

Abstract
The analysis of extracellular vesicles (EV) in blood samples is under intense investigation and holds the potential to deliver clinically meaningful biomarkers for health and disease. Technical variation must be minimized to confidently assess EV‐associated biomarkers, but the impact of pre‐analytics on EV characteristics in blood samples remains minimally explored. We present the results from the first large‐scale EV Blood Benchmarking (EVBB) study in which we systematically compared 11 blood collection tubes (BCT; six preservation and five non‐preservation) and three blood processing intervals (BPI; 1, 8 and 72 h) on defined performance metrics (n = 9). The EVBB study identifies a significant impact of multiple BCT and BPI on a diverse set of metrics reflecting blood sample quality, ex‐vivo generation of blood‐cell derived EV, EV recovery and EV‐associated molecular signatures. The results assist the informed selection of the optimal BCT and BPI for EV analysis. The proposed metrics serve as a framework to guide future research on pre‐analytics and further support methodological standardization of EV studies.
Keywords: anticoagulants, biomarkers, cancer, exosomes, extracellular vesicles, plasma, preservatives, proteomics, RNA sequencing, serum
1. INTRODUCTION
Approximately one third of publications submitted to the EV‐TRACK knowledgebase during the past five years analysed blood samples for extracellular vesicles (EV) (Van Deun et al., 2017), a heterogeneous group of lipid bi‐layer enclosed structures, released by multiple cell types into their surrounding (Hendrix, 2021). The increasing interest in EV analysis from blood samples originates from the observation that the molecular cargo of EV is a spatiotemporal fingerprint of the pathophysiological processes occurring in the cell of origin and includes proteins, nucleic acids, and lipids, enabling multi‐component biomarker detection (Kalluri & LeBleu, 2020). Considering the complexity of blood composition (blood cells, platelets, EV, non‐EV particles, and high abundant soluble proteins), a variety of analytical and pre‐analytical factors hinder accurate EV measurements and thus slow down their maturation into clinical applications (de Wever & Hendrix, 2019; Hendrix, 2021).
At the analytical level, multiple research groups have demonstrated that different methods separate EV with variable efficiency (also referred to as yield or recovery) and specificity (also referred to as purity), influencing EV‐based biomarker discovery in blood samples (Dong et al., 2020; Veerman et al., 2021). The sequential implementation of orthogonal methods including size‐based methods and density‐based methods has been shown to enable EV‐associated biomarker discovery (Dhondt et al., 2020; Karimi et al., 2018; Onódi et al., 2018; Tulkens et al., 2020; Vergauwen et al., 2021; Zhang et al., 2020). The development and wide‐spread implementation of minimal information guidelines (MISEV2014, MISEV2018), reference materials as well as reporting platforms further steered reproducibility (Geeurickx et al., 2021, 2019; Lötvall et al., 2014; Théry et al., 2018; Van Deun et al., 2017; Witwer et al., 2013).
Pre‐analytical factors including blood sample collection, handling, and processing pose an additional source of variability in EV analysis (Coumans et al., 2017; Lacroix et al., 2012), hindering accurate measurement, discovery or validation of EV‐associated biomarkers (de Wever & Hendrix, 2019). The type of blood collection tube (BCT), as well as the blood processing interval (BPI) (i.e. the time between blood collection and processing) potentially impact EV analysis (Witwer et al., 2013). BCT may differentially activate platelets (Yuana et al., 2011), lyse erythrocytes (Ayers et al., 2019) and affect blood cell viability, and thereby influence EV‐based quantification and biomarker discovery. In addition, variable BPI may differentially impact continued release but also EV uptake and processing by blood cells (Matsumoto et al., 2020; Mulcahy et al., 2014), and overall EV stability (Lai et al., 2014). BPI is an increasingly important pre‐analytical factor due to the growing share of clinical trials with biomarker‐based endpoints (Crippa et al., 2020; Mateo et al., 2020), which pose a challenge of complex sample logistics. Biological samples, collected at multiple clinical sites, require transportation to a central laboratory for further processing and biomarker analysis. In addition, samples may be transported in batches instead of individual runs to optimize efficiency, causing further delays in sample processing (Antoniou et al., 2019).
Various BCT are used to collect blood samples for EV analysis. Serum (239 out of 564 experiments; 42%), EDTA (114 out of 564 experiments; 20%) or citrate (68 out of 564 experiments; 12%) BCT are the most frequently reported in the EV‐TRACK knowledgebase and are implemented for a variety of study aims including technological development as well as analysis of EV‐associated molecular signatures and functions (Figure S1). Current guidelines recommend considering both the extent of ex vivo EV release, as well as the envisaged downstream assays when choosing a BCT for EV analysis in blood samples (Coumans et al., 2017; Lacroix et al., 2013; Théry et al., 2018; Witwer et al., 2013).
Only few studies evaluated a limited number of BCT and/or performance metrics for the analysis of blood‐derived EV (Bæk et al., 2016; Buntsma et al., 2021; Connor et al., 2009; Fendl et al., 2016; György et al., 2014; Jayachandran et al., 2012; Kim et al., 2002; Lacroix et al., 2012; Montoro‐García et al., 2012; Wisgrill et al., 2016; Figure S2). In addition, the ability of specialized preservation additives, designed to prevent lysis and apoptosis of blood cells, and thus to stabilize the concentration and population of EV after blood collection, has not been thoroughly evaluated. Finally, no appropriate and quantifiable performance metrics are available to aid the informed selection of the most suitable BCT and BPI for study objectives. There remains an urgent unmet need to set‐up and validate standard operating procedures (SOP) for BCT and BPI, a prerequisite to establish reliable and clinically relevant biorepositories, and thus for EV‐associated biomarker discovery and validation (Coumans et al., 2017).
In the EV Blood Benchmarking (EVBB) study, we evaluated the impact of 11 BCT, including serum, four routinely used anticoagulants in clinical settings (non‐preservation BCT) and six specialized preservation additives (preservation BCT), on nine performance metrics including blood sample quality, EV stability, EV blood cell origins and EV‐associated proteome and small RNA transcriptome signatures (Figure 1). An overview of the selected BCT and their intended use, composition, mode of action and predicted effect on blood cells is provided in Table 1 (Banfi et al., 2007; Das et al., 2017; Fernando, 2017; Fernando & Chao‐Wei, 2016; Horlitz et al., 2020; Mannuß, 2020; Muller et al., 2018). Since sample processing within a short timeframe may be difficult to achieve in a clinical setting, five BCT with favourable performance metrics for EV analysis were additionally evaluated for three clinically relevant sample BPI (Figure 1).
FIGURE 1.

Overview of the study design. The impact of preservation (n = 6) and non‐preservation (n = 5) BCT and BPI during blood storage at room temperature on EV stability (rEV recovery and EV morphology), sample quality (haemolysis, platelet activation, residual platelets and lipoprotein particles (presumably chylomicrons)), ex‐vivo release of blood‐cell derived EV subtypes (activated platelet EV, non‐platelet EV and erythrocyte EV) and EV omics profile (LC–MS/MS proteomics and small RNA sequencing) was evaluated. In the BCT evaluation phase of the study, the impact of 10 BCT on short‐term blood storage (T1) was assessed. Based on these experiments, performance metrics were developed to enable robust and objective comparisons between BCT. In the BPI evaluation phase of the study, a selection of 4 BCT with favourable performance characteristics, and a BCT marketed for EV preservation (*), was additionally evaluated against three clinically relevant sample processing time intervals (T1–T3) to determine the ability of BCT to stabilize EV during blood sample storage over time. The selected time points between blood draw and processing were 1 h (T1) and 8 h (T2) to mimic immediate and same day processing (short‐term stabilizing efficiency), and 72 h (T3) to assess long‐term stabilizing efficiency.
TABLE 1.
Overview of BCT and their intended use, composition, mode of action and predicted effect on blood cells
| Serum | Citrate | EDTA (spray and gel) α | ACD‐A | Paxgene ccfDNA | Streck BCT DNA | Streck BCT RNA | Streck RNA complete BCT | Biomatrica LBgard | Roche cfDNA | |
|---|---|---|---|---|---|---|---|---|---|---|
| Blood sample volume (mL) | 9 | 9 |
Spray: 10 Gel: 8 |
8.5 | 10 | 10 | 10 | 10 | 8.5 | 8.5 |
| Additive volume (mL) | Not applicable | 0.9 | Not applicable | 1.5 | 1.5 | NA | NA | NA | NA | NA |
| Intended use | Biochemistry tests |
1. Coagulation tests 2. Platelet function tests 3. ESR estimation |
1. CBC 2. Hb/Hct 3. Cell morphology |
1. Blood group typing 2. HLA typing 3. Cell conservation |
1. Stabilization of plasma cfDNA 2. Stabilization of nucleated blood cells to prevent gDNA release 3. Stabilization of gDNA in the nuclear fraction |
1. Stabilization of plasma cfDNA 2. Stabilization of CTC 3. Stabilization of nucleated blood cells to prevent gDNA release |
1. Stabilization of plasma cfRNA 2. Stabilization of nucleated blood cells to prevent cellular RNA release |
1. Stabilization of cfRNA and EV in plasma 2. Stabilization of nucleated blood cells and erythrocytes to prevent cellular RNA release and hemolysis |
1. Stabilization of plasma cfDNA, cfRNA and proteins 2. Stabilization of CTC 3. Stabilization of nucleated blood cells to prevent gDNA release |
1. Stabilization of plasma cfDNA 2. Stabilization of nucleated blood cells to prevent gDNA release |
| Anticoagulant | None | Sodium citrate | K2EDTA |
1. Sodium citrate 2. Citric acid 3. (Dextrose) |
K2EDTA | K3EDTA | K3EDTA | Patent pending | Na3DTPA | K3EDTA |
| Effect on hemostasis (mode of action) | Immediate activation of blood coagulation cascade | Weak Ca2+ chelator* |
1. Strong Ca2+ chelator* 2. Dissociates GPIIb‐IIIa complex** |
1. Weak Ca2+ chelator* 2. Extracellular acidosis → Inhibition of Ca2+ influx into platelets*** |
See EDTA | See EDTA | See EDTA | NA | See EDTA | See EDTA |
| Effect on platelets |
Immediate in vitro platelet aggregation, activation (+++) and release of PEV |
1. Time‐dependent change from discoidal to spherical shape (+) 2. Immediate in vitro platelet activation (+) and release of PEV 3. Time‐dependent formation of platelet micro‐aggregates and ↓ platelet count |
1.Time‐dependent hange from discoidal to spherical shape (+++) 2. Immediate in vitro platelet activation (+++) and release of α granule content 3. Potential development of pseudo‐thrombocytopenia through in vitro platelet clumping (0.1%) |
1. Discoidal morphology preserved 2. Inhibition of platelet function |
See EDTA | See EDTA | See EDTA | NA | See EDTA | See EDTA |
| Effect on erythrocytes/ leucocytes |
NA |
Time‐dependent ↑ MCV and osmotic fragility (hemolysis ↑) |
1. Time‐dependent change in erythrocyte morphology and ↑ MCV and osmotic fragility (hemolysis ↑) 2. Time‐dependent change in leukocyte morphology |
Cell viability is extended by dextrose (hemolysis ↓) → Delayed time‐dependent ↑ MCV and osmotic fragility | See EDTA |
1. See EDTA 2. Osmotic erythrocyte shrinking (MCV ↓) β |
1. See EDTA 2. Osmotic erythrocyte shrinking (MCV ↓) β |
NA | See EDTA |
1. See EDTA 2. Osmotic erythrocyte shrinking (MCV ↓) β |
| Preservation solution | Not applicable | Not applicable | Not applicable | Not applicable |
1. Caspase Inhibitor (Q‐VD‐OPh) 2. HMW/LMW Polyethylene glycol (PEG) 3. Dimethyl propionamide (DMPA) |
1. Imidazolidinyl urea (IDU) 2. Glycine |
1. Diazolidinyl urea (DU) 2. Aurintricarboxylic acid (ATA) 3. Glyceraldehyde 4. NaF |
Patent pending |
1. 1‐Hexyl‐3‐methylimidazolium bromide 2. Guanidine hydrochloride 3. Oxalic and tartaric acid 4. LiOH |
No patents |
| Mode of action | Not applicable | Not applicable | Not applicable | Not applicable |
1. Fixation of blood cells and stabilization of extracellular nucleic acids (formaldehyde releaser) 2. Formaldehyde quenching (prevention of DNA crosslinking) |
1. Fixation of blood cells and stabilization of extracellular nucleic acids (formaldehyde releaser) 2. Stabilization of cfRNA (nuclease inhibitor) 3‐4. Metabolic inhibitors (prevention of cfRNA degradation and cell lysis) |
NA |
1. Stabilization of blood cells and extracellular nucleic acids (ionic liquid) 2. Chaotropic agent (protein denaturation) 3. pH buffers 4. Promotion of nucleic acid precipitation |
NA |
Abbreviations: CBC: complete blood count; CTC: circulating tumor cell; ctDNA: circulating tumor DNA; gDNA: genomic DNA; ESR: erythrocyte sedimentation rate; Hb: hemoglobin; Hct: hematocrit; HLA: human leukocyte antigen; HMW: high molecular weight; LMW: low molecular weight; MCV: mean cellular volume; NA: not available; PEV: platelet EV.
α K2EDTA spray coated onto the interior surface of the BCT (Spray: no gel separator / Gel: with gel separator)
β Compared to K2EDTA
Free Ca2+ is essential for platelet activation and aggregation, and is a cofactor in various steps of the coagulation cascade.
Glycoprotein IIb/IIIa (GPIIb‐IIIa) is an integrin complex on platelet membranes, formed through Ca2+‐dependent association of GPIIb and GPIIIa following platelet activation. It plays a role in further platelet activation and in the final stages of platelet aggregation through binding with fibrinogen.
An increased Ca2+ concentration in the platelet cytosol is essential for platelet activation and aggregation.
2. MATERIALS AND METHODS
2.1. Biological sample donors
Venous blood samples from healthy volunteers or cancer patients were collected into blood collection tubes containing different anticoagulants and/or preservatives. For proteomics and RNA sequencing experiments, samples were collected from prostate cancer patients prior to local treatment. All other experiments were performed on healthy donor samples. All donors were non‐smokers. Healthy donors did not take any medication, nor did they suffer from any chronic or acute disease at the time of venipuncture. Cancer patients were not on anticoagulant therapy at the time of sample collection. For all donors, erythrocyte count, haemoglobin, mean cellular volume (MCV), mean cellular haemoglobin (MCH), mean cellular haemoglobin concentration (MCHC), platelet count and leukocyte count were documented by a haematology analyser (XP‐300, Sysmex, Kobe, Japan). Parameters were within the normal range for all included donors. Collection of biological samples was according to the Ethical Committee of Ghent University Hospital approval EC/2015/0260 and in accordance with the guidelines and regulations of the Helsinki Declaration. Participants had given written informed consent. Donor characteristics and clinical chemistry data are summarized in Table S1. Clinical data from cancer patients are summarized in Table S2.
2.2. Biological sample collection and preparation of platelet‐depleted blood plasma
Venipuncture was performed while the donors were fasting. A 21 Gauge straight needle was used for venipuncture of an antecubital vein after applying a light tourniquet. The first few millilitres of blood were collected in a serum tube and discarded. Subsequently, blood was collected in a total of 11 different BCT (Figure 1) each containing specific anticoagulants and/or preservatives: Serum (Vacuette Z serum with separator and clot activator, volume 9 mL, Greiner Bio‐One, Frickenhausen, Germany); Citrate (Vacuette sodium citrate 3.2%, volume 9 mL, Greiner Bio‐One); EDTA spray (Vacutainer K2EDTA, volume 10 mL, BD Biosciences); EDTA gel (Vacuette K2EDTA with separator, volume 8 mL, Greiner Bio‐One); ACD‐A (Vacutainer ACD Solution A, volume 8.5 mL, BD Biosciences), PAXgene ccfDNA (volume 10 mL, PreAnalytiX GmbH, Hombrechtikon, Switzerland); Cell‐Free DNA BCT (volume 10 mL, Streck, La Vista, NE, USA); Cell‐Free RNA BCT (volume 10 mL, Streck); LBgard (volume 8.5 mL, Biomatrica, San Diego, CA, USA); Cell‐Free DNA (volume 8.5 mL, Roche Sequencing Solutions, Pleasanton, CA, USA); RNA complete BCT (volume 10 mL, Streck). The drawing sequence of collection tubes was randomized. Time between start and finish of blood collection was not more than 5 min. To mix anticoagulants with blood, each blood collection tube was inverted by turning BCT vertically for 180° and back to the starting position ten times immediately after collection, as per manufacturer's instructions and European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) guidelines (Simundic et al., 2018). The blood collection tubes were held in a rack in an upright position at room temperature (20°C). For healthy volunteers, venipuncture was performed within the same laboratory as sample preparation and samples were not transported. For cancer patients, venipuncture was performed in the operating theatre and samples were transported on foot to the laboratory within 10 min. Centrifugation was performed exactly 60 min, 8 h or 72 h after collection of the last BCT, as indicated in the results section.
