

Abstract
Background:
The tumor suppressor, protein phosphatase 2A (PP2A), is downregulated in hepatoblastoma. We aimed to examine the effects of two novel compounds of the tricyclic sulfonamide class, ATUX-3364 (3364) and ATUX-8385 (8385), designed to activate PP2A without causing immunosuppression, on human hepatoblastoma.
Methods:
An established human hepatoblastoma cell line, HuH6, and a human hepatoblastoma patient-derived xenograft, COA67, were treated with increasing doses of 3364 or 8385, and viability, proliferation, cell cycle and motility were investigated. Cancer cell stemness was evaluated by real-time PCR and tumorsphere forming ability. Effects on tumor growth were examined using a murine model.
Results:
Treatment with 3364 or 8385 significantly decreased viability, proliferation, cell cycle progression and motility in HuH6 and COA67 cells. Both compounds significantly decreased stemness as demonstrated by decreased abundance of OCT4, NANOG, and SOX2 mRNA. The ability of COA67 to form tumorspheres, another sign of cancer cell stemness, was significantly diminished by 3364 and 8385. Treatment with 3364 resulted in decreased tumor growth in vivo.
Conclusion:
Novel PP2A activators, 3364 and 8385, decreased hepatoblastoma proliferation, viability, and cancer cell stemness in vitro. Animals treated with 3364 had decreased tumor growth. These data provide evidence for further investigation of PP2A activating compounds as hepatoblastoma therapeutics.
Keywords: protein phosphatase 2A, hepatoblastoma, ATUX-3364, ATUX-8385
Introduction
The incidence of hepatoblastoma, the most common primary pediatric hepatic malignancy, has been increasing [1]. Surgical resection remains the mainstay of treatment [2], but a majority of patients present with unresectable disease [3]. Despite slight improvements in prognosis for patients with high-risk disease [4, 5], the chemotherapeutic agent currently employed, cisplatin, results in significant long-term sequelae including nephrotoxicity and ototoxicity [4]. Therefore, it continues to be crucial to investigate novel therapeutic agents that may be used alone or in combination to provide a more efficacious and less toxic regimen.
A potential therapeutic target is protein phosphatase 2A (PP2A). PP2A is a serine/threonine phosphatase that functions as a tumor suppressor, but its activity is reduced in many cancers. PP2A activation has been found to decrease the malignant phenotype in several cancer types [6–8]. Our lab has previously utilized the synthetic sphingosine analog, fingolimod (FTY720), to activate PP2A, and found a decrease in tumor burden of several pediatric solid tumors, including hepatoblastoma [9], neuroblastoma [10], and medulloblastoma [11]. FTY720 is an immunosuppressant [12] mediated by functional antagonism of sphingosine-1-phosphate receptor 1 (S1PR), which forms the basis of its clinical use in multiple sclerosis. To mitigate the issues of immunosuppression, other molecules designed to activate PP2A are being developed. One class of these PP2A activating compounds are tricyclic sulfonamides. These molecules bind directly to the alpha scaffold subunit of PP2A, resulting in conformational changes that promote the assembly of active PP2A holoenzyme complexes [7, 13]. A previous study in neuroblastoma with two other molecules of this class demonstrated a decrease in the malignant phenotype in vitro and in tumor burden in vivo [14]. In the current study, we aimed to evaluate the effects of these newly synthesized tricyclic sulfonamide compounds in models of hepatoblastoma.
2. Methods
2.1. Cells and cell culture
The established human hepatoblastoma cell line, HuH6, was obtained from Thomas Pietschmann (Hannover, Germany) [15], and was maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Corning Inc., Corning, NY) supplemented with 10% fetal bovine serum (HyClone, GE Healthcare Life Sciences, Logan, UT), 2 mmol/L-glutamine (Thermo Fisher Scientific, Waltham, MA), and 1 μg/mL penicillin/streptomycin (Gibco, Carlsbad, CA). The human embryonal hepatoblastoma patient-derived xenograft (PDX), COA67, has been previously described [16]. COA67 was generated at our institution under University of Alabama at Birmingham (UAB) Institutional Review Board (IRB) and Institutional Animal Care and Use Committee (IACUC) approved protocols (IRB-130627006, IACUC-009186, respectively). The PDX was passed serially through mice to maintain the cell line. For experiments, COA67 cells were placed in DMEM/Ham’s F12 (Corning) supplemented with 2 mmol/L 1-glutamine (Thermo Fisher Scientific), 20 ng/mL basic-fibroblast growth factor (bFGF, MilliporeSigma, Billerica, MA), 20 ng/mL epidermal growth factor (EGF, MilliporeSigma), 2% B27 supplement (Gibco), 1 μg/mL penicillin/streptomycin (Gibco), and 2.5 μg/mL amphotericin B (HyClone). Cells were maintained under standard culture conditions at 37 °C and in a humidified atmosphere containing 5% CO2. Cells and PDXs were verified within the last 12 months using short tandem repeat analysis (UAB Genomics Core) and were deemed free of Mycoplasma infection by the Universal Mycoplasma Detection Kit (30–1012K, American Type Culture Collection, ATCC, Manassas, VA).
2.2. Reagents and antibodies
For immunoblotting, the following primary antibodies were used: rabbit monoclonal anti-cleaved PARP (5625) from Cell Signaling (Danvers, MA), rabbit polyclonal anti-CIP2A (ab99518) from Abcam (Cambridge, MA), mouse monoclonal anti-β-actin (A1978) from Sigma Aldrich (St. Louis, MO), mouse monoclonal anti-GAPDH (MAB374) from MilliporeSigma, and rabbit polyclonal anti-I2PP2A (SET, 55201-AP) from Proteintech (Rosemont, IL).
2.3. Synthesis of ATUX-3364 and ATUX-8385
PP2A activators, ATUX-8385 (8385) and ATUX-3364 (3364), were synthesized by a modification of the route described in Ohlmeyer and Zaware in published patent application US 2018-0251456. Structure activity relationships and associated synthetic chemistry will be published elsewhere. The compounds are light sensitive and were therefore stored in the dark in a sealed container.
2.4. Microsome stability
Microsome stability assays were performed by Eurofins. Briefly, 2.5 μL (100 μM in DMSO) of test compounds are added to 197.5 μL of mouse microsome preparation (0.633 mg/mL in phosphate buffer) and mixed gently at 37 °C. The reaction is started by adding 50 μL NADPH (5 mM in phosphate buffer) and mixing at 37 °C. The final reaction microsome concentration is 0.5 mg/mL. The reaction is sampled at 0, 5, 15, 30, and 60 min. Reaction aliquots are quenched by adding to 1:1 methanol–acetonitrile containing analytical standards and analyzed by liquid chromatography with tandem mass spectrometry (LC-MS/MS).
2.5. Protein phosphatase 2A (PP2A) activation
HuH6 (1 × 106) or COA67 (3 × 106) cells were treated with 3364 or 8385 (HuH6: 8 μM, COA67: 2 μM) for 24 h and lysed using HEPES (AC215001000, Thermo Fisher Scientific), MgCl2 (MX0045-4, EM Science, Gibbstown, NJ), KCl (02-003-741, Thermo Fisher Scientific), PMSF (Sigma), dithiothreitol (EC-601, National Diagnostics, Atlanta, GA), and Igepal CA-630 (I7771, Sigma) for 20 min on ice. Lysates were centrifuged at 17 000 rpm for 30 min at 4 °C. A PP2A Immunoprecipitation Phosphatase Assay Kit (17–313, MilliporeSigma) was utilized per manufacturer’s protocol to evaluate the activity of PP2A. Experiments were completed at least in triplicate, and data reported as mean fold change ± standard error of the mean (SEM).
2.6. Immunoblotting
Whole cell lysates were isolated on ice using radioimmunoprecipitation (RIPA) buffer with phosphatase inhibitors (P5726, Sigma), protease inhibitors (P8340, Sigma), and phenylmethanesulfonylfluoride (PMSF, P7626, Sigma) for 60 min. Lysates were centrifuged at 17 000 rpm for 30 min at 4 °C. Pierce BCA Protein Assay (Thermo Fisher Scientific) was used to determine protein concentration and proteins were separated by electrophoresis on sodium dodecyl sulfate polyacrylamide (SDS-PAGE) gels. Antibodies were used according to the manufacturers’ suggested protocol. Expected size of the targeted proteins was confirmed using molecular weight markers (Precision Plus Protein Kaleidoscope, Bio-Rad, Hercules, CA). Immunoblots were developed with Luminata Classico or Crescendo Western horseradish peroxidase substrate (MilliporeSigma). Anti-GAPDH or anti-β-actin served as internal loading controls.
