

Abstract
Periodontitis and caries are driven by complex interactions between the oral microbiome and host factors, i.e. inflammation and dietary sugars, respectively. Animal models have been instrumental in our mechanistic understanding of these oral diseases, although no single model can faithfully reproduce all aspects of a given human disease. This review discusses evidence that the utility of an animal model lies in its capacity to address a specific hypothesis and, therefore, different aspects of a disease can be investigated using distinct and complementary models. As in vitro systems cannot replicate the complexity of in vivo host–microbe interactions and human research is typically correlative, model organisms—their limitations notwithstanding—remain essential in proving causality, identifying therapeutic targets, and evaluating the safety and efficacy of novel treatments. To achieve broader and deeper insights into oral disease pathogenesis, animal model-derived findings can be synthesized with data from in vitro and clinical research. In the absence of better mechanistic alternatives, dismissal of animal models on fidelity issues would impede further progress to understand and treat oral disease.
Keywords: animal models, oral microbiome, periodontitis, dental caries, therapeutic targets, dysbiosis
This review discusses the utility of animal models in addressing specific mechanistic hypotheses related to oral microbiome-driven diseases, leading to new biological concepts and identifying novel targets for therapeutic interventions in human patients.
Introduction
In the context of genetics and environmental exposures of the host, the oral microbiome contributes considerably to oral and systemic health and disease (Barros and Offenbacher 2009, Gomez et al. 2017, Hajishengallis and Lamont 2021). Despite the development of high-throughput omics technologies (genomics, epigenomics, metagenomics, transcriptomics, proteomics, and metabolomics) and their application in human disease research, model organisms remain valuable, if not essential, to understanding the human condition. Studies in human populations and in in vitro cell systems alone, even when complemented with computer modeling, are unlikely to obviate the need for animal experimentation to identify potential mechanisms that underpin dysbiosis, dysfunction, and disease or maintain homeostasis and health. This is in part because human studies are mostly correlative and do not typically address cause-and-effect relationships. Whereas the testing of new drugs in interventional human studies can address causality, this normally cannot occur before supportive evidence on efficacy is obtained from studies in model organisms (Hajishengallis et al. 2015, Nadeau and Auwerx 2019). Moreover, even if human research could become sufficient by itself to enable deep understanding of human biology, animal studies would still be necessary to provide preliminary information on the safety of candidate therapeutic compounds (Graves et al. 2008). In vitro reductionist systems, even when combining microbial and host components, do not currently possess adequate complexity to mimic the cross-interactions of host tissues and recruited immune cells with polymicrobial communities. Of course, recent advances in 3D microfluidic systems (tissue-and organ-on-a-chip models) hold significant promise for the future, especially if they could functionally integrate in a single platform multiple tissues (or organs) including immune cells (Sung et al. 2019, Schmidt et al. 2020, Leung et al. 2022). Nevertheless, infections induce complex and dynamic processes, such as emergency myelopoiesis, and stimulate communication between challenged tissues and organs, including those involved in the production and expansion of different immune lineages, such as the bone marrow and lymph nodes. These aspects will be difficult to be modeled sufficiently even by the most sophisticated in vitro systems. Nevertheless, experiments in animal models have limitations with regard to human physiological relevance, and hence must be complemented by human studies for validation. Figure 1 depicts the utility and advantages of animal model organisms in comparison to in vitro models and human studies.
Figure 1.

Comparison of experimental animals vs. in vitro models and human studies in terms of the indicated parameters. Model organisms represent a point on a spectrum of assay models ranging from the experimentally tractable in vitro systems, through the biological complexity of animals, over to biologically relevant human studies. Perhaps the greatest advantage of animal model studies is their capacity to address mechanistic questions and test cause-and-effect relationships. L, low; H, high.
The utility of validated animal models (Box 1) in microbe-driven oral diseases became evident decades ago and some examples are given here. In pioneering studies in hamsters and other rodents in the 1950s, Keyes and Fitzgerald demonstrated that dental caries is mediated and transmitted (to other hosts) by bacteria. They, moreover, dissected the role of the implicated bacteria (streptococci) from that of genetics and diet (Keyes 1959, Fitzgerald and Keyes 1960) (Fig. 2). Ironically, Keyes and Fitzgerald did not realize that the cariogenic streptococci they had been investigating were similar to a species isolated in 1924 from human carious lesions, named Streptococcus mutans, by J.K. Clarke (Tanzer 1995). In 1976, immunologists at the University of Alabama at Birmingham, essentially the birthplace of the field of Mucosal Immunology, reported that ingestion of killed S. mutans cells via the drinking water induced secretory IgA (S-IgA) antibodies in saliva and milk, and conferred protection from dental caries in rats (Michalek et al. 1976). This pivotal preclinical study not only provided proof-of-concept for a mucosal anticaries vaccine but laid the foundation for establishing the concept of the common, yet compartmentalized, mucosal immune system. In the years that followed, oral immunization of humans with mutans streptococcal antigens was shown to elicit specific salivary S-IgA antibodies that could inhibit oral recolonization by mutans streptococci (Russell et al. 2004), while mucosal immunology nowadays is a major field of Biomedicine and mucosal vaccines against several infectious diseases are either already licensed or in clinical trials (Lavelle and Ward 2022). More than 30 years ago, periodontal researchers showed that implantation of Bacteroides (Porphyromonas) gingivalis into the periodontal microbiota of monkeys initiated periodontal disease (Holt et al. 1988). At the time, this finding was interpreted to suggest specific microbial etiology of periodontitis, a concept that was superseded by the polymicrobial synergy and dysbiosis model of periodontal disease pathogenesis (Lamont et al. 2018). Nevertheless, the landmark study by Holt and colleagues had sparked further interest in P. gingivalis research that over the years contributed critically to what we know today about this keystone pathogen and its interactions with the host and other bacteria. Studies in animal models of periodontitis or dental caries showed that the severity of induced bone loss or carious lesions, respectively, was also dependent upon the genetic background of the host; moreover, intercross breeding experiments showed that susceptibility or resistance to disease is an inherited trait (reviewed by Baker 2005, Werneck et al. 2010). Early studies on antibiotics in oral health showed that systemic tetracyclines suppress periodontal pathogens in humans and inhibit periodontal tissue destruction in rats (Weiner et al. 1979, Slots and Rosling 1983). Intriguingly, moreover, experiments in germ-free diabetic rats showed that tetracyclines could block abnormally elevated collagenolytic activity (Golub et al. 1983). These results led to (i) the realization that tetracyclines could inhibit periodontal tissue breakdown through mechanisms that, in part, are independent from their antimicrobial effects and (ii) the development of new, tetracycline-based drugs that inhibit matrix metalloproteinases with applications in different biomedical fields (Golub and Lee 2020).
Box 1: Animal model validation
Widely accepted criteria for animal model validation include predictive, face, and construct validity. Although these criteria were first applied to neuro-psychiatric diseases (Willner and Mitchell 2002, Nestler and Hyman 2010), they are both useful and relevant to any human disease investigated via model organisms. Predictive validity refers to the precision in predicting whether specific treatments that work in animals can also work efficiently in human patients. Face validity is defined as how well a model mimics the disease phenotype (clinical signs and symptoms, hallmark features) seen in human patients. Construct validity refers to the relevance of the mechanisms that were used to induce disease in model organisms; do such mechanisms reflect those that induce the human disease? In other words, this criterion determines the relevance of the etiologic mechanisms that are used to “construct” the model. As discussed in the main text, a single model is unlikely to replicate a human disease in its entirety and thus may also not satisfy all three criteria. However, a combination of different but complementary models may provide validity across the three criteria of predictive, face, and construct validity. The main text contains several examples of predictive validity for oral disease-related animal models (e.g. success of complement-targeted inhibition for treating periodontal inflammation in humans based on intervention studies in mice). Models of oral diseases (caries, periodontitis) described in this review also have face validity as they mimic the human disease phenotype including hallmark features (e.g. caries models cause cavities and also pain if the lesions extend to and affect the pulp; periodontitis models cause inflammation of the gingiva and loss of the supporting alveolar bone). The same models have construct validity since the mechanisms by which disease is induced in animals are quite similar to those that cause the human disease (e.g. S. mutans in the presence of sugars causes cavities both in animals and humans; dysbiotic polymicrobial communities in the periodontium cause periodontal disease in both animals and humans).
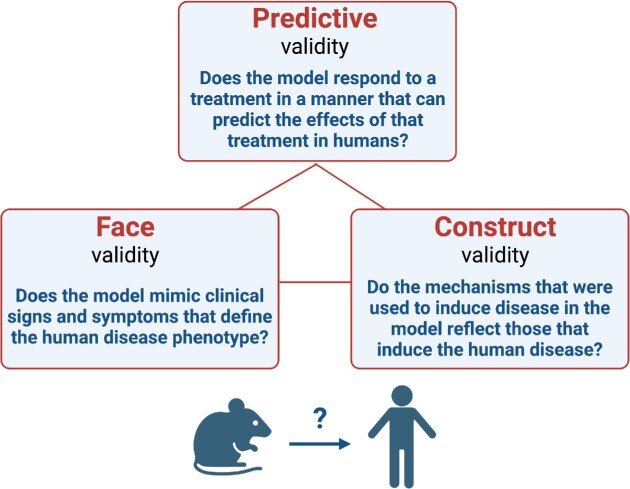
Box figure: criteria for validating animal models.
Figure 2.

The pioneer animal experiments of Keyes and Fitzgerald. The graphically depicted experiments collectively provide unequivocal evidence in support of the bacterial and diet etiology of dental caries. This set of animal model experiments is a classic example that insights gained from animal experimentation can help understand mechanisms and etiology of a human disease. All subsequent observations in human dental caries validated the findings of Keyes and Fitzgerald (Keyes 1959, Fitzgerald and Keyes 1960, Tanzer 1995).
This review discusses the potential utility of animal models in our efforts to understand the impact and mechanisms whereby the oral microbiome–host interactions affect local and systemic health. Although large animal models (e.g. dogs, pigs, and nonhuman primates) have also been used in oral science and have unique advantages, we will primarily focus on the most widely used and economical rodent models, for which there is extensive background information on their immune and physiological systems as well as a wide range of available reagents for analyses (Table 1). In particular, mouse models can be extremely informative in dissecting molecular pathways governing host–microbe interactions due to the availability of numerous genetically engineered strains (e.g. with knock-out or knock-in mutations, gene over-expression, or fluorescently tagged protein expression) (Webster et al. 2020). Moreover, the mouse genome is fully sequenced (http://www.sanger.ac.uk/resources/mouse/genomes/) and essentially all mouse genes have human homologues. Despite their advantages, model organisms also have limitations and thus animal experiments should be designed and interpreted judiciously. After all, a model is only “an approximation or simulation of a real system, i.e. under investigation” (Hajishengallis et al. 2015). This review, therefore, covers also the limitations of model organisms and advocates that broad understanding of oral health and disease requires that animal model-based findings be used in combination with data from in vitro systems and clinical studies (Fig. 3).
Table 1.