Serum was prepared by centrifugation at 1200 × g for 15 min at 20°C. Platelet depleted plasma (PDP) was prepared by two serial centrifugations at 2500 × g for 15 min at 20°C. An Eppendorf 5810 R (Eppendorf, Hamburg, Germany) benchtop centrifuge was used. No brake was applied. After each centrifugation step, serum or blood plasma was transferred to a clean 5 mL polypropylene centrifuge tube (Eppendorf). At least 0.5 cm of serum or blood plasma was left in the tube to avoid contamination with cells. Depletion of platelets in serum or PDP was measured by a haematology analyser (XP‐300, Sysmex) with a detection limit of 1E+7/mL. If residual platelets (>1E+7/mL) were present, one additional centrifugation step was performed. Serum and PDP samples were aliquoted, snap frozen in liquid nitrogen, and stored at −80°C until analysis.
Recombinant EV (rEV) were used as spike‐in controls for rEV recovery experiments (Figure S3); their characteristics and manufacturing were described in detail previously (Geeurickx et al., 2021, 2019). To this end, BCT were opened following sample collection. A fixed volume of whole blood was aliquoted to 4 mL, spiked with 2E+10 rEV, and incubated at room temperature for 60 min, 8 h or 72 h (as indicated in the results section), prior to serum or PDP preparation.
2.3. Haemolysis
Haemolysis was quantified using a haemoglobin colorimetric assay (Haemoglobin Colorimetric Detection Kit, EIAHGBC, Thermo Fisher Scientific, Waltham, MA, USA). The assay was performed according to the manufacturer's instructions.
2.4. Platelet activation
Platelet activation was assessed by measuring plasma concentrations of platelet factor 4 (PF4) and beta thromboglobulin (BTG). These were analysed using enzyme‐linked immunosorbent assay (ELISA) using commercially available kits (PF4 Human ELISA Kit, EHPF4 and NAP‐2/PPBP Human ELISA Kit, EHPPBP, Thermo Fisher Scientific). The assays were performed according to the manufacturer's instructions.
2.5. EV separation
The EV separation workflow is summarized in Figure S3.
For rEV recovery experiments, (r)EV were separated from serum and PDP samples using size‐exclusion chromatography (SEC). To prepare the SEC column, Sepharose CL‐2B (GE Healthcare) was washed three times with PBS buffer. A nylon net with 20 μm pore size (NY2002500, Merck Millipore) was placed on the bottom of a 10 mL syringe (BD Biosciences), followed by stacking of 10 mL washed Sepharose. After loading 2 mL blood plasma on top of the SEC column, individual fractions of 1 mL eluate were collected. EV and rEV containing fractions (4–6) were pooled and used for rEV calculation by fluorescent nanoparticle tracking analysis (fNTA). Of note, as previously reported for blood plasma, SEC separates EV with lower specificity from non‐EV particles (e.g. low density lipoprotein particles and chylomicrons that overlap in size with EV) (Cocozza et al., 2020; Simonsen, 2017; Vergauwen et al., 2021). However, considering that spiked rEV are quantified in fluorescent mode this does not impact the NTA measurements.
For LC–MS/MS and small RNA‐seq, EV were separated from serum and PDP samples using a previously validated sequential biophysical separation protocol, combining SEC and Optiprep density gradient ultracentrifugation (ODG) (Tulkens et al., 2020; Vergauwen et al., 2021). One SEC column processed 2 mL sample and SEC fractions 4–6 were pooled and concentrated to 1 mL using a 10 kDa centrifugal filter device (Amicon Ultra‐2 mL, Merck Millipore). The resulting 1 mL sample was overlaid on top of a discontinuous ODG, prepared by layering 4 mL of 40%, 4 mL of 20%, 4 mL of 10% and 3.5 mL of 5% iodixanol in a 17 mL Thinwall Polypropylene Tube (Beckman Coulter, Fullerton, CA, USA). The ODG was centrifuged 18 h at 100,000 × g and 4°C using a SW 32.1 Ti rotor (Beckman Coulter). Afterwards, gradient fractions of 1 mL were collected from the top of the gradient, with fractions 9 and 10, corresponding to a density of 1.09–1.10 g/mL, being pooled and used for subsequent SEC‐based separation of EV from the iodixanol polymer (Vergauwen et al., 2017). EV‐containing fractions 4–7 were pooled and concentrated to 100 μL using Amicon Ultra‐2 10K filters (Merck Millipore) and stored at −80°C until further downstream analysis. Of note, as previously reported for blood plasma, SEC separates EV with lower specificity from non‐EV particles (e.g. low density lipoprotein particles and chylomicrons that overlap in size with EVs) (Cocozza et al., 2020; Simonsen, 2017; Vergauwen et al., 2021). Therefore, SEC fractions 4–6 are additionally processed by ODG ensuring the preparation of EV with higher specificity, which is fundamental to downstream omics analysis (Vergauwen et al., 2021). Although increased column length or reduced sample volume may increase the specificity of SEC, a combination of two biophysical characteristics (i.e. size and density) results in higher specificity since size only is not sufficient to separate EV from different types of lipoprotein particles (Cocozza et al., 2020; Simonsen, 2017; Vergauwen et al., 2021).
Density of the ODG fractions was assessed using a standard curve of the absorbance values at 340 nm (SpectraMax Paradigm, Molecular Devices, San Jose, CA, USA) of aqueous dilutions of 5, 10, 20 and 40% iodixanol solutions (solutions were diluted 1:1 twice). This standard curve was used to determine the density of fractions collected from a control gradient.
2.6. Nanoparticle tracking analysis (NTA)
Nanoparticle tracking analysis (NTA) was performed using a NanoSight LM10‐HS microscope (NanoSight, Amesbury, UK) equipped with a 45 mW 488 nm laser and an automatic syringe pump system. For conventional NTA, three 30 s videos were recorded of each sample with a camera level of 13, a detection threshold of three and a syringe pump infusion speed of 20. For fluorescent NTA measurements (fNTA), an additional 500 nm longpass filter was used, and the camera level was increased to 16. Temperatures were monitored throughout the measurements. A medium viscosity of 0.929 cP was assumed. Videos were analysed with NTA software v3.3. For optimal measurements, samples were diluted with PBS until particle concentration was within optimal concentration range of the NTA software (3E+8−1E+9). For recovery calculations, the number of fluorescent particles was measured from an rEV stock solution before spiking. All reported size distributions determined with NTA represent the hydrodynamic diameters of the particles in suspension.
2.7. Transmission electron microscopy
ODG fractions were analysed with transmission electron microscopy (TEM). Samples were deposited on Formvar carbon‐coated, glow discharged grids, stained with uranylacetate and embedded in methylcellulose/uranylacetate. These grids were examined using a Tecnai Spirit transmission electron microscope (FEI, Eindhoven, The Netherlands) and images were captured with a Quemasa charge‐coupled device camera (Olympus Soft Imaging Solutions Gmbh, Munster, Germany).
2.8. Protein analysis
Protein concentrations were measured using the fluorometric Qubit Protein Assay (Thermo Fisher Scientific). Sample preparation was done by 1:1 dilution with SDS 0.4%. Protein measurements were performed using the Qubit Fluorometer 3.0 (Thermo Fisher Scientific) according to the manufacturer's instructions.
2.9. Western blotting
Samples were dissolved in reducing sample buffer (0.5 M Tris‐HCl (pH 6.8), 40% glycerol, 9.2% SDS, 3% 2‐mercaptoethanol, 0.005% bromophenol blue) and boiled at 95°C for 5 min. Proteins were separated by SDS‐PAGE (SDS‐polyacrylamide gel electrophoresis), transferred to nitrocellulose membranes (Bio‐Rad, Hercules, CA, USA), blocked in 5% non‐fat milk in PBS with 0.5% Tween‐20 and immunostained. Chemiluminescence substrate (WesternBright Sirius, Advansta, Menlo Park, CA, USA) was added and imaging was performed using the Proxima 2850 Imager (IsoGen Life Sciences, De Meern, The Netherlands).
2.10. Flow cytometry
Flow cytometry (A60‐Micro, Apogee Flow Systems, UK) was used to determine the concentration of EV, lipoprotein particles (presumably chylomicrons) and platelets in serum and PDP. All details about assay controls, calibration, data acquisition, particle characterization, and sample preparation can be found in the completed MIFlowCyt‐EV template (Suppl. Materials 1). In short, we triggered at the side scattering (SSC) detector and measured for 120 s with a flow rate of 3.0 μL/min. The reported concentrations describe the number of particles (1) that exceeded the SSC threshold (de Rond et al., 2018), corresponding to a side scattering cross section of 10 nm3, (2) that were collected during time intervals, for which the count rate was within 25% from the median count rate, (3) with a diameter >200 nm as determined by Flow‐SR (van der Pol et al., 2018), (4) having a refractive index (RI) < 1.42 to omit false positively labelled lipoprotein particles, and (6) that are positive at the corresponding fluorescence detector, per mL of PDP. EV were labelled with CD61‐APC (>150 molecules of equivalent fluorophore [MESF]), lactadherin (Lac)‐FITC (>1000 MESF), and CD235a‐FITC (>825 MESF). In addition to the concentration of labelled EV, the total EV concentration, lipoprotein particle (presumably chylomicron) concentration, and concentration of residual platelets was determined within the detection range of the flow cytometer.
2.11. Antibodies
The following antibodies were used for immunostaining: anti‐CD9 (1:1000, D3H4P, Cell Signalling Technology, Beverly, MA, USA), anti‐Flotillin‐1 (1:1000, 610820, BD Biosciences, Franklin Lakes, NJ, USA), anti‐Syntenin‐1 (1:1000, ab133267, Abcam, Cambridge, UK), anti‐CD81 (1:1000, SC‐166029, Santa Cruz Biotechnology, Dallas, TX, USA), anti‐green fluorescent protein (GFP) (1:1000, MAB3580, Merck Millipore, Billerica, MA, USA), sheep anti‐mouse horseradish peroxidase‐linked antibody (1:3000, NA931V, GE Healthcare Life Sciences, Uppsala, Sweden), donkey anti‐rabbit horseradish peroxidase‐linked antibody (1:4000, NA934V, GE Healthcare Life Sciences).
2.12. Liquid chromatography with tandem mass spectrometry (LC–MS/MS)
Samples were processed for LC‐MS/MS by filter‐aided sample preparation (FASP) (Wiśniewski et al., 2009). Lysates were prepared by mixing samples with SDT‐lysis buffer (2% SDS, 500 mM Tris‐HCL (pH 7.6), 0.5 M DTT) at a 4:1 sample to buffer ratio and incubated at 95°C for 5 min. After clarification of lysates by centrifugation (16,000 × g for 5 min), samples were mixed with 300 μL UA (8 M urea, 0.1 M Tris‐HCl (pH 8.5)) in a Microcon YM‐10 centrifugal filter device (Merck Millipore). Filters were centrifuged twice (14,000 × g for 40 min at 20°C) with the addition of 200 μL UA in between. Proteins were alkylated by addition of 100 μL IAA solution (0.05 M iodoacetamide in UA buffer) and incubated for 30 min at room temperature, followed by centrifugation. This was followed twice by addition of 100 μL UA and twice by addition of 100 μL DB buffer (1 M urea, 0.1 M Tris‐HCl (pH 8.5), with centrifugation in between. Filter units were transferred to new collection tubes and proteins were resuspended in 40 μL DB with Trypsin/Lys‐C mix (Promega, Madison, WI, USA) for overnight proteolytic digestion at 37°C. Digests were collected by addition of 100 μL DB and centrifugation for 15 min at 14,000 × g. This step was repeated once. Collected peptides were acidified with 1% trifluoroacetic acid to a pH of 2–3, followed by desalting with C18‐StageTips (C18 Empore Disks, 3 M, St. Paul, MN, USA). Desalted peptides were vacuum dried, dissolved in 0.1% formic acid and analysed by LC–MS/MS. Peptides were loaded on a nanoflow HPLC system (Easy‐ nLC1000, Thermo Fisher Scientific) coupled to a Q Exactive HF Hybrid Quadrupole‐Orbitrap Mass Spectrometer (Thermo Fisher Scientific) equipped with a nano‐electrospray ionization source. The mobile phase consisted of 0.1% formic acid (solvent A) and acetonitrile/water (95:5 (v/v)) with 0.1% formic acid (solvent B).
The peptides were separated with a 40 min gradient from 8 to 35% of solvent B. Before the end of the run, the percentage of solvent B was raised to 100% in 2 min and kept there for 8 min. Data dependent acquisition was enabled, and higher energy collisional dissociation of top 10 most intense ions (isolation width of 2.0 m/z, dynamic exclusion time of 20 s enabled) from the survey scan over the mass‐to‐charge (m/z) range 300–2000 (140,000 resolution, automatic gain control of 3E6, maximum injection time of 100 ms) was performed with a normalized collision energy of 27%. MS/MS spectra over m/z range 200–2000 were collected (17,500 resolution, automatic gain control of 5E4, maximum injection time of 250 ms). Two repeated runs per sample were performed.
Tandem mass spectra were searched using the MaxQuant software (version v1.5.2.8) against a database containing both reviewed (SwissProt) and unreviewed (TrEMBL) sequences of homo sapiens, including different isoforms, of UniProtKB release 2018_07. Peptide‐spectrum‐match‐ and protein‐level false discovery rates were set at 0.01. Carbamidomethyl (C), as a fixed modification, and oxidation (M) and acetylation of the protein N‐terminus as dynamic modifications were included. A maximum of two missed cleavages was allowed. The LC–MS profiles were aligned, and the identifications were transferred to non‐sequenced or non‐identified MS features in other LC–MS runs (matching between runs). The protein was determined as detected in the sample if its identification had been derived from at least two unique peptide identifications. Filtering for contaminating proteins, reverse identification and identification by site was used. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (Vizcaíno et al., 2014) via the PRIDE (Perez‐Riverol et al., 2019) partner repository with the dataset identifier PXD035199.
2.13. Proteomic data analysis
Samples 2B, 3E and 3I (BCT evaluation phase), as well as 2AT2 and 3BT3 (BPI evaluation phase) were excluded because of inadequate sample quality. Identified proteins were analysed and visualized using Perseus software v1.6.2.2 (Tyanova et al., 2016). Protein intensities were total sum intensity normalized and log2 transformed. Proteins showing valid values in at least 50% of at least one group were selected. Missing values were imputed from the observed normal distribution of intensities. For selected analyses, intensities were transformed to z‐scores. Unsupervised hierarchical clustering heat maps, using 1‐Pearson correlation, were generated using the Morpheus tool. Principle component analysis was performed using Past 4.0 software (Hammer et al., 2001). UpSet plots were generated using the Intervene software package (Khan & Mathelier, 2017; Lex et al., 2014). Pathway enrichment analysis was performed using g:Profiler (Reimand et al., 2007) and data were visualized using Cytoscape and the EnrichmentMap pipeline (Merico et al., 2010), as previously described (Reimand et al., 2019). Gene Set Enrichment Analysis (GSEA) (Java v4.0) (Subramanian et al., 2005) was performed using 1 list representing the human erythrocyte proteome (Kakhniashvili et al., 2004) and 1 list representing genes involved in platelet activation, signalling and aggregation from the Reactome pathway knowledgebase (Fabregat et al., 2018). Differential expression was calculated based on the median expression value for each phenotype. The number of permutations (phenotype) was set to 1000 and FDR q‐values smaller than 0.05 were considered statistically significant.
