

Key Points
-
•
Maturation of embryonic Gata2+/− HSPCs is disturbed by aberrant endothelial gene expression.
-
•
Hematopoietic-specific induction of gfi1b restores the number of embryonic HSCs in gata2b−/− zebrafish.
Visual Abstract
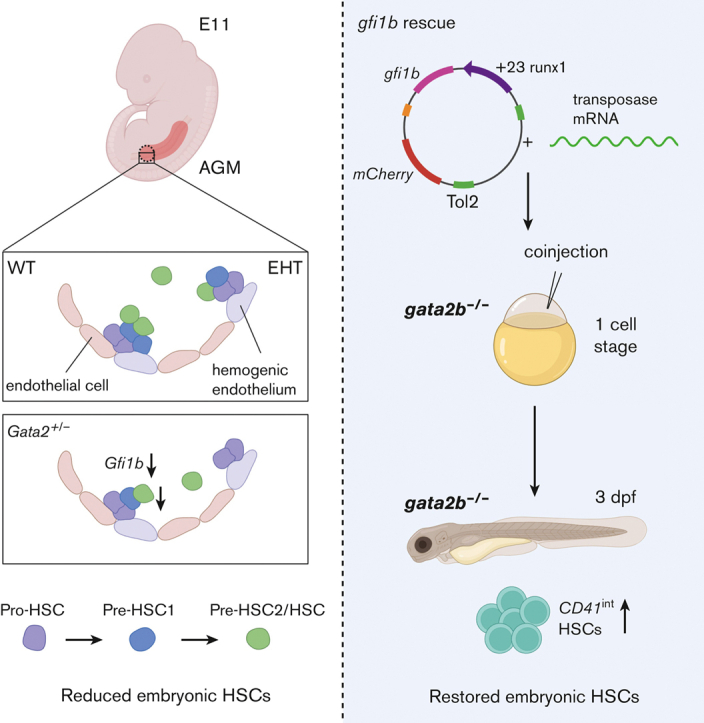
Abstract
The first hematopoietic stem cells (HSCs) are formed through endothelial-to-hematopoietic transition (EHT) during embryonic development. The transcription factor GATA2 is a crucial regulator of EHT and HSC function throughout life. Because patients with GATA2 haploinsufficiency have inborn mutations, prenatal defects are likely to influence disease development. In mice, Gata2 haploinsufficiency (Gata2+/−) reduces the number and functionality of embryonic hematopoietic stem and progenitor cells (HSPCs) generated through EHT. However, the embryonic HSPC pool is heterogeneous and the mechanisms underlying this defect in Gata2+/− embryos remain unclear. Here, we investigated whether Gata2 haploinsufficiency selectively affects a cellular subset undergoing EHT. We showed that Gata2+/− HSPCs initiate, but cannot fully activate, hematopoietic programming during EHT. In addition, due to the reduced activity of the endothelial repressor Gfi1b, Gata2+/− HSPCs cannot repress endothelial identity to complete maturation. Finally, we showed that hematopoietic-specific induction of gfi1b could restore HSC production in gata2b-null (gata2b−/−) zebrafish embryos. This study illustrates the pivotal role of Gata2 in the regulation of the transcriptional network governing HSPC identity throughout the EHT.
Introduction
Hematopoiesis relies on multipotent self-renewing hematopoietic stem cells (HSCs). HSCs originate from the ventral wall of the embryonic dorsal aorta in the aorta-gonad-mesonephros (AGM) region. In the AGM region, definitive hematopoietic stem and progenitor cells (HSPCs) are generated through a transdifferentiation process from a specialized endothelial cell (EC) compartment with hematopoietic potential (hemogenic endothelial cells [HECs]). This process of endothelial-to-hematopoietic transition (EHT) is conserved between mammalian and nonmammalian vertebrates.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 In mice, EHT events occur between embryonic day 10.5 (E10.5) to E12.5. Phenotypic HSPCs emerge from intra-aortic hematopoietic clusters (IAHCs) through EHT and coexpress endothelial markers such as CD31 and hematopoietic markers like cKit.12 Previous studies showed that HSPCs develop through a multistep maturation process within IAHCs and that only a small fraction of IAHC cells become multipotent HSCs.13, 14, 15 Throughout AGM maturation, HSPCs gradually repress the endothelial-specific gene expression and upregulate the expression of hematopoietic-specific genes.16, 17, 18, 19 After their emergence from the AGM, HSCs migrate to the fetal liver and eventually colonize the bone marrow (BM) around birth to maintain the hematopoietic system throughout life.11
The transcription factor GATA2 is one of the key regulators of hematopoietic programming. In mice, Gata2 is required for embryonic HSC generation and survival. Although germ line deletion of Gata2 (Gata2−/−) is lethal at E10, that is, just before the appearance of the first HSCs, Gata2 haploinsufficiency (Gata2+/−) severely reduces the number of embryonic HSPCs, but these heterozygous mice survive to adulthood despite reduced numbers of HSCs.20, 21, 22, 23, 24 Moreover, conditional deletion of Gata2 in HECs does not fully abrogate the formation of IAHCs, but depletes the functional HSCs.20 In addition, HSCs do not survive from conditional deletion of Gata2 after their emergence and become apoptotic.20
The clinical manifestation of germ line heterozygous GATA2 mutations results in GATA2 haploinsufficiency syndrome in patients. Typically, patients present with BM failure and are at a high (80%) risk of developing myelodysplastic syndrome or acute myeloid leukemia before the age of 40.25, 26, 27, 28, 29, 30 Although GATA2 expression is required in HSCs during both embryonic and adult stages, the consequences of embryonic GATA2 haploinsufficiency for disease development remain unexplored.
Despite the requirement for Gata2 during EHT, mechanisms specifically involved in the production and growth of HPSCs in healthy and GATA2 haploinsufficiency states are incompletely understood. Here we sought to understand why GATA2 haploinsufficiency depletes phenotypic HSCs. In this study, we explored how embryonic Gata2 haploinsufficiency affects EHT and the development of the first phenotypic HSCs in mouse AGM. We showed that hematopoietic programming was not abrogated in Gata2+/− E11 HSPCs. However, the maturation of Gata2+/− HSPCs was disturbed and transcriptional profiling showed that Gata2 is a key factor in the gene regulatory network in nascent HSCs. Importantly, we demonstrated that Gata2 regulated Gfi1b to repress endothelial gene expression during HSPC maturation. Ectopic expression of gfi1b restored the number of phenotypic HSCs in gata2b-deficient zebrafish embryos, revealing a previously unidentified role of Gata2 in modulating the transcriptional programming and maturation of HSPCs during EHT.
