

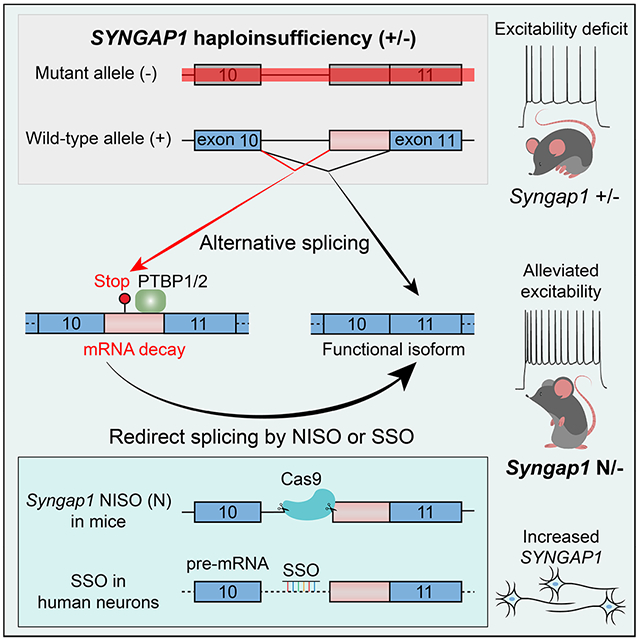
Summary
The Ras GTPase-activating protein SYNGAP1 plays a central role in synaptic plasticity, and de novo SYNGAP1 mutations are among the most frequent causes of autism and intellectual disability. How SYNGAP1 is regulated during development and how to treat SYNGAP1-associated haploinsufficiency remain challenging questions. Here we characterize an alternative 3’ splice site (A3SS) of SYNGAP1 that induces nonsense-mediated mRNA decay (A3SS-NMD) in mouse and human neural development. We demonstrate that PTBP proteins directly bind to SYNGAP1 and promote A3SS inclusion. Genetic deletion of Syngap1 A3SS in mice upregulates Syngap1 protein and alleviates the long-term potentiation and membrane excitability deficits caused by a Syngap1 knockout allele. We further report a splice-switching oligonucleotide (SSO) that converts SYNGAP1 unproductive isoform to the functional form. This study describes the regulation and function of SYNGAP1 A3SS-NMD, the genetic rescue of heterozygous Syngap1 knockout mice, and the development of an SSO to potentially alleviate SYNGAP1-associated haploinsufficiency.
Graphical Abstract
eTOC Blurb:
De novo SYNGAP1 mutations cause autism and intellectual disability. Yang et al. show that PTBP1/2 proteins promote SYNGAP1 mRNA decay through alternative splicing. Redirecting splicing upregulates Syngap1 expression and alleviates neurological deficits in Syngap1 heterozygous knockout mice. These results suggest that redirecting splicing can potentially rescue SYNGAP1 haploinsufficiency.
Introduction
Synaptic transmission and plasticity are fundamental to neuronal functions, and alterations of synaptic protein expression are direct causes of autism, intellectual disability (ID), and epilepsy1,2. De novo loss-of-function (LoF) mutations in SYNGAP1, encoding the synaptic Ras GTPase activating protein3,4, are among the most prevalent causes of ID and autism spectrum disorders5–7. Previous human and mouse studies on SYNGAP1 pathogenic mechanisms converge that de novo mutations are predominantly LoF alleles and lead to SYNGAP1 haploinsufficiency7–9. The development of treatment for SYNGAP1-associated conditions has a far-reaching impact.
Syngap1 was initially identified by sequencing proteins in the post-synaptic density (PSD) or interacting with PDZ domains3,4. Syngap1 suppresses ERK phosphorylation and surface AMPA receptor levels10,11; phosphorylation of Syngap1 by CaM Kinase II rapidly disperses Syngap1 from synaptic spines and triggers AMPA receptor insertion3,12. Homozygous Syngap1 knockout (Syngap1 −/−) mice died within 48 hours after birth; heterozygous Syngap1 knockout led to ~50% reduction of Syngap1 protein, reduced long-term potentiation (LTP), impaired membrane excitability, decreased ability in spatial learning, and reduced cognition primarily due to defects in forebrain excitatory neurons13–16. Loss of Syngap1 in GABAergic cells was also reported to impair cognitive functions17. Mechanistically, Syngap1 heterozygous knockout mice exhibited increased excitatory synaptic transmission in early postnatal development due to enhanced AMPA receptor sensitivity9. The reexpression of Syngap1 in adult mice slightly improved brain functions and behaviors18, implying that upregulating SYNGAP1 protein expression in the brain can potentially alleviate symptoms in human patients with SYNGAP1 haploinsufficiency.
Alternative splicing coupled with nonsense-mediated mRNA decay (AS-NMD), or unproductive splicing, is frequently used by RNA splicing regulators as a negative autofeedback for homeostatic protein expression19,20. AS-NMD is predicted to regulate hundreds of genes in mammalian brains21, but its role in human and mouse brain development has only started to be explored recently22,24. In past years, splice-switching oligonucleotides (SSOs) targeting genes such as SMN2 have been successful in treating neurological disorders25,26, paving the path for SSO-mediated therapy and personalized medicine27. In our efforts to study cell-type-specific alternative splicing, we identified an alternative 3’ splice site (A3SS) of SYNGAP1 intron 10 that led to NMD in mouse and human brain development, hereafter referred to as Syngap1 A3SS-NMD. Alternative splicing was suggested as a potential way to modulate SYNGAP1 protein expression in HEK293 cells28, but it remains unclear to what extent SSO works in human neurons. More importantly, as a safety measure, it is essential to understand the organismal function and dosage effect of Syngap1 A3SS-NMD in vivo before targeting this exon for therapy.
We have characterized Syngap1 A3SS-NMD inclusion in brain development and investigated regulatory mechanisms. We identified intronic sequences required for Syngap1 A3SS-NMD inclusion and genetical deletion of this sequence in mice resulted in skipping of A3SS-NMD and enrichment of the neuronal isoform; therefore, we named this mouse allele Syngap1-NISO (Neuronal ISOform). Notably, we detected decreased A3SS-NMD inclusion and increased Syngap1 protein expression in Syngap1-NISO mouse brains and SSO-treated cerebral organoids. In this study, we investigate the functions of Syngap1 A3SS-NMD in vivo and explore whether the A3SS-NMD exon is a possible target to rescue haploinsufficiency.
Results:
Alternative splicing of mouse Syngap1 intron10 leads to nonsense-mediated mRNA decay
We investigated cell-type-specific alternative splicing during neocortical development at embryonic day 14.5 (E14.5) and identified an A3SS of Syngap1 intron10 (Figure 1A). We examined Syngap1 A3SS in mouse development and found that the inclusion level was high in non-neural tissues and during early brain development but decreased substantially in the adult brain (Figure 1B). RT-PCR of Syngap1 A3SS showed 76% inclusion at E12.5 and 5% inclusion at postnatal day 40 (P40, Figure 1C). Notably, intronic sequences around Syngap1 A3SS are conserved in vertebrates, suggesting a selection pressure during evolution (Figure 1B, bottom track). In cultured primary cortical neurons, Syngap1 A3SS showed 33±5% usage at day1 in vitro (DIV1) and decreased to 5±1% at DIV15 (Figure S1A). These results suggest that the Syngap1 A3SS is included in mouse brain development and remains detectable in differentiated neurons.
Figure 1. Alternative 3’ splice site of mouse Syngap1 inton10 induces nonsense-mediated mRNA decay.

A) Sashimi plots of isolated cortical neurons and apical neural progenitors showing Syngap1 A3SS in embryonic day 14.5 (E14.5) mouse dorsal forebrain. The A3SS exon inclusion ratios are indicated.
B) RNA-Seq results showing that Syngap1 A3SS was enriched in early brain development.
C) RT-PCR results showing that the Syngap1 A3SS was higher in the developing forebrain (76% at E12.5) and remained detectable in adulthood (5% at P40). PSI represents Percent Spliced In. One biological sample per lane.
D) Syngap1 A3SS introduces in-frame stop codons that truncate the protein and/or induce nonsense-mediated mRNA decay.
E) The predicted Syngap1 short protein isoform was not detectable in mouse brain lysates.
F) The Syngap1 A3SS transcripts were enriched in Neuro2a cells treated with cycloheximide (CHX, two biological replicates per condition, p<0.001, unpaired t-test).
G) The Syngap1 A3SS-NMD was upregulated in Neuro2a treated with two siRNAs against Upf1 (adj.p<0.05 for siRNA-1, adj.p<0.01 for siRNA-2, one-way ANOVA). Two biological replicates per condition.
See also Figure S1.
Surprisingly, the Syngap1 A3SS introduces in-frame translational stop codons, which are predicted to truncate the RasGAP domain or cause NMD (Figure 1D). Multiple lines of our results support that Syngap1 A3SS causes NMD (A3SS-NMD): 1) the predicted pre-mature stop-codons are over 50 base pairs away from downstream splice junctions (Figure 1D); 2) an antibody against the N-terminus of Syngap1 recognized the ectopically expressed Syngap1 N-terminal fragment and the endogenous full-length protein, but was unable to detect the truncated isoform from brain lysates (Figure 1E); 3) the Syngap1 A3SS isoform was enriched when NMD was blocked by inhibiting protein translation with cycloheximide (CHX) in Neuro2a cells and in primary neurons (Figure 1F and S1B); and 4) Syngap1 A3SS was upregulated when NMD was blocked by knocking down Upf1 with two different siRNAs in Neuro2a cells (Figure 1G and S1C). These results indicate that Syngap1 A3SS causes NMD while the canonical neuronal splice isoform ensures robust protein expression.
