

Abstract
The immunosuppressant and narrow therapeutic index drug tacrolimus is metabolized mainly via cytochrome P450 (CYP) 3A4 and CYP3A5. For its pharmacokinetics (PK), high inter‐ and intra‐individual variability can be observed. Underlying causes include the effect of food intake on tacrolimus absorption as well as genetic polymorphism in the CYP3A5 gene. Furthermore, tacrolimus is highly susceptible to drug–drug interactions, acting as a victim drug when coadministered with CYP3A perpetrators. This work describes the development of a whole‐body physiologically based pharmacokinetic model for tacrolimus as well as its application for investigation and prediction of (i) the impact of food intake on tacrolimus PK (food–drug interactions [FDIs]) and (ii) drug–drug(−gene) interactions (DD[G]Is) involving the CYP3A perpetrator drugs voriconazole, itraconazole, and rifampicin. The model was built in PK‐Sim® Version 10 using a total of 37 whole blood concentration–time profiles of tacrolimus (training and test) compiled from 911 healthy individuals covering the administration of tacrolimus as intravenous infusions as well as immediate‐release and extended‐release capsules. Metabolism was incorporated via CYP3A4 and CYP3A5, with varying activities implemented for different CYP3A5 genotypes and study populations. The good predictive model performance is demonstrated for the examined food effect studies with 6/6 predicted FDI area under the curve determined between first and last concentration measurements (AUClast) and 6/6 predicted FDI maximum whole blood concentration (Cmax) ratios within twofold of the respective observed ratios. In addition, 7/7 predicted DD(G)I AUClast and 6/7 predicted DD(G)I Cmax ratios were within twofold of their observed values. Potential applications of the final model include model‐informed drug discovery and development or the support of model‐informed precision dosing.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The pharmacokinetics (PK) of tacrolimus exhibit high inter‐ and intra‐individual variability. Tacrolimus is a substrate of cytochrome P450 (CYP) 3A4 and the polymorphically expressed CYP3A5 enzyme, thus highly susceptible to drug–drug(−gene) interactions. In addition, food intake shows a pronounced effect on tacrolimus PK.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study presents the development of a new whole‐body physiologically based pharmacokinetic (PBPK) model for tacrolimus that describes and predicts the influence of food intake and CYP3A perpetrator drugs voriconazole, itraconazole, and rifampicin on tacrolimus PK while incorporating different CYP3A5 activity levels.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The tacrolimus PBPK model helps investigate the underlying mechanisms for the pronounced impact of food intake, CYP3A5 polymorphism, and coadministration of CYP3A perpetrators on tacrolimus exposure and highlights the importance of these individual factors in dosing decisions.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The model can be applied to support model‐informed drug development and a holistic precision dosing approach incorporating a comprehensive set of factors affecting the PK of tacrolimus.
INTRODUCTION
Tacrolimus is the cornerstone of current immunosuppression for the prophylaxis of graft rejection following solid organ transplantation. 1 Data from 2020 show that the calcineurin inhibitor was included in approximately 90% of immunosuppressive regimens for adult and pediatric kidney transplant recipients in the United States. 2
Tacrolimus is classified as a Biopharmaceutics Classification System Class II drug. 3 It demonstrates incomplete absorption across the entire intestine as well as a pronounced intestinal and hepatic first‐pass metabolism after oral intake, 4 , 5 resulting in a low relative bioavailability of 17%–23%. 1 Moreover, in vitro studies have indicated tacrolimus to be a substrate of P‐glycoprotein (P‐gp). 6 Due to extensive binding to erythrocytes, considerably higher tacrolimus whole blood concentrations compared with plasma levels can be observed, with a reported mean blood‐to‐plasma concentration ratio of 15. 7 Tacrolimus is predominately metabolized via cytochrome P450 (CYP) enzymes 3A4 and 3A5, without contribution of its metabolites to the pharmacological effect. 8 , 9
As a narrow therapeutic index drug with high inter‐ and intra‐individual pharmacokinetics (PK) variability, the clinical application of tacrolimus can be challenging. 10 Thus, patients taking tacrolimus are subject to therapeutic drug monitoring to reduce the risk of graft rejection in the event of underdosing as well as to prevent overexposure that might result in nephro‐ and neurotoxicity, infections, and malignancies. 9 Factors potentially influencing the PK of tacrolimus include a pronounced food effect. Comparing the application under fed versus fasted conditions, the rate of tacrolimus absorption is reduced, resulting in a 33% decreased tacrolimus exposure when administered after a standardized breakfast. 11 , 12 Moreover, the PK of tacrolimus is affected by genetic polymorphisms, with the CYP3A5 gene being of particular interest. Following, for example, an oral single dose (SD) administration of tacrolimus, CYP3A5 normal metabolizers (NMs) demonstrate a 35% decreased tacrolimus area under the curve (AUC) compared with a mixed group of intermediate metabolizers (IMs) and poor metabolizers (PMs). 13 Clinical guidelines allow CYP3A5 genotype‐informed dosing. 14
Furthermore, tacrolimus is highly susceptible to drug–drug interactions (DDIs), acting as a victim drug when coadministered with CYP3A perpetrators. Hence, tacrolimus is recommended as a sensitive substrate of CYP3A by the US Food and Drug Administration for use in clinical DDI studies. 15 CYP3A inhibitors cause a significant increase in tacrolimus exposure. Here, azole antifungal agents (e.g., itraconazole, voriconazole) are of specific clinical relevance, as invasive fungal infections are among the most important posttransplant infections requiring frequent concomitant use of tacrolimus and azoles. 16 , 17 , 18 , 19 For instance, pretreatment with voriconazole increases the AUC of tacrolimus by more than fourfold, even reaching sixfold in CYP2C19 PMs that express a reduced voriconazole metabolism. 17 In contrast, pretreatment with the antibiotic agent and CYP3A inducer rifampicin reduces the AUC of tacrolimus by 68%. 5
To improve the safety and efficacy of tacrolimus therapy, factors modulating the PK of tacrolimus, such as food–drug interactions (FDIs) and drug–drug(−gene) interactions (DD[G]Is), should be thoroughly investigated. For this, physiologically based pharmacokinetic (PBPK) modeling allows the study of a drug's PK in different genotypes and interactions with food components or coadministered drugs while covering various (patho‐)physiological characteristics. 20 , 21 By incorporating factors contributing to a drug's interindividual variability, PBPK modeling can be applied to support model‐informed precision dosing. 22 Moreover, PBPK modeling is widely acknowledged for the use in model‐informed drug discovery and development (MID3), demonstrated by a growing number of PBPK applications submitted to regulatory agencies to address specific research questions, mainly related to DDIs. 23
Objectives of the presented study were therefore (a) the development of a whole‐body PBPK model of tacrolimus covering different CYP3A5 activities and clinically relevant formulations as well as the prediction of (b) the influence of food intake on tacrolimus PK and (c) DD(G)Is involving different CYP3A perpetrators, especially azole antifungal agents. The final model will be made publicly accessible (https://github.com/Open‐Systems‐Pharmacology).
METHODS
Software
PK‐Sim® and MoBi® Version 10 (Open Systems Pharmacology Suite, www.open‐systems‐pharmacology.org, 2021) were used for the development of the tacrolimus PBPK model, parameter identification (Levenberg–Marquardt algorithm), and model sensitivity analyses. Published clinical study data were digitized with the help of Engauge Digitizer Version 12.1 (Mitchell et al. 24 ) according to best practices. 25 The compilation of plots as well as calculations of PK parameters and quantitative model performance measures were performed using the R programming language Version 4.2.1 (R Foundation for Statistical Computing).
