

Summary
Most human proteins lack chemical probes, and several large-scale and generalizable small-molecule binding assays have been introduced to address this problem. How compounds discovered in such “binding-first” assays affect protein function, nonetheless, often remains unclear. Here, we describe a “function-first” proteomic strategy that uses size exclusion chromatography (SEC) to assess the global impact of electrophilic compounds on protein complexes in human cells. Integrating the SEC data with cysteine-directed activity-based protein profiling identifies changes in protein-protein interactions that are caused by site-specific liganding events, including the stereoselective engagement of cysteines in PSME1 and SF3B1 that disrupt the PA28 proteasome regulatory complex and stabilize a dynamic state of the spliceosome, respectively. Our findings thus show how multidimensional proteomic analysis of focused libraries of electrophilic compounds can expedite the discovery of chemical probes with site-specific functional effects on protein complexes in human cells.
Graphical Abstract
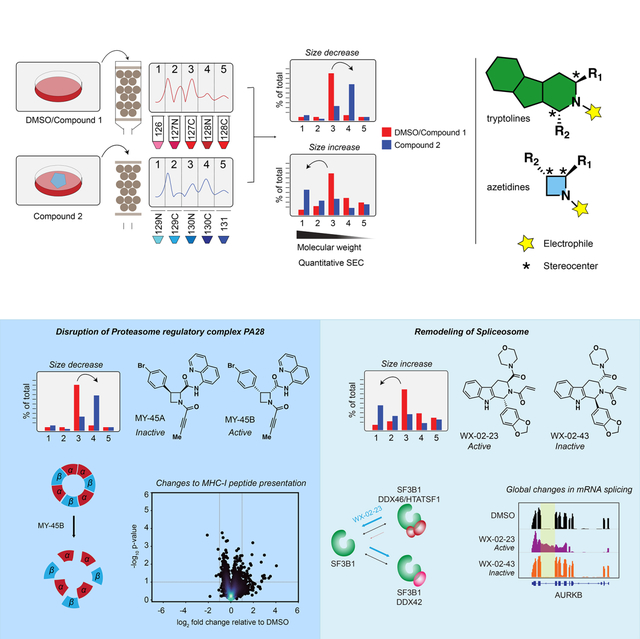
Lazear et al. present a “function-first” proteomic strategy for discovering chemical probes that alter protein complexes in human cells. The authors identify covalent ligands that stereoselectively disrupt the PA28 proteasomal regulatory complex and stabilize a dynamic state of the spliceosome to perturb MHC-I peptide presentation and mRNA splicing, respectively.
Introduction
Chemical probes are vital tools for perturbing proteins and pathways in biological systems and can serve as starting points for novel therapeutics. The discovery of chemical probes has historically relied on the advent of specific functional assays for proteins, which can present a major technical hurdle for proteins that lack readily monitorable biochemical activities. Efforts to expand the druggable proteome have begun to introduce complementary approaches for ligand discovery that leverage binding assays with near-universal applicability to diverse types of proteins.1 Technologies for the discovery of small-molecule binders of proteins include fragment-based screening,2,3 DNA-encoded libraries (DELs),4,5 and chemical proteomics.6–9 These methods have illuminated the broad small-molecule binding potential of proteins from structurally and functionally distinct classes. Nonetheless, whether and how small molecule-protein interactions emanating from “binding-first” assays impact the functions of proteins remain open and important questions – ones that are particularly challenging to address on a global scale when confronted with the divergent and specialized activities performed by proteins in the cell.
In considering ways to relate small-molecule binding events to functional outcomes, we looked to an emerging category of phenotypic assays that provide broad biochemical signatures of cell states. The impact of compounds on global gene and protein expression profiles of cells has, for instance, been assessed using DNA microarrays10,11 and mass spectrometry (MS)-based proteomics,12 respectively. These studies have, however, mostly evaluated compounds with known mechanisms of action, where gene/protein expression profiles serve to augment the understanding of established functional effects on proteins or to reveal potential off-target toxicities. When applied to naïve compounds that lack complementary protein-binding profiles, the problem of relating biochemical signatures to specific protein targets persists.
