

Abstract
Technologies for the labeling, detection, and manipulation of biomolecules have drastically improved our understanding of cell biology. As the myriad of functional roles for RNA in the cell are increasingly recognized, such tools to enable further investigation of RNA are the subject of much interest. RNA-TAG is an enzymatic method for site-specific, covalent labeling of RNA. This methodology makes use of a bacterial tRNA modifying enzyme, tRNA guanine transglycosylase, to incorporate modified substrate analogs into a target RNA, resulting in highly efficient and site-specific RNA labeling. In this chapter, we introduce the underlying principles of the RNA labeling reaction, discuss various applications of RNA-TAG, and present protocols for labeling specific RNA transcripts using this system.
1. Introduction
Once overlooked as a transient messenger between DNA and protein, the role of RNA as an important functional molecule in cell biology has been increasingly recognized. Cellular functions not only rely on the production of canonical ribosomal RNA (rRNA), transfer RNA (tRNA) and messenger RNA(mRNA) molecules, but also functional small nuclear RNAs (snRNAs), microRNAs (miRNAs), and long noncoding RNAs (lncRNAs) (Cech & Steitz, 2014; Cooper, Wan, & Dreyfuss, 2009). Furthermore, the complexity of mammalian mRNAs has emerged as an important aspect of gene regulation and expression, with complex networks of RNA-protein interactions controlling mRNA stability, localization, and expression (Singh, Pratt, Yeo, & Moore, 2015). This growing interest in RNA biology has spurred the demand for new tools to enable further investigation of RNA structure and function.
Functional characterization of biomolecules through their direct conjugation to small molecule probes has proven to be an extremely effective strategy. For example, various methodologies have been developed for protein labeling, enabling advances in our understanding of protein structure and function. These strategies include the use of amine-reactive handles as well as enzymatic labeling approaches, such as SNAP-TAG and other self-labeling proteins, among others (Chen, Howarth, Lin, & Ting, 2005; Gautier et al., 2008; Ho & Tirrell, 2016). While numerous robust tools exist for protein labeling, few tools exist for the site-specific covalent modification of RNA. A commonly used approach employs T4 ligase for 3′ end-labeling with various fluorophores or biotin (Richardson & Gumport, 1983). Recently, other enzymatic methods based on RNA modifying enzymes have been developed. For example, the methyltransferase Tgs has been engineered to enable labeling of the 5′ cap of mRNAs (Schulz, Holstein, & Rentmeister, 2013). Another approach used an archaeal tRNA modifying enzyme to enable labeling of tRNA or RNAs encoding a complete tRNA sequence with an amine-reactive handle (Li et al., 2015). In this chapter, we discuss the use of a bacterial RNA modifying enzyme to label a small, encodable hairpin structure within an RNA of interest using a method called RNA-transglycosylation at guanosine (RNA-TAG). The advantages of this approach include its specific recognition of a unique small molecule nucleobase analog, the ability to directly add functional groups without the need for secondary click reactions, as well as its recognition of a short, 17-nucleotide stem loop that can be encoded at any site within the RNA of interest.
1.1. RNA labeling with RNA-TAG
RNA transglycosylation at guanosine (RNA-TAG) is a technology for RNA labeling that leverages a bacterial tRNA guanine transglycosylase (TGT) enzyme to covalently label an RNA of interest with a small molecule probe (Alexander, Busby, Cole, Zhou, & Devaraj, 2015). In order to achieve this labeling, derivatives of the bacterial small molecule substrate, preQ1, are utilized. A TGT recognition element, consisting of a short (≥17 nucleotide) hairpin that mimics the anticodon stem loop of its native tRNA substrates, is encoded into the RNA of interest in order to facilitate TGT labeling (Fig. 1). In the sections below, substrate recognition by TGT will be discussed. While much crystallographic evidence has been collected using Zymomonas mobilis (Z. mobilis) TGT, E. coli TGT numbering will be used, as this enzyme has been, to date, used more extensively with RNA-TAG.
Fig. 1.
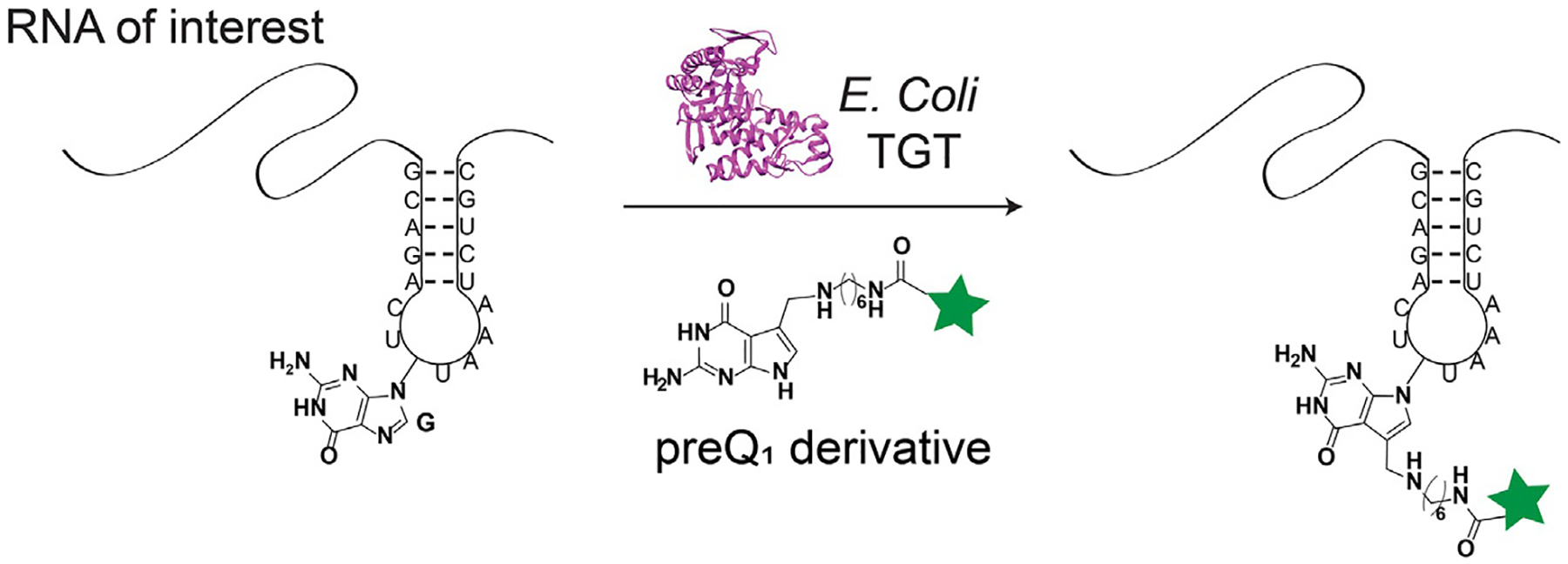
Enzymatic RNA labeling with RNA-TAG. Using bacterial TGT, an RNA of interest containing a TGT recognition hairpin is labeled with a preQ1 derivative.
1.2. RNA modification by tRNA guanine transglycosylase
The tRNA guanine transglycosylases (TGTs) are a well-characterized class of enzymes that are found in archaea, eubacteria and eukaryotes. While most RNA post-transcriptional modifications occur through chemical modification of an existing nucleoside, TGTs are extraordinary in their catalysis of transglycosylation reactions. Bacterial and eukaryotic TGTs recognize and modify four tRNAs, namely tRNAAsn, tRNAAsp, tRNATyr, and tRNAHis, by exchanging a guanine at the wobble position of the anticodon stemloop with a 7-deazaguanine derivative. Bacterial TGTs introduce preQ1 into the tRNA substrate, which is then enzymatically transformed to the hypermodified base queuine. In contrast, eukaryotic TGTs modify their cognate tRNAs directly with queuine that has been salvaged from the environment (Fig. 2). Despite the conservation of queuine modification across kingdoms, its functional role is not fully understood. Previous studies have established potential contributions to invasion and proliferation in bacteria, ribosomal frameshifting in viruses, and roles in development, proliferation, metabolism, cancer, and tyrosine biosynthesis in eukaryotes (Fergus, Barnes, Alqasem, & Kelly, 2015).