Materials and methods
Mouse and zebrafish models
Gata2+/− mice,24 gata2b−/− zebrafish,31 and Tg(CD41:GFP) zebrafish32 have been previously described. All animals were housed and bred in animal facilities at the Erasmus Medical Center, Rotterdam, The Netherlands. Animal studies were approved by the Animal Welfare and Ethics Committees of the Erasmus Medical Center in accordance with legislation in the Netherlands.
Whole-mount immunofluorescence staining
E11 wild-type (WT) and Gata2+/− AGMs were dissected, prepared, and mounted as previously described.33 Primary antibody CD117 (cKit) rat anti-mouse (Invitrogen) was combined with secondary antibody Alexa Fluor-488 goat antirat (Invitrogen) for cKit visualization. CD31 was visualized using biotinylated CD31 rat anti-mouse (BD Biosciences) and Cy5-conjugated Streptavidin (Jackson Immunoresearch) primary and secondary antibodies, respectively. The entire AGM region of each sample was imaged using a Leica SP5 confocal microscope. CD31 and cKit double-positive cells were analyzed using Leica Application Suite X (version 4.3) software.
RNA isolation and sequencing
Cells were sorted in Trizol (Sigma), and total RNA isolation was performed according to the standard protocol using GenElute LPA (Sigma). RNA quality and quantity were assessed using a 2100 Bioanalyzer (Agilent) and RNA 6000 Pico Kit (Agilent). Complementary DNA was prepared using the SMARTer procedure with the SMARTer Ultra Low RNA kit (Clontech) and sequenced on a Novaseq 6000 platform (Illumina).
Gene set enrichment and network analysis
Gene expression values were measured as fragments per kilobase of exon per million fragments mapped, and differential expression analysis was performed using the DESeq2 package in the R environment. Gene set enrichment analysis (GSEA) was performed on the fragments per kilobase of exon per million fragments mapped values using the curated gene sets from the Molecular Signatures Database. GSEA results were used as input for network analysis performed using Cytoscape software.
Assay for transposase-accessible chromatin with sequencing
The cells were processed for library preparation using the previously described protocol by the Delwel group.34 Libraries were quantified using the Qubit and NEBNext Library Quant Kit for Illumina (NEB). The quality of the libraries was determined by visualizing peak distribution using a 2100 Bioanalyzer. The samples were sequenced on a Novaseq 6000 platform. Bigwig files were generated using the bamCoverage tool from deepTools and visualized using Integrative Genomics Viewer software.
Flow cytometry and fluorescence-activated cell sorting
AGM regions were dissected as described before.33 Tissues were incubated with collagenase I (Sigma) in phosphate-buffered saline (PBS) supplemented with 5 IU/mL penicillin, 5 μg/mL streptomycin, and 10% fetal calf serum (FCS) for 45 minutes at 37°C. The cells were stained using the following antibodies: PE-Cy7 anti-mouse CD31, APC rat anti-mouse CD117 (cKit, BD Bioscience), FITC anti-mouse CD41 (Biolegend), PE rat anti-mouse CD43 (BD Bioscience), and Alexa Fluor-700 rat anti-mouse CD45 (BD Bioscience). All antibody incubations were performed in PBS + 10% FCS for 30 minutes on ice. After washing with PBS + 10% FCS at 1000 revolutions per minute for 10 minutes, cell pellets were resuspended in 1:1000 4′,6-diamidino-2-phenylindole in PBS + 10% FCS for live/dead cell discrimination. Fluorescence-activated cell sorted events were recorded, and the cells were sorted using a FACSAria III (BD Biosciences). The results were analyzed and visualized using the FlowJo 7.6.5 software.
CFU assay
Cells were incubated in MethoCult GF M3434 (Stem Cell Technologies) supplemented with 5 IU/mL penicillin and 5 μg/mL streptomycin at 37°C. Colony-forming units (CFUs) were scored after 11 days of culture. The growth of primitive erythroid progenitor cells (burst-forming unit–erythroid) and granulocyte-macrophage progenitor cells (CFU–granulocyte-macrophage, CFU-granulocyte, and CFU-macrophage) were scored using an inverted microscope.
Generation of a gfi1b construct
WT sequences of gfi1b, mCherry, and runx1 +23 enhancer were separately cloned into pJET1.2 vectors using the CloneJet PCR Cloning Kit (Thermo Fisher Scientific). After transformation, the outgrown colonies were selected for DNA isolation. The presence of the insert was confirmed by restriction enzyme digestion using BglII (NEB) followed by agarose gel electrophoresis (1% and 5%). DNA fragments were then used as a polymerase chain reaction template for the Gibson cloning reaction. Fragments were amplified using overhang primers (supplemental Table 1) and purified using a DNA Clean & Concentrator kit (Zymo Research). The pUC19-iTol2 backbone was digested with BamHI-HF (NEB) overnight at 37°C, and NEBuilder HiFi Assembly MasterMix (NEB) was used for the assembly of the fragments. The correct assembly was determined by HindIII restriction enzyme digestion and polymerase chain reaction amplification of the fragments. All transformations were performed using Escherichia coli and by performing heat shock for 30 seconds at 42°C followed by recovery in SOC Outgrowth Medium (NEB) for 1 hour. All colonies were grown in lysogeny broth medium plates supplemented with carbenicillin (50 mg/mL; 1000:1 volume-to-volume ration), and DNA from individual colonies was isolated using a QIAprep Spin Miniprep Kit (Qiagen).
Generation of mRNA transposase
The plasmid with the iTol2 sequence was linearized using the Not1 restriction enzyme (NEB). Linearized DNA was used as a template and RNA synthesis was performed using the HiScribe SP6 RNA Synthesis Kit (NEB) according to the manufacturer's instructions. Messenger RNA (mRNA) was precipitated using 3 M sodium acetate (1:100) and 100% ethanol (3:1) and incubated overnight at −20 °C. Transposase mRNA was verified using 0.7% agarose gel electrophoresis.
Microinjection and embryo selection
The injection needles were prepared using a P-30 Magnetic Glass Microelectrode Vertical Needle Puller (Sutter Instrument). The gfi1b construct and mRNA transposase were coinjected into single cell of WT (CD41:GFP) or gata2b−/− (CD41:GFP) zebrafish embryos at the 1-cell stage using a PV830 Pneumatic PicoPump (WPI). Embryos were anesthetized with 160 mg/L Tricaine (Sigma) for the selection of reporter expression. Reporter expression was assessed using the Leica DMLB fluorescence microscope.
cmyb ISH
After injections, embryos were treated with 0.003% 1-phenyl-2-thiourea (Sigma) at 24 hours postfertilization (hpf) and fixed overnight with 4% paraformaldehyde in PBS containing 3% sucrose at 33 hpf. In situ hybridization (ISH) for cmyb was performed as previously described.31,35 The cmyb probe was a gift from Roger Patient. The results were imaged using an inverted microscope.