Here, we sought to determine whether the integration of two types of global profiling data – biochemical signatures and protein-binding – could facilitate the de novo discovery of chemical probes that produce functional effects in human cells. Aware that small molecules can perturb biochemical features beyond gene/protein expression, we also considered complementary “function-first” signatures of compound action – specifically, global readouts of protein complexation states in cells. Many proteins function as parts of larger homo- or heterotypic complexes,13,14 and small-molecule inhibitors or stabilizers of protein-protein interactions (PPIs) can serve as valuable chemical probes and therapeutic agents.15–17 Several approaches have been introduced for the large-scale mapping of PPIs, including genetic (e.g., yeast two-hybrid18) and biochemical (affinity purification19 or co-fractionation20–24 coupled with MS) methods. Among these options, we viewed co-fractionation-MS as most compatible with devising a streamlined and minimally biased platform for monitoring the effects of small molecules on PPIs in human cells.
Results
A proteomic platform to discover small molecules that alter protein complexes in cells
Chemical probes targeting PPIs can be challenging to discover from conventional compound libraries due to the extensive points of contact required to perturb large protein interfaces.15,25 This problem has been addressed by structure-guided approaches that facilitate the linking of weakly binding fragments into higher-affinity compounds that block PPIs.26 Covalent compounds offer an alternative and possibly more ligand-efficient strategy, where the permanent bonds formed with proteins may be sufficient to disrupt PPIs even at single points of contact.27–31 Previous chemical proteomic studies using cysteine-directed activity-based protein profiling (ABPP) have further demonstrated that electrophilic small molecule-sensitive, or ligandable, cysteines, are found on a wide array of proteins from different structural and functional classes, 6,27,32–34 suggesting that diverse types of protein complexes may be sensitive to covalent compound action.
We established a co-fractionation-MS protocol comparing five size exclusion chromatography (SEC) fractions from two cell-treatment conditions in a single tandem mass tagging (TMT35)-based proteomic analysis (Figure 1A). We found that proteins quantified from soluble lysates of the human prostate cancer cell line 22Rv1 spanned a wide range of molecular weights (Figure S1A>) and showed good correlation with data from previous co-fractionation MS experiments using a much larger number of SEC fractions (40 fractions)20 (Figure 1B). Likewise, we found that the majority of protein complexes (as defined by the CORUM Core Complex database13) identified in our experiments and previous SEC-MS studies20 displayed similar co-elution scores (Figure S1B and Dataset S1), and that the mean elution times for proteins were consistent both within replicate SEC-MS experiments performed on the same cell line and across experiments performed on distinct cell lines (Figure S1C and Dataset S1). On average, each SEC-MS experiment quantified ~3700 total proteins (minimum two unique quantified peptides/protein). We interpret these data to indicate that a five-fraction SEC-MS protocol exhibited sufficient resolution and sensitivity to evaluate the effects of electrophilic compounds on a diverse protein complexes in human cells.
Figure 1.