2.14. RNA isolation and spike‐in RNA addition
RNA was isolated with the miRNeasy Serum/Plasma kit (Qiagen, Hilden, Germany, 217184) according to the manufacturer's instructions. Input volume of 200 μL was used for all samples and total RNA was eluted in 12 μL of RNase‐free water. To allow for a proper BCT comparison, synthetic spike‐in controls were added as previously described (Hulstaert et al., 2021, 2020). In short, per 200 μL biofluid input volume, 2 μL Sequin spike‐in controls (Garvan Institute of Medical Research, Darlinghurst, NSW, Australia) at a dilution of 1/1,300,000 and 2 μL RNA extraction Control (RC) spike‐ins (190 fM) (Integrated DNA Technologies) were added to the lysate. Following RNA isolation, 2 μL of External RNA Control Consortium spike‐in controls (ThermoFisher Scientific, Waltham, MA, USA, 4456740) at a dilution of 1/1,000,000, 2 μL Library Prep Control (LP) spike‐ins (33.8 fM) (Integrated DNA Technologies) (Hafner et al. 2011), 1 μL HL‐dsDNase (ArcticZymes Technologies, Tromsø, Norway, 70800‐202) and 1.6 μL reaction buffer were added.
2.15. Small RNA library preparation and sequencing
Small RNA libraries were prepared starting from 5 μL DNase treated and spike‐in supplemented RNA eluate using a TruSeq Small RNA Library Prep Kit (Illumina, San Diego, CA, USA) according to manufacturer's instructions. After PCR amplification, quality of libraries was assessed using a high sensitivity DNA kit on a Bioanalyser (Agilent, Santa Clara, California, USA) according to manufacturer's instructions. Size selection was performed using 3% agarose dye‐free marker H cassettes on a Pippin Prep (Sage Science, Beverly, Massachusetts, USA) following manufacturer's instructions with a specified collection size range of 125−153 bp. Libraries were further purified and concentrated by ethanol precipitation, resuspended in 10 μL of 10 mM tris‐HCl (pH = 8.5) and quantified using qPCR. Based on KAPA qPCR, equimolar library pools were prepared, quality was assessed (as described above) and the library was further diluted to 4 nM using 10 mM tris‐HCl (pH = 8.5). The pooled library was then sequenced at a final concentration of 1.2 pM on a NextSeq 500 (Illumina) using a high output v2 kit (single‐end, 75 cycles, Illumina). All raw data were submitted to the BioProject database with the dataset identifier PRJNA856490.
2.16. Gene expression analysis
Normalization of RNA sequencing results was performed using DESeq2 (v1.30.1). miRNA showing valid values in at least 50% of at least one group were selected. For selected analyses, intensities were transformed to z‐scores. Unsupervised hierarchical clustering heat maps, using 1‐Pearson correlation, were generated using the Morpheus tool. Principle component analysis was performed using Past 4.0 software (Hammer et al., 2001). UpSet plots were generated using the Intervene software package (v6.0) (Khan & Mathelier, 2017; Lex et al., 2014). For differential expression analysis, FDR q‐values smaller than 0.05 were considered statistically significant.
2.17. Statistical analysis
Data are presented as mean ± SD and were analysed with Graph Pad Prism 6 (GraphPad Software, San Diego, CA, USA) using one‐way RM ANOVA and Dunnet's multiple comparisons test (individual variances computed for each comparison with the golden standard control). Time series analysis was performed using RM two‐way ANOVA and Turkey's multiple comparisons test. P‐values < 0.05 were considered to be statistically significant.
2.18. Performance metrics
To compare BCT and BPI, 9 performance metrics were calculated: rEV recovery (%, measured by fNTA); haemolysis (haemoglobin (Hb) concentration in mg/mL, measured by colorimetric assay); platelet activation (PF4 concentration in ng/mL, measured by ELISA); residual platelets (concentration (mL−1) of CD61+ particles with SSC cross section > 300 nm2, measured by flow cytometry); activated platelet EV (concentration (mL−1) of CD61+Lac+ particles (RI < 1.42), measured by flow‐cytometry); EV released from apoptotic blood cells, defined hereafter as non‐platelet EV (concentration (mL−1) of CD61‐Lac+ particles (RI < 1.42), measured by flow‐cytometry); erythrocyte EV (concentration (mL−1) of CD235+ particles (RI < 1.42); measured by flow‐cytometry) and Pearson similarity of LC‐MS/MS and RNAseq profiles. Individual values for performance metrics were transformed to z‐scores. Unsupervised hierarchical clustering heat maps were generated based on Euclidean distance, as described previously (ExRNAQC Consortium, 2021), using the Morpheus tool.
2.19. EV‐TRACK and MISEV2018
We have submitted all relevant data of our experiments to the EV‐TRACK knowledgebase (EV‐TRACK ID: EV220304) (Van Deun et al. 2017). Adherence to MISEV2018 criteria was summarized in Suppl. Materials 2.
3. RESULTS
3.1. The impact of blood collection tubes on EV performance metrics
3.1.1. Influence of type of BCT on short‐term EV recovery, size, and ultrastructure
To accurately quantify EV recovery, we spiked a known number (2E+10) of fluorescently trackable recombinant EV (rEV) in defined volumes of blood (4 mL) collected in BCT (n = 10) during a single venipuncture from healthy donors (n = 3). Following 60 min incubation at room temperature, serum and platelet‐depleted plasma (PDP) were prepared and EV were separated by SEC. Short‐term (60 min) rEV recovery in EV‐enriched SEC fractions was quantified by fNTA (Geeurickx et al., 2019). The highest rEV recovery was achieved with ACD‐A and citrate, respectively 75% and 72%, while Paxgene ccfDNA showed the lowest rEV recovery (57%) (Figure 2a). rEV size distributions (mode) corresponded to previously described rEV size measurements (Geeurickx et al., 2021, 2019) and did not differ among BCT (Figure 2b).
FIGURE 2.

Impact of BCT type (n = 10) on EV stability a. Recovery rate (%) of recombinant EV (rEV) in EV‐enriched fractions, separated from serum and PDP by SEC, measured by fNTA following 60 min incubation in full blood at room temperature (n = 3). b. Size (nm) of recovered rEV, measured by fNTA (n = 3). Data are depicted as individual values with means. c. Left: NTA of EV‐enriched fractions, separated from serum and PDP by sequential SEC and ODG ultracentrifugation. NTA calculated size distributions are depicted as mean (black line) with standard error (red area) and mean particle size and mode are shown. Right: Transmission electron microscopy of EV‐enriched fractions (scale bar: 100 nm).
To further investigate whether BCT alter EV size and ultrastructure, endogenous EV were separated from unspiked serum and PDP samples (n = 3) through sequential biophysical fractionation (Tulkens et al., 2020; Vergauwen et al., 2021), combining SEC and ODG (Figure S3), and analysed using complementary particle measurements (Figure S4). Size distributions of EV measured by NTA and EV ultrastructure and integrity evaluated by TEM, revealed no BCT induced variations (Figure 2c).
3.1.2. Influence of type of BCT on short‐term sample quality and ex vivo release of blood cell‐derived EV
To evaluate how the type of BCT influences sample quality and ex vivo release of blood cell‐derived EV, blood from healthy donors (n = 10) was collected during a single venipuncture into BCT (n = 10). Donor characteristics are summarized in Table S1. Haematology testing was performed, followed by short‐term preparation of serum and PDP 60 min after the final blood draw (Figure S5 and Table S3). Mean leukocyte, erythrocyte and platelet counts were significantly lower in blood samples collected in citrate (p < 0.001), ACD‐A (p < 0.001) and Paxgene ccfDNA (p < 0.001) compared to EDTA spray. These findings correspond to sample dilution by the additives contained in the respective BCT. Meanwhile, the MCV was significantly higher in Paxgene ccfDNA (p < 0.001), suggesting an osmotic swelling effect by the BCT additive on erythrocytes.
To quantify short‐term (60 min) haemolysis and ex vivo activation of platelets, we calculated and compared fold changes of haemoglobin concentration and soluble platelet activation markers (including PF4 and BTG) for PDP prepared with citrate (3.2% or 0.109 mol/L), a generally recommended BCT for EV analysis (Coumans et al., 2017; Lacroix et al., 2013), versus serum or PDP prepared in other BCT (Figure 3a–c and Table S4). Both haemoglobin concentration (p < 0.001) and soluble platelet activation markers (p < 0.001) were significantly affected by BCT type. PDP obtained in EDTA and Roche cfDNA was significantly more haemolytic, with respectively 1.56‐, 1.47‐ and 2.17‐fold higher mean Hb concentrations in EDTA spray (p = 0.005), EDTA gel (p = 0.005) and Roche cfDNA (p < 0.001) compared to citrate. Levels of PF4 (3352 ± 1222 ng/mL, p < 0.001) and BTG (5151 ± 1683 ng/mL, p < 0.001) were significantly elevated in serum compared to all PDP BCT. In addition, both PF4 and BTG were significantly increased in EDTA spray (resp. 3.24‐ (p = 0.004) and 3.28‐fold (p = 0.002)), EDTA gel (resp. 2.65‐ (p = 0.021) and 2.66‐fold (p = 0.018)), Streck BCT DNA (resp. 15.66‐ (p < 0.001) and 14.55‐fold p < 0.001)), Biomatrica LBgard (resp. 17.03‐ (p < 0.001) and 14.68‐fold (p < 0.001)) and Roche cfDNA (resp. 18.00‐ (p < 0.001) and 14.77‐fold (p < 0.001)). Conversely, PF4 and BTG were significantly lower in ACD‐A (resp. 4.03‐ (p = 0.020) and 3.35‐ (p = 0.007)) and Paxgene ccfDNA (resp. 6.74‐ (p = 0.008) and 3.35‐ fold (p = 0.007)).
FIGURE 3.

Impact of BCT type (n = 10) on sample quality and ex vivo release of blood‐cell derived EV. Analyses were performed on serum and PDP, prepared 60 min after blood draw (n = 10). Data are depicted as individual values with means. The asterisk indicates a statistically significant difference compared to the reference BCT (citrate). a. Haemolysis, quantified as haemoglobin (Hb) concentration (mg/mL), measured by colorimetric assay. b. Platelet activation, quantified as plasma concentration of platelet factor 4 (PF4) (ng/mL), measured by ELISA. c. Platelet activation, quantified as plasma concentration of beta thromboglobulin (BTG) (ng/mL), measured by ELISA. d. Total EV count, quantified as the concentration (mL−1) of particles with a refractive index < 1.42 (RI < 1.42). e. Activated platelet EV count, quantified as the concentration (mL−1) of CD61+Lac+ particles (RI < 1.42). f. Non‐platelet EV count, quantified as the concentration (mL−1) of CD61‐Lac+ particles (RI < 1.42). g. Residual platelet count, quantified as the concentration (mL−1) of CD61+ particles (SSC cross section > 300 nm2). The dotted line indicates the detection limit of a standard haematology analyser. h. Erythrocyte EV count, quantified as the concentration (mL−1) of CD235+ particles (RI < 1.42). i. Lipoprotein particle count, quantified as the concentration (mL−1) of particles with a refractive index > 1.45 (presumably chylomicrons). Particle numbers were measured by flow cytometry.
To quantify short‐term (60 min) release of blood cell‐derived EV subtypes, we measured total EV (RI < 1.42), activated platelet EV (CD61+Lac+; RI < 1.42), non‐platelet EV (CD61‐Lac+; RI < 1.42) and erythrocyte EV (CD235+; RI < 1.42) concentrations by flow cytometry. In addition, residual platelet and lipoprotein particle concentrations (RI > 1.45) were assessed as additional sample quality controls (Figure 3d–i and Table S5). Total EV include endogenous sample EV (activated) platelet EV, erythrocyte EV and EV released from disparate blood cells following sample collection. Phosphatidylserine exposure (Lac+) on cell and EV membranes is a marker of platelet activation (Schoenwaelder et al., 2009) and a hallmark of apoptotic cell death, and immune and endothelial cell activation (Kranich et al., 2020). Non‐platelet EV (CD61‐Lac+; RI < 1.42) therefore represent the EV population that is released ex vivo through blood cell apoptosis, in contrast to EV released from activated blood platelets (CD61+Lac+; RI < 1.42).
Total EV (p < 0.001), activated platelet (p < 0.001) and non‐platelet EV (p < 0.001), erythrocyte EV (p = 0.012) and residual platelet concentrations (p < 0.001) were all significantly affected by BCT type, while lipoprotein particle concentrations were not (p = 0.25). Serum contained the highest number of total EV (5.80E+08 ± 1.60E+08 mL−1), with 3.87‐fold (p < 0.001) higher concentrations compared to citrate PDP (1.50E+08 ± 1.30E+08 mL−1). Similarly, serum contained the highest number of activated platelet EV (3.70E+08 ± 9.60E+07 mL−1), representing 68 ± 9.1% of total EV count in serum (Figure S6). Activated platelet EV concentrations were 77.1‐fold (p < 0.001) higher compared to citrate PDP (4.80E+06 ± 1.50E+06 mL−1), in which 5.2 ± 3.6% of total EV were represented by activated platelet EV. In addition, PDP prepared from Paxgene ccfDNA and Streck BCT RNA contained 1.40‐ (p = 0.004) and 3.13‐fold (p = 0.003) higher activated platelet EV concentrations compared to citrate PDP. EDTA PDP contained significantly lower numbers of activated platelet EV (p = 0.003). The highest numbers of non‐platelet EV were measured in serum and Roche cfDNA, with 1.59‐ (p = 0.043) and 1.79‐fold (p = 0.006) higher concentrations compared to citrate PDP. Residual platelets were still present in serum and PDP prepared from all BCT in concentrations under the detection limit of a standard haematology analyser. Citrate (6.64E+05 ± 5.36E+05 mL−1) and ACD‐A plasma (4.95E+05 ± 3.22E+05 mL−1) contained the lowest numbers of residual platelets. Conversely, PDP prepared from Paxgene ccfDNA and Streck BCT RNA contained 4.07‐ (p < 0.001) and 4.97‐fold (p = 0.001) higher residual platelet concentrations compared to citrate plasma, explaining the release of higher numbers of activated platelet EV in PDP prepared from these BCT. PDP prepared from the remaining BCT, except for EDTA gel, also contained significantly higher numbers of residual platelets compared to citrate plasma, but this did not lead to significantly elevated activated platelet EV levels. Roche cfDNA PDP contained the highest number of erythrocyte EV (3.80E+07 ± 9.40E+06 mL−1), with 1.46‐fold (p = 0.036) higher concentrations compared to citrate plasma (2.60E+07 ± 2.40E+07 mL−1), corresponding to the significant haemolysis observed in these BCT. Conversely, ACD‐A plasma contained 1.86‐fold (p = 0.042) lower numbers of erythrocyte EV (1.40E+07 ± 7.50E+06 mL−1). Finally, flow cytometry data were also used to calculate EV size distributions (RI < 1.42) in serum and PDP across the selected BCT (Figure S7). The decay constant of exponential fits of the size distributions were unaffected by the type of BCT, confirming the observations from the (f)NTA and TEM experiments on spiked rEV and EV separated from plasma (Figure 2).
3.1.3. Influence of type of BCT on EV proteome and transcriptome signatures
To evaluate how the type of BCT influences the EV proteome and transcriptome, blood samples from prostate cancer patients (n = 6) were collected during a single venipuncture into BCT (n = 10). Donor characteristics are summarized in Table S2. EV were separated from serum and PDP through sequential biophysical fractionation, combining SEC and ODG (Figure S3), and analysed by mass spectrometry‐based proteomics (LC–MS/MS) (n = 3) and small RNA sequencing (n = 3).
