

Abstract
Obesity is postulated to independently increase chronic kidney disease (CKD), even after adjusting for type 2 diabetes (T2D) and hypertension. Dysglycemia below T2D thresholds, frequently seen with obesity, also increases CKD risk. Whether obesity increases CKD independent of dysglycemia and hypertension is unknown and likely influences the optimal weight loss (WL) needed to reduce CKD. T2D remission rates plateau with 20–25% WL after bariatric surgery (BS), but further WL increases normoglycemia and normotension. We undertook bidirectional inverse variance weighted Mendelian randomization (IVWMR) to investigate potential independent causal associations between increased BMI and estimated glomerular filtration rate (eGFR) in CKD (CKDeGFR) (<60 mL/min/1.73 m2) and microalbuminuria (MA). In 5,337 BS patients, we assessed whether WL influences >50% decline in eGFR (primary outcome) or CKD hospitalization (secondary outcome), using <20% WL as a comparator. IVWMR results suggest that increased BMI increases CKDeGFR (b = 0.13, P = 1.64 × 10−4; odds ratio [OR] 1.14 [95% CI 1.07, 1.23]) and MA (b = 0.25; P = 2.14 × 10−4; OR 1.29 [1.13, 1.48]). After adjusting for hypertension and fasting glucose, increased BMI did not significantly increase CKDeGFR (b = −0.02; P = 0.72; OR 0.98 [0.87, 1.1]) or MA (b = 0.19; P = 0.08; OR 1.21 [0.98, 1.51]). Post-BS WL significantly reduced the primary outcome with 30 to <40% WL (hazard ratio [HR] 0.53 [95% CI 0.32, 0.87]) but not 20 to <30% WL (HR 0.72 [0.44, 1.2]) and ≥40% WL (HR 0.73 [0.41, 1.30]). For CKD hospitalization, progressive reduction was seen with increased WL, which was significant for 30 to <40% WL (HR 0.37 [0.17, 0.82]) and ≥40% WL (HR 0.24 [0.07, 0.89]) but not 20 to <30% WL (HR 0.60 [0.29, 1.23]). The data suggest that obesity is likely not an independent cause of CKD. WL thresholds previously associated with normotension and normoglycemia, likely causal mediators, may reduce CKD after BS.
ARTICLE HIGHLIGHTS
Obesity is likely not an independent contributor to chronic kidney disease (CKD).
Obesity likely contributes to CKD via effects on glycemia and blood pressure.
Weight loss at thresholds associated with normoglycemia and normotension may reduce CKD.
Introduction
Obesity is a risk factor for chronic kidney disease (CKD), a rising health care burden worldwide that increases morbidity and premature mortality (1,2). Observational studies suggest that obesity may increase CKD, independent of metabolic risk factors such as type 2 diabetes (T2D) and hypertension (3–11). Unlike observational data, Mendelian randomization (MR) is less prone to confounding and can be used to infer causal associations between an exposure and outcome (12). A prior MR study suggested a causal association between increased BMI and CKD but not increased waist-to-hip ratio (a phenotype more strongly associated with metabolic disease) (13). Multivariable MR studies indicate that increased BMI increases CKD, even after adjusting for hypertension and/or T2D, and has been proposed to be an independent causal factor for CKD (14,15). Dysglycemia and elevated fasting glucose (FG) in the non-T2D range have been implicated in glomerular hyperfiltration and associated with CKD in both observational and MR studies (16–19) and may potentially contribute to obesity-associated CKD. Thus, whether obesity is truly an independent causal factor for CKD after adjusting for dysglycemia and hypertension is not known but may inform future strategies to reduce/prevent CKD, including weight loss (WL)–based interventions.
Although WL can potentially reduce/prevent CKD, the optimal amount of WL needed to reduce CKD is unknown and likely depends on the underlying etiology of obesity-associated CKD. Bariatric surgery (BS) is the most efficacious WL intervention and has shown reductions in estimated glomerular filtration rate (eGFR)–based measures of CKD (CKDeGFR) (20,21) in observational studies and microalbuminuria (MA) in people with T2D in a randomized control trial (22). Remission rates of T2D (defined as hemoglobin A1c [HbA1c] <6.5%) after BS plateau after a WL in the 20–25% range (23). WL above this threshold has been associated with an increased likelihood of normalization of glycemia (HbA1c <6%) and achievement of normotension (24–26).
In this study, we undertook bidirectional MR to assess potential causal associations between BMI and CKDeGFR (stage III CKD or worse defined by eGFR <60 mL/min/1.73 m2) (27) and the presence of MA (urinary albumin-to-creatinine ratio [UACR] >25 mg/g in women or >17 mg/g in men) in people of European descent using summary statistics from the largest genome-wide association studies (GWAS). Consistent with prior studies, we found suggestive causal associations between BMI and CKD (14,15) and, therefore, undertook multivariable MR analysis to assess to what extent these potential causal associations are independent of increased FG, T2D, and hypertension. Finally, we assessed the association between WL within 1 year of BS and a 50% decline in eGFR (primary outcome) and CKD hospitalization (secondary outcome).
Research Design and Methods
MR Studies
Cohorts
MR analyses were undertaken in participants of European ancestry using summary statistics from the largest published GWAS. The instruments for glycemic parameters include GWAS of fasting glucose undertaken in people without T2D (i.e., variation in FG in the non-T2D range), as well as GWAS in T2D case and control cohorts (28–34) (Table 1). Informed consent and institutional approval were previously obtained by the individual cohort investigators.
Table 1.
Cohort details
| Trait | Population cohort | Mean age, years | Female, % | Sample size, n | Study participants, n | Control participants, n | PMID |
|---|---|---|---|---|---|---|---|
| BMI | GIANT/UK Biobank | 55.5/56.9* | 54.0/54.2* | 681,275 | 30124842 | ||
| WHR | GIANT/UK Biobank | 55.5/56.9* | 54.0/54.2* | 694,649 | 30239722 | ||
| Hypertension | UK Biobank | 56.9* | 54.2* | 463,010 | 54,358 | 408,652 | GWAS ID: ukb-b-12493a |
| T2D | DIAGRAM/GERA/UK Biobank | 54.1/63.3/56.9* | 50.1/59.0/54.2* | 655,666 | 61,714 | 593,952 | 30054458 |
| FG | MAGIC | 50.9 | 47.7 | 133,010 | 22885924 | ||
| CKD | CKDGen | 54.0 | 50.0 | 625,219 | 64,164 | 561,055 | 31152163 |
| UACR | CKDGen | 57.0 | 54.4 | 547,361 | 31511532 | ||
| MA | CKDGen | 57.0 | 53.0 | 54,116 | 54,116 | 26631737 | |
| eGFR creatinine | CKDGen | 54.0 | 50.0 | 567,460 | 31152163 |
All participants were of European descent. MAGIC, Meta-analyses of Glucose and Insulin-Related Traits Consortium; PMID, PubMed Identifier; WHR, waist-to-hip ratio.
