

Abstract
Puberty is a major maturational event; its mechanisms and timing are driven by genetic determinants, but also controlled by endogenous and environmental cues. Substantial progress towards elucidation of the neuroendocrine networks governing puberty has taken place. However, key aspects of the mechanisms responsible for the precise timing of puberty and its alterations have only recently begun to be deciphered, propelled by epidemiological data suggesting that pubertal timing is changing in humans, via mechanisms that are not yet understood. By integrating basic and clinical data, we provide a comprehensive overview of current advances on the physiological basis of puberty, with a particular focus on the roles of kisspeptins and other central transmitters, the underlying molecular and endocrine mechanisms, and the pathways involved in pubertal modulation by nutritional and metabolic cues. Additionally, we have summarised molecular features of precocious and delayed puberty in both sexes, as revealed by clinical and genetic studies. This Review is a synoptic up-to-date view of how puberty is controlled and of the pathogenesis of major pubertal alterations, from both a clinical and translational perspective. We also highlight unsolved challenges that will seemingly concentrate future research efforts in this active domain of endocrinology.
Introduction
Puberty is a fundamental period when sexual maturity and reproductive capacity are acquired,1 and key somatic, behavioural, and psychological changes occur, leading to an adult phenotype. Its neuroendocrine substrate, defined by full activation of the hypothalamic–pituitary–gonadal (HPG) axis, includes: (1) hypothalamic gonadotropin-releasing hormone (GnRH); (2) pituitary gonadotropins, luteinising hormone and follicle-stimulating hormone, driven by GnRH; and (3) gonadal steroids and peptides, stimulated by gonadotropins,2 all intertwined via feedback loops.
With its origin in early developmental events commencing in utero, normal pubertal timing in humans has a wide interindividual variation (occurring at age 8–13 years in girls and 9–14 years in boys) that is determined by genetic, nutritional, and environmental elements,2 reflecting the complex genetic architecture and regulatory pathways underlying this developmental period. Pubertal onset is indicated by breast development in girls (Tanner stage 2),3 and testicular enlargement (ie, testicular volume >4 mL or testicular length >25 mm) in boys (Tanner stage 2).4 Although this definition remains arbitrary internationally, these indicators are required clinically to orient the diagnosis of pubertal pathology. Precocious puberty is defined as the onset of breast development before age 8 years in girls and increased testicular volume (>4 mL) in boys before age 9 years that is progressive and accompanied by acceleration of bone age and linear growth.5 Delayed puberty is defined as the absence of somatic signs of pubertal development at an age 2 SD higher than the mean (approximately age 14 years for boys and 13 years for girls). Although epidemiological data of precocious and delayed puberty are scarce, multiple aetiologies of pubertal pathology, either congenital or acquired, are known.6,7 Furthermore, trends indicate an advancement in the normal age range of puberty onset at the population level, especially in girls.8,9 Although the reason for earlier pubertal onset is unknown, it can pose risks for disease development later in life.10 In fact, earlier or later puberty, even within the normal limits of maturation in humans, has been linked to increased risk of multiple adverse outcomes in both sexes, including not only gynecological, but also cardiometabolic (eg, hypertension and type 2 diabetes), musculoskeletal, gastrointestinal, and cognitive conditions, and some forms of cancer.10
Because of its importance in human pathophysiology, we have provided an overview of the current knowledge of the molecular basis of normal puberty, its main regulatory pathways, and major deviations.
Neuropeptide control of puberty: kisspeptins and Kiss1 neurons
Considerable efforts have been made to define the neuroendocrine mechanisms for pubertal activation of GnRH neurosecretion; multiple trans-synaptic and glial inputs, of excitatory or inhibitory nature, were shown to contribute to this process.2 Collectively, these studies, conducted in laboratory animals and non-human primates, have revealed the role of different central transmitters in the control of mammalian puberty.2,11,12 Kisspeptins, products of the Kiss1 gene that operate via the surface receptor, Kiss1r (previously known as Gpr54), have emerged as master gatekeepers of puberty, mainly by activating GnRH neurons.13 Indeed, due to their essential roles, Kiss1 neurons, producing kisspeptins in discrete hypothalamic and extra-hypothalamic areas, have drawn extraordinary attention as pivotal elements for the brain’s control of puberty. However, whether kisspeptins are the actual trigger of puberty, or rather operate as an amplifier of the cascade of events leading to full activation of GnRH neurons during pubertal maturation, remains contentious.14
The role of kisspeptins in puberty was disclosed by genetic studies showing that inactivating mutations affecting their receptor (or kisspeptins themselves) caused central hypogonadism and absence of puberty in humans and rodents.15–17 Subsequently, experimental studies showed a complex developmental programme of the hypothalamic Kiss1 system during pubertal maturation, involving not only increased expression of Kiss1 during puberty, but also an increase in the number of Kiss1 neurons and their projections to GnRH neurons in rodents.18,19 Although such changes are likely to affect both Kiss1 neuronal populations located in the arcuate nucleus and the rostral hypothalamus, arcuate nucleus Kiss1 neurons are thought to play a more prominent role because of the conservation of this population across species, including humans,17 and findings on their function as a core component of the GnRH pulse generator.20 A notable amount of arcuate nucleus Kiss1 neurons have been shown to co-express tachykinin, neurokinin B (NKB), and dynorphin (Dyn), and hence are named KNDy (for Kisspeptin, NKB, and Dyn expression) neurons. NKB and Dyn operate in a reciprocal manner to stimulate (NKB) or inhibit (Dyn) kisspeptin output to GnRH neurons (figure 1).21 In addition, fragmentary evidence suggests that a third population of Kiss1 neurons located in the amygdala might also participate in the modulation of puberty in rodents.22
Figure 1: Neuropeptide control of puberty and its metabolic regulation.
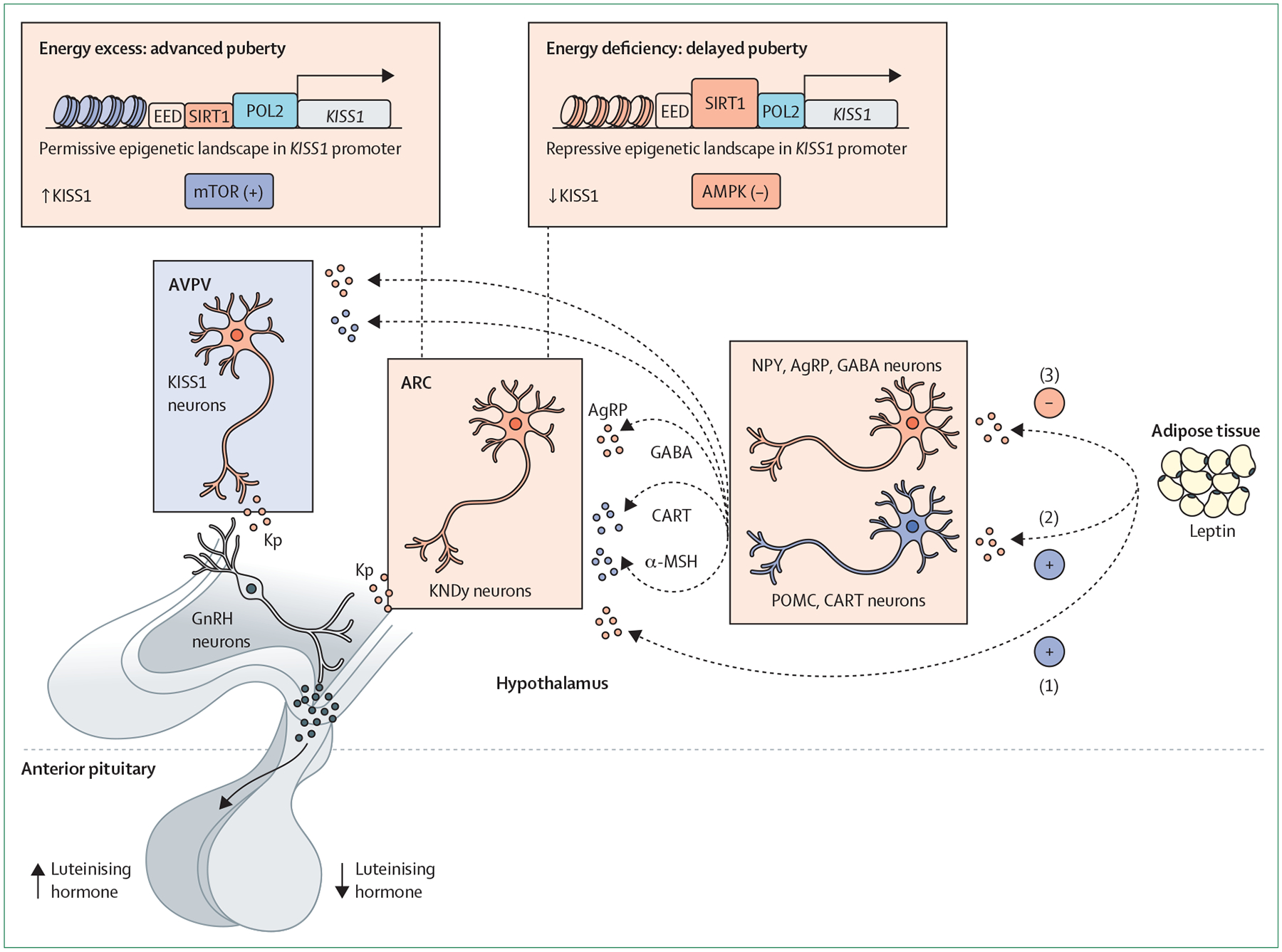
Schematic presentation of populations of Kiss1 neurons at the two main hypothalamic areas, namely the ARC and AVPV, and related hypothalamic neuronal pathways and cellular energy sensors involved in the physiological control of puberty. Related neuronal pathways seemingly include neurons producing NPY and AgRP, and neurons expressing POMC and CART neurons. These pathways participate in transmitting the modulatory actions of key metabolic hormones, such as leptin. Three possible modes of action of leptin are presented: (1) direct excitatory actions on Kiss1 neurons; (2) indirect excitatory actions via POMC neurons; and (3) indirect inhibitory actions on NPY/AgRP neurons. In addition, these circuits convey the influence of conditions of energy deficit, which result in delayed puberty (denoted by low luteinising hormone levels), and situations of energy excess (obesity), that advance pubertal onset (denoted by high luteinising hormone levels). Cellular energy sensors and metabolic mediators involved in this process include mTOR, AMPK and SIRT1. AgRP=agouti-related protein. AMPK=adenosine monophosphate-activated protein kinase. ARC=arcuate nucleus. AVPV=anteroventral periventricular nucleus. CART=cocaine-regulated and amphetamine-regulated transcript. EED=embryonic ectoderm development (component of polycomb repressive complex 2). GABA=γ-aminobutyric acid. GnRH=gonadotropin-releasing hormone. KNDy=kisspeptin, neurokinin B, and dynorphin. Kp=kisspeptin. MSH=melanocyte-stimulating hormone. mTOR=mammalian target of rapamycin. NPY=neuropeptide Y. POL2=RNA polymerase 2. POMC=proopiomelanocortin. SIRT1=sirtuin 1.