Short‐term (60 min) BCT‐induced differences in EV proteomes were assessed by unsupervised hierarchical clustering and principal component analysis (PCA), demonstrating differential clustering of the majority of EV proteomes according to patient instead of BCT, except for EV proteomes prepared from serum and Roche cfDNA PDP (Figure 4a,b and Figure S8). This indicates that EV proteomes from serum and Roche cfDNA PDP do not reflect true biological states, but ex vivo changes induced by the respective BCT. LC‐MS/MS identified 710 unique proteins (Table S6). Of these, 369 (51.97%) ( =% of the total identified proteome over all BCT) were shared among all BCT, including EV‐enriched proteins (ADAM10, ANXA1, ANXA2, ANXA5, CD9, CD63, CD81, FLOT1, FLOT2, HSC70, PDCD6IP (ALIX), SDCBP, TSG101), proteins involved in platelet function (GP1BA, GP9, ITGA2B, ITGB3, MMRN1, PF4) and haemostasis (F2, F5, F8, FGA, FGB, VWF), erythrocyte proteins (ANK1, HBA, HBB, EPB41, EPB42, SLC2A1, SPTA1, SPTB, STOM) and proteins involved in immunity (complement factors, immunoglobulins, HLA class I and II). With 636 (89.58%) and 609 proteins (85.77%), respectively, the highest number of proteins were identified in EV preparations from Roche cfDNA PDP and serum. Furthermore, 30 (4.23%) proteins, including erythrocyte proteins GYPA (CD235a) and GYPC, were uniquely identified in EV preparations from Roche cfDNA PDP (Figure 4c). Differential protein enrichment between EV preparations from serum and Roche cfDNA PDP versus EV preparations from PDP of the other selected BCT using volcano plot analysis, visualizes the relative distribution of multiple relevant proteins (Figure 4d). The enrichment of platelet (activation) proteins in serum, and erythrocyte proteins in Roche cfDNA samples (FDR < 0.05), respectively mirrored the high levels of activated platelet EV and erythrocyte EV in these BCT, confirming previous results (Figure 3). These observations were supported by GSEA, showing significant enrichment of proteins involved in platelet activation, aggregation and signalling in EV preparations from serum, and erythrocyte membrane and cytosolic proteins in EV preparations from Roche cfDNA PDP (FDR < 0.05) (Figure S9). CD81, which is not expressed on platelets or erythrocytes (Uhlen et al., 2015), was equally present in EV samples from all BCT, suggesting minimal influence of platelet activation and haemolysis on this marker. FLOT1 and FLOT2 were significantly enriched in Roche cfDNA samples, while other EV markers such as CD9, CD63, TSG101 and PDCD6IP (ALIX) were underrepresented compared to other BCT. Flotillins are integral proteins of erythrocyte lipid rafts (Salzer & Prohaska, 2001), suggesting their enrichment in erythrocyte EV separated from (haemolytic) plasma (Figure 4d). This was confirmed by western blot analysis of SEC‐based EV preparations from haemolytic and non‐haemolytic PDP samples (Figure 4e). No statistically significant differences in protein enrichment between the remaining BCT could be identified at a false discovery rate (FDR) < 0.05.
FIGURE 4.

Impact of BCT type (n = 10) on EV proteome. Serum and PDP was prepared 60 min after blood draw (n = 3). Mass spectrometry proteomic analysis (LC–MS/MS) was performed on EV‐enriched fractions, separated by sequential biophysical fractionation. Proteomics data were analysed by a. Hierarchical clustering. b. Principal component analysis. Data points are coloured according to BCT. Serum (red) and Roche cfDNA (pink) clusters are indicated by coloured ellipses. c. Upset plot analysis. The total number of identified proteins in a given BCT is represented on the left bar plot. Intersections between BCT are represented by the bottom plot and their occurrence is shown on the top bar plot. d. Volcano plot analysis. Exemplary proteins of interest are highlighted in black. EV protein enrichment in the BCT of interest (n = 1) is compared to EV protein enrichment in the rest of the selected BCT (n = 9). e. The impact of haemolysis on EV‐associated proteins (Flotillin‐1, CD81) in EV‐enriched fractions, separated from plasma by SEC, was evaluated with western blot analysis.
Unsupervised hierarchical clustering and PCA of EV miRNA profiles demonstrated similar patterns as for EV proteomes, with the majority clustering according to patient, while serum and Roche cfDNA samples clustered according to BCT (Figure 5a,b and Figure S10). Small RNA sequencing identified 429 unique miRNA Table S7). Of these, only 100 (23.31%) ( =% of the total identified miRNA over all BCT) were shared among all BCT, including several platelet‐enriched (let‐7, miR‐101, miR‐103a‐3p, miR‐23, miR‐25‐3p, miR‐92a‐3p, miR‐92b‐3p, miR‐140, miR‐26, miR‐21, miR‐191‐5p, miR‐423) (Plé et al., 2012) and erythrocyte‐enriched miRNA (miR‐16‐5p, miR‐92a‐3p, miR‐144‐3p, miR‐451a, miR‐486‐5p) (Doss et al., 2015). The highest numbers of miRNA were identified in EV preparations from Roche cfDNA PDP (223; 51.98%) and serum (250; 58.28%), with 26 (6.06%) and 32 (7.46%) uniquely identified miRNA in each group, respectively (Figure 5c). Volcano plot analysis revealed differential enrichment (FDR < 0.05) of erythrocyte miRNA in EV preparations from Roche cfDNA PDP (Figure 5d), validating the observations from EV proteome data (Figure 4). No miRNAs were found to be differentially expressed between BCT at a false discovery rate (FDR) < 0.05 for the remaining BCT.
FIGURE 5.

Impact of BCT type (n = 10) on EV‐derived small RNA content. Serum and PDP was prepared 60 min after blood draw (n = 3). Small RNA sequencing was performed on EV‐enriched fractions, separated by sequential biophysical fractionation. miRNA expression data were analysed by a. Hierarchical clustering. b. Principal component analysis. Data points are coloured according to BCT. Serum (red) and Roche cfDNA (pink) clusters are indicated by coloured ellipses. c. Upset plot analysis. The total number of identified miRNA in a given BCT is represented on the left bar plot. Intersections between BCT are represented by the bottom plot and their occurrence is shown on the top bar plot. d. Volcano plot analysis. Exemplary miRNA of interest are highlighted. EV miRNA enrichment in the BCT of interest (n = 1) is compared to EV miRNA enrichment in the rest of the selected BCT (n = 9).
3.1.4. Performance metrics for BCT
To enable robust and objective comparisons between BCT, we developed and implemented performance metrics reflecting sample quality (haemolysis, platelet activation, residual platelets), EV recovery (by measuring recovery of spiked rEV), ex vivo release of blood‐cell derived EV subtypes (activated platelet EV, non‐platelet EV, erythrocyte EV) and EV omics profile (Pearson similarity of LC‐MS/MS and RNAseq profiles). A summary plot visualizes all performance metrics after z‐score transformation allowing direct comparison of the contribution of each BCT (Figure 6). For each metric, a higher z‐score indicates a better performance.
FIGURE 6.

Overview of the impact of type of BCT on all EV performance metrics after transforming corresponding values to z‐scores. Higher z‐scores indicate better performance. Rows and columns of the heatmap are clustered according to complete hierarchical clustering based on Euclidean distance.
3.2. The impact of blood processing intervals on EV performance metrics
Since sample processing in the same time interval may be difficult to achieve in a clinical setting, five BCT with favourable short‐term EV performance metrics were additionally evaluated against three clinically relevant BPI to determine the ability of BCT to stabilize EV during blood sample storage over time. The selected BCT were EDTA spray, ACD‐A, Streck BCT DNA, Biomatrica LBgard and Streck RNA complete BCT. EDTA spray was preferred to EDTA gel, since the use of a gel separator did not significantly impact EV performance metrics. ACD‐A outperformed citrate on nearly every performance metric, and was therefore selected as the preferred BCT to obtain citrated blood. Over the course of this study, Streck BCT RNA was discontinued by the manufacturer and replaced by Streck RNA complete BCT. It was included in the time‐series testing, since it is marketed to maintain the draw time concentration of cell‐free RNA and EV. The selected BPI time points between blood draw and processing were 1 h (T1) and 8 h (T2) to mimic immediate and same day processing (short‐term stabilizing efficiency), and 72 h (T3) to assess longer‐term stabilizing efficiency.
3.2.1. Influence of BPI on sample quality and ex vivo release of blood cell‐derived EV
Blood samples from healthy donors (n = 5) were collected during a single venipuncture into the selection of non‐preservation and preservation BCT (n = 5) and processed over 3 BPI (T1‐T3). Donor characteristics are summarized in Table S1. Haematology analysis was performed prior to PDP preparation. Mean leukocyte counts remained stable in all BCT over all time intervals. Also, platelet counts remained stable in all BCT with the exception of Streck RNA complete BCT, in which a significant drop in platelet counts was observed 72 h after sample collection (p < 0.001). While remaining stable in ACD‐A and Streck RNA complete BCT, erythrocyte counts decreased significantly at T3 in EDTA (p = 0.043), Streck BCT DNA (p = 0.009) and Biomatrica LBgard (p = 0.006). MCV increased significantly in all BCT over time (Figure S11 and Table S8). Storage time had a significant impact on sample preparation, due to the inability to prepare PDP at T3 with EDTA, Streck BCT DNA and Biomatrica LBgard using the two step 2500 × g protocol (Figure S11c). Applying a third 2500 × g centrifugation step resulted in successful platelet depletion in plasma obtained from Biomatrica LBgard, while the majority of samples from EDTA and Streck BCT DNA still contained a substantial number of residual platelets (Figure S11d).
After PDP preparation, differences in stabilization efficiency between BCT over time were evaluated using complementary EV performance metrics: rEV recovery, sample quality (haemolysis, platelet activation, residual platelets), EV subtypes (activated platelet EV, non‐platelet EV, erythrocyte EV) and EV omics profiles. All metrics were significantly affected by BPI (p < 0.001) (Figure 7 and Table S9). Spike‐in of rEV in full blood was followed by incubation (1 h, 8 h, 72 h), PDP preparation, EV separation by SEC and measurement of rEV recovery by fNTA, as described earlier. Recovery of rEV decreased significantly from 78.1% to 63,1% in ACD‐A (p = 0.034), and from 68.0% to 54.0% in EDTA (p = 0.048), while no statistically significant differences in rEV recovery could be observed over time for Streck RNA complete BCT. However, with 61.1%, baseline rEV recovery rates were lowest in Streck RNA complete BCT (Figure 7a and Table S9). Sample quality metrics and EV subtypes were measured directly in PDP (Figure 7b–i and Table S9), as described earlier. Mean haemoglobin concentration remained stable at T2, but increased significantly at T3 for all BCT, with mean fold changes ranging from 2.62 to 5.17 (p < 0.001). Preservation BCT had significantly higher concentrations of soluble platelet activation markers compared to ACD‐A and EDTA at T1 and T2. However, BTG and PF4 concentrations remained relatively stable in preservation BCT at T3, with mean fold changes up to 2.37 (p < 0.001). In contrast, long‐term storage (72 h) significantly affected concentrations in non‐preservation BCT, with mean fold changes ranging from 13.54 for EDTA (p = 0.003) to 139.95 for ACD‐A (p = 0.001). Total EV, activated platelet and non‐platelet EV, erythrocyte EV and residual platelet concentrations were all unaffected by short‐term storage (1 h, 8 h), regardless of BCT. After long‐term storage (72 h), the number of total EV was least affected in ACD‐A and Streck RNA complete BCT, with fold changes of 5.63 (p = 0.043) and 6.33 (p = 0.009), respectively, compared to baseline. With mean fold changes ranging from 14.96 to 21.96, total EV counts in the remaining BCT were significantly higher (p < 0.001). Activated platelet and non‐platelet EV counts at T3 were least affected in Streck RNA complete BCT, with fold changes of 20.01 (p = NS) and 3.58 (p = NS), while erythrocyte EV counts were least affected in ACD‐A (p = NS). Conversely, the concentration of EV subtypes after long‐term storage (72 h) was the most significantly affected in EDTA and Streck BCT DNA, with fold changes of 186.11 and 79.27 for activated platelet EV, 21.03 and 24.30 for non‐platelet EV, and 43.93 and 127.03 for erythrocyte EV, respectively (p < 0.001). After long‐term storage (72 h), a large share of the total EV count was represented by activated platelet EV and erythrocyte EV in all BCT (Figure S12). Despite applying an additional 2500 × g centrifugation step for non‐platelet depleted plasma samples, residual platelets remained abundantly present in PDP prepared from EDTA (p < 0.001) and Streck BCT DNA (p = 0.001).
FIGURE 7.

Impact of BPI (n = 3) on EV recovery, sample quality and ex vivo release of blood‐cell derived EV using different BCT (n = 5). Recovery analyses were performed on EV‐enriched fractions, separated from PDP by SEC, following 60 min (T1), 8 h (T2) and 72 h (T3) incubation of spiked rEV in whole blood at room temperature (n = 3). Sample quality and EV subtype analyses were performed on PDP, prepared 60 min (T1), 8 h (T2) and 72 h (T3) after blood draw (n = 5). Data are depicted as individual values with means. The asterisk indicates a statistically significant difference compared to the reference BPI (T1). a. Recovery rate (%) of recombinant EV (rEV), measured by fluorescent nanoparticle tracking analysis. b. Hemolysis, quantified as haemoglobin (Hb) concentration (mg/mL), measured by colorimetric assay. c. Platelet activation, quantified as plasma concentration of platelet factor 4 (PF4) (ng/mL), measured by ELISA. d. Platelet activation, quantified as plasma concentration of beta thromboglobulin (BTG) (ng/mL), measured by ELISA. e. Total EV count, quantified as the concentration (mL−1) of particles with a refractive index < 1.42 (RI < 1.42). f. Activated platelet EV count, quantified as the concentration (mL−1) of CD61+Lac+ particles (RI < 1.42). g. Non‐platelet EV count, quantified as the concentration (mL−1) of CD61‐Lac+ particles (RI < 1.42). g. Residual platelet count, quantified as the concentration (mL−1) of CD61+ particles (SSC cross section > 300 nm2). The dotted line indicates the detection limit of a standard haematology analyser. H. Erythrocyte EV count, quantified as the concentration (mL−1) of CD235+ particles (RI < 1.42).
In summary, rEV recovery, sample quality and ex vivo release of EV subtypes are significantly influenced by increasing BPI, regardless of BCT type.
3.2.2. Influence of BPI on EV proteome and transcriptome
To evaluate how the EV proteome and transcriptome over time is influenced by the type of BCT, blood samples from prostate cancer patients (n = 6) were collected during a single venipuncture into the selection of non‐preservation and preservation BCT (n = 5) and processed over three time intervals (T1–T3). Donor characteristics are summarized in Table S2. After EV separation through sequential biophysical fractionation, samples were analysed by LC–MS/MS (n = 3) and small RNA sequencing (n = 3).
