

Abstract

Diagnosis of infectious agents is increasingly done by the detection of unique nucleic acid sequences, typically using methods such as PCR that specifically amplify these sequences. A largely neglected alternative approach is to use antibodies that recognize nucleic acids. The unique monoclonal antibody S9.6 recognizes DNA–RNA hybrids in a largely sequence-independent manner. S9.6 has been used in several cases for the analysis of nucleic acids. Extending our recent determination of the structure of S9.6 Fab bound to a DNA–RNA hybrid, we have developed reagents and methods for the sensitive detection of specific DNA and RNA sequences. To facilitate the use in diagnostics, we conjugated the S9.6 Fab to the highly active and well-characterized reporter enzyme human-secreted embryonic alkaline phosphatase (SEAP). Two approaches were utilized for conjugation. The first used sortase A (SrtA), which generates a covalent peptide bond between short amino acid sequences added to recombinantly produced S9.6 Fab and SEAP. The second approach was to genetically fuse the S9.6 Fab and SEAP so that the two are produced as a single molecule. Using these two antibody–SEAP proteins, we developed a simplified ELISA format for the identification of synthetic DNA–RNA hybrids, which can be optimized for detecting nucleic acids of pathogens, as well as for other applications. We successfully used this immunosorbent assay, HC-S, to identify DNA–RNA hybrids in solution with high specificity and sensitivity.
Introduction
The unique S9.6 mouse monoclonal antibody was generated in 1986 from mice immunized with a bacteriophage ϕX174 replicative form, a genome-long DNA–RNA hybrid.1 The resulting S9.6 IgG2a binds with high affinity and specificity to DNA–RNA hybrids in a largely sequence-independent manner while also binding with low affinity to double-stranded RNA (dsRNA) and showing no affinity for single-stranded DNA (ssDNA) or ssRNA.1−6 Because of its unique properties, the S9.6 IgG has provided the basis of many applications including in-solution identification of DNA–RNA hybrids,7 microarray-based detection of actively transcribed genomic regions,2,3in vitro and in vivo identification of R-loops (single-stranded genomic DNA annealed to nascent mRNA) in actively transcribing cells,8−10 and label-free miRNA detection assays.11 Protocols using S9.6 IgG to analyze R-loop metabolism include DRIP-seq (DNA–RNA immunoprecipitation followed by high-throughput DNA sequencing)9,12 and DRIPc-seq (DNA–RNA immunoprecipitation followed by cDNA conversion coupled to high-throughput sequencing).10 A conceptually similar use of such antibodies is the commercial clinical lab diagnostic assay for human papillomavirus (HPV) infection that employs a polyclonal DNA–RNA hybrid-specific antibody (rather than a monoclonal one like S9.6 IgG) in a “hybrid-capture” ELISA format.13 These examples point to the potential of S9.6 to serve as a generic approach for the detection of nucleic acids derived from pathogens through hybridization with sequence-specific complementary single-stranded RNA or DNA probes.
While the production of IgG, a large and complex protein with multiple glycosylation sites, can be challenging, it is widely employed commercially for the production of therapeutic agents. More accessible to many research labs are methods for the production of recombinant antigen-binding fragments (Fab) or single-chain fragment variable regions (scFv) of IgG. Fabs are preferred for structural determinations because they are smaller, well-structured, and not glycosylated. Using the sequence data from our previous cloning and characterization of the S9.6 scFv14 and from analysis of the S9.6 Fab, we successfully produced a recombinant S9.6 Fab and determined the crystal structure of its complex with a short DNA–RNA hybrid.6 Having a reliable supply of recombinant S9.6 Fab has allowed us to use this protein in diagnostic tests for the detection of pathogen’s nucleic acids such as DNA–RNA hybrids. To facilitate the use in diagnostics, we sought to conjugate the S9.6 Fab to reporter enzymes.
The recombinant Fab described above was designed to include a C-terminal recognition signal for the sortase A (SrtA) enzyme15 to enable the facile preparation of antibody conjugates. Sortase A is among the most widely used enzymes to generate site-specific covalent bonds;16 other enzymes employed for this purpose include butelase,17 microbial transglutaminase,18 and others.19 To form a site-specific covalent bond, SrtA first binds the 5-amino acid motif LPXTG (sorting signal; SoSi) and cleaves the peptide bond between Thr and Gly to generate a thioacyl–enzyme intermediate. Then, SrtA uses oligoglycine (such as GGG or “G3”) as a nucleophile to replace the thioacyl, forming an amide bond between Thr (from LPXTG) and Gly (from G3).20 SrtA has several biotechnological applications including PEGylation, biorthogonal functional group attachment, probe conjugation, and protein–protein conjugation.4,20,21 SrtA-mediated conjugation in most cases would not be expected to impact the binding activity of antibodies. For instance, the anti-HER2 antibody can be tagged with Cy3 at the C-terminus of its heavy chain using SrtA.22 The recombinant S9.6 Fab used in this work has the heavy-chain C-terminal sequence LPETGGGHHHHHH and is designated as S9.6 Fab-SoSi-His6.
As a reporter enzyme for conjugation to S9.6 Fab-SoSi-His6, we selected human-secreted embryonic alkaline phosphatase (SEAP). SEAP is a well-characterized, highly active enzyme that is commonly used as a reporter of transcriptional activity.23 Due to its high turnover number, SEAP-based assays are approximately 10-fold more sensitive than assays using other reporter enzymes such as horseradish peroxidase and luciferase.23 SEAP is a homodimer of tightly associated, but not disulfide-linked, 54 kDa protein.24 We designate the dimeric form observed under native conditions as SEAPd to distinguish it from the monomeric protein, SEAPm, which is observed under denaturing conditions.
In this study, we utilized two approaches to generate S9.6 Fab-conjugated SEAP proteins. The first approach was to use SrtA as described above to conjugate the purified S9.6 Fab-Sosi-His6 with a modified G3-SEAP. Once this conjugate proved to be useful, we sought to produce a genetically encoded S9.6 Fab–SEAP fusion protein. Using these two antibody–SEAP proteins, we developed a simplified ELISA format for the identification of DNA–RNA hybrids, which can be optimized for detecting nucleic acids of pathogens and for efficient analysis of R-loops.25
Results
S9.6 Fab Production and Preparation of Conjugates to SEAP
In a previous study, we reported the cloning, expression in E. coli, and characterization of the S9.6 scFv.14 For the current study, we used the recombinant S9.6 Fab-SoSi-His6 produced and characterized for the recent work, showing the structure of S9.6 bound to a short DNA–RNA hybrid.6 This S9.6 Fab-SoSi-His6 was purified from ExpiCHO-S culture supernatants using nickel-nitriloacetic acid resin (Ni-NTA) and was found to be mostly in the interchain disulfide-bonded 50 kDa form (Figure 1 and Supporting Figure S1A). Treatment with DTT separated the S9.6 Fab light chain (FabLC, ∼24 kDa) and S9.6 Fab heavy chain (FabHC, ∼25 kDa) peptides (Supporting Figure S1A).
Figure 1.