The underlying mechanisms for activation of the Kiss1 system at puberty are yet to be unfolded but involve epigenetic pathways. Of note, a key component of this process is probably the decline in repressive signals, by the concerted action of miRNAs at the level of Kiss1 neurons, as shown by studies in mice engineered to lack miRNA biosynthesis in Kiss1 cells.23 This phenomenon resembles similar miRNA-based regulatory pathways documented in GnRH neurons, indispensable also for pubertal maturation.24
Multiple neuropeptide pathways cooperate with kisspeptins in the central control of puberty. These include not only NKB, whose genetic inactivation prevents puberty in humans,25 and expression rises in the rodent hypothalamus preceding puberty,26 but involve also other tachykinins such as substance P and neurokinin A, both encoded by Tac1, because Tac1 knock-out mice display delayed puberty.27 Yet, the redundancy between different members of the tachykinin family suggests that some degree of compensation occurs among them. Other central transmitters cooperate with kisspeptin pathways in the control of the reproductive axis at puberty, including prominently melanocortin signalling,28 as supported by human studies published in 2021.29 However, most of these regulatory pathways are not exclusive to puberty and also participate in controlling the adult reproductive axis.
Molecular mechanisms for the control of puberty: emerging roles of epigenetics
Epigenetic processes are enzymatically driven alterations of gene expression induced by post-translational modifications of the chromatin structure or variations in mRNA turnover. These mechanisms are commonly divided into three modes of operation: DNA methylation and hydroxymethylation at the carbon-5 position of cytosines in CpG dinucleotides; histone post-translational modifications that affect chromatin packaging; and non-coding RNAs involved in mRNA degradation and half-life, chromatin condensation, and transcription factor binding.
Although epigenetic mechanisms also operate at other levels of the HPG axis,30 during the past decade, a substantial number of studies have identified epigenetic processes that affect the reactivation of the hypothalamic GnRH pulse generator around puberty in rodents and primates. In the rat arcuate nucleus, the promoter region of the Kiss1 gene has both repressing and activating epigenetic signatures, allowing fast activation or repression of gene expression. Before puberty, arcuate nucleus Kiss1 expression is low, partly due to the presence of the Polycomb Group (PcG) of epigenetic silencers that induce chromatin compaction by trimethylating histone H3 at lysine 27 (H3K27me3) at its 5′ regulatory region,31 and by repressing lysine demethylase 6b (Kdm6b) expression, the enzyme in charge of removing the H3K27me3 repressive mark from gene promoters.32 As puberty progresses, arcuate nucleus Kiss1 expression is enhanced by recruitment of the trithorax group (TrxG) of epigenetic activators to the Kiss1 promoter and enhancer regions, imposing activational H3K4me3 and H3K27Ac post-translational modifications, promoting an open chromatin conformation that enhances access of the transcriptional machinery.33 Similar mechanisms of epigenetic control have been identified in rhesus macaques. During pubertal transition, the chromatin landscape changes at both the Kiss1 and the Tac3 loci in the hypothalamus by removal of the PcG and GATA Zinc Finger Domain Containing 1 (GATAD1) and lysine-specific demethylase 1A (KDM1A) repressors of their promoter sites, after which there is increased binding of the TrxG of transcriptional activators, and heightening of Kiss1 and Tac3 expression due to increased H3K4me3 and recruitment of p300 and CBP acetyltransferases at enhancer sites.34
Messina and colleagues24 showed that GnRH-neuron specific Dicer knock-out produces hypogonadotropic hypogonadism and infertility in mice. With this model, the authors showed that the increase in GnRH expression during the infantile to juvenile transition depends on the integrity of miR-200 and miR-155.24 Similarly, miRNA-mediated suppression of Kiss1 transcriptional repressors, such as Mkrn3, Cbx7, and Eap1, seemingly plays a major role in full pubertal activation and attainment of reproductive capacity in mice, especially in females.23 Some of these epigenetic mechanisms are involved in transmitting metabolic information to the hypothalamic gene networks that control pubertal maturation. Arcuate nucleus Kiss1 neurons react to metabolic challenges with changes in SIRT1 expression. Association of SIRT1 with the PcG alters the chromatin structure of the Kiss1 gene promoter region with changes in H3K27me3 and H3K9-14Ac, thereby modulating pubertal timing.35
Several studies36–40 aimed to identify patterns in DNA methylation or miRNA expression in blood or buccal samples across human puberty. Overall, methylation patterns in peripheral blood accurately predict pubertal development and are enriched in biological pathways related to growth and development of the reproductive system.36–38 The number of differentially methylated CpG islands is higher in females than males, probably related to a more pronounced pubertal transition and hormonal variations due to the initiation of the ovarian cycle.38 DNA methylation changes detected in buccal samples form cohesive networks that correlate with salivary testosterone levels independent of sex.39 Conversely, CpG–miRNA pairs were identified in the imprinted region of 14q32 that are associated with age at menarche, suggesting a causal relationship.40 Methylome profiling in peripheral blood identified 48 zinc finger (ZNF) genes as having hypermethylated CpGs in patients with central precocious puberty and zinc finger protein 57 (ZFP57) was hypomethylated during normal puberty. ZFP57 is a transcriptional repressor involved in maintenance of imprinting regions.41 More research needs to be done to improve our knowledge of peripheral epigenetic markers and their connection with central mechanisms for the control of puberty.
Nutritional and metabolic control of puberty: basic mechanisms and clinical implications
Nutritional and metabolic cues are prominent among the multiple modulators of pubertal timing in humans and other species.42 The concept that body energy reserves have a permissive role in pubertal maturation was scientifically formulated by Frisch and colleagues43 in 1973, proposing the critical weight (fat) mass hypothesis. In humans, the interaction of these physiological axes is observed at two extremes of body energy status: (1)chronic energy deficiency, malnutrition, or anorexia, which can be accompanied by delayed puberty or absence of pubertal progression, and (2) energy excess, or obesity, which can be associated with advanced pubertal onset.42,44 This nutritional effect on pubertal development has been shown in girls,8,45 but might also occur in boys.46
Clinical and experimental studies have identified some key hormonal signals responsible for the coupling of puberty and metabolism. Although extensive recapitulation of these findings is beyond the scope of this Review, the adipose hormone, leptin, can be singled out as the most prominent metabolic modulator of puberty, by signalling sufficient body energy reserves.42 Although initial studies proposed that leptin triggered puberty, subsequent evidence showed that leptin acts rather as a permissive factor for pubertal maturation, especially in girls.42 Hence, threshold leptin levels are needed, but not sufficient per se, to progress through puberty. Other metabolic hormones cooperate with leptin to modulate puberty, including the gut hormone, ghrelin, and the pancreatic hormone, insulin. Elevated ghrelin levels, an index of energy deficit, have been shown to inhibit puberty in rodents.47 By contrast, insulin’s actions are mandatory for puberty to proceed normally, as indicated by human and rodent studies.48 Women with uncontrolled type 1 diabetes can have delayed puberty, and neuronal ablation of the insulin receptor in rodents led to impaired ovarian maturation.48
The interaction between these peripheral hormones and the brain centres controlling puberty takes place primarily in the hypothalamus and probably involves prominent modulation of neuronal pathways upstream of GnRH neurons. Among these pathways, Kiss1 neurons are clearly sensitive to leptin and other metabolic factors, although some of these regulatory actions probably occur indirectly (ie, on afferents of Kiss1 neurons rather than Kiss1 eurons themselves).42 Conditions of energy deficit and leptin deficiency can suppress the hypothalamic Kiss1 system, whereas early-onset obesity in rodent models is linked to premature Kiss1 activation, compatible with partly accelerated puberty onset.42 Cellular energy or metabolic sensors, acting in Kiss1 neurons and their afferents to transduce metabolic information, include the mammalian target of rapamycin (mTOR) and the AMP-activated kinase (AMPK), which operate in a reciprocal manner to centrally modulate energy homoeostasis.49 Using preclinical models, we have documented that brain mTOR signalling is needed for transmission of the permissive effects of leptin on female puberty,50 whereas AMPK activity in Kiss1 neurons participates in the inhibitory actions of subnutrition on pubertal timing (figure 1).51
AMPK probably cooperates with SIRT1, a member of the sirtuin family, in the modulation of pubertal timing. In arcuate nucleus Kiss1 neurons, SIRT1 content increases in conditions of subnutrition and operates as a repressor of the Kiss1 promoter, thereby contributing to pubertal delay. By contrast, conditions of early obesity in female rats result in premature eviction of SIRT1 from the Kiss1 promoter, thus contributing to activation of Kiss1 expression and advanced puberty onset (figure 1).35 However, it should be taken into consideration that changes in Kiss1 RNA expression could be a proxy marker, but might not directly translate into equivalent changes in kisspeptin neurosecretion. Although this mechanism is compatible with enhanced kisspeptin output to GnRH neurons in conditions of obesity, leading to heightened gonadotropin drive to the ovary, we also documented a complementary pathway for advanced puberty due to obesity, involving kisspeptin projections to the paraventricular nucleus, and de novo ceramide synthesis in this nucleus, as putative origin of enhanced sympathetic input to the ovary.52 Thus, obesity might contribute to pubertal acceleration via gonadotropin-dependent and gonadotropin-independent pathways, involving Kiss1 neurons.
