

Abstract
Vitiligo is an autoimmune skin disease caused by melanocyte-targeting autoreactive CD8+ T cells. T regulatory cells (Tregs) have been implicated in restraining vitiligo severity in both mouse models and human patients, however whether they must be present in the skin for their suppressive function is still unclear. We observed an uneven distribution of Tregs within different anatomical locations of mouse skin, which correlated with reduced depigmentation after vitiligo induction. We specifically depleted Tregs in our mouse model of vitiligo and observed increased disease. Next, we found that Tregs contact CD8+ T effector cells in vitiligo lesional skin, and that Treg recruitment to the skin inversely correlated with disease severity, suggesting a critical role for Treg suppression within the skin. When we investigated the signals facilitating Treg migration to the skin, we found that while CXCR3 was dispensable for Treg migration and function in vitiligo, Tregs lacking CCR6 exhibited a reduced capacity to migrate to the skin and suppress depigmentation, despite normal systemic numbers in the skin-draining lymph nodes. Our observations highlight a key role for cutaneous Tregs in disease suppression during vitiligo and identify CCR6 as a chemokine receptor that contributes to Treg migration to the skin.
Keywords: vitiligo, Treg, chemokine, CCR6, CXCR3
INTRODUCTION
Vitiligo is an autoimmune skin disease characterized by epidermal depigmentation resulting from the destruction of melanocytes by autoreactive CD8+ T effector cells (Teffs) (Lang et al. 2001; Ogg et al. 1998; van den Boorn et al. 2009). CD4+ T cells also infiltrate vitiliginous skin (Gross et al. 1987; van den Wijngaard et al. 2000; Strassner et al. 2017), but their role is less clear. Regulatory T Cells (Tregs) are a CD4+ T cell population that may impact vitiligo. Tregs restrain effector T cell function and play a critical role in suppressing a variety of organ-specific autoimmune diseases. Compromised Treg suppression may contribute to pathogenesis during vitiligo, as studies report that Tregs isolated from the blood of patients exhibit decreased suppressive capability (Lili et al. 2012), and reduced expression of suppressive genes like IL10 and CTLA4 (Giri et al. 2020). Additionally, Genome wide association studies (GWAS) identified vitiligo susceptibility loci in genes that support suppressive function in Tregs, such as CTLA4 and IKZF4 (Jin et al. 2016), further suggesting impaired Treg function as a contributing factor to disease.
Decreased Treg frequency has been reported in patients suffering from autoimmune diseases like alopecia areata (Han et al. 2015), multiple sclerosis (Kleinewietfeld and Hafler 2014), and systemic lupus erythematosus (Ohl and Tenbrock 2015). Studies quantifying Tregs in vitiligo lesions have reported no significant differences between patients and healthy controls (Ben Ahmed et al. 2012; Terras et al. 2014), while others reported fewer Tregs in affected skin (Klarquist et al. 2010; Abdallah et al. 2014), and others an increased number (Lili et al. 2012). These conflicting findings highlight the need for mechanistic studies to understand the significance of Treg function in the skin during vitiligo. Previous studies reported that systemic depletion of CD4+ or CD25+ T cells (which include Tregs) in mouse models of vitiligo led to increased disease (Gregg et al. 2010; Chatterjee et al. 2014), however it is still unclear whether Tregs must specifically localize to the skin for their suppressive function in vitiligo.
While we found that the chemokine receptor CXCR3 directs Teff migration to the skin (Rashighi et al. 2014; Richmond et al. 2017), little is known about the signals that govern Treg migration during vitiligo. CCR4/CCL22 signaling can promote T cell migration to the skin, and in a mouse model of vitiligo, overexpression of CCL22 in the skin increased Treg migration and suppressed disease progression (Eby et al. 2015). Signaling between CCR6 and its sole ligand CCL20 can also facilitate lymphocyte migration to the skin. CCL20, expressed at low levels in healthy skin, is upregulated in inflammatory skin diseases like psoriasis, where CCR6/CCL20 signaling contributes the recruitment of skin infiltrating Teffs (Hedrick et al. 2009; Homey et al. 2000; Kim et al. 2014). Furthermore, CCR6 contributes to Treg migration to neonatal skin in mice and sites of inflammation in mouse models of colitis (Kitamura, Farber, and Kelsall 2010), as well as experimental autoimmune encephalomyelitis (EAE) (Yamazaki et al. 2008). Additional studies are needed to define the signaling networks driving lymphocyte migration during vitiligo and how the presence of Tregs in the skin affects disease development. Here, we used our mouse model of vitiligo to reveal a key role for cutaneous Tregs in disease suppression and identify CCR6 as a chemokine receptor that contributes to optimal Treg function through migration to the skin.
RESULTS
T regulatory cells play a suppressive role in a mouse model of vitiligo.
Previous studies with the KRT14-Kitl*4XTG2Bjl (Krt14-Kitl) vitiligo mouse model did not explore the contributions of CD4+ T cells to vitiligo (Harris et al. 2012). To investigate the role of CD4+ T cells, we induced vitiligo in FoxP3-GFP hosts treated with PBS or αCD4-depleting antibody. CD4-depleted mice exhibited nearly 3 times more depigmentation than PBS treated mice (Fig. S1a–b). Using flow cytometry (Fig. S1c for gating strategy), we found that more Teffs accumulated in the SDLNs and skin of CD4-depleted mice (Fig. S1d). This data suggests that CD4+ T cells are dispensable for disease progression after initiation and that CD4-expressing cells may suppress depigmentation by limiting the number of Teffs in the skin during disease. We used flow cytometry to quantify FoxP3+ Tregs in the SDLNs, tail, and ear skin of CD4-depleted mice and observed that the number of Tregs relative to Teffs was decreased at these sites (Fig. S1e). These observations confirmed that CD4 depletion reduces Treg number in mouse models of vitiligo and reinforces results from previously reported studies (Gregg et al. 2010).
CD4 depletion affects other CD4+ cells in addition to Tregs. We induced vitiligo in FoxP3-DTR hosts and treated hosts with PBS or diphtheria toxin (DTX) to specifically and conditionally deplete Tregs during disease (Kim, Rasmussen, and Rudensky 2007). After 7 weeks, DTX treated mice exhibited significantly increased depigmentation (Fig. 1a; Fig S2a). Interestingly, the difference in depigmentation between DTX treated and PBS treated mice was more pronounced in ear skin than in tail skin (Fig. 1b), likely a reflection of the increase in Teffs quantified by flow cytometry (Fig. S2b for gating strategy) in ear skin but not tail skin of DTX treated mice (Fig. 1c–d). These observations specifically confirm Tregs as the CD4+ population responsible for suppression during vitiligo and suggest that the magnitude of Treg suppression may differ depending on the area of skin.
Figure 1: Tregs suppress depigmentation in a mouse model of vitiligo.