Study-specific characteristics were not available for all UK Biobank data and were extrapolated from data available.
Output from Medical Research Council Integrative Epidemiology Unit GWAS pipeline analysis using Phesant-derived variables from UK Biobank, version 2 (https://doi.org/10.5523/bris.pnoat8cxo0u52p6ynfaekeigi). DIAGRAM, Diabetes Genetics Replication and Meta-analysis; GERA, Genetic Epidemiology Research on Aging; CKDGEN, Chronic Kidney Disease Genetics; MRC-IEU, Medical Research Council Integrative Epidemiology Unit.
Primary MR Analyses
For our primary analysis, we undertook bidirectional MR with CKDeGFR and MA as outcomes and BMI as exposure with subsequent multivariable MR adjusted for hypertension, T2D, and FG. P < 0.05 was considered significant for each analysis. The recently published Strengthening the Reporting of Observational Studies in Epidemiology using MR reporting guideline (35) has been incorporated in this article (Supplementary File 1).
Additional Analyses.
We also undertook additional bidirectional MR analysis assessing the effect of BMI as exposures on UACR and eGFR creatinine and cystatin C–based eGFR measures.
MR Assumptions.
First, the instrument is robustly associated with the exposure; thus, we used single nucleotide polymorphisms (SNPs) that were associated with the exposure at genome-wide significance (12). Second, there is no horizontal pleiotropy, meaning that the instrument does not influence the outcome via another pathway other than the outcome (12). Finally, the instrument is not influenced by any confounders (12). For bidirectional MR, we used inverse variance weighted (IVW) MR (IVWMR) and additional sensitivity analyses, including MR-Egger, weighted median, weighted mode, and leave-one-out analyses.
IVWMR was analyzed using meta-analysis of the individual Wald ratio for each SNP. By permitting the IVW to have a nonzero intercept, MR-Egger relaxes the assumption of no horizontal pleiotropy and returns an unbiased causal estimate, in the case of horizontal pleiotropy, provided that the horizontal pleiotropic effects are not correlated with the SNP-exposure effects (instrument strength independent of direct effect assumption) (12,36). The median effect of all SNPs in the instrument was used for analysis using weighted median MR, allowing SNPs with a greater effect on the association to be evaluated by weighting the contribution of each SNP by the inverse variance of its association with the outcome (37). Even if only 50% of the SNPs satisfy all three MR assumptions, the method is robust (37). Finally, SNPs were clustered into groups based on similarity of causal effects for weighted mode MR, with the cluster with the largest number of SNPs deriving the causal effect estimate (38). To assess heterogeneity, Cochran Q test was used, while leave-one-out analysis was conducted to assess whether any MR estimate was biased by a single SNP potentially with horizontal pleiotropic effect (12). F statistics were calculated manually for continuous exposures (12,39,40).
Univariable MR was conducted using the TwoSampleMR package in R (RStudio version 1.3.1073 and R version 4.0.3). Linkage disequilibrium pruning was used to select a proxy (r2 >0.8) if a SNP was not directly matched from the 1000 Genomes Project. The ggplot2 and metaphor packages in R were used to create plots.
We undertook multivariable IVWMR to assess the effect of BMI on CKD independent of hypertension, T2D, and FG (41). Our data indicate a significant correlation between the effects of BMI-associated SNPs on FG and T2D (Spearman rank correlation ρ = 0.37; S = 527,411; P = 7.9 × 10−7). The correlation remained significant after removing four SNPs that were genome-wide significant for both traits (Spearman ρ = 0.32; S = 527,412; P = 2.4 × 10−5) (Supplementary File 5). Given the potential collinearity between the traits, we undertook an analysis adjusted for each glycemic trait individually and in combination with hypertension. Multivariable MR was conducted using the TwoSampleMR, Multivariable MR, and RMultivariable MR packages in R, where the latter two packages assessed heterogeneity via Cochran Q test and strength of the instrument via F statistics (39,41). Plots were generated using plotobject in R.
Overlap Between Exposure and Outcome Cohorts
A total of 456,426 participants from the UK Biobank composed ∼67% of the Genetic Investigation of Anthropometric Traits (GIANT)/UK Biobank GWAS of BMI, 69% of the Diabetes Genetics Replication and Meta-analysis (DIAGRAM)/Genetic Epidemiology Research on Aging (GERA)/UK Biobank GWAS of T2D, and 80% of the Chronic Kidney Disease Genetics (CKDGEN) GWAS of CKD.
Observation Study in a Cohort of Patients Who Underwent BS
Study Design
We conducted a retrospective cohort study using population-level health care administrative databases in Ontario, Canada. The data sources included the Ontario Bariatric Registry, which provides information on patients assessed at bariatric centers and whether they eventually had surgery. Other data sources included the Discharge Abstracts Database, which includes detailed information on all hospital admissions in Ontario; the Registered Persons Database, with demographic information on all Ontario residents; and the Ontario Laboratory Information System to which all community and most hospital-based laboratories have gradually enrolled since 2006 to contribute laboratory test results. These data sets were linked using unique encoded identifiers and analyzed at ICES. We have received institutional ethics approval for the study.
Patients
We identified all people who underwent primary BS between April 2010 and October 2016. Exclusion criteria included residence outside of Ontario, a diagnosis of CKD prior to surgery, eGFR of ≤45 mL/min/1.73 m2 at baseline, last eGFR before index date of ≤0.6× baseline eGFR, no weight recorded at either the 6- or 12-month follow-up visit at the bariatric center, death or left Ontario in the first 12 months after surgery, and no eGFR measurements in the year prior to surgery or after surgery. The final sample size was 5,337 (Supplementary File 4).