Clinical study data
Whole blood concentration–time profiles of tacrolimus in healthy volunteers were gathered from the literature, covering a broad dosing range of tacrolimus administered intravenously and orally in different formulations. Digitized profiles were divided into a training dataset for model development and a test dataset for model evaluation (ratio 1:3). Here, profile assignment to the test and training datasets was accomplished in a nonrandomized fashion to maximize the set of profiles for model evaluation (test dataset) while preferring information‐dense (frequent measurements within a long sampling period) and heterogenous profile data to capture the range of different routes of administration, formulations, and dosage regimens for the training dataset.
PBPK model building
Model development was initiated with an extensive literature search for physicochemical parameters as well as information on the absorption, distribution, metabolism, and excretion of tacrolimus.
For each included study, a representative virtual individual was established based on the mean and mode for age, sex, weight, height, body mass index, and ethnicity of the respective study publication. If demographic information was incomplete, default settings were adopted from the population database accessible in PK‐Sim®. Relative expression of relevant transporters and enzymes in different organs was implemented according to the PK‐Sim® expression database as described in Table S1. A virtual population of 1000 individuals was created for each study population using the respective reported demographic information to visually assess variability based on these characteristics and metabolizing enzymes. If no data were available, an age range of 20–50 years was assumed. Table S1 provides the geometric standard deviations used for the modeled variation of the relevant transporter and enzyme concentrations.
Unknown model parameters and parameters with a high impact on the results of permeability and partition quantitative structure‐activity relationship (QSAR) models of PK‐Sim® were fitted using the training dataset. Oral formulations differing in release kinetics were incorporated via separate Weibull functions (Equation 1), with the time to 50% dissolution and shape as model input parameters. In a stepwise approach, tacrolimus model parameters were first optimized based on intravenous and oral administration of immediate‐release (IR) capsules (Prograf®). Next, the model was adjusted to extended‐release (ER) capsules (Advagraf®) by estimating the respective release model parameters as well as the intestinal permeability, the latter required to account for different permeability behaviors of IR and ER tacrolimus.
| (1) |
where = fraction of administered dose dissolved at time , exp = exponential function, = lag time between drug intake and the start of the dissolution process, b = shape parameter, and a = scale parameter.
Enzyme metabolism was implemented using Michaelis–Menten kinetics (Equation S1). For CYP3A5, different levels of activity were implemented by activity‐specific CYP3A5 catalytic rate constants (kcat). Here, the Michaelis–Menten constant (KM) was fixed for all activity levels to decrease the explorative space of the fitting process. For each study population, the reported fraction of functional *1 allele was used for activity assignment. In the absence of genotype/phenotype information of a study group, CYP3A5 activity was assumed according to the frequency of the *1 allele observed in the respective ethnic group published by Birdwell et al. 14 Assumed relative activities for ethnicities and phenotypes relevant to this work are listed in Table 1. A study investigating CYP3A5 PMs (i.e., lack of CYP3A5 activity) was used to determine CYP3A5 independent metabolism. 13 Genetic variants in CYP3A4 (e.g., CYP3A4*22) were not considered due to missing information in the study cohorts.
TABLE 1.
Frequency of the CYP3A5 *1 allele in different populations, 14 including assumed activity relative to homozygous carriers of the *1 allele.
| Population | Frequency of the CYP3A5 *1 allele, % | Assumed activity, % |
|---|---|---|
| Phenotype | ||
| Normal metabolizer | 100.0 | 100.0 |
| Poor metabolizer | 0.0 | 0.0 |
| Race and Ethnicity | ||
| African American | 60.5 | 60.5 |
| Asian | 25.8 | 25.8 |
| Latin American | 20.2 | 20.2 |
| Caucasian | 7.8 | 7.8 |
Abbreviation: CYP, cytochrome P450.
PBPK model evaluation
The final tacrolimus model was evaluated both graphically and statistically. For visual comparison, predicted whole blood concentration–time profiles were plotted alongside their corresponding observed data points. Goodness‐of‐fit (GOF) plots were generated to examine the deviation of predicted to observed concentration measurements. Moreover, GOF plots were used for comparison of the calculated AUC determined between first and last concentration measurements (AUClast) and maximum whole blood concentration (Cmax) values for all predicted versus observed profiles. Predictions within the twofold range of observed values were considered successful. Statistical evaluation was conducted via calculation of mean relative deviations (MRDs) for all predicted concentration‐time points (Equation 2) as well as geometric mean fold errors (GMFEs) for predicted AUClast and Cmax values (Equation 3).
| (2) |
where = i‐th observed concentration, = predicted concentration corresponding to the i‐th observed concentration, and k = number of observed values.
| (3) |
where = observed AUClast or Cmax value of study i, = corresponding predicted AUClast or Cmax value of study i, and m = number of studies.
Finally, a local sensitivity analysis was performed for each investigated formulation (for details, see Section S2.8.1).
FDI modeling
The effect of food intake on tacrolimus exposure was estimated based on studies providing whole blood concentration–time profiles under both fed and fasted conditions. Altered absorption of tacrolimus due to food intake was implemented by adjusting not only the Weibull parameters (time to 50% dissolution and shape) but also the intestinal permeability to account for presumed binding of tacrolimus to lipoproteins and food components.
FDI modeling was evaluated by comparing predicted to observed whole blood concentration–time profiles under fed and fasted conditions. Furthermore, for each modeled FDI, observed and predicted FDI AUClast and Cmax ratios were calculated (Equation 4) with subsequent comparisons applying the limits proposed by Guest et al. 26 to determine prediction accuracy (including 20% variability).
| (4) |
where PK parameter = AUClast or Cmax, PK parameterfed = AUClast or Cmax of tacrolimus under fed conditions, and PK parameterfasted = AUClast or Cmax of tacrolimus under fasted conditions.
Finally, GMFE values were determined for all predicted and observed FDI AUClast and Cmax ratios according to Equation (3).
DD(G)I modeling
The influence of CYP3A perpetrators on tacrolimus PK was investigated by coupling the tacrolimus model with previously published models of voriconazole, itraconazole, and rifampicin. 27 , 28 The respective model files were downloaded from https://github.com/Open‐Systems‐Pharmacology. PK‐Sim® Version 10 model files for itraconazole and rifampicin were obtained from https://github.com/Open‐Systems‐Pharmacology/Itraconazole‐Model/releases/tag/v1.3 and https://github.com/Open‐Systems‐Pharmacology/Rifampicin‐Model/releases/tag/v1.2. Relevant interaction parameters were incorporated from the literature according to the respective types of interaction reported, that is, induction, competitive inhibition, and mechanism‐based inactivation, as described in Section S4.1. One of the voriconazole–tacrolimus–DDIs was included in the training dataset to inform the contribution of intestinal and intrahepatic tacrolimus metabolism. 16
Evaluation of the modeled DD(G)Is was performed analogously to the investigated FDIs as described in the Methods section (“FDI modeling”).
RESULTS
PBPK model building and evaluation
The tacrolimus PBPK model was built and evaluated using a total of 37 whole blood concentration–time profiles (summarized mean values with variance information if available) obtained from 25 clinical studies for the training and test datasets. Included were five intravenous SD applications of 0.015–0.025 mg/kg body weight (BW) tacrolimus as well as 32 oral administrations of therapeutic doses ranging from 0.5 to 10 mg. IR and ER formulations of tacrolimus accounted for 27 (SD) and five (SD and multiple dose [MD]) of the profiles used, respectively. Information on all profiles, including the frequency of the CYP3A5 *1 allele for each study population, is provided in Table S2.