Illustration for the expression and purification of S9.6 Fab-SoSi-His6 and G3-SEAP followed by SrtA-mediated conjugation. Two batches of ExpiCHO-S cells were used for the expression of proteins. (a) The first contained two cotransfected plasmids expressing S9.6-FabLC and S9.6-FabHC to produce S9.6 Fab, which was purified using a Ni-NTA column. (b) The second batch of cells contained the plasmid expressing G3-SEAP (SEAPd), which was purified through a hydroxyapatite column (Ceramic HTP, BioRad). The purified proteins were then conjugated using sortase A (SrtA) to generate Fab1–SEAPd and Fab2–SEAPd products. The conjugate products were then sequentially purified using Ni-NTA and hydroxyapatite column.
To make it suitable for sortase-mediated conjugation, SEAP was modified to contain an N-terminal Gly3 (GGG) sequence to produce G3-SEAP (sequence provided in Supporting File 1). A pcDNA3.4 plasmid encoding G3-SEAP was constructed, transiently expressed in ExpiCHO-S cells, and the G3-SEAP protein was purified by several chromatographic steps (Figure 1 and Supporting Figure S1B). While in our previous work, each liter of ExpiCHO-S cells yielded 10–12 mg of S9.6 Fab-SoSi-His6, each liter of the G3-SEAP cultures produced about 6 mg of G3-SEAP. The purified G3-SEAP protein was confirmed by mass-spectrometry exact-mass analysis to have the expected molecular weight (Supporting File 2) and was shown to be partially glycosylated with nonmature intermediate glycans, presumably on Asn in the sequence WNR26 (Supporting File 2).
A catalytically enhanced sortase enzyme, SrtA (Supporting Figure S1C), was used to conjugate S9.6 Fab-SoSi-His6 (the SoSi-His6 being on only the FabHC) and G3-SEAP. The reaction mixture was analyzed by size-exclusion chromatography coupled with multiangle light scattering (SEC-MALS) (Figure 2). This analysis showed product peaks consistent with successful conjugation: a peak of 195 kDa for the attachment of Fabs to both monomers in the SEAP dimer (Fab2–SEAPd) and a peak of 150 kDa for the attachment of only one Fab to the SEAP dimer (Fab1–SEAPd). We also observed peaks for the reactants dimeric SEAP (SEAPd, 110 kDa) and Fab (47.2 kDa). Although this reaction used a 3-fold molar excess of Fab, its modest efficiency led to the incomplete conjugation of both SEAP monomers.
Figure 2.

Confirmation of conjugation reaction by size-exclusion chromatography multiangle light scattering (SEC-MALS): elution profile and molecular weights of components from SrtA-mediated conjugation reaction as determined by SEC-MALS. The calculated molecular weights are for S9.6 Fab-SoSi-His6 (∼47.2 kDa), SEAPd (110 kDa), Fab1–SEAPd (∼150 kDa), and Fab2–SEAPd (∼195 kDa).
While both the Fab2–SEAPd and Fab1–SEAPd conjugates can be used in diagnostics, we focused on purifying the former. Hydroxyapatite chromatography proved effective at separating Fab2–SEAPd from Fab1–SEAPd (Figure 3A). The unconjugated starting materials eluted in fractions i and ii along with some Fab1–SEAPd (seen as Fab and Fab1–SEAPm on the SDS gel (Figure 3B)), which were separated from the desired Fab2–SEAPd product in fraction iii. This product has an S9.6 Fab conjugated to each of the G3-SEAP monomers. Thus, on a denaturing but nonreducing SDS gel, each SEAPm protein (54 kDa) is covalently linked to an S9.6 Fab (50 kDa), giving a band of ∼ 105 kDa (Figure 3B). The absence of an unconjugated SEAPm at 54 kDa shows that the product is Fab2–SEAPd, free of its precursor components S9.6 Fab-SoSi-His6 and G3-SEAP (SEAPm). The reduction of Fab2–SEAPd with DTT separated the FabLC from FabHC–SEAPm, confirming a site-specific conjugation of the S9.6 Fab-SoSi-His6 HC with SEAP (Figure 3C). In purifications of fraction ii from Figure 2B, we also obtained the purified Fab1–SEAPd (Supporting Figure S2A,B). These final purified conjugates were again examined by SEC-MALS and shown to be homogeneous and of the correct masses (Figure 3D). This method was also used to confirm that SEAP exists only as a dimer, SEAPd (Figure 3E).
Figure 3.

SrtA-mediated conjugation of purified S9.6 Fab-SoSi-His6 and G3-SEAP: (A) Elution profile of Ni-NTA-purified conjugation reaction run on a hydroxyapatite column. (B, C) Coomassie-stained SDS polyacrylamide gels with a reference ladder [lane L] and fractions from Figure 2A; (B) nonreduced and denatured proteins from fraction i contain SEAP (represented as denatured SEAPm); fraction ii includes a mixture of Fab1–SEAPm, SEAPm, and S9.6 Fab-SoSi-His6; and fraction iii includes purified Fab2–SEAPd (shown as denatured Fab1–SEAPm). (C) Reduced and denatured proteins from fraction ii and fraction iii confirm the site-specific conjugation of G3-SEAP to S9.6 FabHC (shown as FabHC–SEAPm). Denatured SEAP (as SEAPm of 54 kDa) and S9.6 FabLC (24 kDa) have resolved to their expected molecular sizes. (D, E) Confirmation of purified conjugates by SEC-MALS: elution profiles and molecular weights of purified (D) Fab1–SEAPd (145 kDa) and Fab2–SEAPd (195 kDa) proteins and (E) SEAPd (110 kDa) protein.
Once the sortase conjugate proved useful (to be shown below), we proceeded to construct a genetically encoded S9.6 Fab–SEAP fusion protein, where SEAP was fused to the C-terminus of the S9.6 FabHC, so its structure would closely mimic that of the SrtA-generated conjugate. Because SEAP and immunoglobulins are secreted from human cells, we anticipated that the fusion protein could be made effectively in the same ExpiCHO-S system, and this proved true (as described in Experimental Procedures). While the genetic fusion protein was easily purified with Ni-NTA resin to yield a homogeneous Fab2–SEAPd product, the yields were only about 1 mg per L of ExpiCHO-S cells. A mass-spectrometric analysis of the S9.6 Fab–SEAP fusion protein confirmed the mass of the S9.6 FabLC and identified a high level of glycosylation in the S9.6 FabHC–SEAPm polypeptide of the genetic fusion protein (Supporting File 2). Analysis on an SDS gel showed the expected Fab1–SEAPm (105 kDa), which separated upon reduction to the FabHC–SEAPm (80 kDa) and FabLC (25 kDa) (Figure 4).
Figure 4.