Fig. 2.
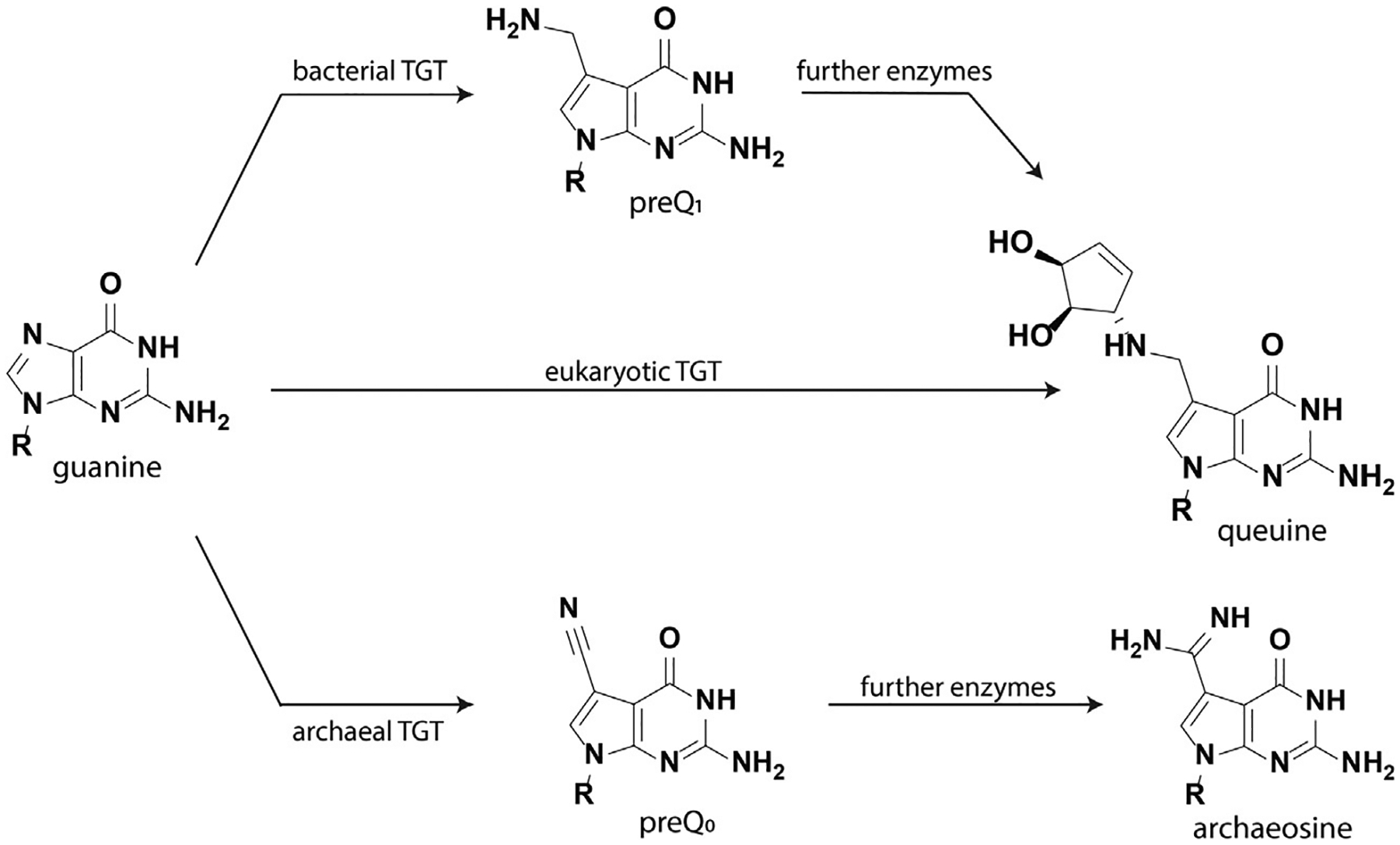
Nucleobase structures recognized by TGT enzymes in bacteria, eukaryotes, and archaea.
TGTs are also observed in archaea, though their function differs significantly from bacterial and eukaryotic TGTs. Sharing approximately 20–25% of sequence identity with bacterial TGTs, archaeal TGTs recognize and modify position 15, a site within the D-arm in the majority of tRNAs (Stengl, Reuter, & Klebe, 2005). An alternative nucleobase, preQ0, is inserted, which is further modified to form archaeosine; it is thought that this modification stabilizes tRNA structure at high temperatures (Gregson et al., 1993).
1.3. RNA recognition by bacterial TGT
Previous biochemical studies of E. coli TGT have established that the entire tRNA structure is not required for recognition of an RNA substrate. Curnow and coworkers initially identified that ECY-A1, a truncated 17 nucleotide RNA hairpin derived from the anticodon stem loop of tRNATyr, was recognized by E. coli TGT (Curnow, Kung, Koch, & Garcia, 1993). Extension of this RNA hairpin by 4 base pairs (ECYMH) was found to improve reaction kinetics at 37 °C, most likely due to increased thermal stability (Curnow & Garcia, 1995). RNA mutational studies of truncated hairpins have demonstrated the importance of the U33G34U35 loop sequence (Table 1) (Curnow & Garcia, 1995; Nakanishi et al., 1994).
Table 1.
Kinetics values of select RNA substrates as reported by Curnow and Garcia (1995).
| Substrate name (Curnow & Garcia, 1995) | Stem loop sequence | Km, μM | Vmax, μMs−1 mg−1 | Vmax/Km, s−1 mg−1 |
|---|---|---|---|---|
| ECY2 (unmodified tRNATyr) | …GCAGACUGUAAAUCUGC… | 2.0 | 3.0 | 1.5 |
| ECY-A1 | GCAGACUGUAAAUCUGC | 13.3 | 0.48 | 0.04 |
| ECYMH | GGGAGCAGACUGUAAAUCUGCUCCC | 6.9 | 2.2 | 0.3 |
| SCDMH | GGCGGCGCUUGUCGCGUGCCGCC | 11.4 | 2.7 | 0.24 |
| SCDMH-U33C | GGCGGCGCUCGUCGCGUGCCGCC | 22.4 | 0.06 | 0.003 |
| SCDMH-G34A | GGCGGCGCUUAUCGCGUGCCGCC | N.D. | N.D. | N.D. |
| SCDMH-U35C | GGCGGCGCUUGCCGCGUGCCGCC | N.D. | N.D. | N.D. |
N.D. = no detectable activity.
Crystallographic studies of bacterial TGT derived from Z. mobilis in complex with a hairpin RNA substrate have also provided insight into how TGT recognizes its RNA substrate (Xie, Liu, & Huang, 2003). In agreement with the biochemical data, conserved interactions are observed with U35 and G34 nucleobases. With respect to U35, Arg286 donates two hydrogen bonds to O2, and Arg289 donates a hydrogen bond to O4. Further interactions such as a cation-pi interaction with Lys52 and hydro-phobic interactions with Val282 strengthen recognition of U35. U33 also makes hydrogen bonding interactions with Lys264 and Asp267. However, these residues are not strictly conserved, and it was hypothesized that instead, U33 plays an important role in the formation of an unusual zig-zag conformation in the loop region. Because there are few base-specific interactions between TGT and its RNA substrate, this conformational change is necessary for optimal binding of TGT through surface and charge complementarity. Crystallographic evidence has also supported the functional significance of a TGT homodimer, where the first unit is catalytic and the second unit plays a role in recognition and proper orientation of the bound tRNA (Stengl et al., 2007).
In order to target an RNA of interest for TGT-mediated labeling, it is necessary to encode an RNA substrate into that RNA of interest. Due to its short (17 nucleotide) length, the ECY-A1 hairpin has been most commonly used; an unstructured spacer is typically used on either side of the hairpin to promote proper folding of the target hairpin (Alexander et al., 2015; Ehret, Zhou, Alexander, Zhang, & Devaraj, 2018; Zhang, Zhou, Busby, Alexander, & Devaraj, 2018; Zhou, Alexander, & Devaraj, 2017). The extended hairpin, ECYMH, has also been successfully utilized, and may be preferred to increase labeling efficiency. Based on the previous literature, it is likely that other RNA hairpin structures, containing the “UGU” sequence and capable of assuming the appropriate binding conformation, may also serve as potential substrates.