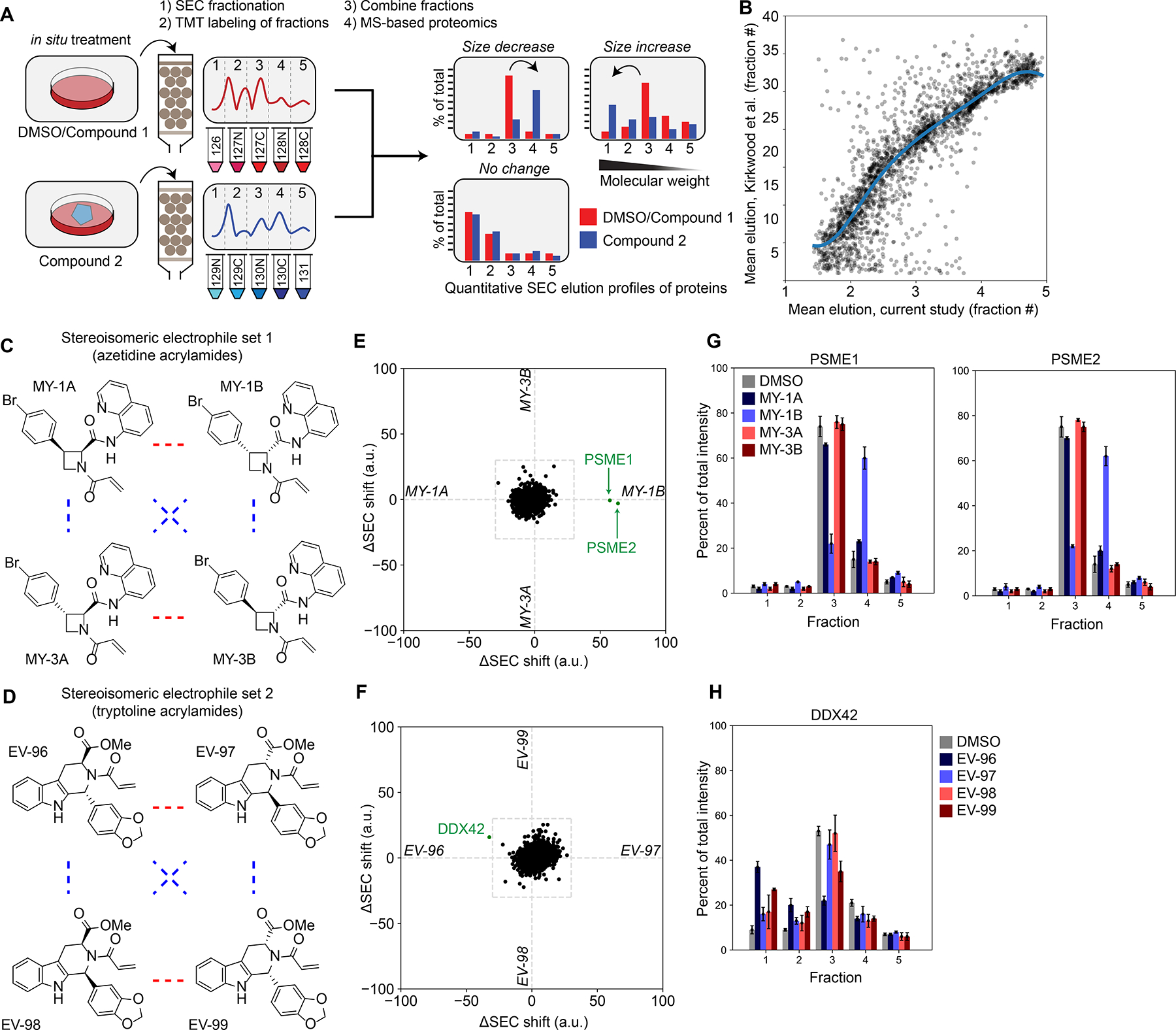
A proteomic platform to discover small-molecule modulators of protein-protein interactions in human cells. A. An SEC-MS proteomic screen to determine the effects of electrophilic compounds on protein complexes in human cells (see Methods section for more details).
B. Comparison of the mean elution times of proteins from SEC-MS experiments performed in this study (x-axis) versus previously20 (y-axis). Each dot represents a protein detected in both experiments. Data are mean elution times (weighted average) from n = 2–11 independent experiments. Support vector regression line displayed in blue.
C, D. Structures for the azetidine (C) and tryptoline (D) acrylamide sets of stereoisomeric electrophilic compounds. Red lines represent enantiomers and blue lines correspond to diastereomers.