Variables
By comparing the lower of weights recorded at the 6- and 12-month follow-up visit with the baseline weight, we calculated the percentage of WL achieved, categorized into <20%, 20 to <30%, 30 to <40%, and ≥40%. The primary outcome was a 50% reduction from baseline in eGFR. Reduction of eGFR of this magnitude is associated with an increased risk of end-stage CKD and death and has been used in clinical trials (42–44). eGFR was calculated using the Chronic Kidney Disease Epidemiology Collaboration 2021 equation (45):
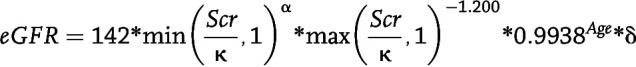 |
where Scr = serum creatinine value (mg/dL), κ = 0.7 for females and 0.9 for males, α = −0.241 for females and −0.302 for males, Age = age of person in years at time of test, and δ = 1.012 for females and 1 for males.
The secondary outcome was first hospitalization for CKD based on the following hospital diagnostic codes: ICD-9 (4030, 4031, 4039, 4040, 4041, 4049, 585,586, 5888, 5889, 2504) and ICD-10 (E102, E112, E132, E142, I12, I13, N08, N18, N19). Outcome ascertainment started 1 year after surgery and ended 31 March 2020.
Baseline characteristics ascertained for each patient included age, sex, income (defined ecologically based on neighborhood-level median household income, divided into quintiles), rurality of residence (46), and prior history of diabetes (47). In addition, the following comorbidities were ascertained from the Ontario Bariatric Registry: hypertension, dyslipidemia, and vascular disease (coronary artery disease, cerebrovascular disease, or heart failure) (48).
Statistical Analysis
Baseline characteristics of the cohort, stratified by the exposure variable, were compared using ANOVA for continuous variables and χ2 tests for categorical variables. Multiple imputation, using five imputed data sets, was used to impute the random missing values for baseline characteristics (49): the number of missing values is included in Table 4. Cox proportional hazards models were fit for each primary and secondary outcome, adjusting for the baseline characteristics and baseline eGFR. The Cox proportional hazards model assumptions were verified with weighted Schoenfeld residuals. Modeling with restricted cubic splines (50) indicated a nonlinear relationship between WL and the primary outcome and linear relationship for the secondary outcome. We therefore assessed outcomes by WL category for the primary outcome. For the secondary outcome, we undertook analyses using WL as a continuous variable. For the primary outcome, patients were censored at the last available eGFR result in the Ontario Laboratory Information System. For the secondary outcome, patients were censored on death or the end of follow-up. We also undertook additional Fine-Gray model analyses with death as a competing risk factor for the secondary outcome (51) but not the primary outcome (as everyone included in the analyses had an eGFR measurement and were alive). Analyses were repeated among the subgroup of patients who had a history of diabetes prior to surgery.
Table 4.
Baseline characteristics according to maximum WL categories for patients undergoing BS in the observational study cohort
| Maximum WL within 1 year postsurgery, % body weight | ||||||
|---|---|---|---|---|---|---|
| <20% | 20 to <30% | 30 to <40% | ≥40% | Missing value, n | P | |
| Participants, n | 426 | 1,894 | 2,184 | 833 | ||
| Age, years | ||||||
| Mean (SD) | 48.81 (9.62) | 47.35 (10.28) | 45.58 (10.10) | 43.41 (9.66) | 0 | <0.0001 |
| Median (IQR) | 50 (42–56) | 48 (40–55) | 46 (38–53) | 43 (37–50) | <0.0001 | |
| Sex, n (% female) | 330 (77.5) | 1,531 (80.8) | 1,865 (85.4) | 739 (88.7) | <0.0001 | |
| Income quintile, n (%) | ||||||
| 1 | 101 (23.7) | 428 (22.6) | 509 (23.3) | 184 (22.1) | 14 | 0.047 |
| 2 | 79 (18.5) | 439 (23.2) | 513 (23.5) | 183 (22.0) | ||
| 3 | 97 (22.8) | 424 (22.4) | 419 (19.2) | 201 (24.1) | ||
| 4 | 76 (17.8) | 346 (18.3) | 435 (19.9) | 158 (19.0) | ||
| 5 | 72 (16.9) | 255 (13.5) | 298 (13.6) | 106 (12.7) | ||
| Rural residence, n (%) | ||||||
| No | 375 (88.0) | 1,640 (86.6) | 1,819 (83.3) | 692 (83.1) | 0 | 0.003 |
| Yes | 51 (12.0) | 254 (13.4) | 365 (16.7) | 141 (16.9) | ||
| Baseline BMI, kg/m2 | ||||||
| Mean (SD) | 48.32 (8.21) | 48.95 (8.25) | 48.92 (7.57) | 50.68 (7.88) | 9 | <0.0001 |
| Median (IQR) | 47 (42–53) | 47 (43–54) | 48 (43–53) | 49 (45–55) | <0.0001 | |
| Diabetes, n (%) | ||||||
| No | 193 (45.3) | 969 (51.2) | 1,363 (62.4) | 585 (70.2) | 0 | <0.0001 |
| Yes | 233 (54.7) | 925 (48.8) | 821 (37.6) | 248 (29.8) | ||
| Hypertension, n (%) | ||||||
| Missing data | 3 (0.7) | 4 (0.2) | 3 (0.1) | 0 (0.0) | 10 | <0.0001 |
| No | 172 (40.4) | 871 (46.0) | 1,125 (51.5) | 478 (57.4) | ||
| Yes | 251 (58.9) | 1,019 (53.8) | 1,056 (48.4) | 355 (42.6) | ||
| Hyperlipidemia, n (%) | ||||||
| Missing data | 4 (0.9) | 4 (0.2) | 4 (0.2) | 2 (0.2) | 14 | <0.0001 |
| No | 237 (55.6) | 1,143 (60.3) | 1,458 (66.8) | 599 (71.9) | ||
| Yes | 185 (43.4) | 747 (39.4) | 722 (33.1) | 232 (27.9) | ||
| Vascular composite, n (%) | ||||||
| Missing data | 4 (0.9) | 5 (0.3) | 3 (0.1) | 2 (0.2) | 14 | 0.0292 |
| No | 370 (86.9) | 1,716 (90.6) | 1,976 (90.5) | 761 (91.4) | ||
| Yes | 52 (12.2) | 173 (9.1) | 205 (9.4) | 70 (8.4) | ||
| Baseline serum creatinine, μmol/L | ||||||
| Mean (SD) | 68.57 (15.46) | 66.06 (13.37) | 65.73 (12.89) | 65.53 (12.60) | 0 | 0.0004 |
| Median (IQR) | 67 (58–76) | 65 (57–74) | 65 (57–72) | 64 (57–72) | 0.0066 | |
| Baseline eGFR, mL/min/1.73 m2 | ||||||
| Mean (SD) | 98.27 (16.79) | 101.36 (15.40) | 102.26 (15.32) | 103.15 (15.52) | 0 ≥ | <0.0001 |
| Median (IQR) | 101 (88–110) | 104 (91–113) | 105 (94–113) | 106 (93–115) | <0.0001 | |
| Days from baseline creatinine measurement to surgery | ||||||
| Mean (SD) | 111.30 (107.40) | 118.13 (108.33) | 133.76 (110.07) | 146.44 (110.78) | <0.0001 | |
| Median (IQR) | 64 (19–190) | 79 (21–211) | 115 (26–229) | 143 (32–240) | <0.0001 | |
Cox proportional hazards models were fit for each primary and secondary outcome, adjusting for the baseline characteristics and baseline eGFR. Multiple imputation was used to impute missing values. ANOVA tests were used for continuous variables, and χ2 tests were used for categorical variables.