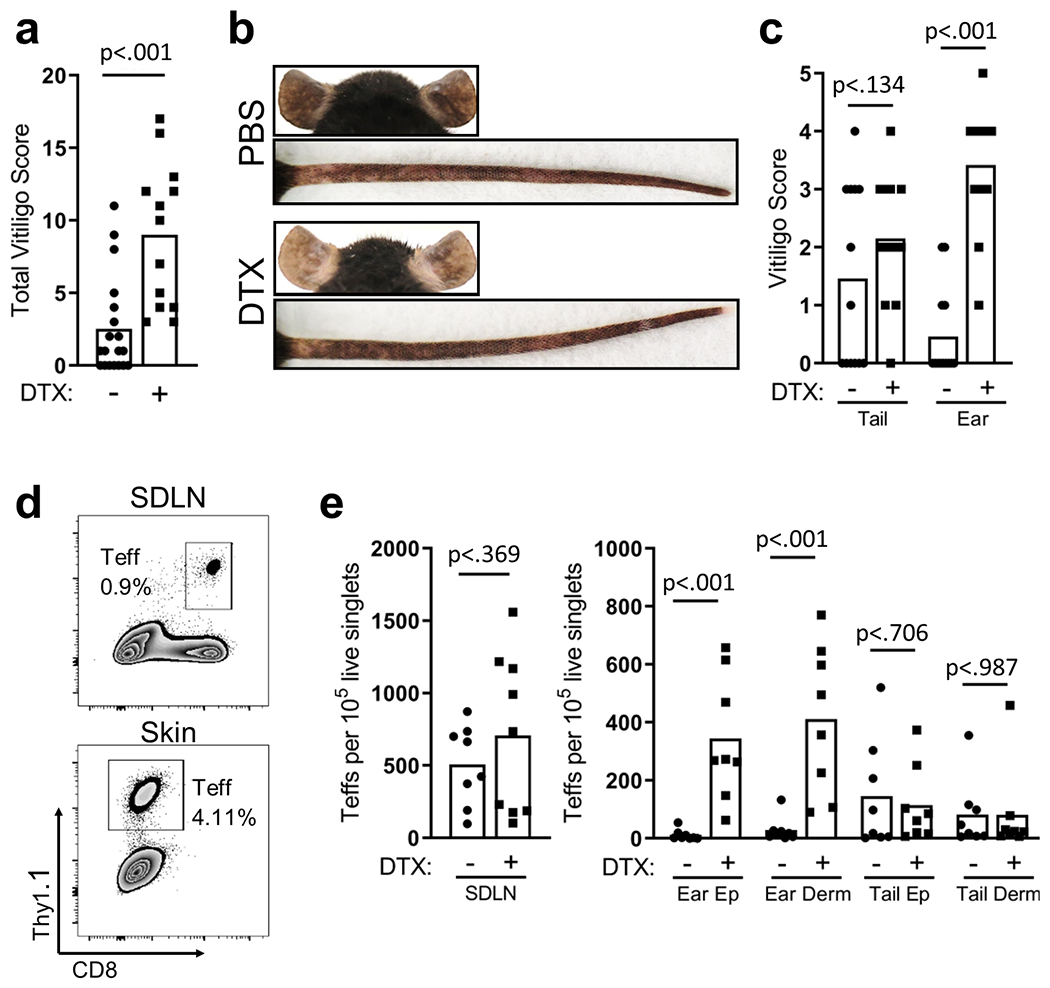
(a) Total vitiligo scores from FoxP3-DTR mice treated with DTX or vehicle; (b) Representative images of ear and tail skin from FoxP3-DTR mice treated with PBS or DTX; (c) disease scores in the ear and tail (7 weeks post disease induction, N=12, two-tailed unpaired t test). (d) Sample flow plots and (e) quantification of Teffs in SDLN and skin of FoxP3-DTR mice treated with DTX (7 weeks post disease induction, N=8, two-tailed unpaired t test).
Cutaneous Treg number inversely-correlates with the extent of depigmentation.
To explore why some anatomical locations were more susceptible to disease after Treg depletion, we used flow cytometry to quantify FoxP3+ Tregs in FoxP3-GFP mice in the absence of vitiligo induction (Fig. S3a for gating strategy). In the skin, the majority of Tregs reside in the dermis and more Tregs reside in the ear compared to the tail (Fig. S3b). This increased number of Tregs likely establishes a more immunosuppressive environment in ear skin, resulting in decreased depigmentation during vitiligo. To investigate this, we compared the extent of depigmentation between tail skin and ear skin in a large number of mice (N = 177 mice) induced with vitiligo, all using the same induction protocol, and observed that ear skin exhibits significantly less depigmentation than tail skin in our experiments (Fig. 2a). This correlation may highlight a role specifically for skin-resident Tregs in disease suppression.
Figure 2: Treg number in the skin correlates with disease severity.
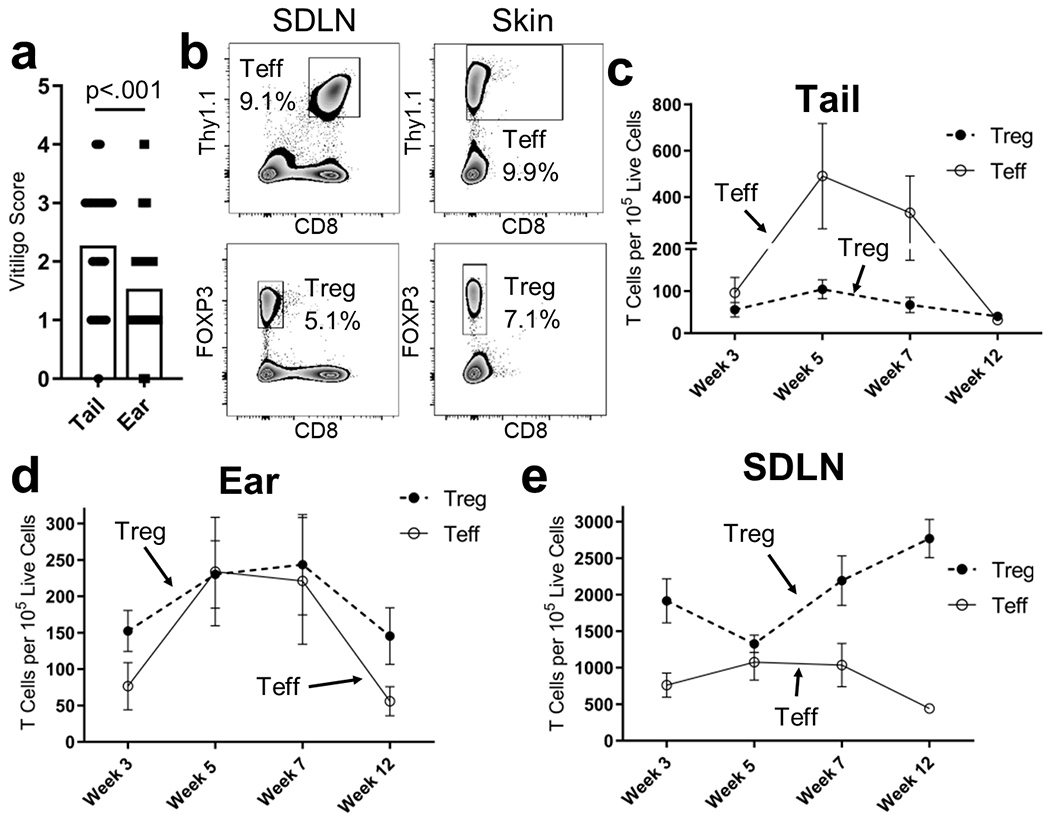
(a) Vitiligo scores of the ears and tails of mice with a total score of at least 5 (N=177, two-tailed unpaired t test). (b) Sample flow plots and quantification of Teffs and Tregs in (c) tail skin, (d) ear skin, or (e) SDLNs in FoxP3-GFP mice with vitiligo at the indicated week after disease induction (N=10, bars represent S.E.M).
To investigate how cutaneous Treg number relates with patterns of depigmentation, we used flow cytometry to compare the number of Tregs and Teffs in the SDLNs and skin of FoxP3-GFP mice as disease progresses (Fig. 2b). We found more Tregs in the dermis than the epidermis (Fig. S3b), and at all weeks, more Tregs accumulated in the ear than in the tail (Fig. 2c–d). In SDLNs we observed a decrease in the number of Tregs at week 5, however by week 7, the number of Tregs begins to increase again (Fig. 2e). Interestingly, this decrease at week 5 occurs during the peak of disease, weeks 5 to 7, and correlates with an increase in cutaneous Treg number (Fig. 2c–d). We compared Treg to Teff migration and observed that while there are a comparable number of Teffs and Tregs in ear skin at the peak of disease, Teffs outnumber Tregs in tail skin (Fig. 2c–d). Thus, the resulting Teff/Treg ratio is significantly higher in tail skin than in ear skin (Fig. S3c) and we observed a statistically significant correlation between Teff/Treg ratios and the severity of depigmentation on ears and tails (Fig. S3d). Since we observed that ear skin is less susceptible to disease than tail skin, these observations further support that the number of Tregs in the skin relative to the number of Teffs influences disease severity and suggest that Tregs suppress Teffs in the skin during vitiligo.
Tregs directly contact Teffs in the skin during vitiligo.
Considering the importance of Tregs in the skin, we investigated whether Tregs and Teffs in lesional skin interact. We induced vitiligo with RFP+ Teffs in FoxP3-GFP hosts and examined lesional ears using en-face fluorescent and confocal microscopy. We observed that GFP+ Tregs and RFP+ Teffs cluster together in ear skin (Fig. 3a–b), and that Tregs directly contact Teffs, at times extending projections to touch neighboring cells (Fig. 3c–d). These observations suggest that Tregs interact with Teffs upon migration to lesional skin and further support a role for Treg suppression in the skin during vitiligo.
Figure 3: Tregs directly contact Teffs in the skin during vitiligo.
