

Abstract
The isolation and purification of protactinium from uranium materials is essential for 231Pa–235U radiochronometry, but separating Pa from uranium-niobium alloys, a common material in the nuclear fuel cycle, is challenging due to the chemical similarity of Pa and Nb. Here we present three resin chromatography separation techniques for isolating Pa from U and Nb which were independently developed by three different laboratories through ad hoc adaptations of standard operating procedures. Our results underscore the need for and value of purification methods suitable for a diversity of uranium-based materials to ensure the operational readiness of nuclear forensics laboratories.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10967-023-08928-y.
Keywords: Radiochronometry, Nuclear forensics, Uranium alloy, 231Pa/235U, Actinide separations, Protactinium
Introduction
When nuclear material is found outside of regulatory control, questions surrounding its source, destination, and suspected use are of immediate concern. In this context, the material’s production date or age is a key forensic signature that may provide insights on its origins and history for law enforcement and nuclear security investigations. To assess the age of nuclear material, nuclear forensic analysts apply a technique known as radiochronometry, in which the abundances of specific radionuclides and their decay products are measured to approximate the time elapsed since the material’s last chemical purification. The daughter-parent isotope pairs 230Th–234U and 231Pa–235U are the most common chronometers used to estimate the age for bulk uranium materials and can be applied to materials made since the advent of the nuclear age (circa 1940s). Over the past two decades, as radiochronometry has become more routine amongst nuclear forensics laboratories, these chronometers have played pivotal roles in the analysis of several nuclear smuggling incidents worldwide [1–7].
While advances in analytical instrumentation, sample purification approaches, calibration methods, and error quantification have led to increasingly accurate and precise isotopic measurements, caution must be exercised in the interpretation of calculated ages for forensic investigations. The suitability of a radiochronometric age as a proxy for the true date of production depends on two conditions: (1) the material was completely purified of progeny isotopes at the time of production, and (2) the material has remained a closed system, with no parent or progeny isotopes gained or lost since its production. Such conditions are not always met in practice. For example, incomplete initial purification or post-production addition of daughter products has been implied in some uranium materials manufactured at the industrial scale, causing measured apparent ages to be biased old by several months or years relative to the true or known production age [2, 8–13]. In some cases, multiple chronometers applied to the same material have yielded repeatably different or “discordant” ages, indicating that the degree to which ideal model conditions are satisfied within a single sample can vary by chronometer [14, 15]. For these reasons, measured ages of produced nuclear materials are customarily referred to as “model ages” to emphasize the assumptions required to interpret age estimates as authentic records of a sample’s history.
These examples highlight the importance of applying multiple chronometers to the same sample to enhance the descriptive power of model ages, especially for materials with higher levels of impurities or complex production histories. If model ages from different chronometers agree within uncertainty or are “concordant,” then they can be more confidently interpreted as true production ages, since age agreement between multiple unique progeny nuclides in a perturbed system would require an exceptional coincidence. While discordant model ages are more difficult to accurately interpret, their occurrence counteracts inaccurate conclusions that may be drawn from a single chronometer in the absence of other radiochronometric data [16]. In addition, discordance may also provide more nuanced information about a sample’s history; the observation that discordance is more common in materials with impurities and complex production histories suggests that these aspects have an influence on progeny nuclide fractionation [14, 15]. Thus, with further research, characteristic patterns of discordance could be harnessed as diagnostic signatures of specific uranium manufacturing processes involved in an unknown material’s production, such as bomb reduction, metal casting, remelting, recycling, or alloying with other elements [17, 18].
To this end, here we present new analytical methods that were developed for an investigation of 231Pa–235U and 230Th–234U model age discordance in uranium-niobium metal alloys. This work represents the first published attempt to analyze U–Nb metal alloys for radiochronometry and was conducted as part of a multi-laboratory collaboration between the Atomic Weapons Establishment (AWE), Lawrence Livermore National Laboratory (LLNL), and Los Alamos National Laboratory (LANL), as described in Higginson et al. [18]. For this collaboration, AWE distributed to each laboratory a set of depleted uranium metals which included two U–Nb metal alloys cast by vacuum induction melting and vacuum arc remelting, two common industrial-scale uranium casting processes [19]. U–Nb metal alloys possess properties that are more desirable for various applications compared to their pure uranium metal counterparts, such as oxidation and corrosion resistance and improved ductility for shaping into specific geometries [20–22]. These samples were selected because documentation of their casting history was available [18]. We refer the reader to the companion article by Higginson et al. (2022) for a detailed description of the U–Nb metal alloy production histories and discussion of model age discordance in cast uranium metals.
The analysis of the depleted U–Nb metal alloys posed a technical challenge, particularly for 231Pa–235U radiochronometry. First and foremost, because Pa and Nb are transition metal homologues with similar chemical properties and behavior in various acidic aqueous solutions [23], standard operating procedures for Pa isolation designed for pure uranium materials are ineffective for the removal of Nb. Preparing highly purified analytical aliquots is essential for accurate and precise isotopic measurements by mass spectrometry, as the presence of excessive matrix elements can introduce effects that distort peak shapes, create isobaric interferences, and compromise external mass bias corrections. Second, due to the low 235U content of these depleted U–Nb samples (~ 0.2–0.3 atom percent 235U), larger sample aliquot masses are needed for Pa assay determination. Because the same sample aliquot is processed for both Pa and Th assay, the concentration of 231Pa is often the controlling factor on aliquot size, as it is typically far lower than 230Th concentrations due to the half-lives of 235U and 234U (~ 700 million years compared to ~ 250,000 years, respectively). Therefore, these depleted U–Nb metal alloys require aliquot sizes that are ~ 1–3 orders of magnitude larger than those needed for highly-enriched uranium materials, increasing the amount of Nb for removal and complicating separation schemes optimized for smaller aliquot sizes (Fig. 1). Lastly, the immersion of certain uranium metal alloys like U–Nb in specific acids can pose an explosive risk if not handled properly [24], emphasizing the need to approach the dissolution of the U–Nb metal alloys with care.
Fig. 1.

(a) The ease of applying 231Pa–235U radiochronometry to a uranium sample depends on its 235U content. The samples analyzed in this study are depleted uranium, containing ~ 0.2 atom percent 235U. (b) For a 1 mg sample aliquot, the cumulative ingrowth of 231Pa in depleted uranium samples is insufficient for precise measurements via mass spectrometry, assuming standard instrument operating conditions. (c) Sample aliquot size must be increased to compensate for the lack of 231Pa in depleted uranium samples. The amount needed is 10- to 100-fold greater than the amount required for more enriched samples. (d) The challenge of applying 231Pa–235U radiochronometry to U–Nb alloys lies in isolating ultra-trace level amounts of Pa from weight-percent abundances of Nb and U.
The combination of these factors—the depleted uranium isotopic composition, significant presence of Nb, and potential digestion hazards of these samples—required the development of methods that could reliably dissolve gram-sized subsamples and separate ultra-trace levels of Pa from weight-percent levels of Nb (i.e., a starting atomic ratio of Nb/Pa ≈ 108–109; Fig. 1d). In this paper, we describe each laboratory’s approach to this technical challenge. Although each sample digestion and Pa separation scheme was independently developed and tailored to the instrumentation, facilities, and workflow of each laboratory at the time of this study, ultimately, all three methods were viable and produced 231Pa–235U model ages that were internally self-consistent by laboratory.