Advantages and disadvantages of rodent models in comparison to large experimental animals.
| Advantages |
| • Moderate costs; easily maintained and genetically manipulated |
| • Short reproduction times; several generations can be studied in relatively brief time periods |
| • Numerous genetically engineered strains and wide range of immunological reagents for analyses available (especially for mice) |
| • Extensive literature on their immune and physiological systems |
| • Established/validated models for a variety of human diseases |
| Disadvantages |
| • Microbiome differs more than that of large animals (e.g. nonhuman primates) as compared to the human microbiome |
| • Anatomy and physiology and disease features not as close to humans as big animals |
| • More inbred than large animals; decreased variability in results but also decreased translatability of findings |
Figure 3.

Animal models are not used or interpreted in isolation. Work in model organisms informs and is being informed by clinical research and in vitro systems as indicated in this feed-forward cycle of knowledge generation. For instance, a clinical observation (e.g. protein “A” is negatively correlated with protein “D”) may prompt a hypothesis that can be tested in an animal model (often with the help of in vitro assays) leading to the conclusion that protein “A” activates a signaling pathway that inhibits protein “D.” If the inhibition of protein “D” in the animal model protects from a disease, then protein “D” can be identified as a candidate therapeutic target. This in turn can be tested for validation in an intervention trial, paving the way to clinical development for the treatment of human patients. One of the greatest contributions of experimental animals, especially mouse models with a plethora of knock-out, knock-in or conditional mutations, is the capacity for testing causality, which cannot be typically addressed in human studies that are mostly correlative.
Advantages and limitations of animal models
Animal models are not without pitfalls. Knowing their limitations enables the researchers to design better and valid studies that are relevant to the human condition. Well-designed animal model-based studies have been extremely helpful in uncovering cause-and-effect relationships and for identifying new targets for disease treatment as discussed below.
Specific hypothesis testing vs. disease replication
It is often said that no animal model can faithfully reproduce human disease in its entirety. This is generally true. However, whether an animal model is appropriate and useful should not be decided on the basis of its fidelity to all aspects of a given human disease. The critical question regarding the appropriateness of a given model is the following: Can the model be used to address a specific hypothesis? Two instructive examples are given here. The injection of bacteria into a tissue is obviously meaningless and irrelevant to the study of bacterial invasion but, at the same time, suitable to study the host inflammatory response upon bacterial invasion. No animal or human develops microbe-driven inflammatory bone loss in the calvaria similar to periodontal disease. However, the calvarial model (injection of microbial or inflammatory stimuli into the connective tissue that overlies the calvarial bone) is relevant to study mechanisms by which diverse microbial or host-derived stimuli, such as specific cytokines, regulate osteoclastogenesis and bone resorption (Boyce et al. 1989, Graves et al. 2001). Although no single model can recapitulate all aspects of a human disease, different models can be employed to study discrete components of the disease process by addressing different but complementary hypotheses (Graves et al. 2008) (Fig. 4). Despite their limitations, animal models are typically superior to in vitro or clinical studies in addressing mechanistic hypotheses (Fig. 1). Moreover, animal experiments are invaluable in testing the potential of candidate therapeutic compounds.
Figure 4.

Animal models and hypothesis testing. Although animal models cannot faithfully reproduce a given human disease in its entirety, their utility can be decided on whether they can address a specific hypothesis related to an aspect of the disease. In that manner, the combination of different models (from the same or different species) investigating distinct and complementary hypotheses can lead to information which, when appropriately synthesized, may provide new knowledge on the human disease and prompt targeted clinical research.
Consistent with the notion that model organisms lie on a spectrum of assay systems spanning from the experimentally tractable in vitro systems to the biological complex and relevant clinical studies (Fig. 1), information derived from model organisms should not be used in isolation but rather synthesized with findings from in vitro cellular and bacterial assays and human clinical data to obtain broader insights into disease pathogenesis (Fig. 3). Ideally, important treatment concepts derived from animal studies should be followed by targeted validation in clinical studies, which would pave the way to novel therapies for human diseases (Fig. 3).
Are mouse models relevant to the human condition and capable of leading to novel drug development?
It has been argued that mouse models do not have sufficient predictive validity, i.e. precision in predicting whether specific treatments that worked in mice can also work efficiently in human patients (Seok et al. 2013, Pound and Bracken 2014, FitzGerald et al. 2018), although such claims have been countered by others (Dirnagl 2014, Takao and Miyakawa 2015, Nadeau and Auwerx 2019). Nadeau and Auwerx cited examples of genes first characterized in mice that were subsequently shown to be relevant for the human condition, such as, Lepob and Ubp1, which regulate, respectively, body weight and blood pressure in both mice and humans (Nadeau and Auwerx 2019). Parabiotic experiments among different strains of genetically obese (ob/ob or db/db) and normal mice were fundamental in the quest for the ultimate discovery and functional characterization of the hormone leptin (the ob gene product) and the leptin receptor (the db product) (Nadeau and Auwerx 2019). Obviously, such pivotal parabiotic experiments would be impossible, let alone unethical, in humans, indicating that major scientific discoveries are often critically dependent on animal experimentation.
Mouse models have also played a critical role in drug discovery, as shown by the following examples. TNF-α overexpression in mice resulted in chronic inflammatory polyarthritis, which displayed features of human rheumatoid arthritis and was treatable by an anti-TNF-α monoclonal antibody (Keffer et al. 1991). This landmark discovery in 1991 foreshadowed and facilitated the successful development of anti-TNF-α therapy for the treatment of rheumatoid disease patients (Radner and Aletaha 2015). The discovery of immune checkpoints and their inhibition in cancer, which importantly resulted in new treatment modalities and the 2018 Nobel Prize in Physiology or Medicine, would not have been possible without fundamental knowledge derived from mouse model experiments in the laboratories of James P. Allison and Tasuku Honjo (Wolchok 2018).
In the context of oral disease, the demonstration that mice lacking the complement component C3 are protected against experimental periodontitis (Maekawa et al. 2014a) led to a phase 2a clinical trial in which a locally administered C3-targeted drug (AMY-101) inhibited periodontal inflammation in human patients (Hasturk et al. 2021). Work in a mouse model of aggressive periodontitis associated with leukocyte adhesion deficiency type-1 (LAD-1) implicated the dysregulation of the IL-23–IL-17 inflammatory axis as the driver of this condition (Moutsopoulos et al. 2014). This discovery was harnessed in the successful application of IL-23-based treatment (using the monoclonal antibody ustekinumab) of human LAD1 periodontitis (Moutsopoulos et al. 2017). Plasminogen-deficient mice spontaneously develop extravascular fibrin deposits leading to neutrophil-mediated periodontal inflammation and bone loss; consistently, polymorphisms in the plasminogen-encoding gene (PLG) are associated with increased prevalence of human periodontal disease (Silva et al. 2021). Mice deficient in the secreted homeostatic protein DEL-1 develop spontaneous inflammatory bone loss associated with high levels of IL-17 in the periodontal tissue (Eskan et al. 2012). Subsequent work showed that DEL-1 and IL-17 are reciprocally negatively regulated, and their balance is important for homeostatic immunity (Maekawa et al. 2015, Kourtzelis et al. 2019). This DEL-1–IL-17 balance principle (Hajishengallis and Chavakis 2019) was later found to be relevant in humans in the context of COVID-19-related and especially Kawasaki disease-related hyperinflammation (Consiglio et al. 2020). Specifically, autoantibodies to DEL-1 (EDIL3), which were particularly overexpressed in Kawasaki disease, were shown to be inversely related to IL-17 concentrations (Consiglio et al. 2020), suggesting neutralization of the anti-inflammatory function of DEL-1, and hence unrestrained production of IL-17. These examples underscore the potential of mouse models to lead to treatments for human disease or to new biological concepts that are relevant to the human condition.
Mice and humans have distinct microbiomes but common features of dysbiosis
There are striking compositional differences in the microbiotas of oral and other mucosal sites between mice and humans (Ley et al. 2005, Dutzan et al. 2018, Li et al. 2019, Payne et al. 2019). The human oral microbiome includes over seven hundred different bacterial species (besides numerous fungal, viral, fungal, and protozoan species) (Zhang et al. 2022). A relatively limited number of microbial species comprise the oral microbiome of experimental mice, which typically do not harbor species that are found in humans (Payne et al. 2019, Abusleme et al. 2020). This discrepancy in the composition of mouse and human microbial communities, however, does not mean that mice are inappropriate to model human dysbiotic inflammatory diseases, such as periodontitis. This is because the emergence of dysbiosis (vs. homeostasis maintenance) is determined not by specific individual bacteria but by the collective output of community action, which in turn is shaped by both interspecies interactions and host genetic and environmental variables (Lamont et al. 2018). In other words, what matters is not so much the microbial roster but the encoded collective gene pool and its interaction with the host environment.
Accordingly, compositional changes (associated with the overgrowth of Gram-negative anaerobes) in the periodontal microbiome of mice subjected to ligature-induced periodontitis (LIP) (Box 2) were sufficient to drive the expansion of T helper 17 (Th17) cells, which in turn mediated inflammatory bone loss (Dutzan et al. 2018). Similarly, the dysbiotic community associated with human periodontitis is overwhelmingly enriched in Gram-negative anaerobic bacteria (Diaz et al. 2016). The clinical relevance of the observation that Th17 cells drive inflammatory bone loss in the LIP model was confirmed in humans, since individuals with defective Th17 cell development were shown to have diminished periodontal inflammation and bone loss (Dutzan et al. 2018). Further studies in the LIP model demonstrated that complement acts upstream of Th17 cell expansion. Specifically, complement activation by the dysbiotic microbiome leads to induction of cytokines (IL-6, IL-23) that are required for the expansion of pathogenic Th17 cells (Wang et al. 2022). The involvement of complement in experimental mouse periodontitis (Maekawa et al. 2014a) was validated in a complement C3-targeted intervention trial in patients with periodontal inflammation (Hasturk et al. 2021). Thus, although mice and humans harbor a different oral microbiome, mice constitute an appropriate study model in which dysbiosis of a community enriched in Gram-negative anaerobes drives periodontitis through the activation of complement and Th17, as occurs in the human disease.
