estimated pulmonary hypertension resolution is 34% at 1 year and and 43% at 3 years after diagnosis of pulmonary hypertension (Figure, B). Resolution of pulmonary hypertension was more likely in patients diagnosed with pulmonary hypertension at age <6 months (54% vs 29% in those diagnosed at age >6 months; P = .002) and had a pretricuspid shunt (65% vs 38% in those without a pretricuspid shunt; P = .02). There was no association between pulmonary hypertension resolution and other forms of CHD or repair status of CHD, or with diagnosed respiratory comorbidities or prematurity. The significance of WSPH group reflects higher resolution rates in groups 1 and 3 vs groups 2 and 5, which showed no resolution in the small number of patients (Table IV), as a pairwise comparison found no difference in resolution rate between groups 1 and 3 (P = .17).
In addition, we sought to assess correlates of pulmonary hypertension severity using a composite outcome measure of clinical characteristics reflecting more severe pulmonary hypertension in the cohort with Down syndrome (see Methods). This composite endpoint was met in 54 patients with Down syndrome (34%), with some meeting more than 1 outcome; 15 (9.5%) were categorized as Pediatric Panama or WHO functional class IV, 30 (19.0%) were hospitalized for pulmonary hypertension, 14 (8.9%) were treated with a continuous prostacyclin, and 12 (7.6%) died. Sixteen patients of the 54 (10.1%) met the composite endpoint at time of pulmonary hypertension diagnosis. The median follow-up was 3.55 years (IQR, 0.84–6.67 years).
We next assessed which clinical comorbidities contributed to development of the composite outcome of severe pulmonary hypertension. Univariate analysis revealed a higher hazard ratio (HR) of severe pulmonary hypertension in patients with aspiration, tracheostomy, treatment with gastroesophageal reflux medications, and male sex (Table V; available at www.jpeds.com).
Table V.
Univariate Cox regression analysis of the composite outcome of severe pulmonary hypertension in Down syndrome (left-truncated delayed entry to risk set methodology)
| Predictors | All Down syndrome | Severe pulmonary hypertension outcome | No severe pulmonary hypertension outcome | HR (95% CI) | P value |
|---|---|---|---|---|---|
|
| |||||
| Number of patients | 158 | 54 | 104 | ||
| Race, n (%) | .27 | ||||
| White | 87 (62.1) | 32 (66.7) | 55 (59.8) | 3.06 (0.41–22.98) | |
| Black | 22 (15.7) | 8 (16.7) | 14 (15.2) | 2.31 (0.28–19.07) | |
| Asian | 21 (15.0) | 7 (14.6) | 14 (15.2) | 1.47 (0.18–12.31) | |
| Other | 10 (7.1) | 1 (2.1) | 9 (9.8) | Reference | |
| Hispanic ethnicity, n (%) | 31 (21.2) | 11 (22.4) | 20 (20.6) | 1.14 (0.57–2.29) | .71 |
| Male sex, n (%) | 85 (53.8) | 36 (66.7) | 49 (47.1) | 1.97 (1.10–3.53) | .02 |
| Primary WSPH, n (%) | |||||
| Group 1 | 110 (69.6) | 35 (64.8) | 75 (72.1) | 0.64 (0.19–2.22) | .22 |
| Group 2 | 2 (1.3) | 1 (1.9) | 1 (1.0) | 2.18 (0.19–24.64) | |
