

Abstract
Sickle Cell Disease (SCD) is one of the most inherited hematologic diseases affecting humans. Clinically, there is a progressive multiorgan failure and increased mortality in severe cases. The highest prevalence is in West Africa, India, the Mediterranean region, and Middle East countries. Hydroxyurea was the primary drug available for SCD and remains first-line therapy for patients with SCD. Three additional drug therapies, L-glutamine, Voxelotor, and Crizanlizumab, have been approved as adjunctive agents. However, none of these treatments are curative. Effective cell-based therapies are available, such as red blood cell (RBC) exchange and the only curative therapy is hematopoietic stem cell transplantation (HSCT). Gene-editing now shows promise in treating SCD and the β-thalassemias. Recent clinical trials have proven that this therapeutic strategy is effective, however costly. Despite the availability of safe and effective drug treatments, questions focusing on the overall value of these drugs exist in light of rising healthcare costs including hospitalizations and medical interventions. Herein, we report a cost-effective evaluation that can guide future efforts in making decisions towards HSCT as cell therapy treatment in SCD patients.
Keywords: Sickle cell disease, fetal hemoglobin, hematopoietic stem cell transplant
Introduction
Sickle Cell Disease (SCD) was identified by Herrick in 1910 [1] and then characterized biochemically and molecularly by Ingram in 1958 [2]. SCD arises as a result of a single missense mutation, leading to a replacement of glutamic acid by valine in the sixth position of the β-globin chain of hemoglobin. This swap on the protein level, converts HbA into the so-called sickle hemoglobin (HbS). In hypoxic conditions, HbS normally polymerizes resulting in the formation of deoxygenated hemoglobin fibrils, due to hydrophobic interactions between the valines in the adjacent HbS molecules, which in turn interact with the cytoskeleton and distort the natural biconcave disc shape of the red blood cell (RBC) creating the irreversible characteristic sickle or sliver moon-shaped cell.
Studies on examining DNA variants within the β-globin gene have confirmed that the HbS mutation occurred independently in several different populations in Central Africa, Central West Africa, African West coast, Arabian Peninsula and India and the presence of falciparum malaria has served as a selective factor in increasing its prevalence [3]. This would suggest that there is genetic pressure or genomic architecture that supports the single base change. Over the generations, the HbS gene has been reached high frequencies in regions with past or present history of malaria endemicity. However, population migration has played a major role in distributing HbS gene even to non-malaria endemic regions. Worldwide, between 300,000-400,000 individuals are born annually with SCD [4].
The extent of HbS polymerization is the primary determinant of the severity of SCD [5]. Clinically, SCD is characterized by two main pathologic events: hemolysis and recurrent acute vaso-occlusive crises (VOC). Over time, individuals with this condition experience numerous other life-threatening comorbidities throughout their lifetimes. Acute comorbidities, which can occur at any age, include VOC, stroke, acute chest syndrome (ACS), acute renal failure, priapism, splenic sequestration and retinopathy. Chronic comorbidities such as skin ulcers, pulmonary hypertension, diastolic heart dysfunction, kidney disease, and osteonecrosis increase with age [6]. Painful VOC crisis are the most common manifestation of SCD and remain the most common reason for presenting to the emergency department and hospitalization. VOC has previously been described to evolve along four distinct phases starting from a low-intensity pain and exacerbating with the development of worsening symptoms such as chronic disabling arthritis due to osteonecrosis affecting the joints, progressive retinopathy, chronic renal failure, increased risks for strokes, and shortened lifespan [7]. Chronic inflammatory processes associated with SCD and originated from a combination of membrane damage of erythrocytes carrying HbS and increased intestinal permeability are the main triggers of VOC development and aggression [8].