Hierarchical clustering and PCA of LC‐MS/MS data distinguished 3 defined sample groups based on BCT and BPI (Figure 8a,b). T1 samples from all BCT clustered together with T2 samples from ACD‐A, EDTA and Streck RNA complete BCT, while T3 samples from ACD‐A and Streck RNA complete BCT clustered together with T2 samples from the remaining preservation BCT. T3 samples from EDTA, Streck BCT DNA and Biomatrica LBgard formed a completely distinct sample group. This indicates that time‐induced ex vivo changes in EV proteomes are influenced by the type of BCT. LC‐MS/MS identified 851 unique proteins (Table S11). The number of identified proteins per BCT increased with increasing BPI, reflecting in‐vitro release of blood‐derived EV subtypes. With 567 (66.63%), 619 (72.74%) and 624 proteins (73.33%), respectively, the highest number of proteins was identified in T3 samples from EDTA, Streck BCT DNA and Biomatrica LBgard. Furthermore, 221 (25.97%) proteins were uniquely identified in this group of samples (Figure 8c). Volcano plot analysis was used to assess the evolution of protein enrichment in BCT over different time points (Figure 8d). No statistically significant differences in protein enrichment could be demonstrated between T1 and T2 samples for all BCT, and between T1 and T3 samples for ACD‐A and Streck RNA complete BCT, at FDR < 0.05. In contrast, T3 samples from EDTA, Streck BCT DNA and Biomatrica LBgard were enriched in platelet (activation) proteins (GP1BA, GP1BB, GP9, ITGA2B, ITGB3, PF4, PPBP, THBS1), erythrocyte proteins (ANK1, HB, EPB41, EPB42, GYPA, SLC2A1, SLC4A1, SPTA1, SPTB, STOM) and a distinct set of EV‐proteins (CD9, CD81, FLOT1, FLOT2) (FDR < 0.05), reflecting the significant increase in blood‐cell derived EV in these BCT over time, as described in the previous experiments (Figure 7). These observations were supported by GSEA, showing significant enrichment of proteins involved in the biological function of platelets and erythrocytes in T3 plasma samples (FDR < 0.05) (Figure S13). In addition, EV preparations from EDTA PDP at T3 were enriched in pan‐leukocyte marker PTPRC (CD45), suggesting time‐dependent release of leukocyte‐derived EV in EDTA plasma, but not in plasma derived from preservation BCT. Correlation analysis based on protein intensities revealed a time‐dependent decrease in similarity within patient samples compared to T1 for all BCT, with mean Pearson's r coefficients down to 0.64–0.79 at T2 and 0.43–0.60 at T3, demonstrating changes in the blood EV proteome with increasing BPI (Figure S14).
FIGURE 8.

Impact of BPI (60 min = T1, 8 h = T2 and 72 h = T3 after blood draw) (n = 3) on EV proteomes using different BCT (n = 5). Mass spectrometry proteomic analysis (LC‐MS/MS) was performed on EV‐enriched fractions, separated by sequential biophysical fractionation. Proteomics data were analysed by a. Hierarchical clustering. b. Principal component analysis. Data point are coloured according to BCT. Time points are indicated by a unique symbol. Clusters of interest are indicated by ellipses. c. Upset plot analysis. The total number of identified proteins in a BCT at a given BPI is represented on the left bar plot. Intersections are represented by the bottom plot and their occurrence is shown on the top bar plot. d. Volcano plot analysis. Exemplary proteins of interest are highlighted.
Hierarchical clustering and PCA of small RNAseq data distinguished two defined sample groups based on time interval between sample collection and processing, in which T3 samples clustered separately from T1 and T2 samples (Figure 9a,b). RNA seq identified 224 unique miRNA (Table S11). In line with the LC‐MS/MS data, the number of identified miRNA per BCT increased with increasing BPI, with the highest numbers of miRNA being identified in EDTA, Streck BCT DNA and Biomatrica LBgard. Only 35 (15.63%) miRNA were shared among all BCT (Figure 9c). Volcano plot analysis was used to assess the evolution of miRNA enrichment in BCT over different time points (Figure 9d). No statistically significant differences in miRNA enrichment could be demonstrated between T1 and T2 samples for all BCT at FDR < 0.05. However, volcano plot analysis revealed differential enrichment (FDR < 0.05) of platelet (let‐7, miR‐101, miR‐25‐3p, miR‐92a‐3p, miR‐423) and erythrocyte miRNA (miR‐16‐5p, miR‐92a‐3p, miR‐144‐3p, miR‐451a, miR‐486‐5p) in plasma EV from T3 samples compared to T1 samples from all BCT (Figure 9d). As with EV proteomes, the EV miRNA profile demonstrated changes with increasing storage times for all BCT, with mean Pearson's r coefficients down to 0.81–0.91 at T2 and 0.59–0.71 at T3 based on correlation analysis within patient samples at T1 (Figure S15).
FIGURE 9.
Impact of BPI (60 min = T1, 8 h = T2 and 72 h = T3 after blood draw) (n = 3) on EV small RNA content using different BCT (n = 5). Small RNA sequencing was performed on EV‐enriched fractions, separated by sequential biophysical fractionation. miRNA expression data were analysed by a. Hierarchical clustering. b. Principal component analysis. Data points are coloured according to BCT. Time points are indicated by a unique symbol. Clusters of interest are indicated by ellipses. c. Upset plot analysis. The total number of identified miRNA in a BCT at a given BPI is represented on the left bar plot. Intersections are represented by the bottom plot and their occurrence is shown on the top bar plot. d. Volcano plot analysis. Exemplary miRNA of interest are highlighted.
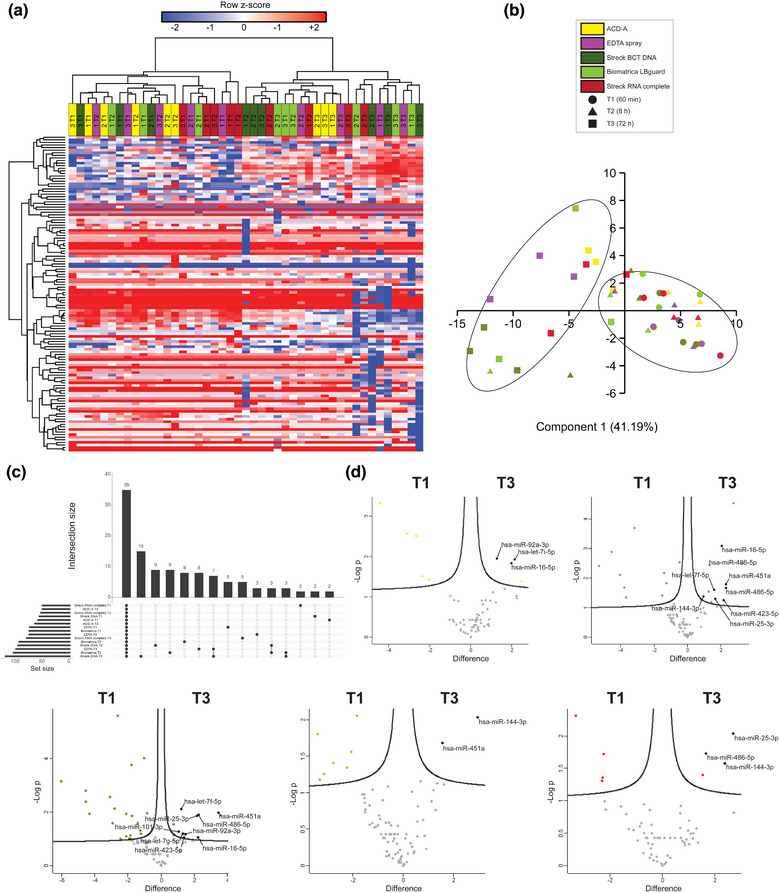
3.2.3. Performance metrics for BPI
To enable robust and objective comparisons between BPI, we developed and implemented performance metrics reflecting sample quality (haemolysis, platelet activation, residual platelets), EV recovery (by measuring recovery of spiked rEV), ex vivo release of blood‐cell derived EV subtypes (activated platelet EV, non‐platelet EV, erythrocyte EV) and EV omics profile (Pearson similarity of LC‐MS/MS and RNAseq profiles). A summary plot visualizes all performance metrics after z‐score transformation allowing direct comparison of the contribution of each BPI (Figure 10). For each metric, a higher z‐score indicates a better performance.
FIGURE 10.
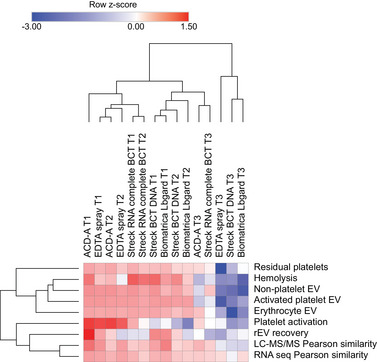
Overview of the impact of BPI using different BCT on all EV performance metrics after transforming corresponding values to z‐scores. Higher z‐scores indicate better performance. Rows and columns of the heatmap are clustered according to complete hierarchical clustering based on Euclidean distance.
4. DISCUSSION
In the EVBB study, we comprehensively compared BCT (n = 11) and BPI (n = 3) in the first large‐scale benchmarking study with the aim to establish improved routines for EV analysis in blood. We demonstrate that the type of BCT and duration of BPI substantially impact EV analysis. To enable this evaluation, we developed and implemented performance metrics reflecting sample quality, EV recovery, ex vivo release of blood‐cell derived EV subtypes and EV‐associated molecular signatures (Figures 6 and 10). Based on these findings, we provide general recommendations for researchers guiding the informed selection of BCT and BPI (Box 1). The EVBB study contributes to methodological standardization for blood EV studies presenting performance metrics that can serve as a framework for newly developed BCT, additional BPI and by extension other pre‐analytical factors.
Box 1. General considerations concluded from the EVBB study towards the selection of BCT and BPI for EV analysis in blood.
|
The use of citrate and ACD‐A plasma associated with the most favourable short‐term performance metrics across all investigated BCT (Figure 6), confirming results from smaller scale studies (György et al., 2014; Lacroix et al., 2012). ACD‐A has previously demonstrated to stabilize EV levels in blood samples for up to 1 h even after agitation and storage at 37°C (György et al., 2014). Citrate has been recommended for studies on blood plasma EV due to its inhibitory effect on the release of activated platelet‐derived EV (Coumans et al., 2017). Our study demonstrated the lowest number of activated platelet EV to be present in EDTA PDP, despite higher levels of soluble platelet activation markers PF4 and BTG compared to citrate and ACD‐A PDP (Figure 3 and Tables S4–S, 5). Similar observations were made for preservation BCT, which contain EDTA as an anticoagulant, in which activated platelet EV levels remained low despite high levels of platelet activation. These observations are supported by recent research, comparing concentrations of activated platelet EV between citrate and EDTA PDP following incubation with thrombin receptor‐activating peptide‐6 (TRAP‐6) to activate platelets. Upon platelet activation, activated platelet EV levels were significantly lower in EDTA compared to citrate PDP. In addition, activated platelet EV concentrations were unaffected by pneumatic tube transport of blood samples in EDTA, but increased in citrate. Levels of erythrocyte and leukocyte EV were similar between both BCT and EV levels were similarly affected following a freeze‐thaw cycle (Buntsma et al., 2021). Unsurprisingly, the performance of serum BCT was significantly affected by platelet activation and ex vivo release of activated platelet EV (Figure 6), representing op to 80% of the total EV count (Figure S6). Similar observations were made in a previous study, demonstrating elevated levels of (activated) platelet EV in serum compared to blood plasma collected with citrate, ACD‐A or EDTA (Palviainen et al., 2020). For preservation BCT, the use of Streck BCT DNA and Biomatrica LBgard was associated with favourable short‐term performance metrics, demonstrating that these BCT can be used for EV analysis. Conversely, the performance of Roche cfDNA was significantly affected by haemolysis (Figure 6), an issue that was reported before (ExRNAQC Consortium, 2021; Zhao et al., 2019).
Performance metrics 8 h following blood sample collection remained relatively unaffected for all of the investigated BCT. Long‐term stability 72 h after sample collection could not be demonstrated using either of the investigated BCT (Figure 10). Surprisingly, preservation BCT did not outperform non‐preservation BCT for multiple metrics, even though the investigated storage time was substantially shorter than advertised for the preservation BCT. Streck RNA Complete BCT, which is specifically marketed to maintain the draw time concentration of EV and extracellular RNA for up to 7 days at room temperature, outperformed other preservation BCT but did not significantly outperform the non‐preservation ACD‐A BCT. In addition, platelet activation, haemolysis and EV levels were significantly higher in some of the preservation BCT compared to non‐preservation BCT, even at baseline (Figure 7 and Table S9). These results are in line with previously published research on extracellular RNA analysis, describing instability of performance metrics over time for all tested preservation BCT (ExRNAQC Consortium, 2021). Similarly, a recent pilot study reported significant ex vivo blood‐cell derived EV release in Streck BCT DNA and Roche cfDNA, only 72 h after sample collection (Heatlie et al., 2020). Finally, long‐term storage had a significant impact on the ability to deplete blood plasma from platelets using the two step 2500 g protocol, especially for EDTA and Streck BCT DNA (Figure S11c,d). Decreased platelet sedimentation rate due to viscous drag might be explained because of elevated blood plasma protein concentration following platelet activation and cell lysis (Késmárky et al., 2008). In addition, the blood plasma volume that could be prepared from blood samples decreased significantly following long‐term storage (Figure S16), further supporting BCT performance instability over time. In line with previous smaller scale studies, ACD‐A and EDTA showed favourable performance metric profiles for EV analysis in blood plasma, which are relatively stable for up to 8 h following sample collection (Buntsma et al., 2021). Although preservation BCT are suitable for EV analysis, their use is not associated with increased stability over prolonged periods of time.
Table 1 summarizes the composition, mode of action and predicted effect on blood cells of the BCT investigated in this work, and can help explain the underlying mechanisms through which BCT affect EV performance metrics. Ca2+ ions play an important role in EV biogenesis and release from platelets (Freyssinet & Toti, 2010). Consequently, the use of citrate PDP, which is a weak chelator of Ca2+, resulted in limited ex vivo generation of activated platelet EV. However, the use of EDTA PDP resulted in even lower levels of platelet‐derived EV, despite higher levels of soluble platelet activation markers, as a consequence of strong Ca2+ chelation inhibiting ex vivo EV release. Similarly, local acidosis in blood collected from ACD‐A BCT inhibits store‐operated Ca2+ influx into platelets, further impeding platelet function and EV release (Marumo et al., 2001). In addition, ACD‐A contains dextrose, extending cell viability ex vivo and thereby limiting haemolysis and release of erythrocyte EV (Mollison, 2000). Several preservation BCT contain formaldehyde‐releasing substances to stabilize cells and extracellular nucleic acids through cell membrane stabilization and inhibition of nucleases (Das et al., 2017; Fernando, 2017; Fernando & Chao‐Wei, 2016). However, it has been reported that formaldehyde induces elevation of intracellular Ca2+ and platelet activation, which is only partially inhibited by Ca2+ chelation (Wong et al., 2006). In general, the mechanism of action through which individual components of proprietary stabilization solutions impact BCT performance metrics remains to be elucidated.
Unbiased assessment of the performance of BCT using integrative omics analyses on blood‐derived EV is currently missing in the scientific literature. To date, only one study evaluated EV miRNA content from different BCT, finding similar RNA profiles between citrate and ACD‐A (György et al., 2014). To evaluate the impact of BCT on the EV proteome and miRNA profile, blood‐derived EV were prepared at three different time points upon blood collection to assess potential changes in EV content due to blood storage at room temperature. We demonstrated that sample quality and ex vivo release of blood‐cell derived EV subtypes are reflected in the downstream EV protein and miRNA content. After short‐term blood storage, no major differences in EV omics profile could be demonstrated between the investigated BCT, with the exception of samples significantly affected by haemolysis and substantially elevated levels of activated platelet‐ and erythrocyte EV (Figures 4, 5, 6). Similar as for EV levels, long‐term stability of EV proteomes and miRNA profiles 72 h after sample collection could not be demonstrated (Figures 8, 9, 10). In addition, the number of identified proteins and miRNAs in EV preparations from blood plasma increased over time. Similarly, EV protein and miRNA profiles demonstrated instability starting from 8 h after sample collection based on correlation analysis with baseline samples (Figure S14–S, 15). In conclusion, the studied BCT are not suitable for EV omics analyses beyond the processing intervals indicated in the current guidelines. We encourage further research to support the development of BCT that preserve the draw time concentration of EV and their corresponding molecular signatures for prolonged time periods to enable clinical implementation.