Box 2: Ligature-induced periodontitis model
Ligature-induced periodontitis (LIP) is currently the most widely used model of experimental periodontitis. In LIP, ligature placement in molar teeth generates a subgingival biofilm-retentive milieu leading to inflammation, dysbiosis, and bone loss in conventional (but not germ-free) mice (Abe and Hajishengallis 2013, Jiao et al. 2013, Shin et al. 2015, Xiao et al. 2017, Tsukasaki et al. 2018, Kourtzelis et al. 2019, Kitamoto et al. 2020); dysbiotic alterations can be investigated by 16S rRNA gene sequencing and bioinformatic analysis (Abusleme et al. 2017, 2020, Dutzan et al. 2018, Kittaka et al. 2020, Williams et al. 2020, Hoare et al. 2021, Johnstone et al. 2021). Although LIP is sometimes referred to as an acute model, it should be noted that only the first 12–24 h represent an acute phase with pronounced recruitment of neutrophils peaking at 12 h (Shin et al. 2015). In essence, LIP represents an accelerated model of periodontitis with early activation of complement and recruitment of neutrophils, followed by macrophage accumulation and T cell activation/expansion, and with accumulation of B-cell-lineage cells in later stages (Abe et al. 2015, Shin et al. 2015, Dutzan et al. 2018, Marchesan et al. 2018, Tsukasaki et al. 2018, Kourtzelis et al. 2019, Kitamoto et al. 2020, Li et al. 2020, Wang et al. 2022), i.e. similar stages to those in human periodontitis (Kornman et al. 1997, Hajishengallis and Korostoff 2017). Although LIP can activate osteoclastogenesis and bone loss as early as 3 days postligation (Abe and Hajishengallis 2013, Abe et al. 2014), maintaining the ligatures for extended periods (e.g. 21 days) bestows chronicity in the pathologic process (Sima et al. 2014, Li et al. 2022). In fact, chronic periodontitis is unlikely a linear, constant pathologic process, which would not explain the often-observed rapid progression and periodic remission of disease. This is best described by the “episodic burst model,” i.e. periodontitis comprises an episodic series of brief acute insults (bursts) separated by periods of remission (Goodson et al. 1982, Socransky et al. 1984, Gilthorpe et al. 2003, Graves et al. 2011). Thus, even the acute phase of LIP is consistent with the chronic nature of human periodontitis. Ligature removal eliminates dysbiosis and the model can be used to study inflammation resolution and tissue repair (Coimbra et al. 2015, Nagai et al. 2020, Yuh et al. 2020). Concepts first shown in the LIP model, such as the causal involvement of complement and Th17 in inflammatory periodontal bone loss, were validated in human studies (Dutzan et al. 2018, Hasturk et al. 2021). For additional information, the reader is referred to a comprehensive review of LIP studies in mice (Lin et al. 2021).
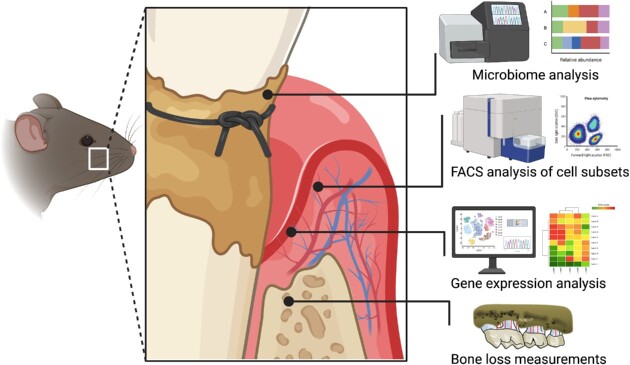
Box figure: ligature-induced periodontitis model and relevant analyses.
With aging, the human microbiome undergoes significant changes that correlate with elevated low-grade systemic inflammation (inflammaging) (Bosco and Noti 2021). Potential causality in this association was addressed by a study in which young and old germ-free were colonized with “young” or “aged” microbiota, i.e. via cohousing, respectively, with young (10–16 weeks of age) or old (18–22 months of age) conventional mice. Young germ-free mice that acquired an aged, but not young, microbiota developed systemic inflammation, suggesting that aging-associated dysbiotic alterations to the microbiota contribute to increased systemic inflammatory burden (Thevaranjan et al. 2017). However, aging by itself is also a contributory factor since old germ-free mice displayed increased systemic inflammation even if they were colonized with “young” microbiota (Thevaranjan et al. 2017).
“Humanized” mice colonized with human microbiome
The shortcoming of human microbiome studies to typically reveal correlations (rather than causality) between microbiome composition and disease phenotypes could also be addressed through the use of germ-free mice that are colonized with human microbiota (“humanized” mice). Such experiments have the potential to link colonization with microbiotas from diseased (but not healthy) individuals to disease phenotypes in mice; such observations can increase confidence in causality of observations made in humans. For instance, experiments in lean germ-free mice inoculated with human gut microbiota from obese or lean twins showed that adiposity is transmissible from humans to mice via microbiota transfer. Specifically, mice receiving “obese” microbiota displayed increased total body and fat mass, whereas those receiving “lean” microbiota maintained normal weight (Ridaura et al. 2013). A caveat that should be borne in mind is that germ-free mice do not have a fully competent immune system; for instance, they have incomplete development of the gut-associated lymphoid tissues (Round and Mazmanian 2009), and thus may not be properly activated upon inoculation with a human microbiota.
A human oral microbiota-associated (HOMA) mouse model was developed by inoculating human saliva into germ-free mice resulting in an oral microbiota, which was distinct from that of conventional mice, while appearing to cluster with the human donor’s oral microbiota, as determined by principal component analysis (Li et al. 2019). To facilitate periodontal disease development in germ-free mice that receive exogenous microbial pathogens, their molar teeth can be ligated prior to microbial inoculation (Xiao et al. 2017, Marchesan et al. 2018). However, the use of mice harboring microbes of human origin is not without pitfalls. This is mostly because the coevolution between a given host and its microbiota may have resulted in species that fill host-specific niches (Kibe et al. 2005, Koskella and Bergelson 2020). Thus, a substantial proportion of human-derived microbes fail to colonize mouse tissues (Fritz et al. 2013, Arrieta et al. 2016). For instance, Faecalibacterium and Bifidobacterium, human symbiotic species with anti-inflammatory properties, could not be transferred to germ-free mice (Lundberg et al. 2020). Moreover, those species that do colonize may impact the mouse host in a different manner as compared to the human host they have coevolved with (Fritz et al. 2013, Arrieta et al. 2016). In this regard, host immune maturation and ability to clear pathogen infection is defective in germ-free mice colonized with human fecal commensal microbiota relative to germ-free mice colonized with mouse fecal commensal microbiota (Chung et al. 2012).
Clinically relevant lessons learned from oral disease models
Since the classic studies by Keyes and Fitzgerald in rodent models that promoted our understanding of the etiology of human dental caries (Keyes 1959, Fitzgerald and Keyes 1960, Tanzer 1995) (Fig. 2), a lot of other concepts underlying oral diseases were elucidated by studies in model organisms, some of which are discussed below.
Dysbiotic alterations rather than mere increase in microbial load drive periodontal disease
Although the importance of bacteria in human periodontal disease pathogenesis is now intuitively understood, hard evidence that the bacteria are required for the induction of periodontal inflammation and bone loss was obtained only after relevant studies in LIP-subjected germ-free or conventional rats (Rovin et al. 1966). The placement of a ligature by itself failed to induce recruitment of inflammatory cells in germ-free hosts, in contrast to conventional animals where the combination of the local microbiota and the ligature led to recruitment of neutrophils followed by fibroblast proliferation and lymphocyte accumulation (Rovin et al. 1966). Consistently, years later, LIP-subjected germ-free mice did not display bone loss unless colonized with bacteria (Jiao et al. 2013, Xiao et al. 2017), whereas treatment of LIP-subjected mice with antibiotics inhibits innate and adaptive mechanisms of inflammation and bone loss (Dutzan et al. 2018, Wang et al. 2022). In other words, inflammatory bone loss is driven by the ligature-associated microbiota and is not a “consequence of a ligature-associated trauma to the periodontal tissues” (Jiao et al. 2013, Marchesan et al. 2018).
Ligature placement in conventional mice causes an increase in bacterial biomass and compositional changes in the microbial community of ligated sites (relative to unligated/healthy sites) and these alterations are associated with inflammatory bone loss (Dutzan et al. 2018). In the same study, the use of different antibiotics, alone or in combinations, revealed that dysbiotic alterations to the periodontal microbial community, rather than a mere augmentation of the total microbial burden, acted as the driver of Th17 cell expansion and bone loss (Dutzan et al. 2018). This conclusion was based on findings that those antibiotic treatments, which inhibited Th17 expansion and bone loss, also caused shifts in the relative abundance of microbial species within the community without necessarily decreasing the total microbial load or targeting high-abundance microbial species (Dutzan et al. 2018). A standardized reanalysis of microbiome 16S rRNA sequencing datasets from nine recent studies utilizing the LIP model revealed comparable microbial richness and diversity among the different studies and concluded that the LIP model is associated with characteristic shifts in periodontal microbiota structure, with Streptococcus sp. overrepresented in health and Enterococcus sp., Faecalibaculum sp., Adlercreutzia sp., and Bifidobacterium sp. dominating in disease (Arce et al. 2022). Consistently, a study that examined longitudinally the development of dysbiosis in the LIP model demonstrated an initial high abundance of Streptococci that declined during the course of the disease and a progressive increase of disease-associated species such as Enterococci (Ribeiro et al. 2022). These findings in the mouse LIP model are consistent with clinical observations that the periodontitis-associated microbiota is characterized by bacterial outgrowth and a dysbiotic shift relative to the microbiota of periodontally healthy sites (Curtis et al. 2020). However, in human periodontitis, it is not practical to distinguish the effects of nonspecific biofilm accumulation from those resulting from shifts in the composition of microbial communities. The LIP model, therefore, has provided a causal inference that dysbiotic changes to the microbiome may also be the trigger of Th17 cell accumulation and bone loss in the human disease.
Similar conclusions, i.e. that dysbiosis drives periodontal disease, were also obtained using the oral gavage model of experimental mouse periodontitis, first introduced by Baker et al. (1999). Repeated inoculation of P. gingivalis by oral gavage (e.g. three times within a week) led to its colonization of the oral cavity and development of inflammatory bone loss 6 weeks after the last inoculation (Baker et al. 1999, Baker 2000). This finding as well as similar observations in nonhuman primates orally inoculated with P. gingivalis (Holt et al. 1988) were interpreted as evidence for specific etiology in periodontitis. However, it was later shown that P. gingivalis causes periodontitis in mice by remodeling a eubiotic oral microbial community into a dysbiotic one, which can actually cause disease after transfer to germ-free mice (Hajishengallis et al. 2011, Payne et al. 2019). Porphyromonas gingivalis by itself could colonize but could not induce bone loss in germ-free mice (Hajishengallis et al. 2011).
