

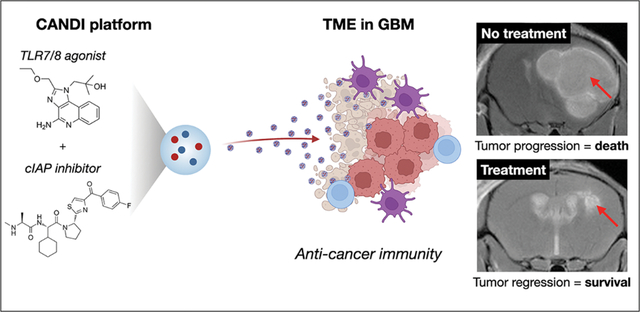
Abstract
Myeloid cells are abundant, create a highly immunosuppressive environment in glioblastoma and thus contribute to poor immunotherapy responses. Based on the hypothesis that small molecules can be used to stimulate myeloid cells to elicit anti-tumor effector functions, we developed a synthetic nanoparticle approach to deliver dual NF-kB pathway-inducing agents into these cells via systemic administration. Synthetic, cyclodextrin-adjuvant nanoconstructs (CANDI) with high affinity for tumor-associated myeloid cells were dually loaded with a TLR7 and 8 (Toll-like receptor, 7 and 8) agonist (R848) and a cIAP (cellular inhibitor of apoptosis protein) inhibitor (LCL-161) to dually activate these myeloid cells. Here we show that CANDI: i) readily enters the glioblastoma tumor microenvironment and accumulates at high concentrations, ii) is taken up by tumor-associated myeloid cells, iii) potently synergizes payloads compared to monotherapy, iv) activates myeloid cells, v) fosters a “hot” tumor microenvironment with high levels of T effector cells, and vi) controls the growth of murine GBM as mono- and combination therapies with anti-PD1. Multi-pathway targeted myeloid stimulation via the CANDI platform can efficiently drive anti-tumor immunity in GBM.
Keywords: nanoparticles, targeting, macrophage, glioblastoma, IL-12, IFNG
Graphical Abstract
Introduction
High-grade glioma (Glioblastoma multiforme, GBM) is the central nervous system’s most common and deadly primary cancer. Standard-of-care therapies include surgical resection with chemoradiation, FDA-approved treatments such as intracavitary chemotherapy wafers, bevacizumab, and alternating electrical fields[1] with median survival times of approximately 15 months from diagnosis[2]. Numerous promising therapeutic modalities (targeted kinase inhibitors, anti-angiogenic treatments, immunotherapies) have been tested in advanced clinical trials without lasting success.
There has been a resurgence of interest in therapeutic modalities that stimulate the immune system against tumor cells. However, immune checkpoint inhibitors (ICI) have generally not been as successful against GBM as they have been for other cancers[3]. Although there is still interest in ICI therapy alone in a neo-adjuvant setting[4], the neurooncology community has been trying to explain its limited efficacy. One accepted explanation is that the GBM microenvironment is dominated by profound myeloid immunosuppression and continuous changes and adaptation to therapies[5–7]. This immunosuppression inhibits effective immune surveillance by altering the ability of anti-tumor effector immune cells (T cells, macrophages, NK cells) to infiltrate and destroy the GBM.
Interleukin-12 (IL-12) has emerged as an attractive cytokine for cancer therapy because it has potent anti-cancer effects[8,9]. IL-12 has been described as a “master” immune-activating cytokine and has a critical role in cancer immune surveillance[9–12]. It is a potent immunostimulatory cytokine that orchestrates a Th1 response with activation of CD8+ T cells, CD4+ Th cells, B cells, and NK cells. One primary downstream effector of IL-12 is IFNG produced by T and NK cells. IFNG is also a potent immune-activating cytokine required for cancer immune surveillance[13]. Signals such as IFNG alter macrophage cells from an immune-suppressive (M2) to a pro-inflammatory (M1) state[6]. IFNG also possesses anti-angiogenic effects.
Clinical trials of systemic IL-12 were undertaken several decades ago but were terminated because the cytokine, systemically administered as a recombinant soluble protein, was poorly tolerated[14]. As a result, attention has turned to increasing local concentrations of IL-12 more selectively, bypassing systemic side effects[15]. This has been attempted by using viral vectors[16,17], lipid nanoparticles containing mRNA for IL-12[18], activating the canonical NF-kB pathway[19–21] or the non-canonical NF-kB pathway through inhibition of the cellular inhibitor of apoptosis protein-1(cIAP)[22–24]. While these therapeutic methods have shown some efficacy, the IL-12 induction is modest, temporally limited, and lack durable response. In the future, the goal would thus be to activate tumor-associated myeloid cells more potently to jumpstart an effective, long lasting immune response.
We reasoned that IL-12 super-production in tumor-associated myeloid cells, including dendritic cells (DC), macrophages (MF), and microglia (MG), could be accomplished by combinatorial activation of both the canonical and non-canonical NF-kB pathways, which signal through distinct mechanisms. Multi-pathway modulation also has the potential for drug synergism, lower drug needs, reduced toxicity and higher thresholds for developing compensatory escape mechanisms. Since tumor associated myeloid cells are highly phagocytic, we used a synthetic nanoparticle approach to co-deliver a TLR7/8 agonist and cIAP antagonist to increase their IL-12 induction and make them tumoricidal[25]. Here we show that this dual myeloid cell modulation approach - facilitated by a novel cyclodextrin-adjuvant nanoconstruct dual immunotherapy (CANDI) platform is remarkably efficient in controlling murine GBM, an otherwise treatment-resistant tumor type.
Results
The rationale for dual myeloid stimulation
Based on the observation that the highest IL-12-producing cells in tumor microenvironments are dendritic cells (DC) in the DC3 cluster[26], we hypothesized that small molecules targeting each of these pathways respectively could increase IL-12 production in the tumor microenvironment since the production is primarily regulated by canonical and non-canonical NF-kB pathways, which signal through distinct mechanisms (Fig. 1). The challenge was to selectively deliver small molecules affecting these pathways in tumor-associated myeloid cells. The latter contributed 25–80% of cellular mass of the GBM[27] and create a highly immunosuppressive environment. Unfortunately, systemic administration of myeloid-targeted small molecules by themselves have either often had dose limiting solubility issues, limited efficacy or high toxicity[28].
Fig. 1: Overview of strategy.
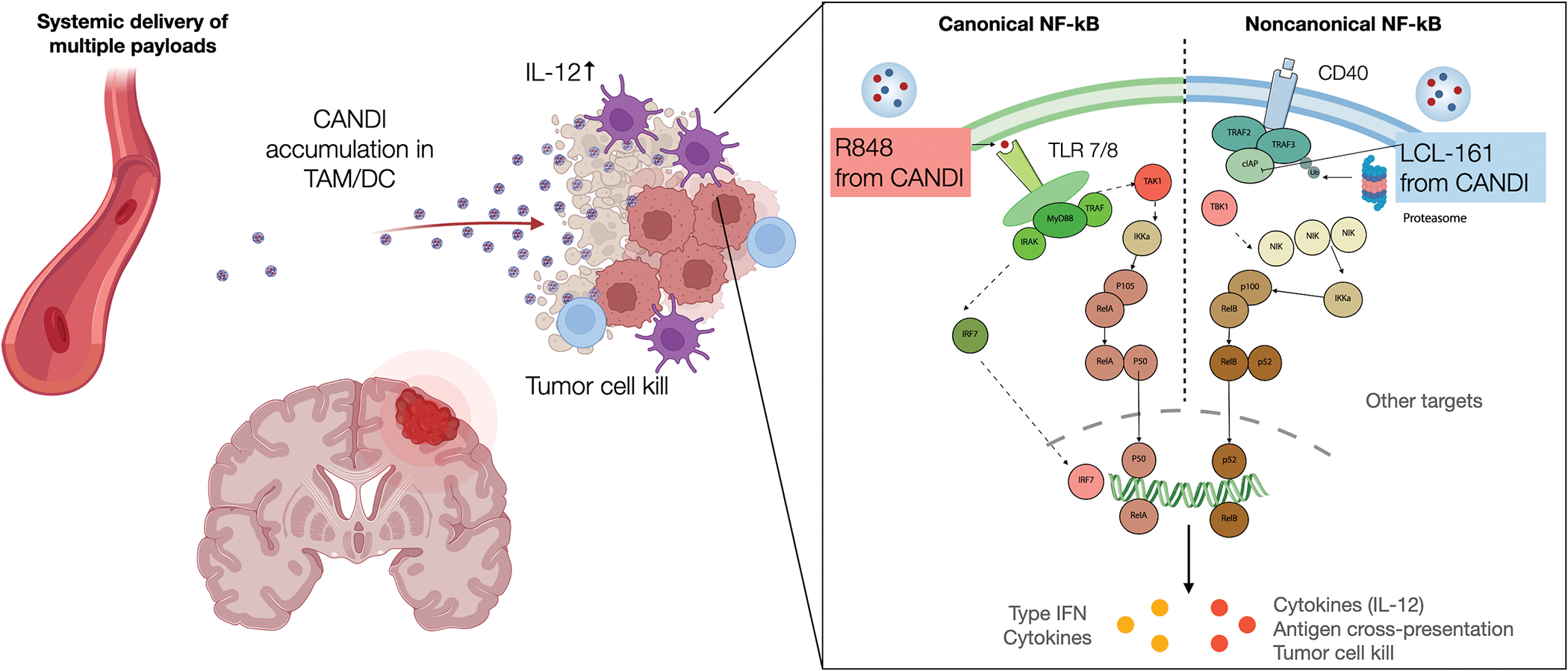
Carbohydrate-based nanoparticles (CANDI) injected systemically were used to selectively deliver several small molecules to myeloid cells to affect their function in brain tumors. Specifically, we used the TLR7/8 agonist R848 to stimulate the canonical NF-kB pathway and the cIAP inhibitor LCL-161 to stimulate the noncanonical NF-kB pathway in myeloid cells. This is important because compensatory mechanism of cells often results in resistant phenotypes. In this research, one readout of myeloid cell stimulation was IL-12 production which was much higher for CANDI combination treatment than with monotherapy (See Fig. 3).
We hypothesized that carbohydrate-based nano-formulations could be used for myeloid cell targeting. Linear, long-chain dextrans are often used as vascular and intravital macrophage makers[29] but lack the ability to carry therapeutic payloads. Alternatives are cyclodextrin-modified polymers but their overall myeloid-targeting efficacy is often low. Prior research had explored a nanoconstruct created entirely out of cross-linked cyclodextrins[19] but the loading efficiency was limited and the preparation was unstable and often precipitated upon drug loading. We thus performed screening experiments to create a more stable preparation capable of multiple drug encapsulations, and combination drug encapsulations. This was achieved by less/more cross linking and a slightly larger particle size. The resultant therapeutic cyclodextrin-adjuvant nanoconstruct for dual immunotherapy (CANDI) was then prepared by loading nanoparticles with dual NF-kB modulators; specifically, R848 as a TLR7/8 agonist and LCL-161 as a cIAP inhibitor (Figs. 1–2).
Fig. 2: CANDI drug delivery.