Method development
After AWE identified uranium materials satisfying the criteria for the interlaboratory investigation [18], subsamples (~ 0.5–1.0 g) of two unique depleted U–Nb alloys, Alloy-1 and Alloy-2, were prepared and distributed to each participating laboratory (Fig. 2). All laboratories subsequently digested these subsamples to create one homogeneous sample solution per alloy, which were then used for 231Pa and 235U concentration measurements by isotope dilution mass spectrometry (IDMS) using 233Pa and 233U isotope tracers. A variety of factors influenced the development of procedures at each laboratory uniquely: the 4- to 6-month timeline to complete the interlaboratory comparison, supply chain issues, and laboratory space occupancy limits imposed in response to the COVID-19 pandemic. To accommodate these factors, instead of developing entirely new separation schemes from scratch, all laboratories modified their existing standard operating procedures to accommodate the U–Nb matrix. In the following subsections, we describe each laboratory’s approach to these non-routine samples and report the separation schemes ultimately applied to determine 231Pa–235U model ages for Alloy-1 and Alloy-2. The implications of the model age results are further discussed in Higginson et al. (2022) [18].
Fig. 2.

Optical microscopy images of the two depleted U–Nb alloy subsamples involved in this study, (a) Alloy-1 (0.54 g) and (b) Alloy-2 (0.62 g), from Higginson et al. (2022) [18]. Both subsamples exhibit a colorful interference surface oxide layer that is characteristic of U–Nb alloys [22] as well as tool marks consistent with known sampling procedures (e.g., indentations from the use of clamps or pliers, saw marks). Black rectangles are 1 mm scale bars
Atomic Weapons Establishment
Like those at other laboratories, the standard operating procedures for sample digestion and radiochronometry at AWE were independently developed, tested, and optimized on uranium materials of high purity [25]. For such routine materials, samples are readily digested in nitric acid (HNO3) and then redissolved in concentrated hydrochloric acid (HCl) to create a primary sample solution. Because both 230Th–234U and 231Pa–235U model ages are generally desired for a single sample, AWE’s standard operating procedure obtains both 230Th and 231Pa assays from the same aliquot of primary solution. An aliquot containing at least 109 atoms of the 230Th and 231Pa daughter isotopes is spiked with the appropriate amounts of 229Th and 233Pa tracer. This spiked sample aliquot then undergoes a combined U, Th, and Pa separation through a stack of three columns attached to a vacuum-assisted flow chamber, which increases the flow rate of column elutions compared to gravity. Each of the three columns contains a distinct extraction chromatography resin that adsorbs one of the three elements of interest: Pa is adsorbed to the TK400 resin (TrisKem International) in the first column; U is adsorbed to the AG1-X8 anion exchange resin in the second column; and Th is adsorbed to the UTEVA resin in the last column. Each column is then separated from the stack and used in a second column purification step with the same resin for further removal of contaminants. For example, for additional purification of the Pa fraction, the TK400 resin column with the adsorbed Pa is subsequently stacked on top of another TK400 resin column. The combined use of a vacuum-assisted flow chamber and this stacked column technique, which eliminates time-consuming evaporative dry down steps, makes the execution of this purification scheme relatively rapid. Purified sample aliquots of U, Pa, and Th can be ready for mass spectrometric analysis within several hours [25]. Additionally, the elimination of dry down stages reduces Pa loss due to hydrolysis [23], maximizing Pa chemical recovery.
AWE modified these standard operating procedures for the U–Nb alloys. For sample digestion, instead of readily dissolving the sample in HNO3, the sample was first treated with aqua regia. Once the initial vigorous reaction subsided, the sample solution was then heated on a hotplate for several hours. Next, the solution was removed from the hotplate and further treated with concentrated sulfuric acid heated below its boiling point. Sulfuric acid was gradually added in 1- to 2-mL increments until the solution no longer exhibited a cloudy green appearance. The solution was then heated until it produced dense white fumes instead of the characteristically deep red nitrogen dioxide vapors. After cooling and checking the solution for undissolved residues, the solution was then completely dried down and redissolved in concentrated HCl (~ 11–12 M) for subsequent aliquoting.
For Pa separations of the U–Nb alloys, AWE modified the standard procedure by supplementing the TK400 resin column with a ZR resin (TrisKem International) column. Although the TK400 resin is excellent for isolating Pa in bulk U materials due to its high retention of Pa and low retention of U and Th in high HCl concentrations [26], TK400 resin also retains Nb in these conditions. Therefore, an additional step is necessary to separate Pa from Nb. AWE determined the distribution coefficients (Dw) of Pa and Nb in ZR resin with 5.5 M HCl and varying concentrations of HF, from 0.001 M up to 1 M. While both Pa and Nb strongly adsorb to ZR resin in HCl, in mixed HCl–HF solutions, Pa forms fluoride complexes more easily than Nb that do not adsorb to the ZR resin [27, 28]. The addition of 0.1 M HF to 5.5 M HCl reduced the Dw of Pa from > 104 to 1 while the Dw of Nb remained high at ~103. Under these conditions, Pa readily elutes while Nb remains adsorbed to the resin (Fig. 3).
Fig. 3.
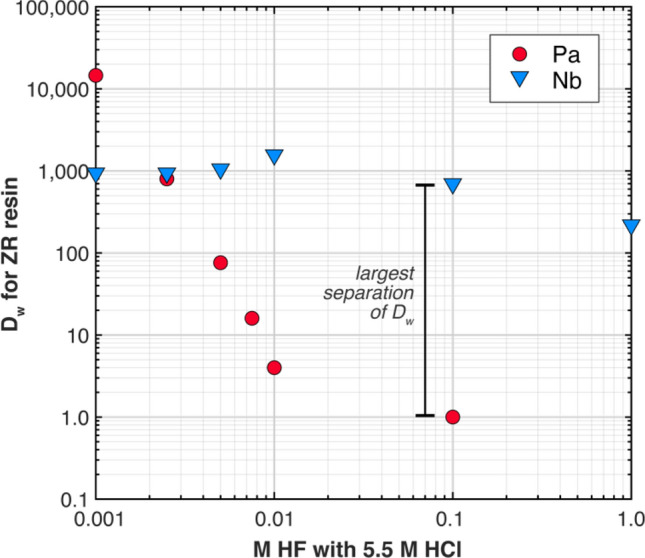
Measured distribution coefficients (Dw) for Pa (red circles) and Nb (blue triangles) on ZR resin indicate that a good separation between Pa and Nb can be achieved with an eluent of 5.5 M HCl + 0.1 M HF
As a result of these adsorption experiments, AWE added a column of ZR resin to their separation scheme for Pa purifications (Fig. 4). After the Pa and Nb are retained on the TK400 resin column of the triple stack sequence, the TK400 column is then removed and placed on top of a ZR resin column. Both Pa and Nb are then transferred to the ZR resin with 5.5 M HCl, which is then stacked on top of an AG1-X8 column for the final separation step. Pa is readily eluted from the ZR resin with 5.5 M HCl + 0.1 M HF while Nb is retained on the resin, and any remnant U is removed from the Pa elution with the AG1-X8 column. To remove potential organics stripped from resins, the Pa elution was dried down with concentrated HNO3 (~15–16 M) and hydrogen peroxide (H2O2) before mass spectrometry.