SYNGAP1 unproductive splicing is functionally conserved in humans
To determine whether human SYNGAP1 is regulated by A3SS in cortical development, we compared RNA-Seq data between the ventricular zone (VZ, enriched for neural progenitors) and cortical plate (CP, enriched for postmitotic neurons) of the gestational week 16 (GW16) fetal human brains29. SYNGAP1 A3SS showed higher inclusion in the VZ than in the CP (Figure 2A). This was further confirmed using RT-PCR with microdissected VZ and CP samples from multiple postmortem fetal dorsal forebrains (Figure 2B). We analyzed the human SYNGAP1 RNA-Seq reads and confirmed the inclusion of premature stop codons (Figure S2A). We further examined human SYNGAP1 A3SS inclusion during induced pluripotentstem cell (iPSC)-to-neuron differentiation and found SYNGAP1 A3SS remained 23±2% in NGN1/2-induced neurons at day4 (Figure 2C and S2B). CHX treatment in iPSCs and iPSC-derived neurons significantly increased SYNGA1 A3SS transcripts (Figure 2D–2E). Although the human SYGNAP1 transcript was also detectable in non-neural tissues (GTEx), the SYNGAP1 A3SS-NMD provides an orthogonal mechanism to ensure neural-specific expression of SYNGAP1 protein. Multiple sequence alignment of mammalian SYNGAP1 intron10 showed that the premature stop codon (TGA) introduced by the A3SS was highly conserved even though the positions of alternative 3’ splice sites varied among different species (Figure 2F). These results indicate that the SYNGAP1 A3SS-NMD is functionally conserved in human neural development.
Figure 2. Human SYNGAP1 A3SS induces nonsense-mediated mRNA decay in neural development.

A) RNA-Seq results showing SYNGAP1 A3SS in the laser microdissected cortical plate (CP) and ventricular zone (VZ) of gestational week 16 (GW16) fetal brains29.
B) RT-PCR results showing that SYNGAP1 A3SS was enriched in fetal cortical development.
C) RT-PCR results showing that SYNGAP1 A3SS levels significantly decreased in iPSCs during NGN1/2-induced neuronal differentiation. Two biological replicates per condition, p<0.001, one-way ANOVA.
D) SYNGAP1 A3SS was enriched in iPSCs after CHX treatment. p<0.001 by t-test, three biological replicates.
E) SYNGAP1 A3SS ratio was increased in iPSC-derived neurons after CHX treatment. p<0.001 by t-test, three biological replicates.
F) Sequence alignment showing that the premature stop codon (TGA) in SYNGAP1 A3SS is conserved in mammals while the splice acceptor sites (AG) are variable. The AG sites were annotated according to RNA-Seq results of corresponding species in NIH Genome Data Viewer.
G-H) RT-PCR results of SYNGAP1 mini-gene constructs (G) transfected in Neuro2a cells showing that human SYNGAP1 intronic mutations identified in autism and ID patients led to intron 10 retention (c.1676 +5 G>A, NM_006772.2) or abnormal A3SS inclusion (c.1677 −2_1685del, p<0.001 by t-test, three biological replicates in H) .
See also Figure S2.
Human mutations causing SYNGAP1 unproductive splicing are associated with ID and autism
To understand whether SYNGAP1 A3SS-NMD is functional in humans, we focused on two previously reported SYNGAP1 mutations in intron10: these mutations were identified in patients with typical ID and autistic features, yet the effects of these mutations on SYNGAP1 expression were not well understood7,30. We constructed wild-type and mutant SYNGAP1 mini-genes (spanning exon9 to exon12), introduced the constructs into Neuro2a cells, and found that the c.1676+5 G>A (NM_006772.2) mutation almost completely disrupted the splice donor and induced high intron10 retention (Figure 2G–2H). The pathogenic c.1677-2_1685del mutation disrupted the canonical splice acceptor of intron 10 and significantly increased SYNGAP1 A3SS usage (Figure 2G–2H). Thus, human mutations that lead to SYNGAP1 A3SS-NMD or intron10 retention could result in autism and intellectual disability, suggesting that the SYNGAP1 A3SS-NMD transcript is not functional in vivo.
SYNGAP1 A3SS-NMD is promoted by PTBP proteins
Alternative pre-mRNA splicing is regulated by RNA sequence in cis and splicing regulator proteins in trans31–33. We analyzed the flanking sequences of SYNGAP1 A3SS-NMD and identified CUCUCU sequences that resemble binding motifs of PTBP1 and PTBP2 (Figure S3A). PTBP proteins are master splicing regulators in mice and humans: PTBP1 is highly expressed in neural progenitors and non-neural tissues (GTEx), and PTBP2 is expressed in immature and differentiating neurons22,32,34. Across different developmental stages in the human dorsal forebrain35 (from gestational week 4 to elderly adults), SYNGAP1 A3SS-NMD inclusion ratios showed the same decreasing trend as PTBP1/2 mRNA levels (Figure S3B). We used shRNAs to knock down Ptbp1 and Ptbp2 in Neuro2a cells and found that the Syngap1 A3SS isoform was suppressed in double knockdown samples (Figure 3A–3B). Conversely, ectopic expression of PTBP1 in primary mouse cortical neurons decreased Syngap1 protein levels (Figure 3C and S3C). These results suggest that PTBP1 and PTBP2 suppress Syngap1 protein translation by promoting A3SS-NMD.
Figure 3. SYNGAP1 A3SS-NMD is regulated by PTBP proteins.

A) Western blot results showing shRNA knockdown of Ptbpl and Ptbp2 in Neuro2a cells. Two different shRNAs were used for each of Ptbpl and Ptbp2.
B) RT-PCR results showing that loss of Ptbp1/2 promoted splicing of the canonical/productive Syngap1 isoform.
C) Immunofluorescence staining of primary cortical neurons (DIV5) showing that ectopic expression of PTBP1 (red, mCherry) decreased Syngap1 protein level (green).
D) CLIP-Seq analyses showing Ptbp1, Ptbp2 and U2af65 binding peaks in the A3SS region.
E) Mini-gene constructs of the human SYNGAP1 showing deletion of predicted splicing elements, and RT-PCR results showing their effects on A3SS insertion. Noticeably, the intronic element #1 and predicted upstream U2AF65-PTBP binding site #1 were required for A3SS inclusion; the predicted U2AF65-PTBP binding site #2 was required for canonical splicing and A3SS skipping. Two biological replicates for each condition.
F) EMSA assay showing that PTBP1 protein had a higher affinity to the PTBP binding site #2 RNA probe.
G) Current working model: in neural progenitors and differentiating neurons, PTBP proteins bind to site#2 in A3SS (red) and suppress the canonical/neuronal 3’ splice site; in neurons, PTBP proteins are turned down/off, site #2 is exposed for splicing machinery (U2AF65) recognition and promotes neuronal isoform expression.
See also Figure S3.
To determine whether Syngap1 is a direct binding target of Ptbp proteins, we re-analyzed RNA crosslinking immunoprecipitation and sequencing (CLIP-Seq) datasets of Ptbp1 in mouse ESCs and NPCs36 and Ptbp2 CLIP-Seq results in E18.5 neocortices37. We identified Ptbp1 and Ptbp2 CLIP tags in the Syngap1 intron10 between the canonical and the alternative 3’ splice sites (Figure 3D). Ptbp proteins bind to CU-rich motifs and can redirect splicing by competing with U2af65/U2af238, a core splicing factor that binds to the polypyrimidine tract for 3’ splice site recognition. We re-analyzed CLIP-Seq data of U2af65 from E18.5 neocortices and found that CLIP-Seq tags of U2af65 and Ptbp1 overlapped with each other on Sygnap1 intron 10 (Figure 3D), suggesting that Ptbp1/2 may compete with U2af65 and regulate the 3’ splice site.
To identify the specific sequences and potential PTBP1 binding sites required for human SYNGAP1 A3SS-NMD inclusion/skipping, we created a series of mini-gene constructs in which conserved DNA sequences and predicted PTBP1 binding sites were deleted (Figure 3E, S3A, and Methods). We identified a deep intronic element and a potential U2AF65-PTBP binding site #1 (Site #1) required for SYNGAP1 A3SS inclusion. Additionally, we identified a fragment within the A3SS-NMD exon that is required for the canonical/productive 3’ splice site usage of SYNGAP1 intron10. After serial deletion, we narrowed it down to a 30-base pair CUCUCU-rich polypyrimidine region, designated potential U2AF65-PTBP potential binding site #2 (Site #2, Figure 3E and S3A). Noticeably, U2AF65/U2AF2 CLIP-Seq tags were concentrated near both splice acceptor sites in HEK293 cells (Figure S3A).
To examine the affinity of PTBP1 to the two predicted U2AF65-PTBP binding sites #1 and #2, we performed an electrophoretic mobility shift assay (EMSA) with PTBP1 protein and two sets of RNA probes (Figure 3F). When mixed with increasing amounts of in vitro translated PTBP1 protein, Cy5-labeled probes (hot) for both Sites #1 and #2 shifted significantly; in parallel, unlabeled cold probes successfully competed with their corresponding hot probes, suggesting that PTBP1 can bind to both Sites #1 and #2 (Figure 3F). Interestingly, cold probe#2 showed more potent competition effects to hot probe#1 than cold probe#1 (Figure 3F, left); and consistently, cold probe#1 showed weaker competition than cold probe#2 to hot probe#2 (Figure 3F, right). These results suggest the model that PTBP proteins bind to SYNGAP1 A3SS and compete with the splicing machinery, likely through U2AF65, for the 3’ splice site definition of intron10: in neural progenitors, PTBP1 protein is expressed, binds to U2AF65-PTBP binding site #2, and suppresses the canonical/productive SYNGAP1 splice site; whereas in neurons, PTBP proteins are turned down (Figure S3B), resulting in exposure of the SYNGAP1 canonical splice site and increased expression of the neuronal SYNGAP1 isoform (Figure 3G).