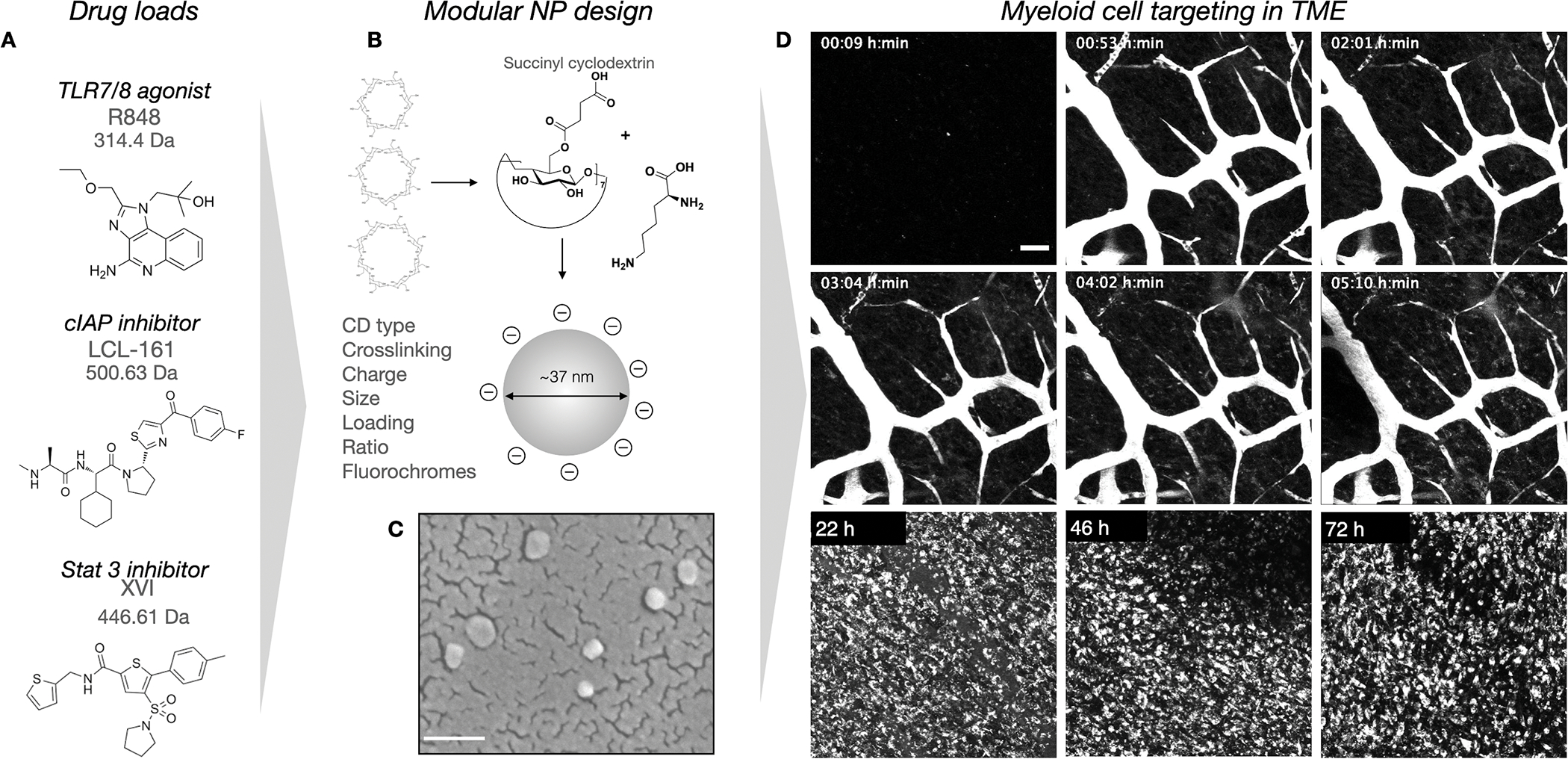
A. A challenge today is to direct multiple small molecules to tumor associated dendritic cells and macrophages at sufficiently high concentrations with eliciting systemic toxicities. B. CANDI consist of succinyl-lysine cross-linked cyclodextrins to yield negatively charged nanoparticles with ~27 nm in diameter in unloaded form and ~37nm in loaded form. The cyclodextrin units serve as reservoirs for small molecule loading. C. Scanning electron microscopy image of CANDI nanoparticles sputter-coated with gold (~8 μm layer, scale bar = 100 nm). D. Serial intravital imaging of tumor showing the distribution of fluorescently labeled CANDI (1 mg CANDIAF647; scale bar = 50 μm) as a function of time. Initially, CANDI is primely located intravascularly but faintly appears in cellular compartments around 2–3 h. At 22–72 h, all CANDI is associated with myeloid cells.
Synthesis and characterization of nanoparticles
Fig. 2 and S1 summarize the lead nanoparticle synthesis, fluorescent labeling, and particle characterization. The empty nanoconstruct (CANDIE) was synthesized by slow cross-linking of succinyl-β- cyclodextrin with lysine using EDC/NHS chemistry. The reaction conditions were chosen to result in nanoparticles with an average size of ~27 ± 4 nm (DLS) and negative zeta potential (−9.6 mW in PBS) for the most efficient in vivo circulation and myeloid cell delivery. Particles appeared monodisperse and homogenous by scanning electron microscopy (Fig. S3) as well as transmission electron microscopy. We also prepared fluorescent versions to track CANDIE in vivo by incorporating Pacific blue (PB) or Alexa Fluor 647 (AF647) into the nanoparticles.
Once prepared and characterized, we focused on drug loading and measuring release kinetics in vitro. The ~1,500 cyclodextrin units in each NP serve as a host for small molecules, such as the chosen R848 and LCL-161 through host-guest interactions. To measure small molecule loading, we implemented a phenolphthalein test. In its deprotonated form, phenolphthalein exhibits high absorption at 550 nm. Upon interaction with the cyclodextrin units in CANDI, phenolphthalein (P) undergoes a spirocyclization reducing its absorption at 550 nm. Conversely, phenolphthalein is released from CANDI when the therapeutic payloads are added, increasing absorption. This competitive assay allowed us to optimize the drug loading of CANDI. Complete signal loss was observed immediately after loading indicator P into CANDIE. We tested different loading ratios to assess binding affinity. Approximately 21 ± 5% and 57 ± 9% signal recovery were measured when 0.04 mg R848 and 0.1 mg LCL-161 where individually loaded per mg of CANDIE. Combined in the same ratio and loaded, 85 ± 4% signal recovery was gained (Fig. S2). Interestingly, we noticed rapid (<1 min) displacement of the phenolphthalein indicator in all reactions. We discovered a slight increase in CANDI size (~10 nm) with optimal loading, which was reversible with drug elution and quantifiable using dynamic light scattering of dialyzed samples (Fig. S2).
We next performed detailed characterization of CANDI to corroborate the presence of both pharmaceuticals payloads in the polymer (Fig. S4–S7). With an optimized method for high-pressure chromatography, we were able to confirm loading and elution of both drugs by retention time and mass spectrometry. This same analysis was used to study the kinetics of release. A standard deviation curve was generated from the chromatograph to quantify counts to particle concentration. Particles where subjected to short rapid centrifugation cycles eluting ~50 μL of media, which where analyzed and quantified. The maximum half-life of the compounds subjected to PBS at 37 °C was 0.5± 0.32 h and 1.4 ± 0.5 h. In these conditions, ~80% of R848 and LCL-161 were released from the particles after 18 h at 37 °C in PBS (Fig. S2).
Screening for IL-12 induction in myeloid cells
We designed an in vitro cell screening assay using primary MF and DC derived from IL-12-eYFP reporter mice to determine the optimum dosing regimen for activating myeloid cells. Cells expressing IL-12 co-express eYFP[30], providing a valuable visual indicator of myeloid cell activation, which can be resolved using high throughput image screening. A similar approach was previously used to identify LCL-161 as an activator of IL-12 in DC[23]. Using either Flt3L-derived DC or M-CSF-derived macrophages, we performed a dose titration of nanoparticles containing R848 solely, LCL-161 solely, or combined R848 and LCL-161. We found that single dosing of macrophages and DC with either R848 or LCL-161 could induce modest levels of IL-12, similarly to previous reports[23].
Interestingly, however, we found that the CANDI combination nanoparticle could induce IL-12 production at substantially higher levels than the additive effects of either single agent (Fig. 3) We speculate that R848, which is a known potent stimulator of Type 1 interferon, primes cells to up-regulate non-canonical NF-kB signaling pathways. Type 1 interferon signals are needed to activate dendritic cells which trigger anti-tumor CD8 T cell responses[31]. We have prior shown that loss of interferon signaling in tumor antigen-presenting cells (CD11c+) renders cancer immunotherapy ineffective and note that components of the non-canonical NF-kB signaling pathway are down-regulated, indicating a lack of priming in this pathway. CANDI combination therapy provides both interferon stimulus and non-canonical NF-kB pathway agonism in one package. Finally, we found that even a low dose of CANDI containing 62.5 ng LCL-161 and 1.75 ng R848 was highly effective for IL-12 induction in MF and DC (Fig. 3).
Fig. 3: Screening data to determine optimal therapeutic efficacy measured by IL-12 induction.
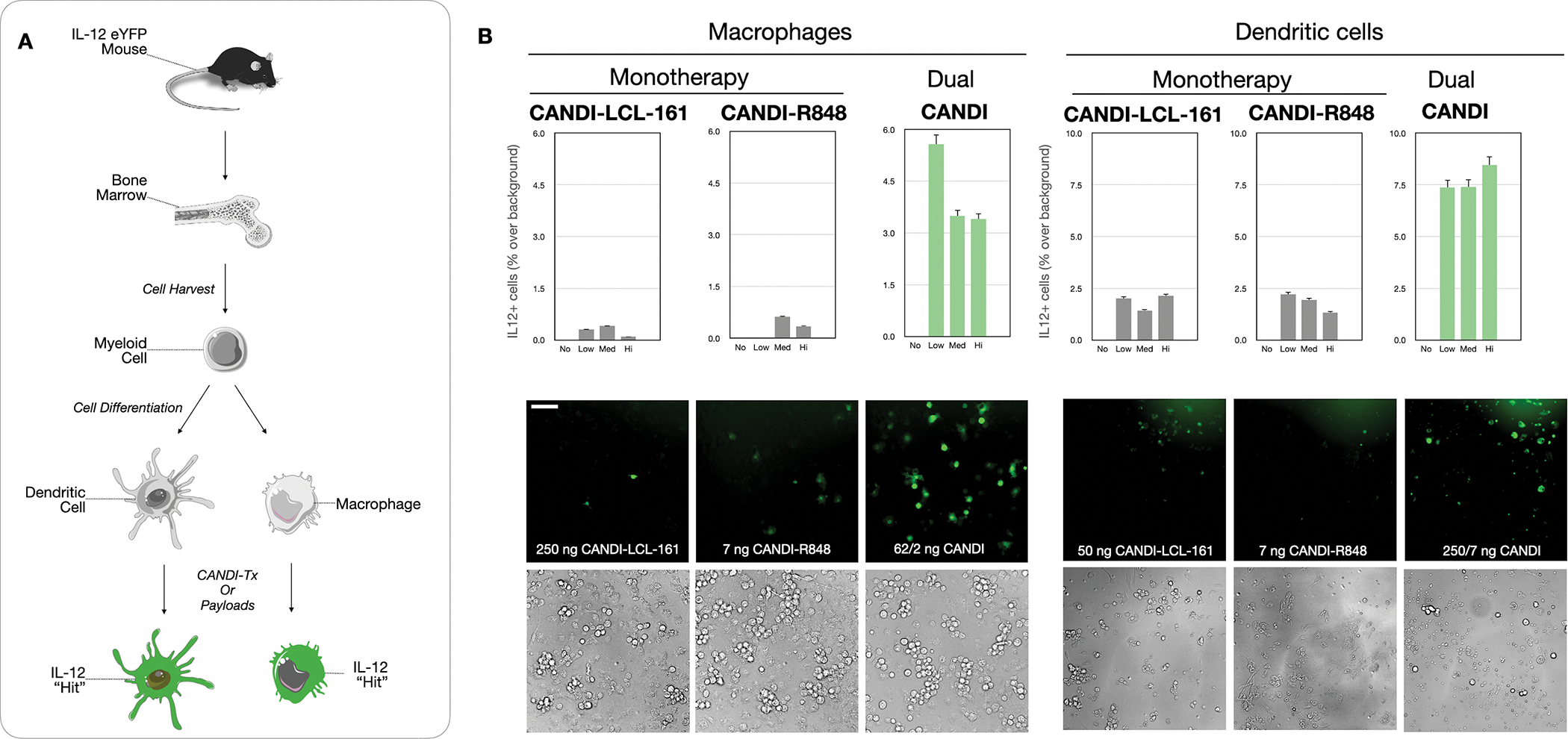
A. Schematic diagram of myeloid cell purification from the bone marrow of IL-12-eYFP mice. Myeloid precursor cells were differentiated into dendritic cells (DC) or macrophages (MF) and then used for screens with different CANDI formulations. B. Summary of screening results in DC and MF using different dosages of nanoparticles (No: negative control; Low: 62.5 ng/1.75 ng CANDI-LCL-161-R848; Med: 125 ng/3.5 ng CANDI-LCL-161-R848; Hi: 250 ng/7 ng CANDI-LCL-161-R848). In the DC example, 250/7 ng CANDI refers to CANDI loaded with 250 ng of R848 and 7 ng of LCL-161. Note that IL-12 induction is always highest in the dually loaded CANDI preparation (P < 0.001 for MF and 0.01 for DC) and much higher than either mono-preparation alone (N = 3 replicates, scale bar = 50 μm).
Pharmacokinetics and spatial distribution of CANDI
Fig. 4 summarizes the pharmacokinetics of CANDI in mouse tumor models bearing intracranially implanted CT2A glioma tumors. The choice of this primary model was based on its resemblance to human GBM[32]. The blood clearance after IV administration showed a biphasic behavior with a fast component of 0.08 h (~5 min) and a slow component of 3.2 h (R2 = 0.99). At 24 h after injection, 99% of the nanoparticle is cleared from circulation. Organ biodistribution was examined by fluorescence reflectance imaging 24 h after injection (Fig. 4B). Interestingly, CANDI accumulation was ~68-fold higher in GBM compared to normal brain (541% ID/g tumor vs. 8% ID/g tissue in the normal brain; Fig. 4B–C). As a consequence of CANDI accumulation within GBM, we also found that tumor-draining cervical lymph nodes likewise retained significant levels of CANDI. As expected, other reticuloendothelial cell-containing organs also showed CANDI, such as the liver (28% ID/g tissue) and spleen (41% ID/g tissue) but at far lower concentrations.
Fig. 4: Pharmacokinetics of CANDI in GBM-bearing mice.