Stroke is the main neurological comorbidity in SCD and unfortunately, is one of the few complications seen more often in children than in adults [9]. In children with a severe phenotype of SCD, ~10% have documented stroke, and approximately 20 to 35% have silent cerebral infarcts. Parallel studies established that approximately 11% of patients with SCD will go on to develop a clinically apparent stroke by the age of 20 years, and 24% by the age of 45 years [10]. Strokes may be complicated by impaired cognition and an overall decrease in mental acuity. Silent infarcts, which do not manifest overtly but can accumulate over time, have been shown to cause neurocognitive deficits including severe headache, altered mental status, slurred speech, seizures, and partial paralysis in cases of overt stroke, in school-aged children and adults [10]. Some important ways that SCD manifests in the respiratory system are ACS, caused by infections and/or a blockage of blood flow to the chest and resulting in lung injury, breathing difficulty, low oxygen to the rest of the body. Repeated episodes can cause pulmonary arterial hypertension from the increased pulmonary vascular resistance and diastolic heart dysfunction. ACS is one of the most common causes of hospitalization for children and adults with SCD and is the root cause for more than 25% of premature deaths in sickle cell disease [11]. Multiple studies have estimated the mortality of the disease and found that 50% of patients died before the fifth decade, and most of those who died did not have overt chronic organ failure but during an episode of acute pain, ACS, or stroke [12]. Prospective follow-up made it possible to determine the incidence (approximately 13 per 100 patient-years), risk factors, presentation, and prognosis of the ACS [13]. Patients with SCD are at high risk for developing chronic kidney disease during their lifetime. It is possible that this progressive loss of kidney function is triggered by anemia, hemolysis, inflammation, infections and nonsteroidal pain medications. Acute kidney injury accounts for between 4% and 10% of hospitalized individuals with SCD [14]. Acute renal injury frequently co-occurs more in patients experiencing ACS (13.6%) than pain crises (2.3%). Progressive end-organ damage to the kidneys may result in adult patients becoming dialysis dependent. However, renal failure does coincide in approximately 75% of painful crisis episodes that involve multi-organ failure.
Occlusive events in the liver lead to intrahepatic cholestasis that presents with increased bilirubin levels and increased alkaline phosphatase, significant jaundice, and an enlarged, tender liver upon examination. Priapism is also a common complication that occurs amongst males, where 35% of all men and boys experience a painful erection that lasts for more than 4 hours and remains a major source of distress for male patient. Other comorbidities include infection and aplastic anemia brought on by parvovirus B-19. Ophthalmologic issues such as sickle cell retinopathy, can lead to vision loss if left uncontrolled in the adult. Leg ulcers and avascular necrosis of bone, most commonly in the femoral head and end with the need for total hip replacement, may limit SCD patients in their social interactions. Unfortunately, all abovementioned comorbidities, may contribute to clinical depression, estimated to be present in at least 20% of the sickle population. Typical ageing disorders further complicate the complex disease process and with the increase in lifespan of SCD patients, there is an increase in comorbidities.
In socio-economical resources-poor countries, it is reported that more than 90% of children with SCD do not survive to adulthood. However, in high-income industrialized countries, the current life expectancy for people living with SCD, is about 40-60 years. Indeed, SCD is considered a public health problem with a high prevalence among people who in many cases make up the poorest groups in society and have less access to health. Low socioeconomic and educational levels directly affect the quality of life of patients with SCD. Emphasis must be placed on implementing clinical and social strategies that specify early diagnosis, ongoing monitoring, and disease-specific therapies, by country. Furthermore, there is need to establish, clinical and socio-economic indicators that especially enhance the development of targeted treatment programs for patients with SCD in their resident region.
This review outlines the current availability of U.S. Food and Drug Administration (FDA)-approved drugs and curative cell therapies, aims to provide further information that would help to evaluate the cost of SCD treatments and clarifies the potential cost effectiveness evaluation among these treatments in making decisions towards hematopoietic stem cell transplant (HSCT) as cell therapy treatment in SCD patients.
SCD treatment
Comorbidities and premature death are well documented in persons with SCD and summarized in Table 1. Treatment goals at most hospitals are driven by the management of the acute complications due to a VOC event. However, treatment is most often dictated by the country where the patient resides, where those in wealthy countries are provided curative therapies and those in less-wealthy countries being restricted to symptom management rather than curative therapy [15].
Table 1.