Deletion of Syngap1 A3SS-NMD in mice upregulates Syngap1 protein expression
We sought to understand whether Syngap1 A3SS-NMD is required for mouse development by genetically deleting this NMD event without affecting the protein-coding isoform (Figure 4A). Based on the results above (Figure 3E), we deleted the intronic elements and the splice acceptor site required for A3SS-NMD inclusion with CRISPR/Cas9 and created a mouse strain named Syngap1-NISO (standing for Neuronal ISOform. N allele, 268 bp deletion, Figure 4A and S4A). We also established a Shorter NISO allele (S allele, 160 bp deletion) where the intronic elements were deleted and the alternative 3’ splice site remained intact (Figure 4A and S4A). The Syngap1-NISO heterozygotes (Syngap1 N/+, and S/+) and homozygotes (Syngap1 N/N, and S/S) were born at expected Mendelian ratios and appeared indistinguishable from their littermates.
Figure 4. Genetic deletion of Syngap1 A3SS-NMD increases Syngap1 protein in the neocortex.

A) CRISPR deletion of Syngap1 A3SS-NMD in mice to generate the Syngap1 NISO (N) allele (chr17:26959184-26959451, mm10) and the short NISO allele (S, chr17:26959185-26959353, mm10).
B-C) RT-PCR results showing that Syngap1 A3SS-NMD exon11 was included in wild-type controls (+/+), decreased in Syngap1-NISO heterozygotes (N/+), and excluded in Syngap1-NISO homozygotes (N/N) in P1 (B) and P10 (C) dorsal cortices.
D) Western-blot results showing Syngap1 protein levels in P2 cortices.
E) Quantification of Syngap1 Western bot signals in (D) relative to Gapdh showing that Syngap1 levels were significantly increased in Syngap1 N/+ (32±9%, p<0.005) and Syngap1 N/N (58±6%, p<0.001, one-way ANOVA, Dunnett’s multiple comparison test) when compared with wild-type (+/+).
F) fEPSP recordings of adult Syngap1 N/+ and N/N mice. Numbers of animals and slices for each genotype were: +/+ (n=6 animals, 11 slices), N/+ (n=6, 13), N/N (n=5, 11). *, adj.p = 0.0223.
G-I) Barnes maze test results showing similar performance between wild-type (+/+, n=6) and mutants (N/+, n=7; N/N, n=6). (G) All genotypes show improvement in the primary latency to the exit zone over the course of training. H) Comparisons of primary latency to the exit zone during the probe trial shows that Syngap1 mutant mice performed similarly to wild-type mice. (I) Heat maps and a box plot showing entry probability to the exit zone I and each false exit during the probe trial. Syngap1 N/+ and Syngap1 N/N animals were not significantly different in behavior from wild-type (+/+) mice (adj.p > 0.05). The greatest entry probability for each genotype corresponds to the exit zone.
See also Figure S4.
We confirmed the depletion of Syngap1 A3SS-NMD intron10 in Syngap1 N/N animals with RT-PCR amplification of total RNA extracted from P1 and P10 neocortices (Figure 4B–4C). Importantly, the inclusion of Syngap1 A3SS decreased in Syngap1-NISO heterozygotes (N/+, Figure 4B–4C). Similarly, the Syngap1 A3SS significantly decreased in neocortices of P1 S/+ and S/S mutants (Figure S4A–S4C). We focused on the N allele thereafter because it led to the complete depletion of Syngap1 A3SS-NMD in the neocortex (Figure 4B–4C). Western blot on cortical lysates uncovered that Syngap1 protein levels were increased by 32±9% in Syngap1 N/+ (adj.p<0.005) and by 58±6% in Syngap1 N/N samples at P2 (adj.p<0.001, one-way ANOVA, Dunnett’s multiple comparisons) when compared with wild-type controls (+/+, Figure 4D–4E), suggesting that one N allele can increase the protein level to 1.6 times of one wild-type allele. Syngap1 protein was also significantly increased by the Syngap1 N/+ (96±22%, adj.p=0.018) and N/N (153±25%, adj.p=0.004) in P2 hippocampi when compared to wild-type (p<0.005, one-way ANOVA, Figure S4D–S4E). No Syngap1 protein was detected in E18.5 Syngap1 N/+ heart or lung tissues (Figure S4G). These results indicate that suppressing Syngap1 A3SS-NMD can increase Syngap1 protein in the developing mouse brain.
Syngap1 has been shown to suppress AMPA receptor insertion at the postsynaptic membrane; Syngap1 heterozygous knockout mice showed enhanced synaptic transmission and displayed defects in learning9. Our Syngap1-NISO allele contradicts the effects of Syngap1 knockout alleles: the NISO allele increases Syngap1 protein expression, while the knockout allele decreases. Thus, the newly established Syngap1-NISO mouse model provides an opportunity to address whether the Syngap1 A3SS-NMD is required to express LTP and influences learning and/or memory in mice.
We generated input-output curves to evaluate basal synaptic transmission and examined the paired-pulse ratio of the field excitatory postsynaptic potentiation (fEPSP) to evaluate neurotransmitter release probability in adult wild-type, Syngap1 N/+ and N/N mice. Neither the input-output relationship nor the paired-pulse ratio was significantly different across the genotypes (Figure S4I–S4J). We measured the LTP of fEPSP in Syngap1 N/+ and Syngap1 N/N adult mouse hippocampi (Figure 4F). Recordings from each of the wild-type (n=8), Syngap1 N/+ (n=7), and Syngap1 N/N (n=5) genotypes showed that while LTP in wild-type and Syngap1 N/+ mutants were comparable, the magnitude of LTP was moderately but significantly reduced in Syngap1 N/N mutants (Figure 4F). These data suggest that synaptic plasticity was affected in Syngap1 N/N but not Syngap1 N/+ adults.
We next assessed performance in the Barnes maze (Figure 4G–4I) to determine spatial learning and memory abilities in Synga1 N/+ and N/N mice39. Six or more adult male animals for each genotype were trained using an abbreviated Barnes maze protocol. There were three consecutive days of training followed by a fourth-day probe trial where the exit hole was closed. The primary latency to exit decreased throughout training in all genotypes (Figure 4G). When compared to the wild-type, neither Syngap1 N/+ nor N/N mutant mice showed differences in primary latency to exit or entry probability to exit during the probe trial (Figure 4H–4I). We also performed Rotarod assays to assess motor learning ability, and neither Syngap1 N/+ nor Syngap1 N/N animals showed a difference in the latency to fall when compared to wild-type mice (Figure S4H). Together, these results suggest that genetic deletion of Syngap1 A3SS-NMD does not overtly impair spatial learning and memory or motor learning behaviors.
The NISO allele alleviates LTP and membrane excitability deficits caused by a Syngap1 knockout allele
Increased Syngap1 protein by the Syngap1-NISO allele suggests that it may alleviate or rescue the Syngap1 haploinsufficiency in mice. Emx1-Cre drives Cre expression in excitatory neurons in the dorsal forebrain and is sufficient to induce Syngap1 haploinsufficient phenotypes in Syngap1 fl/+; Emx1-Cre (cKO/+) mice15. We validated the Syngap1 conditional knockout allele (Figure S5A) in which Syngap1 exon6-7 were floxed9,13, and confirmed that the Syngap1 protein level decreased in adult Syngap1 cKO/+ cortices (Figure S5B). We combined the Syngap1-NISO allele with the Syngap1 floxed allele and generated Syngap1 fl/N; Emx1-Cre (cKO/N) and control animals (Figure S5C). We assessed basal synaptic transmission and neurotransmitter release probability in cKO/+ and cKO/N mice and did not find a significant difference in the input-output curve or paired-pulse ratio between genotypes (Figure S5E–S5F). We then evaluated LTP of the fEPSP in the hippocampi and found the LTP was impaired in Syngap1 cKO/+ mice and rescued in Syngap1 cKO/N animals (Fig. 5A), indicating that deletion of the Syngap1 A3SS-NMD alleviated the LTP deficits caused by the Syngap1 knockout allele.
Figure 5. Genetic deletion of Syngap1 A3SS-NMD alleviates LTP and membrane excitability deficits caused by a conditional Syngap1 knockout allele.

A) fEPSP recordings showing that the N allele alleviates the LTP deficit in P35-40 Syngap1 cKO mice. Numbers of animals and slices for each genotypes are: WT (replotted from Figure 4F), cKO/+ (n=5, 12), and cKO/N (n=6, 12). ****adj.p < 0.0001.
B) Typical traces from L2/3 pyramidal neurons in S1 cortex in WT (top row), cKO/+ (middle row) and cKO/N mice (bottom row). Left to right: traces at current steps ~200pA (left), ~400pA (middle), and ~600pA (right).
C) Spike count (number of evoked spikes) per current step for WT (n=8), cKO/+ (n=8) and cKO/N mice (n=7). The average ± SD is shown for all data points. P=0.0003, F (2, 177)=8.420 for genotype, two-way ANOVA.
D) Dot plots showing the maximal spike frequency obtained from the same recordings. p=0.0007 by one-way ANOVA. Each dot represents one animal. The average ± SD is shown for all data points.
See also Figure S5.