Fig. 4.

Comparison of AWE’s (a) standard operating procedure for Pa separations on pure U metals [25] and (b) the modified procedure for depleted U–Nb alloys. The columns are Rockbourne R1010 medium columns and are stacked on top of a vacuum-assisted flow chamber. The red color of the eluates represents solutions of purified Pa, the element of interest. This procedure has also been successfully executed using pre-packed resin cartridges. To highlight the differences between the two procedures, certain steps such as resin cleaning, column conditioning, and acid washes have been abridged or omitted from this schematic; please refer to the Higginson et al. (2018) [25] and the Supplementary Materials for complete descriptions of the standard and modified procedures
While the total Pa recovery of pure uranium metals through the standard operating procedure is consistently greater than 90%, the Pa recovery of the U–Nb alloys using this modified procedure was reduced and variable, ranging between 20 and 50%. Based on further separation experiments, some Pa loss was traced to the final AG1-X8 column, for unknown reasons. Despite the incomplete and variable recovery of Pa, the amount was sufficient for mass spectrometric analysis via peak jumping routines. A more detailed description of AWE’s modified Pa separation procedure for U–Nb alloys is available in the Supplementary Materials.
Lawrence Livermore National Laboratory
Prior to this study, LLNL had limited experience with performing 230Th–234U and 231Pa–235U radiochronometry on uranium alloys. Sample digestion and U, Th, and Pa separation procedures for bulk uranium were originally developed and optimized for pure uranium metals, oxides, and other pure compounds [10, 29]. Although these standard protocols were mostly untested for other sample matrices, LLNL’s approach to method development began with applying standard operating procedures to the U–Nb alloys and observing departures from ideal sample behavior and isotope recovery. Modifications to standard techniques were then made as needed.
The first irregularity in sample behavior occurred during sample digestion. LLNL aimed to create a single primary solution for Alloy-1 and Alloy-2 by dissolving in their entirety the pieces received from AWE (Fig. 2). In LLNL’s standard procedure [29], a metal sample is first acid leached to remove surface contaminants and oxidation layers, and then subsequently acid digested in concentrated HNO3. Typical metal samples dissolve readily in this initial acid solution. This primary solution is then diluted to 4 M HNO3. HF is then added to achieve a concentration of 0.05 M HF in order to stabilize Th and Pa in the solution [10]. The sample solution is then heated overnight at 120 °C to ensure complete dissolution. Although this procedure results in a primary solution with excellent chemical homogeneity for pure uranium materials, after performing this procedure with the U–Nb alloys, undissolved solids remained in the sample vial (Fig. 5). Since such solids did not previously form in solutions of dissolved pure uranium metals, these solids were thought to be a Nb-based compound. To dissolve these solids, HF was incrementally added to the primary solution and heated after each addition until no solids were evident upon visual inspection. Ultimately, complete dissolution of both U–Nb alloy samples was achieved with a primary solution matrix of 4 M HNO3 + 0.3 M HF (28 mL total solution volume); this acid matrix represents a six-fold increase in HF concentration compared to the primary solutions prepared using the standard protocol for pure uranium materials.
Fig. 5.

Undissolved solids formed during the sample digestion of (a) Alloy-1 and (b) Alloy-2. These photographs were taken after adding HNO3 but before adding any HF. The solids were at first gray in color, but eventually turned white after several hours of refluxing (see Fig. 10 and [30])
The next difficulty in processing the depleted U–Nb samples emerged when preparing the sample aliquots for the chemical separation and isolation of Th and Pa. Because both 230Th–234U and 231Pa–235U model ages are generally desired for a single sample, LLNL’s standard operating procedure obtains both 230Th and 231Pa assays from the same aliquot of primary solution [29]. Aliquot sizes are determined based on estimates of sample 231Pa and 230Th content and instrument sensitivity at the time of expected analysis (Fig. 1a–c). Based on information about the production dates and feedstocks of Alloy-1 and Alloy-2 gathered by AWE [18], LLNL calculated that an aliquot representing ~ 30–50 mg of original sample material was required. This aliquot size is one to two orders of magnitude larger than the size used for most routine analyses (Fig. 1b, c). Given the weight-percent level of Nb in these samples, this aliquot size also contained ~ 2–3 mg of Nb, compared to several picograms of 231Pa (Fig. 1d).
The presence of mg-level amounts of Nb created immediate obstacles for Pa and Th separations. Standard procedure dictates that the sample aliquot be dried down and fully redissolved in 9 M HCl for loading onto AG 1-X8 resin (100–200 mesh size; [29]), but a white precipitate formed in this acid matrix. The white solids were again hypothesized to be Nb-bearing precipitates, although this hypothesis was not verified by analysis of the precipitate material. Physical removal of these solids was considered, but at the time, it was unknown if the Nb precipitates also incorporated or even isotopically fractionated Pa, a cause for concern given the shared chemical properties of Nb and Pa. Because of this potential for Pa loss or fractionation, a separation scheme that did not involve physical removal of the Nb precipitates was desired.
To this end, because precipitates formed in both pure HCl and HNO3 acids, LLNL considered the use of other inorganic reagents. The sample loading solution would need to fully dissolve the sample and create anionic complexes with different adsorption behavior on a chromatography resin to enable chemical separation. Due to pandemic-related delays in supply chains, only resins already in-hand were viable for experimentation (e.g., AG 1-X8, UTEVA, TEVA). In consulting the actinide separation literature, the use of a pure HF solution as the sample loading solution appeared promising, especially after confirming that such solutions fully dissolve the sample when heated to 120 °C. Previously published separation experiments using Dowex 1, a strongly basic anion exchange resin recognized as equivalent to AG 1 [31], showed that Pa, Nb, and U strongly adsorb in low molarity HF solutions as anionic fluoride complexes (Fig. 6a; [32–34]). To then separate the adsorbed Pa from Nb and U, other previous experiments suggested that a mixed HCl–HF solution of high HCl and very low HF concentration may yield a sufficient separation (Fig. 6b; [34, 35]). In 9 M HCl and < 0.05 M HF acid, Nb and U strongly adsorb to the resin as strong anionic chloride or chloro-fluoro complexes, whereas Pa is essentially not adsorbed [34].
Fig. 6.