Dysbiosis and inflammation are reciprocally reinforced in a feed-forward loop
According to the “ecological plaque hypothesis,” environmental factors (e.g. inflammation, available nutrients, redox potential and pH) drive the selective expansion of oral species that act as pathobionts in periodontitis or caries (Marsh 2003). For instance, S. mutans and other cariogenic and aciduric species thrive in the presence of sucrose (at the expense of nonaciduric/noncariogenic commensals) and cause carious lesions through the secretion of lactic acid (Lamont et al. 2018). In the setting of periodontitis, it could be reasoned that degraded collagen and heme-containing compounds released from inflammatory tissue lesions can be used, respectively, as sources of amino acids and iron by periodontitis-associated proteolytic and asaccharolytic bacteria (Hajishengallis 2014, Diaz et al. 2016). Such a nutritionally favorable environment would enable selective expansion of inflammophilic bacteria at the expense of species that cannot capitalize on or endure inflammation, hence compatible with periodontal health. Studies in mice and other animals has lent support to this concept by showing that anti-inflammatory treatments not only inhibit periodontal tissue destruction but also reduce the periodontal microbial load and reverse dysbiosis (Hasturk et al. 2007, Lee et al. 2016, Abe et al. 2012, Eskan et al. 2012, Maekawa et al. 2014a). Accordingly, the lack of sufficient periodontal inflammation in complement C3-deficient mice resulted in a failure to maintain a high microbial load in the periodontium as seen in wild-type littermate controls (Maekawa et al. 2014a). Consistent with the animal studies, in vitro generated oral multispecies biofilms supplemented with serum, hemoglobin or hemin, display selective outgrowth of inflammophilic pathobionts that moreover upregulate virulence genes, which further promote their ability to exploit an inflammatory environment (e.g. genes encoding proteases, proteins involved in hemin transport and hemolysins) (Herrero et al. 2018). Controlled microbiome transfer experiments showed that an oral dysbiotic microbiota is not simply the outcome of a nutritionally conducive inflammatory environment, but also a direct cause of inflammatory bone loss. Specifically, a dysbiotic oral microbial community could be transferred from conventional to cohoused germ-free mice that subsequently developed periodontitis (Payne et al. 2019).
Taken together, these findings from in vitro and in vivo models support the notion that dysbiosis and inflammation are reciprocally reinforced in a self-sustained feed-forward loop that drives the pathogenesis and chronicity of periodontal disease (Hajishengallis et al. 2023). This concept has important implications for the treatment of human periodontitis. It suggests that the vicious cycle that drives the disease can be abrogated by either targeting dysbiosis or inflammation. In the latter regard, blocking inflammation would also negatively affect microbial growth and dysbiosis. This expectation is consistent with an early clinical study revealing faster biofilm accumulation at sites of gingival inflammation; the same sites additionally exhibited faster development of a complex microbiota (“bacterial flora”) (Hillam and Hull 1977). Many years later, a metagenomic study of human periodontitis showed a positive correlation between bacterial biomass and clinical gingival inflammation (Abusleme et al. 2013). Moreover, a recent study correlated subgingival microbiome profiles and local proresolving lipid mediators, further implying that inflammation plays a role in mediating compositional shifts of the subgingival microbiota (Lee et al. 2021). The ability of periodontitis-associated subgingival biofilms to capitalize on inflammation for growth is also supported by an in situ community-wide transcriptomic study that showed increased expression of proteolysis-involved genes and genes associated with peptide transport and acquisition of iron in pathogenic vs. healthy biofilms (Duran-Pinedo et al. 2014). The above-discussed collective findings represent a good example that clinically relevant mechanistic insights into human disease require a combination of findings from in vitro systems, animal models, and clinical observations.
Importance of interspecies and interkingdom interactions in community virulence
Interactions and mechanisms described solely in in vitro systems may be questioned as to their biological significance, a concern that can be addressed in in vivo experiments using appropriate animal models. In vitro experiments had shown that specific adhesin-mediated interactions between P. gingivalis and the early colonizer Streptococcus gordonii promote the development of P. gingivalis biofilms on a streptococcal substrate (Lamont et al. 2002). Whether this in vitro coadhesion increases the in vivo virulence of P. gingivalis was tested later in the oral gavage model of experimental mouse periodontitis. Oral inoculation of mice with both S. gordonii and P. gingivalis induced synergistically more bone loss than that induced by P. gingivalis alone, whereas this synergy was abolished by a peptide that blocks the coadhesion of P. gingivalis and S. gordonii (Daep et al. 2011). Streptococcus gordonii was thus characterized as an accessory pathogen, i.e. a microbe, i.e. intrinsically a commensal but, in a certain context, can enhance the virulence of pathogens by assisting the latter in their colonization or metabolic activities (Hajishengallis and Lamont 2016). It should be noted that oral inoculation of mice with human periodontitis-associated bacteria (P. gingivalis, Fusobacterium nucleatum, Tannerela forsythia, and so on) should not be applied for the entire experimental period. In the original model, Baker et al. (1999) applied the inoculum three times at 2-day intervals to establish infection and examined bone loss 6 weeks later. Thus, the induced bone loss was the result of P. gingivalis colonization of the periodontal tissues. In some studies, however, periodontal pathogens were orally inoculated for the entire experimental period (e.g. the inoculum was applied twice a week for 4 weeks or even at 3-day intervals for 1 or more months). However, this approach is quite artificial since it views periodontitis as the result of repetitive exogenous microbial insult rather than representing a local microbe-driven disease. This point should be taken into consideration regardless of whether oral inoculation with human pathogens follows the original Baker model or the human bacterial inoculum is applied onto sites that have been ligated (combination of LIP and oral gavage model).
Interestingly, P. gingivalis cannot initiate growth from a low-cell-density inoculum (105 cells per ml). In vitro culture studies, including in an open flow chemostat system, showed that P. gingivalis can overcome this handicap by “borrowing” a diffusible growth-promoting molecule released from Veillonella parvula, an early colonizer symbiont, but not from a number of other commensal species examined (Hoare et al. 2021). In a modified version of the mouse LIP model where human bacteria were placed on ligatures via a single inoculation, V. parvula enabled a low-cell-density inoculum of P. gingivalis to colonize the periodontal tissues and to increase bone loss. Under the same conditions with the only exception being the absence of V. parvula, P. gingivalis failed to colonize (Hoare et al. 2021). These findings thus suggest that V. parvula can act as an accessory pathogen. The concept that this interspecies interaction promotes the growth of P. gingivalis could be tested for human relevance via bioinformatic analysis of the subgingival microbiome, e.g. by determining whether the presence of V. parvula is associated with increased P. gingivalis colonization of the subgingival plaque.
Besides the established role of S. mutans in early childhood caries (ECC), clinical studies have additionally shown an association of Candida albicans with ECC, which becomes quite severe in children coinfected with S. mutans and C. albicans (Xiao et al. 2016, 2018, Garcia et al. 2021). Work taking advantage of in vitro and in vivo models has provided a mechanistic explanation of these clinical associations. Specifically, glucosyltransferase enzymes released by S. mutans bind to mannans on the surface of C. albicans and utilize sucrose to form adhesive glucans. The in situ generated glucans promote S. mutans–C. albicans coadhesion and embed these organisms in a glucan matrix that promotes mixed-biofilm accumulation (Falsetta et al. 2014, Hwang et al. 2017). The importance of this cross-kingdom interaction was demonstrated in a caries model in rats fed a sucrose-rich diet. Animals coinfected with S. mutans and C. albicans displayed abundant biofilm formation with significantly increased viable counts of both S. mutans and C. albicans, as well as more extensive carious lesions as compared to those in animals infected with either species alone (Falsetta et al. 2014, Hwang et al. 2017). Therefore, the use of an appropriate in vivo model has explained that the association of this bacterial–fungal coinfection with severe ECC is likely due to synergistic interactions (between the two organisms) that lead to enhanced glucan formation and colonization in tooth-associated biofilms with increased virulence. This pathogenic cross-kingdom interaction is disrupted in vitro by Lactiplantibacillus plantarum (Zeng et al. 2022) which, moreover, can reduce the colonization of both S. mutans and C. albicans and their capacity to induce carious lesions in rats placed on a sucrose-rich diet (Zhang et al. 2020). The result form this animal experiment may prompt future clinical studies to determine whether ECC could be treated with these probiotic Lactobacilli.
Mechanisms linking the oral microbiome to systemic pathology or health
Relative to periodontally healthy individuals, periodontitis patients have elevated serum concentrations of proinflammatory mediators (e.g. IL-1, IL-6, fibrinogen, and C-reactive protein) and blood neutrophil counts (Genco and Van Dyke 2010, Bokhari et al. 2012, D’Aiuto et al. 2013, Schenkein et al. 2020), as well as increased risk of systemic comorbidities (Genco and Sanz 2020). In great part, periodontitis-associated systemic inflammation can be attributed to the fact that oral microbes and their products can access the circulation via the ulcerated periodontal pocket epithelium. Although the resulting bacteremias are transient, they are also quite frequent, being instigated not only by professional dental care (probing, scale and root planing, and tooth extractions), but also by daily activities, including toothbrushing and chewing hard foods (e.g. apple) (Hajishengallis and Chavakis 2021). About half (43%–52%) of bloodborne bacterial species were found to be derived from the oral cavity, according to a method that purified genomic DNA exclusively from intact bacterial cells (Emery et al. 2021).
In line with the clinical findings that associate periodontitis to low-grade systemic inflammation, mice and rats subjected to LIP display increased serum concentrations of proinflammatory cytokines (Anbinder et al. 2016, Matsuda et al. 2016, Kitamoto et al. 2020, Li et al. 2022). These findings suggested that the LIP model could be employed to understand causal mechanisms that link periodontitis-associated systemic inflammation to increased susceptibility to inflammatory comorbidities. In that regard, despite strong epidemiological evidence that periodontitis is associated with increased risk of systemic comorbidities (e.g. cardiovascular disease and arthritis) (Genco and Sanz 2020), the underlying mechanisms remain poorly understood.
A recent study in mice showed that LIP-associated systemic inflammation induced epigenetic inflammatory memory in hematopoietic stem and progenitor cells (HSPC) that were imprinted with a myeloid differentiation bias. This memory was evident upon future inflammatory challenges, since the epigenetically rewired or “trained” HSPC gave rise to increased production of neutrophils and monocytes, which exhibited enhanced inflammatory responsiveness (Li et al. 2022). Moreover, this epigenetically based memory was transmissible by transplantation of HSPC from LIP-subjected mice to naïve recipients, which developed increased joint inflammation and pathology upon collagen antibody-induced arthritis (CAIA), as compared to CAIA-subjected mice transplanted with control (untrained) HSPC (Li et al. 2022). The induction of inflammatory memory was critically dependent on IL-1 signaling in HSPC. Indeed, transplantation of bone marrow from donors with HSPC-specific deletion of IL-1 receptor did not enhance the susceptibility of recipient mice to arthritis (Li et al. 2022). This study has two important implications for the human condition:
First, it implies that, in prospective clinical studies, clinician scientists may need to study the potential impact of inflammatory memory in the donor’s HSPC, in other words, to interrogate whether transplantation of bone marrow from donors, with or without recent history of inflammatory disorders, could influence outcomes in recipients of therapeutic bone marrow transplantation. Second, it suggests that systemic inhibition of IL-1 signaling (by blocking the cytokine or its receptor) might represent a novel therapeutic approach for holistic treatment of inflammatory comorbidities. In that regard, it is tempting to speculate that the success of antibody-mediated neutralization of IL-1β in the CANTOS trial for the treatment of atherosclerosis (Ridker et al. 2017) might, in part, be attributed to inhibition of IL-1-dependent induction of inflammatory memory in the bone marrow. These considerations underscore the potential of basic research in the mouse model to inform future targeted studies in human patients.