Statistics
Statistical analysis was performed using the GraphPad Prism 8.0.1 software. Normally distributed data were analyzed using an unpaired t test; otherwise, the Mann-Whitney test was used. The significance cutoff was set at P < .05.
Results
E11 Gata2+/− HSPCs undergo incomplete EHT
To explore the mechanisms leading to diminished HSPCs in Gata2+/− embryos, we sorted CD31+cKit+ HSPCs from E11 WT and Gata2+/− AGMs and performed RNA sequencing (Figure 1A). We found significant differences in principal component analysis (PCA) between the transcriptomic signatures of WT and Gata2+/− HSPCs (Figure 1B). To understand the biological processes affected by the differentially expressed genes between the 2 genotypes, we performed GSEA using curated gene sets and compared our data to a previously published data set showing upregulated and downregulated gene sets found in EC, HEC, and HSPC compartments during EHT.36 A hematopoietic-specific gene set Hematopoiesis stem cell (upregulated in HECs and HSPCs compared with ECs) was overrepresented in Gata2+/− HSPCs compared with WT, indicating that there was no defect in the initiation of hematopoietic programming in these cells (Figure 1C). Surprisingly, an endothelial-specific gene, set Vegfa targets, which were downregulated in HSPCs compared with ECs and HECs, was also enriched in Gata2+/− HSPCs (Figure 1D). The upregulation of both hematopoietic and endothelial signatures of Gata2+/− HSPCs suggests that these cells can initiate EHT, but are not able to complete it.
Figure 1.

E11 Gata2+/− HSPCs exhibit aberrant hematopoietic and endothelial transcriptome. (A) Sorting strategy for CD31+cKit+ cells from E11 WT (left) or Gata2+/− (right) embryos. (B) PCA of the E11 WT (green) and Gata2+/− (orange) HSPCs. Dots represent the transcriptome of CD31+cKit+ cells from individual embryos (WT, n = 4; Gata2+/−, n = 3). Gene sets upregulated in Gata2+/− HSPCs compared with WT HSPCs in GSEA for Hematopoiesis stem cell (C) and Vegfa targets (D). (E) Network analysis comparing E11 WT and Gata2+/− HSPCs. Red dots show upregulated gene sets and blue dots show downregulated gene sets in Gata2+/− HSPCs compared with those in WT. (F) Comparison of the fragments per kilobase of exon per million fragments mapped (FPKM) values of endothelial (Kdr, Cxcl12, Sox17, and Ramp2) and hematopoietic (Gfi1b, Notch1, Hey1, Hes1, Cdkn1c, Igf2, Myb, and Gpr56)–specific genes between WT and Gata2+/− HSPCs. (G) Comparison of open chromatin between CD31+cKit+ cells isolated from individual E11 WT (N = 3, green) or Gata2+/− (N = 4, orange) embryos visualized using Integrative Genomics Viewer software. Accessible chromatin for Gfi1b and its +16 and +17 distal enhancer regions was analyzed. The peak range was set to minimum = 0 and maximum = 174 for all samples. The tool bar was 10 kb long. Error bars represent standard error of the mean. ∗P < .05, ∗∗P < .01.
To further investigate the transcriptomic differences between WT and Gata2+/− HSPCs, we performed network analysis using Cytoscape software (Figure 1E). In this analysis, dots represent gene sets, lines connect gene sets sharing the same genes, and gene sets associated with the same biological processes form clusters indicated by bigger circles. Earlier studies showed that HSPCs are highly proliferative during EHT.37 Strikingly, many gene sets related to Cell cycle, DNA repair, Transcription, and Translation networks were downregulated in Gata2+/− HSPCs, indicating that these processes are abrogated (Figure 1E). Conversely, gene sets related to Cell junction organization, Integrin signaling, Notch signaling, and Extracellular matrix formation were significantly upregulated in Gata2+/− HSPCs (Figure 1E).
These results suggest that the inability of some embryonic Gata2+/− HSPCs to complete EHT is due to a defective switch from endothelial to hematopoietic programming, which possibly prevents their maturation into HSCs.
Endothelial repressor transcription factor Gfi1b is downregulated in E11 Gata2+/− HSPCs
Because GSEA and network analysis both indicated that endothelial and hematopoietic genes were enriched in Gata2+/− HSPCs, the expression of genes defining these transcriptomic signatures was investigated. Endothelial-specific genes such as Kdr, Cxcl12, Sox17, and Ramp2 were significantly upregulated in Gata2+/− HSPCs (Figure 1F), along with an aberrant hematopoietic transcriptome. Although some genes indicating hematopoietic programming such as Notch1 and its targets Hey1 and Hes1, Cdkn1c and Igf2 were upregulated, other hematopoietic-specific genes such as Myb and Gpr56 (Adgrg1) were significantly downregulated in Gata2+/− HSPCs (Figure 1F). These results support the hypothesis that Gata2+/− HSPCs can initiate hematopoietic programming, but cannot fully gain hematopoietic characteristics due to impaired switching from endothelial-specific to hematopoietic-specific transcriptional programs. Notably, Gfi1b expression was significantly reduced in the Gata2+/− HSPCs (Figure 1F). Gfi1b is known to be responsible for the loss of endothelial identity during EHT and its expression is essential for the formation of IAHCs from HECs.38,39 Gfi1b is directly activated by Gata2 through the hematopoietic-specific +16 and +17 kb distal enhancer regions at E11.5 mouse embryos.40 To further investigate whether Gata2 haploinsufficiency causes Gfi1b downregulation due to the reduced activity in the enhancer regions of Gfi1b, we sorted CD31+cKit+ HSPCs from E11 WT and Gata2+/− AGMs and performed assay for transposase-accessible chromatin with sequencing to determine chromatin accessibility. Strikingly, both +16 and +17 kb distal enhancer regions of Gfi1b were less accessible in Gata2+/− HSPCs than in WT HSPCs, indicating that Gata2 regulates Gfi1b expression through these distal enhancer regions during EHT (Figure 1G) or that the cells that express Gfi1b are not present at levels similar to those in the controls.