The metabolism of tacrolimus was implemented via CYP3A4 and CYP3A5, with KM values adopted from the literature and kcat values optimized during the model‐building process. For a CYP3A5 NM, the contribution of CYP3A4 and CYP3A5 to the overall metabolism accounted for 67% and 33%, respectively. A total of 71% of CYP3A metabolism occurred intestinally in a CYP3A5 NM compared with 59% in a CYP3A5 PM. Moreover, weak mechanism‐based inactivation of CYP3A4 and CYP3A5 by tacrolimus was incorporated using published inhibition data. Initial Weibull starting parameters for IR and ER formulations were derived from literature dissolution profiles according to Langenbucher et al. 29 , 30 , 31 During parameter optimization steps, the respective shape values for both formulations and the time to 50% dissolution for ER tacrolimus were adjusted. Furthermore, separate intestinal permeabilities were optimized for IR and ER tacrolimus. A detailed overview of all tacrolimus model parameters is provided in Table S3. A summary of the key modeling assumptions, including the resulting modeling decisions, is available in Table S4. The tacrolimus PBPK model file can be found in Appendix S2.
Overall, the final tacrolimus model showed good descriptive (training dataset) and predictive (test dataset) performance. A representative sample of whole blood concentration–time profiles from both the training and test datasets is provided in Figure 1. Semilogarithmic and linear plots of all predicted and observed profiles are listed in Sections S2.1–S2.2.
FIGURE 1.
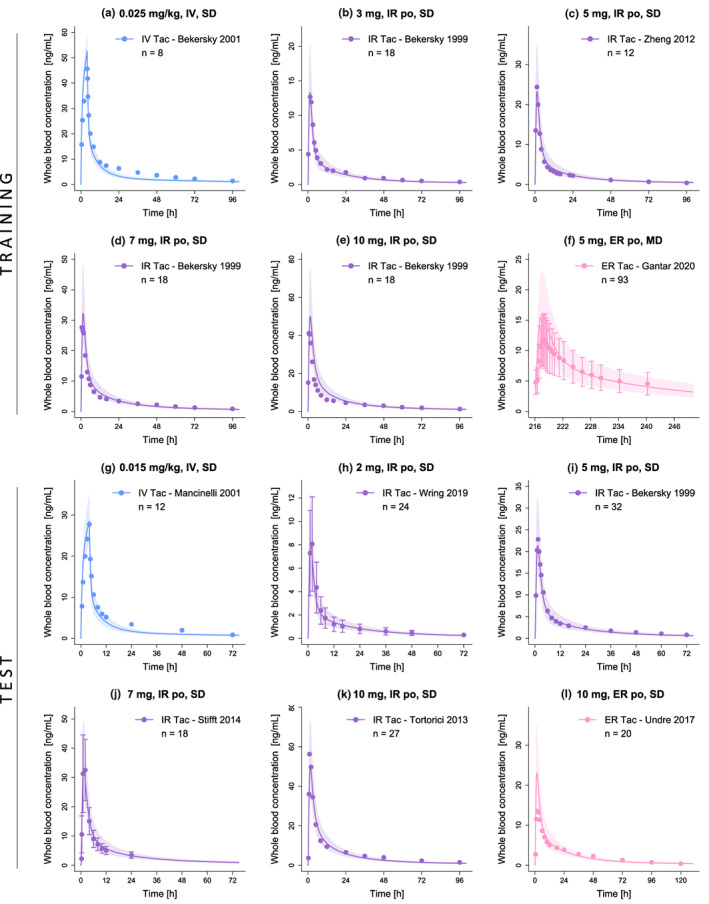
Representative plots of whole blood concentration–time profiles of tacrolimus. Stratified by training (a–f) and test (g–l) dataset, solid lines and ribbons represent population predictions (n = 1000; geometric mean and geometric standard deviation), whereas corresponding observed data are shown as dots (± standard deviation, if available). 13 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 Detailed information on all investigated profiles is provided in Table S2. ER, extended‐release; IR, immediate‐release; IV, intravenous; MD, multiple dose; n, number of participants; po, oral; SD, single dose; Tac, tacrolimus.
Figure 2 displays GOF plots of predicted versus observed concentration measurements as well as AUClast and Cmax values stratified by the training and test datasets. A total of 96% of all predicted concentration measurements as well as 37/37 of predicted AUClast and 37/37 of predicted Cmax values were simulated within twofold of their corresponding observed data. The good descriptive and predictive performance of the model could be further demonstrated by the calculated mean MRD of 1.43 as well as mean GMFEAUClast and GMFECmax values of 1.21 and 1.18, respectively. All individual MRD and GMFE values are listed in Tables S5 and S6.
FIGURE 2.
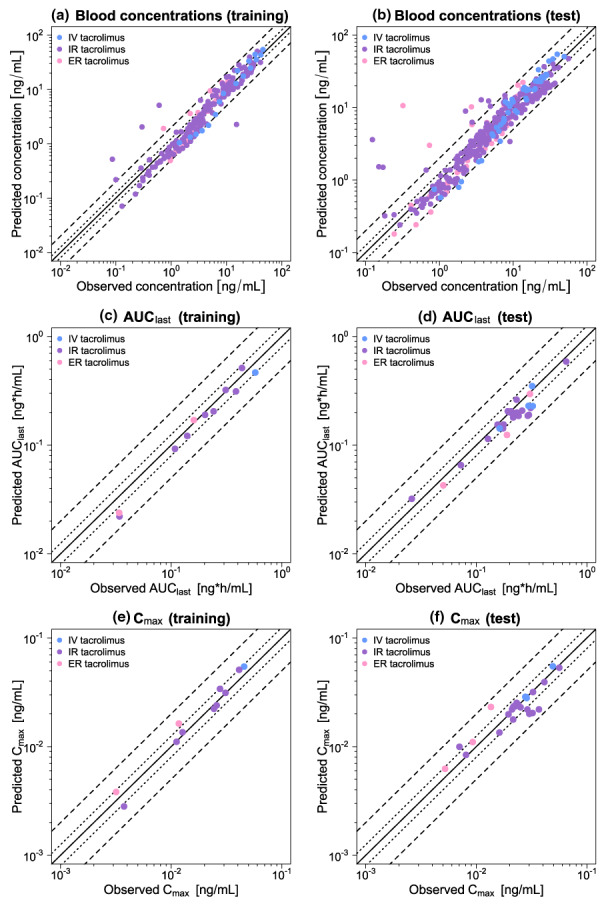
Goodness‐of‐fit plots of the final tacrolimus model. Stratified by training (left column) and test dataset (right column), predicted whole blood concentration measurements (a–b) as well as AUClast (c–d) and Cmax (e–f) values are plotted against corresponding observed data. The solid line represents the line of identity, whereas dotted lines indicate 1.25‐fold and dashed lines twofold deviation from the respective observed value. Detailed information on all investigated profiles is provided in Table S2. AUClast, area under the curve determined between first and last concentration measurements; Cmax, maximum whole blood concentration; ER, extended‐release; IR, immediate‐release; IV, intravenous.
Local sensitivity analyses were performed for SD administrations of intravenous (0.025 mg/kg BW), oral IR (10 mg), and oral ER tacrolimus (10 mg), respectively. The AUClast of intravenous tacrolimus was demonstrated to be most sensitive to perturbations of the acid dissociation constant (literature value), whereas for oral IR and ER tacrolimus, the lipophilicity (optimized) showed the greatest impact on AUClast. A detailed assessment of all sensitivity analyses can be found in Section S2.8.2.