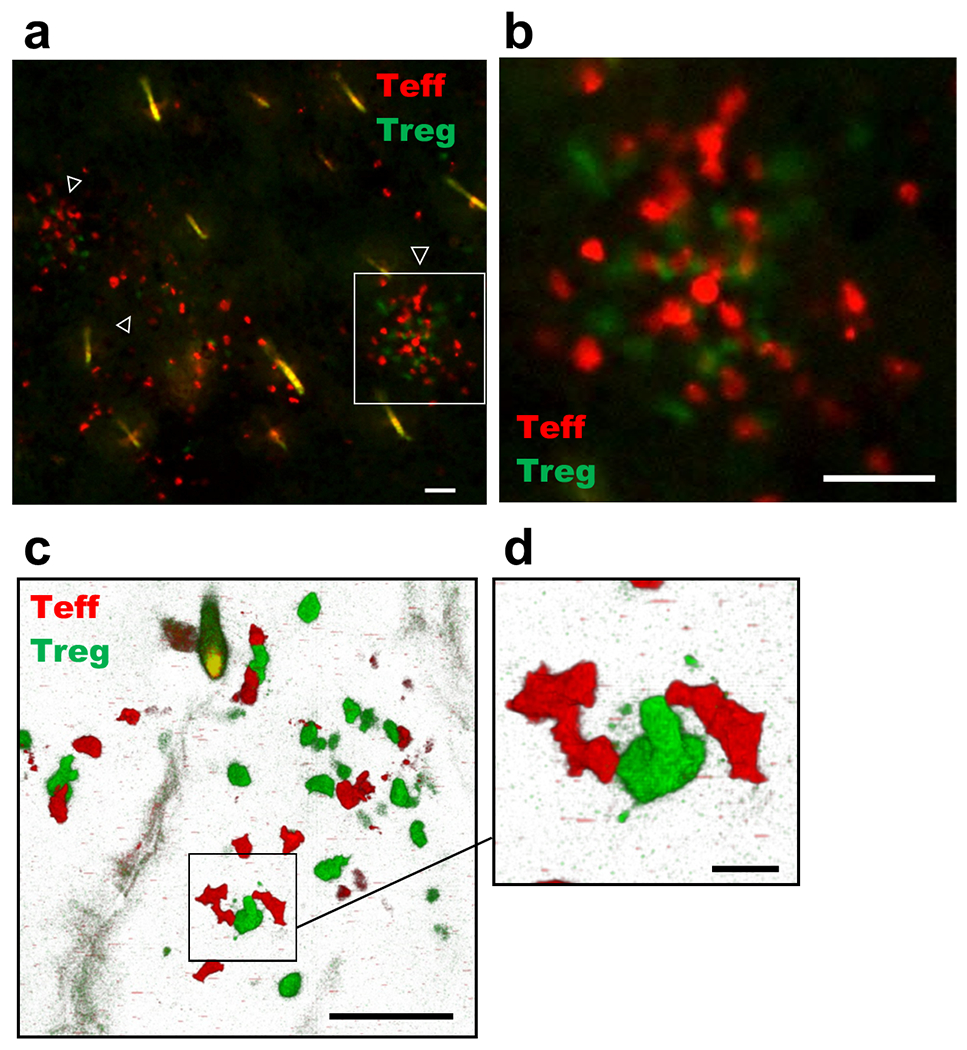
(a) Representative en-face florescent microscopy image depicting GFP+ Tregs and RFP+ Teffs in lesional mouse ear skin; white arrows highlight clusters of Tregs and Teffs, 20x image, bar = 50μM. The area defined by the white box is magnified in (b), 20x image, bar = 50 μM. (c) Representative confocal microscopy image depicting GFP+ Tregs and RFP+ Teffs in lesional mouse ear skin, 40x image; bar = 50μM. The area defined by the black box is magnified in (d), 40x image; bar = 10μM. Imaging experiments were performed 5 weeks post disease induction.
CXCR3 is dispensable for the ability of Tregs to suppress vitiligo.
Previous Treg depletion studies in mouse models of disease systemically reduced Treg number and did not dissect the contributions of Treg suppression in the skin from suppression in secondary lymphoid organs (Gregg et al. 2010; Chatterjee et al. 2014). Considering our data correlating the presence of skin Tregs with decreased disease scores, we investigated the signals driving cutaneous migration to selectively reduce Treg number in the skin. CXCR3 facilitates Teff migration to lesional skin during vitiligo (Rashighi et al. 2014; Richmond et al. 2017), and since we observed that Tregs and Teffs colocalize in the skin, we reasoned that Tregs might also require CXCR3 for recruitment to the skin. To test this, we modified a previously reported mouse model of melanoma immunotherapy in Rag−/− hosts (Antony et al. 2005). We generated Krt14-Kitl×Rag−/− (Rag−/−) mice and adoptively transferred Treg-depleted CD4+ cells and Tregs. After 4 weeks we used flow cytometry to quantify Tregs in ear skin and tail skin and observed that in most areas, Treg numbers in our Rag−/− mice were comparable to Treg numbers in our FoxP3-GFP mice (Fig. S4a). The only exception was the ear dermis, where we observed increased Treg number in Rag−/− hosts.
We induced vitiligo in Rag−/− hosts with WT Tregs, CXCR3−/− Tregs, or no Tregs. We observed that ear skin was more susceptible to the lack of Tregs compared to other sites, consistent with our studies in immunocompetent hosts. Hosts that received CXCR3−/− Tregs had minimal ear depigmentation, suggesting that Tregs do not require CXCR3 to suppress Teff-mediated depigmentation (Fig. S4b–c). We used flow cytometry to quantify the number of Teffs in Rag−/− hosts (Fig. S4d for gating strategy) and found comparable numbers of Teffs in the ear skin of mice receiving CXCR3−/− or WT Tregs (Fig. S4e). Rag−/− hosts that received no Tregs had the most Teffs in ear skin (Fig. S4e). Additionally, CXCR3−/− Tregs did not exhibit a reduced ability to migrate to ear skin compared to WT Tregs, suggesting CXCR3 is dispensable for Treg migration during vitiligo (Fig. S4f). To directly compare the migratory capabilities of CXCR3−/− and WT Tregs, we induced vitiligo in Rag−/− hosts and transferred equal numbers of CXCR3−/− and WT Tregs into hosts to create a competitive transfer model. After mice developed depigmentation, we used flow cytometry to quantify Tregs (Fig. S4g for gating) and observed that the absence of CXCR3 did not reduce the ability of Tregs to populate SDLNs (Fig. S4h). Similarly, comparable numbers of CXCR3−/− and WT Tregs migrated to ear skin (Fig. S4h). These results suggest CXCR3 is dispensable for Treg suppression and migration to the skin and suggest that treatments targeting the CXCL10/CXCR3 axis to block Teff mediated depigmentation will not affect Treg suppression.
Tregs in skin express CCR6 during vitiligo.
We looked for other chemokine receptors that might help to promote Treg recruitment and function during vitiligo. GWAS studies identified CCR6 as a vitiligo susceptibility gene (Spritz 2013) and CCR6 has been reported to mediate Treg migration into neonatal mouse skin during development (Scharschmidt et al. 2017), as well as areas of inflammation in other models of disease (Yamazaki et al. 2008). We used suction blistering to isolate cells from the skin of vitiligo patients and, using flow cytometry, (Fig. S5a for gating) we observed a comparable proportion of CCR6-expressing Tregs in non-lesional and lesional skin (Fig. S5b). We used flow cytometry in FoxP3-GFP mice to investigate whether Tregs express CCR6 in our model (Fig. S5c for gating). In mice without vitiligo, a greater proportion of Tregs express CCR6 in ear (Fig. S5d) and tail skin (Fig. S5d) than in SDLNs. This apparent upregulation in the skin indicates CCR6 may contribute to cutaneous Treg migration in the steady state. To investigate whether Tregs upregulate CCR6 in our mouse model of vitiligo, we induced disease in Foxp3-GFP mice and used flow cytometry to measure CCR6 expression by FoxP3+ Tregs. An increased percentage of Tregs express CCR6 in the SDLNs, ear skin, and tail skin (Fig. S5d) when vitiligo is induced, suggesting that CCR6 may contribute to Treg migration to the skin or function within the skin during disease.
CCL20 is the sole known ligand for CCR6, so we performed RT-PCR to compare mRNA expression levels of CCL20 in the skin of mice with or without vitiligo (Fig. S6). In tail and ear skin of mice with vitiligo we observed less than a 2-fold increase in CCL20 compared to mice without vitiligo, and CCL20 appears to be upregulated more in tail skin than in ear skin (Fig. S6). While this difference is statistically significant, it is unclear whether this is biologically significant.