2.7. Viability and proliferation
To examine viability, alamarBlue assay (Thermo Fisher Scientific) was used. CellTiter 96 Aqueous One Solution Cell Proliferation assay (Promega, Madison, WI) was used to investigate proliferation. These assays are cell-based and detect changes in dye turnover. Cells treated with no drug serve as controls. HuH6 or COA67 (1.5 × 104 cells for viability, 5 × 103 for proliferation) cells were plated in 96-well plates and treated with increasing doses of 3364 or 8385 for 24 h. AlamarBlue or CellTiter 96 dye (10 μL) was added for viability and proliferation assays, respectively. A microplate reader (Epoch Microplate Spectrophotometer, BioTek Instruments, Winooski, VT) measured absorbance at 570 nm for alamarBlue and at 490 nm for CellTiter 96, using 600 nm as a reference. Experiments were completed with at least three biologic replicates, and data reported as fold change ± SEM.
2.8. Cell cycle
To evaluate cell cycle, HuH6 cells (5 × 105) were synchronized overnight in media with 4% FBS. HuH6 cells were treated with 3364 (6 μM) or 8385 (8 μM) for 24 h in standard media with 10% FBS. COA67 cells (3 × 106) were plated and treated with 3364 or 8385 (4 μM) and treated for 24 h. Cells were washed with phosphate-buffered saline (PBS) and fixed on ice for 30 min with 100% ethanol. Following a second PBS wash, cells were stained with 200 μL of propidium iodide (PI, Invitrogen, Waltham, MA), RNAse A (0.1 mg/mL, Qiagen, Germantown, MD), and 0.1% TritonX (Active Motif, Carlsbad, CA). FACSCalibur™ Flow Cytometer (BD Biosciences, Franklin Lakes, NJ) was used for evaluation and data analysis completed with FlowJo software (FlowJo, LLC, Ashland, OR). Experiments were repeated with three biologic replicates and data reported as mean percentage cells in phase ± SEM.
2.9. Motility
Since HuH6 cells propagate as a monolayer, migration was examined using wound healing (scratch) assay. HuH6 cells (5 × 104) were plated in 12-well plates. Once cells reached 80% confluence, a sterile 200 μL pipette tip was used to make a standard scratch in the cell layer, and cells were washed with 1 mL PBS, and treated with 3364 (6 μM) or 8385 (8 μM). Photographs of the plates were obtained at 0, 12, 24, 36, and 48 h. ImageJ MRI Wound Healing Tool (http://imagej.nih.gov/ij/) [17] quantified the open wound area, and data reported as fold change of the open area ± SEM. For COA67 cells which do not grow as a monolayer, migration was assessed using modified Boyden chambers. Micropore inserts with 8 μM pores (Corning) were utilized in 24 well plates. The insert bottoms were coated with fibronectin (10 μg/mL, Qiagen). Cells were treated for 24 h with 3364 (0, 3 μM) or 8385 (0, 3 μM), plated (6 × 104 cells), and allowed to migrate through the membrane for 72 h. Insert membranes were fixed using 4% paraformaldehyde, stained with 1% crystal violet for 15 min, photographed, and migration quantified with ImageJ (http://imagej.nih.gov/ij/). Scratch assays were reported as fold change area remaining open ± SEM.
Cell invasion assays were performed with the HuH6 and COA67 cells using micropore inserts. For the HuH6 cells, the insert bottom was coated with collagen I (10 μg/mL, MP Biomedicals, Santa Ana, CA) and for COA67 cells with fibronectin (10 μg/mL, Qiagen). An additional layer of Matrigel (1 mg/mL, 50 μL, BD Biosciences) was coated on the tops of the inserts. Treatment doses were the same as those utilized in migration assays. HuH6 cells (3 × 104) were allowed to invade for 24 h and COA67 cells (6 × 104) for 72 h. Modified Boyden chamber migration and invasion assays were reported as mean fold change of numbers of migrated or invaded cells ± SEM. All motility experiments were completed with at least three biologic replicates.
2.10. qPCR
HuH6 cells were treated with 3364 or 8385 (0, 8 μM) for 4 h. A RNeasy kit (Qiagen) was utilized to extract total cellular RNA according to the manufacturer’s protocol. iScript cDNA Synthesis kit (Bio-Rad) was used to synthesize cDNA with 1 μg of RNA used in a 20 μL reaction. SsoAdvanced SYBR Green Supermix (Bio-Rad) was utilized according to the manufacturer’s protocol for quantitative real-time PCR (qPCR). Primers specific for octamer-binding transcription factor 4 (Oct4), homeobox protein Nanog, sex determining region Y-box 2 (Sox2), and β-actin were utilized (Invitrogen). Amplification was performed using an Applied Biosystems CFX96 Real-Time System or C1000 Touch Thermal Cycler (Applied Biosystems). qPCR was performed with 10 ng cDNA in 20 μL reaction volume with cycling conditions as previously described [18, 19]. β-actin was utilized as an internal control. Gene expression was calculated using the ΔΔCT method [20] and reported as mean fold change in mRNA abundance ± SEM. Experiments were repeated with at least three biologic replicates.
2.11. Tumorsphere formation
Capacity to form tumorspheres was evaluated in COA67 cells only, as HuH6 cells propagate in culture as attached cells and do not readily form spheres. COA67 cells were plated in non-adherent conditions in a 96-well plate with a decreasing number of cells in each row of 12 wells (1000, 500, 100, 50, 20, or 1 cell per well) and treated with either 3364 or 8385 (0, 2 μM), incubated for one week, and each well was examined for tumorsphere formation. The number of wells with sphere formation were counted by an investigator blinded to the treatment groups, and data was analyzed using the online extreme limiting dilution analysis (ELDA) software (http://bioinf.wehi.edu.au/software/elda/, accessed February 22, 2002).
2.12. Animal statement
The University of Alabama at Birmingham Institutional Animal Care and Use Committee (UAB IACUC-09064) approved all animal experiments, and the studies were conducted within institutional, national, and NIH guidelines.
2.13. In vivo tumor growth
HuH6 cells (2.5 × 106) in 25% Matrigel (BD Biosciences) were injected into the right flank of 6-week-old female athymic nude mice (Fredricks, Charles River, Wilmington, MA). Calipers were used to measure tumors and volumes calculated by the formula (width2 × length)/2; where width was the smaller measurement. Fourteen days after injection, animals were randomized to treatment groups using GraphPad (https://www.graphpad.com/quickcalcs/randomize1/). Two experiments were performed. The first was a preliminary evaluation of 3364 and 8385 in HuH6. Animals were randomized to three groups (n=7, each) to receive 100 μL of either vehicle [N,N-dimethylacetamide (DMA, 271012, Sigma) and Kolliphor HS 15 (Solutol, 42996, Sigma)], 8385 (50 mg/kg in DMA and Solutol) or 3364 (50 mg/kg in DMA and Solutol) twice daily for 21 days by oral gavage. The choice of dosage and timing for this initial pilot study was based on results of previous bioavailability studies of similar compounds [14]. Based on the results of this pilot experiment, a second in vivo study was performed with the same parameters except: (i) the 8385 group was eliminated, (ii) the 3364 dosage was increased to 75 mg/kg po bid (n=4), and (iii) the experimental group was compared to controls from the pilot experiment (n=7). Employing controls from the pilot experiment allowed adherence to the three R’s of animal usage [21]. Animals were weighed daily, and 3364 doses were strictly adjusted based on the weight of each animal each day. At the completion of the experiments at 21 days, or when animals reached IACUC parameters for euthanasia, they were humanely euthanized in their home cages with CO2 and cervical dislocation.
2.14. Statistical analysis
All in vitro experiments were performed with at least three biologic replicates. For PDX cells, biologic replicates were represented by PDX tumors from at least three different animals. Data reported as the mean ± SEM [22]. ANOVA or student’s t-test was utilized, and statistical significance was defined as p≤0.05.