That said, is there evidence that periodontitis can cause inflammatory memory in humans? A number of studies have shown that patients with periodontitis have, in their peripheral blood, increased counts of myeloid cells which also exhibit hyper-responsive phenotypes upon ex vivo stimulation, thus implying the presence of inflammatory memory (reviewed by Irwandi et al. 2022). Intriguingly, this hyper-responsiveness is maintained even after successful periodontal treatment or even full-mouth tooth extraction (Fokkema et al. 2003, Radvar et al. 2008, Ling et al. 2015), which further supports the notion for generation and persistence of inflammatory memory. Moreover, the operation of a periodontitis-bone marrow inflammatory axis in the above discussed preclinical model (Li et al. 2022) is consistent with clinical studies utilizing positron emission tomography/computed tomography with 2-deoxy-2-[fluorine-18]fluoro-D-glucose (18F-FDG-PET/CT). These imaging studies have positively correlated periodontal inflammation with increased hematopoietic tissue activity in the bone marrow, as well as with inflammation in the arterial wall and elevated risk of prospective cardiovascular events (Ishai et al. 2019, Van Dyke et al. 2021).
Conversely, systemic disorders, such as diabetes, can increase the severity of periodontitis and cause dysbiotic alterations to the oral microbiome (Wu et al. 2015, Teles et al. 2021). In this context, studies in diabetic mice provided mechanistic explanations of how diabetes exacerbates periodontitis, including elevated generation of proinflammatory advanced glycation end products, exaggerated TNF-α inflammatory responses leading to bone loss, and defective resolution of inflammation (Wu et al. 2015). Moreover, diabetes causes IL-17-dependent alterations to the oral microbiota, which thereby becomes particularly pathogenic, as evidenced by transfer experiments to germ-free mice (Xiao et al. 2017). Therefore, whereas human studies have established associations between diabetes and several alterations in the periodontium, mechanistic studies in mice (including the use of mutant strains and specific inhibitors) provided evidence of causality linking these parameters.
Humans swallow at least 1 l of saliva daily which contains > 1011 bacteria (Gibbons and Houte 1975, Humphrey and Williamson 2001). However, oral bacteria are typically poor colonizers of the intestine, unless intestinal homeostasis is disrupted by pathological conditions, including inflammatory bowel disease and liver cirrhosis (Schirmer et al. 2018, Schmidt et al. 2019, Yachida et al. 2019, Chen et al. 2021). In settings such as pediatric Crohn’s disease or ulcerative colitis, bacteria of oral origin (e.g. Fusobacteriaceae and Veillonellaceae) are highly abundant (Gevers et al. 2014, Schirmer et al. 2018). These correlations, however, do not prove that oral bacterial species participate in the pathogenesis of these disorders. Causality was again addressed in animal models. Consistent with the clinical findings, studies in mice demonstrated significant ectopic colonization of the mouse intestine by oral bacteria but only under conditions that compromise gut homeostasis, such as, pre-existing intestinal inflammation or genetic deficiency of IL-10, which predisposes to colitis (Kitamoto et al. 2020). Ectopic oral bacteria were shown to cause intestinal inflammation by inducing IL-1β in macrophages and activating Th17 cells, alter the composition of the gut microbiota, and undermine gut barrier function; these effects collectively lead to exacerbation of colitis and induction of endotoxemia, systemic inflammation and changes in the serum metabolome (Kato et al. 2018, Kitamoto et al. 2020, Imai et al. 2021, Tsuzuno et al. 2021, Yamazaki et al. 2021, Xing et al. 2022). In conclusion, these preclinical studies in mice not only linked ectopic colonization of oral bacteria to exacerbation of intestinal inflammation, but also provided mechanistic insights into the so-called oral–gut–liver axis, which can worsen hepatic inflammation and steatosis thereby potentially exacerbating nonalcoholic fatty liver disease (Imai et al. 2021, Albuquerque-Souza and Sahingur 2022).
Besides being a major cariogenic organism, S. mutans is also implicated in cases of infective endocarditis, which often develops in individuals with underlying heart disease (Lemos et al. 2019). A possible route by which S. mutans can contribute to infective endocarditis was demonstrated in a rat caries model, where the animals were also subjected to heart valve injury. Specifically, after the development of severe carious lesions, S. mutans could translocate to the injured heart tissue via the exposed pulp, which is rich in blood vessels (Nomura et al. 2020). Rats with increased numbers of carious teeth with exposed pulp had a significantly increased rate of S. mutans detection in the heart tissue, as compared to rats with decreased numbers of teeth affected in that manner (Nomura et al. 2020). Strains of S. mutans that express collagen-binding protein (Cnm) have also been associated with IgA nephropathy (Ito et al. 2019). A recent study in rats provided inference of causality for this association by showing that S. mutans-induced severe dental caries (extending to the pulp) caused IgA nephropathy-like glomerulonephritis in rats. The disease manifestation (mesangial cell proliferation, increased mesangial matrix in the glomerulus, IgA, and complement C3 deposition) was significantly more pronounced in animals infected with a Cnm-positive strain as compared to a Cnm-negative strain, which essentially had no effect on glomerulonephritis despite causing rampant caries (Naka et al. 2021).
Oral microbial communities use nitrate reductases to reduce dietary nitrate to nitrite, thereby contributing to the generation of nitric oxide, i.e. important for the host’s systemic health, being involved in metabolic and cardiovascular regulation, including the lowering of blood pressure (Lundberg et al. 2018). In this regard, twice-daily treatment of human volunteers with the antiseptic chlorhexidine via mouthwash resulted in significant increase in systolic blood pressure after 7 days of treatment, whereas recovery in viable bacterial counts from chlorhexidine use led to enrichment in nitrate-reducing bacteria on the tongue (Tribble et al. 2019). Moreover, a higher relative abundance of nitrate‐reducing oral bacteria was associated with lower plasma glucose and insulin resistance as well as reduced cardiometabolic risk among diabetes mellitus-free adults (Goh et al. 2019, 2022). These human studies were greatly facilitated at the conceptual level by early studies in animal models: several studies in different animal models, including rats and mice, established the vasodilating effects of nitrite as a source of nitric oxide (reviewed by Lundberg et al. 2018). Oral administration of nitrite to rats or mice conferred protection against hepatic and cardiac ischemia/reperfusion injury (Shiva et al. 2007). Rats topically treated in the mouth with the bactericidal agent povidone–iodine prior to nitrate supplementation, failed to increase nitric oxide levels and to alleviate stress-induced injury in the gastric mucosa (Miyoshi et al. 2003). Consistently, gastric nitric oxide generation is negligible in germ-free rats even after receiving a dietary load of nitrate (Sobko et al. 2004). Thus, studies in model organisms not only address causality, but also provide a conceptual framework upon which targeted clinical studies can be designed.
Surrogate models
Models do not have to mimic a disease to be helpful and an example was given above with the calvarial model, which can be used to understand mechanisms that regulate osteoclastogenesis (Graves et al. 2008), an essential feature of bone loss disorders including periodontitis and arthritis. Moreover, various models where pathogens are injected subcutaneously into the dorsum (“abscess” model), pouches previously injected with air (“airpouch” model), implanted titanium-coil chamber (“chamber” model), or the peritoneal cavity (“peritonitis” model) have been productively used to investigate potential virulence factors of oral pathogens and their immune evasion strategies (Genco et al. 1991, Burns et al. 2006, Graves et al. 2008, Hajishengallis et al. 2008, Singh et al. 2011, Wang et al. 2013, Maekawa et al. 2014b). All these models allow easy recovery of samples for analysis as well as accurate and quantitative assessment of parameters that would be practically impossible to determine in the oral mucosa. For instance, the abscess model was used to establish the spatial organization of Aggregatibacter actinomycetemcomitans in a cross-feeding interaction with S. gordonii. Specifically, 3D image analysis revealed that A. actinomycetemcomitans maintains a “safe” distance (at least 4 µm) from streptococci that enables it to obtain lactate as a carbon source, while minimizing its exposure to inhibitory concentrations of streptococcal hydrogen peroxide (Stacy et al. 2014). The airpouch model was used in early studies that established the proresolving function of lipoxins. Specifically, lipoxins were shown to downregulate inflammatory cytokines/chemokines and inflammatory cell recruitment in response to different stimuli including P. gingivalis (Hachicha et al. 1999, Pouliot et al. 2000), leading to the development of an entire field of proresolving lipid mediators with potential for the treatment of human inflammatory disease (Serhan et al. 2022). The chamber model has been instrumental in demonstrating the ability of P. gingivalis to exploit complement and TLR2 signaling to evade killing by phagocytes while promoting their inflammatory responses (Burns et al. 2006, 2010, Maekawa et al. 2014b, Makkawi et al. 2017), activities that promote the dysbiosis of the periodontal microbiota (Lamont et al. 2018). The highly quantitative thioglycollate-induced peritonitis model was used to dissociate the antineutrophil recruitment activity of the secreted homeostatic protein DEL-1 from its ability to promote the phagocytosis of apoptotic neutrophils (efferocytosis) by macrophages (Kourtzelis et al. 2019). This is because at 72 h postthioglycollate administration, endogenous neutrophils are cleared, hence clearly allowing the demonstration that i.p. administered DEL-1 enhances the uptake of coadministered fluorescently labeled apoptotic neutrophils by resident macrophages, thereby promoting inflammation resolution (Kourtzelis et al. 2019). Although DEL-1 protects against periodontitis in both mice and nonhuman primates (Eskan et al. 2012, Shin et al. 2015), it would not be practically feasible to distinguish its anti-inflammatory from its proresolving activities by assaying the periodontium.
Conclusions and perspectives
From the above discussion of the relevant literature, it can be concluded that the use of animal-based disease models has overall offered mechanistic understanding of oral diseases and has identified potential therapeutic approaches, some of which are in different stages of clinical development (Koo et al. 2017, Balta et al. 2021, Hajishengallis et al. 2021). No single model can faithfully mimic a human disease in its entirety and thus the utility of a given model lies in its capacity to test a specific hypothesis. Accordingly, the use of distinct models can address different but complementary hypotheses involving discrete components of the pathologic process, thus providing a more comprehensive understanding of the disease than obtained from the use of a single model (Fig. 4). Knowledge of the limitations associated with specific models is of great importance, as it can enable the design of improved studies that can be as relevant as possible to the human condition. Moreover, key findings regarding mechanistic and therapeutic concepts derived from disease models should be followed, whenever possible, by targeted validation in human studies.
This review has focused predominantly on rodent and especially mouse models of disease, which have distinct advantages but also disadvantages compared to large animals, as outlined above (Table 1). Whether such models could be replaced in the future by sophisticated in vitro reductionist systems remains uncertain, despite recent advances in tissue- or organ-on-a-chip models. For instance, as outlined earlier in the review, interorgan communication and the dynamics of the immune system are, at least, formidable challenges for in vitro simulation. As in vitro systems are unlikely to fully replicate the complexity of in vivo models and human research is typically correlative, animal disease models remain essential in biomedical research.