FDI modeling
The effect of food intake on the PK of tacrolimus was modeled and evaluated using four FDI studies, with kilocalories ingested ranging from 600 to 1000 and IR tacrolimus administered 0.33 to 1.5 h after the start of each meal. 11 , 12 , 32 , 33 Two whole blood concentration–time profiles of tacrolimus under fed conditions (668 and 849 kilocalories, 0.33 h after food intake) were used to adjust the Weibull parameters (time to 50% dissolution and shape) as well as the intestinal permeability. 11 Compared with parameters for fasted administrations, a 3.3‐fold increased time to 50% dissolution (63.0 vs. 18.9 min), a higher shape parameter (0.94 vs. 0.08, both parabolic curves), and a 90% decreased intestinal permeability (3.79 × 10−7 vs. 3.42 × 10−6 cm/s) were estimated for tacrolimus applications under fed conditions. Information on all included studies is provided in Table S8. The FDI model file is included in Appendix S2.
Figure 3a–f displays the predicted versus observed whole blood concentration–time profiles of IR tacrolimus under fed and fasted conditions, with Figure 3g,h showing the corresponding predicted versus observed FDI AUClast and FDI Cmax ratios. Overall, the FDI model demonstrated good predictive performance regarding the effect of food intake on tacrolimus exposure, as 6/6 of predicted FDI AUClast and 6/6 of predicted FDI Cmax ratios were within the limits proposed by Guest et al., 26 with respective GMFE values averaging 1.21 and 1.19. A list of all individual GMFE values as well as semilogarithmic and linear plots of all predicted and observed whole blood concentration–time profiles are available in Sections S3.2–S3.5.
FIGURE 3.
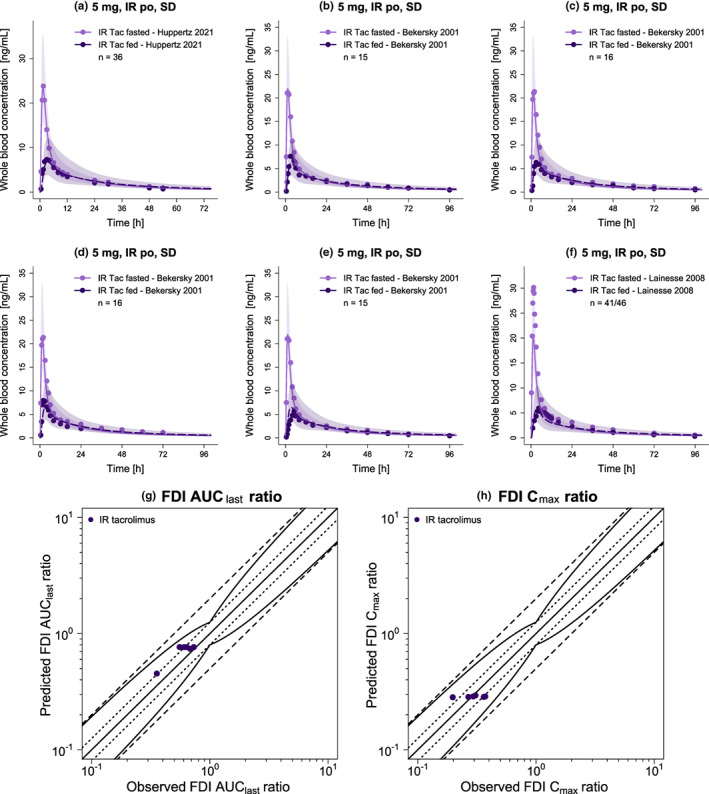
Evaluation of the modeled FDIs. Presented are predicted whole blood concentration–time profiles (a–f) of IR tacrolimus under fed and fasted conditions alongside corresponding observed data. 11 , 12 , 32 , 33 Dashed (fed) and solid (fasted) lines and ribbons represent population predictions (n = 1000; geometric mean and geometric standard deviation), whereas corresponding observed data are shown as dots. Predicted versus observed FDI AUClast (g) and FDI Cmax (h) ratios are shown with the solid line representing the line of identity and dotted lines indicating 1.25‐fold and dashed lines twofold deviation from the respective observed value, along with the curved lines marking the prediction success limits proposed by Guest et al.,26 including 20% variability. Detailed information on all investigated FDI studies is provided in Table S8. AUClast, area under the curve determined between first and last concentration measurements; Cmax, maximum whole blood concentration; FDI, food–drug interaction; IR, immediate‐release; n, number of participants; po, oral; SD, single dose; Tac, tacrolimus.
DD(G)I modeling
The DD(G)I model, centered around tacrolimus as a CYP3A victim drug, was developed using four DD(G)I studies. For the CYP3A4 mechanism‐based inactivator and CYP3A5 competitive inhibitor voriconazole, two studies investigated its pretreatment effect on the PK of IR tacrolimus, 16 , 17 with one of the studies specifically examining the extent of inhibition in different CYP2C19 phenotypes, that is, NMs, IMs, and PMs. 17 Furthermore, one study assessed the impact of pretreatment with the CYP3A4 and CYP3A5 competitive inhibitor itraconazole on ER tacrolimus, 18 whereas the last study addressed the pretreatment influence of the CYP3A4 competitive inhibitor and inducer rifampicin on the PK of intravenously and orally (IR) administered tacrolimus. 5 In two of the aforementioned studies, the CYP3A4 victim drug midazolam was additionally administered. However, as no effect on the PK of tacrolimus was assumed, midazolam administration was not included in the DD(G)I model. 16 , 18 Figure 4 presents a schematic overview of the modeled DD(G)I network. Further details on all included DD(G)I studies, as well as model parameters of the DD(G)I partners, are provided in Sections S4.2–S4.3. The DD(G)I model file is included in Appendix S2.
FIGURE 4.
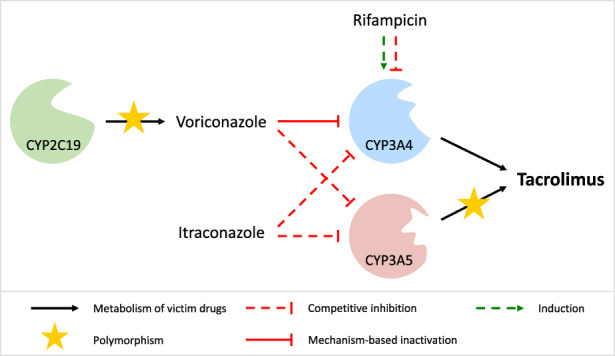
Schematic overview of the modeled drug–drug(−gene) interaction network. While tacrolimus acts as a CYP3A4 and CYP3A5 victim drug, voriconazole, itraconazole, and rifampicin represent CYP3A4 perpetrator drugs. Voriconazole and itraconazole additionally inhibit CYP3A5. For simplicity, the mechanism‐based inactivation of CYP3A4 and CYP3A5 by tacrolimus is not shown. CYP, cytochrome P450.
Figure 5 shows predicted versus observed whole blood concentration–time profiles of tacrolimus administered alone or concomitantly with the respective perpetrator drug. For each DD(G)I, predicted versus observed DD(G)I AUClast and Cmax ratios are displayed in Figure 6, with 7/7 and 6/7 within the limits proposed by Guest et al., 26 respectively. The favorable DD(G)I prediction performance of the model is further demonstrated by low mean GMFE values for the predicted DD(G)I AUClast (1.10) and Cmax ratios (1.41). A list of all individual GMFE values as well as semilogarithmic and linear plots of all predicted and observed whole blood concentration–time profiles are available in Sections S4.4–S4.7.