3.0. Results
3.1. Novel PP2A activators
The compounds used in the current study, ATUX-8385 and ATUX-3364 shown in Fig. S1a, contain a basic piperidine moiety in the central ring constraint to allow for salt formation and provide a larger range of options for formulation of drug, including water solubility. The tricyclic in the compounds is a fluorine substituted carbazole which imparts metabolic stability with respect to oxidative metabolism, which is consistent with the in vitro microsome clearance shown in Fig. S1b. These data indicate high stability with respect to oxidative metabolism. ATUX-8385 and ATUX-3364 are enantiomers.
3.2. Treatment with 3364 and 8385 led to PP2A activation
First we aimed to examine the effects of the tricyclic sulfonamides, 3364 and 8385, on PP2A activation in hepatoblastoma. After 24 h of treatment, PP2A activity was significantly increased in HuH6 cells (Fig. 1a). Similarly, COA67 cells had significantly increased PP2A activity after treatment with either compound (Fig. 1b). Since previous research demonstrated that treatment with PP2A activators altered the expression of cancerous inhibitor of protein phosphatase 2A (CIP2A) and or I2PP2A (SET), two endogenous inhibitors of PP2A [9, 14, 23, 24], we investigated the effects of 3364 and 8385 on these inhibitors. In the current study, HuH6 cells treated with 3364 and 8385 had decreased CIP2A expression (Fig. 1c). In COA67 cells, CIP2A decreased with 3364 but there was no change in CIP2A with 8385 treatment (Fig. 1d). Treatment with 3363 or 8385 did not affect expression of SET in either HuH6 (Fig. 1c) or COA67 (Fig. 1d) cells.
Fig. 1. Effects of ATUX-3364 or ATUX-8385 on PP2A and its endogenous inhibitors.
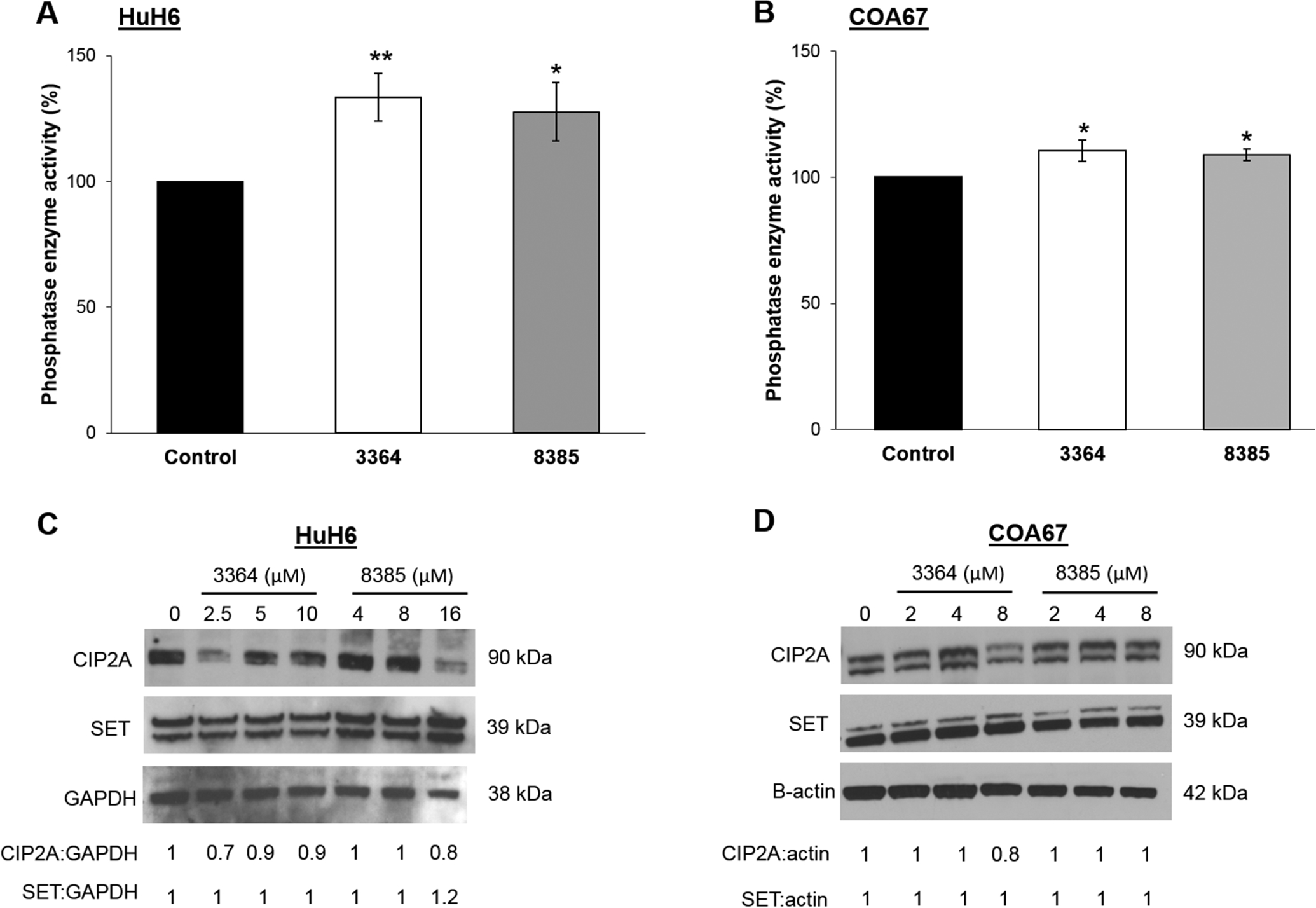
(A) HuH6 or (B) COA67 cells were treated with 3364 or 8385 (HuH6: 8 μM, COA67: 2 μM) for 24 h and PP2A activation measured with a commercially available assay kit. There was a significant increase in PP2A activation following treatment with either compound in HuH6 and COA67 cells. (C, D) Immunoblotting was used to examine the effects of 3364 and 8385 on the endogenous inhibitors of PP2A, including CIP2A and SET. Densitometry was performed and band density reported in relation to that of the housekeeping protein. (C) There was a decrease in CIP2A expression with 3364 and 8385 treatment in HuH6 cells. SET expression did not change with either drug. (D) In COA67 cells, 3364 treatment decreased CIP2A expression, but 8385 did not affect CIP2A expression. Expression of SET was unchanged by 3364 or 8385 treatments. Experiments were repeated with at least three biologic replicates. Data reported as mean fold change ± standard error of the mean (SEM) and compared with two-tailed t-test. *p≤0.05, **p≤0.01.
3.3. Treatment with 3364 and 8385 decreased proliferation
Because other investigators demonstrated decreased cancer cell proliferation with other PP2A activators [9, 10, 25], we investigated the effects of 3364 and 8385 on hepatoblastoma proliferation. After 24 h treatment, HuH6 proliferation was significantly decreased with 3364 (7.5 μM) and 8385 (12.5 μM) (Fig. 2a). Treatment with 4 μM of either compound significantly decreased COA67 proliferation (Fig. 2b).
Fig. 2. ATUX-3364 or ATUX-8385 decreased hepatoblastoma proliferation.