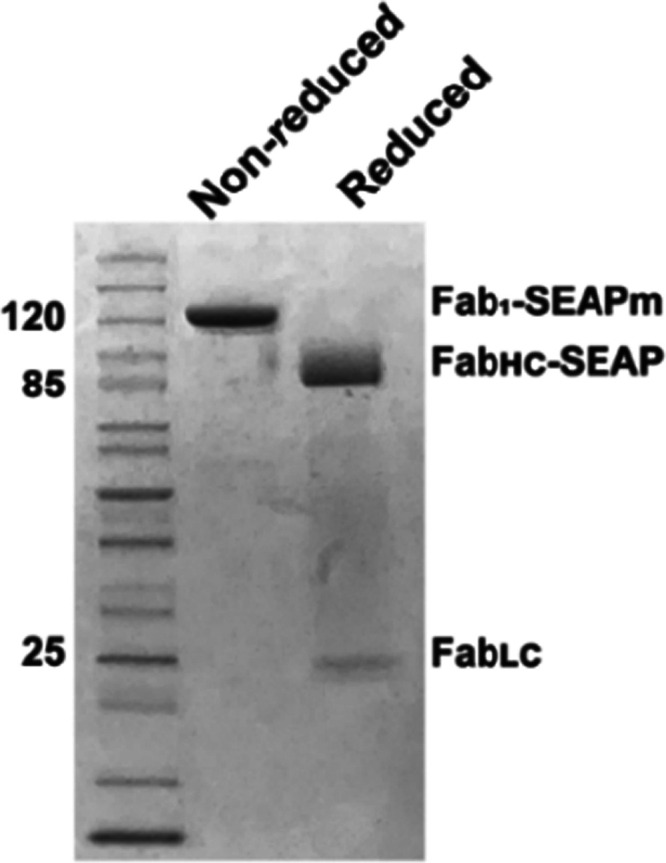
Coomassie-stained SDS polyacrylamide gel analysis of the purified S9.6 Fab–SEAP genetic fusion protein: the nonreduced lane shows the purified S9.6 Fab–SEAP genetic fusion protein as denatured Fab1–SEAPm (114 kDa). The reduced lane shows the FabLC (25 kDa) and FabHC–SEAPm (80 kDa) components of the S9.6 Fab–SEAP genetic fusion protein.
Hybrid-Capture-S ELISA for DNA–RNA Identification Using S9.6 IgG and Fab2–SEAPd
A simplified sandwich ELISA format was developed using S9.6 IgG and Fab2–SEAPd to identify DNA–RNA hybrids in solution. The assay is termed Hybrid-Capture-S (Hybrid-Capture for DNA–RNA (HC) with Sandwich format (HC-S)) (Figure 5A). For the HC-S, the S9.6 IgG purified from hybridoma H8730 (as described in the Experimental Procedures section) was adsorbed to the surface of 96-well plates. These S9.6 IgG-coated plates can be stored at 4 °C for at least 3 months in a dehydrated condition. A 23 bp DNA–RNA hybrid (23DR; see the Experimental Procedures section) was captured onto the surface of the IgG-coated surface. Because the epitope bound by S9.6 is about 6 bp long,6,14 a sufficient portion of the hybrid is expected to remain available to bind the Fab2–SEAPd conjugate. After washing to remove the unbound conjugate, the SEAP substrate is added to generate a signal. As seen in Figure 5B, the Fab2–SEAPd can specifically bind to the DNA–RNA hybrid with a high specificity and affinity of ∼200 nM (Kd). The apparent affinities of Fab2–SEAPd for dsDNA and dsRNA in this assay were found to be >1.5 μM. Also, we observed no affinity of Fab2–SEAPd for ssDNA [23DR-DNA (sense); Table 1] or ssRNA [23DR-RNA (antisense); Table 1] (Supporting Figure S3). The S9.6 Fab2–SEAPd genetic fusion protein showed a 10-fold lower reactivity for the DNA–RNA hybrid (23DR) compared with the Fab2–SEAPd sortase conjugate (Supporting Figure S4). One possible reason for the lower affinity could be the linker present in the fusion protein, which is four amino acids shorter (lacking LPET) than that of the conjugate. To understand how the linker affects the genetic fusion protein activity in HC-S, a genetic fusion variant was created with a linker equivalent to that of the sortase conjugate (S9.6 Fab2–LPET–SEAPd.) Although this modification led to increased activity over the original fusion protein, the linker-equivalent fusion protein still yielded 4-fold less signal for the DNA–RNA hybrid than the sortase conjugate (Figure 5C).
Figure 5.

HC-S ELISA for the identification of DNA–RNA hybrid in solution. (A) Pictorial representation of the molecular interactions in the HC-S ELISA. The DNA–RNA hybrid is captured on S9.6 IgG-coated plates and subsequently detected using Fab2–SEAPd with chemiluminescence from the cleaved substrate as the readout. Image was created using Biorender.com. (B) The HC-S ELISA can specifically detect 100 pmole of DNA–RNA hybrid with as low as 5 pmole of Fab2–SEAPd (* indicates p > 0.05) in 100 μL of reaction. (C) Specific binding of 10 pmole of DNA–RNA hybrid, dsDNA, and dsRNA with 100 pmole of S9.6 Fab–LPET–SEAP genetic fusion and Fab2–SEAPd proteins in 100 μL of reaction measured in chemiluminescent units (A.U.). (D, E) HC-S ELISA is time- and temperature-dependent. The line graphs represent the signal output (as chemiluminescence in A.U. × 104) from HC-S ELISA tested at the mentioned assay times with different concentrations of DNA–RNA hybrids and 10 pmole of Fab2–SEAPd at (D) 4 °C and (E) 25 °C. * indicates p > 0.05 for statistical significance calculated using Student’s t-test.
Table 1. List of Primers and Oligonucleotides.
| primer | sequence (5′–3′) | reference |
|---|---|---|
| SEAP-For | GGTCCATAGTATCATCCCAGTTGAGG | this study |
| SEAP-Rev | TAAGCTTATCAACCCGGGTGCGC | this study |
| pcDNA3.4-Fwd | GCACCCGGGTTGATAAGCTTAAGGGTTCG | this study |
| pcDNA3.4-Rev | CTGGGATGATACTATGGACCCCTGT | this study |
| HC-SEAP_Fwd | TGGACAAGAAGATTTCCGCAGGCGGAGGTATCATCCCAGTTGAGGAGG | this study |
| HC-SEAP_Rev | TGGTGGTGATGGTGATGACCACCCGGGTGCGCGGCGTC | this study |
| pcDNA3.4_HC_Fwd | GGTCATCACCATCACCAC | this study |
| pcDNA3.4_HC_Rev | TGCGGAAATCTTCTTGTC | this study |
| pcDNA3.4-LPET-F | GAAACAGGCGGAGGTATCATCCCA | this study |
| pcDNA3.4-LPET-R | TGGAAGTGCGGAAATCTTCTTGTCCAC | this study |
| 23DR-DNA (sense) | CGGTGTGGTCGCTGTAATCAGAG | this study |
| 23DR-RNA (antisense) | rCrUrCrUrGrArUrUrArCrArGrCrGrArCrCrArCrArCrCrG | this study |
| 23DR-DNA (antisense) | CTCTGATTACAGCGACCACACCG | this study |
| 23DR-RNA (sense) | rCrGrGrUrGrUrGrGrUrCrGrCrUrGrUrArArUrCrArGrArG | this Study |
| FAM-23DR-DNA (sense) | /56-FAM/CGGTGTGGTCGCTGTAATCAGAG | this study |
The binding of Fab2–SEAPd to the DNA–RNA hybrid was found to be time- and temperature-dependent (Figure 5D,E). The HC-S ELISA using 100 nM of Fab2–SEAPd can detect all tested concentrations of 23DR hybrids up to 100 pmole/well in as little as 1 h at 25 °C (room temperature) (Figure 5D). The Fab2–SEAPd seems to lose binding of the ligand with time at 25 °C (Figure 5E).