Gata2 haploinsufficiency impairs HSPC maturation during EHT
The effects of Gfi1b and incomplete suppression of endothelial identity as well as the multistep maturational process in Gata2+/− HSPCs were examined. Previous research showed that HSPCs develop within IAHCs through pro-HSC (CD31+cKit+CD41loCD43−CD45−) to pre-HSC type I (pre-HSC1 or CD31+cKit+CD41loCD43+CD45−) to pre-HSC type II (pre-HSC2) and HSC (CD31+cKit+CD41loCD43+CD45+) transitions.13, 14, 15 In this maturation hierarchy, only the most mature compartment (pre-HSC2/HSCs) contains transplantable HSCs and can produce hematopoietic colonies in CFU-culture (CFU-C).15
Because HSC maturation requires activation of Gfi1b and consequential downregulation of endothelial genes,38,39 we investigated whether Gata2 haploinsufficiency blocks a specific stage of HSC maturation during EHT. To test this, we dissected WT and Gata2+/− AGMs from E11 embryos and performed flow cytometry. Using antibody combinations for CD31, cKit, CD41, CD43, and CD45 (supplemental Figure 1A), the number of pro-HSC, pre-HSC1, and pre-HSC2/HSC cells per in E11 WT and Gata2+/− AGMs were analyzed (Figure 2A). We found that the numbers of E11 pro-HSCs were comparable between WT and Gata2+/− AGMs, indicating that the first step of HSPC formation was not hampered in Gata2+/− embryos (Figure 2B). In contrast, the numbers of pre-HSC1 cells were moderate, and pre-HSC2/HSC cells were more prominently reduced in E11 Gata2+/− embryos, suggesting that Gata2+/− HSPCs cannot complete HSC maturation (Figure 2B).
Figure 2.

HSPC maturation within IAHCs is impaired in Gata2+/− during EHT. (A) Gating strategy to determine HSPC maturation of E11 WT and Gata2+/− AGMs. Representative images of pro-HSC, pre-HSC1, and pre-HSC2 gating obtained from the WT (left) or Gata2+/− (right) AGMs. (B) Quantification of the number of pro-HSC, pre-HSC1, and pre-HSC2 populations in E11 WT (n = 11) or Gata2+/− (n = 13) AGMs. (C) Unbiased heatmap of the transcriptomic signatures of pro-HSC, pre-HSC1, and pre-HSC2 populations from 3 independent E11 WT or 3 independent Gata2+/− AGMs. (D) PCA showing the transcriptome of each sample obtained by RNA sequencing. Black arrows indicate Gata2+/− samples. The green arrow indicates the maturation trajectory based on the transcriptome of WT HSPCs. FPKM values of Gata2 (E), Gfi1b (F), and Notch1 (G) depicted for each stage of maturation and compared between the WT and Gata2+/−. Error bars represent standard error of the mean. Color code for samples according to genotype: WT samples in the shades of green and Gata2+/− samples in the shades of orange. Color code for maturation steps: pro-HSCs, light gray; pre-HSC1, gray; pre-HSC2, dark blue. ∗P < .05.
To investigate whether the maturation of pro-HSCs into pre-HSCs was impaired or delayed in Gata2+/− AGMs, flow cytometry analysis was performed on E12 and E13 AGMs dissected from WT and Gata2+/− embryos. At E12, the numbers of pre-HSC2/HSCs were higher than those at E11 in both WT and Gata2+/−, indicating that HSPCs were still actively undergoing maturation at this stage (supplemental Figure 1B, comparing with Figure 2B). Both pre-HSC1 and pre-HSC2 cells were significantly reduced in Gata2+/− compared with WT AGMs at E12 (supplemental Figure 1B). The numbers of pro-HSC, pre-HSC1, and pre-HSC2/HSC cells were markedly reduced in both WT and Gata2+/− AGMs at E13 compared to those at E11 or E12 (supplemental Figure 1B-C, comparing with Figure 2B); however, Gata2+/− AGMs still contained fewer HSPCs at E13 than WT AGMs (supplemental Figure 1C).
Next, we investigated whether the functionality of Gata2+/− HSCs is altered at E11. Earlier CFU-C studies showed that Gata2+/− HSPCs produce fewer hematopoietic colonies than WT,20 but it remains unclear whether this was due to a reduction in the number of pre-HSC2/HSCs or due to a reduction in their potential to generate CFUs. To address this, we sorted pro-HSC, pre-HSC1, and pre-HSC2/HSC populations from E11 WT and Gata2+/− AGMs, and performed a CFU-C (day 11) assay to quantitatively assess the subset hematopoietic potential. Colony counts normalized to the number of cells plated per dish revealed that the functionality of pre-HSC2/HSCs was preserved in WT and Gata2+/− AGMs, as this population from both genotypes produced similar type and number of hematopoietic colonies (supplemental Figure 1D). In addition, the pro-HSC and pre-HSC1 populations of both WT and Gata2+/− AGMs did not form any colonies after 11 days of culture, confirming that these cells had not yet acquired HSC potential (data not shown).
These results indicate that Gata2+/− HSPCs are arrested in pro-HSC to pre-HSC maturation during EHT. The ability of Gata2+/− AGMs to still produce a few pre-HSC2/HSCs indicates that HSC generation is not fully blocked, but allows at least some haploinsufficient cells to maintain a CFU potential similar to that of WT pre-HSC2/HSCs.
Pre-HSC2/HSCs are marked by unique transcriptomic signatures
To investigate the molecular basis of the effects of Gata2+/− on the pro-HSC stage of maturation, RNA sequencing was performed on the pro-HSC, pre-HSC1, and pre-HSC2/HSC compartments from E11 WT and Gata2+/− AGMs. Differentially expressed genes between the WT and Gata2+/− HSPC subtypes are shown in supplemental Materials 1-3. Heatmaps comparing the transcriptomic signatures of each cell population from WT and Gata2+/− AGMs showed that the overall pro-HSC and pre-HSC1 compartments of both WT and Gata2+/− had comparable transcriptomes that dramatically change during pre-HSC2/HSC maturation (Figure 2C). PCA confirmed that in both WT and Gata2+/−, the transcriptome of pre-HSC2/HSCs clustered separately from the pro-HSC and pre-HSC1 compartments (Figure 2D). Pre-HSC2/HSCs from both genotypes uniquely expressed Ptprc (CD45) were marked by the specific expression of Hdc, Fgl2, Slc45a, Padi2, Rab44, and Fcgr3 (Figure 2C), and Flt4, Ptprm, Itga1, and Sox17 expression was extinguished in pre-HSC2/HSCs of both WT and Gata2+/−, indicating that mature HSCs acquire a unique transcriptomic signature (Figure 2C).
Gata2+/− HSPCs fail to repress endothelial programming throughout HSC maturation
Flow cytometry analysis showed that HSPC maturation in Gata2+/− was predominantly impaired during the pro-HSC to pre-HSC1 transition (Figure 2B; supplemental Figure 1B). Comparison of the transcriptomes of WT and Gata2+/− by heatmap analysis and PCA showed that the transcriptome profiles of WT pre-HSC1 cells were distributed between pro-HSC and pre-HSC2/HSCs states. However, Gata2+/− pre-HSC1 cells were transcriptionally closer to a pro-HSC–like state (Figure 2C-D). Although some endothelial-specific genes, such as Sox7 and Flt4, were expressed in both WT and Gata2+/− pre-HSC1 cells (Figure 2C), other endothelial-specific genes, such as Igfbp2, Vwa1, and Gja4, were upregulated in Gata2+/− compared with WT pre-HSC1 cells (supplemental Figure 2A-C; supplemental Material 2).