We do not claim that our results are universally applicable. Our study increases the awareness about the impact of BCT and BPI on EV analysis, providing the community with performance metrics that can be used to assess newly developed BCT, additional BPI, but also other important pre‐analytic variables such as storage and processing temperature, transport conditions and freeze‐thaw cycles. We recommend that BCT and BPI should be transparently reported and well‐defined at the start of the study. Particularly BPI is crucial to monitor since none of the tested BCT support long‐term stability of EV in blood. The EV field will surely benefit from future systematic and comprehensive studies comparing BCT and BPI, including new emerging BCT compatible with extended BPI. The EVBB study is performed monocentric; interlaboratory validation of the results is warranted and may reveal subtle differences between BCT that could not be addressed in this study with a defined sample size. Finally, the flow cytometer used to measure blood cell‐derived EV subtypes has a detection limit of 10 nm3, which corresponds to an EV diameter of 157 ± 10 nm. As a consequence, flow cytometry results in our study cannot reliably be extrapolated to smaller EV subpopulations or lipoprotein particles (<200 nm). We used Flow‐SR because it: 1) determines size independent of the particle refractive index; and 2) identifies and differentiates EV and lipoprotein particles based upon the refractive index (i.e. label free). Furthermore, in contrast to fNTA, flow cytometry determines the concentration of (blood cell‐derived) EV directly in diluted blood plasma or serum and within a calibrated, known size and fluorescence range.
In conclusion, the EVBB study serves as a framework to guide the informed selection and steer the development/optimization of BCT and BPI for EV analysis.
AUTHOR CONTRIBUTIONS
Bert Dhondt: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Validation; Visualization; Writing – original draft; Writing – review & editing. Cláudio Pinheiro: Data curation; Formal analysis; Investigation; Software; Writing‐original draft; Writing‐review & editing. Edward Geeurickx: Investigation; Resources; Validation; Writing – review & editing. Joeri Tulkens: Investigation; Validation; Visualization; Writing – review & editing. Glenn Vergauwen: Investigation; Validation; Visualization; Writing – review & editing. Edwin van der Pol: Data curation; Resources; Software; Writing – original draft; Writing – review & editing. Rienk Nieuwland: Resources; Writing — review & editing. Anneleen Decock: Methodology; Writing – review & editing. Ilkka Miinalainen: Investigation; Resources; Writing – review & editing. Pekka Rappu: Data curation; Investigation; Resources; Software; Writing – review & editing. Gary Schroth: Resources; Writing – review & editing. Scott Kuersten: Resources; Writing – review & editing. Jo Vandesompele: Methodology; Resources; Writing – review & editing. Pieter Mestdagh: Methodology; Resources; Writing – review & editing. Nicolaas Lumen: Funding acquisition; Project administration; Resources; Supervision; Writing – review & editing. Olivier De Wever: Conceptualization; Funding acquisition; Methodology; Project administration; Resources; Supervision; Writing – original draft; Writing – review & editing. An Hendrix: Conceptualization; Funding acquisition; Methodology; Project administration; Resources; Supervision; Writing – original draft; Writing – review & editing.
CONFLICT OF INTEREST STATEMENT
Edward Geeurickx, Jo Vandesompele, Pieter Mestdagh, Olivier De Wever and An Hendrix are inventors on the patent application covering the rEV technology (WO2019091964). The remaining authors declare no competing interests.
Supporting information
Supplementary figure 1. Analysis of 564 EV‐TRACK knowledgebase1 records on blood‐derived EV. a. The most frequently used blood collection tubes for EV research. b. Study aims in blood‐derived EV research.
Supplementary figure 2. The number of BCT and performance metrics evaluated by other BCT performance studies and this study.
Supplementary figure 3. Illustrative overview of EV separation and analysis. After blood sample collection, serum or PDP is prepared by serial centrifugation. Hematology analysis is performed on whole blood. Particle analysis (EV subtypes, lipoprotein particles (presumably chylomicrons), residual platelets) is performed by flow cytometry on processed blood (serum or PDP) without additional EV separation. For rEV recovery analysis, a crude plasma EV preparation is obtained by size‐exclusion chromatography (SEC). For integrative omics analysis (LC‐MS/MS and small RNAseq), EV are separated from serum or PDP by the orthogonal implementation of SEC and ODG. * rEV spike‐in in whole blood samples is only performed for rEV recovery analysis.
Supplementary figure 4. Characterization of EV preparations separated from plasma using SEC (left) and SEC + ODG (right). a. NTA of EV‐enriched fractions. NTA calculated size distributions are depicted as mean (black line) with standard error (red area) and total particle number, mean particle size and mode are shown for each EV‐enriched fraction. b. Western blot analysis for EV‐ (Flotillin‐1) and non‐EV (Apolipoprotein A1) associated proteins. c. Transmission electron microscopy of EV‐enriched fractions (scale bar: 200 nm). White arrows indicate EV and black arrows indicate lipoprotein particles. d. Technical evaluation of methodological repeatability of SEC+ODG separation of citrate plasma EV (PEV) by mass spectrometry‐based proteomic analysis (LC‐MS/MS) of 5 technical replicates (each replicate indicated by number). LC‐MS/MS data from EV‐enriched fractions are compared by correlation analysis. Data are provided as Pearson's r coefficients.
Supplementary figure 5. Impact of BCT type (n=10) on hematology testing of full blood samples prior to sample preparation (n=10). Data are depicted as individual values with means. The asterisk indicates a statistically significant difference compared to the reference BCT (EDTA). a. Leukocyte (103/μL), erythrocyte (106/μL) and platelet (103/μL) count. b. Mean cellular volume (fL).
Supplementary figure 6. Impact of BCT type (n=10) on the ratio of activated platelet EV (CD61+Lac+; RI<1.42) to total EV (RI<1.42), measured by flow cytometry (n=10). Data are depicted as individual values with means.
Supplementary figure 7. Impact of BCT type (n=10) on EV (RI<1.42) size distribution, measured by flow cytometry (n=10). Data are provided as the decay constant (Mu) of the size distribution slope. Data are depicted as individual values with means.
Supplementary figure 8. Principal component analysis of EV proteome (LC‐MS/MS) data impacted by BCT type. Data points are colored according to biological replicate. Serum (red) and Roche cfDNA (pink) clusters are indicated by colored ellipses.
Supplementary figure 9. GSEA of EV proteome (LC‐MS/MS) data impacted by BCT type. a. The serum EV proteome is enriched in a phenotype corresponding to platelet activation, signaling and aggregation b. The Roche cfDNA EV proteome is enriched in a phenotype corresponding to the erythrocyte membrane and cytosol. Significance threshold was set at FDR < 0.05. The heat map corresponds to the enrichment of phenotypic proteins in individual samples.
Supplementary figure 10. Principal component analysis of EV miRNA (small RNA seq) data impacted by BCT type. Data points are colored according to biological replicate. Serum (red) and Roche cfDNA (pink) clusters are indicated by colored ellipses.
Supplementary figure 11. Impact of BCT type (n=5) on hematology testing of full blood samples prior to sample preparation (n=5) after 60 min (T1), 8h (T2) and 72h (T3) (BPI evaluation). Data are depicted as individual values with means. The asterisk indicates a statistically significant difference compared to the reference BPI (T1). a. Leukocyte (103/μL), erythrocyte (106/μL) and platelet (103/μL) count. b. Mean cellular volume (fL). c. Residual platelet (103/μL) count on hematology testing after plasma preparation.
Supplementary figure 12. GSEA of EV proteome (LC‐MS/MS) data (BPI evaluation). a. The EV proteome at T3 is enriched in a phenotype corresponding to platelet activation, signaling and aggregation compared to T1. b. The EV proteome at T3 is enriched in a phenotype corresponding to the erythrocyte membrane and cytosol compared to T1. Significance threshold was set at FDR < 0.05. The heat map corresponds to the enrichment of phenotypic proteins in individual samples.
Supplementary figure 13. Impact of BCT type (n=5) on the ratio of a. Activated platelet EV (CD61+Lac+; RI<1.42) to total EV (RI<1.42), b. Non‐platelet EV (CD61‐Lac+; RI<1.42) to total EV (RI<1.42) and c. Erythrocyte EV (CD235+; RI<1.42) to total EV (RI<1.42), measured by flow cytometry (n=5) in plasma 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation). Data are depicted as individual values with means.
Supplementary figure 14. Impact of BCT type (n=5) on the EV proteome, analyzed by LC‐MS/MS (n=3) in EV‐enriched fractions prepared from plasma 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation). The EV proteome at T2 and T3 is compared to the EV proteome at T1 by correlation analysis. Data are provided as Pearson's r coefficients and are depicted as individual values with means.
Supplementary figure 15. Impact of BCT type (n=5) on the EV miRNA profile, analyzed by small RNAseq (n=3) in EV‐enriched fractions prepared from plasma 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation). The EV miRNA profile at T2 and T3 is compared to the EV miRNA profile at T1 by correlation analysis. Data are provided as Pearson's r coefficients and are depicted as individual values with means.
Supplementary figure 16. Impact of BCT type (n=5) on the volume of plasma 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation).
Supplementary table 1. Donor characteristics. Clinical chemistry data were obtained by hematology analysis of EDTA blood 60 min after venipuncture. Data are depicted as mean values with standard deviation. Abbreviations: MCV (mean cellular volume), MCH (mean cellular hemoglobin), MCHC (mean cellular hemoglobin concentration).
Supplementary table 2. Patient characteristics. Abbreviations: RP: radical prostatectomy, (s)PLND: (salvage) pelvic lymph node dissection, PSA: prostate specific antigen.
Supplementary table 3. Impact of BCT type (n=10) on hematology testing of full blood samples prior to sample preparation (n=10). Data are depicted as mean values with standard deviation.
Supplementary table 4. Impact of BCT type (n=10) on sample quality. Analyses were performed on serum and platelet‐depleted plasma (PDP), prepared 60 min after blood draw (n=10). Data are depicted as mean values with standard deviation.
Supplementary table 5. Impact of BCT type (n=10) on ex‐vivo release of blood‐cell derived EV subtypes. Analyses were performed on serum and platelet‐depleted plasma (PDP), prepared 60 min after blood draw (n=10). Data are depicted as mean values with standard deviation.
Supplementary table 6. List of proteins detected by mass spectrometry (LC‐MS/MS) across all samples in the BCT evaluation phase.
Supplementary table 7. List of miRNA detected by small RNA sequencing across all samples in the BCT evaluation phase.
Supplementary table 8. Impact of BCT type (n=5) on hematology testing of full blood samples prior to sample preparation (n=5), 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation). Data are depicted as mean values with standard deviation.
Supplementary table 9. Impact of BCT type (n=5) on rEV recovery, sample quality and ex‐vivo release of blood‐cell derived EV (n=5), 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation). Data are depicted as mean values with standard deviation.
Supplementary table 10. List of proteins detected by mass spectrometry (LC‐MS/MS) across all samples in the BPI evaluation phase.
Supplementary table 11. List of miRNA detected by small RNA sequencing across all samples in the BPI evaluation phase.
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
We thank Sofie De Geyter (Laboratory of Experimental Cancer Research, Department of Human Structure and Repair, Ghent University, Ghent, Belgium), and Najat Hajji and Chi Hau (Laboratory of Experimental Clinical Chemistry, Amsterdam UCM, location AMC, University of Amsterdam, Amsterdam, The Netherlands) for experimental support. We also thank the proteomics and electron microscopy core facilities, supported by biocenter Finland and, respectively, the University of Turku (Finland) and University of Oulu (Finland). This work was supported by the Fund for Scientific Spearheads of Ghent University Hospital, Concerted Research Actions from Ghent University, “Stichting tegen Kanker,” the Fund for Scientific Research‐Flanders, and “Kom op tegen Kanker (Stand up to Cancer), the Flemisch cancer society”.
Dhondt, B. , Pinheiro, C. , Geeurickx, E. , Tulkens, J. , Vergauwen, G. , Van Der Pol, E. , Nieuwland, R. , Decock, A. , Miinalainen, I. , Rappu, P. , Schroth, G. , Kuersten, S. , Vandesompele, J. , Mestdagh, P. , Lumen, N. , De Wever, O. , & Hendrix, A. (2023). Benchmarking blood collection tubes and processing intervals for extracellular vesicle performance metrics. Journal of Extracellular Vesicles, 12, e12315. 10.1002/jev2.12315
REFERENCES
- Antoniou, M. , Kolamunnage‐Dona, R. , Wason, J. , Bathia, R. , Billingham, C. , Bliss, J. M. , Brown, L. C. , Gillman, A. , Paul, J. , & Jorgensen, A. L. (2019). Biomarker‐guided trials: Challenges in practice. Contemporary Clinical Trials Communications, 16, 100493. 10.1016/j.conctc.2019.100493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers, L. , Pink, R. , Carter, D. R. F. , & Nieuwland, R. (2019). Clinical requirements for extracellular vesicle assays. Journal of Extracellular Vesicles, 8, 00. Preprint at 10.1080/20013078.2019.1593755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bæk, R. , Søndergaard, E. K. L. , Varming, K. , & Jørgensen, M. M. (2016). The impact of various preanalytical treatments on the phenotype of small extracellular vesicles in blood analysed by protein microarray. Journal of Immunological Methods, 438, 11–20. [DOI] [PubMed] [Google Scholar]
- Banfi, G. , Salvagno, G. L. , & Lippi, G. (2007). The role of ethylenediamine tetraacetic acid (EDTA) as in vitro anticoagulant for diagnostic purposes. Clinical Chemistry and Laboratory Medicine, 45, 565–576. [DOI] [PubMed] [Google Scholar]
- Buntsma, N. C. , Gąsecka, A. , Roos, Y. B. W. E. M. , van Leeuwen, T. G. , van der Pol, E. , & Nieuwland, R. (2021). EDTA stabilizes the concentration of platelet‐derived extracellular vesicles during blood collection and handling. Platelets, 33(5), 764–771. 10.1080/09537104.2021.1991569 [DOI] [PubMed] [Google Scholar]
- Cocozza, F. , Grisard, E. , Martin‐Jaular, L. , Mathieu, M. , & Théry, C. (2020). SnapShot: Extracellular vesicles. Cell, 182, 00. [DOI] [PubMed] [Google Scholar]
- Connor, D. E. , Exner, T. , Ma, D. D. F. , & Joseph, J. E. (2009). Detection of the procoagulant activity of microparticle‐associated phosphatidylserine using XACT. Blood Coagul Fibrinolysis, 20, 558–64. [DOI] [PubMed] [Google Scholar]
- Coumans, F. A. W. , Brisson, A. R. , Buzas, E. I. , Dignat‐George, F. , Drees, E. E. E. , El‐Andaloussi, S. , Emanueli, C. , Gasecka, A. , Hendrix, A. , Hill, A. F. , Lacroix, R. , Lee, Y. , van Leeuwen, T. G. , Mackman, N. , Mäger, I. , Nolan, J. P. , van der Pol, E. , Pegtel, D. M. , Sahoo, S. , … Nieuwland, R. (2017). Methodological guidelines to study extracellular vesicles. Circulation Research, 120(10), 1632–1648. 10.1161/circresaha.117.309417 [DOI] [PubMed] [Google Scholar]