SCD Major complications and manifestations
| Main comorbidities of SCD | Pathological manifestations |
|---|---|
| Acute pain and chronic pain syndrome | Opioid addiction, negative interference with patients’ daily function, depression and anxiety |
| Functional asplenia | Overwhelming infection |
| Splenic sequestration | Sudden pallor, anemia, abdominal pain and fatigue |
| Acute chest syndrome (ACS) | Fever, pain and tightness in chest, fast breathing, cough and shortness of breath |
| Right upper quadrant syndrome | Acute right upper quadrant pain, nausea, fever, tender hepatomegaly and jaundice |
| Cerebrovascular disease and stroke | Severe cerebral damage, weakness and paralysis |
| Neurocognitive deficits | Lower intelligence, visuo-motor impairments, executive dysfunction and memory dysfunction |
| Retinopathy | Vision loss and blindness |
| Priapism | Irreversible erectile dysfunction, Peyronie’s disease and mental morbidity |
| Chronic lung disease | Cyanosis, palpitations, dyspnea, edema, syncope, fatigue, chest pressure and pain |
| Chronic kidney disease | Anemia, hematuria, impaired urinary concentration, albuminuria, glomerular damage and end-stage renal disease |
| Pulmonary hypertension | Right-sided heart failure, chest pain and dyspnea |
| Skin ulcers | Chronic wounds, thrombosis, gangrene and amputation |
| Osteonecrosis | Fractures, Limited range of motion, limping and severe arthritis |
To select targeted therapies for SCD, different international societies have provided evidence-based recommendations by using the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) approach. For instance, the American Society of Hematology (ASH) stated that the decision-making on specific treatment programs should be individualized case-by-case. Moreover, while the preoperative transfusions are based on genotype, total baseline hemoglobin, risks of surgery, complications with prior transfusions, and disease severity, other treatments such as RBC exchange are considered depending on the clinical indication, patient age, baseline and target HbS hemoglobin, venous access preferences (particularly if central access is required), iron overload and iron chelation status, and availability of compatible red cells [16]. In the case of patients with recurrent episodes of ACS, frequent pain, or other complications (e.g., overt stroke), ASH suggests HLA-matched related HSCT rather than the standard of care (HU/transfusion) [17].
Drug therapy for the management of SCD
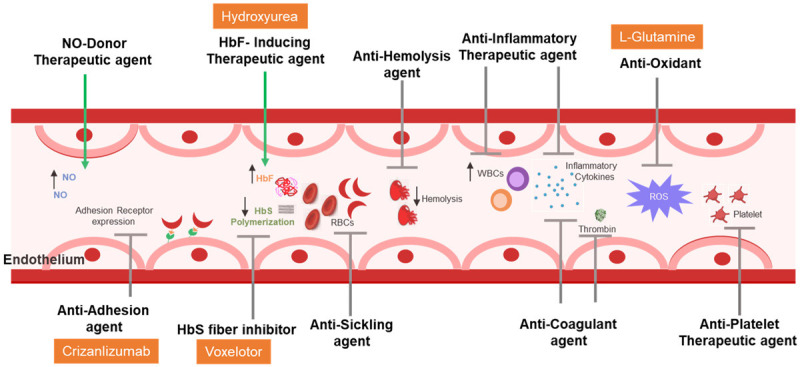
Despite an improved understanding of the pathophysiology of SCD, available drug treatments remain limited (Table 2). Hydroxyurea (HU) remained the only FDA-approved therapy for SCD for decades. HU has shown to increase the levels of fetal hemoglobin (HbF), and modify the severity of SCD. This can be an effective strategy as long as the levels of gamma globin are within a range that no longer presents a possibility of sickling due to polymerization of the mutant globin [18]. Till date, HU appears to be the best available therapeutic agent, for children and adults with SCD, due to its ease of oral administration, safety profile, in vivo efficacy for increasing percentage HbF and decreasing blood viscosity and proven clinical efficacy for preventing VOC events, anemia, hemolysis and chronic organ dysfunction. However, over the past five years, there has been progress in the development of drug therapies with recent approvals of L-glutamine in 2017 [19], Crizanlizumab, and Voxelotor, both in 2019, by the FDA. L-glutamine, an amino acid, was shown to reduce oxidative stress of red blood cells, thus reducing endothelial adhesion and subsequently the number of pain crises. Crizanlizumab, a P-selectin inhibitor reduces adhesion between the endothelium and endothelial cells, platelets, sickled red blood cells, and leukocytes resulting in decreasing the grade of inflammation. Finally, Voxelotor prevents HbS polymerization by increasing hemoglobin’s levels and affinity for oxygen and reducing therefore markers of hemolysis [20]. Based on current knowledge of SCD pathophysiology, there are multiple possible molecular approaches to treat the disease and its comorbidities (Figure 1). Pharmaceutical and clinical research studies aiming to discover novel drugs and their effect in improving outcomes in SCD are needed [21].