| Group 3 | 33 (20.9) | 12 (22.2) | 21 (20.2) | 0.81 (0.22–3.05) | |
| Group 5 | 7 (4.4) | 1 (1.9) | 6 (5.8) | 0.41 (0.04–4.29) | |
| Group 1′ | 2 (1.3) | 2 (3.7) | 0 (0) | 3.51 (0.52–23.87) | |
| Group 1″ | 4 (2.5) | 3 (5.6) | 1 (1.0) | Reference | |
| Age at diagnosis ≤6 mo, n (%) | 82 (51.9) | 29 (53.7) | 53 (51.0) | 1.12 (0.64–1.93) | .70 |
| Gestational age, wk, mean ± SD | 35.9 ± 3.7 | 35.3 ± 4.2 | 36.3 ± 3.3 | 0.97 (0.91–1.04) | .40 |
| Premature (<37 wk of gestational age), n (%) | 68 (49.3) | 25 (50.0) | 43 (48.9) | 1.20 (0.67–2.16) | .54 |
| Aspiration, n (%) | 40 (30.8) | 21 (48.8) | 19 (21.8) | 2.39 (1.29–4.44) | .006 |
| Reflux medications, n (%) | 50 (38.2) | 25 (58.1) | 25 (28.4) | 2.34 (1.25–4.39) | .008 |
| Nissen, n (%) | 19 (13.8) | 8 (17.8) | 11 (11.8) | 1.38 (0.63–2.99) | .42 |
| NJ/J tube, n (%) | 6 (4.5) | 3 (7.1) | 3 (3.3) | 1.55 (0.47–5.11) | .47 |
| Tracheomalacia, n (%) | 19 (13.9) | 9 (20.0) | 10 (10.9) | 2.06 (0.98–4.33) | .06 |
| Tracheal stenosis, n (%) | 5 (3.6) | 3 (6.7) | 2 (2.2) | 1.68 (0.51–5.56) | .40 |
| Upper airway lesions, n (%) | 2 (1.5) | 1 (2.3) | 1 (1.1) | 0.99 (0.13–7.55) | .99 |
| Recurrent respiratory exacerbations, n (%) | 43 (34.7) | 14 (35.9) | 29 (34.1) | 1.29 (0.66–2.53) | .45 |
| Tonsillectomy and/or adenoidectomy, n (%) | 37 (25.0) | 10 (19.6) | 27 (27.8) | 0.91 (0.41–1.99) | .81 |
| Tracheostomy, n (%) | 14 (9.5) | 12 (24.0) | 2 (2.0) | 3.38 (1.69–6.74) | <.001 |
| Other ENT procedure, n (%) | 18 (12.2) | 7 (14.0) | 11 (11.3) | 1.24 (0.55–2.82) | .61 |
| Apnea, n (%) | 19 (90.5) | 7 (87.5) | 12 (92.3) | 1.69 (0.16–17.88) | .66 |
| Hypothyroidism, n (%) | 4 (19.0) | 2 (25.0) | 2 (15.4) | 1.19 (0.11–12.37) | .89 |
| CHD, n (%) | 149 (94.3) | 50 (92.6) | 99 (95.2) | 1.04 (0.36–2.97) | .95 |
| CHD repair, n (%) | 101 (63.9) | 33 (61.1) | 68 (65.4) | 0.84 (0.48–1.48) | .55 |
| Pretricuspid shunt, n (%) | 23 (14.6) | 10 (18.5) | 13 (12.5) | 1.00 (0.48–2.06) | 1.00 |
| Posttricuspid shunt, n (%) | 110 (69.6) | 32 (59.3) | 78 (75.0) | 0.79 (0.42–1.47) | .46 |
| Complex defect, n (%) | 12 (7.6) | 6 (11.1) | 6 (5.8) | 1.60 (0.67–3.84) | .29 |
| Pulmonary vein stenosis, n (%) | 3 (17.6) | 0 (0) | 3 (27.3) | 0.00 (0.00–0.00) | NE |
NE, nonestimable.
We also performed univariate analyses of hemodynamic measurements obtained from the subset of patients with Down syndrome who had cardiac catheterization performed close to the time of pulmonary hypertension diagnosis (n = 79). Only mean right atrial pressure (RAP) was associated with the hazard of the composite failure outcome (HR, 1.29 per mmHg; 95% CI, 1.04–1.61; P = .02; 69 patients with 23 events). Mean RAP was 7.6 ± 2.3 mmHg for those who met the outcome vs 6.9 ± 2.3 mmHg for those who did not meet the outcome. We also compared the hazard of severe pulmonary hypertension for patients with mean RAP above the median (>7.2 mmHg) vs at or below the median. For those with mean RAP above the median in this cohort, the HR was 2.81 (95% CI, 1.17–6.73; P = .02).