1.4. Nucleobase recognition and catalytic mechanism
More than 170 RNA modifications have been identified in nature, with most modifications carried out through enzymatic functionalization of existing nucleobases (Frye, Haranda, Behm, & He, 2018). TGTs are therefore unusual in their removal and replacement of a nucleobase with a different, functionalized, nucleobase, making them an excellent candidate for catalyzing RNA labeling with synthetic analogs. The mechanism of catalysis by bacterial TGT has been studied extensively, and has been shown to follow a ping-pong mechanism (Fig. 3) (Goodenough-Lashua & Garcia, 2003). A highly conserved catalytic aspartate residue (Asp264) acts as a nucleophile to break the glycosidic linkage of the guanosine, forming a TGT-RNA covalent intermediate (Xie et al., 2003). Exchange of the excised guanine in the binding pocket for preQ1 then allows for nucleophilic attack of the ribose by N9 of preQ1, reforming a glycosidic bond. While TGT can catalyze the reversible exchange of guanine, insertion of preQ1 is irreversible (Farkas, Jacobson, & Katze, 1984).
Fig. 3.
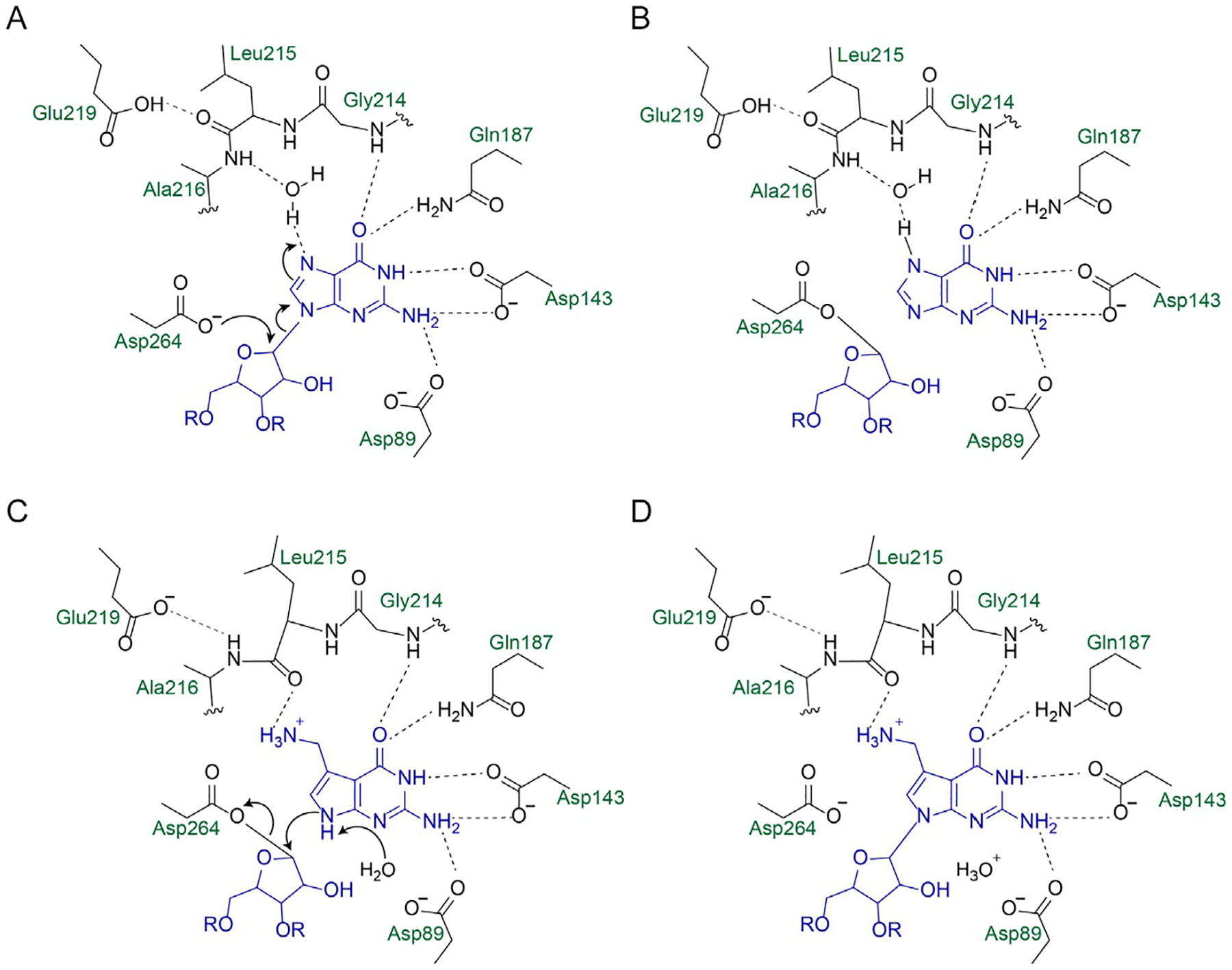
Catalytic mechanism and binding interactions of bacterial TGT with guanine and preQ1. E. coli numbering is used. (A) Asp264 acts as a nucleophile to attack the anomeric carbon of ribose 34, breaking the N–C glycosidic linkage. (B) A covalent TGT-RNA covalent intermediate is formed. (C) preQ1 replaces guanine in the binding pocket, assisted by a conformational change of the Leu215/Ala216 peptide bond. (D) N9 of preQ1 acts as a nucleophile to reform a glycosidic bond with ribose 34.
The binding pocket of TGT responsible for nucleobase recognition has been studied extensively though biochemical and crystallographic studies. Asp89 is a strictly conserved residue involved in recognition of guanine through hydrogen bonding at N2; it is also thought to play a role as a general base in catalysis (Xie et al., 2003). Asp143 plays a key role in recognizing the Watson-Crick face of guanine and preQ1 through hydrogen bonding interactions to N1 and N2 (Todorov & Garcia, 2006; Todorov, Tan, Nonekowski, Garcia, & Carlson, 2005). Additional interactions with Ser99, Gln187, and the backbone of Gly214 also contribute to binding of guanine (Stengl et al., 2005; Xie et al., 2003). PreQ1 recognition is mediated through hydrogen bonding interactions of the carbonyl group in the Leu215/Ala216 peptide bond with the aminomethyl moiety of preQ1, which is most likely in a protonated form at physiological pH (Hoops, Park, Garcia, & Townsend, 1996). It has been hypothesized that this amide bond changes conformation, to present either the NH or carbonyl functionality for hydrogen bonding, to mediate alternative binding of guanine and preQ1 (Stengl et al., 2005). This conformational change, mediated by Glu219 in bacterial TGT, can explain the differential substrate specificities of archaeal TGTs. In crystal structures of the archaeal TGT from Pyrococcus horikoshii, this peptide bond is only observed in the NH-presenting form, consistent with its recognition of preQ0 instead of preQ1 (Ishitani et al., 2002, 2003). Recent biochemical and structural studies of human TGT have also uncovered the basis for the preferential recognition of queuine in eukaryotic systems. The substitution of Val217 for a glycine in human TGT enlarges the binding pocket, allowing for the binding of the cyclopentadiol moiety of queuine (Johannsson, Neumann, & Ficner, 2018). Based on biochemical evidence, it also seems likely that Cys145, which is replaced with a valine in human TGT, plays a role in the differential nucleobase recognition (Chen et al., 2011).
Structure-activity studies examining the recognition of preQ1 have shown that there is a strong impact of the aminomethyl group on bacterial TGT binding affinity (Hoops, Townsend, & Garcia, 1995). While native substrates lacking the aminomethyl substituent, such as guanine and preQ0, are recognized with only a slightly reduced binding affinity (5–6-fold), other 7-deazaguanine derivatives have dramatically reduced binding (Table 2). This evidence suggests that the exocyclic amine plays a large role in bacterial TGT recognition of preQ1, which is further supported by the crystallographic evidence of hydrogen bonding of this group with the peptide backbone (Xie et al., 2003). Because the pKa of preQ1’s exocyclic amine is in the range of 10 (Hoops et al., 1996), it is likely that this amine is protonated at physiological pH, and it has been postulated that charge-assisted binding is also possible (Stengl & Klebe, 2007). Thus, successful preQ1 substrates for use with RNA-TAG have all been modified through alkylation of this exocyclic amine. Both PEG and alkyl linkers have been utilized to append various probes to the preQ1 scaffold, with the use of an alkyl linker improving both binding affinity and enzyme turnover significantly (Zhou et al., 2017). A variety of probes, including BODIPY, Cy5, Cy7, thiazole orange, biotin, photocleavable groups, and tetrazine, have been successfully incorporated into RNA by appending them to preQ1 in this manner.