WT and Gata2+/− pre-HSC2/HSCs did not show striking differences in the transcriptome (Figure 2C; supplemental Material 3), which probably explains the preserved functionality in CFU-C assays. Expression of some endothelial-specific genes, such as Sox17, Kdr, and Flt1, was silenced during pre-HSC1 to pre-HSC2/HSC maturation in both WT and Gata2+/− cells (Figure 2C), whereas others, such as Cxcl12, Col3a1, and Aplnr, were upregulated in Gata2+/− pre-HSC2/HSCs compared with WT. (Figure 2C; supplemental Figure 2D-F).
These differences indicate that the mature pre-HSC1 and pre-HSC2/HSC compartments of Gata2+/− are transcriptionally distinguishable from those of WT, suggesting that Gata2+/− HSPCs incompletely repress endothelial programming through maturation from pro-HSC to pre-HSC1 and pre-HSC2/HSC. However, some Gata2+/− HSPCs are still capable of undergoing complete AGM maturation, despite carrying endothelial transcriptomic signatures. To test if the pre-HSC2/HSCs that were generated in Gata2+/− embryos still expressed endothelial proteins, we performed flow cytometry analysis for the endothelial markers Flk1 and VE-Cadherin. The percentage of Flk1 (not shown) and VE-Cadherin expressing cells was significantly increased in Gata2+/− CD31+ and CD31+cKit+ cells compared with WT controls. Furthermore, upon maturation of CD31+cKit+CD45− cells into CD31+cKit+CD45+ cells, Flk1 (not shown) and VE-Cadherin were significantly downregulated during maturation (supplemental Figure 3F). However, the few Gata2 haploinsufficient HSPCs that were generated still had increased levels of VE-Cadherin compared with the WT controls (supplemental Figure 3E-F).
Gata2+/− HSPCs incompletely activate hematopoietic programming throughout HSC maturation
Because some hematopoietic transcriptomic signatures were upregulated, whereas others were downregulated in Gata2+/− HSPCs (Figure 1F), we investigated whether this observation was due to a reduced number of mature pre-HSC2/HSCs within HSPCs. To explore this, the expression levels of hematopoietic-specific cKit, CD44, and Myb genes were examined at the individual stages of maturation. For both WT and Gata2+/−, the expression of these genes increased throughout maturation (supplemental Figure 2G-I). However, levels were reduced in Gata2+/− HSPCs compared with WT in all maturation stages, confirming that hematopoietic transcriptional programming is hampered in all Gata2+/− HSPCs (supplemental Figure 2G-I). Strikingly, the most significant reduction was observed in the Gata2+/− pro-HSCs (supplemental Figure 2G-I; supplemental Material 1).
Hematopoietic-specific genes, such as Vav1, Rac2, and Mpo, were expressed in WT HSPCs at all stages and their levels gradually increased throughout maturation (supplemental Figure 2J-L). The expression of these genes was downregulated most prominently in pro-HSCs and only activated later during pre-HSC1 and pre-HSC2/HSCs maturation in Gata2+/− cells (supplemental Figure 2J-L), indicating a direct effect of Gata2 haploinsufficiency on the onset of hematopoietic programming.
Expression of the transcription factor Runx1 showed slight, but not a significant reduction upon Gata2 haploinsufficiency (data not shown). Notably, previous studies have placed Gata2 both upstream and downstream of Runx1-controlled transcription, illustrative of the complex interplay between these transcriptional regulators.31,41
Hence, Gata2 likely plays a crucial role in HSPCs undergoing EHT by downregulating endothelial identity and upregulating hematopoietic transcriptional programming to promote complete transition to the HSC-like state.
Gfi1 and Gfi1b are downregulated in Gata2+/− HSPCs during pro-HSC to pre-HSC maturation
Previous studies have shown that Gata2 is upregulated in HSPCs compared with ECs and HECs during EHT.42 By comparing the expression levels of Gata2 among pro-HSC, pre-HSC1, and pre-HSC2/HSC compartments, we found that Gata2 expression levels were highest in the most mature compartment (pre-HSC2/HSC) of HSPCs (Figure 2E) and confirmed that Gata2+/− HSPCs in all maturation stages had reduced Gata2 expression compared with WT (Figure 2E).
We next examined whether the expression of Gfi1b, a known repressor of endothelial gene expression, was altered in Gata2+/− embryos. Gfi1b expression levels were reduced in Gata2+/− pro-HSCs, whereas pre-HSC1 and pre-HSC2/HSC compartments showed only a marginal reduction in Gfi1b expression levels compared with WT (Figure 2F). These results suggest that Gata2 haploinsufficiency impairs Gfi1b activation, predominantly in the pro-HSC stage of HSPC maturation.
Previous studies showed that Gfi1, a highly homologous interaction partner of Gfi1b, is also required for IAHC formation during EHT.38,39 CD45+ pre-HSC2/HSCs coexpress Gfi1b and Gfi1.39 Therefore, we examined whether Gfi1 expression was altered in Gata2+/− HSPCs throughout maturation. Strikingly, Gfi1 expression was completely abolished in Gata2+/− pre-HSC1 cells (supplemental Figure 2M).
Notch signaling is upregulated in Gata2+/− HSPCs
Notch signaling is essential for EHT and HSPC maturation, and Notch1 and its targets, such as Hes1, are also required for the initiation of EHT in the AGM.43,44 However, AGM HSCs become Notch-independent throughout maturation, with continued high Notch1 activity, eventually blocking HSC maturation.44 Because our E11 gene set analysis showed upregulated Notch signaling in Gata2+/− HSPCs, we explored the expression levels of individual key Notch signaling mediators. Both Notch1 and Hes1 were enriched in Gata2+/− HSPCs compared with WT (Figure 2G; supplemental Figure 2N), whereas the expression level of Notch2 was comparable between WT and Gata2+/− HSPCs throughout maturation (supplemental Figure 2O). In accordance with Gata2 being a known downstream target of Notch1, these results suggest a possible feedback regulation of Notch signaling by Gata2 during EHT and HSPC maturation.