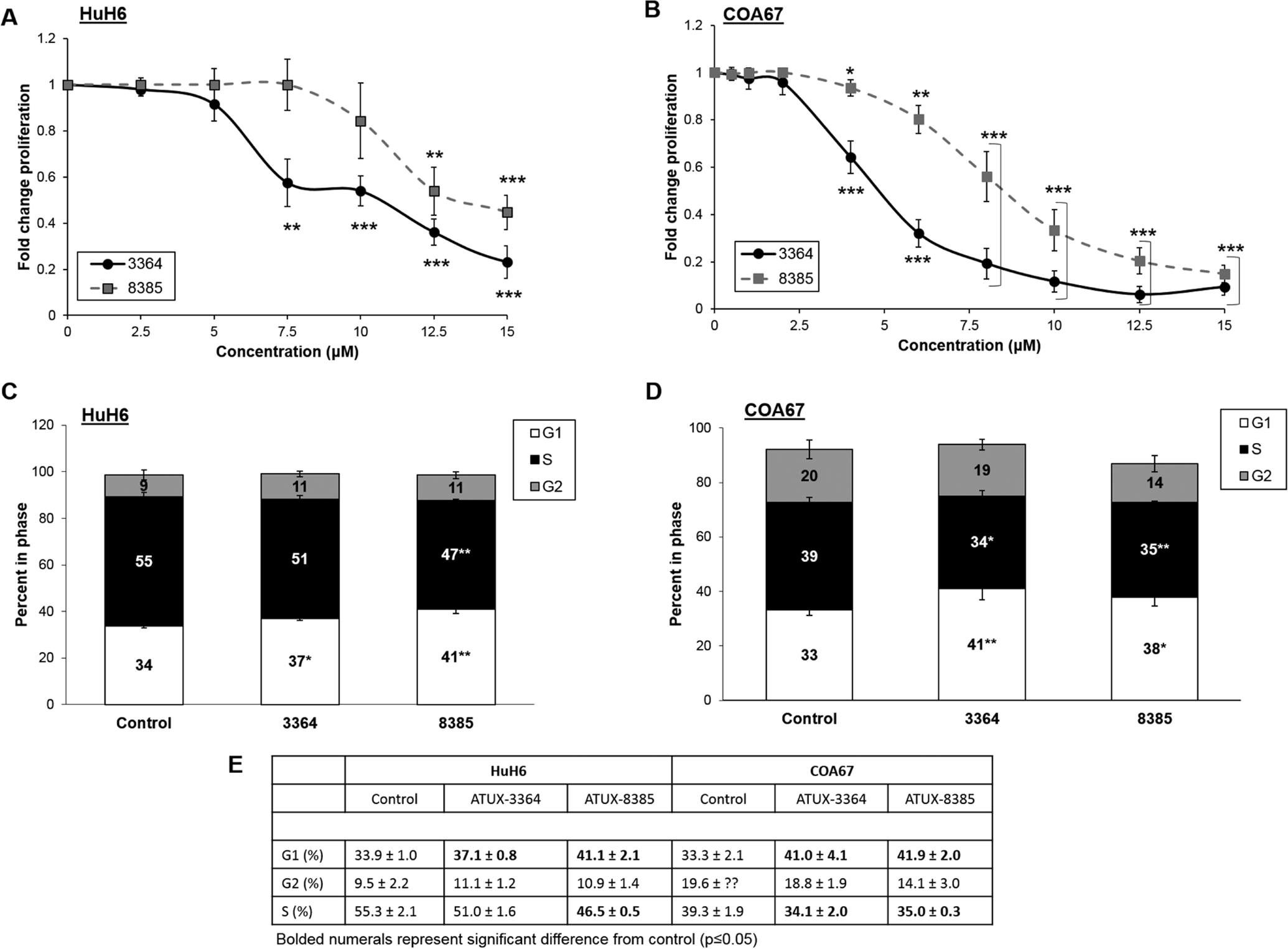
HuH6 or COA67 cells (5 × 103 cells) were plated into 96-well plates and treated for 24 h with increasing doses of 3364 or 8385. (A) In HuH6 cells, 3364 and 8385 significantly decreased proliferation. (B) In COA67 cells proliferation was significantly decreased after treatment with either compound. (C) HuH6 cells were serum starved overnight and treated for 24 h with 3364 (6 μM) or 8385 (8 μM). There was a significant increase in the percentage of cells in G1 phase following treatment with either compound. There was an associated significant decrease in S phase following 8385 treatment with a trend toward significance for cells treated with 3364, indicating a lack of progression through the cell cycle. (D) Similarly, following 24 h treatment of 3364 or 8385 (4 μM), COA67 cells had a significant increase in percentage of cells in G1 phase and decrease in S phase, indicating a lack of progression through the cell cycle. (E) Cell cycle data presented in tabular form. Significant values (p≤0.05) depicted by bold font. Proliferation data reported as mean fold change ± SEM, cell cycle data reported as the mean percent cells in phase ± SEM. Data evaluated with two-tailed t-test. Experiments were repeated with at least three biologic replicates. *p≤0.05, **p≤0.01, ***p≤0.001.
To continue investigating proliferation, cell cycle analysis was performed following 24 h treatment of 3364 or 8385. There was a significant increase in the percentage of HuH6 cells in G1 phase following treatment with 3364 (6 μM, 37.1 ± 0.8 % vs. 33.9 ± 1.0 %, treated vs. control, respectively, p≤0.05) or 8385 (8 μM, 41.1 ± 2.1 % vs. 33.9 ± 1.0 %, treated vs. control, respectively, p≤0.01). Treatment with 8385 (8 μM, 46.5 ± 0.5 % vs. 55.3 ± 2.1 %, treated vs. control, respectively, p≤0.01) resulted in significant decrease in percentage of HuH6 cells in S phase while 3364 (6 μM, 51.0 ± 1.6 % vs. 55.3 ± 2.1 %, treated vs. control, respectively, p≤0.07) resulted in a downward trend that did not reach significance (Fig. 2c,e). COA67 cells had diminished progression through the cell cycle (Fig. 2d,e) as shown by an increase in percentage of cells in G1 phase following treatment with 3364 (41.0 ± 4.1 % vs. 33.3 ± 2.1 %, treated vs. control, respectively, p≤0.01) or 8385 (41.9 ± 2.0 % vs. 30.0 ± 1.7 %, treated vs. control, respectively, p≤0.001) and a decrease in percentage of cells in S phase following treatment with 3364 (34.1 ± 2.0 % vs. 39.3 ± 1.9 %, treated vs. control, respectively, p≤0.05) or 8385 (35.0 ± 0.3 % vs. 39.3 ± 1.9 %, treated vs. control, respectively, p≤0.01). Cell cycle data are presented in tabular form in Fig. 2e. These findings indicate decreased progression through the cell cycle following treatment with the tricyclic sulfonamide compounds.
3.4. Treatment with 3364 and 8385 decreased viability and increased apoptosis
In addition to proliferation, we examined the effects of 3364 and 8385 on hepatoblastoma viability. Treatment with either compound significantly decreased viability in HuH6 (Supplementary Fig. S2a) and COA67 (Supplementary Fig. S2b) cells.
To assess whether decreased viability was due to apoptosis, immunoblotting for cleaved PARP was performed. Expression of cleaved PARP was increased with 3364 and 8385 in HuH6 (Supplementary Fig. S3a) and in COA67 (Supplementary Fig. S3b) cells, indicating apoptosis.
3.5. Treatment with 3364 and 8385 decreased motility
Migration and invasion are key to development of cancer metastases [26], and previous investigators documented decreased cell motility with PP2A activation [9–11, 27, 28]prompting investigation of effects of 3364 and 8385 on motility. A wound healing assay was utilized to examine HuH6 motility. Following 48 h treatment with 3364 (Fig. 3a) or 8385 (Fig. 3b), there was a significantly greater open area of the scratch wound compared to that of control HuH6 cells, signifying decreased ability to migrate. Motility in COA67 cells was evaluated with modified Boyden chamber assays. Treatment with 3364 or 8385 significantly decreased COA67 cell migration (Fig. 3d). Invasion was investigated using Transwell inserts. Treatment with 3364 or 8385 led to significantly decreased HuH6 invasion after 24 h (Fig. 3e). The ability of COA67 cells to invade was significantly decreased after 72 h treatment with 3364 or 8385 (Fig. 3f). Representative photomicrographs of wound assays are provided in Fig. 3c. Photomicrographs of migration and invasion inserts are provided below graphs in Fig. 3d–f.
Fig. 3. ATUX-3364 or ATUX-8385 decreased hepatoblastoma motility.