Table 2.
FDA-approved Drug for the management of SCD
| FDA-approved drug (FDA-approval date) | In vivo Mechanism of action | Treatment-related Benefits | Treatment-Related Adverse Events | General features |
|---|---|---|---|---|
| Hydroxyurea (1998) | HbF induction | ↓Painful acute VOC events | Headache | Single-agent inexpensive orally administered once-daily dosing |
| ↓WBC and platelets | ↓Inflammation | Gastrointestinal symptoms including Abdominal discomfort and nausea | ||
| ↓LDH and bilirubin levels | ↓Hemolysis | Anorexia | ||
| ↓surface expression of adhesion receptors | ↓Mortality Ameliorates anemia | Skin hyperpigmentation | ||
| ↑NO synthesis | Preserves splenic function | Neutropenia | ||
| Maintains excellent growth development and sexual maturation | Reticulocytopenia | |||
| Prevents chronic organ dysfunction | ||||
| L-glutamine (2017) | ↑NAD redox potential in sickle red blood cells through increasing the availability of reduced glutathione | ↓Chronic pain | Headache | Single-agent |
| ↓ROS and oxidative damage in sickle red blood cells | ↑Time to pain crisis | Nausea | orally administered | |
| ↑NO synthesis | ↓Daily narcotic use | Constipation | twice-daily dosing | |
| ↓Episodes of acute chest syndrome | Loss of taste | |||
| Chest and musculoskeletal pain Fatigue | ||||
| Cough | ||||
| Rare renal and hepatic impairment | ||||
| Crizanlizumab (2019) | P-selectin inhibitor, | ↓VOC rate episodes | Nausea | Monthly intravenous infusion |
| Blocks adhesion of neutrophils, activated platelets and sickle red blood cells to the endothelial surface of the blood vessels | Arthralgia | |||
| Back pain | ||||
| Fever | ||||
| Pruritus (pruritus and vulvovaginal pruritus) | ||||
| Pain in the extremity | ||||
| Voxelotor (2019) | ↑Hemoglobin’s affinity for oxygen | ↓Hemolysis | Headache | Orally administered once-daily dosing |
| ↓HbS polymerization | ↓Anemia | Gastrointestinal symptoms | ||
| Improves red blood cell deformability | Arthralgia | |||
| ↓Whole blood viscosity | Fatigue | |||
| Skin rash | ||||
| Fever |
Figure 1.
Possible molecular approaches for drug therapy.
Cell therapy for the management of SCD
Parallel to the new medications being developed, no single pharmacologic therapy has provided a complete suppress of the adverse outcomes of SCD. However, therapeutic approaches involving “cell” and/or “stem cell” potential are well studied and reported as curative therapies in SCD (Table 3). While these cell treatments are considered as optimal therapeutic applications, they remain subject to considerable technological and regulatory challenges and are likely to be costly.
Table 3.