Published distribution coefficients (Kd) of U, Pa, and Nb with strongly basic anion exchange resin using (a) pure HF solution and (b) mixed HCl–HF solutions. A higher Kd indicates stronger adsorption. To separate two analytes, a difference in Kd of at least 100 is generally desired, in which the desorbing element has a Kd of less than 10 [31]. (a) Solids lines are from Faris [32] using Dowex 1-X10 resin in F− form whereas dotted lines are from Caletka and Krivan [33] using Dowex 1-X8 resin in Cl− form. (b) In 9 M HCl, Pa is very weakly adsorbent at every HF molarity, whereas U strongly adsorbs at every HF molarity. Nb strongly adsorbs at very low molarity HF. For > 0.05 M HF in 9 M HCl, Kd for Pa is less than 2, indicating no practical Pa adsorption to the resin. These data are extracted from Kim et al. [34] and Faris [35]. No information was found for the behavior of Th under these conditions in the previous literature
With the aim of exploiting these chemical behaviors to separate and isolate Pa, LLNL carried out a series of column separation experiments testing 1 M HF as the sample load matrix, AG 1-X8 as the anion exchanger, and various high [HCl]-low [HF] acids as the eluent that elutes Pa. These initial experiments focused on (1) determining whether the Pa, Nb, and U fluoride complexes adsorbed to the resin would behave like their chlorofluoro-complex counterparts in high [HCl]-low [HF] acids, allowing for the separation of Pa from Nb and U, and (2) characterizing the adsorption behavior of Th under such conditions. In the interest of conducting multiple experiments quickly and conserving the finite Alloy-1 and Alloy-2 sample solutions, in these initial trials, LLNL created synthetic sample solutions containing ng-levels of U, Th, and Nb from elemental standards and pg-levels of 233Pa milked from an in-house 237Np source [29]. For each column separation experiment, the recoveries of U, Th, and Nb were tracked by concentration measurements of 1 mL elution fractions made on an Agilent 8900 triple quadrupole ICP-MS. Eluted sample volumes were diluted in a 2% HNO3 + 0.005 M HF solution containing 1 ppb of indium to make internal corrections for instrument signal intensity drift. Isotope concentrations were calculated using a linear calibration curve based on measurements of certified external standards in a matching uranium matrix. The recovery of 233Pa was measured via gamma spectroscopy by comparing the peak area around the characteristic 312 keV gamma ray emission line of a collected elution fraction relative to that of a control solution containing 233Pa equal to the amount in the synthetic sample.
These initial trials showed that Pa, Nb, U, and Th are all retained on AG 1-X8 in 1 M HF, replicating earlier experiments in the literature and, to the authors’ best knowledge, revealing previously undocumented Th adsorption behavior. After loading the synthetic samples onto the column, pure 9 M HCl was the most effective eluent of the mixed HCl–HF solutions tested for Pa elution, producing the greatest separation between Pa and Nb while also fully eluting both Pa and Th in the same fraction with minimal U. This Pa behavior was surprising because when Pa is introduced to AG 1-X8 in pure 9 M HCl, it forms Pa chloride complexes that are instead strongly retained on the resin [36]. One possible explanation for this difference in Pa behavior may be related to the speciation of the Pa that is adsorbed to the resin. When Pa is introduced to the resin in 1 M HF, it is adsorbed to the resin as Pa fluoride complexes rather than Pa chloride complexes. When 9 M HCl is applied as an eluent, the Cl− counterions may have a greater affinity for the resin than Pa fluoride complexes, thereby causing Pa to elute. This scenario contrasts the Pa chloride complexes that would have formed if the sample were introduced in highly concentrated HCl, which do not readily exchange with Cl− counterions, as evidenced by their strong adsorption to the resin in HCl [36]. Another explanation is that residual HF from the sample load is mixing with the 9 M HCl to produce a mixed HCl–HF solution with very low HF concentration, which would cause Pa to desorb (Fig. 6b). The exact mechanism for this widely known and well-replicated Pa adsorption behavior in mixed HCl–HF solutions remains unclear, but the behavior is nonetheless advantageous for Pa separations. Regardless of the mechanistic underpinnings of Pa sorption behavior, these experimental results provide a practical means for reliably removing undesirable Nb and U from our analytes of interest, Pa and Th.
After determining that Pa and Th can be separated from U and Nb by loading the sample onto AG 1-X8 in 1 M HF and eluting with a 9 M HCl wash, the next experiments involved testing this separation scheme on the actual depleted U–Nb samples. Since the synthetic samples contained ~ 108 times less U compared to the ~ 30–50 mg aliquot sizes needed for 231Pa–235U radiochronometry, the scalability of the method required testing. Standard operating procedures typically use 1 mL of AG 1-X8 resin and 1 mL for the dissolved sample loading solution. However, these volumes were optimized for processing sub-mg-sized aliquots of U, raising concerns that much larger aliquot sizes could saturate or exceed the available ion exchange sites. Indeed, based on a comparison of evaporation products following two trials in which 33 mg sample aliquots were loaded onto 1 mL and 3 mL volumes of AG 1-X8 resin, the 1 mL resin bed exhibited evidence of resin saturation or capacity exceedance (Fig. 7). Thus, to accommodate the larger aliquot sizes needed for Alloy-1 and Alloy-2, the volumes of both the AG 1-X8 resin and sample loading solution were increased from 1 to 3 mL.
Fig. 7.

Observations of column bleed-through (a) and evaporative dry down products (b–c) for experiments using 1 mL and 3 mL of AG 1-X8 resin, in which the 1 M HF dissolved sample load is followed by washes of 9 M HCl. These observations suggest that the exchange capacity of 1 mL of resin was exceeded for the sample load (~33 mg of a U–Nb sample).
After scaling up the resin and sample loading solution volumes, LLNL tested the repeatability of the separation scheme on actual depleted U–Nb samples. In five separate trials, the recoveries of Pa, Nb, U, and Th were tracked in the same way as in the initial experiments, in which each 1 mL elution fraction was collected and analyzed. Strictly for the purposes of tracking Th yields, the samples were also doped with an additional ~20 ng of 232Th from an in-house standard prior to column loading, compensating for the intrinsically low Th content of the samples which would be difficult to measure otherwise due to ICP-MS detection limits. In these trials, the recovery of Pa was very consistent, with more than 95% of the Pa recovered within the first five 1 mL washes of 9 M HCl. Less than 1% of Nb and less than 0.1% of U was recovered in these elution fractions, indicating excellent separation. In addition, we observed that the elution peak for Nb occurred after the Pa elution peak by 2 mL (Fig. 8). The repeatability and near completeness of Pa recovery is likely related to the Pa fluoride complexes formed in the HF sample loading solution which are resistant to hydrolysis and precipitation [37].
Fig. 8.

Elution profiles of Pa (a, b), Th (c, d), Nb (e), and U (f) from five column separation experiments on Alloy-1 and Alloy-2, plotted as the cumulative volume of eluted acid versus the recovery of each element within each eluted fraction. All sample loads, sequence of acid eluents, and experimental conditions across the five tests were kept constant, varying only the sample used and the amount of time that the sample was refluxed on a 120 °C hotplate before column loading (see legend). Pa recovery was only measured for three experiments using Alloy-2
Although Pa recovery was consistent, Th recovery was much more variable, both in terms of total Th recovery and the fraction containing the peak of Th elution (Fig. 8c, d). We do not fully understand this behavior, but one explanation may be related to the amount of time that the dissolved sample was heated prior to column loading. We noticed that amongst experiments involving the same sample, Alloy-1 and Alloy-2, the aliquots that were heated for longer periods of time at 120 °C exhibited slower rates of Th elution compared to aliquots heated for shorter periods of time. We speculate that the formation of mixed Th chloro-fluoro-complexes may have complicated the retention and elution behaviors of Th, complications which are exacerbated the longer the aliquot is heated or in HF solution. Alternatively, microprecipitation of ThF4, a highly insoluble white precipitate, may decrease yields by causing Th to adhere to the resin, column or vial walls, or frits [38, 39]. Based on these observations, to maximize Th recovery, we recommend that the sample loading solution be heated only for the minimum time needed to fully dissolve the sample, not to exceed 20 min at 120 °C.