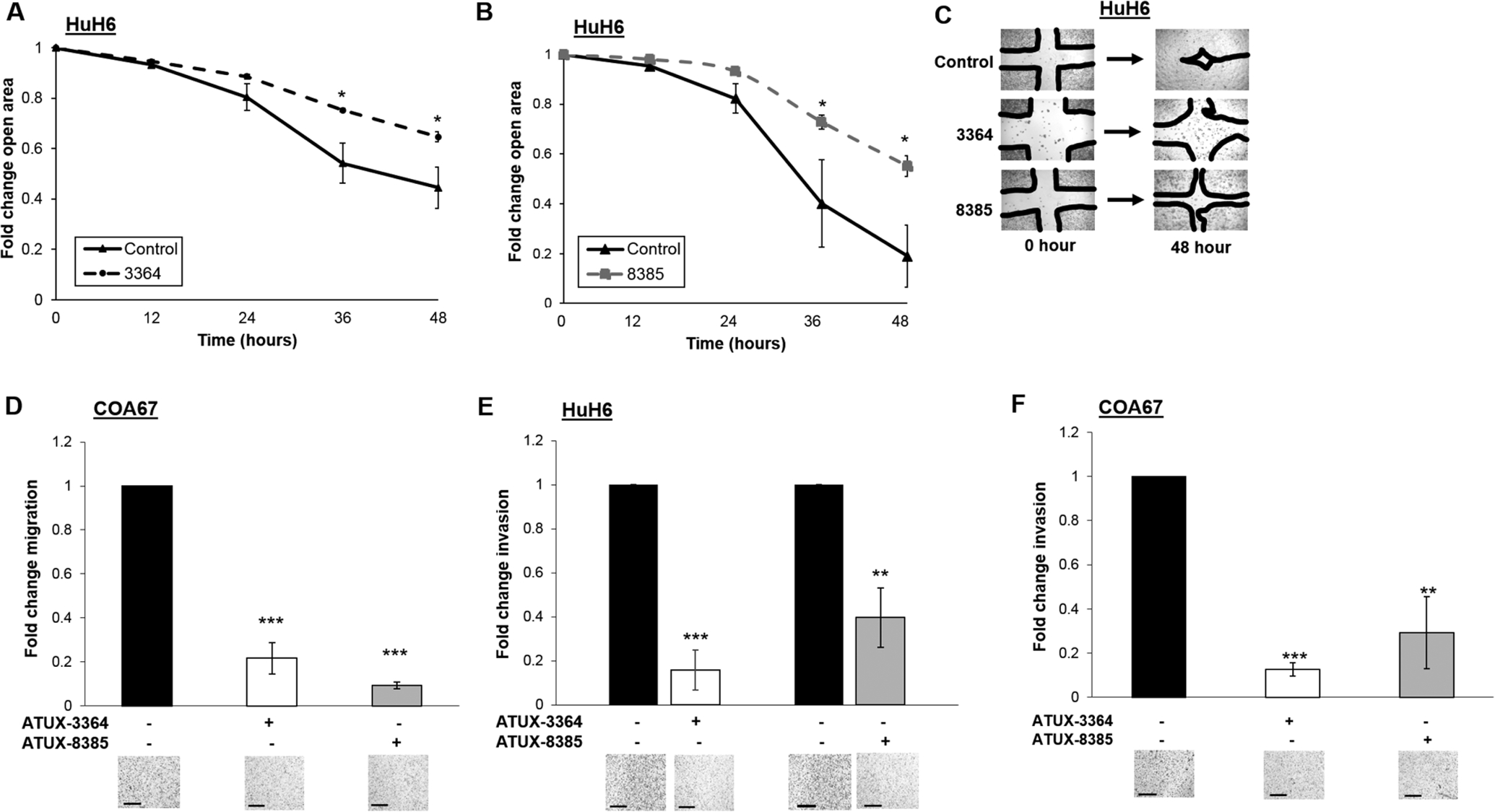
(A) To assess migration, HuH6 (5 × 103) cells were plated in 12 well plates. Once cells reached 80% confluence, a standard scratch was made with a 200 μL pipette tip. Cells were treated with 3364 (0, 6 μM) or (B) 8385 (0, 8 μM) and pictures were taken every 12 h. ImageJ analysis software was used to quantify the open area. Cells treated with 3364 and 8385 had significantly increased open area remaining at 48 h. (C) Representative pictures of scratch wound in control and treated cells at 0 and 48 h. (D) Because COA67 cells do not adhere in cell culture, modified Boyden chamber assays were used to examine migration. Cells were treated for 24 h with 3364 or 8385 (0, 3 μM), plated into the inserts (6 × 104 cells), and allowed to migrate through the membrane for 72 h. Treatment with either 3364 or 8385 resulted in decreased COA67 migration. Representative images are shown below the graph. (E) To investigate invasion, modified Boyden chamber assay experiments were performed in similar fashion with the addition of a layer of Matrigel to the top of the insert. HuH6 cells were treated for 24 h with 3364 (0, 6 μM) or 8385 (0, 8 μM), and then plated (3 × 104 cells) onto the inserts and allowed to invade for 24 h. There was a significant decrease in invasion following treatment with 3364 or 8385. (F) COA67 cells were treated for 24 h with 3364 or 8385 (0, 3 μM), plated (6 × 104 cells) onto inserts and allowed to invade for 72 h. Treatment resulted in a significant decrease in COA67 invasion. Representative photomicrographs of migration and invasion inserts are provided below the graphs. Data from wound healing assay reported as mean fold change open area ± SEM; data from modified Boyden chamber studies reported as mean percent area of membrane with cells present ± SEM. Data were compared using two-tailed t-test. Experiments were repeated with at least three biologic replicates. Scale bars represent 300 μm. *p≤0.05, **p≤0.01, ***p≤0.001.
3.6. Treatment with 3364 and 8385 led to decreased stemness
Since cancer cell stemness is thought to be responsible for hepatoblastoma disease progression, resistance to therapy, and recurrence [29] and other investigators have shown that PP2A activation decreased cancer stemness [30, 31], we examined the effects of 3364 and 8385 on hepatoblastoma cell stemness. qPCR was utilized to examine the abundance of mRNA of OCT4, NANOG, and SOX2, three cancer stem cell markers [32–35], following 4 h treatment with 3364 or 8385. Treatment of HuH6 with either compound (8 μM) resulted in a significant decrease in abundance of mRNA of these stemness markers (Fig. 4a). To further evaluate stemness, we utilized COA67 cells to assess tumorsphere formation, a phenotypic indication of cancer stemness. Following treatment with either 3364 or 8385 (2 μM), the ability of COA67 cells to form tumorspheres was significantly decreased (Fig. 4b), indicating a less cancer stem cell-like phenotype.
Fig. 4. ATUX-3364 and ATUX-8385 decreased stemness.
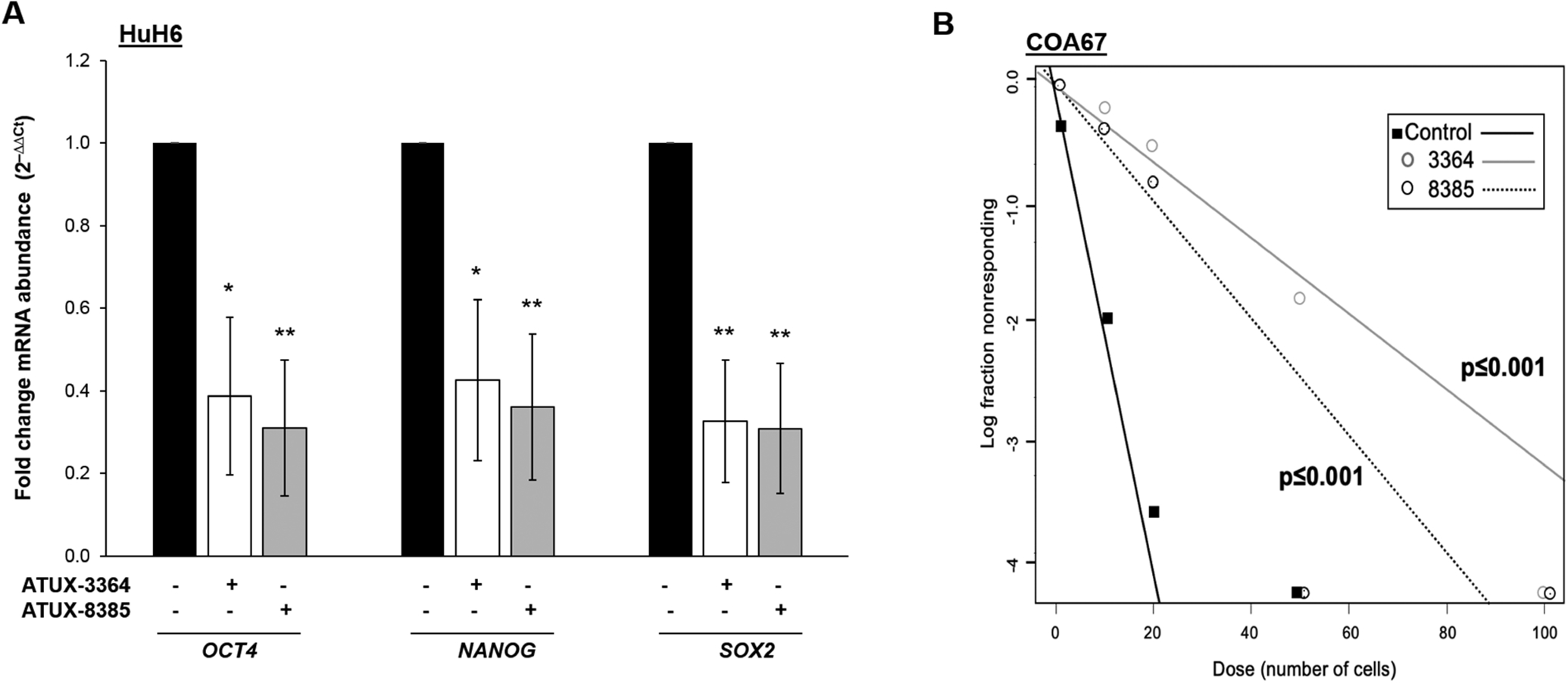
qPCR was used to investigate three stemness markers, OCT4, NANOG, and SOX2, following 3364 or 8385 treatments. (A) HuH6 cells were treated with either 3364 or 8385 (0, 8 μM) for 4 h. The abundance of mRNA of the three stemness markers was significantly decreased in the treated cells. mRNA expression was normalized to β-actin and calculated as fold change versus the control untreated cells using the ΔΔCt method [20]. (B) A tumorsphere formation assay was used to further assess the effects on stemness. COA67 cells were plated at increasing cell concentrations per well in non-adherent conditions (100 to 1 cells). Cells were treated with 3634 or 8385 (0, 2 μM) and after 1 week, the number of wells with spheres were counted. The findings were analyzed using an ELDA online analysis software (http://bioinf.wehi.edu.au/software/elda/, accessed February 22, 2002). Treatment with 3364 or 8385 significantly reduced tumorsphere formation in COA67 cells. Experiments were repeated with at least three biologic replicates. *p≤0.05, **p≤0.01, ***p≤0.001.