Cell therapy for the management of SCD
| Cell therapies in SCD | In vivo Mechanism of Action | Cell Therapy Related - Benefits | Cell Therapy Related-Limitations | |
|---|---|---|---|---|
| RBC exchange | ↓HbS concentration and blood viscosity | Prevents or mitigates neurological disease and the associated comorbidities | Alloimmunization by repeated RBC transfusion | |
| ↓the burden of sickled cells | Risk of iron overload | |||
| Allogeneic HSCT | HLA-matched sibling donor transplant | Restores normal hematopoiesis | Mitigates progressive organ dysfunction | Limitation of suitable donor |
| 97% Overall survival in | Conditioning regimens dependence | |||
| 10-15 year follow-up | Patient with end-organ damage usually excluded | |||
| Lower rates of healthcare utilization | Transplantation related toxicity | |||
| HLA-matched unrelated donor | Restores normal hematopoiesis | Mitigates progressive organ dysfunction | Lack of comprehensive donor registries | |
| Lower rates of healthcare utilization | Time for search process, coordination of donor | |||
| High rates of graft rejection | ||||
| Conditioning regimen-dependence | ||||
| Outcomes related to age of donor and recipient | ||||
| Transplantation related toxicity | ||||
| Haploidentical donor | Myeloablative conditioning regimens required | Larger donor pool | High rates of graft rejection | |
| Available to most patients | Follow up less than 5 years | |||
| Improvement in the intensity of the conditioning regimens | ||||
| ↓Transplantation-related toxicity and graft failure | ||||
| Autologous Hematopoietic Stem Cell gene-based therapy | Gene addition | CD34+ HSCs are genetically modified by adding a therapeutic β-globin gene with lentiviral transduction | Robust β-globin expression in erythroid cells | Toxicity of conditioning |
| Risk of insertional oncogenesis and long-term high-level expression | ||||
| Reduces CD34+ HSCs engraftment ability | ||||
| Transplantation related - toxicity: | ||||
| Gene editing | CD34+ HSCs are genetically modified by CRISPR - based editing of a repressor protein | HbF induction | Minimal transplantation related - toxicity | |
| Avoiding insertional mutagenesis | ||||
RBC exchange is a standard common practice for treating SCD-related complications, where RBCs from a healthy matched donor are infused while the patient’s RBCs are simultaneously removed. This provides both an oxygen carrying capacity and removal of cells able to initiate an occlusive event thereby decreasing the likelihood of sickling and VOCs. An RBC exchange is preferred to a simple RBC transfusion to minimize the clinical complications of the increased viscosity of just adding more RBCs [22]. Repeated RBC exchange leads to sensitization of the patient to non-ABO antigens present on RBC so care in matching is key to long-term use of this option. Minimization of RBC donors for individual patients is an effective strategy often employed [23].
HSCT to replace the source of defective RBCs with marrow from a healthy donor is now a common therapy. The risks associated with this therapy are those that are associated with other allogenic transplants. The mortality of transplants is decreasing as new studies of graft failure, graft versus host disease (GvHD), and cohorts of transplants are performed by age of the recipient. New strategies for immunosuppressive therapy and increased options to manage GvHD are reducing the morbidity and mortality. Current improvements in allogenic transplant with the introduction of engineered grafts where the α/β T-cell fraction is depleted are proving to be much less toxic. The mortality reduction following HSCT also includes recent advances in immunosuppressive therapy and supportive care. The long-term survival of β-thalassemia patients that have undergone a HSCT was shown to be greater than 90% [24], and this has illuminated the utility and safety of this therapy for SCD. Clinical trials that were conducted in children with SCD in Europe and the US showed greater than 90% long-term survival [25]. A major limitation in the use of HSCT for the treatment of SCD is the fact that a matched sibling donor is available to less than 15% of patients who are otherwise suitable candidates for transplantation [26]. In an effort to increase the availability of sources of hematopoietic stem cells for transplantation, clinical trials are being conducted to evaluate cord blood transplantation in the treatment of SCD [9]. To date, very few transplantation procedures have been performed in adults with SCD because of concerns that the morbidity and mortality of HSCT is significantly higher in adults than in children. The use of nonmyeloablative regimens prior to HSCT in an effort to reduce morbidity and mortality have been associated with a very high graft rejection rate [27]. Currently, HSCT is the only approved curative therapy for SCD, and the major challenge is to make it more widely available to patients with a severe disease phenotype. A major limitation to the use of this therapy is the availability of HLA-matched siblings to be donors. The use of unrelated donors with a matched HLA is much less common, primarily due to the risk of GvHD as high as 19% in the first 100 days for acute GvHD and 29% over 3 years for chronic GvHD [28]. Haploidentical donors, where the donor is a parent or sibling are also an alternative approach that has a high graft survival rate and a decreased risk of GvHD, when the graft is manipulated to remove the α/β T-cells. Several recent studies have shown that the age or recipient, at transplant, is a significant variable in successful engraftment while minimizing complications from GvHD the risks decrease when the patient is less than 13 years old [28].