Thus, when animal models are viewed as tools by which to address specific hypotheses relevant to a given aspect of a human disease, they can be used productively for generation or substantiation of new knowledge (Table 2). Whereas animals and humans generally harbor different microbiomes, dysbiosis has more to do with the collective protein expression profile of the community rather than its particular species composition, hence mechanisms and outcomes of dysbiosis can reliably be investigated in model organisms. Our understanding of the human condition is greatly enhanced by synthesizing findings and evidence from different experimental systems, i.e. in vitro models, model organisms and human clinical studies (Fig. 3).
Table 2.
Utility, limitations, and importance of animal models in oral science.
| Essential for testing or discovering: |
| • Cause-and-effect relationships |
| • New biological concepts |
| • Preclinical safety and efficacy of candidate drugs |
| • Biological significance of in vitro findings |
| • Identification of novel potential virulence factors |
| • Identification of novel potential therapeutic targets |
| Limitations: |
| • Do not faithfully replicate all aspects of human disease |
| • Need to confine their use to addressing hypotheses related to specific disease aspects |
| • More than one model may be required to obtain adequate insight |
| Contributions to the understanding and treatment of oral diseases: |
| • Etiologic role of bacteria and sucrose in dental caries |
| • Etiologic role of bacteria and host response in periodontitis |
| • Laying the foundation for mucosal immunization against pathogens that colonize or invade via mucosal surfaces |
| • Dysbiosis rather than individual pathogens cause periodontitis |
| • Mechanistic underpinnings substantiating the epidemiological association of periodontitis with inflammatory comorbidities |
| • Genetic basis of host susceptibility/resistance to caries or periodontitis |
| • Discovery that tetracyclines inhibit tissue breakdown independently of antimicrobial action: anticollagenolytic effects |
| • Identification of IL-23 blockade for the treatment of human LAD1 periodontitis |
| • Identification of complement C3 as therapeutic target in human periodontitis (phase 2a trial) |
The predictive validity of animal models for the identification of drug targets to treat human patients depends greatly on rigorous control for variables including husbandry and environmental conditions, such as, diet, activity, and stress but importantly also the microbiome. In that regard, comparing wild-type and gene knockout mice raised separately may introduce microbiome differences that are not necessarily related to the genetic defect. Thus, to ensure that differences between wild-type controls and mice with gene deletions/alterations are due to genetic (and not environmental) differences, researchers should use as wild-type controls only littermates of mutant mice. As mice are the main preclinical tool for oral basic and mechanistic studies, including the study of microbiome and its effects on health and disease, it becomes imperative that scientists develop a comprehensive bioinformatics tool offering oral microbe taxonomic and genomic information, analogous to the Human Oral Microbiome Database (https://www.homd.org/). Such complete and curated resource would help unambiguously identify species and prevent inconsistency in findings generated from different research groups.
In conclusion, although mice and other model organisms do not flawlessly approximate the human condition, if the experimental questions are framed to fit the model system, they can provide productive insights into human biology and disease. By appreciating both their strengths and limitations, scientists can use animal models to address meaningful hypotheses on basic biological concepts, test causality of associations established in clinical research, and identify potential therapeutic targets for the treatment of human diseases.
Acknowledgement
The figures were created using Biorender.com.
Conflict of interest statement
G.H. is the inventor of a patent that describes the use of complement inhibitors in periodontal disease (“Methods of Treating or Preventing Periodontitis and Diseases Associated with Periodontitis;” patent number 10668135).
Funding
Research in the author’s laboratory is supported by NIH/NIDCR grants DE028561, DE029436, DE026152, and DE031206.
References
- Abe T, AlSarhan M, Benakanakere MRet al. The B cell-stimulatory cytokines BLyS and APRIL are elevated in human periodontitis and are required for B cell-dependent bone loss in experimental murine periodontitis. J Immunol. 2015;195:1427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T, Hajishengallis G. Optimization of the ligature-induced periodontitis model in mice. J Immunol Meth. 2013;394:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T, Hosur KB, Hajishengallis Eet al. Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J Immunol. 2012;189:5442–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T, Shin J, Hosur Ket al. Regulation of osteoclast homeostasis and inflammatory bone loss by MFG-E8. J Immunol. 2014;193:1383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abusleme L, Dupuy AK, Dutzan Net al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013;7:1016–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abusleme L, Hong BY, Hoare Aet al. Oral microbiome characterization in murine models. Bio Protoc. 2017;7:e2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abusleme L, O'Gorman H, Dutzan Net al. Establishment and stability of the murine oral microbiome. J Dent Res. 2020;99:721–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque-Souza E, Sahingur SE. Periodontitis, chronic liver diseases and the emerging oral-gut-liver axis. Periodontol 2000. 2022;89:125–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anbinder AL, Moraes RM, Lima GMGet al. Periodontal disease exacerbates systemic ovariectomy-induced bone loss in mice. Bone. 2016;83:241–7. [DOI] [PubMed] [Google Scholar]
- Arce M, Endo N, Dutzan Net al. A reappraisal of microbiome dysbiosis during experimental periodontitis. Mol Oral Microbiol. 2022;37:180–95. [DOI] [PubMed] [Google Scholar]
- Arrieta MC, Walter J, Finlay BB. Human microbiota-associated mice: a model with challenges. Cell Host Microbe. 2016;19:575–8. [DOI] [PubMed] [Google Scholar]
- Baker PJ, Dixon M, Evans RTet al. CD4(+) T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect Immun. 1999;67:2804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker PJ. Genetic control of the immune response in pathogenesis. J Periodontol. 2005;76:2042–6. [DOI] [PubMed] [Google Scholar]
- Baker PJ. The role of immune responses in bone loss during periodontal disease. Microbes Infect. 2000;2:1181–92. [DOI] [PubMed] [Google Scholar]
- Balta MG, Papathanasiou E, Blix IJet al. Host modulation and treatment of periodontal disease. J Dent Res. 2021;100:798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros SP, Offenbacher S. Epigenetics: connecting environment and genotype to phenotype and disease. J Dent Res. 2009;88:400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhari SAH, Khan AA, Butt AKet al. Non-surgical periodontal therapy reduces coronary heart disease risk markers: a randomized controlled trial. J Clin Periodontol. 2012;39:1065–74. [DOI] [PubMed] [Google Scholar]
- Bosco N, Noti M. The aging gut microbiome and its impact on host immunity. Genes Immun. 2021;22:289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce BF, Aufdemorte TB, Garrett IRet al. Effects of interleukin-1 on bone turnover in normal mice. Endocrinology. 1989;125:1142–50. [DOI] [PubMed] [Google Scholar]
- Burns E, Bachrach G, Shapira Let al. Cutting edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296–300. [DOI] [PubMed] [Google Scholar]
- Burns E, Eliyahu T, Uematsu Set al. TLR2-dependent inflammatory response to Porphyromonas gingivalis is MyD88 independent, whereas MyD88 is required to clear infection. J Immunol. 2010;184:1455–62. [DOI] [PubMed] [Google Scholar]
- Chen B-d, Jia X-m, Xu J-yet al. An autoimmunogenic and proinflammatory profile defined by the gut microbiota of patients with untreated systemic lupus erythematosus. Arthr Rheumatol. 2021;73:232–43. [DOI] [PubMed] [Google Scholar]
- Chung H, Pamp Sünje J, Hill Jonathan Aet al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149:1578–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coimbra LS, Steffens JP, Alsadun Set al. Clopidogrel enhances mesenchymal stem cell proliferation following periodontitis. J Dent Res. 2015;94:1691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consiglio CR, Cotugno N, Sardh Fet al. The immunology of multisystem inflammatory syndrome in children with COVID-19. Cell. 2020;183:968–81.e967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MA, Diaz PI, Van Dyke TE. The role of the microbiota in periodontal disease. Periodontol 2000. 2020;83:14–25. [DOI] [PubMed] [Google Scholar]
- D’Aiuto F, Orlandi M, Gunsolley JC. Evidence that periodontal treatment improves biomarkers and CVD outcomes. J Clin Periodontol. 2013;40:S85–105. [DOI] [PubMed] [Google Scholar]
- Daep CA, Novak EA, Lamont RJet al. Structural dissection and in vivo effectiveness of a peptide inhibitor of Porphyromonas gingivalis adherence to Streptococcus gordonii. Infect Immun. 2011;79:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz PI, Hoare A, Hong BY. Subgingival microbiome shifts and community dynamics in periodontal diseases. J Calif Dent Assoc. 2016;44:421–35. [PubMed] [Google Scholar]
- Dirnagl U. Modeling immunity and inflammation in stroke. Stroke. 2014;45:e177–8. [DOI] [PubMed] [Google Scholar]
- Duran-Pinedo AE, Chen T, Teles Ret al. Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis. ISME J. 2014;8:1659–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzan N, Kajikawa T, Abusleme Let al. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med. 2018;10:eaat0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery DC, Cerajewska TL, Seong Jet al. Comparison of blood bacterial communities in periodontal health and periodontal disease. Front Cell Infect Microbiol. 2021;10:577485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskan MA, Jotwani R, Abe Tet al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol. 2012;13:465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsetta ML, Klein MI, Colonne PMet al. Symbiotic relationship between Streptococcus mutans and Candida albicans synergizes virulence of plaque biofilms in vivo. Infect Immun. 2014;82:1968–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzGerald G, Botstein D, Califf Ret al. The future of humans as model organisms. Science. 2018;361:552–3. [DOI] [PubMed] [Google Scholar]
- Fitzgerald RJ, Keyes PH. Demonstration of the etiologic role of streptococci in experimental caries in the hamster. J Am Dent Assoc. 1960;61:9–19. [DOI] [PubMed] [Google Scholar]
- Fokkema SJ, Loos BG, Hart AAMet al. Long-term effect of full-mouth tooth extraction on the responsiveness of peripheral blood monocytes. J Clin Periodontol. 2003;30:756–60. [DOI] [PubMed] [Google Scholar]
- Fritz JV, Desai MS, Shah Pet al. From meta-omics to causality: experimental models for human microbiome research. Microbiome. 2013;1:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia BA, Acosta NC, Tomar SLet al. Association of Candida albicans and Cbp(+) Streptococcus mutans with early childhood caries recurrence. Sci Rep. 2021;11:10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genco CA, Cutler CW, Kapczynski Det al. A novel mouse model to study the virulence of and host response to Porphyromonas (Bacteroides) gingivalis. Infect Immun. 1991;59:1255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genco RJ, Sanz M. Clinical and public health implications of periodontal and systemic diseases: an overview. Periodontol 2000. 2020;83:7–13. [DOI] [PubMed] [Google Scholar]
- Genco RJ, Van Dyke TE. Prevention: reducing the risk of CVD in patients with periodontitis. Nat Rev Cardiol. 2010;7:479–80. [DOI] [PubMed] [Google Scholar]
- Gevers D, Kugathasan S, Denson Lee Aet al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons RJ, Houte JV. Bacterial adherence in oral microbial ecology. Annu Rev Microbiol. 1975;29:19–44. [DOI] [PubMed] [Google Scholar]