Affinity Measurement of S9.6 Fab–SEAP Proteins Using Fluorescence Polarization (FP)
To measure the affinity of the Fab2–SEAPd SrtA conjugate and genetic fusion proteins for DNA–RNA hybrids, we labeled the 5′ end of the DNA strand of the 23 bp nucleic acid hybrid (23DR) with FAM (fluorescein) and monitored the increase of fluorescence polarization (FP) induced by the Fab2–SEAPd proteins. The Fab2–SEAPd SrtA conjugate was bound to the labeled DNA–RNA hybrid to induce a robust change in fluorescence polarization, giving an apparent Kd of 12 nM, more than 32-fold higher than that of S9.6 Fab alone (392 nM) (Figure 6), possibly because of the bivalent nature of Fab2–SEAPd. The binding affinity measurement of the S9.6 Fab2–SEAPd genetic fusion protein yielded an apparent Kd of 24 nM. The binding of SEAP alone to DNA–RNA hybrid was not detectable (N.D.) in this assay, suggesting no nonspecific interaction between the G3-SEAP enzyme and the DNA–RNA hybrids.
Figure 6.
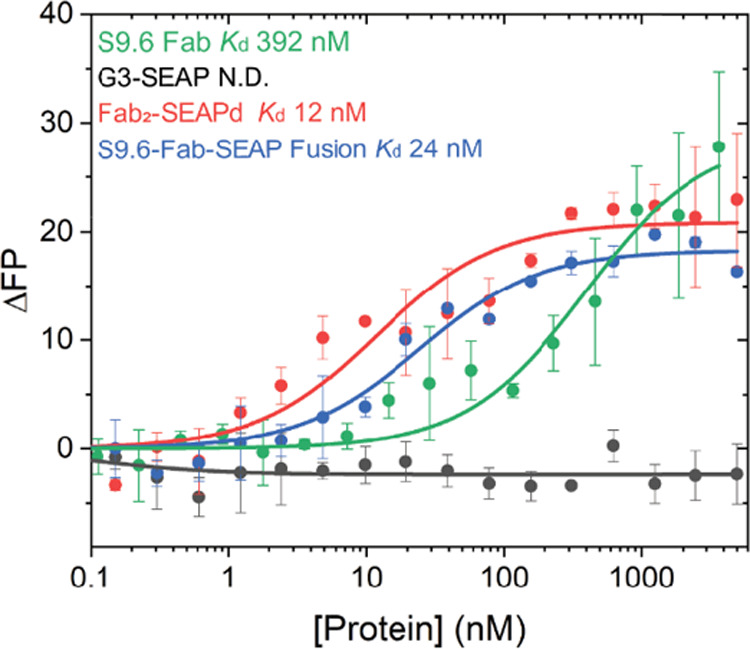
Affinity measurement using fluorescence polarization (FP) assay: 5′-FAM labeled DNA–RNA hybrid was used to measure FP-based affinity for S9.6 Fab (green), G3-SEAP (black), Fab2–SEAPd (red), and S9.6 Fab–SEAP genetic fusion protein (blue). N.D. indicates no interaction detected. Samples were run separately, and the resulting profiles are overlaid with their respective Kd measurements included.
Sensitivity Limit of HC-S ELISA
After demonstrating its high specificity for DNA–RNA hybrids, we sought to understand the sensitivity limit of the sandwich ELISA developed using S9.6 IgG and Fab2–SEAPd. With a saturating amount of S9.6 IgG (5 μg/mL) and 1 μg of Fab2–SEAPd (as described in Experimental Procedures and from Figure 5B), in a 100 μL reaction, the HC-S can specifically detect DNA–RNA hybrids at levels of femtomole (5 fmole, equivalent to 2 × 109 copy number) to picomole (100 pmole, equivalent to 20 × 1013 copy number) (Figure 7). The lowest amount of 5 fmole could yield a maximum of 25% signal readout when compared with the highest amount of the ligand (100 pmole). Using a Fab2–SEAPd concentration of >1 μg/100 μL in HC-S ELISA leads to a higher background and thus a poor signal-to-noise ratio, which limits hybrid detection to the femtomole level.
Figure 7.
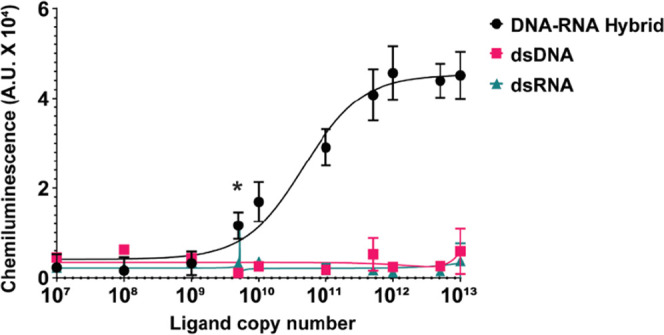
HC-S ELISA can measure femtomoles of DNA–RNA hybrids: the HC-S ELISA with 10 pmole of Fab2–SEAPd can specifically detect up to 5 femtomoles (equivalent to 2 × 109 copy number) of the DNA–RNA hybrid (23 bp) in a 100 μL reaction. The y-axis indicates the signal output captured for the copy number of each hybrid DNA–RNA, dsDNA, and dsRNA shown on the x-axis. * Indicates p > 0.05 for statistical significance calculated using Student’s t-test.
Discussion
Sortase A (SrtA)-mediated antibody–enzyme conjugation has some advantages compared to conventional methods of chemical conjugation. Conventionally, antibodies and enzymes are chemically linked using biorthogonal functional groups such as glutaraldehyde,27 periodate oxidation,28 and m-maleimidobenzoyl-N-hydroxysuccinimide ester.29 These methods depend on the amino acid composition of the antibody, and linkage occurs randomly on several side-chain residues. This method of chemical conjugation can have adverse effects on the antibody’s antigen-binding properties and the stability of the enzyme, antibody, or both. Most recently, antibody–probe conjugates were made in a cell using strain-promoted azide–alkyne cycloaddition (SPAAC), a click chemistry method.30 Though SPAAC is highly selective and can be done in physiological conditions, this method requires prior modification of the protein of interest with either an azide or an alkyne moiety, which can be technically challenging and time-consuming. Another frequently employed method is the expression of a recombinant antibody–enzyme protein, but this approach also has limitations. Cells would need to produce the peptide, perform post-translational modifications on it, fold it, and secrete a fully active antibody-conjugated enzyme. This process introduces opportunities for error at each step. Reporter proteins such as SEAP are bulky and homopolymerize,24,31 which may further impact the overall yield of genetic fusion proteins. SrtA-mediated conjugation can overcome a few of these limitations by site-specific conjugation of fully active conjugants, antibodies, and reporter enzymes. Thus, we aimed to create an antibody–enzyme conjugate from S9.6 Fab and SEAP using SrtA-mediated conjugation and compared its effectiveness to the genetically encoded S9.6 Fab–SEAP fusion protein.