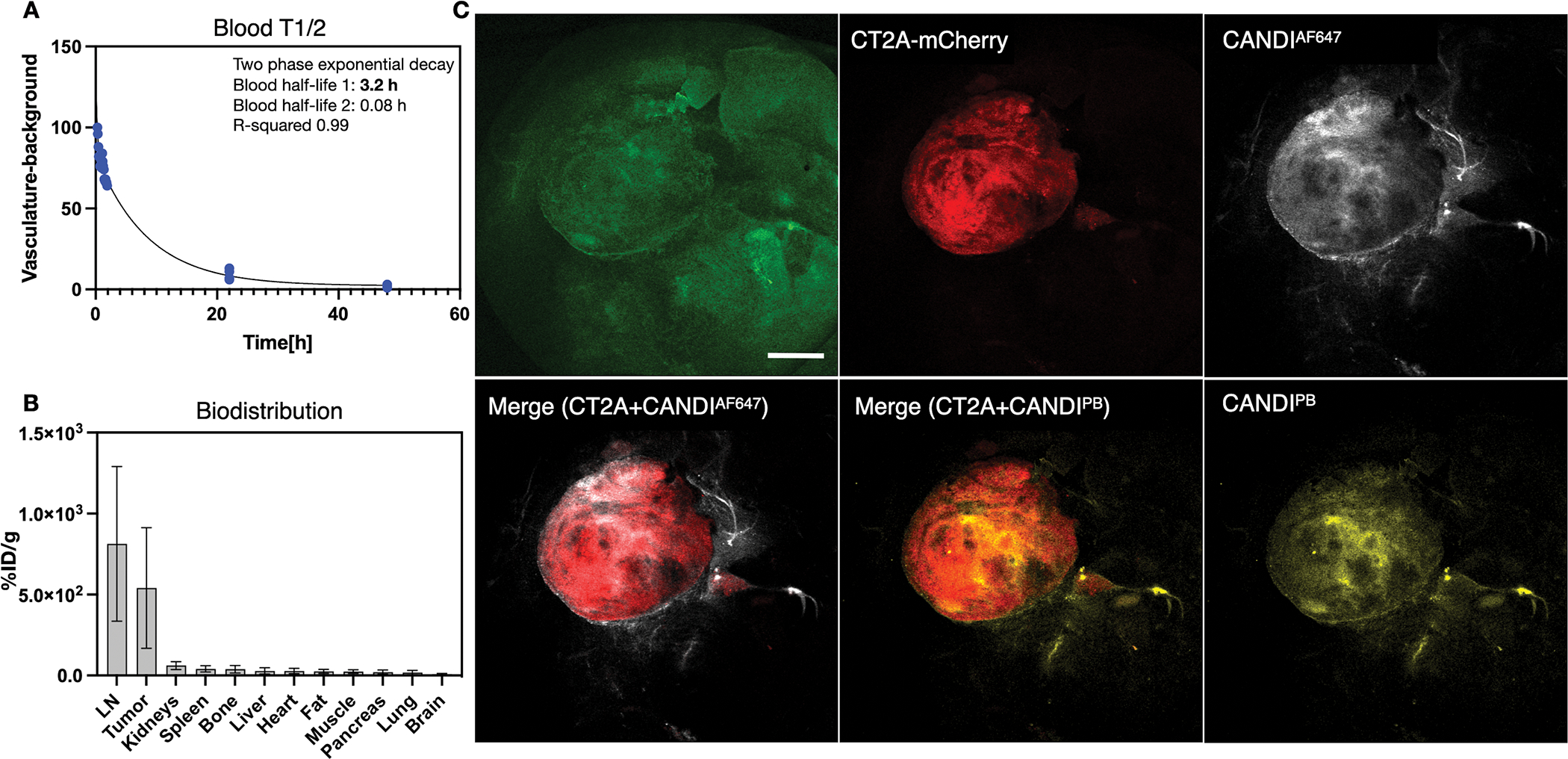
A. Blood half-life (T1/2) determination showed a biphasic decay with a fast component of 5 minutes and a slow component of 3.2 hours (R2 = 0.99). B. The tissues with the highest accumulation of CANDI were GBM and lymph nodes. Note the logarithmic y-axis to show the display of lower tissue concentrations in other organs. Note that GBM/brain ratio is roughly 68:1, presumably because the malignancy alters the blood-brain-barrier and nanoparticles are small enough to efficiently accumulate in phagocytic cells. At the cellular level, the high uptake in GBM (> 500%ID/g tissue) and LN is due to the uptake of CANDI into macrophages and dendritic cells (see Figs. 6–8 for confirmation and quantitation C. Low magnification fluorescence images of resected brains (green autofluorescence) containing CT2A-mCherry tumors (red) and CANDIAF647 (white) or CANDIPB (yellow; scale bar = 1 mm). The left merge image represents the combination of CT2A-mCherry tumors (red) and CANDIAF647 (white). The right merge image represents the combination of CT2A-mCherry tumors (red) and CANDIPB (yellow). Note the high tumoral accumulation of CANDI (for additional example, see Fig. S9; N = 27 mice).
To confirm the kinetics of GBM growth and CANDI accumulation, we performed serial intravital microscopy using CT2A-mApple tumors implanted into the brain of BL6 mice that had a cranial window chamber. Fig. 5 summarizes one of these temporally acquired imaging sets showing rapid tumor growth 2 weeks after implantation. On day 12, we administered CANDIAF647 intravenously and performed serial imaging over several hours to days to demonstrate accumulation in tumors (Fig. S8). Immediately after tail vein injection of the fluorescent nanoparticle, its distribution in the brain was confined to tumor neovasculature. Within hours, CANDIE extravasation could be identified, and cell-associated CANDI became more prominent. Twenty-four hours after injection (Fig. S8), all CANDI signals were cell-associated, with no appreciable signal remaining in vessels. The cellular signal persisted for several days of observations (up to 3 days). We also performed dose studies to determine whether there was an effect of dose on cellular distribution. Fig. S8 shows that different doses resulted in similar cellular uptake patterns across tumors.
Fig. 5: Serial intravital imaging of GBM development.
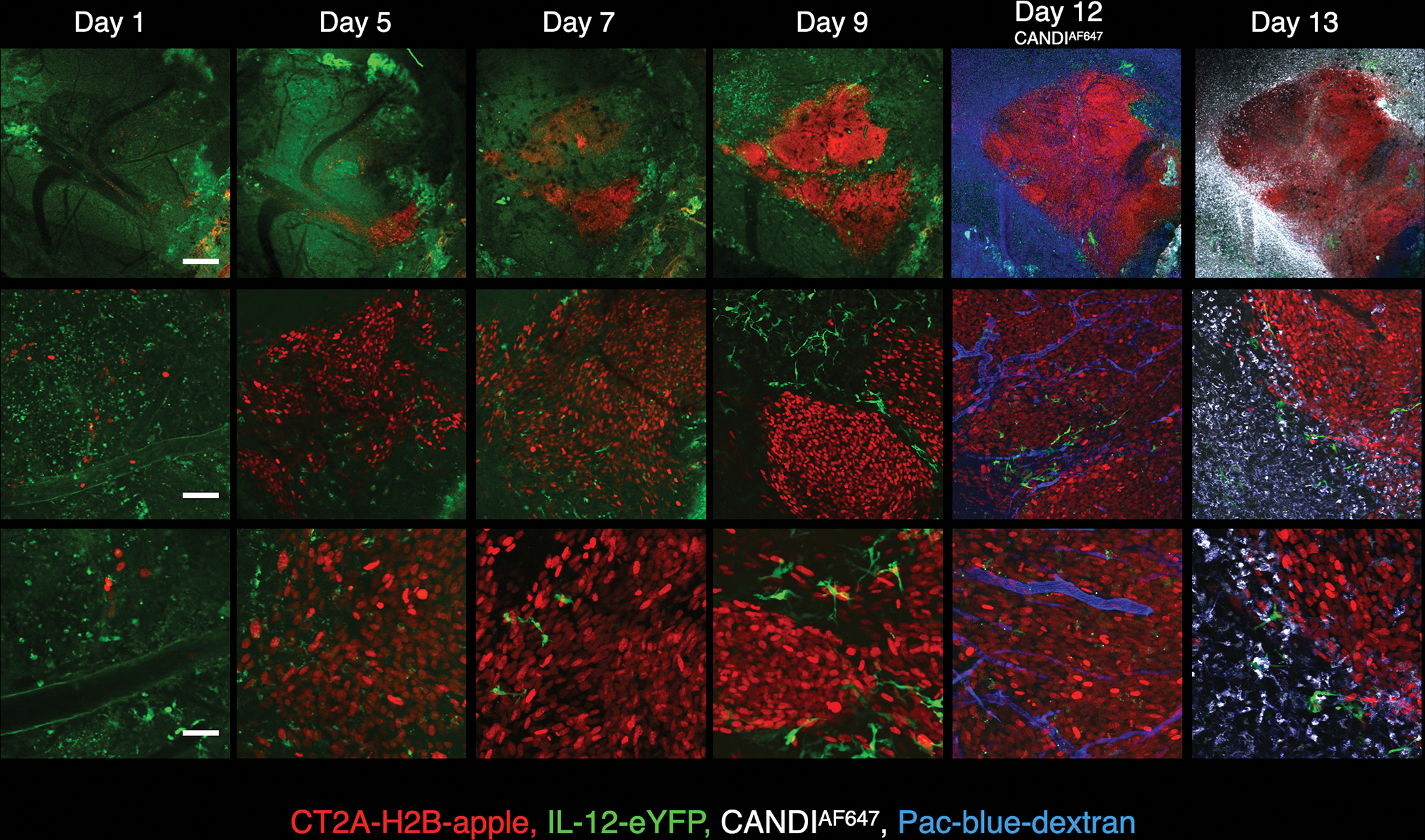
Intravital imaging of brain window chamber implanted into IL-12-eYFP mice. Imaging was performed 1, 5, 7, 9, 12, and 13 days after implantation of CT2A-H2B-mApple tumor cells (red). Shown are three different magnifications: 4x objective (scale bar = 500 μm); 20x objective (scale bar = 100 μm); and 40x objective (scale bar = 50 μm). Note the progressive growth of tumors over the two weeks. IL-12-eYFP positive cells are seen at higher magnifications in the tumor periphery in rapidly patrolling cells, presumably DC (see Movie 2 for kinetics). CANDIAF647 (10 mg/kg) was administered via tail vein on day 12 and can be seen in tumor-associated cells a day later.
We finally investigated which cell types CANDI accumulated in following intravenous administration. Twenty-four hours after injection, GBM tumors were removed and cells were processed for flow cytometry. Fig. 6 and Fig. S9 show that CANDI in GBM was primarily associated with macrophages (56% ID within tumor microenvironment), DC (25% ID), and a much lower amount of other immune cells (3% ID). Overall, >90% of CANDI in GBM was associated with immune cells, and only a minor fraction was associated with tumor cells (~7%). These findings were corroborated by multiplexed immunohistochemistry (Fig. 6A, Fig. S11–12). Furthermore, we performed intravital imaging in Mer-TK-GFP mice (Fig. 6B and Movie 1). In these mice[33], all host myeloid cells contain GFP (especially macrophages, monocytes, dendritic cells, microglia) but not tumoral or other host cells, such as lymphocytes. Our data show clear co-localization of CANDIE with virtually all Mer-TK positive cells in the tumor microenvironment.
Fig. 6: Intratumoral distribution of CANDI.
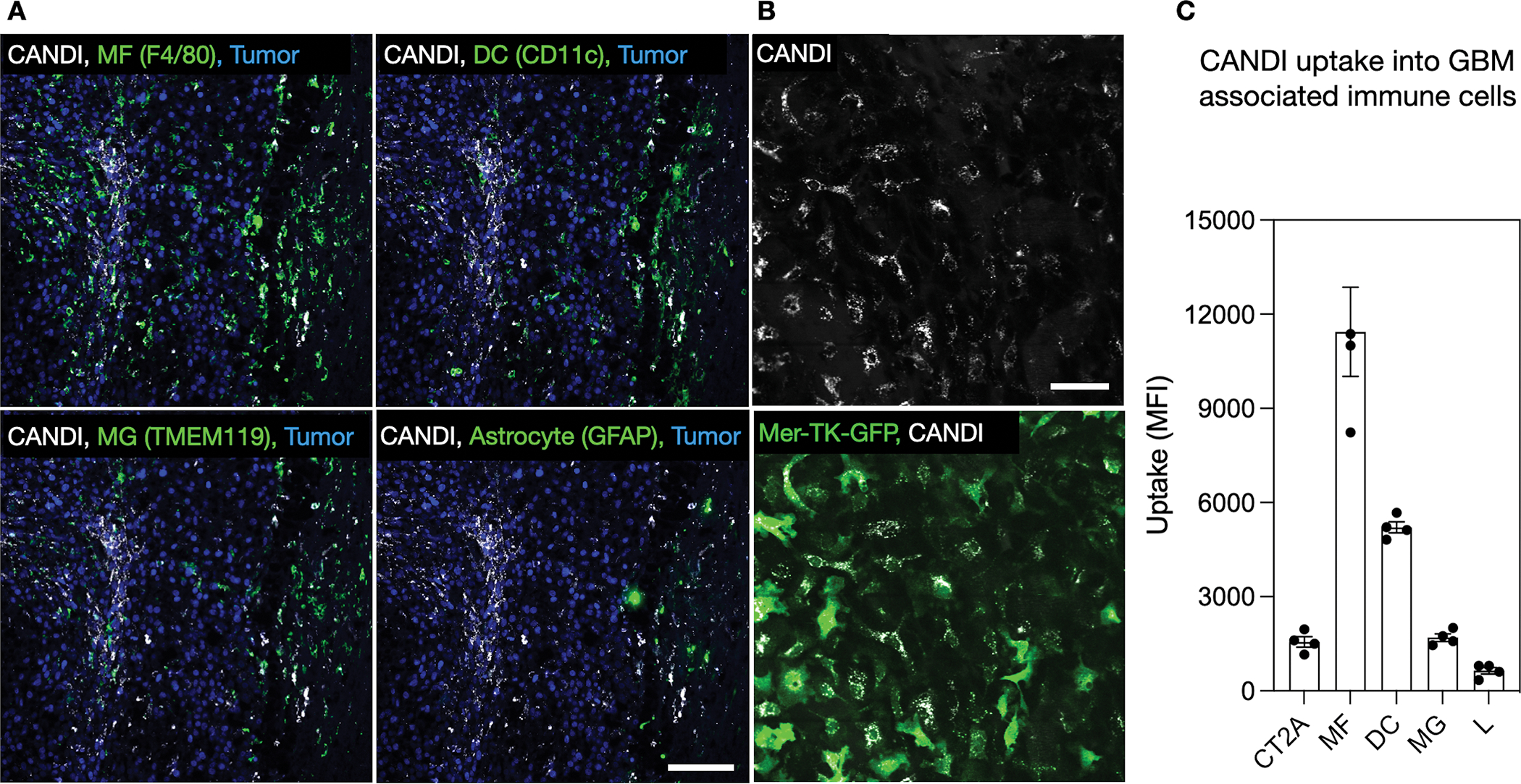
A. Immunofluorescence of CANDIAF647 (white) distribution in tumor-associated macrophages (F4/80+), dendritic cells (CD11c+), microglia (TMEM119+), and astrocytes (GFAP+). Specific immune cell markers are shown in green, and tumor cells with H2B-mApple are shown in blue. Scale bar = 100 μm. B. IV injection of CANDI (white) in tumor-bearing Mer-TK-GFP mice. Note co-localization of CANDI with Mer-TK positive cells. Scale bar = 50 μm. C. Flow cytometry of GBM tissue showing prominent accumulation of CANDI in macrophages (MF) and dendritic cells (DC) and to a much lesser degree in microglia (MG), tumor cells (CT2A), and lymphocytes (L); data from 4 GBM bearing mice; for detailed flow analysis see Fig. S10). The data points are plotted as single measurements and bars represent means and SD (N = 4).