Data and Resource Availability
All data are included in the article.
Results
MR Studies
Primary Outcome
MR Analyses of BMI as Exposure and CKD (eGFR <60 mL/min/1.73 m2) as Outcome.
Consistent with previous data, MR suggested that increased BMI increases CKD (b = 0.134; P = 1.64 × 10−4; odds ratio [OR] 1.143 [95% CI 1.066, 1.226]), which remained significant after adjusting for T2D and hypertension (b = 0.124; P = 0.011; OR 1.132 [1.028, 1.246]). However, the effect was not significant after adjusting for hypertension and FG (b = −0.02; P = 0.72; OR 0.98 [0.87, 1.10) (Table 2, Fig. 1, and Supplementary File 2). Mediation analysis indicates hypertension mediated ∼40% and elevated glucose ∼45% of the effect of obesity and in combination, likely entirely mediated the effect of increased BMI on CKD (Fig. 1).
Table 2.
Univariable and multivariable MR analyses of BMI adjusted for hypertension, T2D, and FG as exposure and CKD or MA as outcome
| Method | Β | SE | P | Egger intercept | P Egger | Cochran Q | Q df | PQ | I 2 | F | OR (95% CI) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariable MR analysis (exposure: BMI, 446 SNPs; outcome: CKD) | |||||||||||
| MR-Egger | 0.095 | 0.092 | 0.306 | 0.001 | 0.644 | 612.243 | 444 | 1.89E-07 | 27.480 | 97.437 | 1.099 (0.917, 1.317) |
| Weighted median | 0.121 | 0.058 | 0.035 | 97.437 | 1.129 (1.008, 1.264) | ||||||
| IVW | 0.134 | 0.036 | 1.64E-04 | 612.538 | 445 | 2.14E-07 | 27.351 | 97.437 | 1.143 (1.066, 1.226) | ||
| Simple mode | −0.125 | 0.162 | 0.440 | 97.437 | 0.882 (0.642, 1.213) | ||||||
| Weighted mode | 0.078 | 0.098 | 0.427 | 97.437 | 1.081 (0.892, 1.308) | ||||||
| Multivariable MR analysis (exposure: BMI adjusted for hypertension and T2D, 340 SNPs; outcome: CKD) | |||||||||||
| IVW | 0.124 | 0.049 | 0.011 | 575.703 | 380 | 3.31E-10 | 33.994 | 16.082 | 1.132 (1.028, 1.246) | ||
| Multivariable MR analysis (exposure: BMI adjusted for hypertension and FG, 167 SNPs; outcome: CKD) | |||||||||||
| IVW | −0.021 | 0.059 | 0.725 | 240.787 | 180 | 1.67E-03 | 25.245 | 12.119 | 0.980 (0.873, 1.099) | ||
| Univariable MR analysis (exposure: BMI, 441 SNPs; outcome: MA) | |||||||||||
| MR-Egger | 0.477 | 0.182 | 0.009 | −0.004 | 0.190 | 444.000 | 439 | 0.424 | 1.126 | 97.437 | 1.611 (1.127, 2.303) |
| Weighted median | 0.340 | 0.122 | 0.005 | 97.437 | 1.405 (1.104, 1.786) | ||||||
| IVW | 0.255 | 0.069 | 2.14E-04 | 445.741 | 440 | 0.415 | 1.288 | 97.437 | 1.291 (1.128, 1.478) | ||
| Simple mode | 0.446 | 0.324 | 0.170 | 97.437 | 1.562 (0.827, 2.949) | ||||||
| Weighted mode | 0.416 | 0.202 | 0.040 | 97.437 | 1.516 (1.020, 2.255) | ||||||
| Multivariable MR analysis (exposure: BMI adjusted for hypertension and T2D, 338 SNPs; outcome: MA) | |||||||||||
| IVW | 0.141 | 0.090 | 0.117 | 383 | 378.000 | 0.418 | 1.318 | 16.232 | 1.151 (0.965, 1.372) | ||
| Multivariable MR analysis (exposure: BMI adjusted for hypertension and FG, 164 SNPs; outcome: MA) | |||||||||||
| IVW | 0.194 | 0.110 | 0.080 | 168 | 177.000 | 0.675 | −5.382 | 13.524 | 1.214 (0.977, 1.507) | ||
Figure 1.


Univariable and multivariable MR analysis. The exposure is BMI adjusted for hypertension, T2D, and FG. The outcome is CKD (eGFR <60 mL/min/1.73 m2) as a binary outcome. A: Scatter plot showing the SNPs associated with BMI against SNPs associated with CKD (vertical and horizontal gray lines around points show 95% CI for five different MR association tests. B: Funnel plot of the effect size against the inverse of the SE for each SNP. C: Multivariable MR analyses of increased BMI adjusted for covariates on CKD as the outcome.
MR Analyses of BMI as Exposure and MA as Outcome.
Univariable MR indicated that increased BMI increases MA (b = 0.255; P = 2.14 × 10−4; OR 1.291 [95% CI 1.128, 1.478]). This finding was not significant after adjustment for hypertension and T2D (b = 0.141; P = 0.117; OR 1.151 [0.965, 1.372), consistent with prior data. Mediation analysis indicated that hypertension mediates ∼11% and T2D ∼48%, with a combined estimate of ∼52% of the effect of increased BMI on MA. Similarly, multivariable MR suggested that elevated FG is a likely contributor to BMI-associated MA. The causal association between increased BMI and MA was not significant after adjustment for hypertension and FG (b = 0.19; P = 0.08; OR 1.21 [0.98, 1.51]) (Table 2, Fig. 2, and Supplementary File 2). In combination, hypertension and increased FG are estimated to mediate ∼20% of the effect of increased BMI on MA (Fig. 2).