Syngap1 has been reported to maintain membrane excitability in L2/3 pyramidal neurons of mouse primary somatosensory cortex (S1)16. As an independent electrophysiological test for the efficacy of our rescue strategy, we performed whole-cell patch-clamp recordings on L2/3 pyramidal neurons in the mouse S1 cortex. Current steps of increasing amplitude (500ms) resulted in an increase in the number of evoked spikes (Fig. 5B–5C). Consistent with the earlier report, we observed a lower number of evoked spikes in the Syngap1 cKO/+ mice (n=8) as compared to WT mice (n=8; two-way ANOVA, genotype: F2,177=8.420; p=0.0003, pairwise adj.p=0.0022). The difference in evoked spike output stayed constant over the range of injected current amplitudes (genotype x current amplitude: F16,117=0.1349, p>0.9999). In Syngap1 cKO/N mice (n=7), the excitability deficit was restored, resulting in no significant difference as compared to WT mice (adj.p=0.9306), but a significant difference as compared to the Syngap1 cKO/+ mice (adj.p=0.0011). In addition to the findings on the overall excitability, we also observed that the maximal spike firing frequency (measured from the interval of the first two evoked spikes during the current injections) was lower in Syngap1 cKO/+ mice (n=8) as compared to WT mice (n=8; WT: 221.43±46.85 Hz; cKO/+: 140.73±21.04 Hz; adj.p=0.0004). In Syngap1 cKO/N mice (n=7), the higher maximal spike frequency was restored (183.99±30.95 Hz; compared to WT: adj.p=0.1197; compared to mutant: adj.p=0.0646 by one-way ANOVA, Fig. 5D). Individual action potential parameters, including overall amplitude, threshold and width at half amplitude, did not differ between the genotypes (Figure S5G–S5I). These results indicate that deletion of the Syngap1 A3SS-NMD alleviated the membrane excitability deficit caused by Syngap1 haploinsufficiency in mice.
SSOs suppress SYNGAP1 A3SS-NMD in human iPSC-derived neurons
SSOs have been successfully developed to treat neurological disorders such as spinal muscular atrophy26, attempted for personalized medicine27, and suggested to treat haploinsufficient human diseases40. Increased Syngap1 protein levels due to Syngap1 A3SS deletion in mice and the conserved SYNGAP1 A3SS in human brains led us to determine whether the SYNGAP1 productive isoform can be upregulated in human cells through SSO-mediated suppression of SYNGAP1 A3SS-NMD. SSOs with (2’-MOE) chemistry have been successfully developed to redirect splicing, manipulate gene expression, and treat diseases41. To identify SSOs that suppress human SYNGAP1 A3SS-NMD, we performed a rationale design by targeting the following sequences (Figure 6A): 1) critical splice elements identified through serial deletion of the human SYNGAP1 mini-gene (Figure 3); 2) predicted splicing regulatory sequences; 3) predicted stem-loop structures and conserved sequences that overlap with experimentally identified splice elements (Figure 6A and S6A).
Figure 6. The lead SSO upregulates SYNGAP1 expression in human iPSCs and iPSC-derived neurons.
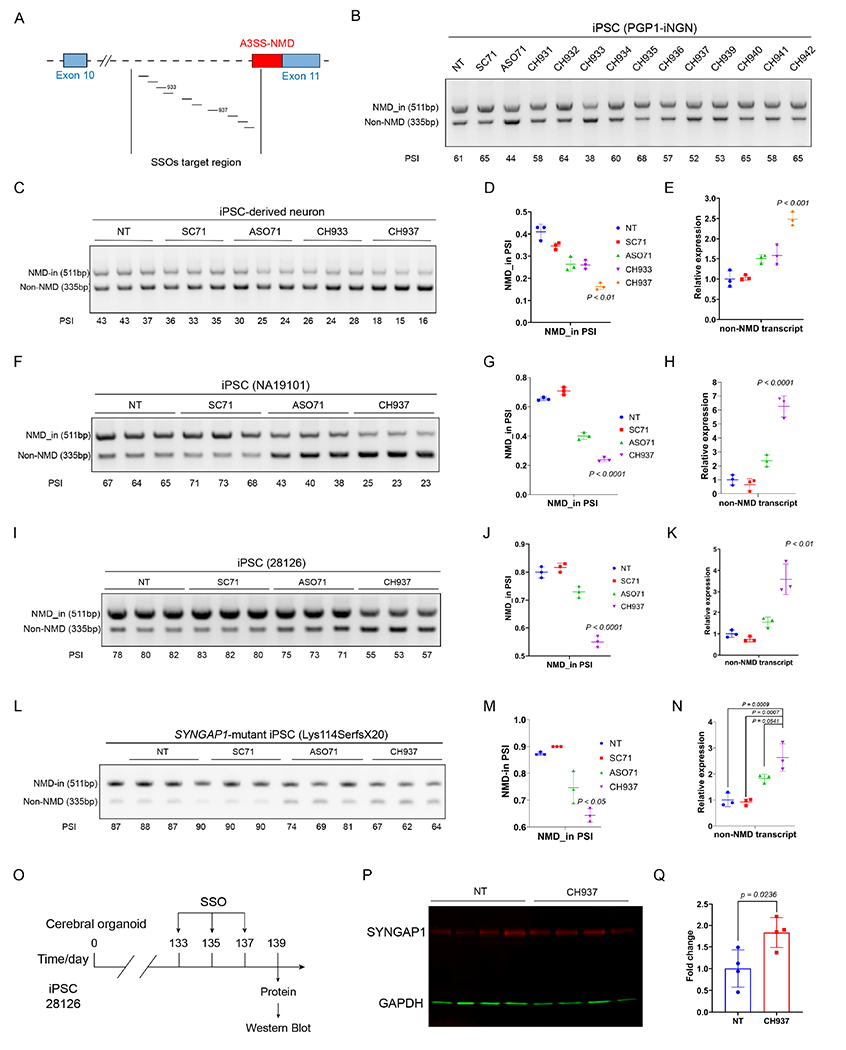
A) Schematic illustration of the SSO design targeting the SYNGAP1 A3SS.
B) RT-PCR results showing the screening of SSOs in iPSCs (PGP1-iNGN). One biological sample per lane.
C-E) Identification of the lead SSO in iPSC-derived neurons. RT-PCR results (C) and quantification (D) showing that CH937 suppresses SYNGAP1 A3SS in iPSC-derived neurons. Q-PCR results (E) showing that the productive SYNGAP1 transcript was upregulated in CH937-treated human iPSC-derived neurons.
F-K) The lead SSO suppresses SYNGAP1 A3SS in two additional human iPSC lines. RT-PCR results (F, I) and quantification (G, J) showing that CH937 suppressed SYNGAP1 A3SS in human iPSCs (NA19101 and 28126). Q-PCR results (H, K) showing that the CH937 significantly increased the productive SYNGAP1 transcript levels in human iPSC lines.
L-N) The lead SSO suppresses SYNGAP1 A3SS-NMD in SYNGAP1 patient-derived iPSCs. RT-PCR results (L) and quantification (M) showing that CH937 suppressed SYNGAP1 A3SS in SYNGAP1 patient-derived iPSCs (333del, Lys114SerfsX20). Q-PCR results (N) showing that the CH937 significantly increased the productive SYNGAP1 mRNA level.
O-Q) Application of CH937 to human iPSC-derived cerebral organoids (O) led to increased SYNGAP1 protein expression (83%±28%, P-Q). p<0.05 by unpaired t-test.
See also Figure S6.
We synthesized eleven SSOs using a phosphorothioate backbone with 2’-MOE modified residues and tested them in human iPSCs (Figure 6A–6B). SSOs CH933 and CH937 most efficiently suppressed A3SS-NMD inclusion in a human iPSC line (PGP1-iNGN, Figure 6B and S6A–S6C). We delivered SSOs CH933 and CH937, a scrambled control, and a previously reported ASO7128 into human iPSC-derived neurons and found that CH937 was the most effective in decreasing the A3SS-NMD inclusion (Figure 6C–6D). We further measured the productive/functional SYNGAP1 transcript using Q-PCR primers specific to the non-NMD isoform and CH937 induced a 2.5-fold increase of functional SYNGAP1 mRNA (Tukey’s multiple comparisons test, adj.p<0.001, Figure 6E). In contrast, the ASO71 developed in HEK293 cells was less efficient in upregulating SYNGAP1 transcript in human iPSC-derived neurons (1.5 fold, adj.p=0.029, Figure 6E). These results indicate that the lead SSO CH937 can effectively suppress human A3SS-NMD and increase the productive SYNGAP1 isoform in human iPSCs and iPSC-derived neurons.
We further examined the effects of the SSO CH937 in two additional control human iPSC lines (NA19101 and 28126) and confirmed that CH937 was more effective than ASO71 in decreasing the SYNGAP1 A3SS-NMD inclusion (Figure 6F–6K). Importantly, CH937 significantly increased the functional SYNGAP1 transcript to 6.3-fold and 3.6-fold of non-treated controls in NA19101 and 28126, respectively (Figure 6H, 6K). Next, we delivered CH937 to a SYNGAP1 patient-derived iPSC line harboring a heterozygous frame-shift mutation (Lys114SerfsX20) and found that the SSO CH937 decreased SYNGAP1 A3SS-NMD inclusion (Figure 6L–6M) and significantly increased functional SYNGAP1 transcript to 2.6-fold of non-treated controls (Figure 6N). These results support that the lead SSO CH937 can effectively suppress SYNGAP1 A3SS-NMD and increase functional SYNGAP1 isoform levels in SYNGAP1 patient-derived iPSCs.
To determine whether suppressing SYNGAP1 A3SS-NMD can upregulate SYNGAP1 protein, we determined the effects of SSO CH937 in iPSC-derived brain organoids. Cerebral organoids were induced from human iPSCs (28126) following an established protocol42, transfected with CH937 at post-induction days 133, 135, and 137, and harvested at day 139 for protein analysis (Figure 6O–6P). CH937 significantly increased SYNGAP1 protein when compared with control organoids (83%±28%, p<0.05 by unpaired t-test). We further tested SSO CH937 in cerebral organoids derived from another iPSC line (21792) at post-induction days 174, and again observed significantly increased SYNGAP1 protein when compared with control oligonucleotides (Figure S6E–6G, increased by 51%±10%, adj.p=0.0051 by one-way ANOVA). These results suggest that the SSO CH937 can upregulate SYNGAP1 protein in human iPSC-derived cerebral organoids.