3.7. PP2A activation decreased tumor growth in vivo
The in vitro data revealed that 3364 and 8385 significantly affected the malignant phenotype of human hepatoblastoma cells, leading us to pursue in vivo investigations. Since these compounds had not been utilized for hepatoblastoma previously, we began with a pilot study comparing tumor growth in animals treated with 3364 or 8385 at 50 mg/kg/bid to those treated with vehicle. Treatment with 8385 had no significant effect on tumor growth compared to vehicle-treated animals (Supplementary Fig. S4a). Animals treated with 3364 had smaller tumor volumes that trended towards significance when compared to vehicle treated animals (Supplementary Fig. S4b). Therefore, a follow up experiment was undertaken, increasing the dosage of 3364 to 75 mg/kg bid. This decision was founded on previous experiments with SMAPs that were well tolerated in doses up to 100 mg/kg bid [13]. The animals treated with 3364 at the increased dose had significantly decreased relative tumor growth compared to vehicle treated animals from the pilot study (Fig. 5a). Tumor growth for individual animals is provided in Fig. 5b. The weights of the animals were not affected by the treatment (Fig. 5c).
Fig. 5. Treatment with ATUX-3364 decreased tumor growth in vivo.
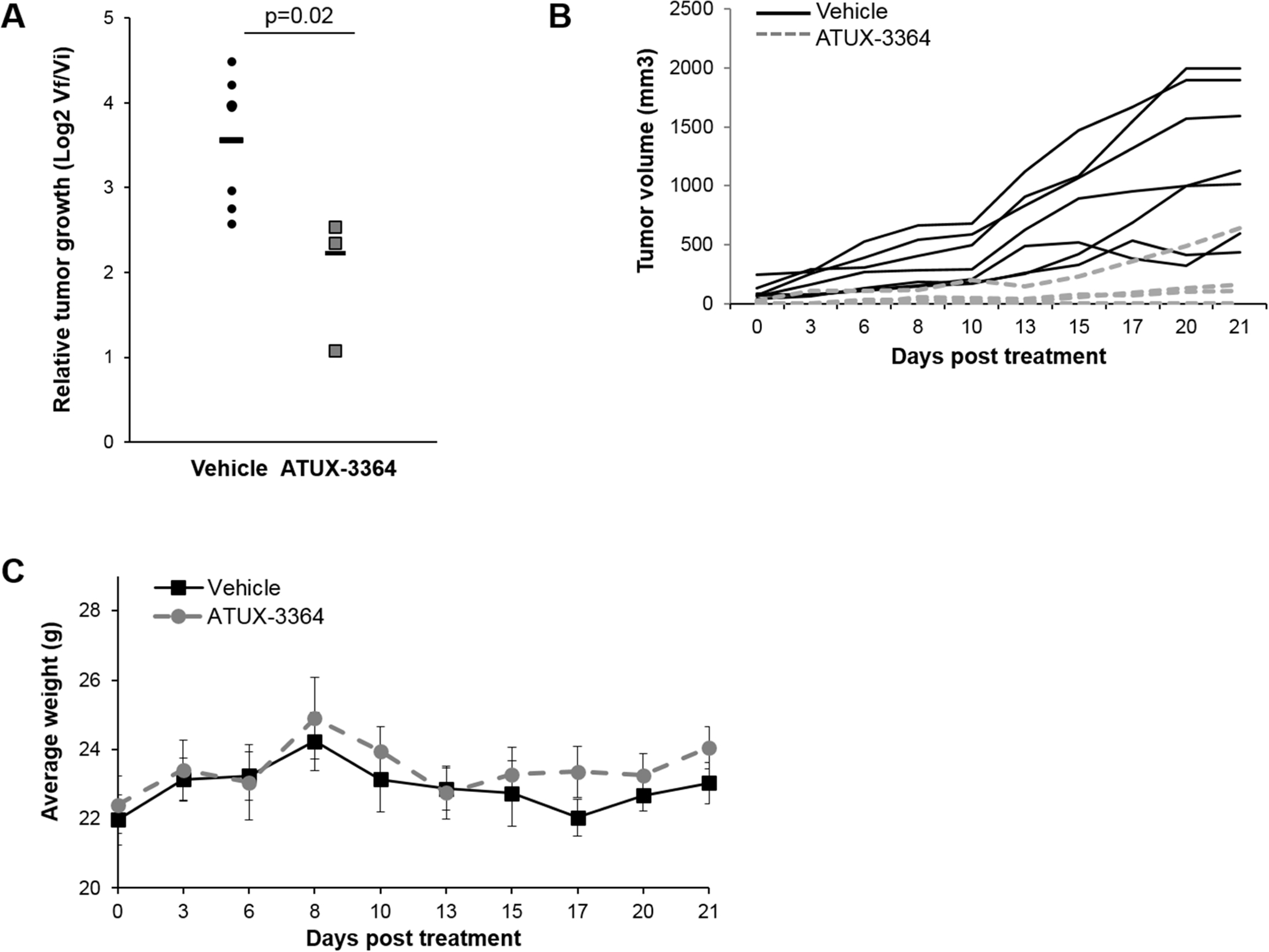
(A) HuH6 cells (2.5 × 106) in 25% Matrigel were injected into the right flank of 6-week-old female athymic nude mice. Fourteen days after injection, animals were randomized to receive either vehicle or 3364 (75 mg/kg po bid) via oral gavage (n = 7 for vehicle, n = 4 for 3364) for 21 days. Tumors were measured with calipers and volumes calculated by the formula (width2 × length)/2; where width was the smaller measurement at least s3 times per week. Animals were weighed daily for dosing adjustments. Animals treated with 3364 had decreased relative tumor growth compared to vehicle treated animals. Student’s t-test was use for comparison. (B) Graph of individual tumor growth displaying trends between vehicle treated (solid black lines) and ATUX-3364 treated (grey dashed lines) animals. (C) The weights of the animals were not affected by the drug treatment.
4.0. Discussion
In the current study, we used two novel tricyclic sulfonamides designed to activate PP2A and demonstrated a decrease in the malignant phenotype of hepatoblastoma in vitro as well as in vivo. Previously we demonstrated that PP2A activation using the sphingosine analog, FTY720, decreased hepatoblastoma tumor growth [9]. Similar effects were noted in neuroblastoma using novel tricyclic sulfonamide small activators of PP2A (SMAPs) [14], providing the rationale for the current study. We utilized the established human hepatoblastoma cell line HuH6 for these investigations. Despite being the most common primary liver cancer in children, HuH6 is the only fully characterized hepatoblastoma cell line that is publicly available for investigation [36]. HuH6 cells are of the embryonal subtype. There are questions that the embryonal and fetal subtypes may not always respond similarly to interventions. To partially mitigate this limitation, we utilized a human hepatoblastoma patient-derived xenograft (PDX), COA67, which is from a mixed hepatoblastoma with both fetal and embryonal subtypes. COA67 has been previously shown by our lab to re-capitulate the histochemistry and genetics of the patient tumor from which it was derived [16].