CCR6−/− Tregs exhibit reduced migration to the skin and disease suppression in a mouse model of vitiligo.
To investigate whether Tregs require CCR6 to migrate to skin and suppress depigmentation, we induced vitiligo in Rag−/− hosts with either no Tregs, WT Tregs, or CCR6−/− Tregs. Hosts without Tregs exhibited extensive depigmentation compared to hosts that received WT Tregs (Fig. 4a–b). In contrast, host that received CCR6−/− Tregs exhibited over 3-fold more depigmentation than hosts with WT Tregs, an increase comparable to hosts without Tregs (Fig. 4a–b). We used flow cytometry to quantify T cells in Rag−/− hosts (Fig. S7a for gating strategy). We observed comparable numbers of Teffs in the SDLNs and skin in hosts of all groups (Fig. 4c). Interestingly, while the number of Tregs in the SDLNs was comparable among groups, we observed a strong trend toward fewer CCR6−/− Tregs than WT Tregs in the skin (Fig. 4d). This likely explains the increased depigmentation that we observed in hosts receiving CCR6−/− Tregs. To directly compare the ability of CCR6−/− Tregs and WT Tregs to migrate to the skin during vitiligo, we induced a competitive adoptive transfer model of disease with CCR6−/− Tregs and WT Tregs. We used flow cytometry to quantify Tregs (Fig. S7b for gating strategy) and observed that the proportion of WT Tregs to CCR6−/− Tregs in SDLNs was comparable to the proportion at input (Fig. 4e). However, the proportion in skin was greatly reduced, suggesting that CCR6−/− Tregs have an impaired capacity to migrate to the skin in our vitiligo model (Fig. 4e). CCR6−/− Tregs have been reported to possess normal suppressive capability in vitro (Turner et al. 2010). Similarly, we observed that CCR6−/− Tregs and WT Tregs exhibited a comparable ability to suppress αCD3/αCD28-induced Teff cytokine expression (Fig. S7c). Together these observations reveal that reducing Treg migration to the skin leads to increased depigmentation. These results highlight the crucial contribution of Treg-mediated suppression within the skin to controlling depigmentation and suggest that CCR6 is an additional chemokine receptor that governs Treg migration to the skin during vitiligo.
Figure 4: Trees require CCR6 for optimal suppression and migration to the skin in a mouse model of vitiligo.
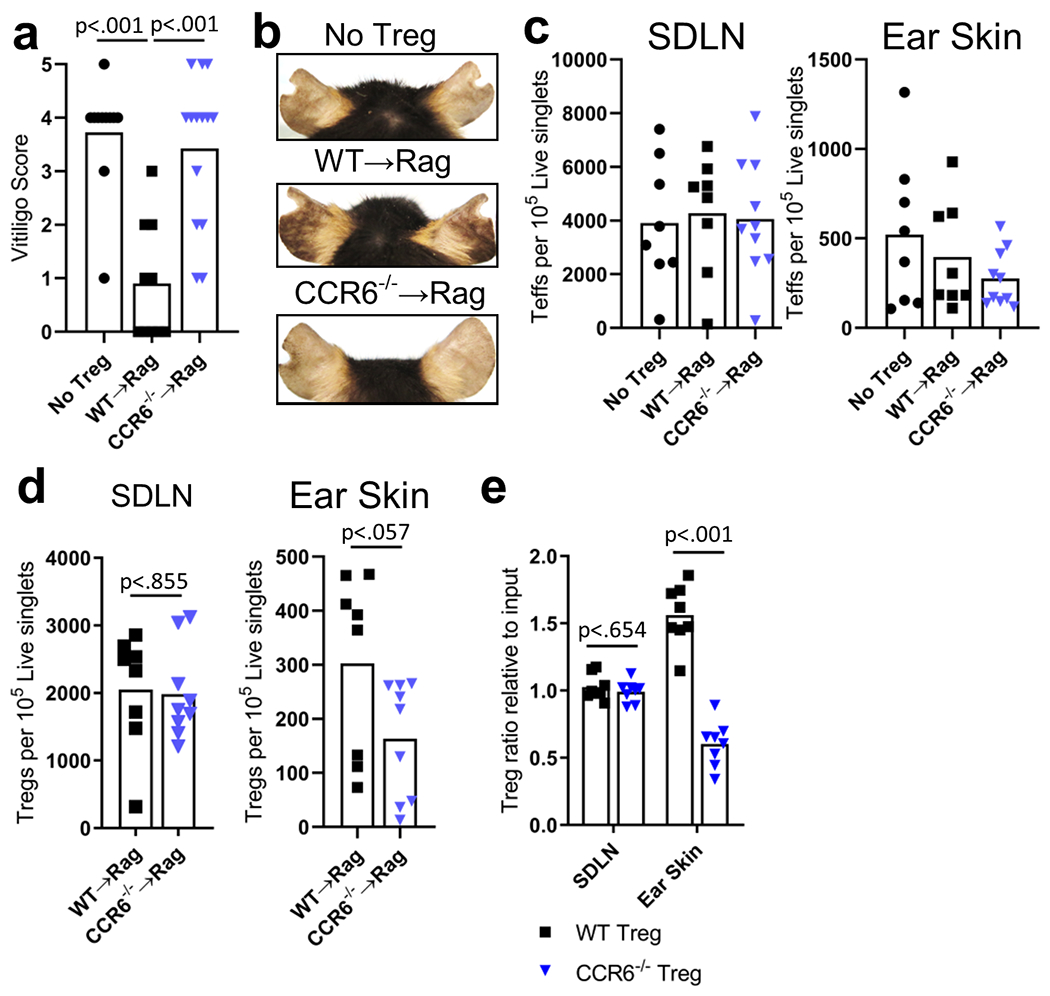
(a) Ear vitiligo scores and (b) representative images of Rag−− mice ears that received no Tregs, WT or CCR6−− Tregs (5 weeks post disease induction, N=14, two-tailed unpaired t test). Quantification of (c) Teffs and (d) Tregs in the SDLNs and ear skin of Rag−− mice (5 weeks post disease induction, N=8 No Treg, WT→Rag, N=9 CCR6−−→Rag two-tailed unpaired t test). (e) Quantification of WT and CCR6−− Tregs in the SDLNs and ear skin of Rag−− mice in a competitive Treg-adoptive transfer model of vitiligo (5 weeks post disease induction, N=8, two-tailed unpaired t test).
DISCUSSION
Here we report that Tregs suppress depigmentation in a mouse model of vitiligo and areas of skin heavily populated by Tregs exhibit reduced depigmentation. Eliminating CCR6 on Tregs reduces their ability to migrate to skin, but not SDLNs, and subsequently abrogates their ability to suppress disease. These results suggest that Treg number and function in the skin are required for disease suppression. Previous studies in patients evaluating whether cutaneous Tregs function normally or are present in sufficient number to counterbalance Teffs have reported conflicting results. The answer to these questions could lead to therapies that re-establish homeostasis in the skin by increasing Treg number, but few animal studies have investigated how solely manipulating cutaneous Treg number correlates with disease severity (Eby et al. 2015; Chatterjee et al. 2014; Miao et al. 2018).
Several studies performed in mouse models of vitiligo revealed that a CD4+CD25+ T cell population plays a suppressive role during disease. These studies implicated Tregs, but used CD4 and CD25 depleting antibodies that preferentially target Tregs, but these strategies can affect activated T cells as well (Gregg et al. 2010; Chatterjee et al. 2014). We specifically depleted Tregs by targeting FoxP3-expressing cells to demonstrate that Tregs specifically suppress depigmentation during vitiligo. It is worth noting that Teffs can still drive vitiligo even though Tregs are present, but that vitiligo severity is increased when Tregs are depleted. This suggests that even though spots are forming, Tregs are still actively restraining Teffs and prevent depigmentation from being as extensive as it could be in their absence.
The increase in depigmentation resulting from Treg depletion was more pronounced on ear skin than tail skin. More Tregs are in ear skin compared to other sites at baseline, and this likely promotes a more immunosuppressive environment in the ear. This could explain why the extent of depigmentation is usually greater on tail skin in our model compared to ears, and why ears are the skin site most affected by Treg depletion. In humans, areas of skin with higher hair follicle density (scalp and face) harbor more Tregs (Sanchez Rodriguez et al. 2014), however vitiligo commonly presents on the face (Ezzedine et al. 2011). It is unclear what role Treg density plays in the distribution of vitiligo in humans.