FIGURE 5.
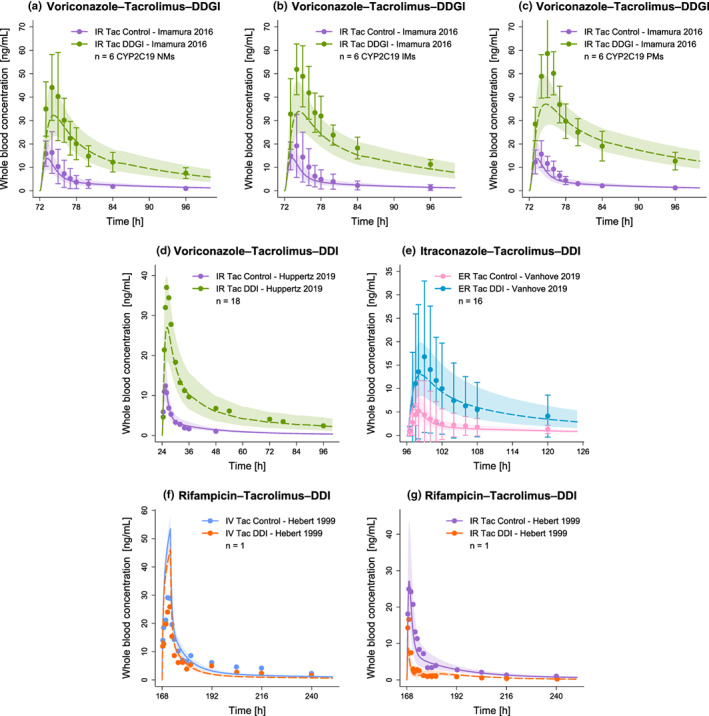
Evaluation of the modeled DD(G)Is (Part I). Presented are predicted whole blood concentration–time profiles of tacrolimus without (Control) and with (DD[G]I) intake of the respective perpetrator drug (voriconazole [a–d], itraconazole [e], rifampicin [f–g]) alongside their corresponding observed data. 5 , 16 , 17 , 18 Solid (Control) and dashed (DD[G]I) lines and ribbons represent population predictions (n = 1000; geometric mean and geometric standard deviation), whereas corresponding observed data are shown as dots (± standard deviation, if available). Detailed information on all investigated DD(G)I studies is provided in Table S10. CYP, cytochrome P450; DDGI, drug–drug−gene interaction; DDI, drug–drug interaction; ER, extended‐release; IM, intermediate metabolizer; IR, immediate‐release; IV, intravenous; n, number of participants; NM, normal metabolizer; PM, poor metabolizer; Tac, tacrolimus.
FIGURE 6.
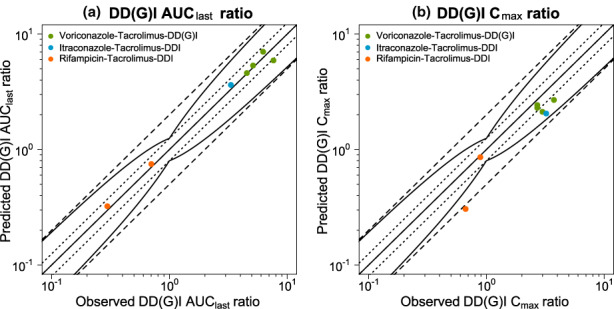
Evaluation of the modeled DD(G)Is (Part II). Predicted versus observed DD(G)I AUClast (a) and DD(G)I Cmax (b) ratios are shown with the solid line representing the line of identity, dotted lines indicating 1.25‐fold and dashed lines twofold deviation from the respective observed value, along with the curved lines marking the prediction success limits proposed by Guest et al. 26 including 20% variability. Detailed information on all investigated DD(G)I studies is provided in Table S10. AUClast, area under the curve determined between first and last concentration measurements; Cmax, maximum whole blood concentration; DD(G)I, drug–drug(−gene) interaction; DDI, drug–drug interaction.
DISCUSSION
In the present work, a whole‐body PBPK model for tacrolimus was built and evaluated allowing the successful description and prediction of whole blood concentration–time profiles over a broad dosing range of intravenously (0.015–0.025 mg/kg BW, SD) and orally (0.5–10 mg, SD and MD, IR and ER) administered tacrolimus. Subsequently, the final model was applied to predict the effect of food intake on tacrolimus PK as well as DD(G)Is involving tacrolimus as a CYP3A victim drug in subjects with varying CYP3A5 and CYP2C19 activity levels.
The simulated total bioavailability for IR tacrolimus ranges from 12% to 18% depending on the administered dose, coinciding with reported total bioavailability values of (18 ± 5)% in healthy individuals. 1 The three determining factors for fraction absorbed, fraction escaping gut wall metabolism (Fg), and fraction escaping first‐pass liver metabolism (Fh) are also well reflected in the model: (1) consistent with literature data, the developed model simulates incomplete intestinal absorption of tacrolimus, with 48% of the applied dose absorbed 1 ; (2) as intravenous profiles are reasonably predicted, we conclude that the hepatic clearance is adequately described, and consequently Fh should be well depicted in the model; (3) the successful prediction of metabolic DDIs suggests a well‐described relationship between the fraction absorbed and Fg by the model.
Numerous studies have demonstrated the predominant involvement of CYP3A enzymes in the metabolism of tacrolimus, with CYP3A4 and CYP3A5 being of particular importance, resulting in their inclusion in the model. 8 To account for interindividual variability caused by the genetic polymorphism of the CYP3A5 gene, CYP3A5 activity was incorporated for each study population based on the frequency of the functional *1 allele, either determined by genotyping or assumed based on the mode of the respective study participants' race and ethnicity. 14 However, as CYP3A5 genotyping had not been performed in the majority of included studies, this lack of information might have led to biases in the modeling and estimation processes. Here, a larger and complete PK dataset on specified CYP3A5 genotypes might be required to further improve and refine the genotype‐specific modeling of tacrolimus exposure. The same applies to CYP3A4 variants, which have not been considered so far.
To predict the release kinetics of IR and ER tacrolimus, separate Weibull parameter values were used in the absorption model. In addition, it became apparent that different intestinal permeability values had to be optimized for IR and ER tacrolimus to adequately describe the observed data, with a slightly higher permeability value estimated for IR compared with ER tacrolimus (3.42 × 10−6 vs. 1.91 × 10−6 cm/s). For ER tacrolimus, the adjusted permeability was within one order of magnitude of the published in vitro value (6.58 × 10−6 cm/s). 30 No reference permeability could be found in the literature for IR tacrolimus. The underlying cause for the observed differences in intestinal permeability for IR and ER formulations is not clear yet, but it is reasonable to assume that different pharmaceutical excipients contained in IR and ER tacrolimus formulations might be the driving factor. For example, only IR tacrolimus capsules include the disintegrant croscarmellose sodium, which has been reported to significantly increase the membrane permeability in the rat small intestine in vitro. 1 , 34 Here, dedicated studies addressing oral tacrolimus formulations would be of interest to investigate possible permeability‐modulating effects. During model development, besides implementing different intestinal permeabilities for IR and ER tacrolimus, alternative approaches covering different solubilities, supersaturation, or release modeling via particle dissolution were tested. However, these approaches were not adopted because no support for their application could be found in the literature and the prediction of the observed data did not improve.