Table 2.
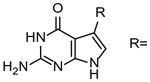
Kinetic parameters of various nucleobase substrates.
| Compound | Km, μM | kcata (×10−3) s−1 | kcat/Kma (×10−3) s−1 μM−1 | Ref. |
|---|---|---|---|---|
| Guanine | 2.2 | 61a | 28 a | Hoops et al. (1995) |
|
||||
| −CH2NH2 (preQ1) | 0.39 | 45a | 115a | Hoops et al. (1995) |
| −CN (preQ0) | 2.35 | 72a | 31 a | Hoops et al. (1995) |
| −CH2OH | 23.0 | 45a | 2.0a | Hoops et al. (1995) |
| −H | 172 | 45a | 0.3a | Hoops et al. (1995) |
| −CH3 | 255 | 45a | 0.2a | Hoops et al. (1995) |
| −CH2OCH3 | 57 | 47a | 0.8a | Hoops et al. (1995) |
| −CH2N(CH3)2 | 75 | 43a | 0.6a | Hoops et al. (1995) |
| −CONH2 | 26 | 69a | 2.7a | Hoops et al. (1995) |
| −CO2CH3 | 87 | 73a | 0.8a | Hoops et al. (1995) |
| −COCH3 | 26 | 73a | 2.8a | Hoops et al. (1995) |
| −CHO | 22 | 73a | 3.3a | Hoops et al. (1995) |
| −CO2H | 126 | 21a | 0.2a | Hoops et al. (1995) |
| −(PEG)3-thiazole orange | 9.8 | 1.6 | 0.2 | Alexander et al. (2015) |
| −C6H12-thiazole orange derivative | 1.6 | 26.7 | 16.7 | Zhou et al. (2017) |
kcat were calculated from reported Vmax values using the reported enzyme concentration (50 nM), and reaction volume (400 μL) (Hoops et al., 1995).
1.5. Key differences in TGT systems for applications in mammalian models
RNA contributes greatly to the complexity of eukaryotic systems, with various controls of gene expression including alternative splicing, polyadenylation, protein binding sites, and microRNAs; therefore, many potential applications of RNA-TAG include its use in the context of mammalian cells. Accordingly, it is important to consider the ways in which bacterial and eukaryotic TGT modifications compare in order to understand where its use may be appropriate. As previously discussed, the small molecule selectivity for bacterial and mammalian TGTs differ; preQ1 is the preferred substrate for bacterial TGT, while queuine is the preferred substrate for mammalian TGT (Table 3) (Chen et al., 2011). Furthermore, while it has been shown that a minimal stem loop substrate is accepted by bacterial TGTs, the mammalian TGT requires the full tRNA structure for recognition (Grosjean, Edqvist, Stråby, & Giegé, 1996).
Table 3.
Kinetic parameters for E. coli and human TGTs with heterocyclic substrates.
| Km, μM | kcat (×10−3) s−1 | kcat/Km (×10−3)s−1 μM−1 | |
|---|---|---|---|
| Guanine | |||
| E. coli | 0.35 | 6.29 | 18.0 |
| Human | 0.41 | 5.86 | 14.2 |
| PreQ1 | |||
| E. coli | 0.05 | 9.57 | 191 |
| Human | 132 | 8.23 | 0.062 |
| Queuine | |||
| E. coli | N.D. | N.D. | N.D. |
| Human | 0.26 | 8.22 | 31.6 |
Data from Chen, Y. C., Brooks, A. F., Goodenough-Lashua, D. M., Kittendorf, J. D., Showalter, H. D., & Garcia, G. A. (2011). Evolution of eukaryal tRNA-guanine transglycosylase: Insight gained from the heterocyclic substrate recognition by the wild-type and mutant human and Escherichia coli tRNA-guanine transglycosylases. Nucleic Acids Research, 39, 2834–2844. https://doi.org/10.1093/nar/gkq1188.
Importantly, preQ1 and queuine modification is irreversible, so queuine-modified tRNA present in mammalian systems would not be expected to be a substrate for further modification by TGTs (Biela et al., 2013; Farkas et al., 1984). Mitochondrial tRNAs have also been found to be queuine-modified (Suzuki & Suzuki, 2014). However, in certain cancer cells, it has been shown that tRNA is under-modified by queuine; therefore optimization may be necessary for use in these cells (Pathak, Jaiswal, & Vinayak, 2005). Furthermore, queuine modification is not observed in yeast tRNAs (Walden, Reyniers, Hiatt, & Farkas, 1982). Studies by our lab have shown that, in E. coli cells that have a deletion of a key preQ1 biosynthetic enzyme (ΔQueC), treatment with preQ1-biotin allows for the labeling and enrichment of tRNA, presumably through the expression of native E. coli TGT (data not shown). Accordingly, there may be potential applications of this technology in the study of tRNA and tRNA modifications in some systems.
1.6. Applications
RNA-TAG has been utilized in a variety of contexts, demonstrating its utility in both the study and manipulation of RNA in biological systems. These applications include fixed cell RNA imaging, affinity purification of RNA, control of translation in mammalian cells, and labeling of modified RNA (modRNA) for therapeutic applications.
The localization of RNA within cells is a key aspect of gene expression that is highly regulated in complex organisms, and the misregulation of RNA localization has been implicated in various neurological and muscle disorders (Chin & Lécuyer, 2017). Fixed cell imaging of RNA is a commonly utilized tool, especially with the popular fluorescence in situ hybridization (FISH) technique. Using RNA-TAG, the direct labeling and imaging of an RNA of interest has been demonstrated using a variety of fluorophores, including Cy7, BODIPY, and thiazole orange derivatives (Alexander et al., 2015; Zhou et al., 2017). In these studies, the targeted RNA (mCherry) was expressed with the ECY-A1 sequence, or “TAG,” appended in the 3’ UTR. This mCherry-TAG transcript could be successfully labeled by treating formaldehyde-fixed CHO cells with E. coli TGT enzyme and the appropriate preQ1-fluorophore derivative. Importantly, controls that lacked the mCherry-TAG transcript, or lacked treatment with E. coli TGT, showed much lower levels of fluorescence. These results indicate that the desired mCherry-TAG transcript was labeled selectively by E. coli TGT, even in the presence of other mammalian RNA species, such as tRNA (Alexander et al., 2015). This technique was further advanced by the development of fluorogenic preQ1-thiazole orange derivatives, in which the bulky substituents reduced the intercalating abilities of thiazole orange such that fluorescence was increased upon covalent incorporation by TGT, thus allowing wash-free imaging (Zhou et al., 2017). In addition, affinity purification of RNA has been demonstrated by labeling an RNA of interest with preQ1-biotin, also in the presence of total RNA from CHO cells (Alexander et al., 2015).
RNA-TAG has also been applied to the manipulation of RNA within mammalian cells, specifically through light-activated control of translation (Zhang et al., 2018). PreQ1 derivatives bearing a coumarin-based photocleavable linker were used to label TGT recognition elements in the 5′ UTR of a capped, polyadenylated RNA of interest. This labeled, mature mRNA was transfected into HeLa cells, and reduced protein production was observed in RNAs labeled with the photocleavable group. Furthermore photoirradiation, which is expected to cleave the photocleavable group, allowed for the activation of translation in these cells.
This method has also been shown to tolerate modified RNA (modRNA) transcripts. Several RNA modifications, such as 5-methylcytosine (5-mC) and pseudouridine (Ψ), have been shown to increase RNA stability, decrease immune response, and increase the translation capacity of RNAs (Karikó et al., 2008; Uchida, Kataoka, & Itaka, 2015); these attributes make modRNA a useful tool for RNA gene therapy approaches. Using RNA-TAG, RNA transcripts with these modified nucleobases could be labeled with high efficiencies (Ehret et al., 2018). Furthermore, the use of preQ1-tetrazine, which could then be used in subsequent tetrazine-mediated click chemistry, was also demonstrated (Ehret et al., 2018).