To achieve this aim, first, we made the S9.6 IgG from hybridoma and expressed S9.6 Fab-SoSi-His6 and G3-SEAP in ExpiCHO-S cells. Next, the purified S9.6 Fab carrying a sorting signal (SoSi; LPETGG) was conjugated to the G3-SEAP using sortase A (SrtA) in a site-specific manner to generate Fab1–SEAPd and Fab2–SEAPd. The SrtA-mediated conjugation allows conjugants to remain in their active form after the site-specific conjugation. Thus, the SrtA-mediated conjugation of free S9.6 Fab-SoSi-His6 and G3-SEAP shows potential to be used as a general method for antibody–enzyme conjugation.
Because previous reports showed that enzymes of the alkaline phosphatase family [E.C 3.1.3.1] are present as metalloenzymes in a homodimer conformation,32 we anticipated that S9.6 Fab–SEAP is present in a dimeric form too. This would result in a bivalent S9.6 Fab comparable in function to S9.6 IgG. We reasoned that the bivalent form may have greater avidity and apparent affinity for DNA–RNA hybrids, making it more useful in future applications, which led us to isolate this reaction product for further study (Figure 2).
The S9.6 IgG and Fab2–SEAPd were used to develop our protocol for HC-S, an immunosorbent assay for identification of DNA–RNA hybrids in solution. In contrast to our previous report,14 the HC-S format revealed a moderate affinity of ∼200 nM for DNA–RNA hybrid. Fab2–SEAPd may have a higher Kd (∼200 nM) than S9.6 scFv (0.6 nM)14 due to competition for the epitopes between Fab2–SEAPd and S9.6 IgG in the HC-S format. The bivalency in Fab2–SEAPd and the affinity calculated with the activity of the SEAP enzyme could also contribute to the contrasting results for two different S9.6 elements (Fab2–SEAPd and scFv) in the two studies. However, the affinity of Fab2–SEAPd (Kd 12 nM) and Fab–SEAP fusion proteins (Kd 24 nM) for DNA–RNA hybrid calculated with fluorescence polarization is in accordance with the previous study.14 The bivalency in these antibodies can form complexes with the DNA–RNA hybrid in a manner that results in two different sizes or shapes of complexes, leading to a biphasic curve in the fluorescence polarization assay. Additionally, earlier reports have suggested that the binding affinity of S9.6 scFv may vary with the length of the DNA–RNA hybrid.14 Previous studies have shown that S9.6 Fab binds to dsRNA with a 5-fold lower affinity compared with the DNA–RNA hybrids.14 However, in this study, we have shown that Fab2–SEAPd can have a >5% cross-reactivity with dsRNA, ssRNA, dsDNA, and ssDNA with signal levels 10- to 50-fold less than against DNA–RNA hybrids. The S9.6 Fab fusion protein showed high background in the blank wells containing no nucleic acid, indicating a nonspecific interaction with the S9.6 IgG-coated wells. The S9.6 Fab–SEAP fusion protein also generated a 10-fold less signal for DNA–RNA hybrid when compared with Fab2–SEAPd (Supporting Figure S4). Contrary to the results obtained with HC-S ELISA, the fluorescence polarization assay suggested that the Fab component of Fab2–SEAPd and S9.6 Fab–SEAP genetic fusion proteins has 10- to 20-fold higher affinity for DNA–RNA hybrid than S9.6 Fab alone.6 Furthermore, SEAP activity is similar among G3-SEAP and all variants of Fab–SEAP (Supporting Figure S5A). One possible explanation for the discrepancy in affinity between the variants observed in two different assays could be the four amino acid shorter linkers present in the fusion protein (lacking LPET) compared to the conjugate. This may cause steric hindrance between the S9.6 IgG capture antibody and S9.6 Fab–SEAP genetic fusion protein. Therefore, the affinity of the fusion protein may be higher when measured using fluorescence polarization since this effect is not present. This hypothesis seems plausible as the longer linker in S9.6 Fab2–LPET–SEAPd led to a 6-fold increased activity over the original fusion protein. Another possible explanation could be higher glycosylation levels in the fusion protein. Intact mass analysis of G3-SEAP and S9.6 Fab–SEAP genetic fusion suggests that G3-SEAP was partially glycosylated with nonmature intermediates, but the genetic fusion protein contained a mature glycosyl group (Supporting File 2). Based on this result, the original fusion, linker-equivalent fusion, and sortase-conjugated proteins were deglycosylated using PNGase F and used in HC-S; however, deglycosylation only marginally increased the activity of the linker-equivalent genetic fusion protein and had no effect on the original genetic fusion protein or the sortase conjugate (Supporting Figure S5B,C). Therefore, part of the discrepancy in the hybrid-capture activity between SrtA-mediated conjugation products and the genetically fused protein is accounted for by these factors, but the full discrepancy remains unexplained.
The HC-S ELISA format can identify the presence of femtomoles of DNA–RNA hybrid in a reaction. This assay is robust and can be adopted for high-throughput screening of R-loops in the total nucleic acid extract from a cell. Additionally, it could be used for the high-throughput identification of miRNAs as oncogenic markers. Specific DNA probes against a miRNA of interest can be used to generate DNA–RNA hybrids for robust and convenient detection using the HC-S format. This assay is primarily limited by its limit of detection (LOD) in the nM range, which is high in comparison to the pM–fM range that can be achieved using a bioanalyzer or qRT-PCR (Agilent). The high LOD observed in the HC-S ELISA could be due to competition for 6 b.p. epitopes between Fab2–SEAPd and S9.6 IgG. Thus, increasing the size of DNA–RNA hybrids could achieve a lower LOD. Moreover, another potential use of HC-S ELISA could be the detection of pathogen nucleic acids through hybridization with sequence-specific complementary single-stranded RNA or DNA probes. However, this application would require thorough standardization of a method to enrich pathogen-specific DNA or RNA from the total nucleic acid pool, which carries preexisting R-loops in infected tissues. A detailed investigation is needed to prove the usefulness of this assay in clinical settings.
Conclusions
In this study, we successfully conjugated the reporter enzyme SEAP to the antigen-binding fragment (Fab) of the S9.6 monoclonal antibody. The recombinant S9.6 Fab–SEAP led us to develop an immunosorbent assay, HC-S, to identify DNA–RNA hybrids in solution. As the HC-S does not require the use of any secondary antibody, S9.6 Fab–SEAP can be directly applied as a molecular beacon to report the presence of DNA–RNA hybrid in solution. The full protocol can be realized in less than 3 h.
Experimental Procedures
Expression Plasmid for G3-SEAP
The coding sequence for SEAP was PCR-amplified from the Addgene plasmid # 11578933 using SEAP-Fwd and SEAP-Rev primers (Table 1). The SEAP-Fwd primer added a sequence encoding the Gly3 sequence needed for sortase conjugation. The pcDNA3.4 plasmid was PCR-amplified from pcDNA3.4–S9.6 FabHC6 using the primers pcDNA3.4-Fwd and pcDNA3.4-Rev (Table 1). These two amplicons were assembled using the NEBuilder HiFi DNA Assembly Cloning Kit (NEB) to create the pcDNA3.4–G3-SEAP vector (Table 2). Following transfection, the clone was sequence-confirmed by Psomagen. The G3-SEAP encoded by this plasmid does not contain a His6 tag.