A multivariable model for the composite outcome of pulmonary hypertension severity (in the full cohort with Down syndrome, not only those with catheterization) identified tracheostomy (HR, 3.29, 95% CI, 1.61–6.69; P < .001) and gastroesophageal reflux medications (HR, 2.08; 95% CI, 1.11–3.90; P = .02) as independent correlates for severe pulmonary hypertension. A minority (18%) of patients with Down syndrome who had neither feature met the severe pulmonary hypertension composite endpoint, and a majority (80%–88%) of those with tracheostomy, with or without gastroesophageal reflux, had severe pulmonary hypertension (Table VI; available at www.jpeds.com). A sensitivity analysis restricted to the cohort of patients enrolled as incident cases also identified tracheostomy as the factor most strongly associated with the severe pulmonary hypertension composite endpoint.
Table VI.
Percentage of patients with Down syndrome with composite outcome of severe pulmonary hypertension by risk profile
| Features |
||||
|---|---|---|---|---|
| Reflux medications | Tracheostomy | Severe pulmonary hypertension, % (n/N) | Number of features | Severe pulmonary hypertension, % (n/N) |
|
| ||||
| No | No | 18 (14/76) | 0 | 18 (14/76) |
| No | Yes | 80 (4/5) | 1 | 47 (22/47) |
| Yes | No | 43 (18/42) | ||
| Yes | Yes | 88 (7/8) | 2 | 88 (7/8) |
Pulmonary Hypertension in Children with Down Syndrome: Results from the Pediatric Pulmonary Hypertension Network 140.e3 Registry
Medical Therapy for Pulmonary Hypertension
We determined whether medical therapy for pulmonary hypertension was prescribed differently in patients with Down syndrome compared with those without Down syndrome recorded in the registry. We assessed the use of any pulmonary hypertension medication, monotherapy vs combination therapy, and the individual classes of pulmonary hypertension medications: phosphodiesterase type V inhibitors, endothelin receptor antagonists, prostacyclins, and calcium channel blockers. Because patients without Down syndrome tended to have a higher PVRi than those with Down syndrome (among those who underwent cardiac catheterization), we assessed pulmonary hypertension medication use controlling for (ie, stratified by) PVRi category: <5, 5–10, and >10 WU*m2 (the highest value observed during follow-up) in the 82 patients with Down syndrome and 692 patients without Down syndrome with PVRi data available (Table VII).
Table VII.
Pulmonary hypertension medication use in patients with Down syndrome vs patients without Down syndrome, overall and stratified by maximum PVRi
| Maximum PVRi, WU × m2* |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Overall |
<5 |
5–10 |
>10 |
||||||
| Medications | Down syndrome | Non-Down syndrome | Down syndrome | Non-Down syndrome | Down syndrome | Non-Down syndrome | Down syndrome | Non-Down syndrome | Adjusted P† |
|
| |||||||||
| Number of patients | 158 | 1317 | 26 | 193 | 29 | 223 | 27 | 276 | .25 |
| Pulmonary hypertension medication used, n (%) | |||||||||
| Never | 22 (13.9) | 282 (21.4) | 7 (26.9) | 45 (23.3) | 5 (17.2) | 66 (29.6) | 5 (18.5) | 60 (21.7) | |
| Ever | 136 (86.1) | 1035 (78.6) | 19 (73) | 148 (76.7) | 24 (82.8) | 157 (70.4) | 22 (81.5) | 172 (62.3) | .37 |
| Mode, n (%)‡ | |||||||||
| Monotherapy | 54 (39.7) | 395 (38.1) | 12 (63.1) | 63 (42.6) | 8 (33.3) | 50 (31.8) | 8 (36.4) | 81 (47.1) | |
| Combination therapy | 82 (60.2) | 640 (61.8) | 7 (36.8) | 85 (57.4) | 16 (66.6) | 107 (68.2) | 14 (63.6) | 135 (78.5) | |
| Phosphodiesterase type 5 inhibitor | 112 (82.4) | 886 (85.6) | 13 (68.4) | 125 (84.5) | 19 (79.2) | 134 (85.3) | 17 (77.3) | 166 (96.5) | .75 |
| Endothelin receptor antagonist | 55 (40.4) | 405 (39.1) | 7 (36.8) | 47 (31.8) | 11 (45.8) | 73 (46.5) | 12 (54.5) | 91 (52.9) | .22 |
| Prostacyclin | 44 (32.3) | 277 (26.7) | 5 (26.3) | 27 (18.2) | 8 (33.3) | 47 (29.9) | 7 (31.8) | 60 (34.9) | .25 |
| Calcium channel blocker | 11 (8.0) | 94 (9) | 1 (5.2) | 10 (6.8) | 4 (16.7) | 11 (7.0) | 2 (9.1%) | 34 (19.8) | .72 |
Excludes 76 patients with Down syndrome and 625 patients without Down syndrome with no available PVRi measurement.