Discussion:
Alternative splicing coupled with NMD selectively removes unproductive transcripts and has been reported to regulate the expression of neuronal genes21,23,43. The therapeutic potential of targeting AS-NMD with SSOs has evoked unprecedented enthusiasm, and SSO screens in cultured cells have been reported in recent studies28,40,44. However, whether it is safe to target naturally occurring AS-NMD for therapy remains a crucial question. Specifically, we need to understand whether such an AS-NMD exon is required for normal organismal functions and whether it is safe to suppress or completely block such an AS-NMD exon in vivo.
Since presenting our SYNGAP1 A3SS-NMD results at the 2019 Society for Neuroscience meeting (Chicago), we have further explored the function and therapeutic potential of the SYNGAP1 A3SS using mouse genetics and human iPSCs. Syngap1 is a synaptic protein barely detected in non-neuronal tissues4. In contrast, the SYNGAP1 transcript is detectable in non-neural tissues in mice and humans (Figures 1 and GTEx), where the Syngap1 A3SS-NMD inclusion is nearly constitutive (Figure 1B). These observations suggest that A3SS-NMD provides an orthogonal mechanism in non-neuronal cells to suppress excess or leaky SYNGAP1 expression which would waste cellular resources and interfere with Ras signaling. Our results support that heterozygous genetic deletion of Syngap1 A3SS in mice upregulated SYNGAP1 protein during brain development. While homozygous deletion of mouse Syngap1 A3SS led to a modest reduction in the magnitude of LTP, it did not overtly impact behavioral performance in the Barnes maze or Rotarod assays. We were unable to detect Syngap1 protein in E18.5 heart and lung tissues from Syngap1 N/+ animals (Figure S4G), nor did we find significant cortical neurogenesis defect in Syngap1 N/N mice (Figure S4F). Our results suggest that the Syngap1 A3SS is moderately required during neural development, and its functions in non-neural tissues remain to be further explored.
Heterozygous deletion of mouse Syngap1 A3SS alleviated the LTP deficits caused by a heterozygous Syngap1 knockout allele, suggesting that suppressing A3SS-NMD may alleviate Syngap1 haploinsufficiency in vivo. A previous study has reported that Syngap1 heterozygous deletion reduced membrane excitability, as tested via trains of injected current pulses of rising amplitude16. Here, we adapted this experimental protocol to capture a cell-autonomous physiological parameter that differs from LTP. We found that the maximal spike firing frequency – which, as a result of spike frequency adaptation occurs at the beginning of the current pulse45 and is typically determined from the interval of the first two spikes – was lower in Syngap1 heterozygous knockouts than in wild-type mice. Both the general excitability and the maximal spike frequency defects were alleviated by the heterozygous deletion of Syngap1 A3SS.
Furthermore, our lead SSO CH937 suppressed SYNGAP1 A3SS in human iPSCs and iPSC-derived neurons and significantly increased the functional SYNGAP1 isoform. The SSO CH937 also significantly increased SYNGAP1 protein expression in cerebral organoids induced from two different human iPSC lines. Despite these encouraging results, several key questions remain to be further addressed: 1) it is important to understand at what time and in which brain regions the rescuing effects of Syngap1 upregulation are sufficient to rescue haploinsufficiency. Specifically, it is critical to understand whether deleting or suppressing the A3SS-NMD exon at postnatal stages can rescue the heterozygous knockout allele. It is equally important to systematically characterize neural circuits and mouse behaviors because SYNGAP1 patients display pleiotropic symptoms7. 2) it is important to understand whether the SSO can effectively upregulate Syngap1 protein in vivo and rescue the heterozygous knockout mouse. While the Syngap1 NISO mouse model and the lead SSO CH937 suggest that SYNGAP1 A3SS is a promising target, rigorous further in vivo studies are required to understand its therapeutic potential fully.
SYNGAP1 protein directly interacts with PSD-95 in the postsynaptic density, and remarkably, both genes undergo unproductive splicing that is promoted by PTBP1 and PTBP2 in early neural development. While PTBP proteins promote SYNGAP1 A3SS-NMD inclusion, they suppress the inclusion of a coding exon in PSD-95/DLG4 and lead to NMD43. Previous studies showed that protein levels of SYNGAP1 and PSD-95 exhibit a near stoichiometric ratio in the PSD and the appropriate protein ratio is critical for the formation of SYNGAP1-PSD-95 liquid-like droplets46. Strikingly, two AS-NMD events are co-regulated by the PTBP1/2 proteins for equilibrated protein expression. De novo mutations in PSD-95 have been reported to cause synaptopathy47. It is interesting to speculate on the functions of the PSD-95 AS-NMD exon and its potential as a therapeutic target.
Hundreds of genes have been reported to undergo AS-NMD during cortical development, and chromatin regulators were highly enriched21. Many of the AS-NMD exons are regulated by PTBP and RBFOX proteins, and mutations in the host genes are frequently associated with neurodevelopmental disorders such as autism and epilepsy22,23,32,44,48,49. Although SSOs have been reported as promising ways to target AS-NMD and treat diseases such as the Dravet Syndrome40, the biological functions of such AS-NMD events are undetermined. Furthermore, the dosage effect of their host genes requires rigorous studies in animal models and human neurons to fully explore their therapeutic potential. Genetic studies of AS-NMD exons will illuminate their organismal functions and further the development of therapeutic targets and strategies.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xiaochang Zhang (xczhanq@uchicago.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The RNA-Sequencing results shown in Figure 1A have been uploaded to NCBI (PRJNA930469: SRR23308049, SRR23308050). The microscopy data reported in this paper will be shared by the lead contact upon request.
This study does not generate new code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Mouse protocols were reviewed and approved by the University of Chicago Institutional Animal Care and Use Committee. Animals were housed under 12 hours light/dark cycles and supplied with food and water ad libitum. Guide RNAs (SG321 and SG322) flanking the designed Syngap1 deletion region were selected with the CRIPSOR online tool. Guide RNAs, tracrRNA, and Cas9 protein were purchased from IDT. Guide RNAs were annealed with tracrRNA, mixed with Cas9 protein in the injection buffer, and injected into C57BL/6 mouse zygotes by the Transgenic Core (University of Chicago). Founder mice were PCR screened for the deletion and positive founders were bred with C57BL/6 (Charles River) to obtain positive F1s, which were further mated with C57BL/6 for positive F2s. F2s and later generations were used in this study. The Syngap1-NISO allele is genotyped using primers SG245F-SG331R-SG202R, and the expected product sizes are: wild-type allele is 405bp (and weak 761bp), and the NISO N allele is 493 bp. The Syngap1 conditional knockout (cKO, Jax#029303) and Emx1-Cre (Jax#005628) mice were obtained from the Jackson Lab and genotyped following providers’ protocols.
Primary cell cultures
Primary hippocampal neurons or cortical neurons (from E18.5 CD1 mouse embryos or neonatal pups) were isolated with Papain (Worthington) and cultured in vitro. Primary neurons were plated onto poly D-lysine coated coverslips and cultured following standard protocols (Neurobasal medium supplemented with GlutaMax, N2, B27, and 1μM AraC during DIV1-DIV3).
Human iPSCs
iPSC lines, including PGP1-iNGN51, 28126, 21792 (Yoav Gilad lab at The University of Chicago), and NA19101 (Marcelo A. Nobrega lab at The University of Chicago), were grown in Essential 8 (Thermo Fisher, A1517001) with penicillin-streptomycin (100U/mL, Thermo Fisher, 15140122) in 10-cm dishes coated with GelTrex (Thermo Fisher, A1413301) in an incubator at 37 °C with 5% CO2. Instead of Essential 8, SYNGAP1-mutant (Lys114SerfsX20, Simons Foundation) iPSCs were grown in StemFlex (Thermo Fisher, A3349401). Rock inhibitor Y-27632 dihydrochloride (10 μM, Tocris, 1254) was added into culture media for 24 hours after passage. For neuron induction, PGP1-iNGN iPSCs were grown in Essential 8 supplemented with doxycycline (1μg/mL, Sigma-Aldrich, D9891) and penicillin-streptomycin (100 U/mL) for 4 days. To analyze the NMD-transcript of SYNGAP1, iPSCs cells and iPSC-derived neurons were treated with cycloheximide (200 μg/mL dissolved in DMSO) for 5 hours in 12-well plates with 1 mL culture media in each well, followed by RNA extraction and analyses.
Cerebral organoids
Induction of brain organoids was performed according to published protocols42. Briefly, 3×106 human iPSCs (28126 and 21792) were seeded per AggreWell 800 well (STEMCELL Technologies, 34815) in Essential 8 supplemented with Y-27632 dihydrochloride (10 μM) (Tocris, 1254) and penicillin-streptomycin (100 U/mL). 24 hours later, iPSC spheroids were transferred into ultra-low attachment 6-well plates and then incubated in Essential 6 supplemented with dorsomorphin (2.5 μM) (Sigma-Aldrich, P5499) and SB-431542 (10 μM) (Tocris, 1614) for 6 days to induce neural spheroids. Then the neural spheroids were incubated in Neurobasal A medium (Thermo Fisher Scientific, 10888022) supplemented with B-27 without vitamin A (1:50) (Thermo Fisher Scientific, 12587010), GlutaMax (1:100) (Thermo Fisher Scientific, 35050-061), epidermal growth factor (EGF) (20ng/mL) (R&D Systems, 236-EG), basic fibroblast growth factor (bFGF) (20ng/ml) (R&D Systems, 233-FB) and penicillin-streptomycin (100 U/mL) for 19 days, after which the EGF and bFGF were replaced by brain-derived neurotrophic factor (BDNF) (20ng/mL) and NT-3 (20ng/mL) for 18 days. From the 43rd day, brain organoids were incubated in Neurobasal A media supplemented with B-27 without vitamin A (1:50), GlutaMax (1:100) and penicillin-streptomycin (100 U/mL) for long-term culture.