Both IAHCs and SBCs are diminished in Gata2+/− AGMs
Our results indicated that HSPC maturation within IAHCs was inhibited in Gata2+/− AGMs. A recent study reported that all IAHCs express Gata2 but that Gata2-expressing CD27+ single bulging cells (SBCs) in 1- to 2-cell clusters are HSCs.45 To clarify whether Gata2 haploinsufficiency exclusively diminishes HSPC maturation within IAHCs or has an effect on these SBCs, we dissected AGMs from E11 WT and Gata2+/− embryos and performed whole-mount immunofluorescence staining for CD31 and cKit (Figure 3A-C). In whole WT and Gata2+/− AGMs, we quantified the number of CD31+cKit+ SBCs and the CD31+cKit+ cells located in IAHCs (Figure 3D, IAHC; Figure 3E, SBC; Figure 3G). We also assessed the number of single CD31+cKit+ cells that were not attached to the endothelium or IAHCs and were found in the aortic lumen (Figure 3F-G). Our results showed that the number of CD31+cKit+ cells within all AGM compartments decreased in E11 Gata2+/− embryos, with significant reductions in both IAHCs and SBCs (Figure 3G). Furthermore, because CD27 is expressed in both SBCs and some IAHCs, and is also a marker for multipotent HSPCs,45 we examined the expression level of CD27 throughout the maturation stages. CD27 expression levels were greatly reduced in the Gata2+/− pre-HSC1 compartment (Figure 3H), suggesting that Gata2 haploinsufficiency does not exclusively affect HSC maturation within IAHCs. Additionally, it reduced the HSC potential within the AGM HSPC pool, thus indicating a broader function of Gata2 during EHT.
Figure 3.

Both IAHCs and SBCs are depleted in Gata2+/− AGMs. (A) Illustration of the AGM region dissected for analysis of E11 WT or Gata2+/− embryos. Representative images of CD31+cKit+ cells obtained by confocal imaging of E11 WT AGM (B), Gata2+/− AGM (C), IAHCs (D), SBC (E), and aortic lumen (AL) (F) cell. Scale bars, 50 μm. (G) Quantification of CD31+cKit+ cells located in IAHCs, as SBCs, and AL cell within E11 WT (n = 3) and Gata2+/− (n = 4) AGMs. (H) FPKM values of CD27 depicted for each stage of the maturation and compared between WT and Gata2+/−. Error bars represent standard error of the mean. ∗P < .05, ∗∗P < .01.
Hematopoietic-specific gfi1b induction restores embryonic HSCs in gata2b−/− zebrafish
To test whether the effect of Gata2 haploinsufficiency on EHT can be overcome by inducing Gfi1b expression, we took advantage of the previously described gata2b−/− zebrafish model.31 In addition, in zebrafish, we detected increased blood vessel morphogenesis gene expression in HSCs (CD41int) of gata2b+/− embryos at 48 hpf compared with WT (Figure 4A-B). Because in gata2b+/− embryos, the numeric differences of HSPCs are very low, we assessed definitive HSPCs as marked by cmyb whole-mount ISH at 33 hpf and CD41int cell number at 3 days postfertilization from homozygous deleted embryos as these markers are significantly reduced in gata2b−/− embryos.31 To test the effect of ectopic gfi1b expression on gata2b−/− HSPCs, we injected a HEC tissue specific gfi1b expression cassette into WT and gata2b−/− CD41:GFP embryos at 1-cell stage. We found that, upon gfi1b induction, cmyb mRNA levels were normalized in gata2b−/− HSPCs at 33 hpf (Figure 4C-D). In addition, the number of CD41int HSCs in gata2b−/− zebrafish was higher in gfi1b-expressing embryos than in uninjected controls at 3 days postfertilization (Figure 4E-G).
Figure 4.

gfi1b induction restores the number of embryonic HSCs in gata2b−/− zebrafish. (A) Gene set enrichment plot depicting the expression of genes involved in blood vessel morphogenesis, highly enriched in gata2b+/−CD41int cells at 48 hpf compared with WT. (B) Expression data of endothelial markers from RNA sequencing results of CD41int HSCs of WT (green) and Gata2b+/− (orange) HSCs at 48 hpf in 3 biological replicates. Significance is shown as ∗adjusted P < .05 and ∗∗adjusted P < .001. (C) Representative images of 3 different staining intensities of cmyb whole-mount in situ expression detecting HSPCs along the dorsal aorta of 33 hpf zebrafish embryos. High cmyb expression is depicted in red, intermediate cmyb expression is depicted in green, and low cmyb expression is depicted in yellow. (D) Quantification of cmyb signal intensity analyzed at 33 hpf using ISH and compared between uninjected (WT, n = 2; gata2b−/−, n = 16) and injected (WT, n = 18; gata2b−/−, n = 16) WT and gata2b−/− embryos. Representative images of the number of CD41int HSCs compared between the uninjected (n = 5) and injected (n = 4) groups of WT (E), and uninjected (n = 5) and injected (n = 6) groups of gata2b−/− embryos (F). (G) The proportion of CD41int HSCs compared between the uninjected and injected groups of WT and gata2b−/− embryos. The dots represent individual samples and each sample contains 4 pooled embryos. dpf, days postfertilization; FACS, fluorescence-activated cell sorting; FDR, false discovery rate; GFP, green fluorescent protein; NES, normalized enrichment score.
Collectively, these results show that ectopic gfi1b expression can rescue the embryonic phenotype of gata2b−/− zebrafish, and indicate that Gata2 regulates Gfi1b expression during EHT.
Discussion
In this study, we aimed to assess the role of Gata2 in the generation of phenotypic HSCs via EHT. Although HSPCs do not completely switch off their endothelial gene expression and remain expressing endothelial markers, such as CD31, throughout EHT and maturation, moderate repression of endothelial programming is essential for maturation and thus for establishing HSC fate.16, 17, 18, 19 We showed that Gata2 haploinsufficiency does not completely abrogate hematopoietic programming during EHT, but reduces the ability of HSPCs to complete HSC maturation in the AGM.
Gata2+/− HSPCs were predominantly blocked during pro-HSC to pre-HSC maturation and functional HSCs (pre-HSC2/HSCs) were significantly reduced at both E11 and E12 AGMs. We showed that phenotypic Gata2+/− HSCs (pre-HSC2/HSCs) not only failed to repress endothelial genes, such as Cxcl12 and Col3a1, they also incompletely activated hematopoietic genes, such as cKit and Myb, during AGM maturation. Together, these results imply that Gata2 acts as a mediator in gene regulation of endothelial and hematopoietic transcriptional programs during EHT. Previous studies have shown that Gata2−/− embryos do not form IAHCs in the AGM, implying that Gata2 expression is required for EHT.20,24 Our results extend these observations by showing that Gata2 plays multiple roles during EHT and is a critical novel regulator of HSPC maturation.