Figure 2.


Univariable and multivariable MR analysis. The exposure is BMI adjusted for hypertension, T2D, and FG. The outcome is MA as a binary outcome. A: Scatter plot showing the SNPs associated with BMI against SNPs associated with MA (vertical and horizontal gray lines around points show 95% CI for five different MR association tests. B: Funnel plot of the effect size against the inverse of the SE for each SNP. C: Multivariable MR analyses of increased BMI adjusted for covariates on MA as the outcome.
Reverse MR Analyses of CKD and MA as Exposure on BMI as Outcome.
We did not find evidence that CKD defined by eGFR <60 mL/min/1.73 m2 (b = 0.009; P = 0.495) or presence of MA (b = 0.003; P = 0.835) increased BMI (Supplementary File 3).
Additional Analyses
Univariable MR indicated that increased BMI decreases UACR (b = −0.031 ± 0.011; P = 0.004) (Table 3). Multivariable MR after adjustment for hypertension and T2D (b = −0.071; P = 3.9 × 10−7) and hypertension and FG (b = −0.061; P = 0.001) indicated that BMI decreases UACR. Univariable MR indicated that increased BMI reduces cystatin C–based eGFR (b = −0.05; P = 6.8 × 10−16), which remained significant after adjustment for hypertension and T2D (−0.05; P = 1 × 10−8) and hypertension and FG (b = −0.03; P = 0.03) (Table 3). Univariable MR suggested that increased BMI did not affect creatinine-based eGFR (b = −0.0004; P = 0.8) (Table 3). We did not find MR evidence that creatinine- or cystatin C–based measures of eGFR or UACR impacts BMI (Supplementary File 3).
Table 3.
Univariable and multivariable MR analyses of BMI adjusted for hypertension, T2D, and FG as exposure and UACR, eGFR defined using creatinine, and eGFR defined using cystatin C as continuous outcome
| Method | Β | SE | P | Egger intercept | P Egger | Cochran Q | Q df | PQ | I 2 | F | OR (95% CI) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariable MR analysis (exposure: BMI, 446 SNPs; outcome: UACR) | |||||||||||
| MR-Egger | 0.051 | 0.028 | 0.068 | −0.001 | 0.001 | 1,194.343 | 444 | 4.64E-70 | 62.825 | 97.437 | 1.052 (0.996, 1.111) |
| Weighted median | −0.027 | 0.012 | 0.029 | 97.437 | 0.973 (0.950, 0.997) | ||||||
| IVW | −0.031 | 0.011 | 0.004 | 1,221.872 | 445 | 1.23E-73 | 63.580 | 97.437 | 0.969 (0.949, 0.990) | ||
| Simple mode | −0.058 | 0.047 | 0.217 | 97.437 | 0.944 (0.861, 1.035) | ||||||
| Weighted mode | −0.038 | 0.032 | 0.236 | 97.437 | 0.962 (0.904, 1.025) | ||||||
| Multivariable MR analysis (exposure: BMI adjusted for hypertension and T2D, 340 SNPs; outcome: UACR) | |||||||||||
| IVW | −0.071 | 0.014 | 3.86E-07 | 969.480 | 380 | 3.53E-53 | 60.804 | 16.080 | 0.932 (0.907, 0.958) | ||
| Multivariable MR analysis (exposure: BMI adjusted for hypertension and FG, 167 SNPs; outcome: UACR) | |||||||||||
| IVW | −0.061 | 0.019 | 0.001 | 510.937 | 180 | 1.87E-33 | 64.771 | 12.119 | 0.941 (0.906, 0.976) | ||
| Univariable MR analysis (exposure: BMI, 446 SNPs; outcome: eGFR defined using creatinine) | |||||||||||
| MR Egger | 0.004 | 0.005 | 0.439 | 0.000 | 0.355 | 1,429.722 | 444 | 0.000 | 68.945 | 97.437 | 1.004 (0.994, 1.015) |
| Weighted median | 0.004 | 0.002 | 0.102 | 97.437 | 1.004 (0.999, 1.008) | ||||||
| IVW | −4.24E-04 | 0.002 | 0.835 | 1,432.480 | 445 | 0.000 | 68.935 | 97.437 | 1.000 (0.996, 1.004) | ||
| Simple mode | 0.008 | 0.008 | 0.345 | 97.437 | 1.008 (0.992, 1.024) | ||||||
| Weighted mode | 0.006 | 0.004 | 0.148 | 97.437 | 1.006 (0.998, 1.015 | ||||||
| Univariable MR analysis (exposure: BMI, 469 SNPs; outcome: eGFR defined using cystatin C) | |||||||||||
| MR Egger | −0.05 | 0.018 | 0.0014 | 0.00 | 0.04 | 1,334.57 | 444 | 2.36E-4 | 63.48 | 97.437 | |
| Weighted median | −0.06 | 0.01 | 1.09E-7 | 97.437 | |||||||
| IVW | −0.05 | 0.007 | 6.8E-16 | 1,384.91 | 445 | 2.41E-4 | 63.31 | 97.437 | |||
| Simple mode | −0.06 | 0.022 | 0.017 | 97.437 | |||||||
| Weighted mode | −0.06 | 0.016 | 0.0013 | 97.437 | |||||||
| Multivariable MR analysis (exposure: BMI adjusted for hypertension and T2D, 338 SNPs; outcome: eGFR defined using cystatin C) | |||||||||||
| IVW | −0.05 | 0.09 | 1E-8 | 1,288.98 | 380 | 6.44E-7 | 61.0 | 16.08 | 0.951 (0.797, 1.13) | ||
| Multivariable MR analysis (exposure: BMI adjusted for hypertension and FG, 152 SNPs; outcome: eGFR defined using cystatin C) | |||||||||||
| IVW | −0.03 | 0.12 | 0.014 | 1,093.29 | 6.45E-7 | 64.51 | 64.51 | 14.93 | 0.97 (0.767, 1.23) | ||
Observation Studies in Patients Undergoing BS
The total sample size was 5,337. Baseline demographic features are listed in Table 4. Older age, male sex, rural location, presence of T2D, hypertension, hyperlipidemia, and vascular disease were associated with reduced WL at 1 year after BS, as well as more frequent measurement of creatinine and a consequently shorter interval between baseline measurement and surgery. The median follow-up was 4.63 (interquartile range [IQR] 3.35–5.81) years for the primary outcome and 5.53 (IQR 4.23–6.45) years for the secondary outcome.