Chiral agents are therapies which combine two enantiomers and are given as a mixture, examples of which include common chemotherapeutics such as leucovorin [37]. Recently, it’s been thought that an individual enantiomer of a chiral compound could have varying metabolisms, potencies, and bioavailability [37, 38]. For example, only the (6S)-leucovorin stereoisomer is active [37]. In the current study, the two PP2A activators, ATUX-3364 and ATUX-8385, are enantiomers. Although they were similarly effective in decreasing the malignant phenotype in vitro, there were some notable differences. For example, ATUX-3364 had a lower LD50 and IC50 and more of an effect on invasion than ATUX-8385. However, ATUX-8385 appeared to affect the expression of stemness markers and tumorsphere formation. The in vivo investigations also demonstrated differences in response between ATUX-3364 and −8385. We hypothesize that these differences may reflect differences in clearance rates by amine conjugation-elimination or rates of excretion of unchanged compound between the enantiomers [39]. There are a couple of other findings which may partially explain the differences seen between the two compounds in the in vivo studies. The immunoblotting findings showed that 3364 decreased CIP2A at lower concentrations than 8385. This decrease in the endogenous inhibitor of PP2A may contribute to the improved response in vivo. In addition, 3364 treatment resulted in slightly better PP2A activation compared to 8385, possibly resulting in a more profound effect on tumor growth.
In the pilot in vivo study, we investigated ATUX-3364 versus AUTX-8385 with 50 mg/kg bid which was the dose that had been tolerated in previous investigations [14]. Animals treated with 3364 had decreased tumor growth while those treated with 8385 showed minimal effect. Therefore, efforts were focused on 3364. Previous studies showed that tricyclic sulfonamide compounds were well tolerated by rodents in doses up to 100 mg/kg bid [13], prompting the use of a higher dose of 3364. Animals bearing HuH6 tumors and treated with 75 mg/kg bid had significantly decreased tumor growth compared to control animals from the pilot study. There were no changes in animal weights at the increased dosage which serves as a surrogate for the lack of potential dose-related toxicities.
Two endogenous inhibitors of PP2A, SET (I2PP2A) and CIP2A, have been studied as drivers of the suppression of PP2A in cancer [40–45]. The tricyclic sulfonamides used in the current study were designed to activate PP2A through direct binding of the alpha subunit and subsequent conformational change [7, 14, 39] and not to directly target SET or CIP2A. Both compounds did result in a significant increase in PP2A activation, but there was also some effect on CIP2A expression. These findings are like those seen previously. Activation of PP2A with FTY720 decreased expression of CIP2A in HuH6 cells without affecting expression of SET [9]. The same effects were seen with FTY720 treatment of medulloblastoma patient-derived xenografts [11]. In colorectal cancer, CIP2A expression decreased while there was no change in SET expression following FTY720 treatment [25]. Previous investigations with SMAPs in neuroblastoma demonstrated a more marked decrease of CIP2A expression compared to minimal change in expression of SET [14]. These data suggest that the changes in the endogenous inhibitors of PP2A may vary based on compound and cell line and defining the mechanism behind these changes and the significance will be the subject of future investigations, considering that a dual mechanism of action toward increasing PP2A activation may prove to be a beneficial attribute for the compound.
Targeting cancer stem cells has become a popular subject for cancer therapeutics, due to the belief that this population of cells, with their ability for self-renewal and resistance to therapy, may fuel disease relapse [46]. In this study, treatment with 3364 or 8385 resulted in a decrease of stemness markers in HuH6 cells as well as decreased tumorsphere formation in the COA67 cells. Other investigators used a novel PP2A activator, OSU-2S, in leukemia and found a decrease in stem and progenitor cell populations [47]. FTY720 decreased the breast cancer stemness markers, Nanog, Oct 3/4, and Sox2 [31]. A study by Stafman et al. showed that cisplatin-induced cancer cell stemness and cisplatin resistance in hepatoblastoma was abrogated following PP2A activation [9], suggesting that PP2A activation could decrease chemotherapeutic resistance. These findings provide an avenue for future studies to investigate synergy between PP2A activating molecules and current chemotherapeutics.
5.0. Conclusion
In this study, we examined two novel PP2A activators, ATUX-3364 and ATUX-8385, in hepatoblastoma and found that treatment decreased proliferation, viability, motility, and cancer cell stemness in the established hepatoblastoma cell line, HuH6, and human hepatoblastoma PDX, COA67. In addition, treatment with 3364 resulted in decreased tumor growth in vivo. These findings provide support for future investigation of PP2A activators as potential therapeutics for hepatoblastoma, and efforts should focus on continuing to develop PP2A activating compounds with improved bioavailability and stability.
Supplementary Material
Acknowledgements
We would like to thank Sagar Hanumanthu and UAB Comprehensive Flow Cytometry Core for his assistance with flow cytometry.
Funding
This project was made possible by funding from the National Cancer Institute of the National Institutes of Health under award numbers T32 CA229102 (LVB, JRJ), 5T32GM008361 (CHQ), P30 AR048311 and P30 AI027767 to the Flow Cytometry Core, and CA013148 to the UAB Genomics Core. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Other funding sources include Dixon grant (SCH), and Starr Fund-Vince Lombardi Cancer Foundation, Rally Foundation for Childhood Cancer Research, Destiny StrongER Foundation, Open Hands Overflowing Hearts, Sid Strong Foundation, Elaine Roberts Foundation, and Kaul Pediatric Research Foundation (EAB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
MO is the inventor of ATUX-3364 and ATUX-8385 and the CEO of Atux Iskay, LLC.
The other authors declare no competing financial interests in relation to the work described.
References
- 1.Feng J, Polychronidis G, Heger U, Frongia G, Mehrabi A, Hoffmann K. Incidence trends and survival prediction of hepatoblastoma in children: a population-based study. Cancer Communications 2019;39(1): 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kremer N, Walther AE, Tiao GM. Management of hepatoblastoma: an update. Curr Opin Pediatr 2014;26(3):362–9. [DOI] [PubMed] [Google Scholar]
- 3.Feng J, He Y, Wei L, Chen D, Yang H, Tan R, Chen Z. Assessment of Survival of Pediatric Patients With Hepatoblastoma Who Received Chemotherapy Following Liver Transplant or Liver Resection. JAMA Network Open 2019;2(10):e1912676–e1912676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zsiros J, Brugieres L, Brock P, Roebuck D, Maibach R, Zimmermann A, et al. Dose-dense cisplatin-based chemotherapy and surgery for children with high-risk hepatoblastoma (SIOPEL-4): a prospective, single-arm, feasibility study. Lancet Oncol 2013;14(9):834–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perilongo G, Shafford E, Maibach R, Aronson D, Brugières L, Brock P, et al. Risk-adapted treatment for childhood hepatoblastoma. final report of the second study of the International Society of Paediatric Oncology--SIOPEL 2. Eur J Cancer 2004;40(3):411–21. [DOI] [PubMed] [Google Scholar]