Despite the fact that HSC numbers were reduced in Gata2+/− AGMs, and their transcriptome was distinct from that of WT HSCs, CFU assays showed that their functionality was preserved. These results revised the observations from previous studies, where it was shown that the colony-forming ability of Gata2+/− AGM HSPCs is reduced20 and now clarify that this is due to impaired HSC maturation in Gata2+/− AGMs and not to a reduction in HSC functionality. However, CFU assays are limited when testing the repopulating ability and lymphoid potential of HSCs. Therefore, transplantation studies are required to elucidate the true potential of embryonic Gata2+/− HSCs. It is expected that these cells will be less robust when transplanted competitively with WT AGM HSCs, as has been previously shown for BM HSCs.22
Gfi1b was downregulated and hematopoietic distal enhancers of Gfi1b were less active in Gata2+/− HSPCs. Because Gfi1b expression is required to repress endothelial programming in HSPCs and for the formation of IAHCs during EHT, we propose that Gata2 activates Gfi1b through its +16 and +17 distal enhancer regions to repress the endothelial identity of HSPCs. Restoring the number of HSCs in gata2b−/− zebrafish embryos upon ectopic gfi1b expression confirmed that Gfi1b acts downstream of Gata2, and showed that this regulatory mechanism is essential for the generation of embryonic HSCs. Although previous studies have provided in silico and experimental evidence suggesting that Gata2 and Gfi1b regulate each other,40 to our knowledge, this study is the first experimental proof of the phenotypic consequences of disruption of this regulatory mechanism during EHT.
Importantly, we found increased activity of Notch signaling in Gata2+/− HSPCs. Previous studies in mice and zebrafish have shown that Notch signaling is an upstream activator of Gata2.41,46 Because the expression of Gata2 is reduced in Gata2+/− HSPCs, increased Notch signaling could be activated as a compensatory mechanism in these cells. Therefore, our results suggest a possible feedback regulation of Notch signaling through Gata2 expression. Because it has been shown that downregulation of Notch signaling is required during HSPC maturation,44 we cannot exclude the possibility that impaired HSPC maturation in Gata2+/− AGMs is influenced by high Notch activity. Although inducing gfi1b can sufficiently rescue the embryonic phenotype in gata2b−/− embryos, the contribution of Notch signaling requires further investigation.
Finally, we showed that many gene sets related to Cell cycle, Proliferation, and DNA repair were downregulated in Gata2+/− HSPCs throughout all maturation stages (supplemental Figure 4A-C). Although decreased proliferative signatures are hallmarks of impaired HSPC maturation, whether this influences the genome stability of Gata2+/− HSPCs remains to be explored. However, this could explain the observation that, although Gata2+/− HSPCs are blocked in their differentiation after the pro-HSC stage, they do not expand at this stage. Because HSCs generated through EHT during embryonic stages are the source of the adult HSC pool, further studies are needed to understand the influence of prenatal GATA2 haploinsufficiency on HSC fitness after birth and throughout adulthood.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgments
The authors thank the members of the de Pater laboratory for helpful discussions. The authors thank the Experimental Animal Facility of the Erasmus Medical Center for animal husbandry and the Erasmus Optical Imaging Center for confocal microscopy services.
This work is supported by the European Hematology Association (junior nonclinical research fellowship) (E.d.P.), the Dutch Cancer Foundation KWF/Alpe d’HuZes (SK10321) (E.d.P.), and the Daniel den Hoed Foundation for support of the Cancer Genome Editing Center (I.P.T.).
Authorship
Contribution: E.d.P. and C.K. conceived the study; C.K., E.G., D.V., H.d.L., J.Z., M.H., M.N.D.t.B., and E.B. performed the experiments; C.K., E.G., D.V., R.H., M.A.S., and E.d.P. analyzed the results; I.P.T. provided resources; C.K. and E.d.P. wrote the manuscript; and I.P.T. and E.D. revised the manuscript.
Footnotes
Sequencing data reported in this article have been deposited in the ArrayExpress database (accession numbers E-MTAB-12570, E-MTAB-12571, E-MTAB-12565, and E-MTAB-12577).
Data are available on request from the corresponding author, Emma de Pater (e.depater@erasmusmc.nl).
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Bertrand J Y, Chi N C, Santoso B, Teng S, Stainier DYR, Traver D. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature. 2010;464(7285):108–111. doi: 10.1038/nature08738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boisset J C, van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature. 2010;464(7285):116–120. doi: 10.1038/nature08764. [DOI] [PubMed] [Google Scholar]
- 3.Ciau-Uitz A, Patient R. Gene regulatory networks governing the generation and regeneration of blood. J Comput Biol. 2019;26(7):719–725. doi: 10.1089/cmb.2019.0114. [DOI] [PubMed] [Google Scholar]
- 4.de Bruijn MFTR, Ma X, Robin C, Ottersbach K, Sanchez M J, Dzierzak E. Hematopoietic stem cells localize to the endothelial cell layer in the midgestation mouse aorta. Immunity. 2002;16(5):673–683. doi: 10.1016/s1074-7613(02)00313-8. [DOI] [PubMed] [Google Scholar]
- 5.Dieterlen-Lievre F. On the origin of haemopoietic stem cells in the avian embryo: an experimental approach. J Embryol Exp Morphol. 1975;33(3):607–619. [PubMed] [Google Scholar]
- 6.Ivanovs A, Rybtsov S, Ng E S, Stanley E G, Elefanty A G, Medvinsky A. Human haematopoietic stem cell development: from the embryo to the dish. Development. 2017;144(13):2323–2337. doi: 10.1242/dev.134866. [DOI] [PubMed] [Google Scholar]
- 7.Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature. 2010;464(7285):112–115. doi: 10.1038/nature08761. [DOI] [PubMed] [Google Scholar]
- 8.Medvinsky A, Dzierzak E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell. 1996;86(6):897–906. doi: 10.1016/s0092-8674(00)80165-8. [DOI] [PubMed] [Google Scholar]
- 9.Müller A M, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity. 1994;1(4):291–301. doi: 10.1016/1074-7613(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 10.North T E, de Bruijn MFTR, Stacy T, et al. Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity. 2002;16(5):661–672. doi: 10.1016/s1074-7613(02)00296-0. [DOI] [PubMed] [Google Scholar]