- Gilthorpe MS, Zamzuri AT, Griffiths GSet al. Unification of the “burst” and “linear” theories of periodontal disease progression: a multilevel manifestation of the same phenomenon. J Dent Res. 2003;82:200–5. [DOI] [PubMed] [Google Scholar]
- Goh CE, Bohn B, Marotz Cet al. Nitrite generating and depleting capacity of the oral microbiome and cardiometabolic risk: results from ORIGINS. J Am Heart Assoc. 2022;11:e023038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh CE, Trinh P, Colombo PCet al. Association between nitrate-reducing oral bacteria and cardiometabolic outcomes: results From ORIGINS. J Am Heart Assoc. 2019;8:e013324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub LM, Lee HM, Lehrer Get al. Minocycline reduces gingival collagenolytic activity during diabetes. Preliminary observations and a proposed new mechanism of action. J Periodontal Res. 1983;18:516–26. [DOI] [PubMed] [Google Scholar]
- Golub LM, Lee HM. Periodontal therapeutics: current host-modulation agents and future directions. Periodontol 2000. 2020;82:186–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez A, Espinoza JL, Harkins DMet al. Host genetic control of the oral microbiome in health and disease. Cell Host Microbe. 2017;22:269–78.e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson JM, Tanner AC, Haffajee ADet al. Patterns of progression and regression of advanced destructive periodontal disease. J Clin Periodontol. 1982;9:472–81. [DOI] [PubMed] [Google Scholar]
- Graves DT, Fine D, Teng YTet al. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol. 2008;35:89–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves DT, Oates T, Garlet GP. Review of osteoimmunology and the host response in endodontic and periodontal lesions. J Oral Microbiol. 2011;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves DT, Oskoui M, Volejnikova Set al. Tumor necrosis factor modulates fibroblast apoptosis, PMN recruitment, and osteoclast formation in response to P. gingivalis infection. J Dent Res. 2001;80:1875–9. [DOI] [PubMed] [Google Scholar]
- Hachicha M, Pouliot M, Petasis NAet al. Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor necrosis factor 1alpha-initiated neutrophil responses and trafficking: regulators of a cytokine-chemokine axis. J Exp Med. 1999;189:1923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Chavakis T. DEL-1-regulated immune plasticity and inflammatory disorders. Trends Mol Med. 2019;25:444–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. 2021;21:426–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Hasturk H, Lambris JDet al. C3-targeted therapy in periodontal disease: moving closer to the clinic. Trends Immunol. 2021;42:856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Korostoff JM. Revisiting the Page & Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later. Periodontol 2000. 2017;75:116–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Lamont RJ, Graves DT. The enduring importance of animal models in understanding periodontal disease. Virulence. 2015;6:229–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Lamont RJ, Koo H. Oral polymicrobial communities: assembly, function, and impact on diseases. Cell Host Microbe. 2023;31;528–38.. 10.1016/j.chom.2023.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Lamont RJ. Dancing with the stars: how choreographed bacterial interactions dictate nososymbiocity and give rise to keystone pathogens, accessory pathogens, and pathobionts. Trends Microbiol. 2016;24:477–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Lamont RJ. Polymicrobial communities in periodontal disease: their quasi-organismal nature and dialogue with the host. Periodontol 2000. 2021;86:210–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Liang S, Payne MAet al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Wang M, Liang Set al. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci USA. 2008;105:13532–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol. 2014;29:248–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- , Hasturk H, Hajishengallis G, Lambris JD, et al., Forsyth Institute Center for Translational Research . Phase IIa clinical trial of complement C3 inhibitor AMY-101 in adults with periodontal inflammation. J Clin Invest. 2021;131:e152973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A, Goguet-Surmenian Eet al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. 2007;179:7021–9. [DOI] [PubMed] [Google Scholar]
- Herrero ER, Fernandes S, Verspecht Tet al. Dysbiotic biofilms deregulate the periodontal inflammatory response. J Dent Res. 2018;97:22034517752675. [DOI] [PubMed] [Google Scholar]
- Hillam DG, Hull PS. The influence of experimental gingivitis on plaque formation. J Clin Periodontol. 1977;4:56–61. [DOI] [PubMed] [Google Scholar]
- Hoare A, Wang H, Meethil Aet al. A cross-species interaction with a symbiotic commensal enables cell-density-dependent growth and in vivo virulence of an oral pathogen. ISME J. 2021;15:1490–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt SC, Ebersole J, Felton Jet al. Implantation of Bacteroides gingivalis in nonhuman primates initiates progression of periodontitis. Science. 1988;239:55–57. [DOI] [PubMed] [Google Scholar]
- Humphrey SP, Williamson RT. A review of saliva: normal composition, flow, and function. J Prosthet Dent. 2001;85:162–9. [DOI] [PubMed] [Google Scholar]
- Hwang G, Liu Y, Kim Det al. Candida albicans mannans mediate Streptococcus mutans exoenzyme GtfB binding to modulate cross-kingdom biofilm development in vivo. PLoS Pathog. 2017;13:e1006407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai J, Kitamoto S, Kamada N. The pathogenic oral-gut-liver axis: new understandings and clinical implications. Expert Rev Clin Immunol. 2021;17:727–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwandi RA, Chiesa ST, Hajishengallis Get al. The roles of neutrophils linking periodontitis and atherosclerotic cardiovascular diseases. Front Immunol. 2022;13:915081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishai A, Osborne MT, El Kholy Ket al. Periodontal disease associates with arterial inflammation via potentiation of a hematopoietic-arterial axis. JACC Cardiovasc Imaging. 2019;12:2271–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Misaki T, Naka Set al. Specific strains of Streptococcus mutans, a pathogen of dental caries, in the tonsils, are associated with IgA nephropathy. Sci Rep. 2019;9:20130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Darzi Y, Tawaratsumida Ket al. Induction of bone loss by pathobiont-mediated Nod1 signaling in the oral cavity. Cell Host Microbe. 2013;13:595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone KF, Wei Y, Bittner-Eddy PDet al. Calprotectin (S100A8/A9) is an innate immune effector in experimental periodontitis. Infect Immun. 2021;89:e0012221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Yamazaki K, Nakajima Met al. Oral administration of Porphyromonas gingivalis alters the gut microbiome and serum metabolome. mSphere. 2018;3:e00460–00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keffer J, Probert L, Cazlaris Het al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyes PH. Dental caries in the Syrian bamster. VIII. The induction of rampant caries activity in albino and golden animals. J Dent Res. 1959;38:525–33. [DOI] [PubMed] [Google Scholar]
- Kibe R, Sakamoto M, Yokota Het al. Movement and fixation of intestinal microbiota after administration of human feces to germ-free mice. Appl Environ Microbiol. 2005;71:3171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto S, Nagao-Kitamoto H, Jiao Yet al. The intermucosal connection between the mouth and gut in commensal pathobiont-driven colitis. Cell. 2020;182:447–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittaka M, Yoshimoto T, Schlosser Cet al. Alveolar bone protection by targeting the SH3BP2-SYK axis in osteoclasts. J Bone Miner Res. 2020;35:382–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo H, Allan RN, Howlin RPet al. Targeting microbial biofilms: current and prospective therapeutic strategies. Nat Rev Microbiol. 2017;15:740–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornman KS, Page RC, Tonetti MS. The host response to the microbial challenge in periodontitis: assembling the players. Periodontol 2000. 1997;14:33–53. [DOI] [PubMed] [Google Scholar]
- Koskella B, Bergelson J. The study of host-microbiome (co)evolution across levels of selection. Philos Trans R Soc Lond B Biol Sci. 2020;375:20190604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtzelis I, Li X, Mitroulis Iet al. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol. 2019;20:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont RJ, El-Sabaeny A, Park Yet al. Role of the Streptococcus gordonii SspB protein in the development of Porphyromonas gingivalis biofilms on streptococcal substrates. Microbiology. 2002;148:1627–36. [DOI] [PubMed] [Google Scholar]
- Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. 2018;16:745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavelle EC, Ward RW. Mucosal vaccines—fortifying the frontiers. Nat Rev Immunol. 2022;22:236–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-T, Teles R, Kantarci Aet al. Resolvin E1 reverses experimental periodontitis and dysbiosis. J Immunol. 2016;197:2796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CT, Li R, Zhu Let al. Subgingival microbiome and specialized pro-resolving lipid mediator pathway profiles are correlated in periodontal inflammation. Front Immunol. 2021;12:691216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JA, Palmer SR, Zeng Let al. The biology of Streptococcus mutans. Microbiol Spectr. 2019;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CM, de Haan P, Ronaldson-Bouchard Ket al. A guide to the organ-on-a-chip. Nat Rev Methods Primers. 2022;2:33. [Google Scholar]
- Ley RE, Bäckhed F, Turnbaugh Pet al. Obesity alters gut microbial ecology. Proc Natl Acad Sci. 2005;102:11070–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Ge Y, Cheng Let al. Oral bacteria colonize and compete with gut microbiota in gnotobiotic mice. Int J Oral Sci. 2019;11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Feng Y, Tang Het al. A simplified and effective method for generation of experimental murine periodontitis model. Front Bioeng Biotechnol. 2020;8:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang H, Yu Xet al. Maladaptive innate immune training of myelopoiesis links inflammatory comorbidities. Cell. 2022;185:1709–27.e1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Niimi H, Ohsugi Yet al. Application of ligature-induced periodontitis in mice to explore the molecular mechanism of periodontal disease. Int J Molec Sci. 2021;22:8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling MR, Chapple IL, Matthews JB. Peripheral blood neutrophil cytokine hyper-reactivity in chronic periodontitis. Innate Immun. 2015;21:714–25. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Carlström M, Weitzberg E. Metabolic effects of dietary nitrate in health and disease. Cell Metab. 2018;28:9–22. [DOI] [PubMed] [Google Scholar]
- Lundberg R, Toft MF, Metzdorff SBet al. Human microbiota-transplanted C57BL/6 mice and offspring display reduced establishment of key bacteria and reduced immune stimulation compared to mouse microbiota-transplantation. Sci Rep. 2020;10:7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa T, Abe T, Hajishengallis Eet al. Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J Immunol. 2014a;192:6020–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa T, Hosur K, Abe Tet al. Antagonistic effects of IL-17 and D-resolvins on endothelial Del-1 expression through a GSK-3beta-C/EBPbeta pathway. Nat Commun. 2015;6:8272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa T, Krauss JL, Abe Tet al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe. 2014b;15:768–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkawi H, Hoch S, Burns Eet al. Porphyromonas gingivalis stimulates TLR2-PI3K signaling to escape immune clearance and induce bone resorption independently of MyD88. Front Cell Infect Microbiol. 2017;7:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesan J, Girnary MS, Jing Let al. An experimental murine model to study periodontitis. Nat Protoc. 2018;13:2247–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh PD. Are dental diseases examples of ecological catastrophes?. Microbiology. 2003;149:279–94. [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Kato T, Takahashi Net al. Ligature-induced periodontitis in mice induces elevated levels of circulating interleukin-6 but shows only weak effects on adipose and liver tissues. J Periodont Res. 2016;51:639–46. [DOI] [PubMed] [Google Scholar]