The chemokine signals required by Tregs for skin migration could potentially be modulated to treat vitiligo. Eby et al. demonstrated that overexpressing the chemokine CCL22 in the skin of vitiligo-prone mice increased Treg number in the skin and decreased depigmentation (Eby et al. 2015). This approach was not durable however, as mice depigmented two weeks after treatment ceased. Effector Tregs can express molecules typically associated with conventional helper CD4+ T cells in response to the prominent cytokine milieu in an area of inflammation (Cretney, Kallies, and Nutt 2013). While vitiligo skin is dominated by a Th1 signature and CXCR3 regulates Teff migration during vitiligo (Rashighi et al. 2014; Richmond et al. 2017), Tregs do not require CXCR3 to suppress depigmentation in vitiligo in our mouse model of vitiligo. This data suggests that treatment strategies aimed at inhibiting Teff migration by targeting CXCL9 or CXCL10 would not directly affect Treg suppressive activity. Our studies with CCR6−/− Tregs functionally demonstrated that reducing Treg number in the skin while minimally affecting numbers in SDLNs results in increased depigmentation. While this result does not eliminate the possibility that some Treg suppression occurs in SDLNs, it highlights a specific role for skin Tregs in suppression of disease in our mouse models. Previous studies in mouse models of vitiligo systemically depleted Tregs and were unable to investigate whether Tregs suppress disease in the SDLNs or in the skin. Our observations support strategies to increase Treg number or function in the skin of patients as a treatment for vitiligo.
While we determined that CCR6 was necessary for migration in our mouse model, we did not observe a pronounced upregulation of CCL20 in the skin during vitiligo. In Th17-driven skin diseases like psoriasis, CCL20 is upregulated by keratinocytes (Homey et al. 2000; Kim et al. 2014; Hedrick et al. 2009), which are likely the key cell types that express this chemokine in all diseases, including healthy skin and vitiligo lesions. CCL20 is constitutively expressed at low levels in all skin (Charbonnier et al. 1999; McCully and Moser 2011), and thus Tregs may upregulate CCR6 to promote optimal migration to the skin during vitiligo.
While Tregs required CCR6 for optimal migration to the skin, CCR6−/− Tregs were not completely absent from the skin. This suggests that there are other chemokine receptors, possibly CCR4 (Eby et al. 2015; Klarquist et al. 2010), that contribute to Treg migration during vitiligo and that a network of signals likely coordinate Treg migration. More work needs to be done to completely determine the signals that support Treg migration to the skin.
CCR6 can also facilitate the migration of Th17 effector cells, which characteristically express the receptor (Annunziato et al. 2007; Izcue et al. 2008), and a role for CCR6/CCL20 in driving skin inflammation in psoriasis has been reported (Furue et al. 2020). In psoriatic skin, tissue infiltrating T cells express high levels of CCR6, facilitating their colocalization with CCL20-producing keratinocytes (Hedrick et al. 2009; Homey et al. 2000; Kim et al. 2014). CCR6 has also been reported to contribute to Treg migration into neonatal skin during development (Scharschmidt et al. 2017) and into sites of inflammation in other models of disease (Yamazaki et al. 2008; Kitamura, Farber, and Kelsall 2010). In addition, GWAS studies identified CCR6 as a vitiligo susceptibility gene, and thus CCR6 polymorphisms may increase risk of disease by modulating Treg migration to the skin.
Understanding how Tregs migrate to skin could improve the strategies that we use to treat vitiligo. Several studies reported a mild elevation of IL17 in the serum and lesional skin of vitiligo patients and correlated these observations to disease severity (Bassiouny and Shaker 2011; Singh et al. 2016). Based on this, some have argued that FDA approved treatments for psoriasis, psoriatic arthritis, and rheumatoid arthritis that target type 17 signaling pathways, such as IL-17 and IL-23 inhibitors, may also be effective for treating vitiligo (Singh et al. 2016). However, treatment with IL-17 or IL-23 antibodies do not reverse vitiligo, but actually induce or exacerbate disease (Frisoli, Essien, and Harris 2020; Speeckaert, Mylle, and van Geel 2019; Méry-Bossard et al. 2017). In addition, our recent single-cell RNA sequencing studies identified a strong type 1 signature in vitiligo patient skin in the absence of an increase in type 17 cytokines (Gellatly et al. 2021). Indeed, our observations from this study suggest that targeting type 17 signaling could exacerbate vitiligo by compromising Treg recruitment to the skin. In fact, strategies that stimulate CCR6-driven Treg migration to the skin could be an effective therapeutic strategy that could be explored in future studies.
MATERIALS AND METHODS
Mice
All mice were obtained from The Jackson Laboratory unless otherwise indicated. KRT14-Kitl*4XTG2Bjl (Krt14-Kitl) mice were a gift from B. J. Longley (University of Wisconsin). The following strains were bred to Krt14-Kitl mice to use as hosts in a vitiligo mouse model: FoxP3-GFP mice (B6.Cg-Foxp3tm1Kuch/J), FoxP3-DTR mice (B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J), and Rag−/− mice (B6.129S7-Rag1tm1Mom/J strain). The Krt14-Kitl allele was bred heterozygous on all mice used in experiments. Thy1.1+Thy1.2+ mice generated by crossing C57BL/6J mice with Thy1.1+ (B6.PL-Thy1a/CyJ mice), CD45.1 mice (B6.SJL-Ptprca Pepcb/BoyJ), CCR6−/− mice (B6.129P2-Ccr6tm1Dgen/J) and CXCR3−/− mice (B6.129P2-Cxcr3tm1Dgen/J) were used as CD4+ T cells or Treg donors. PMEL mice (B6.Cg Thy1a/CyTg(TcraTcrb)8Rest/J) and PMELRFP mice generated by crossing PMEL mice with RFP mice (B6.Cg-Tg(CAG-mRFP1)1F1Hadj/J) we used as Teff donors. All mice were bred on a C57BL/6J background and maintained in pathogen-free facilities at the University of Massachusetts Medical School (UMMS). Procedures were approved by the UMMS Institutional Animal Care and Use Committee and in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Vitiligo induction
We induced vitiligo as described previously (Harris et al. 2012). Teffs were isolated from the spleens of PMEL TCR transgenic mice using a Miltenyi Biotec CD8 isolation kit (Bergisch Gladbach, Germany) according to the manufacturer’s instructions. 106 purified Teffs were injected intravenously into 8- to 12-week-old hosts irradiated with 500 rads 1 day before transfer. Hosts were also infected with 106 plaque-forming units (PFU) of rVV-hPMEL through intraperitoneal (i.p.) injection (N. Restifo, National Cancer Institute, NIH) on the same day of adoptive transfer.
Rag−/− vitiligo induction
We adoptively transferred 106 Teffs, 106 CD4s and either no Tregs or 105 Tregs (WT, CXCR3−/− or CCR6−/− Tregs) into 8-12-week-old Rag−/− hosts infected i.p. with 106 PFU of rVV-hPMEL on the same day of adoptive transfer. Hosts were irradiated with 500 rads 1 day before transfer. In competitive Rag−/− vitiligo models, vitiligo was induced by adoptive transfer of 106 Thy1.1+ Teffs, 106 CD45.1+ CD4+ T cells, and 5.0×104 Thy1.1+Thy1.2+ WT and 5.0×104 Thy1.2+ KO Tregs (CXCR3−/− or CCR6−/−) into 8-12-week-old Rag−/− hosts irradiated with 500 rads 1 day before transfer, and hosts were infected i.p. with 106 PFU of rVV-hPMEL on the same day of adoptive transfer. Teffs were isolated using a Miltenyi Biotec CD8 isolation kits. Treg-depleted CD4+ T cells were isolated from the spleens of C57BL/6J or CD45.1 mice through negative selection using Miltenyi Biotec CD4+CD25+ isolation kits (Bergisch Gladbach, Germany). Tregs were isolated from the spleens of C57BL/6J mice, CXCR3−/− mice, or CCR6−/− mice through positive selection using Miltenyi Biotec CD4+CD25+ isolation kits.
CD4 depletion and Treg depletion by DTX treatment
For CD4 depletion experiments vitiligo was induced in Krt14-Kitl×FoxP3-GFP mice as previously described. After 2 weeks, mice were treated once weekly i.p. with 0.2mg of GK1.5 CD4 depleting antibody (αCD4) (BioXcell; West Lebanon, NH; Cat: BE0003-1) or PBS. After 5 weeks of treatment, a blinded observer quantified depigmentation and tissues were processed for flow cytometry. For DTX depletion experiments vitiligo was induced in Krt14-Kitl×FoxP3-DTR mice as previously described. After 2 weeks, mice were treated with 50μg/kg of diphtheria toxin (DTX) (Sigma-Aldrich; Darmstadt, Germany; Cat: D0564) or PBS every other day for 2 weeks. At 7 weeks post disease induction, a blinded observer quantified depigmentation and tissues were processed for flow cytometry.