Whereas Kiss1 neurons are a major integratory hub for the metabolic control of puberty, other neuronal pathways also play a prominent role, including neurons producing glutamate or pituitary adenylate-cyclase activating polypeptide from the ventral premammillary nucleus,53,54 and leptin-sensitive melanocortin producing neurons from the arcuate nucleus;28 relevance of the second pathway is reinforced by recent human data supporting a fundamental role of melanocortin signalling via MC3-receptors in the integration of nutritional state, rate of growth, and puberty in humans.29 Variants in this receptor were associated with a delay in sexual maturation, similar to that found in mice lacking Mc3r.29 Indeed, pathways from the ventral premammillary nucleus and melanocortin pathways are known to transmit leptin actions and interact with Kiss1 neurons (figure 1). For instance, MC3-receptors are expressed in Kiss1 neurons, supporting the relevance of this pathway involving leptin, hypothalamic anorexigenic proopiomelanocortin (POMC) and Kiss1 neurons, operating via MC3R signalling. Our studies show that GnRH neurons are also equipped with energy-sensing mechanisms, involving AMPK and the G-protein-coupled receptor kinase, GRK2, that probably contribute to suppression of puberty onset in conditions of energy insufficiency.55,56
Central precocious puberty
Precocious puberty is defined as the development of secondary sexual characteristics before the age of 8 years in girls and 9 years in boys, and it has a clear female predominance.7 The premature activation of pulsatile hypothalamic GnRH secretion leads to central precocious puberty, the most common form of premature sexual development. In the past decade, genetic and epigenetic causes of central precocious puberty have been documented by the identification of rare pathogenic gene variants, causing perturbation of specific hypothalamic pathways.7,57 We have schematically presented typical examples in a neuroendocrine context in this Review (figure 2).
Figure 2: Monogenic causes of central precocious puberty.
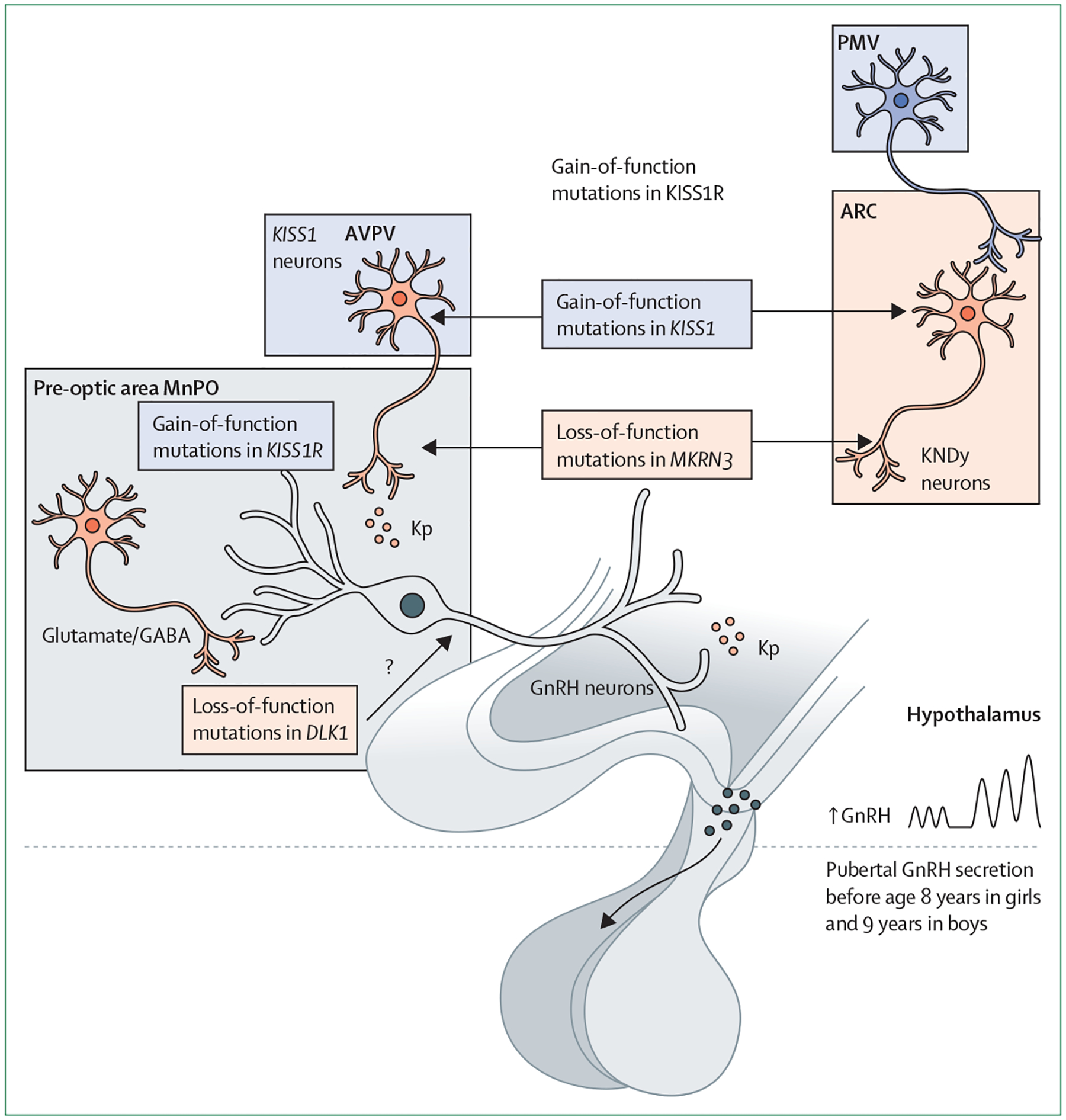
Central precocious puberty is a hypothalamic disorder with multiple aetiologies, including congenital and acquired conditions. Schematic representation of the hypothalamic regions and paradigmatic monogenic causes of central precocious puberty in humans are shown. These monogenic forms include gain-of-function mutations in KISS1, gain-of-function mutations of KISS1R, loss-of-function mutations in MKRN3, and loss-of-function mutations in DLK1. Schematic presentation of the main sites of alteration of hypothalamic circuits due to the above mutations is provided. ARC=arcuate nucleus. AVPV=anteroventral periventricular nucleus. GABA=γ-aminobutyric acid. GnRH=gonadotropin-releasing hormone. KNDy=kisspeptin, neurokinin B, and dynorphin. Kp=kisspeptin. ME=median eminence. MnPO=median preoptic nucleus. PMV=ventral pre-mammillary nucleus.
In 2008, a rare heterozygous activating mutation of KISS1R (p.Arg386Pro) was identified in a girl with central precocious puberty.58 This mutation, located in the C-terminal tail of the receptor, led to prolonged activation of intracellular signalling pathways in response to kisspeptin in mammalian cells.59 A rare kisspeptin variant, p.Pro74Ser, was subsequently identified in the heterozygous state in a boy who developed sporadic central precocious puberty at the age of 1 year.60 The capacity to stimulate signal transduction was prolonged for p.Pro74Ser mutant kisspeptin compared with wild type, suggesting that this variant might be more resistant to degradation.
MKRN3 is an important neuroendocrine player in the control of pubertal timing, acting as an upstream inhibitor of GnRH secretion.61–63 The gene encoding MKRN3 (MKRN3) is located within the maternally imprinted Prader-Willi syndrome critical region on chromosome 15q11.2. The role of MKRN3 in the pathogenesis of central precocious puberty was first shown in 2013, when whole exome sequence analysis was done in several families with central precocious puberty.61 15 individuals (eight girls and seven boys) from five unrelated families carried loss-of-function MKRN3 mutations, characterising a monogenic familial central precocious puberty with autosomal dominant inheritance and exclusively paternal transmission.61 To date, approximately 59 different mutations in the MKRN3 gene have been identified in children with central precocious puberty.
A systematic review and meta-analysis evaluating 857 patients with sporadic or familial central precocious puberty from 14 studies showed a pooled overall MKRN3 mutation prevalence of 9.0% in quantitative analyses.62 Interestingly, higher prevalence was found in males, familial cases, and in non-Asian countries.62 In 2021, a multi-ethnic cohort of 716 children with central precocious puberty revealed 71 cases with different types of loss-of-function MKRN3 mutations.63 Patients with more severe MKRN3 mutations (ie, frameshift, nonsense, or promoter mutations) had greater bone age advancement and higher basal luteinising hormone levels at the time of presentation compared with patients with missense mutations. Central precocious puberty due to loss-of-function mutations of MKRN3 is clinically indistinct from idiopathic central precocious puberty; however, the type of genetic defect might affect the severity of the phenotype.63
In mice, Mkrn3 mRNA levels in the mediobasal hypothalamus are high before puberty, suggesting a potential action as a suppressor of GnRH secretion.61 Hypothalamic Mkrn3 expression declines before puberty, mediated by miR-30 microRNA, which binds to a highly conserved region of the Mkrn3 mRNA 3′-UTR.64 Abreu and colleagues65 showed that Mkrn3 is expressed in Kiss1 neurons of the mouse hypothalamic arcuate nucleus, whereas cell reporter assays documented that MKRN3 represses human KISS1 and TAC3 gene promoter activities, in a manner dependent upon its ubiquitin ligase activity. This activity is reduced by pathogenic mutations affecting the RING finger domain of the protein.65 In addition, interaction of MKRN3 with several proteins, including some implicated in pubertal timing, has been identified in vitro.66–68 Loss of Mkrn3 in mice can cause central precocious puberty, with early pubertal onset, as shown by advanced timing of preputial separation and vaginal opening in male and female Mkrn3-deficient mice, respectively.67 By contrast, persistence of peripubertal hypothalamic Mkrn3 expression resulted in delayed puberty in female mice.69
In 2017, a complex genetic defect (14 kb deletion associated with a 269 bp insertion) involving another maternally imprinted gene, Delta-like homologue 1 (DLK1, located on chromosome 14q), was identified in a family with central precocious puberty.70 In the past 3 years, new, rare frameshift mutations of DLK1 in girls with central precocious puberty or precocious menarche (age <9 years) were identified.71,72 In all reported cases, familial segregation analysis was consistent with the known maternal imprinting of DLK1. To date, 7 distinct deleterious defects in DLK1 have been identified in cases of central precocious puberty, all located in the extracellular region, which contains EGF-like domains. Notably, metabolic conditions, such as overweight or obesity and insulin resistance, were more prevalent in individuals with central precocious puberty associated with DLK1 mutations than in people with idiopathic central precocious puberty, suggesting that DLK1 is a new factor linking reproduction and metabolism.71 DLK1 is a non-canonical ligand of the Delta-Notch evolutionarily conserved signalling pathway, which controls a broad range of developmental processes, including cell fate determination, terminal differentiation, and proliferation; however, the potential mechanisms by which DLK1 deficiency leads to human central precocious puberty remains unknown (panel 1).