Table 2. List of Clones and Expression Vectors Used in This Study.
| plasmid and synthetic nucleic acid | relevant characteristics | reference |
|---|---|---|
| pcDNA3.4 TOPO | constitutive mammalian expression vector | Thermo Fisher |
| pAK400–ScFv | S9.6 VL and VH coding sequences | (14) |
| pcDNA3.4–S9.6 FabLC | mammalian expression vector for S9.6 Fab light chain | (6) |
| pcDNA3.4–S9.6 FabHC | mammalian expression vector for S9.6 Fab heavy chain | (6) |
| pcDNA3.3 ss–FLAG–AP–hNDP | expresses and secretes human-secreted alkaline phosphatase tags | Addgene #115789 |
| pcDNA3.4–G3-SEAP | mammalian expression vector for G3-SEAP | this study |
| pcDNA3.4–S9.6 FabHC–SEAP | mammalian expression vector for the S9.6 FabHC–SEAP fusion protein | this study |
| pET30b–5M SrtA | bacterial expression vector for sortase transpeptidase | (34) |
S9.6 Fab Proteins (S9.6 Fab-SoSi-His6 and S9.6 Fab–SEAP fusion protein)
The preparation of the S9.6 Fab-SoSi-His6 protein used for structure determination was previously described in detail.6 Briefly, pcDNA3.4 plasmids encoding the heavy and light chains were transfected into ExpiCHO-S cells and the secreted Fab was purified on Ni+2-nitriloacetic acid (Ni-NTA) agarose resin. For the expression of fusion proteins of S9.6 Fab and SEAP, the SEAP sequence was excised from Addgene plasmid #11578933 and was inserted into pcDNA3.4–S9.6 FabHC using the NEBuilder HiFi DNA Assembly Cloning Kit (NEB) to create pcDNA3.4–S9.6 FabHC–SEAP and pcDNA3.4–S9.6 FabHC–LPET–SEAP. For purification, a His6 tag was added to the C-terminus of SEAP. For expression of the Fabs, these plasmids were combined with the original S9.6 FabLC plasmid. Transfection and purification were as above for the Fab-SoSi-His6. The primers used for the HiFi-based assembly are listed in Table 2.
Purification of G3-SEAP
G3-SEAP was harvested from the culture supernatant using hydroxyapatite with a gradient between buffer A (5 mM potassium phosphate, 100 mM NaCl, pH 7.0) and buffer B (500 mM potassium phosphate, 100 mM NaCl, pH 7.0). The purified protein was analyzed by SDS-PAGE on 4–20% tris–glycine polyacrylamide gels (Invitrogen). The SEAP enzyme catalytic activity in G3-SEAP and variants of Fab2–SEAPd proteins was measured using the Tropix CDP-Star Chemiluminescent substrate (Roche) using the manufacturer’s protocol.
Purification of S9.6 IgG and SrtA Transpeptidase
S9.6 IgG purification followed standard methods as previously described.2 Briefly, the HB-8730 hybridoma (ATCC, Manassas, VA) that produces the S9.6 IgG was grown in DMEM supplemented with 10% fetal bovine serum, 2 mM glutamax, 1 mM sodium pyruvate, and 50 μg/mL of gentamicin (Life Technologies, Carlsbad, CA) until the pH of the medium fell below 7.0. The S9.6 IgG was purified from culture supernatants using HiTrap Protein G columns (GE Healthcare Life Sciences, Piscataway, NJ).
The sortase A (SrtA) variant having five mutations to enhance catalytic activity was expressed in E. coli from Addgene plasmid #51140 using standard methods. This plasmid, pet30b–5M SrtA, was a gift from Hidde Ploegh.34 The protein contains a C-terminal His6 tag, which was used for its purification on Ni-NTA affinity resin as described above.
SrtA-Mediated S9.6 Fab–SEAP Conjugation
The purified SrtA transpeptidase was used to conjugate the S9.6 Fab-SoSi-His6 with G3-SEAP. The protocol used the dependence of the enzyme on >10 mM Ca+2 for activity. First, 150 μg of purified SrtA enzyme was activated with 150 mM of DTT at room temperature to reduce the cysteines necessary for the reaction. Since DTT can reduce the S9.6 Fab into FabHC and FabLC components, the activated SrtA enzyme was diluted to minimize the reducing effect of DTT or this was removed by gel filtration. S9.6 Fab-SoSi-His6 and G3-SEAP were used either in equimolar concentrations or with excess S9.6 Fab to generate the product with each SEAPm attached to a Fab. These reactants were concentrated to 1 mL and then mixed with 0.75 mL of 20× reaction buffer (200 mM HEPES, pH 7.5, 4 M NaCl) and 3.75 mL of 20% glycerol, and the reaction volume was increased to 15 mL with water. The mixture was concentrated using Amicon Ultra-15 (Ultracel-10K) centrifugal filters to 1 mL. The activated SrtA was added to the concentrated reaction at a molar ratio of 1:200, and CaCl2 was added to a final concentration of 20 mM to induce activity. The reaction was incubated at room temperature for 10 min and then quenched by diluting the reaction with dilution buffer (1× reaction buffer, 10% glycerol, 1.0 mM EDTA) to 15 mL. The conjugation reaction was again concentrated in the same centrifugal filter to remove the G3–His6 reaction product. The SrtA conjugation followed by concentration was repeated twice more. Conjugated products were sequentially purified by Ni-NTA agarose and hydroxyapatite columns. First, the Ni-NTA agarose was used to remove unconjugated S9.6 Fab-SoSi-His6 from the reaction. Finally, the conjugated Fab2–SEAPd in the Ni-NTA eluate was purified using hydroxyapatite with a gradient between buffer A (5 mM potassium phosphate, 100 mM NaCl, pH 7.0) and buffer B (500 mM potassium phosphate, 100 mM NaCl, pH 7.0). The (byproduct) Fab1–SEAPd was purified from other hydroxyapatite column fractions using a 24 mL Superdex-200 gel filtration column.
Nucleic Acid Hybrid Formation
The dsDNA, dsRNA, and DNA–RNA hybrids (all 23 bp long) were generated using the respective oligos as listed in Table 2. Briefly, 1 μL of 10 μM of respective complementary strands of dsDNA, dsRNA, and DNA–RNA hybrid was mixed in equimolar ratios in a hybridization buffer (25 mM tris–Cl pH 8.0, 150 mM NaCl, and 1 mM MgCl2). The nucleic acid mixture was denatured at 95 °C for 3 min, and the reaction was transferred to a boiling water bath, in which the reaction was gradually cooled to room temperature, allowing the strands to hybridize.
Protein Deglycosylation Using PNGase F
Deglycosylation of S9.6 Fab–SEAP glycoprotein variants was performed using the manufacturer’s protocol except for the buffer used. Briefly, 20 μg of glycoprotein was mixed with 10 U of PNGase F (NEB #P0704S) and 50 mM of ammonium bicarbonate buffer (pH 7.8). The reaction was incubated at 37 °C for 24 h, and the deglycosylated proteins were directly used for HC-S ELISA.