Movie 1: Brain window with CT2A-H2B-apple GBM in IL-12-eYFP and MerTk-GFP mice.
Note the accumulation of CANDI in MerTk positive cells. IL-12-induced cells are both mobile and stationary following CANDI treatment.
Finally, we performed PK/PD modeling to estimate the comparative advantages of encapsulating small molecules in CANDI for TAM delivery (Fig. S18). The modeling was performed for human PK data, as those had been published for the small molecules[34–36]. As can be seen, CANDI encapsulation resulted in an approximately 5–6 orders of magnitude higher tumoral concentrations of the small molecules at 1 day after systemic administration. Furthermore the local GBM drug concentrations remained stable for several days.
Therapeutic efficacy
We next determined therapeutic efficacy of the three different treatment regimens CANDIE control (group 1; N = 18 mice), CANDI (group 2; N = 25 mice), and CANDI + a-PD1 (a-PD1, group 3; N = 16 mice). CT2A tumor cells (50,000 per mouse) were implanted intracranially on day 0, followed by serial bioluminescent imaging (BLI) (days 13 and 21) and MR imaging (days 13 and 23) to monitor tumor growth with systemic intravenous administration of drugs (day 14, 17 and 20). Animals were sacrificed after 22 days, and residual tumor sizes were determined at necropsy. Immune cell infiltration was measured by multiplexed FAST imaging. Additional animal cohorts were used for survival analysis (N = 23).
Fig. 7 summarizes the therapeutic efficacy of the different treatment groups as determined by MR imaging and survival data. At the time of sacrifice, the CT2A tumor volumes were 162.7 ± 31.3 mm3 in the empty CANDIE control group, 11.4 ± 3.8 mm3 in the CANDI group, and 3.1 ± 1.9 mm3 in the CANDI + a-PD1 (a-PD1) group. The differences between group 2 and group 1 (P = 0.0005) and group 3 and 1 (P = 0.0003) were highly statistically significant. The difference between groups 2 and 3 was insignificant (P = 0.0665). Similar findings were observed by bioluminescence imaging (Fig. S13) and MR imaging (Fig. S14). There were also statistically significant differences in the survival study, with a 30% probability of survival beyond 60 days in the CANDI group, 43% in the CANDI + a-PD1 group, and 0% in the CANDIE control group (P < 0.0001). Treated animals did not show significant body weight loss (compared to untreated tumor-bearing mice), and liver histology did not reveal any hepatotoxicity (Fig. S15).
Fig. 7: Therapeutic efficacy.
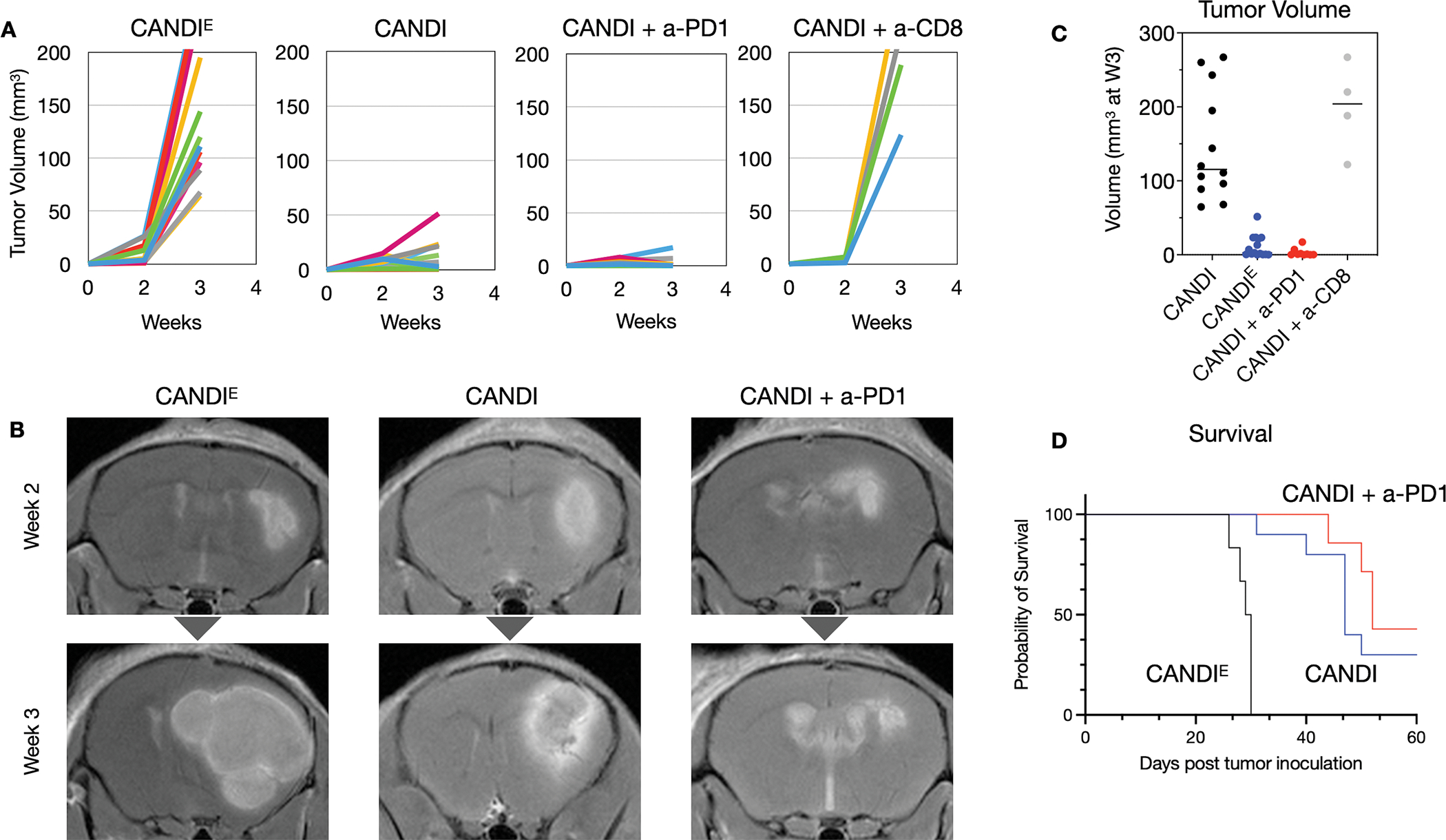
A. CT2A GBM-bearing mice were treated with either empty control CANDIE (N = 18), CANDI (0.5 mg LCL-161, 0.2 mg R848; N = 25), CANDI + a-PD1 (500 μg LCL-161, 200 μg R848, 200 μg a-PD1 antibody; N = 16) or CANDI with a-CD8 (500 μg LCL-161, 200 μg R848, 200 μg a-CD8; N = 4). Animals were serially imaged (MRI and BLI) until the time of sacrifice (23 days). Note the significant growth retardation with CANDI and CANDI + a-PD1, an effect that was abolished by CD8 depletion. This data and the data from Fig. S12 show that CANDI therapy functions through anti-tumor T cell production of IFNG. B. Shown are representative T1W MR images following administration of gadolinium. Note the substantial therapeutic effect of CANDI and its additive effect with a-PD1 antibody. The differences between CANDI and CANDIE (P = 0.0003) were highly statistically significant. See Fig. S13 and Fig. S14 for details on bioluminescence and additional MR imaging. C. Tumor volumes in the 4 different groups as determined at the time of sacrifice. D. Survival graphs for three different treatment groups. All animals injected with CANDIE (without payload) had died by day 30 after tumor implantations, as expected from the MRI (see panel B). The difference between the CANDI group and the control group injected with empty CANDIE was highly significant (P < 0.0001).
Mechanisms of action
We performed additional experiments to shed light on the mechanisms of CANDI-driven anti-tumor effects. First, we determined whether tumor myeloid cell-directed CANDI would increase the expression of IL-12 in the tumor microenvironment. In vivo doses of 5 mg of CANDI nanoparticle translates to 0.2 mg of R848 and 0.5 mg of LCL-161 per mouse. For these experiments, we investigated IL-12-eYFP mice by intravital microscopy. Intravital imaging showed a ~13-fold induction in cell-associated IL-12 within 48 h of treatment, with rapid induction of IL-12 signal following CANDI treatment. Fig. 8A shows a representative example of spatial IL-12 induction in the IVM model, while Movies 2 and 3 provide temporal contexts (Fig. S16). IL-12 itself does not act directly on cancer cells and likely functions as an inflammatory amplifier in anti-tumor immunity. As IL-12 is a known positive regulator of IFNG and cytotoxic lymphocyte responses, which are both potent anti-tumor effector mechanisms, we hypothesized that CANDI would enhance intratumoral levels of IFNG. Finally, we imaged IFNG expression in IFNG-eYFP reporter mice following CANDI administration (Fig. S17 and Movie 4). We show marked IFNG upregulation in the tumor microenvironment (~40-fold) for several days to weeks following IV CANDI administration (Fig. 8B). Pre-treatment samples were completely devoid of IFNG-producing cells, and signal was only significantly detected at 4 days following CANDI treatment. Expression was entirely within rapidly moving, spherical cells, consistent with prior observations that lymphocytes are the main source of IFNG from immunotherapy treatment.
Fig. 8. Effects of CANDI on immune cell status in GBM.