P value from logistic regression of medication use in patients with Down syndrome vs patients without Down syndrome, adjusted for categorical PVRi.
Percentages for monotherapy and combination therapy and the individual medication classes use the number ever used as the denominator.
The majority of patients in the registry were prescribed pulmonary hypertension medications, 86% in the Down syndrome cohort and 79% in the non-Down syndrome cohort. Adjusting for PVRi, there was no difference in the rate of pulmonary hypertension medication use between the 2 cohorts. Prescribing practices of monotherapy vs combination pulmonary hypertension therapy were similar in the 2 cohorts, with combination therapy generally being more common with increasing PVRi (Table VII).
Discussion
Pulmonary hypertension contributes to morbidity and mortality in diverse heart, lung, and systemic diseases, including many children and young adults with Down syndrome. Children with Down syndrome are particularly susceptible, as ~28% develop severe pulmonary hypertension.1,6,27,28 Pulmonary hypertension is the primary predictor of adverse cardiovascular outcome in adults with Down syndrome29; however, underlying factors that modify the risk of pulmonary hypertension in Down syndrome remain poorly understood, given the lack of a comprehensive understanding of the pulmonary hypertension phenotype and longer-term outcomes in children with Down syndrome. To address this need, we evaluated the 158 children with Down syndrome in the PPHNet registry and found a high burden of both pulmonary hypertension severity and comorbid conditions.
We found that the median age of pulmonary hypertension diagnosis and cardiac catherization rate was similar in patients with Down syndrome and those without Down syndrome, although age at catheterization was younger in the former group, possibly related to the high incidence of CHD in our cohort with Down syndrome (95%). Patients with Down syndrome had slightly milder disease at presentation and a median Qp:Qs >1, consistent with a net left-to-right shunt. As such, subjects with Down syndrome were more likely to be classified as having pulmonary hypertension associated with CHD rather than lung disease, different from the larger registry cohort without Down syndrome, in which pulmonary hypertension associated with lung disease was most common. However, respiratory comorbidities, including abnormal airway structure, apnea, aspiration, and others, were identified as relatively common in Down syndrome–associated pulmonary hypertension and not infrequently complicated the course of group 1 disease. It remains challenging to ascertain the relative contribution of different factors to the development of pulmonary hypertension in the cohort with Down syndrome, with a wide variety of respiratory and other diagnoses and a spectrum of CHD severity and repair status. A possible factor in the high proportion of group 1 classification in children with multiple contributors to pulmonary hypertension is the fact that CHD may be more readily diagnosed, and diagnosed at a younger age, than developmental lung disease in patients with Down syndrome. This may be especially true if lung disease is mild in severity, or characterized by symptoms, such as tachypnea, that may be nonspecific or potentially attributable to CHD. The high prevalence of CHD may account, at least in part, for the increased rate of electrocardiography at diagnosis compared with non-Down syndrome patients. The lower rate of chest CT scans in patients with Down syndrome at time of pulmonary hypertension diagnosis may suggest a lack of recognition of potential lung disease by clinicians, a perceived lack of therapeutic benefit for lung imaging at time of pulmonary hypertension diagnosis, difficulty obtaining CT scans owing to such challenges as the need for sedation in a young child, or other reasons. The lower frequency of pulmonary function testing and the 6-minute walk test in patients with Down syndrome vs patients without Down syndrome at the time of pulmonary hypertension diagnosis may reflect a limitation related to age or developmental ability to perform these tests.