In summary, to extract Pa from the U–Nb alloys, LLNL first administered two AG1-X8 column steps with 1 M HF as the sample loading solution before applying the existing standard procedure for Pa purification (Fig. 9). A complete description of LLNL’s modified Pa separation procedure is available in the Supplementary Materials.
Fig. 9.

Comparison of LLNL’s (a) standard operating procedure for Pa separations on pure U metals [29] and (b) the modified procedure for depleted U–Nb alloys. LLNL’s standard operating procedure for Pa separations involves two columns of AG 1-X8, followed by one silica gel column. For depleted U–Nb alloys, LLNL added two additional AG 1-X8 columns to the beginning of the standard operating procedure. In the schematic, the taller columns represent Environmental Express columns (5.5 mL volume) and shorter columns represent Bio-Rad Poly-Prep columns (2 mL volume). In the eluates, colors represent the elution of Th (green) and Pa (red), with changing opacity representing sequential improvements to the purity of each element (i.e., dark red represents a more purified Pa solution). To highlight the differences between the two procedures, certain steps such as resin cleaning, column conditioning, acid washes, and the application of H3BO3 as a fluoride scavenger have been abridged or omitted from this schematic; please refer to Treinen et al. (2018) [29] and the Supplementary Materials of this article for complete descriptions of the standard and modified procedures
Los Alamos National Laboratory
LANL approached sample digestion and Pa separation method development for the U–Nb alloys in the same manner as the other laboratories, applying their standard operating procedures and adjusting protocols only as needed. Due to LANL and LLNL’s shared history as U.S.-based laboratories that have long collaborated on radiochronometry analyses, their standard operating procedures share many common components [29, 40]. The LANL standard procedure for sample digestion involves an initial acid leach to remove surface oxidation, followed by complete sample dissolution in 8 M HNO3 at 130 °C and then subsequent dilution to 4 M HNO3 + 0.005 M HF [40]. When applying this method to the U–Nb alloy samples, the same aforementioned white precipitates formed and remained. In order to dissolve these solids, LANL increased both the acid volume and the HF concentration used during sample digestion. As a result, the primary sample solutions for the U–Nb alloys were approximately twice the volume of those for routine U metals with similar sample mass (125 mL vs. 60 mL) and 20 times the standard concentration of HF (4 M HNO3 + 0.1 M HF vs. 4 M HNO3 + 0.005 M HF). Thus, LANL and LLNL independently arrived at similar strategies for U–Nb alloy sample digestion.
Despite similarities in approach for sample digestion, LANL developed a distinct strategy for Pa separations. Speculating that the white solids observed during initial sample dissolution were a precipitated Nb compound, LANL exploited the formation of these precipitates as a separation mechanism itself. Elemental concentration and gamma spectroscopy measurements of the solids confirmed that the precipitates contained ~ 98% of the total Nb load and < 1% of the loaded Pa [30]. Therefore, physical removal of the solids would effectively remove the vast majority of the Nb with minimal Pa loss. Leveraging this behavior, LANL purposely produced and then physically removed the white precipitates before applying additional chemical separation procedures using chromatography resins. After sample aliquoting and spiking for IDMS, the sample was dried down and redissolved in 9 M HCl to produce the white solids. The solution was then transferred to a conical tube and centrifuged to capture the solid phase at the tube base, such that the supernatant could be decanted and transferred to another vial for further purification (Fig. 10). To minimize Pa loss, the process of centrifuging the sample aliquot and decanting the supernatant was repeated twice, wherein the precipitate is resuspended in a second rinse of 9 M HCl to extract residual sample solution retained within pore spaces. While this physical separation process successfully removed the vast majority of Nb present in each sample aliquot, further purification of Nb from Pa was nonetheless required given the weight-percent levels of Nb in the original U–Nb alloys.
Fig. 10.

Photographs of the U–Nb samples received and processed at LANL. (a) The largest two subsample fragments of Alloy-1 were combined and digested together to create one sample solution for the purposes of the interlaboratory comparison study. (b) A white Nb precipitate forms when an aliquot of the U–Nb sample is dissolved in HNO3 or HCl acid (see bottom of centrifuge tube). Part of LANL’s method for Pa purifications involved physically separating this precipitate by centrifuging and decanting the supernatant
To purify Pa of the remaining Nb, LANL next performed several experiments testing various modifications to their standard separation scheme involving three columns, one with AG1-X8 resin followed by two with silica gel [40]. Like the other laboratories, this standard procedure was optimized for pure uranium materials (oxides, metals, compounds). One experiment replaced the second column with Eichrom DGA resin [41], but initial results were not encouraging enough for further pursuit. Another experiment modified the second column by adding washes of concentrated HCl after sample loading to remove Nb while retaining Pa on the silica gel [42]. The modified silica gel column results provided a better separation but at the expense of Pa recovery. LANL hypothesized that Pa loss occurred due to incomplete sorption of Pa to the silica gel in the presence of Nb, so LANL next performed experiments using increased volumes of silica gel to improve Pa sorption. Despite testing various silica gel volumes ranging from 2 to 6 mL, no improvement in Pa recovery was observed.
Because attempts to reduce Pa loss in the silica gel columns were unsuccessful, LANL refocused their efforts on improving the Pa/Nb separation in the first AG1-X8 resin column of the standard operating procedure. By reducing the amount of Nb eluted with Pa in the first column, the adverse impact of Nb on Pa recovery in subsequent silica gel columns may be mitigated. LANL explored the effect of varying the HF concentration of the elution acid based on a study by Kraus and Moore (1951) that demonstrated good Pa–Nb separation in mixed HCl–HF solutions with anion exchange resin [27]. In this study, the largest separation between Pa and Nb was achieved with 9 M HCl + 0.02 M HF, wherein Pa was eluted while Nb was retained on the resin (Fig. 11). Thus, instead of using 9 M HCl + 0.05 M HF to elute Pa, LANL used 9 M HCl + 0.02 M HF. In addition, the volume of AG1-X8 resin used for the first column was increased from 1 to 3 mL to increase the number of exchange sites for Nb sorption. In summary, LANL’s modifications of their standard operating procedure for U–Nb alloys were as follows: (1) an increase in AG1-X8 resin volume for the first column from 2 to 3 mL; (2) a decrease in the HF concentration of the Pa elution reagent of the first column from 9 M HCl + 0.05 M HF to 9 M HCl + 0.02 M HF; and (3) the addition of concentrated HCl washes to the first silica gel column to further remove Nb (Fig. 12b). A complete description of LANL’s modified Pa separation procedure is available in the Supplementary Materials.
Fig. 11.