The broad scope of applications possible with RNA-TAG demonstrates the breadth and robustness of this labeling technique. A variety of probes, including fluorophores, biotin, click handles, and even photocleavable functionalities can be incorporated into an RNA of interest with high levels of efficiency. The observed selectivity and high efficiencies of this reaction indicate the potential for this technique to be utilized as a tool in various areas of RNA biology.
2. Method
2.1. Definition
Enzymatic covalent labeling of RNA with RNA transglycosylation at guanosine (RNA-TAG).
2.2. Rationale
Few methods exist for the enzymatic, site-specific labeling of RNA. While other approaches allow labeling at the 5′ or 3′ ends of RNA, or labeling of large encodable structures, RNA-TAG enables the labeling of an RNA transcript bearing a short stem loop sequence. Furthermore, RNA-TAG utilizes a unique nucleobase substrate, preQ1, to which probes can be directly attached for enzymatic incorporation into the RNA of interest. The protocols below describe the various biochemical methods needed to execute labeling of a transcript using this technique, including expression and purification of the TGT enzyme, cloning of a plasmid to transcribe an RNA with an appended TAG sequence, and carrying out the labeling reaction.
2.3. Materials, equipment and reagents
Expression and purification of E. coli TGT
Ultra-pure water, nuclease free
Bacterial expression plasmid encoding E. coli TGT (TGT-His, Addgene ID 138201)
BL21 (DE3) competent cells (New England Biolabs)
Phenylmethylsulfonyl fluoride (PMSF)
Tris base (Tris(hydroxymethyl)aminomethane)
Tris hydrochloride
NaCl
HEPES sodium salt
HEPES
Dithiothreitol (DTT)
500 mM EDTA, pH 8.0, nuclease free (Invitrogen)
HisPur™ Ni-NTA Spin Purification Kit, 0.2 mL (Thermo Scientific)
Dialysis cassettes (Slide-a-lyzer MINI, 20 kDa, Thermo Scientific)
Incubator-shaker
Refrigerated microcentrifuge
Probe sonicator
Construction of an RNA expression construct with a TGT recognition element
Ultra-pure water, nuclease free
Plasmid for transcription of RNA with TAG recognition element (pcDNA3.1-(empty)-TAG, Addgene ID 138209)
Isolated total RNA, human (alternatively, Universal Human Reference RNA, Agilent)
- Oligos for RT-PCR amplification of the desired RNA sequence, with flanking restriction sites (examples given in text)
- dNTP mix, 10 mM each (Invitrogen)
- Maxima reverse transcriptase (Thermo Scientific)
- RNAse inhibitor, murine (New England Biolabs)
- Q5 high fidelity 2 × master mix (New England Biolabs)
- PCR cleanup kit (QIAquick PCR cleanup, Qiagen)
- NheI-HF (New England Biolabs)
- BamHI-HF (New England Biolabs)
- Antarctic phosphatase (New England Biolabs)
- T4 ligase (New England Biolabs)
- DH5α competent cells (New England Biolabs)
- Thermocycler
RNA labeling with RNA-TAG
Ultra-pure water, RNAse free
Labeled preQ1 derivative (discussed in Section 2.5)
RNA transcript bearing TGT recognition element
Expressed E. coli TGT enzyme
HEPES sodium salt
HEPES
Dithiothreitol (DTT)
Magnesium chloride, hexahydrate
RNAse inhibitor, murine
100% ethanol
3 M sodium acetate
Water bath, heat block, or thermocycler for 37 °C incubation
Refrigerated microcentrifuge
2.4. Protocols
2.4.1. Expression and purification of E. coli TGT
2.4.1.1. Overview
In this protocol, a method for expression and purification of His-tagged E. coli TGT is briefly described. While this method has been used several times for the successful purification of E. coli TGT, modifications of this general method may be acceptable, including the use of alternative lysis and purification buffers, or alternative lysis techniques. For best results, the use of nuclease free reagents and water is critical, and testing of the resulting purified protein for nuclease contamination is recommended. Purified E. coli TGT should be stored in aliquots at −80 °C, as storage at −20 °C, even as a glycerol stock, is not recommended.
Transform BL21 (DE3) competent cells with the TGT-His plasmid according to the manufacturer’s protocol. Protein can be expressed according to standard laboratory procedures; typically, a culture volume of 200 mL is used, and induction is performed at an ~OD of 0.5–0.8 with the addition of IPTG to 1 mM. Expression of TGT is typically carried out for 4 h at 37 °C.
Harvest the bacterial cells by centrifugation at 10,000 × g for 15 min at 4 °C. Cell pellets can be stored at −20 °C if desired.
- Prepare the following buffers and solutions for protein purification. Typically, commercial nuclease-free water is used, although MilliQ water can be used if it is tests negative for RNAses.
- 100 mM PMSF
- Dissolve 174 mg of PMSF in isopropanol to 10 mL. Solution can be stored at −20 °C.
- 2 M imidazole (included in HisPur™ Ni-NTA Spin Purification Kit).
- 2 × TGT lysis buffer, 50 mL (40 mM Tris pH 7.9, 1 M NaCl)
- 93.7 mg Tris base
- 193.2 mg Tris HCl
-
2.92 g NaClAdjust pH to 7.9, total volume of 50 mL.
- 10 × TGT storage buffer, 50 mL (250 mM HEPES pH 7.3, 20 mM DTT, 10 mM EDTA)
- 1.26 g HEPES sodium salt
- 1.83 g HEPES
- 154.2 mg DTT
-
1 mL 500 mM EDTAAdjust pH to 7.3, total volume of 50 mL. Buffer should be stored at 4 °C for short-term storage.
- Lysis and purification buffers:
Buffer Description Recipe 1 × lysis buffer, 10 mL 20mM Tris pH 7.9, 500mM NaCl, 100μM PMSF, 10mM imidazole 5mL 2 × lysis buffer
10μL 100mM PMSF
50μL 2M imidazole
Adjust to 10mL with H2O1 × wash buffer, 10 mL 20mM Tris pH 7.9, 500mM NaCl, 100μM PMSF, 25mM imidazole 5mL 2 × lysis buffer
10μL 100mM PMSF
125μL 2M imidazole
Adjust to 10mL with H2O1 × dilution buffer, 10 mL 20mM Tris pH 7.9, 500mM NaCl, 100μM PMSF, 350 mM imidazole 5mL 2 × lysis buffer
10μL 100mM PMSF
1.75 mL 2M imidazole
Adjust to 10mL with H2O1 × TGT storage buffer, 500 mL 25mM HEPES, pH 7.3, 2 mM DTT, 1 mM EDTA, 100μM PMSF 50mL 10 × TGT storage buffer
500μL 100 mM PMSF
Adjust to 500 mL with H2O
Resuspend bacterial pellet in 1 × lysis buffer. A volume of 2 mL is typically sufficient when 200 mL of culture is used.
Lyse cells with a probe sonicator using standard conditions. For example, a Branson Sonifier 450 can be used for 4–5 cycles of 30 s each, with 2 min on ice between cycles, with the output control set to 5 (~20 W output) and a duty cycle of 30%. Note: Lysis with a French press also yielded good results (Alexander et al., 2015).
Centrifuge lysate at 10,000 × g for 30 min at 4 °C to remove cellular debris.
Purify TGT-His protein using the buffers described above and the HisPur™ Ni-NTA Spin Purification Kit, according to manufacturer’s instructions.
Verify purification of TGT-His by SDS-PAGE analysis of fractions. A strong band at a molecular weight of ~43 kDa is expected in the elution fractions.
Combine elution fractions containing purified protein; perform dialysis or buffer exchange into 1 × TGT storage buffer.
The concentration of the resulting purified protein can be measured using standard laboratory techniques, such as a BCA assay compatible with DTT (i.e. Pierce™ BCA Protein Assay Kit—Reducing Agent Compatible, Thermo Scientific).