The final tacrolimus model was successfully applied to predict the influence of food intake on IR tacrolimus whole blood levels. Food effects may result from a variety of mechanisms arising from the interaction of postprandial physiology and compounds. For tacrolimus, the influence of different meal compositions on the exposure has been examined by Bekersky et al., however, the underlying mechanisms are still unknown. 11 , 12 , 35 Therefore, the food effect was empirically modeled. The Weibull parameters were adjusted to represent food‐induced changes in release kinetics, for example, due to altered gastric pH. In addition, the intestinal permeability was estimated to account for altered absorption, presumably due to the binding of tacrolimus to lipoproteins and food components, given its pronounced lipophilicity. 36 During model development, other approaches to model the effect were pursued, such as a change in solubility, gastric pH, gastric emptying time, or intestinal/hepatic blood flow. However, these approaches were eventually not retained due to an inadequate description of the observed food effect. In the case of a prolonged gastric emptying time, this observation is consistent with the findings of Kuypers et al. showing no significant difference in the extent of absorption in patients with and without delayed gastric emptying. 37 According to Deng et al., as tacrolimus is a low extraction drug, a change in splanchnic blood flow would not be expected to affect tacrolimus kinetics; hence, the lack of effect of an increase in intestinal/hepatic blood flow is in line with the literature. 35 , 38 The final model describes a more pronounced release and slower absorption under fed conditions. Overall, a higher fraction of the tacrolimus dose is absorbed than under fasting conditions, with ultimately lower exposure due to increased intestinal metabolism, a finding consistent with a hypothesis proposed by Huppertz et al. 12 Unfortunately, due to the administration of 5 mg IR tacrolimus in all food effect studies, the investigation of the FDI lacked diversity in applied doses and formulations. Hence, additional studies examining different doses and oral formulations of tacrolimus would be necessary to increase the prediction confidence and domain extrapolation of the presented FDI model. In general, to pursue a modeling approach that captures the underlying mechanisms of the food effect, a more diverse set of training data and further studies on mechanisms are needed.
Furthermore, DD(G)I modeling involving tacrolimus as a CYP3A victim drug was successfully performed by coupling the developed tacrolimus model with models of the CYP3A perpetrator drugs voriconazole, itraconazole, and rifampicin while considering different CYP3A5 activity levels. 27 , 28 However, a slight underprediction of tacrolimus Cmax values can be observed for each modeled DD(G)I. Here, various approaches were explored to refine DD(G)I predictions. Among others, the inclusion of P‐gp in the model was tested, as tacrolimus has been identified as a P‐gp substrate in vitro and inhibition of P‐gp by itraconazole and rifampicin is described in the literature. 6 , 39 , 40 For the final model, the implementation of P‐gp was eventually discarded due to its insignificant improvements for DD(G)I predictions. Potential reasons for the remaining slight underprediction of tacrolimus Cmax values could be, for example, the involvement of additional, still unknown transporters in the distribution of tacrolimus or a discrepancy between the incorporated expression profiles of CYP3A4 and CYP3A5 and the respective actual in vivo expression in different organs. Overall, depending on the CYP2C19 phenotype, modeled pretreatment with voriconazole increased the total bioavailability of tacrolimus by 2.5‐fold (NMs), 2.7‐fold (IMs), and 3.1‐fold (PMs). In the case of pretreatment with itraconazole and rifampicin, the total bioavailability of tacrolimus increased by 2.4‐fold and decreased by 58%, respectively. All three perpetrators showed no effect on the fraction absorbed of tacrolimus.
Other whole‐body PBPK models of tacrolimus are available in the literature, focusing, for instance, on pregnant populations or DDIs between tacrolimus and Wuzhi capsule ingredients. 41 , 42
Other investigators used a minimal PBPK model of tacrolimus to also investigate the impact of CYP3A5 genotype on tacrolimus PK in addition to other factors such as hematocrit. 43 Due to considerable structural differences between whole‐body and minimal PBPK models, significant differences in the observed study populations, and differing implementation of CYP3A5, a comparison of the two models is difficult. Moreover, due to the minimal overlap of estimated and used model input parameters as well as vast differences in cardinality of training and test datasets, comparing the predictive model performances might not be reasonable.
The presented tacrolimus model provides new important insights by investigating the food effect of tacrolimus as well as clinically relevant DD(G)Is involving CYP3A perpetrators, particularly azole antifungal agents, while considering different CPY3A5 activities. In addition, the model was built and evaluated using a comprehensive set of clinical data, including multiple clinically relevant formulations over wide dosing ranges and a large number of 37 mean whole blood concentration–time profiles obtained from a total of 911 study participants of different ethnicities. However, further studies investigating MD applications of tacrolimus would be beneficial to assess possible tacrolimus accumulation.
To conclude, the developed tacrolimus whole‐body PBPK model demonstrates good descriptive and predictive performance in healthy individuals. Moreover, the model was successfully used to investigate and predict the interaction between tacrolimus PK and food intake as well as the influence of the CYP3A perpetrator drugs voriconazole, itraconazole, and rifampicin on tacrolimus exposure in DD(G)I scenarios. Potential applications of the model include MID3 or the support of precision dosing. Once clinical study data and corresponding PBPK models on additional CYP3A perpetrators become available, the model can be extended to further investigate the role of tacrolimus as a CYP3A victim drug. Here, DD(G)I studies analyzing different CYP3A5 genotypes would be of particular interest providing a more detailed understanding of the CYP3A5 polymorphism and its effects on tacrolimus PK. Finally, the model could be expanded to investigate tacrolimus PK in patients, especially recipients of different types of solid organ transplants.
AUTHOR CONTRIBUTIONS
H.L.H.L., D.F., S.R., D.S., M.S., D.T., S.F., M.v.d.L., D.J.A.R.M., J.J.S., and T.L. wrote the manuscript. H.L.H.L., D.F., S.R., D.S., and T.L. designed the research. H.L.H.L. performed the research. H.L.H.L., D.F., S.R., D.S., and T.L. analyzed the data.
FUNDING INFORMATION
This work is part of the Horizon 2020 INSPIRATION (Qualified Open Systems Pharmacology Modeling Network of Drug–Drug‐Gene Interactions) project. The INSPIRATION project (FKZ 031 L0241) is supported by the German Federal Ministry of Education and Research under the framework of ERACoSysMed (Collaboration on systems medicine funding to promote the implementation of systems biology approaches in clinical research and medical practice). Matthias Schwab was supported in parts by the Robert Bosch Stiftung Stuttgart, Germany, and the Deutsche Forschungsgemeinschaft under Germany‘s Excellence Strategy‐EXC 2180–390900677.
CONFLICT OF INTEREST STATEMENT
Donato Teutonico is an employee of Sanofi and uses Open Systems Pharmacology software, tools, or models in his professional role. Donato Teutonico and Thorsten Lehr are members of the Open Systems Pharmacology Management Team. Sebastian Frechen uses Open Systems Pharmacology software, tools, or models in his professional role and is a member of the Open Systems Pharmacology Sounding Board. All other authors declared no competing interests for this work.
Supporting information
Appendix S1
Appendix S2
ACKNOWLEDGMENT
Open Access funding enabled and organized by Projekt DEAL.
Loer HLH, Feick D, Rüdesheim S, et al. Physiologically based pharmacokinetic modeling of tacrolimus for food–drug and CYP3A drug–drug–gene interaction predictions. CPT Pharmacometrics Syst Pharmacol. 2023;12:724‐738. doi: 10.1002/psp4.12946
REFERENCES
- 1. Astellas Pharma, Inc . Highlights of prescribing information PROGRAF® (tacrolimus). 2021.