Abnormal genetic and epigenetic findings associated with human central precocious puberty have revealed that this paediatric endocrine condition is strongly influenced by epigenetic mechanisms, as shown by the identification of loss-of-function mutations in two imprinted genes (MKRN3, DLK1) and its potential association with other epigenetic syndromes, such as Prader-Willi syndrome, Temple syndrome, and Rett syndrome.7
Desensitisation therapies using GnRH analogues constitute the treatment of choice in patients with central precocious puberty.73 Several analogues are available, including leuprorelin and triptorelin, which can be administered as intramuscular injections every 1 or 3 months (leuprorelin), or every semester (triptorelin). Another therapeutic option is the subcutaneous implantation of the long-acting GnRH analogue, histreline, which is replaced every 1–2 years.73 There is no doubt regarding the therapeutic benefit of these treatments in girls younger than 6 years and boys younger than 9 years. However, this treatment needs to be individualised in girls aged between 6 years and 8 years.74 Genetic and epigenetic discoveries regarding the aetiology of central precocious puberty could provide the basis for additional treatment targets in the future.
Peripheral precocious puberty
Peripheral precocious puberty defines clinical presentations of precocious puberty with an increase in sex steroids without activation of the HPG axis.75 Although epidemiological information is scarce, peripheral precocious puberty is clearly a rare entity, much less frequent than central precocious puberty, with its prevalence (excluding congenital adrenal hyperplasia) estimated at 14 per million of the paediatric population at risk, with a female to male ratio of 4:1.76
The cause of peripheral precocious puberty can be congenital or acquired, with congenital adrenal hyperplasia being the most frequent congenital cause of peripheral precocious puberty in boys (panel 2). Both classic and non-classic congenital adrenal hyperplasia can debut as the appearance of pubarche, axillary hair, penis enlargement, and growth acceleration, with testicular size less than 4 mL. Because thelarche before the age of 8 years is the main indication of possible precocious puberty in girls, congenital adrenal hyperplasia is not included as a possible cause of peripheral precocious puberty, since late-onset forms usually appear as the display of pubic or axillary hair without thelarche before the age of 8 years.77,78
Males and females with McCune-Albright syndrome have somatic gain-of-function mutations in GNAS1 (20q13.2) inducing mosaic Gαs activation, which, in turn, induces an elevated output of intracellular cyclic adenosine monophosphate (cAMP).79 Excessive production of cAMP and, therefore, the clinical spectrum of McCune-Albright syndrome (fibrous dysplasia of bone, café-au-lait skin macules, and hyper-functioning endocrinopathies) is mainly conditioned by the location and extent of tissues in which the GNAS1 mutation is expressed.80 In McCune-Albright syndrome endocrinopathies, gonadotropin-independent gonadal hyperfunction and peripheral precocious puberty are paradigmatic; the over-production of sex steroids in McCune-Albright syndrome, due to excessive cAMP synthesis in the gonads, is more common in girls, with about 85% of all girls with GNAS1 activation displaying oestrogen over-production and peripheral precocious puberty, compared with only 21% of boys with this condition showing excessive testosterone production.80,81
Familial male limited precocious puberty, or testo-toxicosis, is an infrequent form of due to heterozygous constitutively activating mutations of the luteinising hormone receptor gene (LHR) that can occur de novo or be inherited in an autosomal dominant pattern. This condition is distinguished by signs of virilisation before the age of 4 years, with mild testicular enlargement usually being observed.75,82 In boys, peripheral precocious puberty can also be due to mutations in NR0B1, which encodes DAX1. This is an X-linked recessive disorder that mainly presents as congenital adrenal hypoplasia.83
In males, acquired aetiology includes stromal cell tumours in the testes, mainly Leydig cell tumours, adrenal tumours, and extragonadal β-human chorionic gonadotropin-producing germ cell tumours mostly located in the liver, mediastinum, and brain. In girls, this acquired aetiology can include ovarian cysts and ovarian stromal cell tumours, predominantly granulosa cell tumours. Primary hypothyroidism and the exogenous administration of sex steroids are exceptional causes of peripheral precocious puberty in both sexes.75,84,85
The differential diagnosis between peripheral precocious puberty and central precocious puberty can be challenging; data on the emergence and progression of secondary sexual characteristics and the existence of family cases, together with the presence of additional pathology, can help to guide the diagnostic suspicion. The presence of café-au-lait spots and abdominal masses should be excluded. In boys, testicular palpation allows exclusion of testicular asymmetry and evaluation of testicular volume, which might be slightly larger than 4 mL only in familial male limited precocious puberty and some hCG-producing extragonadal tumours. The requirement of additional diagnostic tools that could include basal or stimulated hormonal studies, tumour markers, and radiological tests can be decided.84–86
Treatment of acquired peripheral precocious puberty should focus on the underlying pathology. Experience in treatment of congenital forms, such as McCune-Albright syndrome and familial male limited precocious puberty, is scarce due to the absence of randomised clinical trials, together with the impossibility of recruiting a relevant number of patients in the pilot studies that have been done.75,80,82
Delayed puberty
Delayed puberty is commonly defined as puberty commencing more than 2 SD later than the mean age for the population.6 Cutoffs used in clinical practice are the absence of testicular enlargement (volume <4 mL) in boys by the age of 14 years and no breast development in girls by the age of 13 years.87
The differential diagnosis of delayed puberty can be divided into three main categories: (1) hypergonadotropic hypogonadism, characterised by elevated luteinising hormone and follicle-stimulating hormone concentrations, due to gonadal disorders; (2) transient hypogonadotropic hypogonadism (or functional hypogonadotropic hypogonadism), in which pubertal delay is due to delayed maturation of the HPG axis secondary to underlying adverse conditions; and (3) permanent hypogonadotropic hypogonadism, with constitutively low luteinising hormone and follicle-stimulating hormone concentrations (figure 3). Young people and their parents should be questioned about a history or symptoms of chronic disease, with emphasis on disorders (eg, anorexia, inflammatory bowel disease, celiac disease, or thyroid disease), that could cause temporary delay of puberty (ie, functional hypogonadotropic hypogonadism), as well as medication usage, nutritional status, and psychosocial habits, which could lead also to delayed puberty.
Figure 3: Flowchart for the evaluation of a patient with delayed puberty.
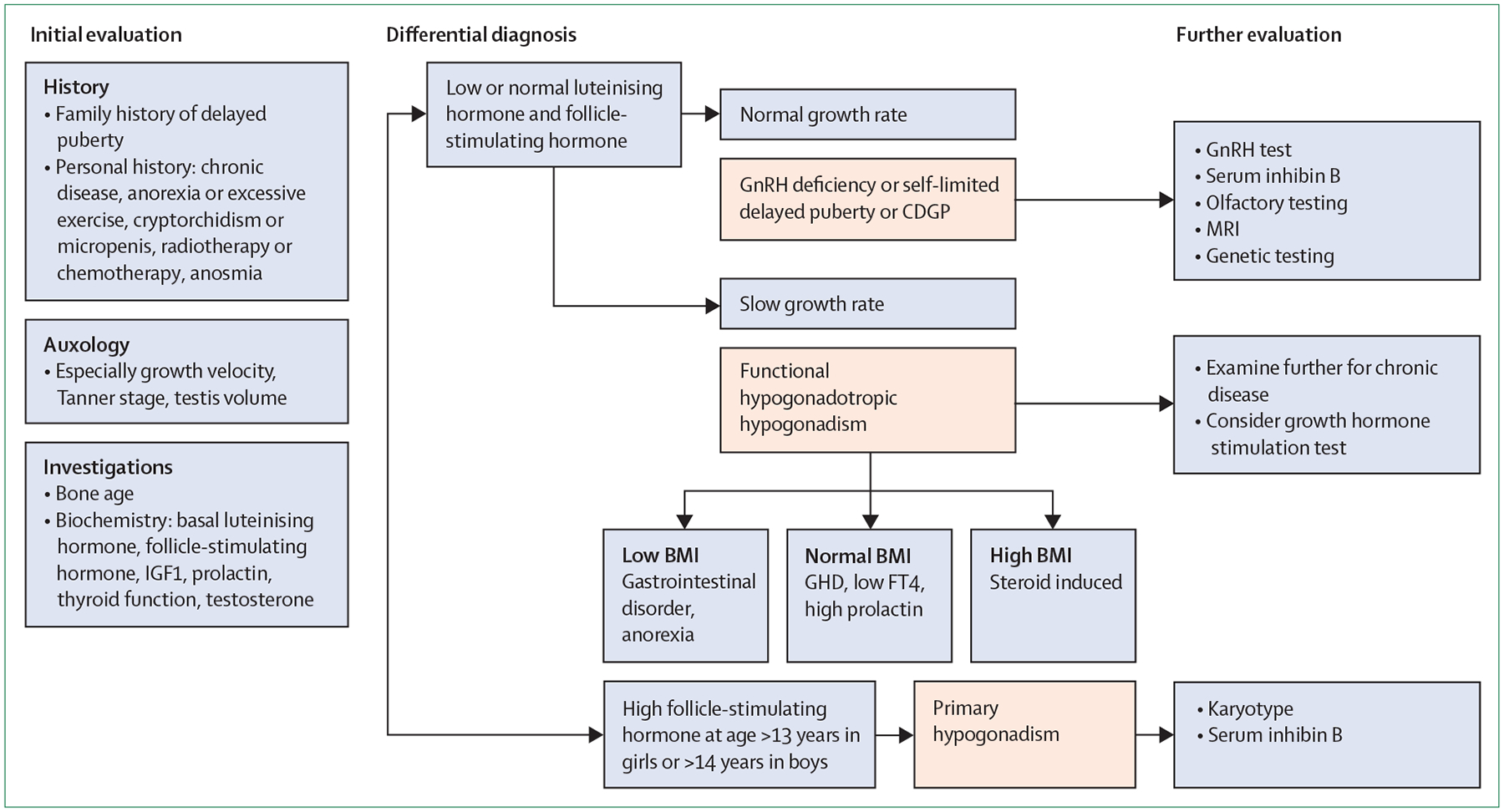
CDGP=constitutional delay of growth and puberty. CHH=congenital hypogonadotropic hypogonadism. FT4=free thyroxine. GHD=growth hormone deficiency. GnRH=gonadotropin-releasing hormone. hCG=human chorionic gonadotropin.