-
Purified protein should be stored as single-use aliquots at −80 °C. E. coli TGT has been observed to have reduced stability at −20 °C, even if stored as a glycerol stock.
Tip: Purified TGT-His often has significant levels of RNAse contamination, most likely due to residual E. coli RNAses. For best results, a second Ni-NTA purification should be performed after buffer exchange, as this tends to reduce the RNAse contamination significantly. Commercial RNAse detection kits (i.e. RNaseAlert™, Invitrogen) can be used to quantify RNAse contamination.
2.4.2. Construction of an RNA expression construct with a TGT recognition element
2.4.2.1. Overview
In this protocol, a method is presented as an example to demonstrate how a TGT recognition element can be incorporated into an RNA of interest using the pcDNA3.1-(empty)-TAG plasmid, Addgene ID 138209. The method described here appends a 25-nucleotide TGT recognition element, ECYMH, to human β-actin mRNA. This method relies on restriction cloning to insert the sequence for the RNA of interest into a plasmid containing the TGT recognition element, as well as the elements necessary for transcription by T7 RNA polymerase. Depending on the desired application, other cloning methods may be preferred; for example, a short TGT recognition sequence (ECY-A1, discussed above) can be inserted into an existing vector using site directed mutagenesis.
| Primer name | Primer sequence (restriction site in bold, gene-specific sequence capitalized) |
|---|---|
| NheI-B-actin-fwd | aagctggctagcACCGCCGAGACCGCG |
| BamHI-B-actin-rev | gttagaggatccCAACTGGTCTCAAGTCAGTGTACAGG |
| B-actin RT | AAGGTGTGCACTTTTATTCAACT |
Obtain RT-PCR primers corresponding to the RNA sequence desired. Here, a gene-specific RT primer was designed to reverse transcribe the human β-actin sequence, and PCR primers were designed to amplify the desired sequence and add NheI and BamHI restriction sites, to facilitate cloning into the pcDNA3.1-(empty)-TAG vector.
- Set up a reverse transcription reaction to synthesize cDNA corresponding to human β-actin mRNA from isolated or purchased total RNA.
Reagent Volume (μL) Ultra-pure water 10.5 Isolated total human RNA, 10ng/μL 1 dNTPs, 10 mM 1 B-actin RT primer, 7.5 mM 2 5 × RT buffer 4 Murine RNAse Inhibitor (40U/μL) 0.5 Maxima RT (200U/μL) 1 Total volume 20 - Incubate reverse transcription reaction on a thermocycler with the following program:
- 50 °C, 30 min
- 85 °C, 5 min
- 4 °C, hold.
- Set up a PCR reaction to amplify the cDNA gene and append restriction sites:
Reagent Volume (μL) Ultra-pure water 19 Q5® high fidelity 2 × master mix 25 NheI-B-actin-fwd, 10 mM 2.5 BamHI-B-actin-rev, 10 mM 2.5 Reverse transcription product 1 Total volume 50 - Incubate PCR reaction on a thermocycler with the following program:
- 98 °C, 30 s
- 35 cycles of:
- 98 °C, 10 s
- 70 °C, 30 s (annealing temperature, Ta, corresponding to the β-actin primers used)
- 72 °C, 1 min
- 72 °C, 2 min
- 4 °C, hold.
Purify PCR product using a PCR cleanup kit (i.e. QIAquick PCR cleanup).
- Set up parallel restriction digests of both the β-actin PCR amplicon and pcDNA3.1-(empty)-TAG vector:
Reagent Amount DNA (PCR amplicon OR vector) 2μg 10 × CutSmart buffer 10μL NheI-HF 2.5μL BamHI-HF 2.5μL Ultra-pure water To 100μL Incubate restriction digest reactions at 37 °C for 1 h.
To restriction digest reaction containing the vector, add 11.5 μL 10 × antarctic phosphatase buffer and 5 μL antarctic phosphatase. Incubate an additional 30 min at 37 °C.
Purify the digested DNA samples using a PCR cleanup kit (i.e. QIAquick PCR cleanup).
- Set up a ligation reaction:
Reagent Amount 10 × T4 ligase buffer 2μg Vector (~5.5 kb) 50ng (15fmol) Insert (~1.8kb) 50ng (45fmol) T4 ligase 1μL Ultra-pure water To 20μL Incubate ligation reaction at room temperature for 10 min.
Transform 1–5 μL of ligation reaction into DH5α competent cells, according to manufacturer’s protocol.
-
Select using ampicillin and sequence the resulting plasmid to verify gene sequence. The following sequencing primers can be used for pcDNA3.1-(empty)-TAG:
CMV-Forward CGC AAA TGG GCG GTA GGC GTG BGH-Reverse TAG AAG GCA CAG TCG AGG Tip: Run-off transcription can be carried out according to standard procedures using this plasmid backbone. Restriction digestion with XbaI is typically performed, followed by a phenol-chloroform extraction and ethanol precipitation. Plasmids purified with typical Miniprep and Maxiprep procedures are often contaminated with RNAse A; therefore, stringent purification is necessary before use in transcription reactions.
2.4.3. RNA labeling with RNA-TAG
2.4.3.1. Overview
In this protocol, a general method is presented for the labeling of an RNA transcript bearing a TGT recognition element. The use of the extended TGT recognition element, ECYMH, and preQ1 derivatives functionalized with alkyl linkers are preferred to achieve high labeling efficiencies (>80%).
- Prepare the following buffers and solutions, which can be stored as aliquots at −20 °C:
- 100 mM DTT, 1 mL
- Dissolve 15.42 mg DTT with 1 mL of nuclease-free water.
- 10 × TGT reaction buffer, 10 mL (1 M HEPES, pH 7.3, 200 mM MgCl2)
- 1.01 g HEPES sodium salt
- 1.46 g HEPES
-
406.6 mg MgCl2 6H2OAdjust pH to 7.3, total volume of 10 mL.
- Dissolve the purified preQ1 derivative in nuclease-free water to make a 1000 μM stock solution; DMSO can also be utilized for probes with low solubility. Probe concentration can be verified by measuring absorbance, if desired. Stock solutions of preQ1 derivatives can be stored at −20 °C for up to 1 year with good results.
-
Calculate the molarity of your RNA transcript, in μM, using the following equation:For example, β-actin-TAG has a molecular weight of 630,595 g/mol, as calculated using OligoCalc (Kibbe, 2007). Therefore, a stock solution of 2240 ng/μL would be 3.55 μM.
-
Calculate the molarity of your TGT enzyme, in μM, using the following equation:For example, a purified protein prep of 1.61 mg/mL would have a concentration of 37.1 μM.
Thaw RNA transcript and TGT enzyme on ice. 10 × TGT reaction buffer, 100 mM DTT, and preQ1 stock solutions can be thawed at room temperature.
-
Set up labeling reactions as follows:
Reagent Final concentration 10 × TGT reaction buffer 1 × (100 mM HEPES, pH 7.3, 20 mM MgCl2) 100mM DTT 5mM RNA transcript 1μM TGT enzyme 1μM preQ1-C6H12-derivative 10μM RNAse inhibitor, murine 1U/μL Typical reaction volumes are 50 μL, but can be adjusted as necessary; our lab has performed reaction sizes varying from 12.5 to 250 μL.
Incubate labeling reactions for 2 h at 37 °C.
Purify the RNA through ethanol precipitation by adding 0.1 vol. 3 M NaOAc and 3 volumes of ice-cold, 100% ethanol. Linear polyacrylamide or glycogen may be added as a carrier, if desired.
Precipitate RNA at −20 °C 1 h to overnight.
Centrifuge at 16,000 × g for 30 min at 4 °C to pellet the RNA.
Carefully decant and discard supernatant, taking care not to disturb the RNA pellet.
Wash pellet with 75% ice cold ethanol, and centrifuge at 16,000 × g an additional 5 min at 4 °C.
Discard supernatant and let pellet air dry.
-
Resuspend labeled RNA sample in nuclease-free water for downstream use.
Tip: Oligo Clean and Concentrator (Zymo Scientific) can be used as an alternative purification method, if desired.
2.5. Precursor techniques
2.5.1. Synthesis of labeled preQ1 derivatives
PreQ1 hydrochloride is available commercially (Sigma Aldrich) or can be synthesized using previously reported procedures (Klepper, Polborn, & Carell, 2005). Derivatization of preQ1 has been reported elsewhere (Ehret et al., 2018).