- Crippa, A. , De Laere, B. , Discacciati, A. , Larsson, B. , Connor, J. T. , Gabriel, E. E. , Thellenberg, C. , Jänes, E. , Enblad, G. , Ullen, A. , Hjälm‐Eriksson, M. , Oldenburg, J. , Ost, P. , Lindberg, J. , Eklund, M. , & Grönberg, H. (2020). The ProBio trial: Molecular biomarkers for advancing personalized treatment decision in patients with metastatic castration‐resistant prostate cancer. Trials, 21(1), 10.1186/s13063-020-04515-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, K. , Fernando, M. R. , & Ryan, W. L. (2017). Blood Collection Device for Stabilizing Cell‐Free Plasma DNA (EP2814981B1). European Patent Office. https://worldwide.espacenet.com/patent/search/family/047755032/publication/EP2814981B1?q=EP2814981B1
- de Rond, L. , Coumans, F. A. W. , Nieuwland, R. , van Leeuwen, T. G. , & van der Pol, E. (2018). Deriving extracellular vesicle size from scatter intensities measured by flow cytometry. Current Protocols in Cytometry, 86, e43. [DOI] [PubMed] [Google Scholar]
- de Wever, O. , & Hendrix, A. (2019). A supporting ecosystem to mature extracellular vesicles into clinical application. EMBO Journal, 38, e101412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhondt, B. , Geeurickx, E. , Tulkens, J. , Van Deun, J. , Vergauwen, G. , Lippens, L. , Miinalainen, I. , Rappu, P. , Heino, J. , Ost, P. , Lumen, N. , De Wever, O. , & Hendrix, A. (2020). Unravelling the proteomic landscape of extracellular vesicles in prostate cancer by density‐based fractionation of urine. Journal of Extracellular Vesicles, 9(1), 1736935. Portico. 10.1080/20013078.2020.1736935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, L. , Zieren, R. C. , Horie, K. , Kim, C. , Mallick, E. , Jing, Y. , Feng, M. , Kuczler, M. D. , Green, J. , Amend, S. R. , Witwer, K. W. , de Reijke, T. M. , Cho, Y. , Pienta, K. J. , & Xue, W. (2020). Comprehensive evaluation of methods for small extracellular vesicles separation from human plasma, urine and cell culture medium. Journal of Extracellular Vesicles, 10(2), Portico. 10.1002/jev2.12044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doss, J. F. , Corcoran, D. L. , Jima, D. D. , Telen, M. J. , Dave, S. S. , & Chi, J.‐T. (2015). A comprehensive joint analysis of the long and short RNA transcriptomes of human erythrocytes. BMC Genomics, 16(1). 10.1186/s12864-015-2156-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ExRNAQC Consortium . (2021). Performance of RNA purification kits and blood collection tubes in the Extracellular RNA Quality Control (exRNAQC) study. bioRxiv, 2021.05.11.442610 10.1101/2021.05.11.442610 [DOI]
- Fabregat, A. , Jupe, S. , Matthews, L. , Sidiropoulos, K. , Gillespie, M. , Garapati, P. , Haw, R. , Jassal, B. , Korninger, F. , May, B. , Milacic, M. , Roca, C. D. , Rothfels, K. , Sevilla, C. , Shamovsky, V. , Shorser, S. , Varusai, T. , Viteri, G. , Weiser, J. , … D'Eustachio, P. (2018). The reactome pathway knowledgebase. Nucleic Acids Research, 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendl, B. , Weiss, R. , Fischer, M. B. , Spittler, A. , & Weber, V. (2016). Characterization of extracellular vesicles in whole blood: Influence of pre‐analytical parameters and visualization of vesicle‐cell interactions using imaging flow cytometry. Biochemical and Biophysical Research Communications, 478, 168–173. [DOI] [PubMed] [Google Scholar]
- Fernando, M. R. (2017). Preservation of cell‐free nucleic acids (EP2398912B1). European Patent Office. https://worldwide.espacenet.com/patent/search/family/042112198/publication/EP2398912B1?q=pn%3DEP2398912B1
- Fernando, M. R. , & Chao‐Wei, C. K. (2016). Preservation of fetal nucleic acids in maternal plasma (EP2228453B1). European Patent Office. https://worldwide.espacenet.com/patent/search/family/042235130/publication/EP2228453B1?q=pn%3DEP2228453B1
- Freyssinet, J. M. , & Toti, F. (2010). Formation of procoagulant microparticles and properties. Thrombosis Research, 125, S46–S48. [DOI] [PubMed] [Google Scholar]
- Geeurickx, E. , Lippens, L. , Rappu, P. , De Geest, B. G. , De Wever, O. , & Hendrix, A. (2021). Recombinant extracellular vesicles as biological reference material for method development, data normalization and assessment of (pre‐)analytical variables. Nature Protocols, 16(2), 603–633. 10.1038/s41596-020-00446-5 [DOI] [PubMed] [Google Scholar]
- Geeurickx, E. , Tulkens, J. , Dhondt, B. , Van Deun, J. , Lippens, L. , Vergauwen, G. , Heyrman, E. , De Sutter, D. , Gevaert, K. , Impens, F. , Miinalainen, I. , Van Bockstal, P.‐J. , De Beer, T. , Wauben, M. H. M. , Nolte‐‘t‐Hoen, E. N. M. , Bloch, K. , Swinnen, J. V. , van der Pol, E. , Nieuwland, R. , … Hendrix, A. (2019). The generation and use of recombinant extracellular vesicles as biological reference material. Nature Communications, 10(1), 285–292. 10.1038/s41467-019-11182-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- György, B. , Pálóczi, K. , Kovács, A. , Barabás, E. , Bekő, G. , Várnai, K. , Pállinger, É. , Szabó‐Taylor, K. , Szabó, T. G. , Kiss, A. A. , Falus, A. , & Buzás, E. I. (2014). Improved circulating microparticle analysis in acid‐citrate dextrose (ACD) anticoagulant tube. Thrombosis Research, 133(2), 285–292. 10.1016/j.thromres.2013.11.010 [DOI] [PubMed] [Google Scholar]
- Hafner, M. , Renwick, N. , Brown, M. , Mihailović, A. , Holoch, D. , Lin, C. , Pena, J. T. G. , Nusbaum, J. D. , Morozov, P. , Ludwig, J. , Ojo, T. , Luo, S. , Schroth, G. , & Tuschl, T. (2011). RNA‐ligase‐dependent biases in miRNA representation in deep‐sequenced small RNA cDNA libraries. RNA, 17(9), 1697–1712. 10.1261/rna.2799511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer, D. A. T. , Ryan, P. D. , Hammer, Ø. , & Harper, D. A. T. (2001). Past: Paleontological statistics software package for education and data analysis. Palaeontologia Electronica, 4, 00. [Google Scholar]
- Heatlie, J. , Chang, V. , Fitzgerald, S. , Nursalim, Y. , Parker, K. , Lawrence, B. , Print, C. G. , & Blenkiron, C. (2020). Specialized cell‐free DNA blood collection tubes can be repurposed for extracellular vesicle isolation: A pilot study. Biopreservation and Biobanking, 18(5), 462–470. 10.1089/bio.2020.0060 [DOI] [PubMed] [Google Scholar]
- Hendrix, A. (2021). The nature of blood(y) extracellular vesicles. Nature Reviews Molecular Cell Biology, 2021 22:4, 22, 243–243. [DOI] [PubMed] [Google Scholar]
- Horlitz, M. , Sprenger‐Haussels, M. , & Schubert, A. (2020). Stabilisation and isolation of extracellular nucleic acids (US10676780B2). U.S. Patent and Trademark Office. https://worldwide.espacenet.com/patent/search/family/047994308/publication/US10676780B2?q=US10676780
- Hulstaert, E. , Decock, A. , Morlion, A. , Everaert, C. , Verniers, K. , Nuytens, J. , Nijs, N. , Schroth, G. P. , Kuersten, S. , Gross, S. M. , Mestdagh, P. , & Vandesompele, J. (2021). Messenger RNA capture sequencing of extracellular RNA from human biofluids using a comprehensive set of spike‐in controls. STAR Protocols, 2(2), 100475. 10.1016/j.xpro.2021.100475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulstaert, E. , Morlion, A. , Avila Cobos, F. , Verniers, K. , Nuytens, J. , Vanden Eynde, E. , Yigit, N. , Anckaert, J. , Geerts, A. , Hindryckx, P. , Jacques, P. , Brusselle, G. , Bracke, K. R. , Maes, T. , Malfait, T. , Derveaux, T. , Ninclaus, V. , Van Cauwenbergh, C. , Roelens, K. , … Mestdagh, P. (2020). Charting extracellular transcriptomes in the human biofluid RNA atlas. Cell Reports, 33(13), 108552. 10.1016/j.celrep.2020.108552 [DOI] [PubMed] [Google Scholar]
- Jayachandran, M. , Miller, V. M. , Heit, J. A. , & Owen, W. G. (2012). Methodology for isolation, identification and characterization of microvesicles in peripheral blood. Journal of Immunological Methods, 375, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakhniashvili, D. G. , Bulla, L. A. , & Goodman, S. R. (2004). The human erythrocyte proteome: analysis by ion trap mass spectrometry. Molecular & Cellular Proteomics, 3, 501–9. [DOI] [PubMed] [Google Scholar]
- Kalluri, R. , & LeBleu, V. S. (2020). The biology, function, and biomedical applications of exosomes. Science, 367, 00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi, N. , Cvjetkovic, A. , Jang, S. C. , Crescitelli, R. , Hosseinpour Feizi, M. A. , Nieuwland, R. , Lötvall, J. , & Lässer, C. (2018). Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cellular and Molecular Life Sciences, 75(15), 2873–2886. 10.1007/s00018-018-2773-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Késmárky, G. , Kenyeres, P. , Rábai, M. , & Tóth, K. (2008). Plasma viscosity: A forgotten variable. Clinical Hemorheology and Microcirculation, 39, 243–246. [PubMed] [Google Scholar]
- Khan, A. , & Mathelier, A. (2017). Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinformatics, 18, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. K. , Song, K. S. , Lee, E. S. , Lee, Y. J. , Park, Y. S. , Lee, K. R. , & Lee, S. N. (2002). Optimized flow cytometric assay for the measurement of platelet microparticles in plasma: pre‐analytic and analytic considerations. Blood Coagulation & Fibrinolysis, 13(5), 393–397. 10.1097/00001721-200207000-00003 [DOI] [PubMed] [Google Scholar]
- Kranich, J. , Chlis, N. , Rausch, L. , Latha, A. , Schifferer, M. , Kurz, T. , Kia, F.‐A. , Simons, M. , Theis, F. J. , & Brocker, T. (2020). In vivo identification of apoptotic and extracellular vesicle‐bound live cells using image‐based deep learning. Journal of Extracellular Vesicles, 9(1), 1792683. Portico. 10.1080/20013078.2020.1792683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix, R. , Judicone, C. , Mooberry, M. , Boucekine, M. , Key, N. S. , & Dignat‐George, F. (2013). Standardization of pre‐analytical variables in plasma microparticle determination: Results of the International Society on Thrombosis and Haemostasis SSC Collaborative workshop. Journal of Thrombosis and Haemostasis, 11(6), 1190–1193. 10.1111/jth.12207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix, R. , Judicone, C. , Poncelet, P. , Robert, S. , Arnaud, L. , Sampol, J. , & Dignat‐George, F. (2012). Impact of pre‐analytical parameters on the measurement of circulating microparticles: towards standardization of protocol. Journal of Thrombosis and Haemostasis, 10(3), 437–446. 10.1111/j.1538-7836.2011.04610.x [DOI] [PubMed] [Google Scholar]
- Lai, C. P. , Mardini, O. , Ericsson, M. , Prabhakar, S. , Maguire, C. A. , Chen, J. W. , Tannous, B. A. , & Breakefield, X. O. (2014). Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano, 8(1), 483–494. 10.1021/nn404945r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lex, A. , Gehlenborg, N. , Strobelt, H. , Vuillemot, R. , & Pfister, H. (2014). UpSet: Visualization of Intersecting Sets. IEEE Transactions on Visualization and Computer Graphics, 20, 1983–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lötvall, J. , Hill, A. F. , Hochberg, F. , Buzás, E. I. , Di Vizio, D. , Gardiner, C. , Gho, Y. S. , Kurochkin, I. V. , Mathivanan, S. , Quesenberry, P. , Sahoo, S. , Tahara, H. , Wauben, M. H. , Witwer, K. W. , & Théry, C. (2014). Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. Journal of Extracellular Vesicles, 3(1), 26913. 10.3402/jev.v3.26913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannuß, S. (2020). Influence of different methods and anticoagulants on platelet parameter measurement. Journal of Laboratory Medicine, 44, 255–272. [Google Scholar]
- Marumo, M. , Suehiro, A. , Kakishita, E. , Groschner, K. , & Wakabayashi, I. (2001). Extracellular pH affects platelet aggregation associated with modulation of store‐operated Ca2+ entry. Thrombosis Research, 104, 353–360. [DOI] [PubMed] [Google Scholar]
- Mateo, J. , Porta, N. , Bianchini, D. , McGovern, U. , Elliott, T. , Jones, R. , Syndikus, I. , Ralph, C. , Jain, S. , Varughese, M. , Parikh, O. , Crabb, S. , Robinson, A. , McLaren, D. , Birtle, A. , Tanguay, J. , Miranda, S. , Figueiredo, I. , Seed, G. , … de Bono, J. S. (2020). Olaparib in patients with metastatic castration‐resistant prostate cancer with DNA repair gene aberrations (TOPARP‐B): a multicentre, open‐label, randomised, phase 2 trial. The Lancet Oncology, 21(1), 162–174. 10.1016/s1470-2045(19)30684-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto, A. , Takahashi, Y. , Chang, H. Y. , Wu, Y. W. , Yamamoto, A. , Ishihama, Y. , & Takakura, Y. (2020). Blood concentrations of small extracellular vesicles are determined by a balance between abundant secretion and rapid clearance. Journal of Extracellular Vesicles, 9, 1696517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico, D. , Isserlin, R. , Stueker, O. , Emili, A. , & Bader, G. D. (2010). Enrichment map: A network‐based method for gene‐set enrichment visualization and interpretation. PLoS One, 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollison, P. L. (2000). The introduction of citrate as an anticoagulant for transfusion and of glucose as a red cell preservative. British J ournal of Haematology, 108, 13–18. [DOI] [PubMed] [Google Scholar]
- Montoro‐García, S. , Shantsila, E. , Orenes‐Piñero, E. , Lozano, M. , & Lip, G. Y. H. (2012). An innovative flow cytometric approach for small‐size platelet microparticles: Influence of calcium. Thrombosis and Haemostasis, 108, 373–383. [DOI] [PubMed] [Google Scholar]
- Mulcahy, L. A. , Pink, R. C. , & Carter, D. R. F. (2014). Routes and mechanisms of extracellular vesicle uptake. Journal of Extracellular Vesicles, 3, 00. Preprint at 10.3402/jev.v3.24641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, R. , Whitney, S. E. , & Wilkinson, S. (2018). Compositions for stabilizing DNA, RNA, and proteins in blood and other biological samples during shipping and storage at ambient temperatures (US9999217B2). U.S. Patent and Trademark Office. https://worldwide.espacenet.com/patent/search/family/045559995/publication/US9999217B2?q=US9999217
- Onódi, Z. , Pelyhe, C. , Terézia Nagy, C. , Brenner, G. B. , Almási, L. , Kittel, Á. , Manček‐Keber, M. , Ferdinandy, P. , Buzás, E. I. , & Giricz, Z. (2018). Isolation of high‐purity extracellular vesicles by the combination of iodixanol density gradient ultracentrifugation and bind‐elute chromatography from blood plasma. Frontiers in Physiology, 9, 10.3389/fphys.2018.01479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palviainen, M. , Saraswat, M. , Varga, Z. , Kitka, D. , Neuvonen, M. , Puhka, M. , Joenväärä, S. , Renkonen, R. , Nieuwland, R. , Takatalo, M. , & Siljander, P. R. M. (2020). Extracellular vesicles from human plasma and serum are carriers of extravesicular cargo—Implications for biomarker discovery. PLoS One, 15(8), e0236439. 10.1371/journal.pone.0236439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol, Y. , Csordas, A. , Bai, J. , Bernal‐Llinares, M. , Hewapathirana, S. , Kundu, D. J. , Inuganti, A. , Griss, J. , Mayer, G. , Eisenacher, M. , Pérez, E. , Uszkoreit, J. , Pfeuffer, J. , Sachsenberg, T. , Yılmaz, Ş. , Tiwary, S. , Cox, J. , Audain, E. , Walzer, M. , … Vizcaíno, J. A. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Research, 47(D1), D442–D450. 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plé, H. , Landry, P. , Benham, A. , Coarfa, C. , Gunaratne, P. H. , & Provost, P. (2012). The repertoire and features of human platelet microRNAs. PLoS ONE, 7(12), e50746. 10.1371/journal.pone.0050746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand, J. , Isserlin, R. , Voisin, V. , Kucera, M. , Tannus‐Lopes, C. , Rostamianfar, A. , Wadi, L. , Meyer, M. , Wong, J. , Xu, C. , Merico, D. , & Bader, G. D. (2019). Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nature Protocols, 14(2), 482–517. 10.1038/s41596-018-0103-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand, J. , Kull, M. , Peterson, H. , Hansen, J. , & Vilo, J. (2007). g:Profiler–a web‐based toolset for functional profiling of gene lists from large‐scale experiments. Nucleic Acids Research, 35, W193–W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer, U. , & Prohaska, R. S. (2001). flotillin‐1, and flotillin‐2 are major integral proteins of erythrocyte lipid rafts. Blood, 97, 00. [DOI] [PubMed] [Google Scholar]