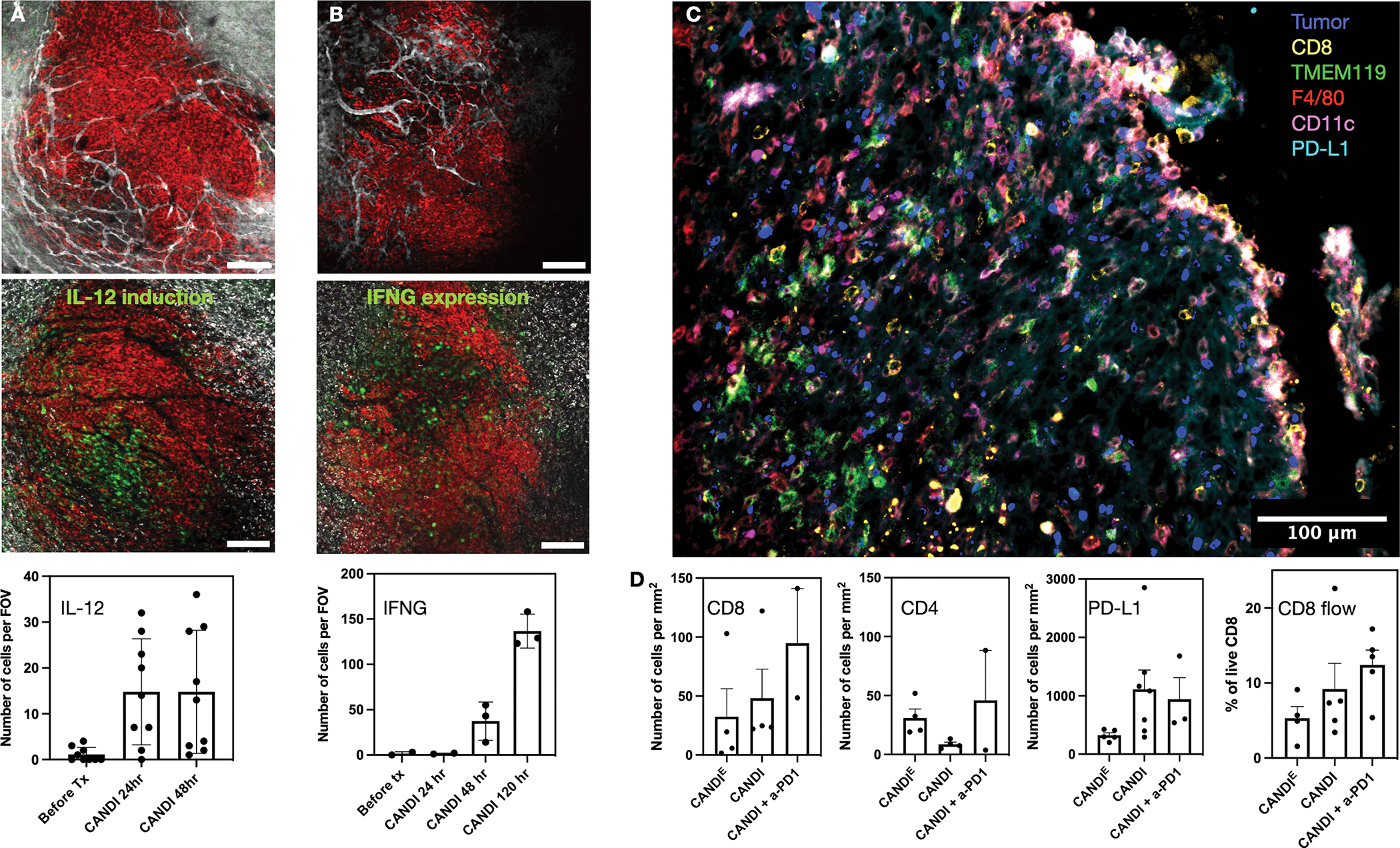
A. Effects of CANDI on IL-12 expression in GBM. Intravital imaging was performed in CT2A (red) brain window model in IL-12-eYFP reporter mice receiving CANDI (white). The top row shows CANDIE distribution at 2 h and the bottom at 48 h after IV administration. After CANDI administration, many more cells are green in the tumor microenvironment (N = 9). Scale bar = 200 μm. B. Effects of CANDI on IFNG expression in T cells. Intravital imaging was performed in CT2A (red) brain window model in IFNG-eYFP reporter mice (GREAT). The top row shows CANDIE distribution at 2 h and the bottom at 96 h after IV administration. Note the substantial increase (>100-fold) of IFNG positive cells over several days after CANDI administration (N = 3). C. Representative multiplexed FAST analysis of CANDI treated GBM section. Note the substantial immune cell infiltration. D. Quantitation of cell subsets in differently treated groups. Note the tendency of increased T-cell and PD-L1 expressing cell numbers upon CANDI and CANDI + a-PD1 treatment due to the dramatic IFNG response. The first three graphs are derived from frozen tissue sections as shown in C and the last graph was obtained by processing GBM for flow cytometry. Graphs present the average values with standard error of the means (N = 4 replicates).
Movie 2: CANDI accumulation in another tumor model (MC38-BFP).
CANDI and FITC-dextran co-localize in tumor-associated cells.
Movie 3: Brain window with CT2A-H2B-apple GBM in IL-12-eYFP mice.
Note the motility of IL-12 expressing cells in the tumor periphery (not treated)
Movie 4: Brain window with CT2A-H2B-apple GBM in GREAT-eYFP mice (IFNG).
Note the induction of IFNG 1–4 days after CANDI treatment.
We also performed flow cytometry and multiplexed histological evaluation (Fig. 8) of untreated and treated CT2A tumors by assaying for different immune cells. This staining panel allowed us to characterize intratumoral immune cells and quantitate their frequency and spatial distribution in different treatment groups. We found that there was a slight increase in T cells (CANDI: 1.5-fold, CANDI + a-PD1: 2.9-fold increase) and PD-L1 positive cells (CANDI: 3.4-fold, CANDI + a-PD1: 2.9-fold increase) in treated GBM as would be expected by the profound local IL-12 and IFNG expression. Interestingly, we did not see statistically significant differences in other immune cell populations across treatment groups (TAM, DC, MHCII, MG, astrocytes). Since we identified IFNG responses induced by CANDI, and we had previously shown that intratumoral IFNG is mainly produced by CD8 T cells[37], we depleted CD8 T cells with a-CD8 in a separate cohort of mice (N = 4). The results showed this cohort’s complete abrogation of therapeutic CANDI effects, underscoring the importance of CD8 cytotoxic T cells for CANDI therapeutic efficacy (Fig. 7). We also performed IL-12 neutralization experiments. We found that this carried an intermediate phenotype, with 50% of animals showing diminished treatment responses. These studies show that CANDI accumulates efficiently in murine GBM and induces potent myeloid cell activation in the tumor microenvironment. This is caused by IL-12 and leads to IFNG stimulation in T cells, and anti-tumor response to CANDI ultimately relies upon CD8 T cells.
Discussion
It is generally accepted that GBM has a poorer response to cancer immunotherapies than other malignancies. The most recognized explanations are that the GBM microenvironment: i) is highly immunosuppressive, ii) that macrophages and myeloid cells dominate the suppressive environment, iii) that the genetic heterogeneity of tumors and their environment continually changes and adapts to the therapies imposed upon it[5], and iv) that the BBB poses additional delivery challenges. Overall, the macrophage/myeloid cell-driven immunosuppression alters the ability of activated anti-tumor immune cells (T cells, macrophages, NK cells) to infiltrate and destroy the relatively antigenically “cold” GBM. Based on these observations, we hypothesized that reprogramming tumor-associated myeloid cells would be a way towards enabling cancer immunotherapy response. Here we show, using a dual-loaded nanocarrier avidly taken up by MF/DCs, that myeloid re-education through a combination of canonical and non-canonical NF-kB signaling can remove myeloid associated immune suppression and drive anti-tumor immunity. Our experimental results show that this synthetic chemical IL-12-inducing therapy can show remarkable efficacy in glioblastoma and synergize with ICI effects. Specifically, we show a marked reduction in tumor growth and a robust survival benefit in an immunotherapy refractory GBM model. Remarkably, we show that CANDI is a specific myeloid targeted therapeutic that enables a potent anti-tumor IFNG response through lymphocytes. We speculate that CANDI activates pathways at the core of antigen presentation to lymphocytes that facilitates myeloid-lymphoid crosstalk within the tumor microenvironment. This tumor immune microenvironment “re-conditioning” makes the tumor permissive for IFNG and response to a-PD1 therapy. Treatment effects depend upon the induction of CD8 T cell immunity by CANDI treatment, establishing that CANDI treatment subverts GBM immune suppression to enable effective cancer immune surveillance. The prolonged IFNG response was an intriguing finding and determining the specificity and phenotypes of CANDI elicited lymphocytes warrants future investigations.
Novelty of CANDI
CANDI represents a cross-linked nanoparticulate drug delivery platform with i) optimized pharmacokinetics and drug delivery to the tumor microenvironment, ii) high myeloid cell targeting capability, iii) high multi-drug loading capacity, v) enhancement of solubility of hydrophobic small molecule drugs, v) extraordinary efficacy in modulating tumor-associated myeloid cells, and vi) a unique ability to secondarily activate tumor lymphocytes. It is well established that macrophages (and other myeloid cells have an affinity for carbohydrates and other nanomaterials[29]. Short-chain carbohydrates (and modifications thereof) offer advantages over other synthetic polymers because they are biocompatible, less likely to elicit adverse reactions, generally do not lead to antibody formation, and have been tested in millions of patients (e.g., dextrans, cyclodextrins). Unfortunately, therapeutic payloads are often on the small side[38]. We reasoned that short-chain cyclodextrins could be assembled into more efficient drug delivery vehicles. Such an approach would be particularly helpful for modulating immune suppressive tumor myeloid cells where there is a dire need for new therapeutics[39]. We first experimented with crosslinked cyclodextrins[19] and showed that relatively high doses could be used to modestly affect the growth of peripheral tumors that are relatively easy to target[19]. During these early experiments, it became clear that tumor-associated macrophages (esp subtypes M2-M9) possess powerful compensatory mechanisms, ultimately leading to low drug activity or reduced efficacy with repeat administrations. Thus, dual and triple targeting strategies had to be developed, all while improving the nanoparticle carrier efficacy. The current version of CANDI bypasses these initial limitations by co-delivering multiple small molecule payloads that activate or inhibit multiple myeloid pathways that are essential for myeloid-lymphocyte communication. One obvious approach was to upregulate IL-12 production as it is a critical mediator in anti-tumor immunity and is predominantly expressed by macrophages and DC. This was done by targeting both axes of the NF-kB pathway (Fig. 1). A less obvious effect was the profound effect that CANDI had on IFNG response as our drug distribution studies clearly showed that CANDI biodistributes to macrophages and DC, not lymphocytes. To accommodate higher and differing small molecule drug payloads into CANDI all while maintaining solubility, we performed a series of optimization experiments exploring the type of cyclodextrin, crosslinking particle size, and charges (Fig. 2; Figs S1–S3). These experiments resulted in CANDI, which differs from the early CDNP versions by having i) enhanced particle stability and loading efficiency (see methods and Fig. S1–3), ii) dual payload loading with matching affinity complexation towards cyclodextrin (Fig. S6) and iii) allow for cargo permeation and release in myeloid cells within GBM. Our results show that the CANDI platform offers attractive solutions for targeted combinatorial immunotherapies. Furthermore, we envision that these novel therapies can be expanded to additional libraries of small-molecule inhibitors that can complex withα, β, or γ, CD units to accommodate other small molecules. For optimal loading, strong inclusion complexes are favored when lipophilic functional handles are present in the payload. Other characteristics including low molecular weight (<500 Da) and an overall neutral charge (Fig. 2) have been firmly established for non-crosslinked cyclodextrins[40,41].
Comparative efficacy
Several different carbohydrate-based nanomaterials with macrophage and dendritic cell targeting capabilities have been described[29]. Many of these materials were initially developed for imaging and thus conceptualized without therapeutic payloads. Recent biological advances have furthermore contributed to a more nuanced understanding of macrophage, dendritic cell, and neutrophil diversity and function[6,39,42]. This has allowed a better handle on which subtypes to target[42] and which to avoid as they elicit toxicity[43]. The question then is, what are the comparative efficacies of different carbohydrate delivery platforms to tumor-associated myeloid cells? While this research field is just beginning to emerge, we compare some published results in Table S3. To our knowledge, CANDI has one of the highest TAM accumulation and increased payload capacity and was designed for multiple small molecule loadings. Fig. S18 models some of the resultant drug delivery capabilities
Mechanism of action
Pharmacokinetics and organ distribution of nanomaterials depend on numerous factors such as size, surface properties (composition, density, charge), opsonization, and particle shape, among others[44]. As a general rule, most clinical carbohydrate materials (~10–300 nm in diameter) accumulate in the liver, spleen, lymph nodes, and bone marrow, as these organs contain high numbers of tissue macrophages[29]. It is interesting to note that CANDI also accumulates in these organs but at a smaller relative amount compared to clinical materials (e.g., MACRIN, Ferumoxytol[29]). A current unknown is the specific molecular mechanism of CANDI uptake into TAM. Prior proof of principle work had excluded the C-type lectin DC-SIGN as the responsible receptor. However, it is well known that macrophages internalize different nanoparticles via phagocytosis and micropinocytosis[45]. The latter mechanism is up-regulated in TAM[46] and is enhanced by Axl/MerTK [46] or inhibited by macropinocytosis inhibitor amiloride[47]. Beyond TAM uptake and release of payloads, it is also possible that CANDI has additional mechanisms of action. For example, macrophages and other phagocytic cells can serve as a cellular slow-release system for encapsulated drugs[48]. Whether this played a role in the current study is hard to ascertain as there are no fluorescent versions of resiquimod or LCL-161 to test this.
The uniqueness of our approach was to stimulate both canonical and noncanonical NF-kB pathways, which resulted in much higher endogenous IL-12 production than monotherapy alone (Fig. S6). Furthermore, the literature demonstrates that canonical and non-canonical NF-kB pathways are complementary immune stimulators with divergent activation mechanisms [49]. We had previously shown that IL-12 induction effects of LCL-161 depend upon the critical non-canonical NF-kB inducing kinase (NIK), arguing for a specific non-canonical NF-kB mechanism of LCL-161 anti-tumor activity. Further, the anti-tumor effects of a different cIAPi, AZD5582, were dependent upon IL-12[26]. Deficiency of NIK in DCs has been shown to impair CD8 T cell immune responses, underscoring the effects of the non-canonical NF-kB pathway in regulating CD8 T cell immunity[50]. The effects of locally increased IL-12 are likely multifactorial. They include i) orchestration of a Th1 response with activation of CD8+ T cells, CD4+ Th cells, B cells, NK cells, ii) alteration of mononuclear cells from an M2 (immune-suppressive) to an M1 (pro-inflammatory) state, iii) anti-angiogenic effects and iv) induction of IFNG production by T cells. The kinetics of IL-12 induction preceding the increase in IFNG suggest a model that myeloid targeted therapies communicate to adaptive immunity and lymphocytes through IL-12 production, which elicits IFNG responses. It is likely that CANDI, which does not activate T cells directly, leverages this signaling loop for therapeutic efficacy. We had previously shown that IL-12:IFNG signaling loops are needed for immunotherapy response[26] in many therapeutic models. Combined, these effects have potent anti-cancer activity confirmed by flow cytometry, multiplexed immunohistochemistry, and different therapeutic efficacy studies.