Fetal cortical tissue
De-identified frozen fetal human cortical tissues were obtained from the NIH Neurobiobank at the University of Maryland (Baltimore, MD), microdissected, and subjected to RNA extraction. The works described here involving de-identified postmortem tissues were reviewed and approved by The University of Chicago Institutional Review Board.
METHOD DETAILS
Molecular cloning
To detect the A3SS with RT-PCR, the human SYNGAP1 alternative exon11 was amplified with primer pairs CH748-CH749, and the mouse Syngap1 A3SS was amplified with primer pairs CH743-CH744.
pCAG-SYNGAP1 (N-terminus): the N-terminal SYNGAP1 coding sequence was amplified using SG020 and SG022 and inserted into pCZ01 using Gibson Assembly (NEB). pCZ01 is a modified pCAG-IG vector described previously22.
pCAG-HA-Flag-PTBP1-IRES-mCherry: PTBP1 cDNA was amplified with CH448-CH422-2, digested with AscI and NotI, and ligated into linearized pCAG-HA-Flag-IRES-mCherry.
For in vitro translation, PTBP1 cDNA was amplified using primer pair SG209-SG210. To knock down Ptbp1/2, shRNA lenti-vectors were used as described previously22.
The SYNGAP1 wild-type mini-gene construct spanning exon9 through exon12: genomic DNA extracted from HEK293FT cells was amplified with primer pairs SG085-SG086 and the purified PCR product was inserted to pCZ01 using Gibson Assembly (NEB).
SYNGAP1 mini-gene deletion constructs:
To delete the deep intronic element, two fragments were amplified using PCR primer pairs SG159-SG086 and SG160-SG085, and inserted into linearized pCZ01 using Gibson Assembly.
To delete predicted U2AF65 binding site #1, two fragments were amplified using PCR primer pairs SG155-SG086 and SG156-SG085, and inserted into pCZ01.
To delete predicted U2AF65 binding site #1 + extended, two fragments were amplified using PCR primer pairs SG095-SG086 and SG098-SG085, and inserted into pCZ01.
To delete the A3SS, two fragments were amplified using PCR primer pairs SG087-SG086 and SG088-SG085, and inserted into pCZ01.
To delete predicted U2AF65 binding site #2, two fragments were amplified using PCR primer pairs SG089-SG086 and SG090-SG085, and inserted into pCZ01.
SYNGAP1 mini-gene with patient mutations:
To introduce the mutation c.1676 +5 G>A (NM_006772.2), two fragments were amplified using PCR primer pairs SG189-SG086 and SG190-SG085, and inserted into pCZ01.
To introduce the mutation c.1677-2_1685del, two fragments were amplified using PCR primer pairs SG191-SG086 and SG192-SG085, and inserted into pCZ01.
Plasmids were transfected into Neuro2a cells (ATCC) with Lipofectamine 2000 (Thermo Fisher), selected by puromycin and total RNA was extracted with Trizol (Sigma). Reverse transcription was performed with random primers following manufacturer’s protocols (Superscript IV, Thermo Fisher). Primers sequences are listed in Table S1.
RT-PCR and Western blot
For RNA extraction, brain tissues, or culture cells were dissolved in TRIzol by firmly pipetting and then subjected to either precipitation or Direct-zol RNA Purification Kit. For Western blotting, protein lysates were extracted with RIPA buffer (Thermo Fisher, PI89901) supplemented with proteinase inhibitors (Sigma-Aldrich, 11836170001). Protein samples were loaded onto SDS-PAGE gels, transferred to PVDF membranes, incubated with primary and secondary antibodies successively (Table S1), and then imaged using the LI-COR Odyssey system (LI-COR, 9142).
EMSA
Cy5 conjugated RNA probes (SG-probe1/2) and unlabeled cold competitors of potential PTBP1 binding sites were synthesized by IDT. PTBP1 protein was produced by TnT SP6 High-Yield Wheat Germ Protein Expression System (Promega). In vitro translated PTBP1 was diluted in the RNA-protein binding solution, incubated with Cy5 probes with or without cold competitor probes, and resolved on 8% TBE gels (Thermo Fisher, EC6215BOX). The gel was directly visualized with a Typhoon imaging system.
Mouse behavioral tests
Barnes maze test was performed following published protocols39,50. Motor coordination and balance of mice (aged around 2 months to 4 months) were examined by Rotarod tests: mice were gently placed on a rotatable rod (Columbus Instruments, ECONOMEX) that was accelerated at 0.2 rpm/second from 5 rpm over a 3-minute period. Time from rotation start to mouse fall in each trial was recorded as the latency to fall. Three trials were performed for each mouse on the test day.
fEPSP recordings
Acute hippocampal slices were prepared from young adult (1-2.5 months) male mice, which were anesthetized with isoflurane and euthanized by rapid decapitation. The brain was rapidly harvested and blocked, rinsed with cold artificial cerebrospinal fluid (aCSF) and mounted for vibratome sectioning. The mounted brain tissue was submerged in aCSF (4°C; equilibrated with 95% O2, 5% CO2) and coronal cortico-hippocampal brain slices (350 μm thick) were prepared. Slices were immediately transferred into a holding chamber containing aCSF equilibrated with 95% O2, 5% CO2 (at 20.5±1°C). Slices were allowed to recover a minimum of one hour prior to the transfer into recording chamber and were used within eight hours following tissue harvest. The composition of aCSF (in mM):118 NaCl, 10 Glucose, 20 sucrose, 25 NaHCO3, 3.0 KCl, 1.5 CaCl2, 1.0 NaH2PO4 and 1.0 MgCl2. The osmolarity of aCSF was 305-315 mOsm and equilibrated, and the pH was 7.42±0.02.
The extracellular recording of the fEPSP was established in aCSF (31.0 ± 2°C, equilibrated with 95% O2 5% CO2) superfused and recirculated over the preparation. The stimulation electrode, a custom constructed bipolar electrode composed of twisted Teflon coated platinum wires (wire diameter: 127 μm, catalog number 778000, AM Systems.), was positioned in the Schaffer Collateral and the recording electrode (1-2 MΩ) was placed into the stratum radiatum of the CA1. The intensity of the electrical current (100-400 μA; 0.1-0.2 ms duration) was set to the minimum intensity required to generate the 50% maximal fEPSP. After 10 minutes of recording the baseline fEPSP, LTP was induced using Theta Burst Stimulation (TBS: four trains of 10 bursts at 5 Hz, each burst was comprised four pulses at 100 Hz). Following stimulation, recordings continued for up to one hour. The fEPSP slope was normalized to baseline values. Recordings were made using either a Multiclamp 700B (Molecular Devices) or using a differential amplifier (AM system).
Whole-cell patch-clamp recordings from neocortical pyramidal neurons
Slices from primary somatosensory cortex (S1; 350μm thick) were prepared from young mice (postnatal day 16-28) after isoflurane anesthesia and decapitation. The procedure was described in detail before45 and is in accordance with the guidelines of the Animal Care and Use Committee of the University of Chicago. The slices were cut on a vibratome (Leica VT1000S) using ceramic blades. To preserve thalamocortical projections, slices were cut at a 55° angle to the right (posterior) of the anterior-to-posterior axis of the brain (see Agmon and Connors, 1991). The slices were cut in a sucrose slicing solution containing the following (in mM): 185 sucrose, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.2 NaH2PO4, 0.5 CaCl2, and 0.5 MgCl2, bubbled with 95% O2 and 5% CO2. Following slicing, the slices were kept in artificial cerebrospinal fluid (ACSF) containing the following (in mM): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 2 CaCl2, 2 MgSO4, 26 NaHCO2, and 10 D-glucose, bubbled with 95% O2 and 5% CO2. The slices were allowed to recover for at least 1h and were subsequently transferred to a submerged recording chamber superfused with ACSF at elevated temperature (28-30°C). Whole-cell patch-clamp recordings were performed under visual control using a 40x water-immersion objective in combination with near-infrared light illumination (IR-DIC) and a Zeiss AxioCam MRm camera mounted on a Zeiss Examiner A1 microscope (Carl Zeiss Microimaging). Patch pipettes (~2.5-4.5 MΩ) were filled with internal saline containing the following (in mM): 9 KCl, 10 KOH, 120 K-gluconate, 3.48 MgCl2, 10 HEPES, 4 NaCl, 4 Na2ATP, 0.4 Na3GTP, and 17.5 sucrose, pH adjusted to 7.25. Patch-clamp recordings were performed in current-clamp mode using an EPC-10 amplifier (HEKA Electronics). Input resistance (Ri) was measured by injection of hyperpolarizing test currents (100pA, 100ms). For all recordings and analyses, we used a blind approach, in which the researcher was ignorant of the mouse genotype. Data obtained from the patch-clamp recordings were analyzed using Pulsefit (HEKA Electronics), Igor Pro (WaveMetrics) and MatLab. For statistical comparison of action potential-related parameters between genotypes, we used the one-way ANOVA. A two-way ANOVA test was used to examine statistical relationships between a) genotypes and b) genotypes x current amplitude in the input (current injection) - output (spike number) measurements.
Primary neurons immunostaining
Primary neurons were transfected by Lipofectamine 2000 (Thermo Fisher) or jetOPTIMUS (Polyplus). For immunostaining, primary neurons were fixed 10 minutes in 4% paraformaldehyde (PFA) at 4°C, rinsed with 1x PBS, and incubated with blocking buffer (1x PBS containing 0.03% Triton X-100 and 5% normal donkey serum) in room temperature for 30 mins, and further incubated with primary antibodies diluted in PBST buffer (1x PBS containing 0.03% Triton X-100) overnight at 4°C. After 3 times washing with 1x PBS, slides were incubated for one hour at room temperature with fluorophore-conjugated secondary antibodies in the dark. Slides were scanned with a Leica SP8 confocal microscope. The antibodies were listed in the table S1.