2.5.2. In vitro transcription with T7 RNA polymerase
Several methods have been reported for transcription mediated by T7 RNA polymerase (Cazenave & Uhlenbeck, 1994), which have been successfully used in conjunction with RNA-TAG labeling (Alexander et al., 2015). Helpful guides for performing transcription reactions can be found online (Sauer Lab, 2006). Furthermore, commercial kits are available for transcription (i.e. MEGAscript T7 transcription kit, Invitrogen).
2.5.3. Procedure for validation of TGT enzyme and preQ1 derivatives
In order to validate newly synthesized preQ1 derivatives or batches of purified TGT protein, a gel shift assay with ECY-A1 can be performed. Due to the small size of ECY-A1 (5138.3 g/mol), a characteristic gel shift is observed on denaturing polyacrylamide gels upon the addition of a labeled preQ1 derivative (Alexander et al., 2015). Typically, labeling reactions are performed in a similar manner as described above for RNA transcripts, using 10 μM ECY-A1, 10 μM TGT, and 20 μM preQ1 derivative. Ethanol precipitation prior to PAGE analysis improves the resolution of this assay. Common RNA stains, such as GelRed, SYBR green, or methylene blue, can be used; the band intensity of a control sample can be compared to the unshifted, unlabeled band intensity in the labeled sample to evaluate labeling efficiency. Depending on which probe is appended to the RNA, fluorescence of the probe-labeled band may be detected, and can potentially interfere if only the labeled band is analyzed.
2.6. Analysis and statistics
Analysis of transcripts labeled with RNA-TAG is dependent on the probe that is being used to label the RNA. For example, degree of labeling with fluorescent reporters has been described using an absorbance assay (Ehret et al., 2018). Another method of analysis is to run transcripts on a denaturing polyacrylamide gel, and detect fluorescent bands; for example, with a bio-molecular imager (i.e. Typhoon). There are also several available methods for biotin detection, for example streptavidin mediated affinity purification followed by denaturing PAGE (Alexander et al., 2015). An alternative method for biotin detection is northern blotting followed by chemiluminescent detection with biotin-HRP (i.e. Chemiluminescent Nucleic Acid Detection Module Kit, Fisher Scientific).
2.7. Troubleshooting and optimization
| Problem | Solution |
|---|---|
| Significant degradation is observed during TGT labeling | TGT enzyme is contaminated with nucleases; test for RNAses and subject enzyme to two rounds of Ni-NTA purification. Alternatively, the concentration of RNAse inhibitor in the labeling reaction can be increased |
| Low labeling efficiency is observed | Validate TGT enzyme and preQ1 derivative using “Procedure for validation ofTGT enzyme and preQ1 derivatives,” described above. Labeling of ECY-A1 should be >90%. Re-express TGT if necessary, minimizing storage time at 4 °C or −20 °C, and storing purified aliquots at −80 °C. Check purity of preQ1 derivative; certain derivatives may be less well accepted by TGT |
| Reaction can be optimized by increasing TGT concentration, preQ1 derivative concentration, or length of incubation. If a negative control is desired, a G → C mutation to the TGT recognition element can be made (Alexander et al., 2015) |
3. Summary
Many investigations in RNA biology can benefit from the covalent, site-specific modification of RNA with RNA-TAG. While other RNA labeling methods require end labeling, the installation of long encoded elements, or the use of reactive compounds, RNA-TAG has several advantages. One key advantage is that the site of modification within an RNA of interest can be selected by insertion of a short 17–25 nucleotide stem loop. Furthermore, RNA-TAG leverages a unique nucleobase-probe conjugate that does not react non-specifically with RNA or other biomolecules and is expected to be selective in mammalian systems. In order to implement RNA-TAG labeling, standard techniques for protein expression and purification from E. coli can be applied to facilitate the purification of E. coli TGT. There are several applicable cloning methods for the insertion of TGT recognition element into a sequence encoding an RNA of interest; however, a straight-forward restriction cloning approach is presented here. Lastly, a general method for RNA labeling with preQ1 derivatives is presented. These methods have been successfully implemented with a wide variety of preQ1 probes, including fluorophores, affinity ligands, and click handles, to demonstrate the utility of this approach in numerous applications. The breadth of this approach suggests that this methodology may hold promise for future applications such as RNA interactome studies, RNA imaging, and RNA manipulation in live cells, making even further insights into RNA biology possible.
Acknowledgment
This work was supported by the National Institutes of Health under award R01 GM123285-01.
References
- Alexander SC, Busby KN, Cole CM, Zhou CY, & Devaraj NK (2015). Site-specific covalent labeling of RNA by enzymatic transglycosylation. Journal of the American Chemical Society, 137(40), 12756–12759. 10.1021/jacs.5b07286. [DOI] [PubMed] [Google Scholar]
- Biela I, Tidten-Luksch N, Immekus F, Glinca S, Nguyen TXP, Gerber HD, … Reuter K (2013). Investigation of specificity determinants in bacterial tRNA-guanine transglycosylase reveals queuine, the substrate of its eucaryotic counterpart, as inhibitor. PLoS One, 8(5). 10.1371/journal.pone.0064240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazenave C, & Uhlenbeck OC (1994). RNA template-directed RNA synthesis by T7 RNA polymerase. Proceedings of the National Academy of Sciences of the United States of America, 91(15), 6972–6976. 10.1073/pnas.91.15.6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech TR, & Steitz JA (2014). The noncoding RNA revolution—Trashing old rules to forge new ones.pdf. Cell, 157(1), 77–94. 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Chen YC, Brooks AF, Goodenough-Lashua DM, Kittendorf JD, Showalter HD, & Garcia GA (2011). Evolution of eukaryal tRNA-guanine transglycosylase: Insight gained from the heterocyclic substrate recognition by the wild-type and mutant human and Escherichia coli tRNA-guanine transglycosylases. Nucleic Acids Research, 39, 2834–2844. 10.1093/nar/gkq1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Howarth M, Lin W, & Ting AY (2005). Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nature Methods, 2(2), 99–104. 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- Chin A, & Lécuyer E (2017). RNA localization: Making its way to the center stage. Biochimica et Biophysica Acta-General Subjects, 1861(11), 2956–2970. 10.1016/j.bbagen.2017.06.011. [DOI] [PubMed] [Google Scholar]
- Cooper TA, Wan L, & Dreyfuss G (2009). RNA and disease. Cell, 136(4), 777–793. 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curnow AW, & Garcia GA (1995). tRNA-guanine transglycosylase from Escherichia coli: Minimal tRNA structure and sequence requirements for recognition. Journal of Biological Chemistry, 270(29), 17264–17267. 10.1074/jbc.270.29.17264. [DOI] [PubMed] [Google Scholar]
- Curnow A, Kung FL, Koch K, & Garcia G (1993). tRNA-guanine transglycosylase from Escherichia coli: Gross tRNA structural requirements for recognition. Biochemistry, 32, 5239–5246. [DOI] [PubMed] [Google Scholar]
- Ehret F, Zhou CY, Alexander SC, Zhang D, & Devaraj NK (2018). Site-specific covalent conjugation of modified mRNA by tRNA guanine transglycosylase. Molecular Pharmaceutics, 15(3), 737–742. 10.1021/acs.molpharmaceut.7b00356. [DOI] [PubMed] [Google Scholar]
- Farkas WR, Jacobson KB, & Katze JR (1984). Substrate and inhibitor specificity of tRNA-guanine ribosyltransferase. BBA-Gene Structure and Expression, 781(1–2), 64–75. 10.1016/0167-4781(84)90124-6. [DOI] [PubMed] [Google Scholar]