The high prevalence of CHD in our registry supports the likelihood that CHD of being a significant contributor to pulmonary hypertension in children with Down syndrome. The distribution of and types of lesions in the PPHNet registry, with VSD and AVSD the most prevalent, are similar to other reports of children with Down syndrome (with and without pulmonary hypertension).10,30,31 We presume the 68% of repaired lesions likely were hemodynamically significant, especially in those with posttricuspid shunts or complex CHD, which could contribute to the development of pulmonary vascular disease.19,32 In the 13% with ASDs, more than one-half underwent repair. Whether an isolated pretricuspid shunt contributes to the development of pulmonary hypertension in Down syndrome remains unclear, but ASDs in isolation are rarely associated with childhood-onset pulmonary hypertension.33,34 We hypothesize that these patients might have had more of a contribution from developmental mechanisms, but the heterogeneity of other clinical factors limits more refined understanding. Additional limitations to understanding these contributions include the fact that only one-half of the patients in the registry had hemodynamic data from cardiac catheterization, and there was limited formal diagnosis of chronic lung disease and lung imaging, because such investigations were not protocolized for the study. We acknowledge that we may have overestimated the percentage of persistent CHD cases in the registry, given that data pertaining to repair status were limited to the time of enrollment and spontaneous closure of defects was not specifically queried.
Growing evidence suggests that children with Down syndrome have insufficient lung development consistent with a lung hypoplasia phenotype.13,14 Bush et al demonstrated that the underdeveloped pulmonary vascular and alveolar phenotype in patients with Down syndrome is similar to that in the lungs of infants with prematurity-associated bronchopulmonary dysplasia, which likely contributes to the increased incidence and severity of pulmonary hypertension.12 This contributed to the inclusion of pulmonary hypertension associated with Down syndrome as part of the group 3 (lung disease) classification in the most recent WSPH.7,19 In our study, airway and lung parenchymal abnormalities and comorbidities were quite common in children with Down syndrome and pulmonary hypertension. More than one-half of the patients with Down syndrome (for whom data were available) were diagnosed with sleep apnea (not further specified as central or obstructive), nearly one-half were born prematurely, and >30% had recurrent respiratory exacerbations. However, few patients in our cohort were explicitly labeled as having chronic lung disease. Although the number was surprisingly low, given that many children with Down syndrome have some form of impaired lung, pulmonary vascular, and/or lymphatic growth, this may be an underestimate owing to limitations in current clinical and imaging markers of developmental lung disease, as well as a lack of protocolized assessment mandated for study subjects. In addition, there remains a need to develop sufficient diagnostic tools and criteria to better identify the presence of and quantify the relative contribution of impaired lung development in children with Down syndrome–associated pulmonary hypertension.
Resolution of pulmonary hypertension was high in our cohort (43% at 3 years), similar to the finding reported for the overall registry for patients with group 1 pulmonary hypertension (42% at 3 years) but lower than that reported for group 3 pulmonary hypertension (60%).20 This may suggest that pulmonary hypertension in Down syndrome is phenotypically closer to group 1 pulmonary hypertension in children, which includes PPHN and CHD-related pulmonary hypertension. Resolution in patients with Down syndrome was associated with diagnosis of pulmonary hypertension at age <6 months and the presence of a pretricuspid shunt (regardless of repair status). The resolution of pulmonary hypertension diagnosed in infancy suggests contributions of developmental components, such as lung immaturity and/or PPHN-like vascular physiology, that improve with age and growth. The association with pulmonary hypertension resolution and the presence of a pretricuspid shunt is less obvious, with no known association between disease resolution and ASD described previously. Although resolution of pulmonary hypertension is possible after CHD repair or correction of impaired ventilation due to sleep apnea, it was perhaps somewhat surprising that resolution was not associated with other recognized, modifiable comorbidities, such as repaired CHD, specific type of CHD, or upper airway intervention. Age at diagnosis may have been a more powerful predictor for resolution, as it may encompass multiple reversible processes. This analysis is limited by relatively small numbers of patients and missing data, especially with respect to neonatal and respiratory comorbidities. Bush et al reported a much higher rate of reversible or “transient” pulmonary hypertension, in 70% of patients with Down syndrome and pulmonary hypertension in their cohort.1 This likely reflects differences in enrollment, as patients with PPHN that resolved early in infancy might not have been referred to a pulmonary hypertension program and would not have been captured in the PPHNet registry.1,20
Despite high overall survival, approximately one-third of patients met the criteria for a composite clinical outcome of pulmonary hypertension severity, which included death, lung transplant, Potts shunt, continuous prostacyclin therapy, hospitalization attributed to pulmonary hypertension after diagnosis, and Pediatric Panama or WHO functional class IV, with hospitalization the most common event. This highlights a subset of children with Down syndrome–associated pulmonary hypertension with severe, persistent disease. Independent correlates of severe pulmonary hypertension included tracheostomy and use of gastroesophageal reflux medications, together implicating significant lung or airway pathology or ongoing insult to the developing lung by aspiration in the more severe phenotype.