Size-Exclusion Chromatography Coupled to Multiangle Light Scattering (SEC-MALS)
To assess the size, homogeneity, and oligomeric nature of various proteins, 75 μL of 45 μM proteins was injected onto a Superdex-200 Increase 10/30 GL column on an Agilent HPLC system equilibrated in 25 mM tris–HCl, pH 7.5, 150 mM NaCl, and 2 mM MgCl2. The HPLC system was coupled to a DAWN HELEOSII detector equipped with a quasi-elastic light scattering module and an Optilab T-rEX refractometer (Wyatt Technology). Data were analyzed using the ASTRA 7.3 software (Wyatt Technology Europe) to determine the molecular weights of the eluting proteins. Data conversion and final analysis were performed with Origin software (OriginLab Corporation, Northampton, MA).
Fluorescence Polarization (FP)
5 nM of 5′-fluorescein-labeled DNA–RNA hybrid (23DR) was titrated with increasing amounts of S9.6 proteins in a buffer consisting of 25 mM tris–HCl (pH 7.5), 50 mM NaCl, and 2 mM MgCl2 in a 96-well plate at 21 °C. FP values were measured in triplicate wells using a BMG CLARIOstar Plus microplate reader with excitation at 482 nm, emission at 530–540 nm, and LP (longpass) 504 dichroic filter settings. Changes in FP (ΔFP) as a function of S9.6 concentrations were fit with the following equation to determine the apparent dissociation constants
Hybrid-Capture Sandwich ELISA for DNA–RNA Hybrid (HC-S)
A sandwich ELISA format was developed for the identification of DNA–RNA hybrids. The assay is termed HC-S (Hybrid-Capture assay for DNA–RNA (HC) with Sandwich format) (Figure 2A). For surface coating with S9.6 monoclonal IgG, 1 μg of S9.6 IgG in 200 μL of 100 mM sodium carbonate–bicarbonate buffer (pH 9.4) was added per well to white flat-bottom polystyrene high-bind microplates (Corning Costar). The plates were sealed and kept at 4 °C for 16 h. The plate was washed three times with wash buffer (25 mM tris–Cl, pH 7.5, 150 mM NaCl, 0.1% BSA, 0.005% Tween-20). The wells were blocked by adding 200 μL of blocking buffer (50 mM tris–HCl, pH 7.0; 150 mM NaCl; 0.05% NaN3; 0.2% bovine serum albumin and 5% NFDM Superblock [Biorad]). The plates were sealed and kept at 4 °C for 16 h. The next day, the plates were washed three times and stored at 4 °C.
The S9.6 IgG-coated plates were incubated with 100 pmole of double-stranded nucleic acid hybrids per well in the binding buffer (50 mM tris–Cl, pH 7.5, 150 mM NaCl, 0.05% Tween-20, and 0.1% BSA) at 4 °C. The plates were washed three times with wash buffer to remove any unbound nucleic acid. S9.6 Fab–SEAP variants were diluted to different concentrations (as mentioned in Figure 2) in a 1× Odyssey LiCOR blocking buffer and incubated at 4 °C for 16 h (unless otherwise specified). Plates were washed three times with a wash buffer. The bound S9.6 Fab–SEAP was measured by adding 50 μL of CDP-Star (Roche), resulting in luminescence, which was captured for 1 s per well using a Victor3 plate reader (PerkinElmer). Chemiluminescence (in arbitrary units, A.U.) as a function of S9.6 concentrations was fit with the following equation to determine the apparent dissociation constant by
Acknowledgments
This research was supported by the intramural research programs of the National Institute of Allergy and Infectious Diseases and The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.2c00609.
Supporting Figures S1–S5: chromatogram and additional control experiments; Supporting File 1: G3-SEAP enzyme protein sequence; and Supporting File 2: extracted ion chromatograms (XICs) with the observed molecular weight of purified proteins (PDF)
Author Contributions
A.B. and S.H.L. conceptualized, designed, and supervised the work. E.W. and A.B. performed cloning; S.H.L. performed transfection in ExpiCHO-S; R.F., S.H.L., E.W., and A.B. performed protein purification; E.W., S.H.L., and A.B. performed sortase A-mediated conjugation; C.B.N. and J.Z. performed experiments with SEC-MALS; and F.P., E.W., M.L.P., and A.B. performed HC-S ELISA. All authors contributed to manuscript preparation.
The authors declare no competing financial interest.
Supplementary Material
References
- Boguslawski S. J.; Smith D. E.; Michalak M. A.; Mickelson K. E.; Yehle C. O.; Patterson W. L.; Carrico R. J. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J. Immunol. Methods 1986, 89, 123–130. 10.1016/0022-1759(86)90040-2. [DOI] [PubMed] [Google Scholar]
- Hu Z.; Zhang A.; Storz G.; Gottesman S.; Leppla S. H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Res. 2006, 34, e52 10.1093/nar/gkl142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutrow N.; Nix D. A.; Holt D.; Milash B.; Dalley B.; Westbroek E.; Parnell T. J.; Cairns B. R. Dynamic transcriptome of Schizosaccharomyces pombe shown by RNA-DNA hybrid mapping. Nat.Genet. 2008, 40, 977–986. 10.1038/ng.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidian M.; Dozier J. K.; Distefano M. D. Enzymatic labeling of proteins: techniques and approaches. Bioconjugate Chem. 2013, 24, 1277–1294. 10.1021/bc400102w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartono S. R.; Malapert A.; Legros P.; Bernard P.; Chedin F.; Vanoosthuyse V. The Affinity of the S9.6 Antibody for Double-Stranded RNAs Impacts the Accurate Mapping of R-Loops in Fission Yeast. J. Mol. Biol. 2018, 430, 272–284. 10.1016/j.jmb.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bou-Nader C.; Bothra A.; Garboczi D. N.; Leppla S. H.; Zhang J. Structural basis of R-loop recognition by the S9.6 monoclonal antibody. Nat. Commun. 2022, 13, 1641 10.1038/s41467-022-29187-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yehle C. O.; Patterson W. L.; Boguslawski S. J.; Albarella J. P.; Yip K. F.; Carrico R. J. A solution hybridization assay for ribosomal RNA from bacteria using biotinylated DNA probes and enzyme-labeled antibody to DNA:RNA. Mol.Cell Probes 1987, 1, 177–193. 10.1016/0890-8508(87)90026-0. [DOI] [PubMed] [Google Scholar]; b Miller C. A.; Patterson W. L.; Johnson P. K.; Swartzell C. T.; Wogoman F.; Albarella J. P.; Carrico R. J. Detection of bacteria by hybridization of rRNA with DNA-latex and immunodetection of hybrids. J. Clin. Microbiol. 1988, 26, 1271–1276. 10.1128/jcm.26.7.1271-1276.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Casebolt D. B.; Stephensen C. B. Monoclonal antibody solution hybridization assay for detection of mouse hepatitis virus infection. J. Clin. Microbiol. 1992, 30, 608–612. 10.1128/jcm.30.3.608-612.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a El Hage A.; French S. L.; Beyer A. L.; Tollervey D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. 10.1101/gad.573310. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Skourti-Stathaki K.; Proudfoot N. J.; Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. 10.1016/j.molcel.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno P. A.; Lott P. L.; Christensen H. C.; Korf I.; Chedin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. 10.1016/j.molcel.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L. A.; Chedin F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2019, 14, 1734–1755. 10.1038/s41596-019-0159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanchetta G.; Carzaniga T.; Vanjur L.; Casiraghi L.; Tagliabue G.; Morasso C.; Bellini T.; Buscaglia M. Design of a rapid, multiplex, one-pot miRNA assay optimized by label-free analysis. Biosens. Bioelectron. 2021, 172, 112751 10.1016/j.bios.2020.112751. [DOI] [PubMed] [Google Scholar]