Gene editing therapy has now been used in a few clinical trials with good results. Correcting the defect in the β-globin gene using gene editing technologies such as CRISPR has now been used [29]. In this scenario, the bone marrow stem cells from the patient are treated in the laboratory with a gene editing method that corrects the single base defect and restores a functional β-globin gene. The cells are then infused back into the patient where they repopulate the marrow and produce “normal” β-globin. Ensuring that the only change made within the genome of the cells is critical to the success of this treatment, ensuring that no other problems are inadvertently introduced with the correction of the β-globin gene. As the editing technology becomes better and methods are in place to ensure there are no off-target effects this will become more widely used.
Gene silencing is also now used to treat SCD. The BCL11A gene encodes a protein that is responsible for the down-regulation of the γ-globin genes shortly after birth and is responsible for the fetal to adult switch of the hemoglobin gene. The treatment uses a short hairpin RNA to silence the BCL11A gene, removing the negative regulation and resulting in an increase in the γ-globin transcription. In a phase 1 study of six patients, bone marrow stem cells (CD34+) were modified with a lentivirus that carried the BCL11A silencing short hairpin RNA. In this small study all six patients showed increased HbF expression (58.9 to 93.6%) at a median follow-up of 18 months [30]. Patients reported no VOC events and other complications related to SCD were reduced in all subjects.
In a first-in-human study, Crispr/Cas9 editing was used to silence the BCL11A gene in two patients with β-thalassemia and sickle cell disease each [31]. Therefore, autologous CD34+ stem cells were edited by electroporation with no evidence of off-target editing. After myeloablation, the patients received the edited CD34+ stem cells. After more than one year, both patients had high levels of allelic editing in bone marrow and blood and an increase in HbF. The patient with β-thalassemia became transfusion independent and VOC were eliminated in the patient with SCD [31].
In any scenario, we consider that the diagnosis and treatment must be aligned with the current international guidelines and evidence-based recommendations, while the safety and efficacy of novel drugs and interventions can be evaluated in the context of formal clinical trials. Moreover, the clinical decisions shall be patient-tailored and based on individual features of SCD.
Conclusions
When looking at cost of treatment in SCD one must factor in the costs of all hospitalizations and medical interventions associated with the disease. These costs include hospitalizations due to pain crisis, procedures used to work up ACS, laboratory tests to examine RBC profile and any laboratory tests and procedures need due to infection. The yearly cost of drug therapies are outlined in Table 4. This clearly highlights that these drugs are a significant expense that do not cure the disease. Additional clinical trials in the area of haploidentical transplant with matched unrelated donors and the use of engineered grafts will continue to decrease the risks associated with GVHD and graft rejection. More clinical trials are urgently needed to identify ways to reduce the toxicity of conditioning regimens. Currently, the most economically viable strategy for curing SCD is HSCT. In light of the decreased mortality associated from allogeneic transplants, 93% survival in some recent studies, the risk benefit calculation becomes more acceptable. Crispr/Cas9 gene editing of autologous CD34+ stem cells has now been shown to be an attractive alternative, however the associated cost is prohibitive for most individuals within the affected geographic regions. Gene therapy, for older patients with SCD, in whom allogeneic transplantation has a high transplant-related morbidity and mortality now have a low risk alternative to HSCT. The clinical trials supporting gene editing therapy show promising results, however the number of subjects in these trials remains small, long-term follow-up is needed. Both HSCT and gene editing fix the stem cell compartment but do not address the problem associated with genetic transmission. Awareness, screening and education are still the most cost effective tools to reduce the overall burden of SCD.
Table 4.
Cost of current treatment
| Drug/treatment | MOA | USD |
|---|---|---|
| Hydroxyurea | Increase HbF | 16,800 |
| L-glutamine | Decreased oxidation | 36,000 |
| Crizanlizumab | P-seletin binding | 100,000 |
| Voxelotor | Decreased hemolysis | 125,000 |
| RBC exchange | Non-SS RBCs | 3,000* |
| Allo-transplant | Corrected source | 145,000^ |
| Gene therapy | BCL11A disruption | 2,700,000^ |
| Gene therapy | CRISPR correct globin | >2,000,000^ |
This is cost per event not per year, RBC exchange may be monthly.
This is one time only cost.