Limitations of this study include the enrollment of subjects from subspecialty centers with unique expertise in carefully diagnosing and treating patients with pulmonary hypertension, which may impair generalizability. That said, there is variability in terms of the patient population, consented enrollment, and clinical care approaches among the centers in the registry. This variation, as well as the relatively small patient numbers at individual sites, likely accounts for the wide range of percentage of patients with Down syndrome with pulmonary hypertension by site. This does reflect the “real world” variability among many of the pediatric pulmonary hypertension centers in North America. In addition, patients in whom pulmonary hypertension resolved before referral to a pulmonary hypertension program, or those who died prior to referral, might not have been enrolled. There are also limitations to the outcomes analysis. By using time of pulmonary hypertension medication discontinuation as a surrogate for disease resolution, we might have overestimated the time to pulmonary hypertension resolution. Similarly, we might have underestimated the incidence of the composite severity outcome because of missing functional class data during follow-up visits. Additional limitations include potential bias in complete capture of prevalent cases during the enrollment period and missing clinical data.
Despite these limitations, we highlight the prevalence of cardiac and noncardiac comorbidities that likely contribute to the development and/or persistence of pulmonary hypertension in children with Down syndrome. Despite these comorbidities, overall outcomes were good, with rates of survival and resolution of pulmonary hypertension similar to those in patients with group 1 pulmonary hypertension who do not have Down syndrome. The high prevalence of CHD in our cohort with Down syndrome suggests a significant contribution from CHD, despite the wide spectrum of severity, that may be influenced by respiratory comorbidities, even in children not primarily classified as having group 3 pulmonary hypertension. This study supports evaluation for modifiable comorbidities in patients with Down syndrome and pulmonary hypertension, specifically highlighting severe lung or airway disease (as indicated by the need for tracheostomy) and gastroesophageal reflux as independent correlates of pulmonary hypertension severity. This underscores the need for additional studies to further elucidate the influence and interaction of comorbidities on the development and progression of pulmonary vascular disease.
Acknowledgments
Supported by a grant from the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health (NIH) (U01 HL12118, to K.M. and S.A.). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI or NIH. The authors declare no conflicts of interest.
We thank the staff at the PPHNet sites, investigators, and patients who contributed to the PPHNet registry. We also thank Rachel Shwalb, MIS and Minmin Lu, MS for their contributions to data management and analysis.
Glossary
- ASD
Atrial septal defect
- AVSD
Atrioventricular septal defect
- CHD
Congenital heart disease
- CT
omputed tomography
- PPHN
Persistent pulmonary hypertension of the newborn
- PPHNet
Pediatric Pulmonary Hypertension Network
- PVR
Pulmonary vascular resistance
- PVRi
Pulmonary vascular resistance indexed to body surface area
- RAP
Right atrial pressure
- VSD
Ventricular septal defect
- WHO
World Health Organization
- WSPH
World Symposium on Pulmonary Hypertension
References
- 1.Bush D, Galambos C, Ivy DD, Abman SH, Wolter-Warmerdam K, Hickey F. Clinical characteristics and risk factors for developing pulmonary hypertension in children with Down syndrome. J Pediatr 2018;202:212–9.e2. 10.1016/j.jpeds.2018.06.031 [DOI] [PubMed] [Google Scholar]