- Michalek SM, McGhee JR, Mestecky Jet al. Ingestion of Streptococcus mutans induces secretory immunoglobulin A and caries immunity. Science. 1976;192:1238–40. [DOI] [PubMed] [Google Scholar]
- Miyoshi M, Kasahara E, Park AMet al. Dietary nitrate inhibits stress-induced gastric mucosal injury in the rat. Free Radic Res. 2003;37:85–90. [DOI] [PubMed] [Google Scholar]
- Moutsopoulos NM, Konkel J, Sarmadi Met al. Defective neutrophil recruitment in leukocyte adhesion deficiency. type I disease causes local IL-17–driven inflammatory bone loss. Sci Transl Med. 2014;6:229ra240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutsopoulos NM, Zerbe CS, Wild Tet al. Interleukin-12 and interleukin-23 blockade in leukocyte adhesion deficiency type 1. N Engl J Med. 2017;376:1141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau JH, Auwerx J. The virtuous cycle of human genetics and mouse models in drug discovery. Nat Rev Drug Disc. 2019;18:255–72. [DOI] [PubMed] [Google Scholar]
- Nagai K, Ideguchi H, Kajikawa Tet al. An injectable hydrogel-formulated inhibitor of prolyl-4-hydroxylase promotes T regulatory cell recruitment and enhances alveolar bone regeneration during resolution of experimental periodontitis. FASEB J. 2020;34:13726–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka S, Wato K, Misaki Tet al. Streptococcus mutans induces IgA nephropathy-like glomerulonephritis in rats with severe dental caries. Sci Rep. 2021;11:5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13:1161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura R, Matayoshi S, Otsugu Met al. Contribution of severe dental caries induced by Streptococcus mutans to the pathogenicity of infective endocarditis. Infect Immun. 2020;88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne MA, Hashim A, Alsam Aet al. Horizontal and vertical transfer of oral microbial dysbiosis and periodontal disease. J Dent Res. 2019;98:1503–10. [DOI] [PubMed] [Google Scholar]
- Pouliot M, Clish CB, Petasis NAet al. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: a role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry. 2000;39:4761–8. [DOI] [PubMed] [Google Scholar]
- Pound P, Bracken MB. Is animal research sufficiently evidence based to be a cornerstone of biomedical research?. BMJ. 2014;348:g3387. [DOI] [PubMed] [Google Scholar]
- Radner H, Aletaha D. Anti-TNF in rheumatoid arthritis: an overview. Wiener Medizinische Wochenschrift. 2015;165:3–9. [DOI] [PubMed] [Google Scholar]
- Radvar M, Tavakkol-Afshari J, Bajestan MNet al. The effect of periodontal treatment on IL-6 production of peripheral blood monocytes in aggressive periodontitis and chronic periodontitis patients. Iran J Immunol. 2008;5:100–6. [DOI] [PubMed] [Google Scholar]
- Ribeiro AA, Jiao Y, Girnary Met al. Oral biofilm dysbiosis during experimental periodontitis. Mol Oral Microbiol. 2022;37:256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FEet al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren Tet al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31. [DOI] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovin S, Costich ER, Gordon HA. The influence of bacteria and irritation in the initiation of periodontal disease in germ-free and conventional rats. J Periodontal Res. 1966;1:193–204. [DOI] [PubMed] [Google Scholar]
- Russell MW, Childers NK, Michalek SMet al. A caries vaccine? The state of the science of immunization against dental caries. Caries Res. 2004;38:230–5. [DOI] [PubMed] [Google Scholar]
- Schenkein HA, Papapanou PN, Genco Ret al. Mechanisms underlying the association between periodontitis and atherosclerotic disease. Periodontol 2000. 2020;83:90–106. [DOI] [PubMed] [Google Scholar]
- Schirmer M, Denson L, Vlamakis Het al. Compositional and temporal changes in the gut microbiome of pediatric ulcerative colitis patients are linked to disease course. Cell Host Microbe. 2018;24:600–10.e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Markus J, Kandarova Het al. Tissue-on-a-Chip: microphysiometry with human 3D models on transwell inserts. Front Bioeng Biotechnol. 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TS, Hayward MR, Coelho LPet al. Extensive transmission of microbes along the gastrointestinal tract. Elife. 2019;8:e42693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AGet al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110:3507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Libreros S, Nshimiyimana R. E-series resolvin metabolome, biosynthesis and critical role of stereochemistry of specialized pro-resolving mediators (SPMs) in inflammation-resolution: preparing SPMs for long COVID-19, human clinical trials, and targeted precision nutrition. Sem Immunol. 2022;101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Maekawa T, Abe Tet al. DEL-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Sci Transl Med. 2015;7:307ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S, Sack MN, Greer JJet al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LM, Doyle AD, Greenwell-Wild Tet al. Fibrin is a critical regulator of neutrophil effector function at the oral mucosal barrier. Science. 2021;374:eabl5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima C, Gastfreund S, Sun Cet al. Rac-null leukocytes are associated with increased inflammation-mediated alveolar bone loss. Am J Pathol. 2014;184:472–82. [DOI] [PubMed] [Google Scholar]
- Singh A, Wyant T, Anaya-Bergman Cet al. The capsule of Porphyromonas gingivalis leads to a reduction in the host inflammatory response, evasion of phagocytosis, and increase in virulence. Infect Immun. 2011;79:4533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slots J, Rosling BG. Suppression of the periodontopathic microflora in localized juvenile periodontitis by systemic tetracycline. J Clin Periodontol. 1983;10:465–86. [DOI] [PubMed] [Google Scholar]
- Sobko T, Reinders C, Norin Eet al. Gastrointestinal nitric oxide generation in germ-free and conventional rats. Am J Physiol Gastrointest Liver Physiol. 2004;287:G993–997. [DOI] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD, Goodson JMet al. New concepts of destructive periodontal disease. J Clin Periodontol. 1984;11:21–32. [DOI] [PubMed] [Google Scholar]
- Stacy A, Everett J, Jorth Pet al. Bacterial fight-and-flight responses enhance virulence in a polymicrobial infection. Proc Natl Acad Sci USA. 2014;111:7819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JH, Wang YI, Narasimhan Sriram Net al. Recent advances in body-on-a-chip systems. Anal Chem. 2019;91:330–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao K, Miyakawa T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2015;112:1167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzer JM. Dental caries is a transmissible infectious disease: the Keyes and Fitzgerald revolution. J Dent Res. 1995;74:1536–42. [DOI] [PubMed] [Google Scholar]
- Teles F, Wang Y, Hajishengallis Get al. Impact of systemic factors in shaping the periodontal microbiome. Periodontol 2000. 2021;85:126–60. [DOI] [PubMed] [Google Scholar]
- Thevaranjan N, Puchta A, Schulz Cet al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe. 2017;21:455–66.e454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribble GD, Angelov N, Weltman Ret al. Frequency of tongue cleaning impacts the human tongue microbiome composition and enterosalivary circulation of nitrate. Front Cell Infect Microbiol. 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukasaki M, Komatsu N, Nagashima Ket al. Host defense against oral microbiota by bone-damaging T cells. Nat Commun. 2018;9:701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuno T, Takahashi N, Yamada-Hara Met al. Ingestion of Porphyromonas gingivalis exacerbates colitis via intestinal epithelial barrier disruption in mice. J Periodont Res. 2021;56:275–88. [DOI] [PubMed] [Google Scholar]
- Van Dyke TE, Kholy KE, Ishai Aet al. Inflammation of the periodontium associates with risk of future cardiovascular events. J Periodontol. 2021;92:348–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ideguchi H, Kajikawa Tet al. Complement is required for microbe-driven induction of Th17 and periodontitis. J Immunol. 2022;209:1370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Jotwani R, Le Jet al. Filifactor alocis infection and inflammatory responses in the mouse subcutaneous chamber model. Infect Immun. 2013;8:1205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster JD, Santagostino SF, Foreman O. Applications and considerations for the use of genetically engineered mouse models in drug development. Cell Tissue Res. 2020;380:325–40. [DOI] [PubMed] [Google Scholar]
- Weiner GS, DeMarco TJ, Bissada NF. Long term effect of systemic tetracycline administration on the severity of induced periodontitis in the rat. J Periodontol. 1979;50:619–23. [DOI] [PubMed] [Google Scholar]
- Werneck R, Mira M, Trevilatto P. A critical review: an overview of genetic influence on dental caries. Oral Dis. 2010;16:613–23. [DOI] [PubMed] [Google Scholar]
- Williams DW, Vuong HE, Kim Set al. Indigenous microbiota protects against inflammation-induced osteonecrosis. J Dent Res. 2020;99:676–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner P, Mitchell PJ. The validity of animal models of predisposition to depression. Behav Pharmacol. 2002;13:169–88. [DOI] [PubMed] [Google Scholar]
- Wolchok J. Putting the immunologic brakes on cancer. Cell. 2018;175:1452–4. [DOI] [PubMed] [Google Scholar]
- Wu Y-Y, Xiao E, Graves DT. Diabetes mellitus related bone metabolism and periodontal disease. Int J Oral Sci. 2015;7:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao E, Mattos M, Vieira GHAet al. Diabetes enhances IL-17 expression and alters the oral microbiome to increase its pathogenicity. Cell Host Microbe. 2017;22:120–8.e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Huang X, Alkhers Net al. Candida albicans and early childhood caries: a systematic review and meta-analysis. Caries Res. 2018;52:102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Moon Y, Li Let al. Candida albicans carriage in children with severe early childhood caries (S-ECC) and maternal relatedness. PLoS ONE. 2016;11:e0164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing T, Liu Y, Cheng Het al. Ligature induced periodontitis in rats causes gut dysbiosis leading to hepatic injury through SCD1/AMPK signalling pathway. Life Sci. 2022;288:120162. [DOI] [PubMed] [Google Scholar]
- Yachida S, Mizutani S, Shiroma Het al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med. 2019;25:968–76. [DOI] [PubMed] [Google Scholar]
- Yamazaki K, Kato T, Tsuboi Yet al. Oral pathobiont-induced changes in gut microbiota aggravate the pathology of nonalcoholic fatty liver disease in mice. Front Immunol. 2021;12:766170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuh DY, Maekawa T, Li Xet al. The secreted protein DEL-1 activates a β3 integrin-FAK-ERK1/2-RUNX2 pathway and promotes osteogenic differentiation and bone regeneration. J Biol Chem. 2020;295:7261–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Fadaak A, Alomeir Net al. Lactobacillus plantarum disrupts S. mutans–C. albicans cross-kingdom biofilms. Front Cell Infect Microbiol. 2022;12:872012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Whiteley M, Lewin GR. Polymicrobial interactions of oral microbiota: a historical review and current perspective. Mbio. 2022;13:e00235–00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Qin S, Xu Xet al. Inhibitory effect of Lactobacillus plantarum CCFM8724 towards Streptococcus mutans- and Candida albicans-induced caries in rats. Oxid Med Cell Longev. 2020;2020:4345804. [DOI] [PMC free article] [PubMed] [Google Scholar]