Flow Cytometry
Ears, tails, and skin-draining lymph nodes were harvested at the indicated times. All flow data were collected with an LSR II and were analyzed with FlowJo software (FlowJo LLC, Ashland, OR). Please see Supplementary Methods for a detailed methods and a list of flow antibodies used.
Imaging Treg-Teff interactions
To visualize interactions between murine Tregs and Teffs, vitiligo was induced in Krt14-Kitl×FoxP3-GFP mice as previously described using RFP expressing Teffs from PMELRFP donors. After 5 weeks, mice were euthanized and lesional ears were excised. Following treatment of ears with Nair to remove hair, the dorsal and ventral sides were separated and mounted on slides for imaging. All confocal imaging was performed on a Leica SP8 confocal microscope.
Suction blistering
Suction blistering was performed as previously described (Strassner et al. 2017). Blisters were induced on the skin and the blister fluid was aspirated through blister roofs using a 1 mL insulin syringe. Skin infiltrating cells within the blister fluid were pelleted at 330×g for 10 minutes and then stained to be analyzed with flow cytometry.
Statistical analyses
All statistical analyses were performed with GraphPad Prism software. Dual comparisons were made with two-tailed unpaired t tests assuming equal variance.
Study subjects
The collection of blister fluid from patients with vitiligo was approved by the Institutional Review Board (IRB) at the University of Massachusetts Medical School (UMMS). All participants gave written informed consent before all procedures. Identification and acquisition of FFPE clinical tissue samples were IRB-approved at UMMS, and all samples were de-identified before use in experiments.
Supplementary Material
Figure S1: CD4 depletion exacerbates depigmentation in a mouse model of vitiligo. (a) Vitiligo scores and (b) representative images of FoxP3-GFP mice treated with PBS or CD4-depleting antibody (αCD4). (c) Gating strategy used to quantify Tregs and Teffs in FoxP3-GFP mice with vitiligo (plots shown represent the SDLNs and ear skin) and quantification of (d) Teffs and (e) Treg/Teff ratio (Treg #/Teff #) in SDLNs and skin (7 weeks post disease induction, N=10, two-tailed unpaired t test).
Figure S2: Tregs suppress depigmentation in a mouse model of vitiligo. (a) Gating strategy used to quantify Teffs in FoxP3-DTR mice with vitiligo 7 weeks post disease induction (plots shown represent the SDLNs and ear skin).
Figure S3: Treg number increases in the skin during vitiligo in a mouse model of disease. (a) Gating strategy used to quantify Tregs in FoxP3-GFP mice with or without vitiligo (plots shown represent the SDLNs and ear skin from mice with no vitiligo). (b) Quantification of Tregs in SDLN, ear and tail skin of FoxP3-GFP mice with or without vitiligo. In mice with vitiligo, Tregs were quantified at the peak of disease, 5 weeks post disease induction (N=12 Mice w/o vitiligo; N=10 vitiligo mice, two-tailed unpaired t test). (c) Quantification of the Teff/Treg ratio in tail and ear skin of mice 5- or 7-weeks post disease induction (N=19, two-tailed paired T test). (d) Linear regression analysis comparing the Teff/Treg ratio in vitiligo skin (x-axis) with the vitiligo score of the skin (y-axis) (N=38).
Figure S4: Tregs do not require CXCR3 to suppress depigmentation in a mouse model of vitiligo. (a) Quantification of Tregs in Rag−/− hosts 5 weeks after receiving WT Tregs or Tregs in FoxP3-GFP mice (WT Mice) (N=4 WT→Rag; N=12 WT mice, two-tailed unpaired t test). (b) Ear vitiligo scores and (c) representative images of Rag−/− mice receiving no Tregs, WT or CXCR3−/− Tregs. (d) Gating strategy and quantification of (e) Teffs and (f) Tregs in the SDLNs and ear skin of Rag−/− mice (5 weeks post disease induction, N>9, two-tailed unpaired t test). (g) Gating strategy and (h) quantification of WT and CXCR3−/− Tregs in the SDLNs and ear skin of Rag−/− mice in a Treg-competitive adoptive transfer model of vitiligo (5 weeks post disease induction, N=10, two-tailed unpaired t test).
Figure S5: Tregs in mouse and human skin express CCR6. (a) Representative flow plots depicting CCR6 expression on Tregs in patient skin. (b) Quantification of the proportion of Tregs expressing CCR6 in patient skin (N=5; two-tailed unpaired t test). (c) Representative flow plots depicting CCR6 expression on Tregs in the ear skin of FoxP3-GFP mice without vitiligo. (d) Quantification of the proportion of Tregs expressing CCR6 in SDLN and skin of FoxP3-GFP mice with vitiligo (5 weeks post disease induction) and FoxP3-GFP mice not induced with vitiligo (no vit) (N=8 no vitiligo; N=10 vitiligo, two-tailed unpaired t test).
Figure S6: CCL20 gene expression in mouse skin during vitiligo. RT-PCR was performed on RNA isolated from the skin of mice with vitiligo and mice not induced with vitiligo. CCL20 expression for mice with vitiligo is shown relative to CCL20 expression in control mice not induced with vitiligo (5 weeks post disease induction, N=10 ear; N=9 tail, two-tailed unpaired t test).
Figure S7: Gating strategy for T cells in a Rag−/− model of vitiligo. CCR6−/− Tregs suppress activation-induced cytokine expression by Teffs. (a) Gating strategy used to quantify Teffs and Tregs in Rag−/− hosts (plots shown represent the ear skin from Rag−/− hosts with WT Tregs). (b) Gating strategy used to quantify WT Tregs and CCR6−/− Tregs (KO) in Rag−/− hosts from competitive transfer experiments. (c) Quantification of IFNγ or TNFα expression by αCD3/αCD28 activated Teffs cultured in the presence of no Tregs, WT Tregs or CCR6−/− Tregs (N=18 No Treg; N=17 WT Treg; N=17 CCR6−/− Treg, two-tailed unpaired T test).
ACKNOWLEDGMENTS
We thank the members of the Harris Lab for technical assistance. Equipment used for flow cytometry and microscopy were maintained by the University of Massachusetts Medical School Flow Cytometry Core. Research reported in this publication was supported by the National Institutes of Health under award number R01AR069114 (J.E.H.). J.P.S was supported by T32 awards AI132152 and GM107000. K.I.E was supported by T32 awards AI095213.
Footnotes
CONFLICT OF INTEREST STATEMENT
JEH is an inventor on patent application #15/851,651, “Anti-human CXCR3 antibodies for the Treatment of Vitiligo” which covers targeting CXCR3 for the treatment of vitiligo. J.E.H. is a scientific founder of Villaris Therapeutics Inc., which develops therapeutic treatments for vitiligo.