How can the approach be paired with other synergistic treatments?
Other combination treatments can further enhance the CANDI approach shown here. We have shown that combination with a-PD1 therapy is additive. This was surprising, as a-PD1 treatment by itself has failed mainly as a primary anti-GBM treatment in humans[3,51], unlike in other peripheral cancers, and the mouse GBM model we used, CT2a, is poorly responsive to single agent a-PD1 treatment. We believe that the CANDI/a-PD1 additive effect is due to activation and re-polarization of the tumor myeloid microenvironment and that pre-existing tumor myeloid suppressor cells inhibit the a-PD1 response.
The current study explored CANDI as a platform for NF-kB combination drug dosing. Logical first combination partners were R848 and LCL-161, as they independently work on the canonical and non-canonical signaling pathways. However, it is also possible to target other signaling nodes. For example, IL-10 is a known inhibitor of IL-12 responses[52] and signals through STAT3. STAT3 is hyperactivated in immune myeloid and cancer cells and plays a central role in inhibiting immune activation regulators[53].
We have performed initial feasibility experiments and shown synergistic STAT3 effects with CANDI (Fig. R1). One can also postulate that IL-10 could be blocked with IL-10R blocking antibodies. Finally, it is possible to pair cIAPi and indoleamine 2,3-dioxygenase (IDO) inhibitors, as IDO can be induced via the non-canonical NF-kB pathway[54]. IDO plays a multifaceted role in immunosuppression and could be targeted by CANDI.
Other likely additive or synergistic effects could occur with low-dose radiation therapy. Prior research has shown that sub-therapeutic radiation bursts can increase nanoparticle tumoral uptake by 4–10-fold[55]. This increase in drug delivery is due to enhanced extravasation (“vascular bursting”) and cellular nanoparticle uptake by GBM[56].
Towards translation
To translate the current strategy, several future studies may be warranted. First, while similarly, sized carbohydrate nanoparticles are currently in phase 1 trials for imaging (NCT04843891), the exact cyclodextrin nanoparticle must undergo formal testing. We have not observed any cellular toxicities in exploratory studies, but more in-depth analyses are warranted. Second, it may be beneficial to optimize drug choices and loadings further. We chose R848 as a proof-of-principle TLR7/8 agonist because of prior studies in other peripheral cancers. R848 also has some undesired effects that have precluded its use as a systemic therapy[21]. Similarly, LCL-161 was chosen because it emerged as one of the top hits in a phenotypic screen[23] and had recently been studied by others[24,25]. Alternative small molecules may emerge to activate both canonical and non-canonical NF-kB pathways more selectively and reduce off-target and undesired side effects. Finally, additional preclinical efficacy studies are warranted to test if combinations of therapies, such as those discussed above, would allow drug potentiation and further improved efficacy. Irrespective of these studies, our data clearly shows effectiveness in modulating the immunosuppressive GBM environment by dual activation of both the canonical and non-canonical NF-kB pathways.
Materials and Methods
Synthesis and characterization of nanoparticles
Materials.
All solvents and reagents were purchased from Sigma-Aldrich, Thermo Fischer, or VWR and used without further purification. MilliQ water obtained from Waters filtration system was used for all experiments. All buffers used were prepared and filtered through a 0.22 μm sterile filter (VWR) before use. Pharmacological drugs R848 and LCL-161 were purchased from Medchem Express and used as received.
Synthesis of CANDI.
The synthesis of cyclodextrin nanoparticles (CANDIE) was adapted from previously reported methods[19,57]. Briefly, succinyl-β-cyclodextrin (Sigma; 250 mg, 1.0 eq. to carboxylate) was dissolved in MES buffer (6 mL, 50 mM, pH = 5) and activated with N-(3-(dimethylamino)propyl)-N′-ethyl carbodiimide hydrochloride (EDC) (Fisher; 1.5 g, 10.0 eq. to carboxylate) and N-hydroxysuccinimide NHS (Sigma; 550 mg, 5.0 eq. to carboxylate) for 30 min at 25 °C. A solution containing L-lysine (Sigma; 70 mg, 0.5 eq to carboxylate) in MES buffer (1.5 mL) was added in a drop-wise matter, and the reaction was allowed to stir for 18 h at 25 °C. Under vigorous stirring, the nanoparticle solution was added drop-wise to an ice-cold absolute ethanol solution (70 mL), yielding a white precipitate decanted and dissolved in water (14 mL). The clear solution was filtered through a 0.22 μm centrifugal filter (VWR) and purified with 10 kDa MWCO centrifugal filters (Amicon; 10,0000 g for 8 min). The clear syrup-like solution was washed with water (x3), filtered, and collected. The CANDIE were sterilized by filtration through a 0.22 μm sterile filter (VWR) and lyophilized for 48 h to yield ~344 mg of CANDIE particle as a fluffy white powder. The CANDIE was stored at −20 °C until further use.
Fluorescent labeling of CANDI.
Lyophilized CANDIE (20 mg) was dissolved in carbonate buffer (1 mL, 0.1 M, pH = 8.5) and Pacific Blue™ Succinimidyl Ester (Fisher; 5 mg/mL in dimethylformamide) or AF647 Succinimidyl Ester (Fisher; 2 mg/mL in dimethylsulfoxide) was added to achieve a final concentration of 50 μM. The reaction was stirred for 2 h at room temperature in a thermocycler (Eppendorf, 550 rpm). The nanoparticles were purified by buffer exchange into water against 10 kDa MWCO centrifugal filters (Amicon; 10,0000 g for 8 min). The filtrate was washed with water (350 μL, 3x), and the final products (CANDIPB & CANDIAF647) were diluted with water (in vitro experiments) or PBS (in vivo experiments) to a final concentration of 50 mg/mL. The final product was sterilized by filtration through a 0.22 μm sterile filter (VWR). The solution was stored at −20 °C until further use.
Drug loading.
Payloads (LCL-161 and R848) were added at a molar ratio of 1.5:1 in a 10% DMSO solution, and these conditions were kept constant (R848 0.2 mg and LCL-161 0.5 mg dissolved in 10 μL DMSO). The solution was resuspended with 100 μL of CANDIE for fluorescent versions of CANDIE at a concentration of 5 mg/mL in PBS, vortexed and mixed until the solution became clear. For in vivo administration, a dose of 5 mg of CANDIE was loaded with 0.5 mg (10 mM) and 0.2 mg (6 mM) of LCL-161 and R848, respectively, in 100 μL PBS, yielding CANDI. Before injection, all solutions were sterilized by filtration through a 0.22 μm sterile filter (VWR). Drug loading analysis was performed by a colorimetric binding assay using phenolphthalein I (Sigma) as an indicator [58]. In brief, a standard curve was made using phenolphthalein in carbonate buffer (100 μL, 200 μM, pH = 10.5) mixed with varying concentrations of CANDIE (100 μL, 0–2.5 mg). Complete loading of the I was determined by loss of absorbance at 550 nm (Tecan). Drug loading was assessed by adding R848 and LCL-161 (1:1.5, 0–10 mM, 5% DMSO) and recovery of the absorbance at 550 nm. All experiments were done in triplicates unless stated otherwise.
Loading ratios.
The amounts and relative ratios of R848/LCL-161 were chosen based on screening experiments in bone marrow-derived macrophages and dendritic cells (Fig. 3). This was done for monotherapies with either agent alone (to determine efficacy and toxicity of either agent alone) and in combination. We considered three ratios of R848/LCL-161 for testing experimentally in cell culture (R848/LCL-161 = 1:65, 1:35 and 1:2.5 ). Serial dilutions were made to confirm IL-12 signal induction without toxicity in macrophages and DCs in vitro (R848/LCL-161 1:35; low = 1.75:62.5 ng, med = 3.14:125 ng, and high 6.28:250 ng, Fig. 3). For in vivo experiments, we selected the ratio with the highest R848 concentration (1:2.5 ratio or 0.2:0.5 mg R848:LCL-161 per 5 mg CANDIE) for optimal loading efficiency (Fig. S2) without causing apparent cytotoxicity.
Nanoparticle characterization and release kinetics.
Particle size and surface charge for all vehicles and loaded CANDI were determined by dynamic light scattering and zeta potential, respectively (Malvern, Zetasizer APS) at 5 mg/mL concentration in PBS buffer (0.1x). For drug release kinetics, the loaded particles. CANDI was subjected to dialysis (Slide-A-Lyzer, 3 k) for 24 h in PBS (0.01x), and the particle diameter was compared by DLS measurements measured in PBS (0.01x). All experiments were done in triplicates unless stated otherwise. Release kinetics were determined by quantifying the ELSD peak signal corresponding to each drug via reverse-phase liquid chromatography-mass spectrometry LCMS (water: acetonitrile 95:5→ 100%, 0.1% Formic Acid, 3 min). In brief, a solution of 5 mg CANDI (100 μL, 0.2 mg R848, 0.5 mg LCL-161) was diluted x5 in PBS. A membrane spinning filter (500 μL, <3k Da, Amicon) was used to collect the payloads (10’000 rcf, 1 min). Each elution volume (20 μL) was analyzed by LCMS and the area under the curve for each peak was quantified using Masslynx software. Standard deviation curves of R848 and LCL-161 were made to quantify molecule concentration to integrated ELSD peaks (area under the curve, Fig. S2, C). All values were normalized to the hypothetical maximum loading efficiency to obtain % of drug released for each condition (t = 0.25, 0.5, 1, 2, 4 and 18 h).
Scanning electron microscopy.
CANDI solution (5 mg) was diluted (30:1) in a H2O:EtOH (1:1) mixture, placed on a silicon wafer, and dried in a desiccator (>5 h) at room temperature. The surface of the sample was sputter coated with ~8 nm gold (MITnano Facility). The sample was loaded on a carbon strip and loaded on a field emission scanning microscope (Gemini 2, Zeiss).
Nanoparticle Tracking Analysis.
Particle count, size distribution and homogeneity for CANDIE and CANDI solutions (1:250 dilution in PBS) were analyzed using a Panalytical NanoSight LN10 (Malvern) nanoparticle characterization system. All nanoparticle tracking analyses (NTA) were done with identical experiment settings. For optimal results, the concentrations were adjusted to obtain ~50 nanoparticles in the field of view.
GBM model
The primary glioma cell line model for this study was CT2A. However, initial feasibility studies in the GL261[59] and 005 models[60] showed similar efficacies. The syngeneic CT2A GBM model is more histologically similar to human GBM[32]. Cells were obtained from Dr. Samuel Rabkin (CT2A) Drs. Bakhous Tannous (CT2A-mCherry-Fluc) and Dr. Katy Yang (CT2A-H2B-mApple). We used CT2A mCherry-luc cells for pharmacokinetics experiments, MRI experiments, bioluminescence imaging, and survival studies. For IVM and histology experiments, we used CT2A H2B-mApple. in separate control experiments, we determined that all CT2A subclones had similar growth rates in vitro and in vivo.
Mouse models
We used various mouse models to study different aspects of drug delivery, IL-12 induction, and therapeutic efficacy. The MGH Institutional Animal Care and Use Committee (IACUC) committee approved all studies. A total of N = 104 mice were used. This includes immunocompetent C57BL/6J wild-type mice for GBM implantations (N = 85, C57BL/6J, Strain Number: 000664, JAX, Bar Harbor, ME), MerTK-GFP[33] mice for co-localization IVM studies (N = 4; Strain Number: 036071, JAX, Bar Harbor, ME), GREAT mice for IVM studies (N = 2; Strain Number: 017580, JAX, Bar Harbor, ME), BATF3KO mice for GBM implantations (N = 2; Strain Number: 013757, JAX, Bar Harbor, ME), and IL-12-eYFP mice for GBM implantations, IVM mechanistic studies and bone marrow harvesting (N = 6; B6.129-IL-12btm1.1Lky/J, Strain Number: 006412, JAX, Bar Harbor, ME).
Tumor Cell implantation
For intracranial tumor implantations, 5 × 104 CT2A cells were diluted in 2 μl sterile PBS (Sigma-Aldrich) and stereotactically implanted into the right basal ganglia of 8–10 week old C57Bl/6J mice (coordinates: 2 mm right lateral of the bregma and 0.5 mm anterior to the coronal suture with an injection depth of 2.5 mm below the dural surface) using a 10 μl Hamilton micro-syringe driven by a fine step stereotactic device (Kopf). CT2A mCherry-luc cells were used for therapeutic efficacy studies, whereas CT2A H2B-mApple cells were utilized for IVM studies and multiplexed FAST-profiling of GBM.
CANDI and a-PD1 Treatments
Mice were split into three treatment cohorts: CANDIE vehicle control (N = 18), CANDI (N = 25) or CANDI + a-PD1 (N = 16). Nanoparticle treatments were administered on days 14, 17, and 20 post-tumor cell inoculation (day 0). Treatments were diluted in 200μL sterile PBS (Sigma-Aldrich) and injected under isoflurane anesthesia via a tail vein catheter. The following doses were administered: CANDIE vehicle control: 5mg CANDI/mouse (200 mg/kg); CANDI: 5 mg CANDI/mouse (200 mg/kg) loaded with 500 μg LCL-161(25 mg/kg) and 200μg R848 (10 mg/kg). In the CANDI + a-PD1 cohort, a-PD1 treatment was administered on days 14, 15, and 16 post tumor cell inoculation. For each treatment, mice received an IP injection of 50 μL sterile PBS (Sigma-Aldrich) containing a dose of 200 μg a-PD1 antibody (InVivoMAb anti-mouse PD1 - CD279 – 5mg, BioxCell). Representative tissues were processed for histopathology at the time of sacrifice.