Splice switching oligonucleotides (SSOs)
SSOs were synthesized by Integrated DNA Technologies (IDT). For SSO transfection, 4×105 cells were seeded in 12-well plates coated with GelTrex (Thermo Fisher, Gibco, A1413301). 200nM SSOs were transfected into cells using TranslT-LT1 Transfection Reagent (Mirus, MIR2300) according to the manufacturer’s instructions. Brain organoids derived from iPSC 28126 were treated by three doses of SSOs (300nM) from day 133 to 137. On day 139, RNA and protein were extracted from brain organoids for PCR and western blot analyses. For brain organoids derived from iPSC 21792, they were treated by five doses of ASOs (200nM) from day 169 to 173. RNA and protein were extracted on day 174. Total RNA was extracted using TRIzol reagent (Thermo Fisher, 15596018) and Direct-zol RNA Purification Kit (Zymo Research, R2060) 24 hours after transfection. cDNA was synthesized by Superscript IV Reverse Transcriptase kit (Thermo Fisher, 18090050). Quantitative PCR (Q-PCR) was performed using SYBR Green PCR Master Mix (Thermo Fisher, 4344463) in QuanStudio Real-Time PCR Systems (Thermo Fisher, ZG11CQS3STD) according to manufacturers’ instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
To identify differential splicing events, we analyzed at least two biological replicates for each genotype using rMATS and filtered by False Discovery Rate (FDR)<0.05 and |differential PSI|>10%. CLIP-Seq data were aligned and mapped to hg38 genome by CLIPSeqTools, using the default parameters. The aligned files were visualized in IGV. CLIP-Seq datasets used in this study: PTBP1_ESC (SRR2121761), PTBP1_NPC (SRR2121762), U2AF65 (ERR208893, ERR208897, GSE83923), PTBP2 (SRR871026, SRR871030). Multiple Sequence Alignment Genome sequences of multiple vertebrates were obtained from NCBI HomoloGene database. Multiple sequence alignment and phylogeny analysis were performed by MAFFT over the conserved region of SYNGAP1 intron10. The splice site usage was validated by deposited RNA-Seq data of non-neuronal tissues from the corresponding species. Comparisons of electrophysiology recordings were assessed by a one-way ANOVA followed by the Bonferroni correction in Prism (GraphPad). Comparisons of mouse behaviors and Q-PCR results were done with Tukey’s multiple comparisons in Prism, and the adjusted p-values were indicated.
Supplementary Material
Table S1. Sequences and coordinates of oligonucleotides.
Related to the following sections in METHOD DETAILS: Molecular cloning, RT-PCR, EMSA, Primary neurons immunostaining, and Splice switching oligonucleotides.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Syngap1 (C terminal) | Thermo-Fisher | Cat# PA1-046, RRID: AB_2287112 |
| Anti-Syngap1 (N terminal) | Thermo-Fisher | Cat# PA5-37909, RRID: AB_2554517 |
| Anti-mCherry | MyBioSource | 12158167001, RRID: AB_2827808 |
| Anti-Ptbp1 | Abcam | Cat# ab5642, RRID: AB_305011 |
| Anti-Ptbp2 | Millipore | Cat# ABE431, RRID: AB_2928034 |
| Anti- GAPDH | Cell Signaling | Cat# 97166S, RRID: AB_2756824 |
| Chemicals, peptides, and recombinant proteins | ||
| Cycloheximide solution | Sigma-Aldrich | Cat# C4859-1ML |
| Doxycycline hyclate, ≥98% (HPLC) | Sigma-Aldrich | Cat# D9891-1G |
| NotI-HF | NEB | Cat# R3189L |
| AscI | NEB | Cat# R0558L |
| Phusion Hot Start II High-Fidelity DNA Polymerase | Thermo-Fisher | Cat# F537S |
| FuGENE HD Transfection Reagent | Promega | Cat# E2311 |
| Lipofectamin 2000 | Thermo-Fisher | Cat# 12566014 |
| Novex 4-12% Tris-Glycine Mini Gels | Thermo-Fisher | Cat# XP04122BOX |
| Novex TBE Gels, 8% | Thermo-Fisher | Cat# EC6215BOX |
| Critical commercial assays | ||
| TnT SP6 High-Yield Wheat Germ Protein Expression System | Promega | Cat# L3260 |
| Quick-RNA MiniPrep | Zymo | Cat# R1054 |
| GeneJET Genomic DNA Purification Kit | Thermo-Fisher | Cat# K0721 |
| TruSeq RNA Library Prep Kit | Illumina | Cat# RS-122-2001 |
| Alt-R Genome Editing Detection Kit | IDT | Cat# 1075931 |
| SuperScript IV | Thermo-Fisher | Cat# 18091050 |
| Gibson Assembly Master Mix | NEB | Cat# E2611S |
| Quick Ligation Kit | NEB | Cat# M2200S |
| Deposited data | ||
| Mouse cortical neurons and neural progenitors RNA-seq | This study | BioProject ID: PRJNA930469 |
| Experimental models: Cell lines | ||
| Neuro2a | ATCC | Cat# CCL-131 |
| SYNGAP1-mutant iPSCs (333del, Lys114SerfsX20) | SFARI | Cat# 15209-x1 |
| iNGN | Benoit Laurent lab, Université de Sherbrooke | N/A |
| NA19101 | Marcelo A. Nobrega lab, The University of Chicago | N/A |
| 28126 | Yoav Gilad lab, The University of Chicago | N/A |
| 21792 | Yoav Gilad lab, The University of Chicago | N/A |
| Primary mouse neurons | This study (from E18.5 CD1 mouse embryos or neonatal pups) | N/A |
| Experimental models: Organisms/strains | ||
| Syngap1 conditional knockout mouse | The Jackson Laboratory | Cat# 029303, RRID: IMSR_JAX:029303 |
| Emx1-Cre mouse | The Jackson Laboratory | Cat# 005628, RRID: IMSR_JAX:005628 |
| Syngap1-NISO mouse | This study | N/A |
| Oligonucleotides | ||
| Nucleotide synthesis | IDT | https://www.idtdna.com/pages |
| Software and algorithms | ||
| STAR 2.7 | PMID: 23104886 | https://github.com/alexdobin/STAR |
| rMATS 3.2.5 | PMID: 25480548 | http://rnaseq-mats.sourceforge.net/rmats3.2.5/ |
| rMAPS2 | PMID: 27174931 | http://rmaps.cecsresearch.org/ |
| CLIPSeqTools | PMID: 26577377 | http://mourelatos.med.upenn.edu/clipseqtools/ |
| MAFFT v7 | PMID: 28968734 | https://mafft.cbrc.jp/alignment/server/ |
| Vertebrate RNA-Seq | NIH Genome Data Viewer | RNA-Seq tracks for each species |
Highlights:
SYNGAP1 alternative 3’ splice site induces nonsense-mediated mRNA decay (A3SS-NMD)
PTBP1/2 proteins suppress SYNGAP1 by promoting A3SS-NMD
Deleting A3SS-NMD alleviates deficits in heterozygous Syngap1 knockout mice
Splice-switching oligonucleotide targeting the A3SS upregulates SYNGAP1 expression
Acknowledgment
The authors would like to thank Profs. Jonathan P. Staley (UChicago), Xiaoxi Zhuang (UChicago), Francois Spitz (UChicago), Yoav Gilad (UChicago), and Benoit Laurent (Université de Sherbrooke) for sharing reagents and valuable comments; thank colleagues in the Department of Human Genetics, the Neuroscience Institute, and the Genomics Core at UChicago for supporting this work; and thank Zhang lab members for technical support and in-depth discussions. R.S.Z. was supported by the Quad Undergraduate Research Scholarship. C.A.P was supported by the 2022 DENDRITES Leadership Alliance Program. A.A.C, A.P., and A.J.G were supported by a grant from the NINDS (R01 NS107421) to A.J.G. The generation of Syngap1-NISO mice was supported by the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH) through Grant Number 5UL1TR002389-04 that funds the Institute for Translational Medicine (ITM). This work was supported by grants from the NIMH (K01 MH109747 and R01 MH130594), and the NIGMS (DP2 GM137423) to X.Z.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests. A provisional patent on splice-switching oligonucleotides described in this study has been filed on behalf of X.Z. and R.Y.