- Gibbons H. R.; Aune T. M. Immunoprecipitation of DNA:RNA Hybrids Using the S9.6 Antibody. Methods Mol. Biol. 2020, 2161, 195–207. 10.1007/978-1-0716-0680-3_14. [DOI] [PubMed] [Google Scholar]
- a Eder P. S.; Lou J.; Huff J.; Macioszek J. The next-generation Hybrid Capture High-Risk HPV DNA assay on a fully automated platform. J. Clin. Virol. 2009, 45, S85–92. 10.1016/S1386-6532(09)70013-7. [DOI] [PubMed] [Google Scholar]; b Schachter J.; Hook E. W. III.; McCormack W. M.; Quinn T. C.; Chernesky M.; Chong S.; Girdner J. I.; Dixon P. B.; DeMeo L.; Williams E.; et al. Ability of the Digene hybrid capture II test to identify Chlamydia trachomatis and Neisseria gonorrhoeae in cervical specimens. J. Clin. Microbiol. 1999, 37, 3668–3671. 10.1128/JCM.37.11.3668-3671.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D. D.; Garboczi D. N.; Singh K.; Hu Z.; Leppla S. H.; Leysath C. E. The sub-nanomolar binding of DNA-RNA hybrids by the single-chain Fv fragment of antibody S9.6. J. Mol. Recognit. 2013, 26, 376–381. 10.1002/jmr.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton-That H.; Liu G.; Mazmanian S. K.; Faull K. F.; Schneewind O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 12424–12429. 10.1073/pnas.96.22.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp M. W.; Antos J. M.; Grotenbreg G. M.; Spooner E.; Ploegh H. L. Sortagging: a versatile method for protein labeling. Nat. Chem. Biol. 2007, 3, 707–708. 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- Nguyen G. K.; Cao Y.; Wang W.; Liu C. F.; Tam J. P. Site-Specific N-Terminal Labeling of Peptides and Proteins using Butelase 1 and Thiodepsipeptide. Angew. Chem., Int. Ed. 2015, 54, 15694–15698. 10.1002/anie.201506810. [DOI] [PubMed] [Google Scholar]
- Deweid L.; Neureiter L.; Englert S.; Schneider H.; Deweid J.; Yanakieva D.; Sturm J.; Bitsch S.; Christmann A.; Avrutina O.; et al. Directed Evolution of a Bond-Forming Enzyme: Ultrahigh-Throughput Screening of Microbial Transglutaminase Using Yeast Surface Display. Chem. – Eur. J. 2018, 24, 15195–15200. 10.1002/chem.201803485. [DOI] [PubMed] [Google Scholar]
- Fairhead M.; Howarth M. Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol. 2015, 1266, 171–184. 10.1007/978-1-4939-2272-7_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antos J. M.; Ingram J.; Fang T.; Pishesha N.; Truttmann M. C.; Ploegh H. L. Site-Specific Protein Labeling via Sortase-Mediated Transpeptidation. Curr. Protoc. Protein Sci./editorial board, John E. Coligan··· [et al.] 2017, 89, 15 13 11–15 13 19. 10.1002/cpps.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp M. W.; Dougan S. K.; Chuang T. Y.; Spooner E.; Ploegh H. L. Sortase-catalyzed transformations that improve the properties of cytokines. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 3169–3174. 10.1073/pnas.1016863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Cohen J.; Song X.; Zhao A.; Ye Z.; Feulner C. J.; Doonan P.; Somers W.; Lin L.; Chen P. R. Improved variants of SrtA for site-specific conjugation on antibodies and proteins with high efficiency. Sci. Rep. 2016, 6, 31899 10.1038/srep31899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T. T.; Sinai P.; Kitts P. A.; Kain S. R. Quantification of gene expression with a secreted alkaline phosphatase reporter system. Biotechniques 1997, 23, 1110–1114. 10.2144/97236pf01. [DOI] [PubMed] [Google Scholar]
- Llinas P.; Stura E. A.; Menez A.; Kiss Z.; Stigbrand T.; Millan J. L.; Le Du M. H. Structural studies of human placental alkaline phosphatase in complex with functional ligands. J. Mol. Biol. 2005, 350, 441–451. 10.1016/j.jmb.2005.04.068. [DOI] [PubMed] [Google Scholar]
- Ramirez P.; Crouch R. J.; Cheung V. G.; Grunseich C. R-loop analysis by dot-blot. J. Vis. Exp. 2021, e62069 10.3791/62069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olczak M.; Szulc B. Modified secreted alkaline phosphatase as an improved reporter protein for N-glycosylation analysis. PLoS One 2021, 16, e0251805 10.1371/journal.pone.0251805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engvall E.; Perlmann P. Enzyme-linked immunosorbent assay, Elisa. 3. Quantitation of specific antibodies by enzyme-labeled anti-immunoglobulin in antigen-coated tubes. J. Immunol. 1972, 109, 129–135. 10.4049/jimmunol.109.1.129. [DOI] [PubMed] [Google Scholar]
- Burkot T. R.; Wirtz R. A.; Lyon J. Use of fluorodinitrobenzene to identify monoclonal antibodies which are suitable for conjugation to periodate-oxidized horseradish peroxidase. J. Immunol. Methods 1985, 84, 25–31. 10.1016/0022-1759(85)90411-9. [DOI] [PubMed] [Google Scholar]
- O’Sullivan M. J.; Gnemmi E.; Morris D.; Chieregatti G.; Simmonds A. D.; Simmons M.; Bridges J. W.; Marks V. Comparison of two methods of preparing enzyme-antibody conjugates: application of these conjugates for enzyme immunoassay. Anal. Biochem. 1979, 100, 100–108. 10.1016/0003-2697(79)90117-9. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- Bowers G. N. Jr.; McComb R. B. Measurement of total alkaline phosphatase activity in human serum. Clin. Chem. 1975, 21, 1988–1995. 10.1093/clinchem/21.13.1988. [DOI] [PubMed] [Google Scholar]
- Mornet E.; Stura E.; Lia-Baldini A. S.; Stigbrand T.; Menez A.; Le Du M. H. Structural evidence for a functional role of human tissue nonspecific alkaline phosphatase in bone mineralization. J. Biol. Chem. 2001, 276, 31171–31178. 10.1074/jbc.M102788200. [DOI] [PubMed] [Google Scholar]
- Lai M. B.; Zhang C.; Shi J.; Johnson V.; Khandan L.; McVey J.; Klymkowsky M. W.; Chen Z.; Junge H. J. TSPAN12 Is a Norrin Co-receptor that Amplifies Frizzled4 Ligand Selectivity and Signaling. Cell Rep 2017, 19, 2809–2822. 10.1016/j.celrep.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Kundrat L.; Pishesha N.; Bilate A.; Theile C.; Maruyama T.; Dougan S. K.; Ploegh H. L.; Lodish H. F. Engineered red blood cells as carriers for systemic delivery of a wide array of functional probes. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 10131–10136. 10.1073/pnas.1409861111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.