DATA AVAILABILITY STATEMENT
No datasets were generated or analyzed during the current study
REFERENCES
- Abdallah M, Lotfi R, Othman W, and Galal R. 2014. ‘Assessment of tissue FoxP3+, CD4+ and CD8+ T-cells in active and stable nonsegmental vitiligo’, Int J Dermatol, 53: 940–6. [DOI] [PubMed] [Google Scholar]
- Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, and Romagnani S. 2007. ‘Phenotypic and functional features of human Th17 cells’, J Exp Med, 204: 1849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, Palmer DC, Chan CC, Klebanoff CA, Overwijk WW, Rosenberg SA, and Restifo NP. 2005. ‘CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells’, J Immunol, 174: 2591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassiouny DA, and Shaker O. 2011. ‘Role of interleukin-17 in the pathogenesis of vitiligo’, Clin Exp Dermatol, 36: 292–7. [DOI] [PubMed] [Google Scholar]
- Ben Ahmed M, Zaraa I, Rekik R, Elbeldi-Ferchiou A, Kourda N, Belhadj Hmida N, Abdeladhim M, Karoui O, Ben Osman A, Mokni M, and Louzir H. 2012. ‘Functional defects of peripheral regulatory T lymphocytes in patients with progressive vitiligo’, Pigment Cell Melanoma Res, 25: 99–109. [DOI] [PubMed] [Google Scholar]
- Charbonnier AS, Kohrgruber N, Kriehuber E, Stingl G, Rot A, and Maurer D. 1999. ‘Macrophage inflammatory protein 3alpha is involved in the constitutive trafficking of epidermal langerhans cells’, J Exp Med, 190: 1755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Eby JM, Al-Khami AA, Soloshchenko M, Kang HK, Kaur N, Naga OS, Murali A, Nishimura MI, Caroline Le Poole I, and Mehrotra S. 2014. ‘A quantitative increase in regulatory T cells controls development of vitiligo’, J Invest Dermatol, 134: 1285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretney E, Kallies A, and Nutt SL. 2013. ‘Differentiation and function of Foxp3(+) effector regulatory T cells’, Trends Immunol, 34: 74–80. [DOI] [PubMed] [Google Scholar]
- Eby JM, Kang HK, Tully ST, Bindeman WE, Peiffer DS, Chatterjee S, Mehrotra S, and Le Poole IC. 2015. ‘CCL22 to Activate Treg Migration and Suppress Depigmentation in Vitiligo’, J Invest Dermatol, 135: 1574–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzedine K, Gauthier Y, Leaute-Labreze C, Marquez S, Bouchtnei S, Jouary T, and Taieb A. 2011. ‘Segmental vitiligo associated with generalized vitiligo (mixed vitiligo): a retrospective case series of 19 patients’, J Am Acad Dermatol, 65: 965–71. [DOI] [PubMed] [Google Scholar]
- Frisoli ML, Essien K, and Harris JE. 2020. ‘Vitiligo: Mechanisms of Pathogenesis and Treatment’, Annu Rev Immunol, 38: 621–48. [DOI] [PubMed] [Google Scholar]
- Furue K, Ito T, Tsuji G, Nakahara T, and Furue M. 2020. ‘The CCL20 and CCR6 axis in psoriasis’, Scand J Immunol, 91: e12846. [DOI] [PubMed] [Google Scholar]
- Gellatly KJ, Strassner JP, Essien K, Refat MA, Murphy RL, Coffin-Schmitt A, Pandya AG, Tovar-Garza A, Frisoli ML, Fan X, Ding X, Kim EE, Abbas Z, McDonel P, Garber M, and Harris JE. 2021. ‘scRNA-seq of human vitiligo reveals complex networks of subclinical immune activation and a role for CCR5 in T(reg) function’, Sci Transl Med, 13: eabd8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri PS, Dwivedi M, Laddha NC, Begum R, and Bharti AH. 2020. ‘Altered expression of nuclear factor of activated T cells, forkhead box P3, and immune-suppressive genes in regulatory T cells of generalized vitiligo patients’, Pigment Cell Melanoma Res. [DOI] [PubMed] [Google Scholar]
- Gregg RK, Nichols L, Chen Y, Lu B, and Engelhard VH. 2010. ‘Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice’, J Immunol, 184: 1909–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, Tapia FJ, Mosca W, Perez RM, Briceno L, Henriquez JJ, and Convit J. 1987. ‘Mononuclear cell subpopulations and infiltrating lymphocytes in erythema dyschromicum perstans and vitiligo’, Histol Histopathol, 2: 277–83. [PubMed] [Google Scholar]
- Han YM, Sheng YY, Xu F, Qi SS, Liu XJ, Hu RM, Miao Y, Huang GQ, and Yang QP. 2015. ‘Imbalance of T-helper 17 and regulatory T cells in patients with alopecia areata’, J Dermatol, 42: 981–8. [DOI] [PubMed] [Google Scholar]
- Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, and Turka LA. 2012. ‘A mouse model of vitiligo with focused epidermal depigmentation requires IFN-gamma for autoreactive CD8(+) T-cell accumulation in the skin’, J Invest Dermatol, 132: 1869–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick MN, Lonsdorf AS, Shirakawa AK, Richard Lee CC, Liao F, Singh SP, Zhang HH, Grinberg A, Love PE, Hwang ST, and Farber JM. 2009. ‘CCR6 is required for IL-23-induced psoriasis-like inflammation in mice’, J Clin Invest, 119: 2317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homey B, Dieu-Nosjean MC, Wiesenborn A, Massacrier C, Pin JJ, Oldham E, Catron D, Buchanan ME, Müller A, deWaal Malefyt R, Deng G, Orozco R, Ruzicka T, Lehmann P, Lebecque S, Caux C, and Zlotnik A. 2000. ‘Up-regulation of macrophage inflammatory protein-3 alpha/CCL20 and CC chemokine receptor 6 in psoriasis’, J Immunol, 164: 6621–32. [DOI] [PubMed] [Google Scholar]
- Izcue A, Hue S, Buonocore S, Arancibia-Carcamo CV, Ahern PP, Iwakura Y, Maloy KJ, and Powrie F. 2008. ‘Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis’, Immunity, 28: 559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Andersen G, Yorgov D, Ferrara TM, Ben S, Brownson KM, Holland PJ, Birlea SA, Siebert J, Hartmann A, Lienert A, van Geel N, Lambert J, Luiten RM, Wolkerstorfer A, Wietze van der Veen JP, Bennett DC, Taieb A, Ezzedine K, Kemp EH, Gawkrodger DJ, Weetman AP, Koks S, Prans E, Kingo K, Karelson M, Wallace MR, McCormack WT, Overbeck A, Moretti S, Colucci R, Picardo M, Silverberg NB, Olsson M, Valle Y, Korobko I, Bohm M, Lim HW, Hamzavi I, Zhou L, Mi QS, Fain PR, Santorico SA, and Spritz RA. 2016. ‘Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants’, Nat Genet, 48: 1418–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, and Rudensky AY. 2007. ‘Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice’, Nat Immunol, 8: 191–7. [DOI] [PubMed] [Google Scholar]
- Kim TG, Jee H, Fuentes-Duculan J, Wu WH, Byamba D, Kim DS, Kim DY, Lew DH, Yang WI, Krueger JG, and Lee MG. 2014. ‘Dermal clusters of mature dendritic cells and T cells are associated with the CCL20/CCR6 chemokine system in chronic psoriasis’, J Invest Dermatol, 134: 1462–65. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Farber JM, and Kelsall BL. 2010. ‘CCR6 marks regulatory T cells as a colon-tropic, IL-10-producing phenotype’, J Immunol, 185: 3295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarquist J, Denman CJ, Hernandez C, Wainwright DA, Strickland FM, Overbeck A, Mehrotra S, Nishimura MI, and Le Poole IC. 2010. ‘Reduced skin homing by functional Treg in vitiligo’, Pigment Cell Melanoma Res, 23: 276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinewietfeld M, and Hafler DA. 2014. ‘Regulatory T cells in autoimmune neuroinflammation’, Immunol Rev, 259: 231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang KS, Caroli CC, Muhm A, Wernet D, Moris A, Schittek B, Knauss-Scherwitz E, Stevanovic S, Rammensee HG, and Garbe C. 2001. ‘HLA-A2 restricted, melanocyte-specific CD8(+) T lymphocytes detected in vitiligo patients are related to disease activity and are predominantly directed against MelanA/MART1’, J Invest Dermatol, 116: 891–7. [DOI] [PubMed] [Google Scholar]
- Lili Y, Yi W, Ji Y, Yue S, Weimin S, and Ming L. 2012. ‘Global activation of CD8+ cytotoxic T lymphocytes correlates with an impairment in regulatory T cells in patients with generalized vitiligo’, PLoS One, 7: e37513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCully ML, and Moser B. 2011. ‘The human cutaneous chemokine system’, Front Immunol, 2: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méry-Bossard L, Bagny K, Chaby G, Khemis A, Maccari F, Marotte H, Perrot JL, Reguiai Z, Sigal ML, Avenel-Audran M, Boyé T, Grasland A, Gillard J, Jullien D, and Toussirot E. 2017. ‘New-onset vitiligo and progression of pre-existing vitiligo during treatment with biological agents in chronic inflammatory diseases’, J Eur Acad Dermatol Venereol, 31: 181–86. [DOI] [PubMed] [Google Scholar]