References:
- 1.Zoghbi HY, and Bear MF (2012). Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 4. 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sudhof TC (2018). Towards an Understanding of Synapse Formation. Neuron 100, 276–293. 10.1016/j.neuron.2018.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen HJ, Rojas-Soto M, Oguni A, and Kennedy MB (1998). A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 20, 895–904. [DOI] [PubMed] [Google Scholar]
- 4.Kim JH, Liao D, Lau LF, and Huganir RL (1998). SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 20, 683–691. [DOI] [PubMed] [Google Scholar]
- 5.Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Cote M, Perreau-Linck E, Carmant L, et al. (2009). Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med 360, 599–605. 10.1056/NEJMoa0805392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, et al. (2020). Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584 e523. 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vlaskamp DRM, Shaw BJ, Burgess R, Mei D, Montomoli M, Xie H, Myers CT, Bennett MF, XiangWei W, Williams D, et al. (2019). SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy. Neurology 92, e96–e107. 10.1212/WNL.0000000000006729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berryer MH, Hamdan FF, Klitten LL, Moller RS, Carmant L, Schwartzentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot-Cerioli M, et al. (2013). Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat 34, 385–394. 10.1002/humu.22248. [DOI] [PubMed] [Google Scholar]
- 9.Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Almonte AG, Miller BH, Wiltgen BJ, Miller CA, et al. (2012). Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723. 10.1016/j.cell.2012.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim MJ, Dunah AW, Wang YT, and Sheng M (2005). Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 46, 745–760. 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 11.Rumbaugh G, Adams JP, Kim JH, and Huganir RL (2006). SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc Natl Acad Sci U S A 103, 4344–4351. 10.1073/pnas.0600084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Araki Y, Zeng M, Zhang M, and Huganir RL (2015). Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron 85, 173–189. 10.1016/j.neuron.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJ, O’Carroll CM, Martin SJ, Morris RG, et al. (2002). SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci 22, 9721–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JH, Lee HK, Takamiya K, and Huganir RL (2003). The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J Neurosci 23, 1119–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ozkan ED, Creson TK, Kramar EA, Rojas C, Seese RR, Babyan AH, Shi Y, Lucero R, Xu X, Noebels JL, et al. (2014). Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron 82, 1317–1333. 10.1016/j.neuron.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michaelson SD, Ozkan ED, Aceti M, Maity S, Llamosas N, Weldon M, Mizrachi E, Vaissiere T, Gaffield MA, Christie JM, et al. (2018). SYNGAP1 heterozygosity disrupts sensory processing by reducing touch-related activity within somatosensory cortex circuits. Nat Neurosci 21, 1–13. 10.1038/s41593-018-0268-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berryer MH, Chattopadhyaya B, Xing P, Riebe I, Bosoi C, Sanon N, Antoine-Bertrand J, Levesque M, Avoli M, Hamdan FF, et al. (2016). Decrease of SYNGAP1 in GABAergic cells impairs inhibitory synapse connectivity, synaptic inhibition and cognitive function. Nat Commun 7, 13340. 10.1038/ncomms13340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Creson TK, Rojas C, Hwaun E, Vaissiere T, Kilinc M, Jimenez-Gomez A, Holder JL Jr., Tang J, Colgin LL, Miller CA, and Rumbaugh G (2019). Re-expression of SynGAP protein in adulthood improves translatable measures of brain function and behavior. Elife 8. 10.7554/eLife.46752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lareau LF, Inada M, Green RE, Wengrod JC, and Brenner SE (2007). Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446, 926–929. 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- 20.Black DL (2003). Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72, 291–336. 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 21.Yan Q, Weyn-Vanhentenryck SM, Wu J, Sloan SA, Zhang Y, Chen K, Wu JQ, Barres BA, and Zhang C (2015). Systematic discovery of regulated and conserved alternative exons in the mammalian brain reveals NMD modulating chromatin regulators. Proc Natl Acad Sci U S A 112, 3445–3450. 10.1073/pnas.1502849112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, Ozawa M, Ma J, Yoshida N, Reiter JF, et al. (2016). Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 166, 1147–1162 e1115. 10.1016/j.cell.2016.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carvill GL, Engel KL, Ramamurthy A, Cochran JN, Roovers J, Stamberger H, Lim N, Schneider AL, Hollingsworth G, Holder DH, et al. (2018). Aberrant Inclusion of a Poison Exon Causes Dravet Syndrome and Related SCN1A-Associated Genetic Epilepsies. Am J Hum Genet 103, 1022–1029. 10.1016/j.ajhg.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin L, Zhang M, Stoilov P, Chen L, and Zheng S (2020). Developmental Attenuation of Neuronal Apoptosis by Neural-Specific Splicing of Bak1 Microexon. Neuron 107, 1180–1196 e1188. 10.1016/j.neuron.2020.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, and Krainer AR (2011). Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126. 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, et al. (2017). Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 377, 1723–1732. 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 27.Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A, Pendergast MK, Goldkind SF, Lee EA, Kuniholm A, et al. (2019). Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N Engl J Med 381, 1644–1652. 10.1056/NEJMoa1813279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim KH, Han Z, Jeon HY, Kach J, Jing E, Weyn-Vanhentenryck S, Downs M, Corrionero A, Oh R, Scharner J, et al. (2020). Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat Commun 11, 3501. 10.1038/s41467-020-17093-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Brauninger M, Lewitus E, Sykes A, Hevers W, Lancaster M, et al. (2015). Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci U S A 112, 15672–15677. 10.1073/pnas.1520760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prchalova D, Havlovicova M, Sterbova K, Stranecky V, Hancarova M, and Sedlacek Z (2017). Analysis of 31-year-old patient with SYNGAP1 gene defect points to importance of variants in broader splice regions and reveals developmental trajectory of SYNGAP1-associated phenotype: case report. BMC Med Genet 18, 62. 10.1186/s12881-017-0425-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darnell RB (2013). RNA protein interaction in neurons. Annu Rev Neurosci 36, 243–270. 10.1146/annurev-neuro-062912-114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vuong CK, Black DL, and Zheng S (2016). The neurogenetics of alternative splicing. Nat Rev Neurosci 17, 265–281. 10.1038/nrn.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raj B, and Blencowe BJ (2015). Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 87, 14–27. 10.1016/j.neuron.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, Shiue L, Ares M Jr., and Black DL (2007). A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 21, 1636–1652. 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindsay SJ, Xu Y, Lisgo SN, Harkin LF, Copp AJ, Gerrelli D, Clowry GJ, Talbot A, Keogh MJ, Coxhead J, et al. (2016). HDBR Expression: A Unique Resource for Global and Individual Gene Expression Studies during Early Human Brain Development. Front Neuroanat 10, 86. 10.3389/fnana.2016.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linares AJ, Lin CH, Damianov A, Adams KL, Novitch BG, and Black DL (2015). The splicing regulator PTBP1 controls the activity of the transcription factor Pbx1 during neuronal differentiation. Elife 4, e09268. 10.7554/eLife.09268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Licatalosi DD, Yano M, Fak JJ, Mele A, Grabinski SE, Zhang C, and Darnell RB (2012). Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev 26, 1626–1642. 10.1101/gad.191338.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sauliere J, Sureau A, Expert-Bezancon A, and Marie J (2006). The polypyrimidine tract binding protein (PTB) represses splicing of exon 6B from the beta-tropomyosin pre-mRNA by directly interfering with the binding of the U2AF65 subunit. Mol Cell Biol 26, 8755–8769. 10.1128/MCB.00893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pitts MW (2018). Barnes Maze Procedure for Spatial Learning and Memory in Mice. Bio Protoc 8. 10.21769/bioprotoc.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, Liu C, Leiser SC, Meena, Aznarez I, et al. (2020). Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med 12. 10.1126/scitranslmed.aaz6100. [DOI] [PubMed] [Google Scholar]
- 41.Kole R, Krainer AR, and Altman S (2012). RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 11, 125–140. 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon SJ, Elahi LS, Pasca AM, Marton RM, Gordon A, Revah O, Miura Y, Walczak EM, Holdgate GM, Fan HC, et al. (2019). Reliability of human cortical organoid generation. Nat Methods 16, 75–78. 10.1038/s41592-018-0255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng S, Gray EE, Chawla G, Porse BT, O’Dell TJ, and Black DL (2012). PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat Neurosci 15, 381–388, S381. 10.1038/nn.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dawicki-McKenna JMF, A. J; Waxman EA; Cheng C; Amado DA; Ranum PT; Lea V. Dungan A,B; Heller EA; French DL; Davidson BL; Prosser BL (2022). Mapping PTBP splicing in human brain identifies targets for therapeutic splice switching including SYNGAP1. Biorxiv. 10.1101/2022.07.15.500219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gill DF, and Hansel C (2020). Muscarinic Modulation of SK2-Type K(+) Channels Promotes Intrinsic Plasticity in L2/3 Pyramidal Neurons of the Mouse Primary Somatosensory Cortex. eNeuro 7. 10.1523/ENEURO.0453-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng M, Shang Y, Araki Y, Guo T, Huganir RL, and Zhang M (2016). Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell 166, 1163–1175 e1112. 10.1016/j.cell.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez-Palmero A, Boerrigter MM, Gomez-Andres D, Aldinger KA, Marcos-Alcalde I, Popp B, Everman DB, Lovgren AK, Arpin S, Bahrambeigi V, et al. (2021). DLG4-related synaptopathy: a new rare brain disorder. Genet Med 23, 888–899. 10.1038/s41436-020-01075-9. [DOI] [PubMed] [Google Scholar]
- 48.Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, Zhang Z, Xue C, Herre M, Silver PA, Zhang MQ, et al. (2014). HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep 6, 1139–1152. 10.1016/j.celrep.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li YI, Sanchez-Pulido L, Haerty W, and Ponting CP (2015). RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res 25, 1–13. 10.1101/gr.181990.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arias-Cavieres A, Khuu MA, Nwakudu CU, Barnard JE, Dalgin G, and Garcia AJ 3rd (2020). A HIF1a-Dependent Pro-Oxidant State Disrupts Synaptic Plasticity and Impairs Spatial Memory in Response to Intermittent Hypoxia. eNeuro 7. 10.1523/ENEURO.0024-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Busskamp V, Lewis NE, Guye P, Ng AH, Shipman SL, Byrne SM, Sanjana NE, Murn J, Li Y, Li S, et al. (2014). Rapid neurogenesis through transcriptional activation in human stem cells. Mol Syst Biol 10, 760. 10.15252/msb.20145508. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequences and coordinates of oligonucleotides.
Related to the following sections in METHOD DETAILS: Molecular cloning, RT-PCR, EMSA, Primary neurons immunostaining, and Splice switching oligonucleotides.
Data Availability Statement
The RNA-Sequencing results shown in Figure 1A have been uploaded to NCBI (PRJNA930469: SRR23308049, SRR23308050). The microscopy data reported in this paper will be shared by the lead contact upon request.
This study does not generate new code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.