Disclosure of conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1.Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch Intern Med. 1910;5:517–521. [PMC free article] [PubMed] [Google Scholar]
- 2.Ingram VM. Abnormal human haemoglobins. I. The comparison of normal human and sickle-cell haemoglobins by fingerprinting. Biochim Biophys Acta. 1958;28:539–545. doi: 10.1016/0006-3002(58)90516-x. [DOI] [PubMed] [Google Scholar]
- 3.El-Hazmi MA, Al-Hazmi AM, Warsy AS. Sickle cell disease in Middle East Arab countries. Indian J Med Res. 2011;134:597–610. doi: 10.4103/0971-5916.90984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, Temperley WH, Williams TN, Weatherall DJ, Hay SI. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381:142–151. doi: 10.1016/S0140-6736(12)61229-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood. 1985;65:183–189. [PubMed] [Google Scholar]
- 6.Ogu UO, Billett HH. Comorbidities in sickle cell disease: adult providers needed! Indian J Med Res. 2018;147:527–529. doi: 10.4103/ijmr.IJMR_1019_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballas SK. The sickle cell painful crisis in adults: phases and objective signs. Hemoglobin. 1995;19:323–333. doi: 10.3109/03630269509005824. [DOI] [PubMed] [Google Scholar]
- 8.Jang T, Poplawska M, Cimpeanu E, Mo G, Dutta D, Lim SH. Vaso-occlusive crisis in sickle cell disease: a vicious cycle of secondary events. J Transl Med. 2021;19:397. doi: 10.1186/s12967-021-03074-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kavanagh PL, Fasipe TA, Wun T. Sickle cell disease: a review. JAMA. 2022;328:57–68. doi: 10.1001/jama.2022.10233. [DOI] [PubMed] [Google Scholar]
- 10.Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, Wethers DL, Pegelow CH, Gill FM. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:288–294. [PubMed] [Google Scholar]
- 11.Khan MI, Patel N, Meda RT, Nuguru SP, Rachakonda S, Sripathi S. Sickle cell disease and its respiratory complications. Cureus. 2022;14:e28528. doi: 10.7759/cureus.28528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 13.Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative study of sickle cell disease. Blood. 1997;89:1787–1792. [PubMed] [Google Scholar]
- 14.Nath KA, Hebbel RP. Sickle cell disease: renal manifestations and mechanisms. Nat Rev Nephrol. 2015;11:161–171. doi: 10.1038/nrneph.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crossley M, Christakopoulos GE, Weiss MJ. Effective therapies for sickle cell disease: are we there yet? Trends Genet. 2022;38:1284–1298. doi: 10.1016/j.tig.2022.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chou ST, Alsawas M, Fasano RM, Field JJ, Hendrickson JE, Howard J, Kameka M, Kwiatkowski JL, Pirenne F, Shi PA, Stowell SR, Thein SL, Westhoff CM, Wong TE, Akl EA. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv. 2020;4:327–355. doi: 10.1182/bloodadvances.2019001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanter J, Liem RI, Bernaudin F, Bolanos-Meade J, Fitzhugh CD, Hankins JS, Murad MH, Panepinto JA, Rondelli D, Shenoy S, Wagner J, Walters MC, Woolford T, Meerpohl JJ, Tisdale J. American Society of Hematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood Adv. 2021;5:3668–3689. doi: 10.1182/bloodadvances.2021004394C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy PS, Cai SW, Barrera L, King K, Badawy SM. Higher hydroxyurea adherence among young adults with sickle cell disease compared to children and adolescents. Ann Med. 2022;54:683–693. doi: 10.1080/07853890.2022.2044509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu HW, Gannon M, Hsu LL. Evaluation of glutamine utilization in patients with sickle cell disease. J Pediatr Hematol Oncol. 2023;45:e52–e55. doi: 10.1097/MPH.0000000000002519. [DOI] [PubMed] [Google Scholar]