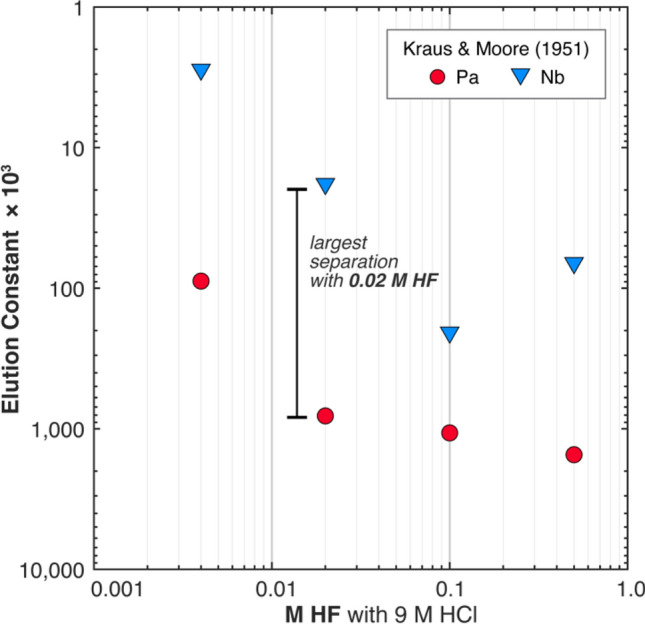
Previous reported data on Pa and Nb separations using strongly basic anion exchange resin with mixed HCl–HF solutions. A higher elution constant indicates weaker adsorption to resin. A solution of 9 M HCl + 0.02 M HF yields the greatest separation between Pa and Nb. All data are from Kraus and Moore (1951) using Dowex 1 in Cl− form [27]. Both Dowex 1 (made by Dow Chemical) and AG 1 (made by Bio-Rad and Eichrom) are monofunctional strongly basic anion exchange resins and are recognized as functionally equivalent [31]
Fig. 12.

Comparison of LANL’s a standard operating procedure for Pa separations on pure U materials [40] and b the modified procedure for depleted U–Nb alloys. LANL’s standard operating procedure for Pa separations involves one column with AG 1-X8, followed by two silica gel columns. For depleted U–Nb alloys, LANL modified the AG 1-X8 and first silica gel column. In the schematic, the taller columns represent Environmental Express columns (5.5 mL volume) and shorter columns represent Bio-Rad Poly-Prep columns (2 mL volume). Like in previous figures, the color of the eluates represents the elution of Th (green) and Pa (red), with changing opacity representing sequential improvements to the purity of each element. To highlight the differences between the two procedures, certain steps such as resin cleaning, column conditioning, acid washes, the application of H3BO3 as a fluoride scavenger have been abridged or omitted from this schematic; please refer to Denton et al. (2020) [40] and the Supplementary Materials of this article for complete descriptions of the standard and modified procedures
With this modified procedure, the Pa recovery of the U–Nb alloy samples was ~ 50–70% lower than the recoveries observed for the pure U materials using the standard procedure. Because the Pa yields for the blanks and quality control reference materials processed using the modified procedure were much higher, LANL concluded that the presence of the Nb was still interfering with the Pa separations, perhaps due to silica gel capacity issues. Further experiments confirmed that most of the Pa loss occurred in the second column of the procedure, with Pa prematurely eluting during the sample loading, rinsing, and washing steps of the first silica gel column before the intended elution of Pa. While further work to improve Pa recovery with this modified procedure is needed, the amount of purified Pa produced from this method was sufficient for high-precision analysis by mass spectrometry. We refer the reader to published follow-up research to optimize this separation chemistry in Engel et al. (2023) [30].
Discussion
All participating laboratories successfully devised viable sample digestion and Pa separation schemes for 231Pa–235U radiochronometry on the depleted U–Nb alloys. While each procedure has specific areas for improvement, each method also offers unique benefits that elevate the advantages of existing laboratory workflows. For example, although Pa recovery was variable (20–80%), AWE’s modified method allows for chemical separations to be completed rapidly (within a day), owing to the lack of evaporative dry down steps and the use of a vacuum-assisted flow chamber [18, 25]. LLNL’s modified method produces comparatively more consistent and complete Pa yields (80–100%; Fig. 8), but the procedure presently requires five columns that are each followed by dry down steps, which is time consuming. LANL’s modified method also has Pa recovery issues (30–50%), but the use of only three columns is less labor and resource intensive. Future research by AWE and LANL should investigate ways to improve the consistency and completeness of Pa recovery, whereas LLNL should examine ways to simplify the scheme (e.g., the last AG1-X8 column may not be necessary given the amount of U already removed by this stage of the purification).
Although each procedure was distinct and tailored for the specific needs, instrumentation, and equipment of each laboratory, the resulting 231Pa–235U model ages were internally reproducible within laboratories. However, when comparing model ages between labs, some differences were observed: in particular, the 231Pa–235U model ages measured by AWE were consistently younger than those determined by LLNL and LANL for both U–Nb alloy samples (Fig. 13). The cause of this discrepancy could be related to several factors, such as differences in sample preparation, spike calibrations, and possible spatial heterogeneity in isotopic composition between the U–Nb alloy subsamples allotted to each laboratory. For instance, regarding sample preparation, both LLNL and LANL applied an initial acid leach to their metal subsamples and added trace HF to their primary digestion solutions, unlike AWE. Previous work suggests that the presence of trace HF in solutions effectively mitigates Th and Pa loss due to sorption onto vial interior surfaces [10]; thus, Pa loss in the primary solutions prepared by AWE may contribute to the offset 231Pa–235U model ages. In terms of subsample heterogeneity, because quenched alloys commonly exhibit grain sizes that can be on the order of millimeters in size [43], Pa concentrations may vary at a scale that is discernible between the mm-scale sample fragments distributed between laboratories.
Fig. 13.

Comparison of 231Pa–235U model ages for a Alloy-1 and b Alloy-2 by the three laboratories. Uncertainties represent expanded uncertainties with a coverage factor of k = 2. Each laboratory produced two replicate analyses from a single sample dissolution of Alloy-1 and Alloy-2. For original data, we refer the reader to data tables contained in Higginson et al. (2022) [18]
Due to the study design, it is unknown which of the aforementioned factors contributes most to the interlaboratory differences in the 231Pa–235U model ages of these U–Nb alloys. Despite this ambiguity, the observed spread is unsurprising, if not expected. In two previous radiochronometry intercomparisons performed on certified reference materials U010 and U850, two well-purified uranium oxides with simpler matrices, 231Pa–235U model ages differed by 1–5% relative to the average age measured across all labs [40, 44]. This difference is comparable in magnitude to the relative differences in 231Pa–235U model ages observed in this study, 2–4% for Alloy-1 and 1–5% for Alloy-2 (relative to the multi-laboratory averages). Given that standard radiochronometry methods were developed using uranium oxide reference materials, the expectation is that interlaboratory model ages on other complex and untested material types would exhibit at least as much variability as that observed in routinely analyzed uranium materials.
In an ideal scenario, a U–Nb metal alloy reference material and a material certified for 231Pa–235U radiochronometry would be employed in this study, but no such materials currently exist. In lieu of better options, each laboratory applied their modified Pa separation scheme to certified reference materials U630 or CRM 125-A. The resulting 231Pa–235U model ages produced by each lab all agreed within uncertainty with the certified model purification dates, suggesting that the new Pa–Nb separation methods are nominally valid for uranium oxides (see Table 1 in the Supplementary Materials). Future and on-going interlaboratory comparisons involving sample material exchanges will include solution aliquots from a homogeneous bulk sample dissolution to control for possible analytical contributions to interlaboratory inconsistencies and evaluate potential sample-to-sample isotopic heterogeneity.