Bone marrow-derived cell screens.
Whole bone marrow was isolated from IL-12 eYFP reporter mice by flushing femurs with sterile PBS with a 28 gauge needle. Red blood cells were lysed using RBC Lysis Buffer (Biolegend), and bone marrow cells were counted using a hemocytometer. Bone marrow cells at a concentration of 1 × 105 were seeded into 96 well plates with imaging glass bottom (iBidi) and cultured in the presence of 20 ng/ml recombinant murine M-CSF (Biolegend) or 300 ng/ml recombinant mouse Flt3L (Biolegend). For macrophages, cultures were differentiated for 5 days, with M-CSF media changed every 2 days. For dendritic cells, cultures were differentiated for 9 days, with Flt3L media change after 4 days. Cells were treated with different nanoparticle amounts, and payloads and cells were examined for IL-12-eYFP signals after 24 h using an automated Olympus screening microscope (BX63). The drug concentrations were as follows in the different treatment groups shown in Fig. 3: Low: 0.5 μg CANDI containing 62.5 ng LCL-161 and 1.57 ng R848; Med: 1 μg CANDI containing 125ng LCL-161 and 3.5 ng R848; Hi: 02 μg CANDI containing 250 ng LCL-161 and 6.28 ng R848; None: negative control.
Flow cytometry
Tissues were isolated, mechanically dissociated using surgical scissors, and digested using Collagenase IV at 0.2 mg/ml in RPMI 1640 at 37 C for 45 minutes with vigorous shaking. After digestion, tissues were filtered through a 40 μM cell strainer and resuspended in protein-free PBS. Cells were stained using AquaAmine Live Dead Fixable viability stain (Thermo Fisher) and then washed with PBS. Cells were then resuspended in FACS buffer (PBS with 2 mM EDTA and 2% Fetal Calf Serum) and stained with Fc block (Biolegend) and fluorochrome-conjugated antibodies. Sample data were acquired using an Attune NxT flow cytometer (Thermo Fisher), and data were analyzed using FlowJo 10 software (TreeStar).
Intravital imaging
Brain window tumor model.
All experiments were approved by the MGH Institutional Animal Care and Use Committee (IACUC) and performed according to MGH IACUC regulations. Cranial windows were implanted with modifications according to established methods[61]. The head of 10–14 week old mice was shaved, animals were immobilized in a stereotactic frame (Kopf, Tujunga, CA), and the skull was sterilized with two cycles of betadine-isopropanol. A large oval skin area was removed, from behind the ears to between the eyes, surrounding the lambda and bregma sutures. The periosteum was pushed to the side, and all tissue on top of the skull was scraped off. The rim of the 5 mm circular section of the head, excluding the lambda and bregma, was sanded down using a Dremel with a burr (Fine Science Tools, 19007–07) and removed to provide an opening to the brain. Using stereotaxic positioning, 2 μl of Optimum (Thermo Fisher Scientific) with 105 CT2A-H2B-mApple cells were injected at about 1 mm depth near the middle of the opening, avoiding vasculature. Gelfoam and saline were used to remove blood during surgery and after injection. A drop of saline and an 8 mm round cover glass were placed onto the opening. Super glue was used to attach only the rim of the cover glass to the skull, avoiding any contact between the adhesive and the brain. Then, dental cement was used to cement the cover glass onto the skull, cover skull areas without skin, and form an elevated rim for water immersion imaging.
Confocal imaging.
All confocal images were collected using a customized Olympus FV1000 confocal microscope (Olympus America). A 2x (XLFluor, NA 0.14), a 4x (UPlanSApo, NA 0.16), and an XLUMPlanFL N 20x (NA 1.0) water immersion objective were used for imaging (Olympus America). CT2A H2B-apple tumor cells, CANDIAF647, and vascular probes were excited sequentially using a 405 nm, a 473 nm, a 559 nm, and a 633 nm diode laser, respectively, in combination with a DM-405/488/559/635 nm dichroic beam splitter. Emitted light was further separated by beam splitters (SDM-473, SDM-560, and SDM-640) and emission filters BA430–455, BA490–540, BA575–620, and BA655–755 (Olympus America). Confocal laser power settings were carefully optimized to avoid photobleaching, phototoxicity, or damage to the brain. All images were processed using Fiji (ImageJ2, Vers.2.3/1.53f).
Blood half-life (t1/2) measurement of the CANDI nanoparticle was performed using confocal imaging. IL-12-eYFP mice (B6.129-IL-12btm1.1Lky/J, Strain Number: 006412, JAX, Bar Harbor, ME) harboring a brain window with 9 to 15 days old CT2A H2B-apple brain tumors were used for quantification. Mice were anesthetized using isoflurane, stabilized using a stereotaxic (Kopf, Tujunga, CA) for motion-free imaging, and a vascular probe was injected to select areas with good vasculature for imaging. Time-series of confocal imaging stacks in multiple locations were initiated before injection of fluorescent CANDIAF647 via tail vein catheter. Z-stacks from various areas were collected over two h and again 24 after injection of CANDIAF647 using identical image acquisition settings. Average fluorescence signal intensity was quantified using six ROIs, each inside the vasculature and outside of the vasculature (background) using Fiji (ImageJ2, Vers.2.3/1.53f). Background fluorescence was subtracted from the average signal inside the vasculature, and the values were analyzed and plotted in GraphPad Prism (San Diego, CA, Version 9.3.1 for Mac).
Biodistribution:
CT2A tumor-bearing mice (N = 4) were injected with a single dose of 5mg CANDIAF647 diluted in 200 μL sterile PBS via tail vein catheter on day 14 post tumor cell inoculation. One control mouse was injected with 200 μL sterile PBS via a tail vein catheter. Whole-body biodistribution was assessed 24 h after injection. Mice were sacrificed via left ventricle heart puncture and perfusion with 0.9% saline solution. Tissues were subsequently harvested and weighed. Fluorescence reflectance imaging was performed using an OV110 (Olympus) small animal imaging system (exposure time: 122 ms, λex = 620–650 nm, λem = 680–710 nm). Mean fluorescence intensity per gram of tissue was measured by selecting ROIs of tissues and background subtraction of the PBS-treated control. All images were processed using Fiji (ImageJ2, Vers.2.3/1.53f). Values for mean fluorescence intensity per gram tissue were then converted to a percentage of injected dose per gram tissue (% iD/g) tissue according to biodistribution data of 64Cu-labeled polyglucose nanoparticles[62].
MR imaging
MRI was performed on an animal 4.7-T MR imaging unit (Bruker Pharmascan) at baseline (day 13) and after treatment (day 22) under respiration-monitored isoflurane anesthesia. Coronal imaging parameters for pre-and post-contrast enhanced T1-weighted imaging were as follows: repetition time (TR) = 700 ms, echo time (TE) = 14 ms, matrix size 256 × 256, and slice thickness 0.5 mm. 12 sections were acquired. Imaging parameters for pre-enhanced T2-weighted imaging were as follows: TR = 4,000 ms, TE = 53.3333 ms, matrix size 256 × 256, slice thickness 0.5 mm. Twelve sections were acquired. Tumor volumes were calculated by using the Horos image-processing software for tumor volumetric data via ROI-based 3D analysis of Gd-DTPA enhanced T1-weighted MR images (Horos, horosproject.org).
Bioluminescence imaging
Bioluminescence imaging was performed using an Ami HTX - Spectral Instruments Imaging instrument at baseline (day 13) and after treatment (day 22). Mice received an intraperitoneal injection of Luciferin (5mg/mouse) and were maintained under respiration-monitored isoflurane anesthesia during imaging. Imaging parameters were as follows: 60 seconds and 0.5 seconds exposure, binning levels 1, 2, 4, and 8. Total flux (p/s) was used for signal quantification.
Multiplexed tissue analysis using FAST-Frozen Sections
FAST reagents.
FAST probes were constructed as a modular linker connecting fluorochromes and antibodies with an embedded TCO for clicking with a Tz-quencher. FAST probes were custom synthesized as described in detail in our previous study[63]. FAST probes were stored as carboxylic acids and activated for antibody labeling with our in situ NHS/TFP activation chemistry. The dTCO-PEG6-CO2H blocking reagent was synthesized from dTCO-PNP and amino-dPEG6-CO2H and characterized by LC-MS. All reagents were obtained from commercial sources at the highest grade available. Fluorophores were purchased from Click Chemistry Tools or Fluoroprobes; BHQ®−3 Amine from LGC Biosearch Technologies (5 or 25 mg aliquots); N-α-Boc-N-ε-Fmoc-Lysine from Chem-Impex; and amino-dPEG®n-carboxylic acids (N = 4,6) from Quanta BioDesign. Dry solvents and coupling reagents were obtained from Sigma Aldrich.
Antibody modifications.
Antibodies without carriers were purchased (Table S1) to be labeled with FAST probes as previously described[64]. A 40k Zeba column (Thermo Fisher) was used to buffer-exchange the antibodies into bicarbonate buffer (pH 8.4). Then antibodies (1–3 mg/ml) were incubated with a 5- to 10-fold molar excess of the FAST probe with 10% DMSO for 25 minutes at room temperature in the dark. After the conjugation reaction, FAST-antibody conjugate was loaded onto another 40k Zeba column equilibrated with PBS for desalting and removing unreacted dye molecules. The absorbance spectrum of the FAST-labeled antibody was measured using a Nanodrop 1000 (Thermo Scientific) to determine the degree of labeling (DOL). The known extinction coefficients of the dye (AF488, AF555, AF647), IgG antibody, and the correction factor for the dye absorbance at 280 nm were used to calculate DOL. The FAST-labeled antibodies were stored protected from light at 4 °C in PBS. Information on the antibodies used for the multiplexed immunofluorescence is summarized in Table S1. Antibodies were tested and validated on positive cell lines, mouse splenocytes, or brain tissue sections of wild-type B6 mice before usage.
Imaging of tissue sections.
Frozen tissue sections were cut to 5 μm and then processed for immunohistochemistry or FAST cycling. Frozen tissue sections were thawed, rehydrated, and blocked with Intercept Blocking buffer (LI-COR) for 30 min before antibody staining. For FAST imaging, tissue sections were incubated with antibodies for 1–2 h and washed with PBS for 10 min three times. After imaging, the fluorescence signal was quenched by incubating tissue sections in 10 Mμ Tz-BHQ3 for 30 min. The quenched signal was imaged. Then tissue sections were incubated with 50 μM of free TCO solution to react with the residual quencher. Antibodies for the subsequent cycle were then added, and the cycles were repeated until all proteins of interest were imaged. Olympus BX-63 system and Metamorph were used for image acquisition. CellProfiler version 4.1.8 was used for the analysis of tissue section images.
Modeling
Pharmacokinetic modeling and equations follow previously described methods[65]. Briefly, an avascular spherical lesion of 0.2 mm diameter was modeled, surrounded by perfused/vascularized tissue in equilibrium with blood concentrations. Reaction rates and modeling parameters followed prior publication details with exceptions noted in the above tables. Lipid partitioning and albumin binding were not included due to their relatively minor impact for these drugs. Systemic PK parameters were fit to published clinical blood PK data. Drug target engagement was approximated from estimated and published dissociation constants, with kon rates representative of other small molecule targeted drugs[65], but target engagement did not substantially impact overall PK at the >10 nM binding affinities considered.
Statistics
GraphPad Prism was used for statistical analysis. Results were expressed as mean ± SEM. Statistical tests included one-way ANOVA followed by Tukey’s or Dunnett’s multiple comparison test. When applicable, the unpaired one-tailed and two-tailed Student’s t-tests using Welch’s correction for unequal variances was used.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs Bakhous Tannous and Samuel Rabkin for CT2A cell lines, Katy Yang for transfection with m-Apple, and Jonathan Carlson for FAST reagents. Greg Wojtkiewicz and Ella Scott assisted with MRI and bioluminescence imaging, Dr. Ursula Winter for NTA analysis, and Dr. JaeJun Kim in the acquisition of SEM. We acknowledge the use of the Harvard Center for Nanoscale Systems for acquisition and analysis XPS, FTIR and TEM. This study was supported in part by 5P01CA069246 (EAC, XOB, RW), 5R33CA202064 (RW), 5U01CA206997 (RW) and R35 CA232103 (XOB). SL was supported by the Studienstiftung. EAH was supported by the Swiss National Science Foundation (SNSF).
Footnotes
CONFLICT OF INTEREST
R.W. is a consultant to ModeRNA, Lumicell, Seer Bioscience, and Accure Health. E.A.C. is an advisor to Advantagene Inc. and DNAtrix Inc. and has an equity interest in DNAtrix. C.S.G. is a consultant to Cellino Biotech. The other authors report no industrial interactions.