- 2.Berger RM, Beghetti M, Humpl T, Raskob GE, Ivy DD, Jing ZC, et al. Clinical features of paediatric pulmonary hypertension: a registry study. Lancet 2012;379:537–46. 10.1016/S0140-6736(11)61621-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marín MJ del C, Rotés AS, Ogando AR, Soto AM, Jiménez MQ, Camacho JLG, et al. Assessing pulmonary hypertensive vascular disease in childhood. Data from the Spanish registry. Am J Respir Crit Care Med 2014;190:1421–9. 10.1164/rccm.201406-1052OC [DOI] [PubMed] [Google Scholar]
- 4.van Loon RLE, Roofthooft MTR, Hillege HL, ten Harkel ADJ, van Osch-Gevers M, Delhaas T, et al. Pediatric pulmonary hypertension in the Netherlands: epidemiology and characterization during the period 1991 to 2005. Circulation 2011;124:1755–64. 10.1161/CIRCULATIONAHA.110.969584 [DOI] [PubMed] [Google Scholar]
- 5.Moledina S, Hislop AA, Foster H, Schulze-Neick I, Haworth SG. Childhood idiopathic pulmonary arterial hypertension: a national cohort study. Heart 2010;96:1401–6. 10.1136/hrt.2009.182378 [DOI] [PubMed] [Google Scholar]
- 6.Weijerman ME, van Furth AM, van der Mooren MD, van Weissenbruch MM, Rammeloo L, Broers CJ, et al. Prevalence of congenital heart defects and persistent pulmonary hypertension of the neonate with Down syndrome. Eur J Pediatr 2010;169:1195–9. 10.1007/s00431-010-1200-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53:1801913. 10.1183/13993003.01913-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Espinola-Zavaleta N, Soto ME, Romero-Gonzalez A, Gómez-Puente L del C, Muñoz-Castellanos L, Gopal AS, et al. Prevalence of congenital heart disease and pulmonary hypertension in Down’s syndrome: an echocardiographic study. J Cardiovasc Ultrasound 2015;23:72–7. 10.4250/jcu.2015.23.2.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benhaourech S, Drighil A, Hammiri AE. Congenital heart disease and Down syndrome: various aspects of a confirmed association. Cardiovasc J Afr 2016;27:287–90. 10.5830/CVJA-2016-019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tandon R, Edwards JE. Cardiac malformations associated with Down’s syndrome. Circulation 1973;47:1349–55. 10.1161/01.CIR.47.6.1349 [DOI] [PubMed] [Google Scholar]
- 11.Cappelli-Bigazzi M, Santoro G, Battaglia C, Palladino MT, Carrozza M, Russo MG, et al. Endothelial cell function in patients with Down’s syndrome. Am J Cardiol 2004;94:392–5. 10.1016/j.amjcard.2004.04.047 [DOI] [PubMed] [Google Scholar]
- 12.Bush D, Abman SH, Galambos C. Prominent intrapulmonary bronchopulmonary anastomoses and abnormal lung development in infants and children with Down syndrome. J Pediatr 2017;180:156–62.e1. 10.1016/j.jpeds.2016.08.063 [DOI] [PubMed] [Google Scholar]
- 13.Schloo BL, Vawter GF, Reid LM. Down syndrome: patterns of disturbed lung growth. Hum Pathol 1991;22:919–23. 10.1016/0046-8177(91)90183-p [DOI] [PubMed] [Google Scholar]
- 14.Cooney TP, Thurlbeck WM. Pulmonary hypoplasia in Down’s syndrome. N Engl J Med 1982;307:1170–3. 10.1056/NEJM198211043071902 [DOI] [PubMed] [Google Scholar]
- 15.Galambos C, Minic AD, Bush D, Nguyen D, Dodson B, Seedorf G, et al. Increased lung expression of anti-angiogenic factors in Down syndrome: potential role in abnormal lung vascular growth and the risk for pulmonary hypertension. PLoS One 2016;11:e0159005. 10.1371/journal.pone.0159005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sánchez O, Domínguez C, Ruiz A, Ribera I, Alijotas J, Cabero L, et al. Angiogenic gene expression in Down syndrome fetal hearts. Fetal Diagn Ther 2016;40:21–7. 10.1159/000441356 [DOI] [PubMed] [Google Scholar]