- 11.Zovein A C, Hofmann J J, Lynch M, et al. Fate tracing reveals the endothelial origin of hematopoietic stem cells. Cell Stem Cell. 2008;3(6):625–636. doi: 10.1016/j.stem.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yokomizo T, Dzierzak E. Three-dimensional cartography of hematopoietic clusters in the vasculature of whole mouse embryos. Development. 2010;137(21):3651–3661. doi: 10.1242/dev.051094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rybtsov S, Batsivari A, Bilotkach K, et al. Tracing the origin of the HSC hierarchy reveals an SCF-dependent, IL-3-independent CD43(-) embryonic precursor. Stem Cell Rep. 2014;3(3):489–501. doi: 10.1016/j.stemcr.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rybtsov S, Sobiesiak M, Taoudi S, et al. Hierarchical organization and early hematopoietic specification of the developing HSC lineage in the AGM region. J Exp Med. 2011;208(6):1305–1315. doi: 10.1084/jem.20102419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taoudi S, Gonneau C, Moore K, et al. Extensive hematopoietic stem cell generation in the AGM region via maturation of VE-cadherin+CD45+ pre-definitive HSCs. Cell Stem Cell. 2008;3(1):99–108. doi: 10.1016/j.stem.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Baron C S, Kester L, Klaus A, et al. Single-cell transcriptomics reveal the dynamic of haematopoietic stem cell production in the aorta. Nat Commun. 2018;9(1):2517. doi: 10.1038/s41467-018-04893-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oatley M, Bölükbası ÖV, Svensson V, et al. Single-cell transcriptomics identifies CD44 as a marker and regulator of endothelial to haematopoietic transition. Nat Commun. 2020;11(1):1–18. doi: 10.1038/s41467-019-14171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swiers G, Baumann C, O’Rourke J, et al. Early dynamic fate changes in haemogenic endothelium characterized at the single-cell level. Nat Commun. 2013;4:1–10. doi: 10.1038/ncomms3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou F, Li X, Wang W, et al. Tracing haematopoietic stem cell formation at single-cell resolution. Nature. 2016;533(7604):487–492. doi: 10.1038/nature17997. [DOI] [PubMed] [Google Scholar]
- 20.de Pater E, Kaimakis P, Vink C S, et al. Gata2 is required for HSC generation and survival. J Exp Med. 2013;210(13):2843–2850. doi: 10.1084/jem.20130751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao X, Johnson K D, Chang Y I, et al. Gata2 cis-element is required for hematopoietic stem cell generation in the mammalian embryo. J Exp Med. 2013;210(13):2833–2842. doi: 10.1084/jem.20130733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ling K W, Ottersbach K, van Hamburg J P, et al. GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. J Exp Med. 2004;200(7):871–882. doi: 10.1084/jem.20031556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodrigues N P, Janzen V, Forkert R, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood. 2005;106(2):477–484. doi: 10.1182/blood-2004-08-2989. [DOI] [PubMed] [Google Scholar]
- 24.Tsai F Y, Keller G, Kuo F C, et al. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371(6494):221–226. doi: 10.1038/371221a0. [DOI] [PubMed] [Google Scholar]
- 25.Dickinson R E, Griffin H, Bigley V, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118(10):2656–2658. doi: 10.1182/blood-2011-06-360313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donadieu J, Lamant M, Fieschi C, et al. French GATA2 study group Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica. 2018;103(8):1278–1287. doi: 10.3324/haematol.2017.181909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hahn C N, Chong C E, Carmichael C L, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012–1017. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu A P, Sampaio E P, Khan J, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118(10):2653–2655. doi: 10.1182/blood-2011-05-356352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ostergaard P, Simpson M A, Connell F C, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome) Nat Genet. 2011;43(10):929–931. doi: 10.1038/ng.923. [DOI] [PubMed] [Google Scholar]
- 30.Spinner M A, Sanchez L A, Hsu A P, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123(6):809–821. doi: 10.1182/blood-2013-07-515528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gioacchino E, Koyunlar C, Zink J, et al. Essential role for Gata2 in modulating lineage output from hematopoietic stem cells in zebrafish. Blood Adv. 2021;5(13):2687–2700. doi: 10.1182/bloodadvances.2020002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma D, Zhang J, Lin H F, Italiano J, Handin R I. The identification and characterization of zebrafish hematopoietic stem cells. Blood. 2011;118(2):289–297. doi: 10.1182/blood-2010-12-327403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yokomizo T, Yamada-Inagawa T, Yzaguirre A D, Chen M J, Speck N A, Dzierzak E. Whole-mount three-dimensional imaging of internally localized immunostained cells within mouse embryos. Nat Protoc. 2012;7(3):421–431. doi: 10.1038/nprot.2011.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ottema S, Mulet-Lazaro R, Erpelinck-Verschueren C, et al. The leukemic oncogene EVI1 hijacks a MYC super-enhancer by CTCF-facilitated loops. Nat Commun. 2021;12(1):1–13. doi: 10.1038/s41467-021-25862-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chocron S, Verhoeven M C, Rentzsch F, Hammerschmidt M, Bakkers J. Zebrafish Bmp4 regulates left-right asymmetry at two distinct developmental time points. Dev Biol. 2007;305(2):577–588. doi: 10.1016/j.ydbio.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Solaimani Kartalaei P, Yamada-Inagawa T, Vink C S, et al. Whole-transcriptome analysis of endothelial to hematopoietic stem cell transition reveals a requirement for Gpr56 in HSC generation. J Exp Med. 2015;212(1):93–106. doi: 10.1084/jem.20140767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zape J P, Lizama C O, Cautivo K M, Zovein A C. Cell cycle dynamics and complement expression distinguishes mature haematopoietic subsets arising from hemogenic endothelium. Cell Cycle. 2017;16(19):1835–1847. doi: 10.1080/15384101.2017.1361569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lancrin C, Mazan M, Stefanska M, et al. GFI1 and GFI1B control the loss of endothelial identity of hemogenic endothelium during hematopoietic commitment. Blood. 2012;120(2):314–322. doi: 10.1182/blood-2011-10-386094. [DOI] [PubMed] [Google Scholar]
- 39.Thambyrajah R, Mazan M, Patel R, et al. GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat Cell Biol. 2016;18(1):21–32. doi: 10.1038/ncb3276. [DOI] [PubMed] [Google Scholar]
- 40.Moignard V, Macaulay I C, Swiers G, et al. Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat Cell Biol. 2013;15(4):363–372. doi: 10.1038/ncb2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobrzycki T, Mahony C B, Krecsmarik M, et al. Deletion of a conserved Gata2 enhancer impairs haemogenic endothelium programming and adult Zebrafish haematopoiesis. Commun Biol. 2020;3(1):71. doi: 10.1038/s42003-020-0798-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eich C, Arlt J, Vink C S, et al. In vivo single cell analysis reveals Gata2 dynamics in cells transitioning to hematopoietic fate. J Exp Med. 2018;215(1):233–248. doi: 10.1084/jem.20170807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robert-Moreno A, Espinosa L, de la Pompa J L, Bigas A. RBPjkappa-dependent Notch function regulates Gata2 and is essential for the formation of intra-embryonic hematopoietic cells. Development. 2005;132(5):1117–1126. doi: 10.1242/dev.01660. [DOI] [PubMed] [Google Scholar]
- 44.Souilhol C, Lendinez J G, Rybtsov S, et al. Developing HSCs become Notch independent by the end of maturation in the AGM region. Blood. 2016;128(12):1567–1577. doi: 10.1182/blood-2016-03-708164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vink C S, Calero-Nieto F J, Wang X, et al. Iterative single-cell analyses define the transcriptome of the first functional hematopoietic stem cells. Cell Rep. 2020;31(6) doi: 10.1016/j.celrep.2020.107627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guiu J, Shimizu R, D'Altri T, et al. Hes repressors are essential regulators of hematopoietic stem cell development downstream of Notch signaling. J Exp Med. 2013;210(1):71–84. doi: 10.1084/jem.20120993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.