Conclusions
This analytical exercise simulated how various nuclear forensics laboratories would approach a request for radiochronometry on non-routine uranium materials in an investigatory context. Each laboratory required several weeks to months of dedicated effort to develop a reliable Pa separation method for the U–Nb alloys. Such lengthy processes are not ideal for law enforcement or nuclear security investigations that require key information in a timely manner. Thus, these results emphasize the need for further development of either ‘universal’ separation techniques applicable to a wide diversity of uranium materials, or more realistically, several methods optimized specifically for various material types. This study also highlights the need for reference materials certified for 231Pa–235U radiochronometry, as well as reference materials of diverse matrices other than uranium oxides. Without such reference materials, interlaboratory disagreements in 231Pa–235U model ages on non-routine uranium metals and alloys will be difficult to diagnose.
Although an investment in method development was required, the successful analysis of the U–Nb metal alloys demonstrates the resilience of each laboratory under time pressure and non-ideal pandemic-related working constraints. Some laboratories have since applied their modified Pa separation scheme for other U–Nb and metal alloys, highlighting the operational benefits of these shared material analysis exercises, especially when researchers are given access to uncommon materials. The proactive development of separation methods suitable for a variety of nuclear materials and reference materials with certified model ages will enhance the efficiency and preparedness of all nuclear forensics laboratories worldwide.
Supplementary Information
Below is the link to the electronic supplementary material.
Author contributions
CYC: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Supervision; Validation; Visualization; Writing—Original draft; Writing—Review and editing. MAH: Conceptualization; Data curation; Formal analysis; Funding acquisition; Methodology; Project administration; Supervision; Validation; Writing—Review and editing. TMKB: Conceptualization; Data curation; Formal analysis; Funding acquisition; Methodology; Project administration; Supervision; Validation; Writing—Review and editing. JSD: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Writing—Review and editing. JD: Conceptualization; Investigation; Methodology; Writing—Review and editing. MAE: Conceptualization; Investigation; Writing—Review and editing. CE: Conceptualization; Investigation. JRE: Conceptualization; Investigation; Writing—Review and editing. AMG: Conceptualization; Funding acquisition; Methodology; Project administration; Supervision; Writing—Review and editing. CG: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Writing—Review and editing. MNM: Conceptualization; Investigation. JMR: Conceptualization; Investigation; Methodology. MES: Conceptualization; Investigation. AMW: Conceptualization; Investigation.
Funding
This work was funded by AWE and the National Nuclear Security Administration NA-83 Office of Nuclear Forensics. Portions of this work were performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344 (LLNL-JRNL-843763) and Los Alamos National Laboratory under Contract 89233218CNA000001 (LA-UR-23-21440).
Declarations
Conflict of interest
All authors have declared that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wallenius M, Mayer K, Ray I. Nuclear forensic investigations: two case studies. Forensic Sci Int. 2006;156:55–62. doi: 10.1016/j.forsciint.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 2.Keegan E, Kristo MJ, Colella M, et al. Nuclear forensic analysis of an unknown uranium ore concentrate sample seized in a criminal investigation in Australia. Forensic Sci Int. 2014;240:111–121. doi: 10.1016/j.forsciint.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Niemeyer S, Hutcheon I (2002) Forensic analysis of a smuggled HEU sample interdicted in Bulgaria. In: Advances in destructive and non-destructive analysis for environmental monitoring and nuclear forensics, proceedings of an international conference, Karlsrube. International atomic energy agency, Vienna, Austria, pp 105–106
- 4.Grant PM, Moody KJ, Hutcheon ID, et al. Nuclear forensics in law enforcement applications. J Radioanal Nucl Chem. 1998;235:129–132. doi: 10.1007/BF02385950. [DOI] [Google Scholar]
- 5.Stanley FE. A beginner’s guide to uranium chronometry in nuclear forensics and safeguards. J Anal At Spectrom. 2012;27:1821–1830. doi: 10.1039/C2JA30182B. [DOI] [Google Scholar]
- 6.Kristo MJ, Gaffney AM, Marks N, et al. Nuclear forensic science: analysis of nuclear material out of regulatory control. Annu Rev Earth Planet Sci. 2016;44:555–579. doi: 10.1146/annurev-earth-060115-012309. [DOI] [Google Scholar]
- 7.Keegan E, Kristo MJ, Toole K, et al. Nuclear forensics: scientific analysis supporting law enforcement and nuclear security investigations. Anal Chem. 2016;88:1496–1505. doi: 10.1021/acs.analchem.5b02915. [DOI] [PubMed] [Google Scholar]
- 8.Kristo MJ, Keegan E, Colella M, et al. Nuclear forensic analysis of uranium oxide powders interdicted in Victoria, Australia. Radiochim Acta. 2015;103:487–500. doi: 10.1515/ract-2014-2363. [DOI] [Google Scholar]
- 9.Varga Z, Wallenius M, Mayer K, Hrnecek E. Alternative method for the production date determination of impure uranium ore concentrate samples. J Radioanal Nucl Chem. 2011;290:485–492. doi: 10.1007/s10967-011-1233-5. [DOI] [Google Scholar]
- 10.Williams RW, Gaffney AM. 230Th–234U model ages of some uranium standard reference materials. Proc Radiochem. 2011;1:31–35. doi: 10.1524/rcpr.2011.0005. [DOI] [Google Scholar]
- 11.Kristo MJ, Tumey SJ. The state of nuclear forensics. Nucl Instrum Methods Phys Res Sect B. 2013;294:656–661. doi: 10.1016/j.nimb.2012.07.047. [DOI] [Google Scholar]
- 12.Eppich GR, Williams RW, Gaffney AM, Schorzman KC. 235U–231Pa age dating of uranium materials for nuclear forensic investigations. J Anal At Spectrom. 2013;28:666. doi: 10.1039/c3ja50041a. [DOI] [Google Scholar]
- 13.Gaffney AM, Hubert A, Kinman WS, et al. Round-robin 230Th–234U age dating of bulk uranium for nuclear forensics. J Radioanal Nucl Chem. 2016;307:2055–2060. doi: 10.1007/s10967-015-4334-8. [DOI] [Google Scholar]
- 14.Kayzar TM, Williams RW. Developing 226Ra and 227Ac age-dating techniques for nuclear forensics to gain insight from concordant and non-concordant radiochronometers. J Radioanal Nucl Chem. 2016;307:2061–2068. doi: 10.1007/s10967-015-4435-4. [DOI] [Google Scholar]