- 17.Bush D, Wolter-Warmerdam K, Wagner BD, Galambos C, Ivy DD, Abman S, et al. Angiogenic profile identifies pulmonary hypertension in children with Down syndrome. Pulm Circ 2019;9:2045894019866549. 10.1177/2045894019866549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffiths M, Yang J, Vaidya D, Nies M, Brandal S, Ivy DD, et al. Biomarkers of pulmonary hypertension are altered in children with Down syndrome and pulmonary hypertension. J Pediatr 2022;241:68–76.e3. 10.1016/j.jpeds.2021.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Haworth S, et al. Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J 2019;53:1801916. 10.1183/13993003.01916-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abman SH, Mullen MP, Sleeper LA, Austin ED, Rosenzweig EB, Kinsella JP, et al. Characterisation of pediatric pulmonary hypertensive vascular disease from the PPHNet Registry. Eur Respir J 2021;59:2003337. 10.1183/13993003.03337-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, et al. Pediatric pulmonary hypertension: guidelines from the American Heart Association and American Thoracic Society. Circulation 2015;132:2037–99. 10.1161/CIR.0000000000000329 [DOI] [PubMed] [Google Scholar]
- 22.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc 1999;94:496–509. 10.1080/01621459.1999.10474144 [DOI] [Google Scholar]
- 23.Matsuura M, Eguchi S. Modeling late entry bias in survival analysis. Biometrics 2005;61:559–66. 10.1111/j.1541-0420.2005.00325.x [DOI] [PubMed] [Google Scholar]
- 24.Betensky RA, Mandel M. Recognizing the problem of delayed entry in time-to-event studies: better late than never for clinical neuroscientists. Ann Neurol 2015;78:839–44. 10.1002/ana.24538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lammers AE, Adatia I, del Cerro MJ, Diaz G, Freudenthal AH, Freudenthal F, et al. Functional classification of pulmonary hypertension in children: report from the PVRI pediatric taskforce, Panama 2011. Pulm Circ 2011;1:280–5. 10.4103/2045-8932.83445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.R Core Team. R: a language and environment for statistical computing: R Foundation for Statistical Computing. Accessed November 1, 2021. https://www.r-project.org/ [Google Scholar]
- 27.Cua CL, Blankenship A, North AL, Hayes J, Nelin LD. Increased incidence of idiopathic persistent pulmonary hypertension in Down syndrome neonates. Pediatr Cardiol 2007;28:250–4. 10.1007/s00246-006-0011-6 [DOI] [PubMed] [Google Scholar]
- 28.McDowell KM, Craven DI. Pulmonary complications of Down syndrome during childhood. J Pediatr 2011;158:319–25. 10.1016/j.jpeds.2010.07.023 [DOI] [PubMed] [Google Scholar]
- 29.Hayes SA, Kutty S, Thomas J, Johnson JT, Yetman AT. Cardiovascular and general health status of adults with Trisomy 21. Int J Cardiol 2017;241:173–6. 10.1016/j.ijcard.2017.03.040 [DOI] [PubMed] [Google Scholar]
- 30.Freeman SB, Taft LF, Dooley KJ, Allran K, Sherman SL, Hassold TJ, et al. Population-based study of congenital heart defects in Down syndrome. Am J Med Genet 1998;80:213–7. [DOI] [PubMed] [Google Scholar]
- 31.Pfitzer C, Helm PC, Rosenthal LM, Berger F, Bauer UMM, Schmitt KRI. Dynamics in prevalence of Down syndrome in children with congenital heart disease. Eur J Pediatr 2018;177:107–15. 10.1007/s00431-017-3041-6 [DOI] [PubMed] [Google Scholar]
- 32.Rabinovitch M, Keane JF, Norwood WI, Castaneda AR, Reid L. Vascular structure in lung tissue obtained at biopsy correlated with pulmonary hemodynamic findings after repair of congenital heart defects. Circulation 1984;69:655–67. [DOI] [PubMed] [Google Scholar]
- 33.Steele PM, Fuster V, Cohen M, Ritter DG, McGoon DC. Isolated atrial septal defect with pulmonary vascular obstructive disease—long-term follow-up and prediction of outcome after surgical correction. Circulation 1987;76:1037–42. 10.1161/01.cir.76.5.1037 [DOI] [PubMed] [Google Scholar]
- 34.Rosenzweig EB, Barst RJ. Congenital heart disease and pulmonary hypertension: pharmacology and feasibility of late surgery. Prog Cardiovasc Dis 2012;55:128–33. 10.1016/j.pcad.2012.07.004 [DOI] [PubMed] [Google Scholar]