Primary Outcome: WL Categories and Primary Outcome of >50% Decline in eGFR
There were 172 events among 5,337 patients. In unadjusted models, WL of 20 to <30% (3.6%), 30 to <40% (2.5%), and ≥40% (3.2%) was associated with a reduction in the primary outcome compared with <20% (5.2%; P = 0.02). After adjustment for covariates, WL was associated with a reduction in the primary outcome, which was significant for the 30 to <40% group (hazard ratio [HR] 0.53 [95% CI 0.32, 0.87]) (Fig. 3) but not the 20 to <30% group (HR 0.72 [0.44, 1.2]) or the ≥40% group (HR 0.73 [0.4, 1.3]). Similar trends were seen in the subgroup of patients with diabetes (n = 2,227 [41.7%]; 20–30% group: HR 0.61 [0.36, 1.05]; 30–40% group: HR 0.4 [0.22, 0.72]; ≥40% group: HR 0.58 [0.28, 1.21]) (Fig. 3).
Figure 3.

Outcomes after BS by WL category (20 to <30%, 30 to <40%, and ≥40%). The <20% WL group is the comparator. Each outcome is presented for the whole cohort and the subgroup with diabetes. Lines indicate 95% CIs. A: Primary outcome (≥50% reduction in eGFR). B: Secondary outcome (CKD hospitalization). C: Secondary outcome with Fine-Gray analysis with death as a competing outcome.
Secondary Outcome: WL and Hospitalization for CKD
There were 53 hospitalizations for CKD. In unadjusted models, WL of 20 to <30% (1.3%), 30 to <40% (0.7%), and ≥40% (0.4%) was associated with a reduction in the secondary outcome compared with <20% (2.6%) (P = 0.0005). After adjustment for covariates, compared with the <20% WL category, increased WL was associated with a progressive reduction in CKD hospitalization, which was significant for 30 to <40% (HR 0.37 [95% CI 0.17, 0.83]) and ≥40% (HR 0.24 [0.07, 0.89]) WL but not 20 to <30% WL (HR 0.60 [0.29, 1.23]). Fine-Gray analysis with death as a competing outcome yielded similar estimates (20 to <30% group: HR 0.63 [0.30, 1.3]; 30 to <40% group: HR 0.39 [0.17, 0.89]; ≥40% group: HR 0.26 [0.07, 0.97]) (Fig. 3).
Similar trends were seen in the diabetes subgroup (20 to <30% group: HR 0.71 [95% CI 0.32, 1.56]; 30 to <40% group: HR 0.38 [0.16, 0.91]; ≥40% group: HR 0.33 [0.09, 1.25]). Fine-Gray analysis with death as a competing outcome yielded similar estimates (20 to <30% group: HR 0.74 [0.33, 1.66]; 30 to <40% group: HR 0.40 [0.16, 0.97]; ≥40% group: HR 0.35 [0.09, 1.33]) (Fig. 3).
Assessment of WL as a continuous variable showed that after adjustment for covariates, each 1% increase in WL was associated with a further reduction in hospitalization for CKD (HR 0.94 [95% CI 0.91, 0.98). Fine-Gray analysis with death as a competing outcome yielded the same conclusion (HR 0.94 [0.91, 0.98]). Analysis of the patients with diabetes was similar with each 1% additional WL associated with an HR of 0.94 (0.91, 0.98). Fine-Gray analysis with death as a competing outcome yielded similar estimates in patients with diabetes (HR 0.95 [0.90, 0.99]).
Discussion
Observational and MR studies have suggested that obesity may be an independent cause of CKD based on an increased risk of CKD after adjusting for T2D and hypertension (3–6,13–15). Findings from our MR analyses suggest that obesity is likely not an independent cause of CKD but, rather, the effect is mediated by hypertension and dysglycemia. Notably, elevated FG in the non-T2D range is likely a significant contributor to both eGFR-based measures of CKD and MA in people with obesity. The mediation analysis findings suggest that increased FG is likely the largest contributor to the creatinine-derived eGFR measure of CKD, while T2D is the biggest contributor to MA. Concordant with MR analysis, WL at thresholds previously associated with normoglycemia and normotension is associated with reduced CKD outcomes after BS. WL of 30 to <40% was associated with significantly reduced primary (50% decline in eGFR) and secondary CKD outcome (hospitalization for CKD), while ≥40% WL was associated with a significantly greater reduction in the secondary outcome. Prior data suggests T2D remission rates plateau with 20–25% WL, but further WL is associated with increased normoglycemia and normotension (23–26,52). The reduction in the primary outcome did not reach statistical significance with ≥40% weight loss, which likely reflects the smaller sample size for this category and hence merits some caution in interpretation. Given the nonlinear relationship between WL and the primary outcome, studies with a larger sample size will likely yield more definitive insights into the relationship between the amount of WL and reduction in eGFR. Subgroup analyses in people with T2D yielded generally similar findings for both the primary and secondary outcome. Whether achieving normoglycemia and normotension via means other than WL/BS also reduces obesity-associated CKD outcomes remains to be determined.
Our MR analyses suggests that increased BMI increases eGFR estimated by cystatin C measurement, even after adjustment for hypertension, increased FG, and/or T2D. However, previous work suggests that increased BMI, diabetes, and inflammation can underestimate eGFR based on cystatin C measurement compared with direct measures of eGFR, thus precluding definitive conclusions from these data (53). Consistent with prior data (14), we also report that increased BMI decreases UACR but with no effect on eGFR. We did not have individual-level data on participants, including medication use. Greater use of renoprotective medications in people with a higher BMI may plausibly explain these findings. Illustrating this, recent MR analyses suggest that increased BMI lowers apolipoprotein B likely because of the confounding effects of cholesterol-lowering medication, an effect not seen when analyzing people not taking statins (54).
The strengths of this study include MR analyses with the largest available GWAS in populations of European descent along with observational data from patients undergoing BS. However, this study has several limitations. We used a creatinine-based diagnosis of CKD, which is an indirect measure of renal function and can be affected by muscle mass. Similarly, cystatin C–based measures of eGFR can be underestimated with increased BMI, diabetes, and inflammation (53). The retrospective observational nature of the BS cohort with potential uncaptured confounders is a major limitation; however, these limitations are less likely with MR analyses. There was a >50% sample overlap between MR population cohorts, which can overestimate the effect size when weak instrument bias is present, although this effect is attenuated by the strength of the instruments (40). The MR analyses was undertaken in European populations and may not translate to other ethnic groups. The greater availability of genetic data from other ethnicities will enable similar analyses in these cohorts. Although we do not have data on ethnicity in our BS cohort, previous data suggests that ∼20% of patients undergoing BS are of non-European ancestry (55). Lack of individual-level data, including medications, is a limitation of our analyses. We also do not have details on the type of BS performed, MA/proteinuria, and longer-term WL.