- 15.Rolison JM, Williams RW. Application of the 226Ra–230Th–234U and 227Ac–231Pa–235U radiochronometers to UF6 cylinders. J Radioanal Nucl Chem. 2018;317:897–905. doi: 10.1007/s10967-018-5955-5. [DOI] [Google Scholar]
- 16.Sturm M, Richter S, Aregbe Y, et al. Evaluation of chronometers in plutonium age determination for nuclear forensics: What if the ‘Pu/U clocks’ do not match? J Radioanal Nucl Chem. 2014;302:399–411. doi: 10.1007/s10967-014-3294-8. [DOI] [Google Scholar]
- 17.Kayzar-Boggs TM, Luitjohan KE, Imhoff SD, et al. Results from a controlled depleted uranium metal casting experiment designed to investigate nuclear forensic radiochronometry signautres. J Radioanal Nucl Chem. 2023 doi: 10.1007/s10967-023-08881-w. [DOI] [Google Scholar]
- 18.Higginson MA, Kayzar-Boggs TM, Chen CY, et al. Establishing discordance as a radiochronometric signature for nuclear forensic investigations: a multi-laboratory intercomparison exercise. J Radioanal Nucl Chem. 2022;331:4799–4815. doi: 10.1007/s10967-022-08428-5. [DOI] [Google Scholar]
- 19.Ripley EB. Melting and casting of uranium. In: Morrell JS, Jackson MJ, editors. Uranium processing and properties. New York: Springer; 2013. pp. 35–69. [Google Scholar]
- 20.Vandermeer RA, Ogle JC, Northcutt WG., Jr A phenomenological study of the shape memory effect in polycrystalline uranium-niobium alloys. Metall Trans A. 1981;12A:733–741. doi: 10.1007/BF02648337. [DOI] [Google Scholar]
- 21.Eckelmeyer KH, Romig AD, Weirick LJ. The effect of quench rate on the microstructure, mechanical properties, and corrosion behavior of U-6 wt pct Nb. Metall Trans A. 1984;15:1319–1330. doi: 10.1007/BF02648560. [DOI] [Google Scholar]
- 22.Lillard JA, Hanrahan RJ., Jr . Corrosion of uranium and uranium alloys. In: Cramer SD, Covino BS Jr, editors. Corrosion: materials. Detroit: ASM International; 2005. [Google Scholar]
- 23.Brown D, Maddock AG. Protactinium. Q Rev Chem Soc. 1963;17:289–341. doi: 10.1039/QR9631700289. [DOI] [Google Scholar]
- 24.Jackson RJ, Johns WL (1970) Explosive nature of uranium-base niobium alloys after immersion in nitric acid. The Dow Chemical Company, Rocky Flats Division
- 25.Higginson M, Gilligan C, Taylor F, et al. Development of rapid methodologies for uranium age dating. J Radioanal Nucl Chem. 2018;318:157–164. doi: 10.1007/s10967-018-6021-z. [DOI] [Google Scholar]
- 26.Jerome SM, Collins SM, Happel S, et al. Isolation and purification of protactinium-231. Appl Radiat Isot. 2018;134:18–22. doi: 10.1016/j.apradiso.2017.07.051. [DOI] [PubMed] [Google Scholar]
- 27.Kraus KA, Moore GE. Anion exchange studies. III. Protactinium in some HCl–HF mixtures: separation of niobium, tantalum, and protactinium. J Am Chem Soc. 1951;73:2900–2902. doi: 10.1021/ja01150a140. [DOI] [Google Scholar]
- 28.Edgington DN. The estimation of thorium and uranium at the submicrogram level in bone by neutron activation. Int J Appl Radiat Isot. 1967;18:11–18. doi: 10.1016/0020-708X(67)90167-6. [DOI] [PubMed] [Google Scholar]
- 29.Treinen KC, Gaffney AM, Rolison JM, et al. Improved protactinium spike calibration method applied to 231Pa–235U age-dating of certified reference materials for nuclear forensics. J Radioanal Nucl Chem. 2018;318:209–219. doi: 10.1007/s10967-018-6149-x. [DOI] [Google Scholar]
- 30.Engel J, Denton JS, LaMont SP, et al. A chromatography chemistry for purifying Pa from U–Nb metal alloys. J Radioanal Nucl Chem. 2023 doi: 10.1007/s10967-022-08747-7. [DOI] [Google Scholar]
- 31.Korkisch J. Handbook of ion exchange resins: their application to inorganic analytical chemistry. Boca Raton: CRC Press; 1989. [Google Scholar]
- 32.Faris JP. Adsorption of the elements from hydrofluoric acid by anion exchange. Anal Chem. 1960;32:520–522. doi: 10.1021/ac60160a019. [DOI] [Google Scholar]
- 33.Caletka R, Krivan V. Behavior of 18 elements in HF and HF-NH 4 media on anion exchanger in various ionic forms. J Radioanal Nucl Chem. 1990;142:359–371. doi: 10.1007/BF02040306. [DOI] [Google Scholar]
- 34.Kim JI, Lagally H, Born H-J. Ion exchange in aqueous and in aqueous-organic solvents: part I. Anion-exchange behaviour of Zr, Nb, Ta and Pa in aqueous HCl–HF and in HCl–HF-organic solvent. Anal Chim Acta. 1973;64:29–43. doi: 10.1016/S0003-2670(00)86889-0. [DOI] [Google Scholar]
- 35.Faris JP. Separation of metal ions by anion exchange in mixtures of hydrochloric acid and hydrofluoric acid. National Technical Information Service: Argonne National Lab; 1978. [Google Scholar]
- 36.Kraus KA, Moore GE. Adsorption of protactinium from hydrochloric acid solutions by anion exchange resin. J Am Chem Soc. 1950;72:4293–4294. doi: 10.1021/ja01165a525. [DOI] [Google Scholar]
- 37.Kirby HW. The radiochemistry of protactinium. Washington, D.C.: National Academy of Sciences National Research Council; 1959. [Google Scholar]
- 38.Hyde EK. The radiochemistry of thorium. Berkeley: Lawrence Berkeley National Laboratory; 1959. [Google Scholar]
- 39.Kmak KN, Shaughnessy DA, Vujic J. Batch and column studies of radium, actinium, thorium and protactinium on CL resin in nitric acid, hydrochloric acid and hydrofluoric acid. J Radioanal Nucl Chem. 2021;328:225–233. doi: 10.1007/s10967-021-07636-9. [DOI] [Google Scholar]
- 40.Denton JS, Treinen KC, Chen Y, et al. International cooperation in age-dating uranium standards for nuclear forensics using the 231Pa/235U radiochronometer. J Radioanal Nucl Chem. 2020;324:705–714. doi: 10.1007/s10967-020-07084-x. [DOI] [Google Scholar]
- 41.Mastren T, Stein BW, Parker TG, et al. Separation of protactinium employing sulfur-based extraction chromatographic resins. Anal Chem. 2018;90:7012–7017. doi: 10.1021/acs.analchem.8b01380. [DOI] [PubMed] [Google Scholar]
- 42.Yoshida M, Numata Y, Seki R, Ikeda N. A new method for separation of niobium from zirconium with silica gel. Radioisotopes. 1990;39:547–552. doi: 10.3769/radioisotopes.39.12_547. [DOI] [Google Scholar]
- 43.Field RD, Thoma DJ. Crystallographic and kinetic origins of acicular and banded microstructures in U–Nb alloys. J Nucl Mater. 2013;436:105–117. doi: 10.1016/j.jnucmat.2013.01.309. [DOI] [Google Scholar]
- 44.Kayzar-Boggs TM, Treinen KC, Okubo A, et al. An interlaboratory collaboration to determine consensus 231Pa/235U model ages of a uranium certified reference material for nuclear forensics. J Radioanal Nucl Chem. 2020;323:1189–1195. doi: 10.1007/s10967-020-07030-x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.