In this section, we aim to highlight the heterogeneity of genetic defects resulting in delayed and disordered puberty, with a special emphasis on congenital hypogonadotropic hypogonadism and the condition called self-limited delayed puberty, also known as constitutional delay.
Although congenital hypogonadotropic hypogonadism and self-limited delayed puberty present with delayed pubertal onset, these two conditions differ in clinical course and requirement for treatment.88 In congenital hypogonadotropic hypogonadism, or Kallmann syndrome (ie, congenital hypogonadotropic hypogonadism with anosmia), hormone therapy is usually needed for the induction of secondary sexual characteristics, whereas in self-limited delayed puberty, puberty will ultimately commence spontaneously. If congenital hypogonadotropic hypogonadism is diagnosed, treatment methods to allow optimisation of future fertility (particularly for boys) can be used—in the form of gonadotropins rather than sex steroids—for induction of puberty, and commenced earlier than the puberty induction regimen used for patients with self-limited delayed puberty.89 Moreover, precise diagnosis can be helpful to facilitate appropriate counselling on likelihood of inheritance of the condition within families.90
In congenital hypogonadotropic hypogonadism, different inheritance patterns, including autosomal dominant, autosomal recessive, and X-linked inheritance have been found. In the past few years, the traditional Mendelian view of congenital hypogonadotropic hypogonadism as a monogenic disorder has been revised after the identification of oligogenic forms in about 20% of patients.91 Furthermore, reversal of congenital hypogonadotropic hypogonadism occurs in about 10–20% of patients with congenital hypogonadotropic hypogonadism, which challenges the view that the condition is lifelong.92
More than 60 genes are associated with congenital hypogonadotropic hypogonadism (table).93 Causal genes have functions in (1) regulating GnRH neuronal development, migration, and maturation (eg, ANOS1, FGFR1, FGF17, FGF8, IL17RD, PROK2, PROKR2, HS6ST1, FEZF1, DCC, NTN1, NDNF, SOX10, DUSP6, FLRT3, SPRY4, KLB, WDR11, and CHD7), (2) GnRH neuronal activity (KISS1, KISS1R, TAC3, and TACR3), and (3) GnRH downstream function (GNRH1, GNRHR, FSHB, and LHB). Importantly, the majority of the known gene defects are associated with either syndromic presentation or other clinical features, which might be helpful for correct diagnosis (table).
Table:
Genes associated with congenital hypogonadotropic hypogonadism or self-limited delayed puberty
| Inheritance | Contributes to oligogenity | Kallmann syndrome | CHH | Reversal | Self-limited delayed puberty | Other phenotypic or syndromic features | Phenotype MIM ID | |
|---|---|---|---|---|---|---|---|---|
| AMH | AD | ·· | Yes | Yes | ·· | Yes | ·· | ·· |
| AMHR2 | AD | ·· | Yes | Yes | ·· | Yes | ·· | ·· |
| ANOS1 (KAL1) | XLR | Yes | Yes | Yes | Yes | Yes | Bimanual synkinesis, aplasia or hypoplasia of olfactory bulbs, unilateral renal aplasia | 308700 |
| AXL | AD | ·· | Yes | Yes | ·· | Yes | ·· | ·· |
| BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, BBS9, BBS10, TRIM32, BBS12 | AR, DR | Yes | ·· | Yes | ·· | ·· | Bardet-Biedl syndrome | 209900 |
| CCDC141 | AD, AR | ·· | Yes | Yes | ·· | Yes | ·· | ·· |
| CHD7 | AD | Yes | Yes | Yes | Yes | Yes | CHARGE syndrome | 612370 |
| CPE | AR | ·· | ·· | Yes | ·· | ·· | BDV syndrome | 114855 |
| DCAF17 | AR | ·· | ·· | Yes | ·· | ·· | Woodhouse-Sakati syndrome, alopecia, diabetes mellitus, mental retardation, deafness | 241080 |
| DCC/NTN1 | AD | Yes | Yes | Yes | ·· | Yes | Synkinesis, mid brain malformation, deafness | ·· |
| DMXL2 | AD, AR | ·· | ·· | Yes | ·· | ·· | PEPNS (polyendocrine polyneuropathy) | 616113 |
| DUSP6 | AD | ·· | Yes | Yes | ·· | ·· | Dental agenesis, syndactyly, color blindness | 615269 |
| EAP1 (IRF2BPL) | AD | ·· | ·· | ·· | ·· | Yes | ·· | 611720 |
| FEZF1 | AR | ·· | Yes | Yes | ·· | Yes | ·· | 616030 |
| FGF17 | AD | Yes | Yes | Yes | ·· | ·· | ·· | 615270 |
| FGF8 | AD | Yes | Yes | Yes | ·· | ·· | Combined pituitary hormone deficiency | 612702 |
| FGFR1 | AD | Yes | Yes | Yes | Yes | Yes | SOD, Hartsfield syndrome, split hand/foot malformation, combined pituitary hormone deficiency, bimanual synkinesis | 147950 |
| FLRT3 | AD | Yes | Yes | Yes | ·· | ·· | ·· | 615271 |
| FSHB | AR | ·· | ·· | Yes | ·· | ·· | ·· | 229070 |
| FTO | AD | ·· | ·· | ·· | ·· | Yes | ·· | 612938 |
| GNRH1 | AR | ·· | ·· | Yes | ·· | ·· | ·· | 614841 |
| GNRHR | AR | Yes | ·· | Yes | Yes | Yes | ·· | 146110 |
| HDAC8 | XLR | ·· | ·· | ·· | ·· | Cornelia de Lange syndrome | 300882 | |
| HESX1 | AD, AR | ·· | Yes | Yes | ·· | ·· | Combined pituitary hormone deficiency, SOD | 182230 |
| HFE | AD, AR | ·· | ·· | ·· | ·· | ·· | Hereditary hemochromatosis | 235200 |
| HS6ST1 | AD | Yes | Yes | Yes | Yes | Yes | ·· | 614880 |
| IGSF1 | XLR | ·· | ·· | Yes | ·· | Yes | Central hypothyroidism, macroorchidism | 300888 |
| IGSF10 | AD | ·· | ·· | ·· | ·· | Yes | ·· | 617351 |
| IL17RD (SEF) | AD, AR or digenic dominant | Yes | Yes | Yes | ·· | Yes | ·· | 615267 |
| KISS1 | AR | ·· | ·· | Yes | ·· | ·· | ·· | 614842 |
| KISS1R | AR | Yes | ·· | Yes | ·· | ·· | ·· | 614837 |
| KLB | AD, complex | Yes | Yes | Yes | Yes | Yes | Obesity, insulin resistance | ·· |
| LEP | AR | ·· | ·· | Yes | ·· | ·· | Obesity, recurrent respiratory infections | 614962 |
| LEPR | AR | Yes | ·· | Yes | ·· | Obesity, recurrent respiratory infections | 614963 | |
| LGR4 | AD | ·· | ·· | ·· | Yes | ·· | 619613 | |
| LHB | AR | ·· | ·· | Yes | ·· | ·· | ·· | 228300 |
| LHX | AR | ·· | ·· | Yes | ·· | ·· | Combined pituitary hormone deficiency, sensorineural deafness (variable) | 221750 |
| NDN, SNRPN | AD, deletion of the paternal copy | ·· | ·· | Yes | ·· | ·· | Prader-Willi syndrome | 176270 |
| NDNF | AD | ·· | Yes | Yes | ·· | Yes | ·· | 616506 |
| NR0B1 (DAX1) | XLR | ·· | ·· | Yes | ·· | ·· | Congenital adrenal hypoplasia | 300200 |
| NRP1 | AD | Yes | Yes | Yes | ·· | ·· | ·· | ·· |
| NRP2 | AD | Yes | Yes | Yes | ·· | ·· | ·· | ·· |
| NSMF (NELF) | AD | Yes | Yes | Yes | Yes | ·· | ·· | 614838 |
| OTUD4, RNF216 | AR | ·· | ·· | Yes | ·· | ·· | Gordon Holmes syndrome | 212840 |
| PCSK1 | AR | ·· | ·· | Yes | ·· | ·· | Obesity, small-intestinal dysfunction, complex endocrinopathies | 600955 |
| PHF6 | XLR | ·· | ·· | Yes | ·· | ·· | Börjeson-Forssman-Lehmann syndrome | 301900 |
| PLXNA1 | AD | Yes | Yes | Yes | ·· | Yes | ·· | |
| PLXNA3 | Complex | Yes | Yes | Yes | ·· | ·· | ·· | ·· |
| PNPLA6 | AR | ·· | ·· | Yes | ·· | ·· | Boucher-Neuhauser, Gordon Holmes, Oliver McFarlane, Lawrence Moon syndromes | 215470 |
| POLR3A | AR | ·· | ·· | Yes | ·· | ·· | Leukodystrophy, oligodontia, ataxia | 607694 |
| POLR3B | AR | ·· | ·· | Yes | ·· | ·· | Leukodystrophy, oligodontia, ataxia | 614381 |
| PROK2 | AD | Yes | Yes | Yes | ·· | ·· | ·· | 610628 |
| PROKR2 | AD | Yes | Yes | Yes | Yes | Yes | Combined pituitary hormone deficiency, synkinesis | 244200 |
| PROP1 | AR | ·· | ·· | Yes | ·· | ·· | Combined pituitary hormone deficiency | 262600 |
| RAB18 | AR | ·· | ·· | Yes | ·· | ·· | Warburg micro syndrome | 614222 |
| RAB3GAP1 | AR | ·· | ·· | Yes | ·· | ·· | Warburg micro syndrome | 600118 |
| RAB3GAP2 | AR | ·· | ·· | Yes | ·· | ·· | Martsolf syndrome | 212720 |
| REV3L/PLXND1 | AD | ·· | Yes | ·· | Yes | ·· | Moebius syndrome | ·· |
| RMB28 | AR | ·· | ·· | ·· | ·· | ·· | Alopecia, neurological defects, and endocrinopathy syndrome | 612079 |
| SEMA3A | AD | Yes | Yes | ·· | ·· | Yes | ·· | 614897 |
| SEMA3F | complex | Yes | Yes | Yes | ·· | ·· | ·· | ·· |
| SEMA3E | AD | ·· | Yes | Yes | ·· | ·· | CHARGE syndrome | 608166 |
| SEMA7A * | DR | Yes | Yes | Yes | ·· | ·· | Pending full validation | ·· |
| SMCHD1 | AD | ·· | ·· | Yes | ·· | ·· | Bosma arhinia microphthalmia syndrome | 603457 |
| SOX10 | AD | ·· | Yes | Yes | ·· | ·· | Waardenburg syndrome type 2E | 611584 |
| SOX2 | AD | ·· | ·· | Yes | ·· | ·· | Optic nerve hypoplasia, CNS abnormalities | 206900 |
| SOX3 | XLR | Yes | ·· | Yes | ·· | ·· | Intellectual disability, craniofacial abnormalities, multiple pituitary hormone deficiency | ·· |
| SPRY4 | AD | Yes | Yes | Yes | ·· | ·· | Hearing loss | 615266 |
| SRA1 | AD | ·· | ·· | Yes | ·· | ·· | ·· | ·· |
| STUB1 | AR | ·· | ·· | ·· | ·· | ·· | Spinocerebellar ataxia | 615768 |
| TAC3 | AR | Yes | Yes | Yes | Yes | Yes | ·· | 614839 |
| TACR3 | AR | Yes | ·· | Yes | Yes | Yes | ·· | 614840 |
| TBC1D20 | AR | ·· | ·· | Yes | ·· | ·· | Warburg micro syndrome | 615663 |
| TCF12 | AD | ·· | Yes | Yes | ·· | ·· | Craniosynostosis 3 | 615314 |
| TUBB3 | AD | ·· | Yes | Yes | ·· | ·· | Congenital fibrosis of the extraocular muscles | 600638 |