- 2. Lentine KL, Smith JM, Hart A, et al. OPTN/SRTR 2020 annual data report: kidney. Am J Transplant. 2022;22:21‐136. doi: 10.1111/ajt.16982 [DOI] [PubMed] [Google Scholar]
- 3. Takagi T, Ramachandran C, Bermejo M, Yamashita S, Yu LX, Amidon GL. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol Pharm. 2006;3:631‐643. doi: 10.1021/mp0600182 [DOI] [PubMed] [Google Scholar]
- 4. Tsunashima D, Kawamura A, Murakami M, et al. Assessment of tacrolimus absorption from the human intestinal tract: open‐label, randomized, 4‐way crossover study. Clin Ther. 2014;36:748‐759. doi: 10.1016/j.clinthera.2014.02.021 [DOI] [PubMed] [Google Scholar]
- 5. Hebert MF, Fisher RM, Marsh CL, Dressler D, Bekersky I. Effects of rifampin on tacrolimus pharmacokinetics in healthy volunteers. J Clin Pharmacol. 1999;39:91‐96. doi: 10.1177/00912709922007499 [DOI] [PubMed] [Google Scholar]
- 6. Saeki T, Ueda K, Tanigawara Y, Hori R, Komano T. Human P‐glycoprotein transports cyclosporin a and FK506. J Biol Chem. 1993;268:6077‐6080. [PubMed] [Google Scholar]
- 7. Venkataramanan R, Swaminathan A, Prasad T, et al. Clinical pharmacokinetics of tacrolimus. Clin Pharmacokinet. 1995;29:404‐430. doi: 10.2165/00003088-199529060-00003 [DOI] [PubMed] [Google Scholar]
- 8. Dai Y, Hebert MF, Isoherranen N, et al. Effect of CYP3A5 polymorphism on tacrolimus metabolic clearance in vitro. Drug Metab Dispos. 2006;34:836‐847. doi: 10.1124/dmd.105.008680 [DOI] [PubMed] [Google Scholar]
- 9. Brunet M, van Gelder T, Åsberg A, et al. Therapeutic drug monitoring of tacrolimus‐personalized therapy: second consensus report. Ther Drug Monit. 2019;41:261‐307. doi: 10.1097/FTD.0000000000000640 [DOI] [PubMed] [Google Scholar]
- 10. Kim EJ, Kim SJ, Huh KH, et al. Clinical significance of tacrolimus intra‐patient variability on kidney transplant outcomes according to pre‐transplant immunological risk. Sci Rep. 2021;11:12114. doi: 10.1038/s41598-021-91630-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bekersky I, Dressler D, Mekki QA. Effect of low‐ and high‐fat meals on tacrolimus absorption following 5 mg single oral doses to healthy human subjects. J Clin Pharmacol. 2001;41:176‐182. doi: 10.1177/00912700122009999 [DOI] [PubMed] [Google Scholar]
- 12. Huppertz A, Bollmann J, Behnisch R, et al. Differential effect of a continental breakfast on tacrolimus formulations with different release characteristics. Clin. Pharmacol. Drug Dev. 2021;10:899‐907. doi: 10.1002/cpdd.924 [DOI] [PubMed] [Google Scholar]
- 13. Zheng S, Tasnif Y, Hebert MF, et al. Measurement and compartmental modeling of the effect of CYP3A5 gene variation on systemic and intrarenal tacrolimus disposition. Clin Pharmacol Ther. 2012;92:737‐745. doi: 10.1038/clpt.2012.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Birdwell KA, Decker B, Barbarino JM, et al. Clinical pharmacogenetics implementation consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin Pharmacol Ther. 2015;98:19‐24. doi: 10.1002/cpt.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drug development and drug interactions: FDA table of substrates, inhibitors and inducers. Accessed April 23, 2022. https://www.fda.gov/drugs/drug‐interactions‐labeling/drug‐development‐and‐drug‐interactions‐table‐substrates‐inhibitors‐and‐inducers
- 16. Huppertz A, Ott C, Bruckner T, et al. Prolonged‐release tacrolimus is less susceptible to interaction with the strong CYP3A inhibitor voriconazole in healthy volunteers. Clin Pharmacol Ther. 2019;106:1290‐1298. doi: 10.1002/cpt.1529 [DOI] [PubMed] [Google Scholar]
- 17. Imamura CK, Furihata K, Okamoto S, Tanigawara Y. Impact of cytochrome P450 2C19 polymorphisms on the pharmacokinetics of tacrolimus when coadministered with voriconazole. J Clin Pharmacol. 2016;56:408‐413. doi: 10.1002/jcph.605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vanhove T, Annaert P, Knops N, de Loor H, de Hoon J, Kuypers DRJ. In vivo CYP3A4 activity does not predict the magnitude of interaction between itraconazole and tacrolimus from an extended release formulation. Basic Clin Pharmacol Toxicol. 2019;124:50‐55. doi: 10.1111/bcpt.13092 [DOI] [PubMed] [Google Scholar]
- 19. Pappas PG, Alexander BD, Andes DR, et al. Invasive fungal infections among organ transplant recipients: results of the transplant‐associated infection surveillance network (TRANSNET). Clin Infect Dis. 2010;50:1101‐1111. doi: 10.1086/651262 [DOI] [PubMed] [Google Scholar]
- 20. Zhuang X, Lu C. PBPK modeling and simulation in drug research and development. Acta Pharm Sin B. 2016;6:430‐440. doi: 10.1016/j.apsb.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Türk D, Fuhr LM, Marok FZ, et al. Novel models for the prediction of drug–gene interactions. Expert Opin Drug Metab Toxicol. 2021;17:1293‐1310. doi: 10.1080/17425255.2021.1998455 [DOI] [PubMed] [Google Scholar]
- 22. Gonzalez D, Rao GG, Bailey SC, et al. Precision dosing: public health need, proposed framework, and anticipated impact. Clin Transl Sci. 2017;10:443‐454. doi: 10.1111/cts.12490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grimstein M, Yang Y, Zhang X, et al. Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration's Office of Clinical Pharmacology. J Pharm Sci. 2019;108:21‐25. doi: 10.1016/j.xphs.2018.10.033 [DOI] [PubMed] [Google Scholar]
- 24. Mitchell BM, Muftakhidinov TW, Jędrzejewski‐Szmek Z. Engauge digitizer sofware. Accessed September 21, 2021. https://merkummitchell.github.io/engauge‐digitizer
- 25. Wojtyniak J‐G, Britz H, Selzer D, Schwab M, Lehr T. Data digitizing: accurate and precise data extraction for quantitative systems pharmacology and physiologically‐based pharmacokinetic modeling. CPT Pharmacometrics Syst Pharmacol. 2020;9:322‐331. doi: 10.1002/psp4.12511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guest EJ, Aarons L, Houston JB, Rostami‐Hodjegan A, Galetin A. Critique of the two‐fold measure of prediction success for ratios: application for the assessment of drug‐drug interactions. Drug Metab Dispos. 2011;39:170‐173. doi: 10.1124/dmd.110.036103 [DOI] [PubMed] [Google Scholar]
- 27. Hanke N, Frechen S, Moj D, et al. PBPK models for CYP3A4 and P‐gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacometrics Syst Pharmacol. 2018;7:647‐659. doi: 10.1002/psp4.12343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li X, Frechen S, Moj D, et al. A physiologically based pharmacokinetic model of voriconazole integrating time‐dependent inhibition of CYP3A4, genetic polymorphisms of CYP2C19 and predictions of drug–drug interactions. Clin Pharmacokinet. 2020;59:781‐808. doi: 10.1007/s40262-019-00856-z [DOI] [PubMed] [Google Scholar]