- 20.Migotsky M, Beestrum M, Badawy SM. Recent advances in sickle-cell disease therapies: a review of voxelotor, crizanlizumab, and L-glutamine. Pharmacy (Basel) 2022;10:123. doi: 10.3390/pharmacy10050123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rai P, Ataga KI. Drug therapies for the management of sickle cell disease. F1000Res. 2020;9 doi: 10.12688/f1000research.22433.1. F1000 Faculty Rev-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziemba Y, Xu C, Fomani KM, Nandi V, Yuan T, Rehmani S, Sachais BS, Appiah-Kubi AO, Aygun B, Louie JE, Shi PA. Safety and benefits of automated red cell depletion-exchange compared to standard exchange in patients with sickle cell disease undergoing chronic transfusion. Transfusion. 2021;61:526–536. doi: 10.1111/trf.16225. [DOI] [PubMed] [Google Scholar]
- 23.Aggarwal G, Tiwari AK, Dhiman P, Arora D, Pabbi SM, Setya D. Red blood cell exchange in sickle cell disease patient with multiple alloantibodies. Asian J Transfus Sci. 2020;14:70–73. doi: 10.4103/ajts.AJTS_36_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucarelli G, Galimberti M, Polchi P, Angelucci E, Baronciani D, Durazzi SM, Giardini C, Nicolini G, Politi P, Albertini F. Bone marrow transplantation in thalassemia. Hematol Oncol Clin North Am. 1991;5:549–556. [PubMed] [Google Scholar]
- 25.Galal A, Asslan M. Allogenic hematopoietic stem cell transplantation in sickle cell disease. Curr Opin Hematol. 2019;26:399–405. doi: 10.1097/MOH.0000000000000545. [DOI] [PubMed] [Google Scholar]
- 26.Walters MC, Patience M, Leisenring W, Eckman JR, Buchanan GR, Rogers ZR, Olivieri NE, Vichinsky E, Davies SC, Mentzer WC, Powars D, Scott JP, Bernaudin F, Ohene-Frempong K, Darbyshire PJ, Wayne A, Roberts IA, Dinndorf P, Brandalise S, Sanders JE, Matthews DC, Appelbaum FR, Storb R, Sullivan KM. Barriers to bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 1996;2:100–104. [PubMed] [Google Scholar]
- 27.Luftinger R, Zubarovskaya N, Galimard JE, Cseh A, Salzer E, Locatelli F, Algeri M, Yesilipek A, de la Fuente J, Isgro A, Alseraihy A, Angelucci E, Smiers FJ, La La Nasa G, Zecca M, Fisgin T, Unal E, Kleinschmidt K, Peters C, Lankester A, Corbacioglu S EBMT Pediatric Diseases, Inborn Errors Working Parties. Busulfan-fludarabine- or treosulfan-fludarabine-based myeloablative conditioning for children with thalassemia major. Ann Hematol. 2022;101:655–665. doi: 10.1007/s00277-021-04732-4. [DOI] [PubMed] [Google Scholar]
- 28.Eapen M, Brazauskas R, Walters MC, Bernaudin F, Bo-Subait K, Fitzhugh CD, Hankins JS, Kanter J, Meerpohl JJ, Bolanos-Meade J, Panepinto JA, Rondelli D, Shenoy S, Williamson J, Woolford TL, Gluckman E, Wagner JE, Tisdale JF. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet Haematol. 2019;6:e585–e596. doi: 10.1016/S2352-3026(19)30154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Germino-Watnick P, Hinds M, Le A, Chu R, Liu X, Uchida N. Hematopoietic stem cell gene-addition/editing therapy in sickle cell disease. Cells. 2022;11:1843. doi: 10.3390/cells11111843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esrick EB, Lehmann LE, Biffi A, Achebe M, Brendel C, Ciuculescu MF, Daley H, MacKinnon B, Morris E, Federico A, Abriss D, Boardman K, Khelladi R, Shaw K, Negre H, Negre O, Nikiforow S, Ritz J, Pai SY, London WB, Dansereau C, Heeney MM, Armant M, Manis JP, Williams DA. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med. 2021;384:205–215. doi: 10.1056/NEJMoa2029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, Foell J, de la Fuente J, Grupp S, Handgretinger R, Ho TW, Kattamis A, Kernytsky A, Lekstrom-Himes J, Li AM, Locatelli F, Mapara MY, de Montalembert M, Rondelli D, Sharma A, Sheth S, Soni S, Steinberg MH, Wall D, Yen A, Corbacioglu S. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N Engl J Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]