| WDR11 | AD | Yes | Yes | Yes | Yes | ·· | Combined pituitary hormone deficiency | 614858 |
AD=autosomal dominant. AR=autosomal recessive. CHARGE=coloboma of the eye, heart defects, atresia choanae, retardation of growth, genital abnormalities, and ear abnormalities. CHH=congenital hypogonadotropic hypogonadism. DR=digenic recessive. XLR=X-linked recessive. MIM=Mendelian Inheritance in Man.
Pending full validation.
The underlying genetic basis of self-limited delayed puberty remains less completely understood. Self-limited delayed puberty is often seen in multiple generations of the same family; 50–75% of patients with self-limited delayed puberty have a positive family history.94 Self-limited delayed puberty and congenital hypogonadotropic hypogonadism might sometimes have an overlapping genetic basis; for instance, homozygous mutations in genes such as GNRHR, HS6ST1, TAC3, and TAC3R, cause congenital hypogonadotropic hypogonadism, whereas heterozygous carriage of the same variants is associated with self-limited delayed puberty.6
To date, at least 24 genes have been identified as contributing to self-limited delayed puberty, including those found in relatives of congenital hypogonadotropic hypogonadism probands and others identified from large cohorts of families segregating exclusively with self-limited delayed puberty.95 As with congenital hypogonadotropic hypogonadism, genes confined to self-limited delayed puberty have functions related to regulation of GnRH neuronal development and migration (eg, IGSF10, LGR4, CCDC141),96 GnRH upstream control,97 or GnRH downstream action and energy metabolism.98
Central to the evaluation process for diagnosing congenital hypogonadotropic hypogonadism or self-limited delayed puberty is the exclusion of differential diagnoses (figure 3).87 The diagnosis of functional hypogonadotropic hypogonadism should be guided by clinical signs and symptoms, because many underlying causes of delayed puberty can be found in this category. First-line biochemical screening tests can include basic blood biochemistry, kidney, liver, and thyroid function tests, as well as screening for coeliac disease. Further testing should be based on the suspected diagnosis by using an approach integrating the patient’s family history, clinical signs and symptoms, auxological data, and biochemistry. At times, several rounds of analyses are needed until the definite underlying cause for delayed puberty is found. Of note, although a variety of clinical and biochemical parameters are available to assist with diagnosis of delayed puberty, none of these can reliably distinguish congenital hypogonadotropic hypogonadism from self-limited delayed puberty in adolescence. Both conditions might present with similar hormonal profiles, defined by low gonadotropin and sex steroid concentrations. Micropenis or the history of cryptorchidism in males are suggestive signs pointing to diagnosis of congenital hypogonadotropic hypogonadism.
As per genetic testing, the first step is to establish the inheritance pattern. Testing should also be guided by the presence of syndromic or other phenotypical features (table).99 Syndromic features might suggest a contiguous gene syndrome for which a karyotype or comparative genomic hybridisation (CGH) array analysis are useful for identifying chromosomal abnormalities. Despite the rapid expansion of knowledge in the area, approximately only 50% of patients receive a precise genetic diagnosis. Although there is overlap in the genetic background of congenital hypogonadotropic hypogonadism and self-limited delayed puberty, the majority of mutations are distinct between these two conditions (table).95,100
Conclusions
The progress in our understanding of the mechanisms controlling puberty is a success story in modern neuroendocrinology. Discoveries on the genetic determinants of puberty are now being translated into clinical practice to allow a better comprehension of the basis of disordered pubertal development, which has a complex genetic landscape that is being revealed by the increasing genetic data linked to clinical information. In parallel, as exemplified by KISS1 and TAC3 mutations, clinical findings have fuelled basic mechanistic studies, using suitable models, which have been instrumental to further expose the neuroendocrine circuitry responsible for pubertal maturation, and its modulation by nutritional and other environmental cues. This bidirectional interaction between basic and clinical research will probably continue in coming years, thereby making it possible for us to have an integral understanding of puberty. Novel tools for sophisticated exploration of pubertal neuroendocrine circuits in vivo in suitable preclinical models, from fibre photometry to genetic editing of key loci in Kiss1 (and related) neurons, will help to deepen our knowledge of the physiological control of puberty. In turn, massive genetic studies in patients with pubertal disorders, coupled to big data (including clinical) analysis, will permit further definition of the pathogenic basis of pubertal alterations. Basic and clinical studies on the environmental determinants of puberty, from nutritional conditions to endocrine disruptors, are needed also and might allow novel insights into key modifiers of pubertal timing and potential effects in long-term health.
Panel 1: Causes of central precocious puberty.
CNS lesions
Congenital
Hypothalamic hamartoma
Arachnoid cyst
Hydrocephalus
Septo-optic dysplasia
Chiari malformations
Myelomeningocele
Acquired
Tumours: low grade gliomas, ependymoma, pinealoma, craniopharyngioma, germinoma
Cranial irradiation
Traumatic brain injury
CNS infection
Granulomatous disease
Intracranial bleeding
Cerebral palsy secondary to perinatal hypoxic–ischemic encephalopathy
No documented CNS lesions
Genetic
KISS1 gain-of-function mutations
KISS1R gain-of-function mutations
MKRN3 loss-of-function mutations
DLK1 loss-of-function mutations
Chromosomal abnormalities
Environmental
International adoption (ie, adoption of a child from another country)
Endocrine-disrupting chemicals
Obesity
Idiopathic
A diagnosis of exclusion
Secondary central precocious puberty
Following treatment of peripheral precocious puberty, after withdrawal of chronic sex hormone exposure
Panel 2: Causes of peripheral precocious puberty.
Boys
Congenital
Congenital adrenal hyperplasia
Familial male limited precocious puberty
McCune-Albright syndrome
NR0B1 mutations
Acquired
Testicular tumour
Adrenal tumour
β-human chorionic gonadotropin secreting tumours (mediastinum, liver, brain)
Exogenous exposure to sex steroids
Primary hypothyroidism (Van Wyk-Grumbach syndrome)
Girls
Congenital
McCune-Albright syndrome
Acquired
Ovarian cyst
Ovarian tumour
Exogenous exposure to sex steroids
Primary hypothyroidism (Van Wyk-Grumbach syndrome)
Search strategy and selection criteria.
We did a non-systematic search on MEDLINE of original articles and reviews, published from Jan 1, 2006, to Sept 1, 2022. We searched multiple terms, including, “puberty”, “kisspeptins”, “neuroendocrine-control”, “genetics”, “epigenetics”, “obesity”, “metabolic homeostasis”, “precocious puberty”, “delayed puberty”, “hypogonadism and mutations”, variably connected by Boolean operators. We filtered searches using our previous knowledge, focusing on clinical and preclinical studies, and addressing mechanistic, neuroendocrine, and clinical aspects of pubertal maturation. We also considered studies on genetics determinants of normal and pathological puberty.
Acknowledgments
The contributions of the members within each author’s laboratory to the work summarised in this Review are cordially appreciated. We acknowledge the assistance of Dr Juan M Castellano in preparation of graphical materials and Dr Julie Chowen in critical read and final style revision of the manuscript. The work from the authors’ laboratories summarised in this Review was supported by grants BFU2017-83934-P and PID2020-118660GB-I00 (to MT-S; Agencia Estatal de Investigación, Spain; co-funded with EU funds from FEDER Program); projects FIS-PI19/00166 (to JA) and PIE14-00005 (to MT-S; Flexi-Met, Instituto de Salud Carlos III, Ministerio de Sanidad, Spain); Centro de Investigación Biomédica en Red Fisiopatología de Obesidad y Nutrición (CIBEROBN; to JA and MT-S; Instituto de Salud Carlos III), Projects P12-FQM-01943 and P18-RT-4093 (MT-S, Junta de Andalucía, Spain); EU research contract GAP-2014-655232 to MT-S; grants 1R01HD084542 and 1R21AG061141 to AL, and R01HD082314 and R21HD098684 to UBK (NIH); and 8P51OD011092 grant for the operation of the Oregon National Primate Research Center (to AL). CIBER is an initiative of Instituto de Salud Carlos III.