In summary, the MR analyses suggest that obesity is likely not an independent causal factor for CKD, with its deleterious renal effects mediated by dysglycemia and hypertension. These data underscore the likely causal role of hyperglycemia below the T2D threshold to obesity-associated CKD. WL at or above thresholds known to improve/remit these cardiometabolic parameters are associated with reduced CKD outcomes after BS, and these findings await confirmation with well-powered prospective studies. Whether achieving normoglycemia and normotension in the absence of WL reduces CKD also awaits independent confirmation.
Article Information
Acknowledgments. The authors thank Lei Sun, PhD (Dalla Lan School of Public Health and Department of Statistical Sciences, University of Toronto), for invaluable guidance in statistical analysis and interpretation. The Ontario Bariatric Registry (OBR) collects baseline, treatment, and postoperative data of patients undergoing bariatric treatment at Bariatric Centres of Excellence across Ontario. The OBR is managed by the Centre for Surgical Invention and Innovation in collaboration with the Population Health Research Institute and the Ministry of Health and Long-Term Care.
Funding. S.D. receives funding from the Canadian Institute for Health Information, the Heart and Stroke Foundation of Canada, Diabetes Canada, and the Banting & Best Diabetes Centre (The DH Gales Family Charitable Foundation new investigator award and a Reuben and Helene Dennis scholar in diabetes research). This study was supported by ICES (formerly the Institute for Clinical Evaluative Sciences), which is funded by an annual grant from the Ontario Ministry of Health.
ICES is an independent, nonprofit research institute whose legal status under Ontario’s health information privacy law allows it to collect and analyze health care and demographic data, without consent, for health system evaluation and improvement. Parts of this material are based on data and/or information compiled and provided by the Canadian Institute for Health Information and the Ontario Ministry of Health. The analyses, conclusions, opinions, and statements expressed herein are solely those of the authors and do not reflect those of the funding or data sources; no endorsement is intended or should be inferred.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. A.N., R.K., Y.G., A.M., B.R.S., A.D.P., and S.D. designed the study. A.N., R.K., A.M., and S.D. wrote the manuscript. S.D., A.D.P., A.N., R.K., Y.G., B.R.S., D.R., M.A., B.L., D.Z.I.C., and M.E.F. reviewed the data and edited the manuscript. S.D. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.21913053.
A.N. and R.K. contributed equally.
References
- 1. Jha V, Modi GK. Getting to know the enemy better-the global burden of chronic kidney disease. Kidney Int 2018;94:462–464 [DOI] [PubMed] [Google Scholar]
- 2. Docherty NG, le Roux CW. Bariatric surgery for the treatment of chronic kidney disease in obesity and type 2 diabetes mellitus. Nat Rev Nephrol 2020;16:709–720 [DOI] [PubMed] [Google Scholar]
- 3. Garofalo C, Borrelli S, Minutolo R, Chiodini P, De Nicola L, Conte G. A systematic review and meta-analysis suggests obesity predicts onset of chronic kidney disease in the general population. Kidney Int 2017;91:1224–1235 [DOI] [PubMed] [Google Scholar]
- 4. Xu H, Kuja-Halkola R, Chen X, Magnusson PKE, Svensson P, Carrero JJ. Higher body mass index is associated with incident diabetes and chronic kidney disease independent of genetic confounding. Kidney Int 2019;95:1225–1233 [DOI] [PubMed] [Google Scholar]
- 5. Vivante A, Golan E, Tzur D, et al. Body mass index in 1.2 million adolescents and risk for end-stage renal disease. Arch Intern Med 2012;172:1644–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hsu C, McCulloch CE, Iribarren C, Darbinian J, Go AS. Body mass index and risk for end-stage renal disease. Ann Intern Med 2006;144:21–28 [DOI] [PubMed] [Google Scholar]
- 7. Jackson EK, Li P. Human leptin has natriuretic activity in the rat. Am J Physiol 1997;272(3 Pt 2):F333–F338 [DOI] [PubMed] [Google Scholar]
- 8. Carlyle M, Jones OB, Kuo JJ, Hall JE. Chronic cardiovascular and renal actions of leptin: role of adrenergic activity. Hypertension 2002;39:496–501 [DOI] [PubMed] [Google Scholar]
- 9. Shankar A, Syamala S, Xiao J, Muntner P. Relationship between plasma leptin level and chronic kidney disease. Int J Nephrol. 2012;2012:269532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wolf G, Chen S, Han DC, Ziyadeh FN. Leptin and renal disease. Am J Kidney Dis 2002;39:1–11 [DOI] [PubMed] [Google Scholar]
- 11. Wolf G, Hamann A, Han DC, et al. Leptin stimulates proliferation and TGF-β expression in renal glomerular endothelial cells: potential role in glomerulosclerosis. Kidney Int 1999;56:860–872 [DOI] [PubMed] [Google Scholar]
- 12. Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin S, Tyrrell J, Thomas EL, et al. Disease consequences of higher adiposity uncoupled from its adverse metabolic effects using Mendelian randomisation. Elife 2022;11:e72452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kjaergaard AD, Teumer A, Witte DR, et al. Obesity and kidney function: a two-sample Mendelian randomization study. Clin Chem 2022;68:461–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu P, Herrington WG, Haynes R, et al. Conventional and genetic evidence on the association between adiposity and CKD. J Am Soc Nephrol 2021;32:127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim GS, Oh HH, Kim SH, Kim BO, Byun YS. Association between prediabetes (defined by HbA1c, fasting plasma glucose, and impaired glucose tolerance) and the development of chronic kidney disease: a 9-year prospective cohort study. BMC Nephrol 2019;20:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Melsom T, Mathisen UD, Ingebretsen OC, et al. Impaired fasting glucose is associated with renal hyperfiltration in the general population. Diabetes Care 2011;34:1546–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Honigberg MC, Zekavat SM, Pirruccello JP, Natarajan P, Vaduganathan M. Cardiovascular and kidney outcomes across the glycemic spectrum: insights from the UK Biobank. J Am Coll Cardiol 2021;78:453–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Emanuelsson F, Marott S, Tybjærg-Hansen A, Nordestgaard BG, Benn M. Impact of glucose level on micro- and macrovascular disease in the general population: a Mendelian randomization study. Diabetes Care 2020;43:894–902 [DOI] [PubMed] [Google Scholar]
- 20. Aminian A, Zajichek A, Arterburn DE, et al. Association of metabolic surgery with major adverse cardiovascular outcomes in patients with type 2 diabetes and obesity. JAMA 2019;322:1271–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dash S, Everett K, Jackson T, et al. Cardiorenal outcomes in eligible patients referred for bariatric surgery. Obesity (Silver Spring) 2021;29:2035–2043 [DOI] [PubMed] [Google Scholar]