- 29. Petan JA, Undre N, First MR, et al. Physiochemical properties of generic formulations of tacrolimus in Mexico. Transplant Proc. 2008;40:1439‐1442. doi: 10.1016/j.transproceed.2008.03.091 [DOI] [PubMed] [Google Scholar]
- 30. Mercuri A, Wu S, Stranzinger S, et al. In vitro and in silico characterisation of tacrolimus released under biorelevant conditions. Int J Pharm. 2016;515:271‐280. doi: 10.1016/j.ijpharm.2016.10.020 [DOI] [PubMed] [Google Scholar]
- 31. Langenbucher F. Linearization of dissolution rate by the Weibull distribution. J Pharm Pharmac. 1972;24:979‐981. doi: 10.1111/j.2042-7158.1972.tb08930.x [DOI] [PubMed] [Google Scholar]
- 32. Bekersky I, Dressler D, Mekki Q. Effect of time of meal consumption on bioavailability of a single oral 5 mg tacrolimus dose. J Clin Pharmacol. 2001;41:289‐297. doi: 10.1177/00912700122010104 [DOI] [PubMed] [Google Scholar]
- 33. Lainesse A, Hussain S, Monif T, et al. Bioequivalence studies of tacrolimus capsule under fasting and fed conditions in healthy male and female subjects. Arzneimittel‐Forschung. 2008;58:242‐247. doi: 10.1055/s-0031-1296500 [DOI] [PubMed] [Google Scholar]
- 34. Takizawa Y, Kishimoto H, Nakagawa M, et al. Effects of pharmaceutical excipients on membrane permeability in rat small intestine. Int J Pharm. 2013;453:363‐370. doi: 10.1016/j.ijpharm.2013.05.055 [DOI] [PubMed] [Google Scholar]
- 35. Deng J, Zhu X, Chen Z, et al. A review of food–drug interactions on oral drug absorption. Drugs. 2017;77:1833‐1855. doi: 10.1007/s40265-017-0832-z [DOI] [PubMed] [Google Scholar]
- 36. Koziolek M, Alcaro S, Augustijns P, et al. The mechanisms of pharmacokinetic food‐drug interactions – a perspective from the UNGAP group. Eur J Pharm Sci. 2019;134:31‐59. doi: 10.1016/j.ejps.2019.04.003 [DOI] [PubMed] [Google Scholar]
- 37. Kuypers DRJ, Claes K, Evenepoel P, Maes B, Vanrenterghem Y. The rate of gastric emptying determines the timing but not the extent of oral tacrolimus absorption: simultaneous measurement of drug exposure and gastric emptying by carbon‐14‐octanoic acid breath test in stable renal allograft recipients. Drug Metab Dispos. 2004;32:1421‐1425. doi: 10.1124/dmd.104.001503 [DOI] [PubMed] [Google Scholar]
- 38. Undre NA, Stevenson P, Schäfer A. Pharmacokinetics of tacrolimus: clinically relevant aspects. Transplant Proc. 1999;31:21 S‐24 S. doi: 10.1016/S0041-1345(99)00788-5 [DOI] [PubMed] [Google Scholar]
- 39. Shityakov S, Förster C. In silico structure‐based screening of versatile P‐glycoprotein inhibitors using polynomial empirical scoring functions. Adv Appl Bioinforma Chem. 2014;7:1‐9. doi: 10.2147/AABC.S56046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reitman ML, Chu X, Cai X, et al. Rifampin's acute inhibitory and chronic inductive drug interactions: experimental and model‐based approaches to drug‐drug interaction trial design. Clin Pharmacol Ther. 2011;89:234‐242. doi: 10.1038/clpt.2010.271 [DOI] [PubMed] [Google Scholar]
- 41. Jogiraju VK, Avvari S, Gollen R, Taft DR. Application of physiologically based pharmacokinetic modeling to predict drug disposition in pregnant populations. Biopharm Drug Dispos. 2017;38:426‐438. doi: 10.1002/bdd.2081 [DOI] [PubMed] [Google Scholar]
- 42. Zhang H, Bu F, Li L, et al. Prediction of drug–drug interaction between tacrolimus and principal ingredients of Wuzhi capsule in chinese healthy volunteers using physiologically‐based pharmacokinetic modelling. Basic Clin Pharmacol Toxicol. 2018;122:331‐340. doi: 10.1111/bcpt.12914 [DOI] [PubMed] [Google Scholar]
- 43. Emoto C, Johnson TN, Hahn D, et al. A theoretical physiologically‐based pharmacokinetic approach to ascertain covariates explaining the large interpatient variability in tacrolimus disposition. CPT Pharmacometrics Syst Pharmacol. 2019;8:273‐284. doi: 10.1002/psp4.12392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bekersky I, Dressler D, Alak A, Boswell GW, Mekki QA. Comparative tacrolimus pharmacokinetics: normal versus mildly hepatically impaired subjects. J Clin Pharmacol. 2001;41:628‐635. doi: 10.1177/00912700122010519 [DOI] [PubMed] [Google Scholar]
- 45. Bekersky I, Dressler D, Mekki QA. Dose linearity after oral administration of tacrolimus 1‐mg capsules at doses of 3, 7, and 10 mg. Clin Ther. 1999;21:2058‐2064. doi: 10.1016/S0149-2918(00)87237-9 [DOI] [PubMed] [Google Scholar]
- 46. Gantar K, Škerget K, Mochkin I, Bajc A. Meeting regulatory requirements for drugs with a narrow therapeutic index: bioequivalence studies of generic once‐daily tacrolimus. Drug Healthc Patient Saf. 2020;12:151‐160. doi: 10.2147/DHPS.S256455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mancinelli LM, Frassetto L, Floren LC, et al. The pharmacokinetics and metabolic disposition of tacrolimus: a comparison across ethnic groups. Clin Pharmacol Ther. 2001;69:24‐31. doi: 10.1067/mcp.2001.113183 [DOI] [PubMed] [Google Scholar]
- 48. Wring S, Murphy G, Atiee G, et al. Clinical pharmacokinetics and drug‐drug interaction potential for coadministered SCY‐078, an oral fungicidal glucan synthase inhibitor, and tacrolimus. Clin Pharmacol Drug Dev. 2019;8:60‐69. doi: 10.1002/cpdd.588 [DOI] [PubMed] [Google Scholar]
- 49. Bekersky I, Dressler D, Colburn W, Mekki Q. Bioequivalence of 1 and 5 mg tacrolimus capsules using a replicate study design. J Clin Pharmacol. 1999;39:1032‐1037. doi: 10.1177/00912709922011791 [DOI] [PubMed] [Google Scholar]
- 50. Stifft F, Vanmolkot F, Scheffers I, van Bortel L, Neef C, Christiaans M. Rectal and sublingual administration of tacrolimus: a single‐dose pharmacokinetic study in healthy volunteers. Br J Clin Pharmacol. 2014;78:996‐1004. doi: 10.1111/bcp.12420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tortorici MA, Parks V, Matschke K, Korth‐Bradley J, Patat A. The evaluation of potential pharmacokinetic interaction between sirolimus and tacrolimus in healthy volunteers. Eur J Clin Pharmacol. 2013;69:835‐842. doi: 10.1007/s00228-012-1407-2 [DOI] [PubMed] [Google Scholar]
- 52. Undre N, Dickinson J. Relative bioavailability of single doses of prolonged‐release tacrolimus administered as a suspension, orally or via a nasogastric tube, compared with intact capsules: a phase 1 study in healthy participants. BMJ Open. 2017;7:1‐7. doi: 10.1136/bmjopen-2016-012252 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Appendix S2