Footnotes
Declaration of interests
UBK has received honoraria for lectures related to puberty from Novartis/Sandoz, National Institutes of Health (NIH), and the universities of Toronto, Vanderbilt, and Chicago. All other authors declare no competing interests.
References
- 1.Parent AS, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr Rev 2003; 24: 668–93. [DOI] [PubMed] [Google Scholar]
- 2.Avendaño MS, Vazquez MJ, Tena-Sempere M. Disentangling puberty: novel neuroendocrine pathways and mechanisms for the control of mammalian puberty. Hum Reprod Update 2017; 23: 737–63. [DOI] [PubMed] [Google Scholar]
- 3.Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child 1969; 44: 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child 1970; 45: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soriano-Guillén L, Corripio R, Labarta JI, et al. Central precocious puberty in children living in Spain: incidence, prevalence, and influence of adoption and immigration. J Clin Endocrinol Metab 2010; 95: 4305–13. [DOI] [PubMed] [Google Scholar]
- 6.Howard SR, Dunkel L. Delayed puberty-phenotypic diversity, molecular genetic mechanisms, and recent discoveries. Endocr Rev 2019; 40: 1285–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brito VN, Canton APM, Seraphim CE, et al. The congenital and acquired mechanisms implicated in the etiology of central precocious puberty. Endocr Rev 2022; published online Aug 5. 10.1210/endrev/bnac020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reinehr T, Roth CL. Is there a causal relationship between obesity and puberty? Lancet Child Adolesc Health 2019; 3: 44–54. [DOI] [PubMed] [Google Scholar]
- 9.Eckert-Lind C, Busch AS, Petersen JH, et al. Worldwide secular trends in age at pubertal onset assessed by breast development among girls: a systematic review and meta-analysis. JAMA Pediatr 2020; 174: e195881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day FR, Elks CE, Murray A, Ong KK, Perry JR. Puberty timing associated with diabetes, cardiovascular disease and also diverse health outcomes in men and women: the UK Biobank study. Sci Rep 2015; 5: 11208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plant TM, Barker-Gibb ML. Neurobiological mechanisms of puberty in higher primates. Hum Reprod Update 2004; 10: 67–77. [DOI] [PubMed] [Google Scholar]
- 12.Terasawa E, Fernandez DL. Neurobiological mechanisms of the onset of puberty in primates. Endocr Rev 2001; 22: 111–51. [DOI] [PubMed] [Google Scholar]
- 13.Sobrino V, Avendaño MS, Perdices-López C, Jimenez-Puyer M, Tena-Sempere M. Kisspeptins and the neuroendocrine control of reproduction: Recent progress and new frontiers in kisspeptin research. Front Neuroendocrinol 2022; 65: 100977. [DOI] [PubMed] [Google Scholar]
- 14.Clarkson J, Boon WC, Simpson ER, Herbison AE. Postnatal development of an estradiol-kisspeptin positive feedback mechanism implicated in puberty onset. Endocrinology 2009; 150: 3214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA 2003; 100: 10972–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med 2003; 349: 1614–27. [DOI] [PubMed] [Google Scholar]
- 17.Pinilla L, Aguilar E, Dieguez C, Millar RP, Tena-Sempere M. Kisspeptins and reproduction: physiological roles and regulatory mechanisms. Physiol Rev 2012; 92: 1235–316. [DOI] [PubMed] [Google Scholar]
- 18.Clarkson J, Herbison AE. Postnatal development of kisspeptin neurons in mouse hypothalamus; sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology 2006; 147: 5817–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bentsen AH, Ansel L, Simonneaux V, Tena-Sempere M, Juul A, Mikkelsen JD. Maturation of kisspeptinergic neurons coincides with puberty onset in male rats. Peptides 2010; 31: 275–83. [DOI] [PubMed] [Google Scholar]
- 20.Clarkson J, Han SY, Piet R, et al. Definition of the hypothalamic GnRH pulse generator in mice. Proc Natl Acad Sci USA 2017; 114: E10216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehman MN. Origins of the ‘KNDy hypothesis’ of GnRH pulse generation. Nat Rev Endocrinol 2022; 18: 521. [DOI] [PubMed] [Google Scholar]
- 22.Adekunbi DA, Li XF, Li S, et al. Role of amygdala kisspeptin in pubertal timing in female rats. PLoS One 2017; 12: e0183596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roa J, Ruiz-Cruz M, Ruiz-Pino F, et al. Dicer ablation in KISS1 neurons impairs puberty and fertility preferentially in female mice. Nat Commun 2022; 13: 4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Messina A, Langlet F, Chachlaki K, et al. A microRNA switch regulates the rise in hypothalamic GnRH production before puberty. Nat Neurosci 2016; 19: 835–44. [DOI] [PubMed] [Google Scholar]
- 25.Topaloglu AK, Reimann F, Guclu M, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet 2009; 41: 354–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navarro VM, Ruiz-Pino F, Sánchez-Garrido MA, et al. Role of neurokinin B in the control of female puberty and its modulation by metabolic status. J Neurosci 2012; 32: 2388–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maguire CA, Song YB, Wu M, et al. TAC1 signaling is required for sexual maturation and responsiveness of GnRH neurons to kisspeptin in the male mouse. Endocrinology 2017; 158: 2319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manfredi-Lozano M, Roa J, Ruiz-Pino F, et al. Defining a novel leptin-melanocortin-kisspeptin pathway involved in the metabolic control of puberty. Mol Metab 2016; 5: 844–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam BYH, Williamson A, Finer S, et al. MC3R links nutritional state to childhood growth and the timing of puberty. Nature 2021; 599: 436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vazquez MJ, Daza-Dueñas S, Tena-Sempere M. Emerging roles of epigenetics in the control of reproductive function: focus on central neuroendocrine mechanisms. J Endocr Soc 2021; 5: bvab152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lomniczi A, Loche A, Castellano JM, et al. Epigenetic control of female puberty. Nat Neurosci 2013; 16: 281–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wright H, Aylwin CF, Toro CA, Ojeda SR, Lomniczi A. Polycomb represses a gene network controlling puberty via modulation of histone demethylase Kdm6b expression. Sci Rep 2021; 11: 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toro CA, Wright H, Aylwin CF, Ojeda SR, Lomniczi A. Trithorax dependent changes in chromatin landscape at enhancer and promoter regions drive female puberty. Nat Commun 2018; 9: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lomniczi A, Wright H, Castellano JM, et al. Epigenetic regulation of puberty via Zinc finger protein-mediated transcriptional repression. Nat Commun 2015; 6: 10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vazquez MJ, Toro CA, Castellano JM, et al. SIRT1 mediates obesityand nutrient-dependent perturbation of pubertal timing by epigenetically controlling Kiss1 expression. Nat Commun 2018; 9: 4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Almstrup K, Lindhardt Johansen M, Busch AS, et al. Pubertal development in healthy children is mirrored by DNA methylation patterns in peripheral blood. Sci Rep 2016; 6: 28657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han L, Zhang H, Kaushal A, et al. Changes in DNA methylation from pre- to post-adolescence are associated with pubertal exposures. Clin Epigenetics 2019; 11: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen S, Refaey H, Mukherjee N, et al. Age at onset of different pubertal signs in boys and girls and differential DNA methylation at age 10 and 18 years: an epigenome-wide follow-up study. Hum Reprod Open 2020; 2020: hoaa006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moore SR, Humphreys KL, Colich NL, et al. Distinctions between sex and time in patterns of DNA methylation across puberty. BMC Genomics 2020; 21: 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huan T, Mendelson M, Joehanes R, et al. Epigenome-wide association study of DNA methylation and microRNA expression highlights novel pathways for human complex traits. Epigenetics 2020; 15: 183–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bessa DS, Maschietto M, Aylwin CF, et al. Methylome profiling of healthy and central precocious puberty girls. Clin Epigenetics 2018; 10: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vazquez MJ, Velasco I, Tena-Sempere M. Novel mechanisms for the metabolic control of puberty: implications for pubertal alterations in early-onset obesity and malnutrition. J Endocrinol 2019; 242: R51–65. [DOI] [PubMed] [Google Scholar]
- 43.Frisch RE, Revelle R, Cook S. Components of weight at menarche and the initiation of the adolescent growth spurt in girls: estimated total water, llean body weight and fat. Hum Biol 1973; 45: 469–83. [PubMed] [Google Scholar]
- 44.Hill JW, Elias CF. Neuroanatomical framework of the metabolic control of reproduction. Physiol Rev 2018; 98: 2349–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martos-Moreno GA, Chowen JA, Argente J. Metabolic signals in human puberty: effects of over and undernutrition. Mol Cell Endocrinol 2010; 324: 70–81. [DOI] [PubMed] [Google Scholar]
- 46.De Leonibus C, Marcovecchio ML, Chiavaroli V, de Giorgis T, Chiarelli F, Mohn A. Timing of puberty and physical growth in obese children: a longitudinal study in boys and girls. Pediatr Obes 2014; 9: 292–99. [DOI] [PubMed] [Google Scholar]
- 47.Tena-Sempere M Ghrelin, the gonadal axis and the onset of puberty. Endocr Dev 2013; 25: 69–82. [DOI] [PubMed] [Google Scholar]
- 48.Codner E, Merino PM, Tena-Sempere M. Female reproduction and type 1 diabetes: from mechanisms to clinical findings. Hum Reprod Update 2012; 18: 568–85. [DOI] [PubMed] [Google Scholar]
- 49.Xu J, Ji J, Yan XH. Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr 2012; 52: 373–81. [DOI] [PubMed] [Google Scholar]
- 50.Roa J, Garcia-Galiano D, Varela L, et al. The mammalian target of rapamycin as novel central regulator of puberty onset via modulation of hypothalamic KISS1 system. Endocrinology 2009; 150: 5016–26. [DOI] [PubMed] [Google Scholar]