- Fergus C, Barnes D, Alqasem MA, & Kelly VP (2015). The queuine micronutrient: Charting a course from microbe to man. 7, 2897–2929. 10.3390/nu7042897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Haranda BT, Behm M, & He C (2018). RNA modifications modulate gene expression during development. Science, 361(September), 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A, Juillerat A, Heinis C, Corrêa IR, Kindermann M, Beaufils F, et al. (2008). An engineered protein tag for multiprotein labeling in living cells. Chemistry and Biology, 15(2), 128–136. 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Goodenough-Lashua DAM, & Garcia GA (2003). tRNA-guanine transglycosylase from E. coli: A ping-pong kinetic mechanism is consistent with nucleophilic catalysis. Bioorganic Chemistry, 31(4), 331–344. 10.1016/S0045-2068(03)00069-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregson JM, Crain PF, Edmonds CG, Gupta R, Hashizume T, Phillipson DW, et al. (1993). Structure of the archaeal transfer RNA nucleoside G*-15 (2-amino-4,7-dihydro-4-oxo-7-β-D-ribofuranosyl-1H-pyrrolo[2,3-d]pyrimidine-5- carboximidamide (Archaeosine)). Journal of Biological Chemistry, 268(14), 10076–10086. [PubMed] [Google Scholar]
- Grosjean H, Edqvist J, Stråby KB, & Giegé R (1996). Enzymatic formation of modified nucleosides in tRNA: Dependence on tRNA architecture. Journal of Molecular Biology, 255, 67–85. 10.1006/jmbi.1996.0007. [DOI] [PubMed] [Google Scholar]
- Ho SH, & Tirrell DA (2016). Chemoenzymatic labeling of proteins for imaging in bacterial cells. Journal of the American Chemical Society, 138(46), 15098–15101. 10.1021/jacs.6b07067. [DOI] [PubMed] [Google Scholar]
- Hoops GC, Townsend LB, & Garcia GA (1995). tRNA-guanine transglycosylase from Escherichia coli: Structure-activity studies investigating the role of the aminomethyl substituent of the heterocyclic substrate PreQ1. Biochemistry, 34, 15381–15387. [DOI] [PubMed] [Google Scholar]
- Hoops GC, Park J, Garcia GA, & Townsend LB (1996). The synthesis and determination of acidic ionization constants of certain 5-substituted 2-aminopyrrolo [2,3-d]pyrimidin-4-ones and methylated analogs. Journal of Heterocyclic Chemistry, 33(3), 767–781. 10.1002/jhet.5570330341. [DOI] [Google Scholar]
- Ishitani R, Nureki O, Fukai S, Kijimoto T, Nameki N, Watanabe M, … Yokoyama S (2002). Crystal structure of archaeosine tRNA-guanine transglycosylase. Journal of Molecular Biology, 318(3), 665–677. 10.1016/S0022-2836(02)00090-6. [DOI] [PubMed] [Google Scholar]
- Ishitani R, Nureki O, Nameki N, Okada N, Nishimura S, & Yokoyama S (2003). Alternative tertiary structure of tRNA for recognition by a posttranscriptional modification enzyme. Cell, 113(3), 383–394. 10.1016/S0092-8674(03)00280-0. [DOI] [PubMed] [Google Scholar]
- Johannsson S, Neumann P, & Ficner R (2018). Crystal structure of the human tRNA guanine transglycosylase catalytic subunit QTRT1. Biomolecules, 8(3), 81. 10.3390/biom8030081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karikó K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, et al. (2008). Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Molecular Therapy, 16(11), 1833–1840. 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbe WA (2007). OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Research, 35(Suppl.2), 43–46. 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepper F, Polborn K, & Carell T (2005). Robust synthesis and crystal-structure analysis of 7-cyano-7-deazaguanine (PreQ0 base) and 7-(aminomethyl)-7-deazaguanine (PreQ1 base). Helvetica Chimica Acta, 88(10), 2610–2616. 10.1002/hlca.200590201. [DOI] [Google Scholar]
- Li F, Dong J, Hu X, Gong W, Li J, Shen J, … Wang J (2015). A covalent approach for site-specific RNA labeling in mammalian cells. Angewandte Chemie-International Edition, 54(15), 4597–4602. 10.1002/anie.201410433. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Ueda T, Hori H, Yamazaki N, Watanabe N, & Okada K (1994). A UGU sequence in the anticodon loop is a minimum requirement for recognition by Escherichia coli tRNA-guanine transglycosylase. Journal of Biological Chemistry, 269, 32221–32225. [PubMed] [Google Scholar]
- Pathak C, Jaiswal YK, & Vinayak M (2005). Hypomodification of transfer RNA in cancer with respect to queuosine. RNA Biology, 2(4), 143–148. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/17114931. [DOI] [PubMed] [Google Scholar]
- Richardson RW, & Gumport RI (1983). Biotin and fluorescent labeling of RNA using T4 RNA ligase. Nucleic Acids Research, 11(18), 6167–6184. 10.1093/nar/11.18.6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer Lab. (2006). In vitro transcription with T7 RNA polymerase. Retrieved from 25 July 2006 website: https://openwetware.org/wiki/Sauer:In_vitro_transcription_with_T7_RNA_polymerase.
- Schulz D, Holstein JM, & Rentmeister A (2013). A chemo-enzymatic approach for site-specific modification of the RNA cap. Angewandte Chemie-International Edition, 52(30), 7874–7878. 10.1002/anie.201302874. [DOI] [PubMed] [Google Scholar]
- Singh G, Pratt G, Yeo GW, & Moore MJ (2015). The clothes make the mRNA: Past and present trends in mRNP fashion. Annual Review of Biochemistry, 84(1), 325–354. 10.1146/annurev-biochem-080111-092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengl B, & Klebe G (2007). Novel leads for selective antibiotics against shigellosis by virtual screening, crystallography and synthesis. Supramolecular Structure and Function, 9, 209–249. [Google Scholar]
- Stengl B, Meyer E. a., Heine A, Brenk R, Diederich F, & Klebe G (2007). Crystal structures of tRNA-guanine transglycosylase (TGT) in complex with novel and potent inhibitors unravel pronounced induced-fit adaptations and suggest dimer formation upon substrate binding. Journal of Molecular Biology, 370, 492–511. 10.1016/j.jmb.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Stengl B, Reuter K, & Klebe G (2005). Mechanism and substrate specificity of tRNA-guanine transglycosylases (TGTs): tRNA-modifying enzymes from the three different kingdoms of life share a common catalytic mechanism. ChemBioChem, 6, 1926–1939. 10.1002/cbic.200500063. [DOI] [PubMed] [Google Scholar]
- Suzuki T, & Suzuki T (2014). A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Research, 42(11), 7346–7357. 10.1093/nar/gku390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorov KA, & Garcia GA (2006). Role of aspartate 143 in Escherichia coli tRNA-guanine transglycosylase: Alteration of heterocyclic substrate specificity. Biochemistry, 45(2), 617–625. 10.1021/bi051863d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorov KA, Tan X-J, Nonekowski ST, Garcia GA, & Carlson HA (2005). The role of aspartic acid 143 in E. coli tRNA-guanine transglycosylase: Insights from muta-genesis studies and computational modeling. Biophysical Journal, 89(3), 1965–1977. 10.1529/biophysj.105.059576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S, Kataoka K, & Itaka K (2015). Screening of mRNA chemical modification to maximize protein expression with reduced immunogenicity. Pharmaceutics, 7(3), 137–151. 10.3390/pharmaceutics7030137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walden T, Reyniers JP, Hiatt V, & Farkas WR (1982). Yeast cells cannot incorporate queuine into their tRNA. Proceedings of the Society for Experimental Biology and Medicine, 170(3), 328–332. 10.3181/00379727-170-41438. [DOI] [PubMed] [Google Scholar]
- Xie W, Liu X, & Huang RH (2003). Chemical trapping and crystal structure of a catalytic tRNA guanine transglycosylase covalent intermediate. Nature Structural Biology, 10(10), 781–788. 10.1038/nsmb0704-678a. [DOI] [PubMed] [Google Scholar]
- Zhang D, Zhou CY, Busby KN, Alexander SC, & Devaraj NK (2018). Light-activated control of translation by enzymatic covalent mRNA labeling. Angewandte Chemie-International Edition, 57(11), 2822–2826. 10.1002/anie.201710917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CY, Alexander SC, & Devaraj NK (2017). Fluorescent turn-on probes for wash-free mRNA imaging via covalent site-specific enzymatic labeling. Chemical Science, 8(10), 7169–7173. 10.1039/c7sc03150e. [DOI] [PMC free article] [PubMed] [Google Scholar]