- 22. Cohen RV, Pereira TV, Aboud CM, et al. Effect of gastric bypass vs best medical treatment on early-stage chronic kidney disease in patients with type 2 diabetes and obesity: a randomized clinical trial. JAMA Surg 2020;155:e200420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meerasa A, Dash S. Weighing in on type 2 diabetes remission. Diabetes Care 2022;45:28–30 [DOI] [PubMed] [Google Scholar]
- 24. Hofsø D, Fatima F, Borgeraas H, et al. Gastric bypass versus sleeve gastrectomy in patients with type 2 diabetes (Oseberg): a single-centre, triple-blind, randomised controlled trial. Lancet Diabetes Endocrinol 2019;7:912–924 [DOI] [PubMed] [Google Scholar]
- 25. Schiavon CA, Bhatt DL, Ikeoka D, et al. Three-year outcomes of bariatric surgery in patients with obesity and hypertension: a randomized clinical trial. Ann Intern Med 2020;173:685–693 [DOI] [PubMed] [Google Scholar]
- 26. Stenberg E, Marsk R, Sundbom M, et al. Remission, relapse, and risk of major cardiovascular events after metabolic surgery in persons with hypertension: a Swedish nationwide registry-based cohort study. PLoS Med 2021;18:e1003817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen TK, Knicely DH, Grams ME. Chronic kidney disease diagnosis and management: a review. JAMA 2019;322:1294–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Teumer A, Tin A, Sorice R, et al.; DCCT/EDIC . Genome-wide association studies identify genetic loci associated with albuminuria in diabetes. Diabetes 2016;65:803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Teumer A, Li Y, Ghasemi S, et al. Genome-wide association meta-analyses and fine-mapping elucidate pathways influencing albuminuria. Nat Commun 2019;10:4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wuttke M, Li Y, Li M, et al.; Lifelines Cohort Study; V. A. Million Veteran Program . A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet 2019;51:957–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scott RA, Lagou V, Welch RP, et al.; Diabetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium . Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 2012;44:991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xue A, Wu Y, Zhu Z, et al.; eQTLGen Consortium . Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun 2018;9:2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pulit SL, Stoneman C, Morris AP, et al.; GIANT Consortium . Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet 2019;28:166–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yengo L, Sidorenko J, Kemper KE, et al.; GIANT Consortium . Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet 2018;27:3641–3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the Reporting of Observational Studies in Epidemiology using Mendelian randomisation (STROBE-MR): explanation and elaboration. BMJ 2021;375:n2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol 2017;46:1985–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sanderson E, Spiller W, Bowden J. Testing and correcting for weak and pleiotropic instruments in two-sample multivariable Mendelian randomization. Stat Med 2021;40:5434–5452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sadreev II, Elsworth BL, Mitchell RE, et al. Navigating sample overlap, winner’s curse and weak instrument bias in Mendelian randomization studies using the UK Biobank. 1 July 2021. [preprint]. medRxiv:21259622v1 [Google Scholar]
- 41. Sanderson E, Davey Smith G, Windmeijer F, Bowden J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int J Epidemiol 2019;48:713–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coresh J, Turin TC, Matsushita K, et al. Decline in estimated glomerular filtration rate and subsequent risk of end-stage renal disease and mortality. JAMA 2014;311:2518–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thompson A, Lawrence J, Stockbridge N. GFR decline as an end point in trials of CKD: a viewpoint from the FDA. Am J Kidney Dis 2014;64:836–837 [DOI] [PubMed] [Google Scholar]
- 44. Heerspink HJL, Stefánsson BV, Correa-Rotter R, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med 2020;383:1436–1446 [DOI] [PubMed] [Google Scholar]
- 45. Delgado C, Baweja M, Crews DC, et al. A unifying approach for GFR estimation: recommendations of the NKF-ASN Task Force on Reassessing the Inclusion of Race in Diagnosing Kidney Disease. Am J Kidney Dis 2022;79:268–288.e1 [DOI] [PubMed] [Google Scholar]
- 46. McLeod L, Buckley G, Sweetman A. Ontario primary care models: a descriptive study. CMAJ Open 2016;4:E679–E688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lipscombe LL, Hwee J, Webster L, Shah BR, Booth GL, Tu K. Identifying diabetes cases from administrative data: a population-based validation study. BMC Health Serv Res 2018;18:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Doumouras AG, Hong D, Lee Y, Tarride J-E, Paterson JM, Anvari M. Association between bariatric surgery and all-cause mortality: a population-based matched cohort study in a universal health care system. Ann Intern Med 2020;173:694–703 [DOI] [PubMed] [Google Scholar]
- 49. Austin PC, White IR, Lee DS, van Buuren S. Missing data in clinical research: a tutorial on multiple imputation. Can J Cardiol 2021;37:1322–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Harrell FE Jr, Lee KL, Pollock BG. Regression models in clinical studies: determining relationships between predictors and response. J Natl Cancer Inst 1988;80:1198–1202 [DOI] [PubMed] [Google Scholar]
- 51. Austin PC, Fine JP. Practical recommendations for reporting Fine-Gray model analyses for competing risk data. Stat Med 2017;36:4391–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barthold D, Brouwer E, Barton LJ, et al. Minimum threshold of bariatric surgical weight loss for initial diabetes remission. Diabetes Care 2022;45:92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stevens LA, Schmid CH, Greene T, et al. Factors other than glomerular filtration rate affect serum cystatin C levels. Kidney Int 2009;75:652–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bell JA, Richardson TG, Wang Q, et al. Effects of general and central adiposity on circulating lipoprotein, lipid, and metabolite levels in UK Biobank: a multivariable Mendelian randomization study. Lancet Reg Health Eur 2022;21:100457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang JC, Tomlinson G, Wnuk S, Sockalingam S, Cram P. Disparities in receipt of bariatric surgery in Canada: an analysis of data from an Ontario bariatric surgery referral center. Med Care 2019;57:723–727 [DOI] [PubMed] [Google Scholar]