- 51.Roa J, Barroso A, Ruiz-Pino F, et al. Metabolic regulation of female puberty via hypothalamic AMPK-kisspeptin signaling. Proc Natl Acad Sci USA 2018; 115: E10758–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heras V, Castellano JM, Fernandois D, et al. Central ceramide signaling mediates obesity-induced precocious puberty. Cell Metab 2020; 32: 951–66.e8. [DOI] [PubMed] [Google Scholar]
- 53.Ratra DV, Elias CF. Chemical identity of hypothalamic neurons engaged by leptin in reproductive control. J Chem Neuroanat 2014; 61–62: 233–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ross RA, Leon S, Madara JC, et al. PACAP neurons in the ventral premammillary nucleus regulate reproductive function in the female mouse. eLife 2018; 7: e35960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Franssen D, Barroso A, Ruiz-Pino F, et al. AMP-activated protein kinase (AMPK) signaling in GnRH neurons links energy status and reproduction. Metabolism 2021; 115: 154460. [DOI] [PubMed] [Google Scholar]
- 56.Perdices-Lopez C, Avendaño MS, Barroso A, et al. Connecting nutritional deprivation and pubertal inhibition via GRK2-mediated repression of kisspeptin actions in GnRH neurons. Metabolism 2022; 129: 155141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roberts SA, Kaiser UB. Genetics in endocrinology: Genetic etiologies of central precocious puberty and the role of imprinted genes. Eur J Endocrinol 2020; 183: R107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teles MG, Bianco SD, Brito VN, et al. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med 2008; 358: 709–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bianco SD, Vandepas L, Correa-Medina M, et al. KISS1R intracellular trafficking and degradation: effect of the Arg386Pro disease-associated mutation. Endocrinology 2011; 152: 1616–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Silveira LG, Noel SD, Silveira-Neto AP, et al. Mutations of the KISS1 gene in disorders of puberty. J Clin Endocrinol Metab 2010; 95: 2276–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abreu AP, Dauber A, Macedo DB, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med 2013; 368: 2467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Valadares LP, Meireles CG, De Toledo IP, et al. MKRN3 mutations in central precocious puberty: a systematic review and meta-analysis. J Endocr Soc 2019; 3: 979–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seraphim CE, Canton APM, Montenegro L, et al. Genotypephenotype correlations in central precocious puberty caused by MKRN3 mutations. J Clin Endocrinol Metab 2021; 106: 1041–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heras V, Sangiao-Alvarellos S, Manfredi-Lozano M, et al. Hypothalamic miR-30 regulates puberty onset via repression of the puberty-suppressing factor, MKRN3. PLoS Biol 2019; 17: e3000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abreu AP, Toro CA, Song YB, et al. MKRN3 inhibits the reproductive axis through actions in kisspeptin-expressing neurons. J Clin Invest 2020; 130: 4486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yellapragada V, Liu X, Lund C, et al. mkrn3 interacts with several proteins implicated in puberty timing but does not influence GNRH1 expression. Front Endocrinol (Lausanne) 2019; 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li C, Lu W, Yang L, et al. MKRN3 regulates the epigenetic switch of mammalian puberty via ubiquitination of MBD3. Natl Sci Rev 2020; 7: 671–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li C, Han T, Li Q, et al. MKRN3-mediated ubiquitination of Poly(A)-binding proteins modulates the stability and translation of GNRH1 mRNA in mammalian puberty. Nucleic Acids Res 2021; 49: 3796–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roberts SA, Naulé L, Chouman S, et al. Hypothalamic overexpression of makorin ring finger protein 3 results in delayed puberty in female mice. Endocrinology 2022; 163: bqac132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dauber A, Cunha-Silva M, Macedo DB, et al. Paternally inherited dlk1 deletion associated with familial central precocious puberty. J Clin Endocrinol Metab 2017; 102: 1557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gomes LG, Cunha-Silva M, Crespo RP, et al. DLK1 Is a novel link between reproduction and metabolism. J Clin Endocrinol Metab 2019; 104: 2112–20. [DOI] [PubMed] [Google Scholar]
- 72.Montenegro L, Labarta JI, Piovesan M, et al. Novel genetic and biochemical findings of dlk1 in children with central precocious puberty: a Brazilian-Spanish study. J Clin Endocrinol Metab 2020; 105: dgaa461. [DOI] [PubMed] [Google Scholar]
- 73.Popovic J, Geffner ME, Rogol AD, et al. Gonadotropin-releasing hormone analog therapies for children with central precocious puberty in the United States. Front Pediatr 2022; 10: 968485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Soriano-Guillén L, Argente J. Central precocious puberty, functional and tumor-related. Best Pract Res Clin Endocrinol Metab 2019; 33: 101262. [DOI] [PubMed] [Google Scholar]
- 75.Haddad NG, Eugster EA. Peripheral precocious puberty including congenital adrenal hyperplasia: causes, consequences, management and outcomes. Best Pract Res Clin Endocrinol Metab 2019; 33: 101273. [DOI] [PubMed] [Google Scholar]
- 76.Atay Z, Yesilkaya E, Erdeve SS, et al. The etiology and clinical features of non-CAH gonadotropin-independent precocious puberty: a multicenter study. J Clin Endocrinol Metab 2016; 101: 1980–88. [DOI] [PubMed] [Google Scholar]
- 77.El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet 2017; 390: 2194–210. [DOI] [PubMed] [Google Scholar]
- 78.Merke DP, Auchus RJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med 2020; 383: 1248–61. [DOI] [PubMed] [Google Scholar]
- 79.Boyce AM, Collins MT. Fibrous dysplasia/McCune-Albright syndrome: a rare, mosaic disease of Gαs activation. Endocr Rev 2020; 41: 345–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spencer T, Pan KS, Collins MT, Boyce AM. The clinical spectrum of McCune-Albright syndrome and its management. Horm Res Paediatr 2019; 92: 347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boyce AM, Chong WH, Shawker TH, et al. Characterization and management of testicular pathology in McCune-Albright syndrome. J Clin Endocrinol Metab 2012; 97: E1782–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yuan X, Chen R, Zhang Y, Yang X, Lin X. Long-term treatment with letrozole in a boy with familial male-limited precocious puberty. Front Endocrinol (Lausanne) 2022; 13: 906852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nagel SA, Hartmann MF, Riepe FG, Wudy SA, Wabitsch M. Gonadotropin- and adrenocorticotropic hormone-independent precocious puberty of gonadal origin in a patient with adrenal hypoplasia congenita due to DAX1 gene mutation—a case report and review of the literature: implications for the pathomechanism. Horm Res Paediatr 2019; 91: 336–45. [DOI] [PubMed] [Google Scholar]
- 84.Cheuiche AV, da Silveira LG, de Paula LCP, Lucena IRS, Silveiro SP. Diagnosis and management of precocious sexual maturation: an updated review. Eur J Pediatr 2021; 180: 3073–87. [DOI] [PubMed] [Google Scholar]
- 85.Soriano-Guillén L, Sarafoglou K, Argente J. Precocious puberty. In: Sarafoglou K, Hoffmann GF, Roth KS, eds. Textbook Pediatric Endocrinology and Inborn Errors of Metabolism. 2nd edn. New York: McGraw Hill, 2017: 643–61. [Google Scholar]
- 86.Bradley SH, Lawrence N, Steele C, Mohamed Z. Precocious puberty. BMJ 2020; 368: l6597. [DOI] [PubMed] [Google Scholar]
- 87.Palmert MR, Dunkel L. Clinical practice. Delayed puberty. N Engl J Med 2012; 366: 443–53. [DOI] [PubMed] [Google Scholar]
- 88.Young J, Xu C, Papadakis GE, et al. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev 2019; 40: 669–710. [DOI] [PubMed] [Google Scholar]
- 89.Raivio T, Wikström AM, Dunkel L. Treatment of gonadotropindeficient boys with recombinant human FSH: long-term observation and outcome. Eur J Endocrinol 2007; 156: 105–11. [DOI] [PubMed] [Google Scholar]
- 90.Maione L, Dwyer AA, Francou B, et al. Genetics in endocrinology: genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol 2018; 178: R55–80. [DOI] [PubMed] [Google Scholar]
- 91.Pitteloud N, Quinton R, Pearce S, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest 2007; 117: 457–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Raivio T, Falardeau J, Dwyer A, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med 2007; 357: 863–73. [DOI] [PubMed] [Google Scholar]
- 93.Boehm U, Bouloux PM, Dattani MT, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism—pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2015; 11: 547–64. [DOI] [PubMed] [Google Scholar]
- 94.Wehkalampi K, Widén E, Laine T, Palotie A, Dunkel L. Patterns of inheritance of constitutional delay of growth and puberty in families of adolescent girls and boys referred to specialist pediatric care. J Clin Endocrinol Metab 2008; 93: 723–28. [DOI] [PubMed] [Google Scholar]
- 95.Saengkaew T, Patel HR, Banerjee K, et al. Genetic evaluation supports differential diagnosis in adolescent patients with delayed puberty. Eur J Endocrinol 2021; 185: 617–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Howard SR, Guasti L, Ruiz-Babot G, et al. IGSF10 mutations dysregulate gonadotropin-releasing hormone neuronal migration resulting in delayed puberty. EMBO Mol Med 2016; 8: 626–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mancini A, Howard SR, Cabrera CP, et al. EAP1 regulation of GnRH promoter activity is important for human pubertal timing. Hum Mol Genet 2019; 28: 1357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Howard SR, Guasti L, Poliandri A, et al. Contributions of function-altering variants in genes implicated in pubertal timing and body mass for self-limited delayed puberty. J Clin Endocrinol Metab 2018; 103: 649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Butz H, Nyírő G, Kurucz PA, Likó I, Patócs A. Molecular genetic diagnostics of hypogonadotropic hypogonadism: from panel design towards result interpretation in clinical practice. Hum Genet 2021; 140: 113–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cassatella D, Howard SR, Acierno JS, et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. Eur J Endocrinol 2018; 178: 377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]