REFERENCES
- [1].Nam JY,de Groot JF. J Oncol Pract 2017, 13, 629. [DOI] [PubMed] [Google Scholar]
- [2].Marenco-Hillembrand L, Wijesekera O, Suarez-Meade P, Mampre D, Jackson C, Peterson J, Trifiletti D, Hammack J, Ortiz K, Lesser E, Spiegel M, Prevatt C, Hawayek M, Quinones-Hinojosa A,Chaichana KL. J Neurooncol 2020, 147, 297. [DOI] [PubMed] [Google Scholar]
- [3].Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, Baehring J, Ahluwalia MS, Roth P, Bähr O, Phuphanich S, Sepulveda JM, De Souza P, Sahebjam S, Carleton M, Tatsuoka K, Taitt C, Zwirtes R, Sampson J,Weller M. JAMA Oncol 2020, 6, 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, Kawaguchi ES, Du L, Li G, Yong WH, Gaffey SC, Cohen AL, Mellinghoff IK, Lee EQ, Reardon DA, O’Brien BJ, Butowski NA, Nghiemphu PL, Clarke JL, Arrillaga-Romany IC, Colman H, Kaley TJ, de Groot JF, Liau LM, Wen PY,Prins RM. Nat Med 2019, 25, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Medikonda R, Dunn G, Rahman M, Fecci P,Lim M. J Neurooncol 2021, 151, 41. [DOI] [PubMed] [Google Scholar]
- [6].Pittet MJ, Michielin O,Migliorini D. Nat Rev Clin Oncol 2022, 19, 402. [DOI] [PubMed] [Google Scholar]
- [7].Zomer A, Croci D, Kowal J, van Gurp L,Joyce JA. iScience 2022, 25, 104570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nguyen KG, Vrabel MR, Mantooth SM, Hopkins JJ, Wagner ES, Gabaldon TA,Zaharoff DA. Front Immunol 2020, 11, 575597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lasek W, Zagożdżon R,Jakobisiak M. Cancer Immunol Immunother 2014, 63, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ,Schreiber RD. Nature 2007, 450, 903. [DOI] [PubMed] [Google Scholar]
- [11].Rodolfo M,Colombo MP. Methods 1999, 19, 114. [DOI] [PubMed] [Google Scholar]
- [12].Trinchieri G. Nat Rev Immunol 2003, 3, 133. [DOI] [PubMed] [Google Scholar]
- [13].Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ,Schreiber RD. Nature 2001, 410, 1107. [DOI] [PubMed] [Google Scholar]
- [14].Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, Ritz J, Sandler AB, Edington HD, Garzone PD, Mier JW, Canning CM, Battiato L, Tahara H,Sherman ML. Clin Cancer Res 1997, 3, 409. [PubMed] [Google Scholar]
- [15].Algazi AP, Twitty CG, Tsai KK, Le M, Pierce R, Browning E, Hermiz R, Canton DA, Bannavong D, Oglesby A, Francisco M, Fong L, Pittet MJ, Arlauckas SP, Garris C, Levine LP, Bifulco C, Ballesteros-Merino C, Bhatia S, Gargosky S, Andtbacka RHI, Fox BA, Rosenblum MD,Daud AI. Clin Cancer Res 2020, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Barrett JA, Cai H, Miao J, Khare PD, Gonzalez P, Dalsing-Hernandez J, Sharma G, Chan T, Cooper LJN,Lebel F. Cancer Gene Ther 2018, 25, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chiocca EA, Yu JS, Lukas RV, Solomon IH, Ligon KL, Nakashima H, Triggs DA, Reardon DA, Wen P, Stopa BM, Naik A, Rudnick J, Hu JL, Kumthekar P, Yamini B, Buck JY, Demars N, Barrett JA, Gelb AB, Zhou J, Lebel F,Cooper LJN. Sci Transl Med 2019, 11, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hewitt SL, Bailey D, Zielinski J, Apte A, Musenge F, Karp R, Burke S, Garcon F, Mishra A, Gurumurthy S, Watkins A, Arnold K, Moynihan J, Clancy-Thompson E, Mulgrew K, Adjei G, Deschler K, Potz D, Moody G, Leinster DA, Novick S, Sulikowski M, Bagnall C, Martin P, Lapointe JM, Si H, Morehouse C, Sedic M, Wilkinson RW, Herbst R, Frederick JP,Luheshi N. Clin Cancer Res 2020, 26, 6284. [DOI] [PubMed] [Google Scholar]
- [19].Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, Kohler RH, Pittet MJ,Weissleder R. Nat Biomed Eng 2018, 2, 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vinod N, Hwang D, Azam SH, Van Swearingen AED, Wayne E, Fussell SC, Sokolsky-Papkov M, Pecot CV,Kabanov AV. Sci Adv 2020, 6, eaba5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bhagchandani S, Johnson JA,Irvine DJ. Adv Drug Deliv Rev 2021, 175, 113803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Koch PD, Pittet MJ,Weissleder R. RSC Chemical Biology 2020, 1, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Koch PD, Rodell CB, Kohler RH, Pittet MJ,Weissleder R. Cell Chem Biol 2020, 27, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Clancy-Thompson E, Ali L, Bruck PT, Exley MA, Blumberg RS, Dranoff G, Dougan M,Dougan SK. Cancer Immunol Res 2018, 6, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Roehle K, Qiang L, Ventre KS, Heid D, Ali LR, Lenehan P, Heckler M, Crowley SJ, Stump CT, Ro G, Godicelj A, Bhuiyan AM, Yang A, Quiles Del Rey M, Biary T, Luoma AM, Bruck PT, Tegethoff JF, Nopper SL, Li J, Byrne KT, Pelletier M, Wucherpfennig KW, Stanger BZ, Akin JJ, Mancias JD, Agudo J, Dougan M,Dougan SK. Sci Transl Med 2021, 13, eabf5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, Freeman GJ, Warren SE, Ong S, Browning E, Twitty CG, Pierce RH, Le MH, Algazi AP, Daud AI, Pai SI, Zippelius A, Weissleder R,Pittet MJ. Immunity 2018, 49, 1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, Brouland JP, Iacobuzio-Donahue CA, Brennan C, Tabar V, Gutin PH, Daniel RT, Hegi ME,Joyce JA. Cell 2020, 181, 1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cassetta L,Pollard JW. Nat Rev Drug Discov 2018, 17, 887. [DOI] [PubMed] [Google Scholar]
- [29].Weissleder R, Nahrendorf M,Pittet MJ. Nat Mater 2014, 13, 125. [DOI] [PubMed] [Google Scholar]
- [30].Reinhardt RL, Hong S, Kang SJ, Wang ZE,Locksley RM. J Immunol 2006, 177, 1618. [DOI] [PubMed] [Google Scholar]
- [31].Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM,Gajewski TF. J Exp Med 2011, 208, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Saha D, Martuza RL,Rabkin SD. Cancer Cell 2017, 32, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mohan JF, Kohler RH, Hill JA, Weissleder R, Mathis D,Benoist C. Proc Natl Acad Sci U S A 2017, 114, E7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S,Cohen RB. J Clin Oncol 2014, 32, 3103. [DOI] [PubMed] [Google Scholar]
- [35].Baggio C, Gambini L, Udompholkul P, Salem AF, Aronson A, Dona A, Troadec E, Pichiorri F,Pellecchia M. J Med Chem 2018, 61, 6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pockros PJ, Guyader D, Patton H, Tong MJ, Wright T, McHutchison JG,Meng TC. J Hepatol 2007, 47, 174. [DOI] [PubMed] [Google Scholar]
- [37].Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, Miller MA, Carlson JC, Freeman GJ, Anthony RM, Weissleder R,Pittet MJ. Sci Transl Med 2017, 9, eaal3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kaittanis C, Bolaender A, Yoo B, Shah N, Ouerfelli O,Grimm J. Nano Lett 2017, 17, 7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Weissleder R,Pittet MJ. Nat Biomed Eng 2020, [Google Scholar]
- [40].Stella Rao, Zannou Zia. Adv Drug Deliv Rev 1999, 36, 3. [DOI] [PubMed] [Google Scholar]
- [41].Haley RM, Gottardi R, Langer R,Mitchell MJ. Drug Deliv Transl Res 2020, 10, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, Choi S, Richards WG, De Rienzo A, Tenen DG, Bueno R, Levantini E, Pittet MJ,Klein AM. Immunity 2019, 50, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Siwicki M, Gort-Freitas NA, Messemaker M, Bill R, Gungabeesoon J, Engblom C, Zilionis R, Garris C, Gerhard GM, Kohl A, Lin Y, Zou AE, Cianciaruso C, Bolli E, Pfirschke C, Lin YJ, Piot C, Mindur JE, Talele N, Kohler RH, Iwamoto Y, Mino-Kenudson M, Pai SI, deVito C, Koessler T, Merkler D, Coukos A, Wicky A, Fraga M, Sempoux C, Jain RK, Dietrich PY, Michielin O, Weissleder R, Klein AM,Pittet MJ. Sci Immunol 2021, 6, eabi7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mitchell MJ, Jain RK,Langer R. Nat Rev Cancer 2017, 17, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Griffiths G, Gruenberg J, Marsh M, Wohlmann J, Jones AT,Parton RG. Adv Drug Deliv Rev 2022, 188, 114403. [DOI] [PubMed] [Google Scholar]
- [46].Li R, Ng TSC, Wang SJ, Prytyskach M, Rodell CB, Mikula H, Kohler RH, Garlin MA, Lauffenburger DA, Parangi S, Dinulescu DM, Bardeesy N, Weissleder R,Miller MA. Nat Nanotechnol 2021, 16, 830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lin XP, Mintern JD,Gleeson PA. Membranes (Basel) 2020, 10, E177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B, Kolishetti N, Pittet M, Lippard SJ, Farokhzad OC,Weissleder R. Nat Commun 2015, 6, 8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sun SC. Nat Rev Immunol 2017, 17, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Katakam AK, Brightbill H, Franci C, Kung C, Nunez V, Jones C, Peng I, Jeet S, Wu LC, Mellman I, Delamarre L,Austin CD. Proc Natl Acad Sci U S A 2015, 112, 14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chuntova P, Chow F, Watchmaker PB, Galvez M, Heimberger AB, Newell EW, Diaz A, DePinho RA, Li MO, Wherry EJ, Mitchell D, Terabe M, Wainwright DA, Berzofsky JA, Herold-Mende C, Heath JR, Lim M, Margolin KA, Chiocca EA, Kasahara N, Ellingson BM, Brown CE, Chen Y, Fecci PE, Reardon DA, Dunn GP, Liau LM, Costello JF, Wick W, Cloughesy T, Timmer WC, Wen PY, Prins RM, Platten M,Okada H. Neuro Oncol 2021, 23, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Saraiva M, Vieira P,O’Garra A. J Exp Med 2020, 217, e20190418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yu H, Pardoll D,Jove R. Nat Rev Cancer 2009, 9, 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tas SW, Vervoordeldonk MJ, Hajji N, Schuitemaker JH, van der Sluijs KF, May MJ, Ghosh S, Kapsenberg ML, Tak PP,de Jong EC. Blood 2007, 110, 1540. [DOI] [PubMed] [Google Scholar]
- [55].Miller MA, Chandra R, Cuccarese MF, Pfirschke C, Engblom C, Stapleton S, Adhikary U, Kohler RH, Mohan JF, Pittet MJ,Weissleder R. Sci Transl Med 2017, 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Erel-Akbaba G, Carvalho LA, Tian T, Zinter M, Akbaba H, Obeid PJ, Chiocca EA, Weissleder R, Kantarci AG,Tannous BA. ACS Nano 2019, 13, 4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ahmed MS, Rodell CB, Hulsmans M, Kohler RH, Aguirre AD, Nahrendorf M,Weissleder R. Bioconjug Chem 2019, 30, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Higuti I, Silva P, Papp V, Okiyama M, Alves de Andrade E, Marcondes A,Nascimento A. Braz Arch Biol Technol. 2004, 47, 837–841. [Google Scholar]
- [59].Berger G, Knelson EH, Jimenez-Macias JL, Nowicki MO, Han S, Panagioti E, Lizotte PH, Adu-Berchie K, Stafford A, Dimitrakakis N, Zhou L, Chiocca EA, Mooney DJ, Barbie DA,Lawler SE. Proc Natl Acad Sci U S A 2022, 119, e2111003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, Martuza RL,Rabkin SD. Proc Natl Acad Sci U S A 2013, 110, 12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mostany R,Portera-Cailliau C. J Vis Exp 2008, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kim HY, Li R, Ng TSC, Courties G, Rodell CB, Prytyskach M, Kohler RH, Pittet MJ, Nahrendorf M, Weissleder R,Miller MA. ACS Nano 2018, 12, 12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ko J, Oh J, Ahmed MS, Carlson JCT,Weissleder R. Angew Chem Weinheim Bergstr Ger 2020, 132, 6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Oh J, Carlson JCT, Landeros C, Lee H, Ferguson S, Faquin WC, Clark JR, Pittet MJ, Pai SI,Weissleder R. Clin Cancer Res 2021, 27, 4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ng TSC, Hu H, Kronister S, Lee C, Li R, Gerosa L, Stopka SA, Burgenske DM, Khurana I, Regan MS, Vallabhaneni S, Putta N, Scott E, Matvey D, Giobbie-Hurder A, Kohler RH, Sarkaria JN, Parangi S, Sorger PK, Agar NYR, Jacene HA, Sullivan RJ, Buchbinder E, Mikula H, Weissleder R,Miller MA. Sci Adv 2022, 8, eabl6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Rodell CB, Ahmed MS, Garris CS, Pittet MJ,Weissleder R. Theranostics 2019, 9, 8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.