- Miao X, Xu R, Fan B, Chen J, Li X, Mao W, Hua S, and Li B. 2018. ‘PD-L1 reverses depigmentation in Pmel-1 vitiligo mice by increasing the abundance of Tregs in the skin’, Sci Rep, 8: 1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg GS, Rod Dunbar P, Romero P, Chen JL, and Cerundolo V. 1998. ‘High frequency of skin-homing melanocyte-specific cytotoxic T lymphocytes in autoimmune vitiligo’, J Exp Med, 188: 1203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohl K, and Tenbrock K. 2015. ‘Regulatory T cells in systemic lupus erythematosus’, Eur J Immunol, 45: 344–55. [DOI] [PubMed] [Google Scholar]
- Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su MW, Zhou Y, Deng A, Hunter CA, Luster AD, and Harris JE. 2014. ‘CXCL10 Is Critical for the Progression and Maintenance of Depigmentation in a Mouse Model of Vitiligo’, Sci Transl Med, 6: 223ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JM, Masterjohn E, Chu R, Tedstone J, Youd ME, and Harris JE. 2017. ‘CXCR3 Depleting Antibodies Prevent and Reverse Vitiligo in Mice’, J Invest Dermatol, 137: 982–85. [DOI] [PubMed] [Google Scholar]
- Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, Yang SH, Anthony BA, Sverdrup FM, Krow-Lucal E, MacKenzie TC, Johnson DS, Meyer EH, Lohr A, Hsu A, Koo J, Liao W, Gupta R, Debbaneh MG, Butler D, Huynh M, Levin EC, Leon A, Hoffman WY, McGrath MH, Alvarado MD, Ludwig CH, Truong HA, Maurano MM, Gratz IK, Abbas AK, and Rosenblum MD. 2014. ‘Memory regulatory T cells reside in human skin’, J Clin Invest, 124: 1027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharschmidt TC, Vasquez KS, Pauli ML, Leitner EG, Chu K, Truong HA, Lowe MM, Sanchez Rodriguez R, Ali N, Laszik ZG, Sonnenburg JL, Millar SE, and Rosenblum MD. 2017. ‘Commensal Microbes and Hair Follicle Morphogenesis Coordinately Drive Treg Migration into Neonatal Skin’, Cell Host Microbe, 21: 467–77 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Lee KM, Vujkovic-Cvijin I, Ucmak D, Farahnik B, Abrouk M, Nakamura M, Zhu TH, Bhutani T, Wei M, and Liao W. 2016. ‘The role of IL-17 in vitiligo: A review’, Autoimmun Rev, 15: 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speeckaert R, Mylle S, and van Geel N. 2019. ‘IL-17A is not a treatment target in progressive vitiligo’, Pigment Cell Melanoma Res, 32: 842–47. [DOI] [PubMed] [Google Scholar]
- Spritz RA 2013. ‘Modern vitiligo genetics sheds new light on an ancient disease’, J Dermatol, 40: 310–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassner JP, Rashighi M, Ahmed Refat M, Richmond JM, and Harris JE. 2017. ‘Suction blistering the lesional skin of vitiligo patients reveals useful biomarkers of disease activity’, J Am Acad Dermatol, 76: 847–55.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terras S, Gambichler T, Moritz RK, Altmeyer P, and Lambert J. 2014. ‘Immunohistochemical analysis of FOXP3+ regulatory T cells in healthy human skin and autoimmune dermatoses’, Int J Dermatol, 53: 294–9. [DOI] [PubMed] [Google Scholar]
- Turner JE, Paust HJ, Steinmetz OM, Peters A, Riedel JH, Erhardt A, Wegscheid C, Velden J, Fehr S, Mittrucker HW, Tiegs G, Stahl RA, and Panzer U. 2010. ‘CCR6 recruits regulatory T cells and Th17 cells to the kidney in glomerulonephritis’, J Am Soc Nephrol, 21: 974–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boorn JG, Konijnenberg D, Dellemijn TA, van der Veen JP, Bos JD, Melief CJ, Vyth-Dreese FA, and Luiten RM. 2009. ‘Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients’, J Invest Dermatol, 129: 2220–32. [DOI] [PubMed] [Google Scholar]
- van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, Tigges B, Westerhof W, and Das P. 2000. ‘Local immune response in skin of generalized vitiligo patients. Destruction of melanocytes is associated with the prominent presence of CLA+ T cells at the perilesional site’, Lab Invest, 80: 1299–309. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, Martin-Orozco N, Kang HS, Ma L, Panopoulos AD, Craig S, Watowich SS, Jetten AM, Tian Q, and Dong C. 2008. ‘CCR6 Regulates the Migration of Inflammatory and Regulatory T Cells’, The Journal of Immunology, 181: 8391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: CD4 depletion exacerbates depigmentation in a mouse model of vitiligo. (a) Vitiligo scores and (b) representative images of FoxP3-GFP mice treated with PBS or CD4-depleting antibody (αCD4). (c) Gating strategy used to quantify Tregs and Teffs in FoxP3-GFP mice with vitiligo (plots shown represent the SDLNs and ear skin) and quantification of (d) Teffs and (e) Treg/Teff ratio (Treg #/Teff #) in SDLNs and skin (7 weeks post disease induction, N=10, two-tailed unpaired t test).
Figure S2: Tregs suppress depigmentation in a mouse model of vitiligo. (a) Gating strategy used to quantify Teffs in FoxP3-DTR mice with vitiligo 7 weeks post disease induction (plots shown represent the SDLNs and ear skin).
Figure S3: Treg number increases in the skin during vitiligo in a mouse model of disease. (a) Gating strategy used to quantify Tregs in FoxP3-GFP mice with or without vitiligo (plots shown represent the SDLNs and ear skin from mice with no vitiligo). (b) Quantification of Tregs in SDLN, ear and tail skin of FoxP3-GFP mice with or without vitiligo. In mice with vitiligo, Tregs were quantified at the peak of disease, 5 weeks post disease induction (N=12 Mice w/o vitiligo; N=10 vitiligo mice, two-tailed unpaired t test). (c) Quantification of the Teff/Treg ratio in tail and ear skin of mice 5- or 7-weeks post disease induction (N=19, two-tailed paired T test). (d) Linear regression analysis comparing the Teff/Treg ratio in vitiligo skin (x-axis) with the vitiligo score of the skin (y-axis) (N=38).
Figure S4: Tregs do not require CXCR3 to suppress depigmentation in a mouse model of vitiligo. (a) Quantification of Tregs in Rag−/− hosts 5 weeks after receiving WT Tregs or Tregs in FoxP3-GFP mice (WT Mice) (N=4 WT→Rag; N=12 WT mice, two-tailed unpaired t test). (b) Ear vitiligo scores and (c) representative images of Rag−/− mice receiving no Tregs, WT or CXCR3−/− Tregs. (d) Gating strategy and quantification of (e) Teffs and (f) Tregs in the SDLNs and ear skin of Rag−/− mice (5 weeks post disease induction, N>9, two-tailed unpaired t test). (g) Gating strategy and (h) quantification of WT and CXCR3−/− Tregs in the SDLNs and ear skin of Rag−/− mice in a Treg-competitive adoptive transfer model of vitiligo (5 weeks post disease induction, N=10, two-tailed unpaired t test).
Figure S5: Tregs in mouse and human skin express CCR6. (a) Representative flow plots depicting CCR6 expression on Tregs in patient skin. (b) Quantification of the proportion of Tregs expressing CCR6 in patient skin (N=5; two-tailed unpaired t test). (c) Representative flow plots depicting CCR6 expression on Tregs in the ear skin of FoxP3-GFP mice without vitiligo. (d) Quantification of the proportion of Tregs expressing CCR6 in SDLN and skin of FoxP3-GFP mice with vitiligo (5 weeks post disease induction) and FoxP3-GFP mice not induced with vitiligo (no vit) (N=8 no vitiligo; N=10 vitiligo, two-tailed unpaired t test).
Figure S6: CCL20 gene expression in mouse skin during vitiligo. RT-PCR was performed on RNA isolated from the skin of mice with vitiligo and mice not induced with vitiligo. CCL20 expression for mice with vitiligo is shown relative to CCL20 expression in control mice not induced with vitiligo (5 weeks post disease induction, N=10 ear; N=9 tail, two-tailed unpaired t test).
Figure S7: Gating strategy for T cells in a Rag−/− model of vitiligo. CCR6−/− Tregs suppress activation-induced cytokine expression by Teffs. (a) Gating strategy used to quantify Teffs and Tregs in Rag−/− hosts (plots shown represent the ear skin from Rag−/− hosts with WT Tregs). (b) Gating strategy used to quantify WT Tregs and CCR6−/− Tregs (KO) in Rag−/− hosts from competitive transfer experiments. (c) Quantification of IFNγ or TNFα expression by αCD3/αCD28 activated Teffs cultured in the presence of no Tregs, WT Tregs or CCR6−/− Tregs (N=18 No Treg; N=17 WT Treg; N=17 CCR6−/− Treg, two-tailed unpaired T test).
Data Availability Statement
No datasets were generated or analyzed during the current study