- 6.Chien W, Sun QY, Lee KL, Ding LW, Wuensche P, Torres-Fernandez LA, et al. Activation of protein phosphatase 2A tumor suppressor as potential treatment of pancreatic cancer. Mol Oncol 2015;9(4):889–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sangodkar J, Perl A, Tohme R, Kiselar J, Kastrinsky DB, Zaware N, et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J Clin Invest 2017;127(6):2081–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tohmé R, Izadmehr S, Gandhe S, Tabaro G, Vallabhaneni S, Thomas A, et al. Direct activation of PP2A for the treatment of tyrosine kinase inhibitor-resistant lung adenocarcinoma. JCI Insight 2019;4(4):e125693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stafman LL, Williams AP, Marayati R, Aye JM, Stewart JE, Mroczek-Musulman E, et al. PP2A activation alone and in combination with cisplatin decreases cell growth and tumor formation in human HuH6 hepatoblastoma cells. PLoS One 2019;14(4):e0214469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams AP, Garner EF, Waters AM, Stafman LL, Aye JM, Markert H, et al. Investigation of PP2A and Its Endogenous Inhibitors in Neuroblastoma Cell Survival and Tumor Growth. Transl Oncol 2019;12(1):84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garner EF, Williams AP, Stafman LL, Aye JM, Mroczek-Musulman E, Moore BP, et al. FTY720 decreases yumorigenesis in group 3 medulloblastoma patient-derived xenografts. Sci Rep 2018;8(1):6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oaks JJ, Santhanam R, Walker CJ, Roof S, Harb JG, Ferenchak G, et al. Antagonistic activities of the immunomodulator and PP2A-activating drug FTY720 (Fingolimod, Gilenya) in Jak2-driven hematologic malignancies. Blood 2013;122(11):1923–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merisaari J, Denisova OV, Doroszko M, Le Joncour V, Johansson P, Leenders WPJ, et al. Monotherapy efficacy of blood-brain barrier permeable small molecule reactivators of protein phosphatase 2A in glioblastoma. Brain Commun 2020;2(1):fcaa002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bownes LV, Marayati R, Quinn CH, Beierle AM, Hutchins SC, Julson JR, et al. Pre-clinical study evaluating novel protein phosphatase 2A activators as therapeutics for neuroblastoma. Cancers (Basel) 2022;14(8):1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillory LA, Stewart JE, Megison ML, Nabers HC, Mroczek-Musulman E, Beierle EA. FAK inhibition decreases hepatoblastoma survival both in vitro and in vivo. Transl Oncol 2013:6(2):206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stafman LL, Mruthyunjayappa S, Waters AM, Garner EF, Aye JM, Stewart JE, et al. Targeting PIM kinase as a therapeutic strategy in human hepatoblastoma. Oncotarget 2018:9(32):22665–22679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9(7):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bownes LV, Williams AP, Marayati R, Quinn CH, Hutchins SC, Stewart JE, et al. Serine-threonine kinase receptor-associated protein (STRAP) knockout decreases the malignant phenotype in neuroblastoma cell lines. Cancers (Basel) 2021;13(13):3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marayati R, Bownes LV, Stafman LL, Williams AP, Quinn CH, Atigadda V, et al. 9-cis-UAB30, a novel rexinoid agonist, decreases tumorigenicity and cancer cell stemness of human neuroblastoma patient-derived xenografts. Transl Oncol 2021;14(1):100893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winer J, Kung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem 1999;270(1):41–9. [DOI] [PubMed] [Google Scholar]
- 21.Fenwick N, Griffin G, Gauthier C. The welfare of animals used in science: how the “Three Rs” ethic guides improvements. Can Vet J 2009;50(5):523–30. [PMC free article] [PubMed] [Google Scholar]
- 22.Tang L, Zhang H, Zhang B. A note on error bars as a graphical representation of the variability of data in biomedical research: choosing between standard deviation and standard error of the mean. J Pancreatol 2019;2(3):69–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu CY, Huang TT, Chen YT, Chen JL, Chu PY, Huang CT, et al. Targeting SET to restore PP2A activity disrupts an oncogenic CIP2A-feedforward loop and impairs triple negative breast cancer progression. EBioMedicine 2019;40:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Wang SW, Zhang DS, Sun Y, Zhu CY, Fei Q, et al. FTY720-induced enhancement of autophagy protects cells from FTY720 cytotoxicity in colorectal cancer. Oncol Rep 2016;35(5):2833–42. [DOI] [PubMed] [Google Scholar]
- 25.Cristóbal I, Manso R, Rincón R, Caramés C, Senin C, Borrero A, et al. PP2A inhibition is a common event in colorectal cancer and its restoration using FTY720 shows promising therapeutic potential. Mol Cancer Ther 2014;13(4):938–47. [DOI] [PubMed] [Google Scholar]
- 26.Stuelten CH, Parent CA, Montell DJ. Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat Rev Cancer 2018;8(5):296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu L, Deng X. Suppression of cancer cell migration and invasion by protein phosphatase 2A through dephosphorylation of mu- and m-calpains. J Biol Chem 2006;281(46):35567–75. [DOI] [PubMed] [Google Scholar]
- 28.Zheng HY, Shen FJ, Tong YQ, Li Y. PP2A inhibits cervical cancer cell migration by dephosphorylation of p-JNK, p-p38 and the p-ERK/MAPK signaling pathway. Curr Med Sci 2018;38(1):115–123. [DOI] [PubMed] [Google Scholar]
- 29.Bahnassy AA, Fawzy M, El-Wakil M, Zekri ARN, Abdel-Sayed A, Sheta M. Aberrant expression of cancer stem cell markers (CD44, CD90, and CD133) contributes to disease progression and reduced survival in hepatoblastoma patients: 4-year survival data. Transl Res 2015;165(3):396–406. [DOI] [PubMed] [Google Scholar]
- 30.Enjoji S, Yabe R, Tsuji S, Yoshimura K, Kawasaki H, Sakurai M, et al. Stemness is enhanced in gastric cancer by a SET/PP2A/E2F1 axis. Mol Cancer Res 2018;16(3):554–563. [DOI] [PubMed] [Google Scholar]
- 31.Hirata N, Yamada S, Yanagida S, Ono A, Kanda Y. FTY720 Inhibits expansion of breast cancer stem cells via PP2A activation. Int J Mol Sci 2021;22(14):7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marayati R, Stafman LL, Williams AP, Bownes LV, Quinn CH, Aye JM, et al. PIM kinases mediate resistance to cisplatin chemotherapy in hepatoblastoma. Sci Rep 2021;11(1):5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.You L, Guo X, Huang Y. Correlation of cancer stem-cell markers OCT4, SOX2, and NANOG with clinicopathological features and prognosis in operative patients with rectal cancer. Yonsei Med J 2018;59(1):35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marayati R, Bownes LV, Quinn CH, Wadhwani N, Williams AP, Markert HR, et al. , Novel second-generation rexinoid induces growth arrest and reduces cancer cell stemness in human neuroblastoma patient-derived xenografts. J Pediatr Surg 2021;56(6):1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang F, Zhang J, Yang H. OCT4, SOX2, and NANOG positive expression correlates with poor differentiation, advanced disease stages, and worse overall survival in HER2(+) breast cancer patients. Onco Targets Ther 2018;11:7873–7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rikhi RR, Spady KK, Hoffman RI, Bateman MS, Bateman M, Howard LE. Hepatoblastoma: a need for cell lines and tissue banks to develop targeted drug therapies. Front Pediatr 2016;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wainer IW. Stereoisomers in clinical oncology: why it is important to know what the right and left hands are doing. Ann Oncol 1993;4 Suppl 2:7–13. [DOI] [PubMed] [Google Scholar]
- 38.Wainer IW, Granvil CP. Stereoselective separations of chiral anticancer drugs and their application to pharmacodynamic and pharmacokinetic studies. Ther Drug Monit 1993;15(6):570–5. [DOI] [PubMed] [Google Scholar]
- 39.Coelho MM, Fernandes C, Remião F, Tiritan ME., Enantioselectivity in drug pharmacokinetics and toxicity: pharmacological relevance and analytical methods. Molecules 2021;26(11):3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richard NP, Pippa R, Cleary MM, Puri A, Tibbitts D, Mahmood S, et al. , Combined targeting of SET and tyrosine kinases provides an effective therapeutic approach in human T-cell acute lymphoblastic leukemia. Oncotarget 2016;7(51):84214–84227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med 2013;5(1):105–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Switzer CH, Cheng RY, Vitek TM, Christensen DJ, Wink DA, Vitek MP. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene 2011;30(22):2504–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khanna A, Pimanda JE, Westermarck J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res 2013:73(22):6548–53. [DOI] [PubMed] [Google Scholar]
- 44.Junttila MR, Puustinen P, Niemelä M, Ahola R, Arnold H, Böttzauw T, et al. CIP2A inhibits PP2A in human malignancies. Cell 2007;130(1):51–62. [DOI] [PubMed] [Google Scholar]
- 45.Laine A, Nagelli SG, Farrington C, Butt U, Cvrljevic AN, Vainonen JP, et al. CIP2A Interacts with TopBP1 and Drives Basal-Like Breast Cancer Tumorigenesis. Cancer Res 2021;81(16):4319–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang N, Yang T, Huang Q, Yu P, Liu C, Chen L, et al. Mechanism of cancer stemness maintenance in human liver cancer. Cell Death Dis 2022;13(4):394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goswami S, Mani R, Nunes J, Chiang CL, Zapolnik K, Hu E, et al. PP2A is a therapeutically targetable driver of cell fate decisions via a c-Myc/p21 axis in human and murine acute myeloid leukemia. Blood 2022;139(9):1340–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.