- Schoenwaelder, S. M. , Yuan, Y. , Josefsson, E. C. , White, M. J. , Yao, Y. , Mason, K. D. , O'Reilly, L. A. , Henley, K. J. , Ono, A. , Hsiao, S. , Willcox, A. , Roberts, A. W. , Huang, D. C. S. , Salem, H. H. , Kile, B. T. , & Jackson, S. P. (2009). Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood, 114(3), 663–666. 10.1182/blood-2009-01-200345 [DOI] [PubMed] [Google Scholar]
- Simonsen, J. B. (2017). What are we looking at? Extracellular vesicles, lipoproteins, or both? Circulation Research, 121, 920–922. [DOI] [PubMed] [Google Scholar]
- Simundic, A.‐M. , Bölenius, K. , Cadamuro, J. , Church, S. , Cornes, M. P. , van Dongen‐Lases, E. C. , Eker, P. , Erdeljanovic, T. , Grankvist, K. , Guimaraes, J. T. , Hoke, R. , Ibarz, M. , Ivanov, H. , Kovalevskaya, S. , Kristensen, G. B. B. , Lima‐Oliveira, G. , Lippi, G. , von Meyer, A. , … Nybo, M. (2018). Joint EFLM‐COLABIOCLI Recommendation for venous blood sampling. Clinical Chemistry and Laboratory Medicine (CCLM), 56(12), 2015–2038. 10.1515/cclm-2018-0602 [DOI] [PubMed] [Google Scholar]
- Subramanian, A. , Tamayo, P. , Mootha, V. K. , Mukherjee, S. , Ebert, B. L. , Gillette, M. A. , Paulovich, A. , Pomeroy, S. L. , Golub, T. R. , Lander, E. S. , & Mesirov, J. P. (2005). Gene set enrichment analysis: A knowledge‐based approach for interpreting genome‐wide expression profiles. Proceedings of the National Academy of Sciences, 102(43), 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J.‐M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , … Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles, 7(1), 1535750. 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulkens, J. , de Wever, O. , & Hendrix, A. (2020). Analyzing bacterial extracellular vesicles in human body fluids by orthogonal biophysical separation and biochemical characterization. Nature Protocols, 15, 40–67. [DOI] [PubMed] [Google Scholar]
- Tyanova, S. , Temu, T. , Sinitcyn, P. , Carlson, A. , Hein, M. Y. , Geiger, T. , Mann, M. , & Cox, J. (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods, 13(9), 731–740. 10.1038/nmeth.3901 [DOI] [PubMed] [Google Scholar]
- Uhlen, M. , Fagerberg, L. , Hallström, B. M. , Lindskog, C. , Oksvold, P. , Mardinoglu, A. , Sivertsson, Å. , Kampf, C. , Sjöstedt, E. , Asplund, A. , Olsson, I. , Edlund, K. , Lundberg, E. , Navani, S. , Szigyarto, C. A. , Odeberg, J. , Djureinovic, D. , Takanen, J. O. , Hober, S. , … Pontén, F. (2015). Tissue‐based map of the human proteome. Science (1979), 347, 1260419–1260419. [DOI] [PubMed] [Google Scholar]
- van der Pol, E. , de Rond, L. , Coumans, F. A. W. , Gool, E. L. , Böing, A. N. , Sturk, A. , Nieuwland, R. , & van Leeuwen, T. G. (2018). Absolute sizing and label‐free identification of extracellular vesicles by flow cytometry. Nanomedicine: Nanotechnology, Biology and Medicine, 14(3), 801–810. 10.1016/j.nano.2017.12.012 [DOI] [PubMed] [Google Scholar]
- Van Deun, J. , Mestdagh, P. , Agostinis, P. , Akay, Ö. , Anand, S. , Anckaert, J. , Martinez, Z. A. , Baetens, T. , Beghein, E. , Bertier, L. , Berx, G. , Boere, J. , Boukouris, S. , Bremer, M. , Buschmann, D. , Byrd, J. B. , Casert, C. , Cheng, L. , … Hendrix, A. (2017). EV‐TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nature Methods, 14(3), 228–232. 10.1038/nmeth.4185 [DOI] [PubMed] [Google Scholar]
- Veerman, R. E. , Teeuwen, L. , Czarnewski, P. , Güclüler Akpinar, G. , Sandberg, A. , Cao, X. , Pernemalm, M. , Orre, L. M. , Gabrielsson, S. , & Eldh, M. (2021). Molecular evaluation of five different isolation methods for extracellular vesicles reveals different clinical applicability and subcellular origin. Journal of Extracellular Vesicles, 10(9). Portico. 10.1002/jev2.12128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergauwen, G. , Dhondt, B. , Van Deun, J. , De Smedt, E. , Berx, G. , Timmerman, E. , Gevaert, K. , Miinalainen, I. , Cocquyt, V. , Braems, G. , Van den Broecke, R. , Denys, H. , De Wever, O. , & Hendrix, A. (2017). Confounding factors of ultrafiltration and protein analysis in extracellular vesicle research. Scientific Reports, 7(1), 10.1038/s41598-017-02599-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergauwen, G. , Tulkens, J. , Pinheiro, C. , Avila Cobos, F. , Dedeyne, S. , De Scheerder, M. , Vandekerckhove, L. , Impens, F. , Miinalainen, I. , Braems, G. , Gevaert, K. , Mestdagh, P. , Vandesompele, J. , Denys, H. , De Wever, O. , & Hendrix, A. (2021). Robust sequential biophysical fractionation of blood plasma to study variations in the biomolecular landscape of systemically circulating extracellular vesicles across clinical conditions. Journal of Extracellular Vesicles, 10(10), Portico. 10.1002/jev2.12122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno, J. A. , Deutsch, E. W. , Wang, R. , Csordas, A. , Reisinger, F. , Ríos, D. , Dianes, J. A. , Sun, Z. , Farrah, T. , Bandeira, N. , Binz, P.‐A. , Xenarios, I. , Eisenacher, M. , Mayer, G. , Gatto, L. , Campos, A. , Chalkley, R. J. , Kraus, H.‐J. , Albar, J. P. , … Hermjakob, H. (2014). ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nature Biotechnology, 32(3), 223–226. 10.1038/nbt.2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisgrill, L. , Lamm, C. , Hartmann, J. , Preißing, F. , Dragosits, K. , Bee, A. , Hell, L. , Thaler, J. , Ay, C. , Pabinger, I. , Berger, A. , & Spittler, A. (2016). Peripheral blood microvesicles secretion is influenced by storage time, temperature, and anticoagulants. Cytometry Part A, 89(7), 663–672. Portico. 10.1002/cyto.a.22892 [DOI] [PubMed] [Google Scholar]
- Wiśniewski, J. R. , Zougman, A. , Nagaraj, N. , & Mann, M. (2009). Universal sample preparation method for proteome analysis. Nature Methods, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- Witwer, K. W. , Buzás, E. I. , Bemis, L. T. , Bora, A. , Lässer, C. , Lötvall, J. , Nolte‐‘t Hoen, E. N. , Piper, M. G. , Sivaraman, S. , Skog, J. , Théry, C. , Wauben, M. H. , & Hochberg, F. (2013). Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. Journal of Extracellular Vesicles, 2(1), 20360. 10.3402/jev.v2i0.20360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, K. , Li, X. , & Ma, Y. (2006). Paraformaldehyde induces elevation of intracellular calcium and phosphatidylserine externalization in platelets. Thrombosis Research, 117, 537–542. [DOI] [PubMed] [Google Scholar]
- Yuana, Y. , Bertina, R. M. , & Osanto, S. (2011). Pre‐analytical and analytical issues in the analysis of blood microparticles. Thrombosis and Haemostasis, 105, 396–408. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Borg, E. G. F. , Liaci, A. M. , Vos, H. R. , & Stoorvogel, W. (2020). A novel three step protocol to isolate extracellular vesicles from plasma or cell culture medium with both high yield and purity. Journal of Extracellular Vesicles, 9, 1791450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Zhao, Y. , Li, Y. , Chen, P. , Li, S. , Luo, J. , & Xia, H. (2019). Performance comparison of blood collection tubes as liquid biopsy storage system for minimizing cfDNA contamination from genomic DNA. Journal of Clinical Laboratory Analytical, 33, e22670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. Analysis of 564 EV‐TRACK knowledgebase1 records on blood‐derived EV. a. The most frequently used blood collection tubes for EV research. b. Study aims in blood‐derived EV research.
Supplementary figure 2. The number of BCT and performance metrics evaluated by other BCT performance studies and this study.
Supplementary figure 3. Illustrative overview of EV separation and analysis. After blood sample collection, serum or PDP is prepared by serial centrifugation. Hematology analysis is performed on whole blood. Particle analysis (EV subtypes, lipoprotein particles (presumably chylomicrons), residual platelets) is performed by flow cytometry on processed blood (serum or PDP) without additional EV separation. For rEV recovery analysis, a crude plasma EV preparation is obtained by size‐exclusion chromatography (SEC). For integrative omics analysis (LC‐MS/MS and small RNAseq), EV are separated from serum or PDP by the orthogonal implementation of SEC and ODG. * rEV spike‐in in whole blood samples is only performed for rEV recovery analysis.
Supplementary figure 4. Characterization of EV preparations separated from plasma using SEC (left) and SEC + ODG (right). a. NTA of EV‐enriched fractions. NTA calculated size distributions are depicted as mean (black line) with standard error (red area) and total particle number, mean particle size and mode are shown for each EV‐enriched fraction. b. Western blot analysis for EV‐ (Flotillin‐1) and non‐EV (Apolipoprotein A1) associated proteins. c. Transmission electron microscopy of EV‐enriched fractions (scale bar: 200 nm). White arrows indicate EV and black arrows indicate lipoprotein particles. d. Technical evaluation of methodological repeatability of SEC+ODG separation of citrate plasma EV (PEV) by mass spectrometry‐based proteomic analysis (LC‐MS/MS) of 5 technical replicates (each replicate indicated by number). LC‐MS/MS data from EV‐enriched fractions are compared by correlation analysis. Data are provided as Pearson's r coefficients.
Supplementary figure 5. Impact of BCT type (n=10) on hematology testing of full blood samples prior to sample preparation (n=10). Data are depicted as individual values with means. The asterisk indicates a statistically significant difference compared to the reference BCT (EDTA). a. Leukocyte (103/μL), erythrocyte (106/μL) and platelet (103/μL) count. b. Mean cellular volume (fL).
Supplementary figure 6. Impact of BCT type (n=10) on the ratio of activated platelet EV (CD61+Lac+; RI<1.42) to total EV (RI<1.42), measured by flow cytometry (n=10). Data are depicted as individual values with means.
Supplementary figure 7. Impact of BCT type (n=10) on EV (RI<1.42) size distribution, measured by flow cytometry (n=10). Data are provided as the decay constant (Mu) of the size distribution slope. Data are depicted as individual values with means.
Supplementary figure 8. Principal component analysis of EV proteome (LC‐MS/MS) data impacted by BCT type. Data points are colored according to biological replicate. Serum (red) and Roche cfDNA (pink) clusters are indicated by colored ellipses.
Supplementary figure 9. GSEA of EV proteome (LC‐MS/MS) data impacted by BCT type. a. The serum EV proteome is enriched in a phenotype corresponding to platelet activation, signaling and aggregation b. The Roche cfDNA EV proteome is enriched in a phenotype corresponding to the erythrocyte membrane and cytosol. Significance threshold was set at FDR < 0.05. The heat map corresponds to the enrichment of phenotypic proteins in individual samples.
Supplementary figure 10. Principal component analysis of EV miRNA (small RNA seq) data impacted by BCT type. Data points are colored according to biological replicate. Serum (red) and Roche cfDNA (pink) clusters are indicated by colored ellipses.
Supplementary figure 11. Impact of BCT type (n=5) on hematology testing of full blood samples prior to sample preparation (n=5) after 60 min (T1), 8h (T2) and 72h (T3) (BPI evaluation). Data are depicted as individual values with means. The asterisk indicates a statistically significant difference compared to the reference BPI (T1). a. Leukocyte (103/μL), erythrocyte (106/μL) and platelet (103/μL) count. b. Mean cellular volume (fL). c. Residual platelet (103/μL) count on hematology testing after plasma preparation.
Supplementary figure 12. GSEA of EV proteome (LC‐MS/MS) data (BPI evaluation). a. The EV proteome at T3 is enriched in a phenotype corresponding to platelet activation, signaling and aggregation compared to T1. b. The EV proteome at T3 is enriched in a phenotype corresponding to the erythrocyte membrane and cytosol compared to T1. Significance threshold was set at FDR < 0.05. The heat map corresponds to the enrichment of phenotypic proteins in individual samples.
Supplementary figure 13. Impact of BCT type (n=5) on the ratio of a. Activated platelet EV (CD61+Lac+; RI<1.42) to total EV (RI<1.42), b. Non‐platelet EV (CD61‐Lac+; RI<1.42) to total EV (RI<1.42) and c. Erythrocyte EV (CD235+; RI<1.42) to total EV (RI<1.42), measured by flow cytometry (n=5) in plasma 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation). Data are depicted as individual values with means.
Supplementary figure 14. Impact of BCT type (n=5) on the EV proteome, analyzed by LC‐MS/MS (n=3) in EV‐enriched fractions prepared from plasma 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation). The EV proteome at T2 and T3 is compared to the EV proteome at T1 by correlation analysis. Data are provided as Pearson's r coefficients and are depicted as individual values with means.
Supplementary figure 15. Impact of BCT type (n=5) on the EV miRNA profile, analyzed by small RNAseq (n=3) in EV‐enriched fractions prepared from plasma 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation). The EV miRNA profile at T2 and T3 is compared to the EV miRNA profile at T1 by correlation analysis. Data are provided as Pearson's r coefficients and are depicted as individual values with means.
Supplementary figure 16. Impact of BCT type (n=5) on the volume of plasma 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation).
Supplementary table 1. Donor characteristics. Clinical chemistry data were obtained by hematology analysis of EDTA blood 60 min after venipuncture. Data are depicted as mean values with standard deviation. Abbreviations: MCV (mean cellular volume), MCH (mean cellular hemoglobin), MCHC (mean cellular hemoglobin concentration).
Supplementary table 2. Patient characteristics. Abbreviations: RP: radical prostatectomy, (s)PLND: (salvage) pelvic lymph node dissection, PSA: prostate specific antigen.
Supplementary table 3. Impact of BCT type (n=10) on hematology testing of full blood samples prior to sample preparation (n=10). Data are depicted as mean values with standard deviation.
Supplementary table 4. Impact of BCT type (n=10) on sample quality. Analyses were performed on serum and platelet‐depleted plasma (PDP), prepared 60 min after blood draw (n=10). Data are depicted as mean values with standard deviation.
Supplementary table 5. Impact of BCT type (n=10) on ex‐vivo release of blood‐cell derived EV subtypes. Analyses were performed on serum and platelet‐depleted plasma (PDP), prepared 60 min after blood draw (n=10). Data are depicted as mean values with standard deviation.
Supplementary table 6. List of proteins detected by mass spectrometry (LC‐MS/MS) across all samples in the BCT evaluation phase.
Supplementary table 7. List of miRNA detected by small RNA sequencing across all samples in the BCT evaluation phase.
Supplementary table 8. Impact of BCT type (n=5) on hematology testing of full blood samples prior to sample preparation (n=5), 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation). Data are depicted as mean values with standard deviation.
Supplementary table 9. Impact of BCT type (n=5) on rEV recovery, sample quality and ex‐vivo release of blood‐cell derived EV (n=5), 60 min (T1), 8h (T2) and 72h (T3) following sample collection (BPI evaluation). Data are depicted as mean values with standard deviation.
Supplementary table 10. List of proteins detected by mass spectrometry (LC‐MS/MS) across all samples in the BPI evaluation phase.
Supplementary table 11. List of miRNA detected by small RNA sequencing across all samples in the BPI evaluation phase.
Supporting Information
Supporting Information
Supporting Information