E, F. Protein size shift scores (arbitrary units, a.u.) plotted for SEC-MS experiments performed with proteomes from 22Rv1 cells treated with the indicated compounds (20 μM, 3 h). X-axis represents comparison of protein size shifts caused by MY-1A and MY-1B (E) or EV-98 and EV-99 (F) (in each case, difference in SEC shifts from DMSO vs. compound, see Equation 4 in Methods). Y-axis represents comparison of protein size shifts caused by MY-3A and MY-3B (E) or EV-96 and EV-97 (F). Data are average values from n = 2–11 independent experiments.
G. SEC elution profiles for PSME1 and PSME2 from 22Rv1 cells treated with azetidine acrylamides (20 μM, 3 h). Data are average values ± SEM from n = 2–11 independent experiments.
H. SEC elution profile for DDX42 from 22Rv1 cells treated with tryptoline acrylamides (20 μM, 3 h). Data are average values ± SEM from n = 2–11 independent experiments.
We exposed 22Rv1 cells to two structurally distinct sets of four stereoisomeric electrophilic compounds (azetidine acrylamides36 (Figure 1C) and tryptoline acrylamides32 (Figure 1D); 20 μM, 3 h), which were constructed based on principles of diversity-oriented synthesis (DOS)37 and found previously to stereoselectively engage cysteine residues on diverse classes of proteins in human T-cells32,36. Here, we sought to identify proteins showing stereoselective shifts in SEC migration (size shifts), which could then be correlated with stereoselective changes in cysteine reactivity. We quantified stereoselective size shift scores by calculating the Euclidean distance between individual protein elution profiles across the five SEC fractions collected from cells treated with enantiomeric compound pairs, where a score of >30 was considered of potential interest (see Methods). An additional comparison to DMSO-treated cells enabled determination of which of the two enantiomeric compounds caused the observed protein size shift(s). The migration profiles and abundances of most proteins were not affected in cells treated with enantiomeric pairs of electrophilic compounds (Figure S1D, E and Dataset S1). However, stereoselective size shifts were observed for individual proteins in cells treated with the azetidine acrylamide MY-1B (2) (Figure 1E) and the tryptoline acrylamide EV-96 (5) (Figure 1F).
MY-1B caused two proteins – PSME1 and PSME2 – to shift from a higher molecular weight (MW) fraction 3 to a lower MW fraction 4 (Figure 1E, G). PSME1 and PSME2 form the heptameric PA28 proteasomal regulatory complex38,39 (Figure S1F) that modulates antigenic peptide processing by the proteasome.40–42 The coordinated size shifts observed for PSME1 and PSME2 suggested that MY-1B may disrupt the PA28 complex. In contrast, subunits of the 20S core proteasomal complex were unaffected in SEC migration by MY-1B (Figure S1G). The major stereoselective effect of tryptoline acrylamides was an EV-96-induced size shift for the RNA helicase DDX42 from a lower MW fraction 3 to a higher MW fraction 1 (Figures 1F and 1H), suggesting that EV-96 may promote the assembly of DDX42 into a larger protein complex.
Covalent ligands disrupt the PA28 complex by engaging C22 of PSME1
To determine the mechanistic basis for changes in protein migration caused by the stereoisomeric electrophilic compounds, we performed cysteine-directed ABPP, initially focusing on the azetidine acrylamides. Proteomes from DMSO- or compound-treated cells (20 μM compound, 1 h) were exposed to the broad-spectrum cysteine-reactive probe iodoacetamide-desthiobiotin (IA-DTB), and probe-labeled cysteines enriched and quantified by multiplexed (TMT) MS analysis.32 Cysteines showing a substantial loss (> 60%) in IA-DTB labeling in cells treated with one or more of the azetidine acrylamides were considered to have been covalently engaged or liganded. Of ~8,800 quantified cysteines, 43 were liganded by MY-1B, and a much smaller subset (seven cysteines) were stereoselectively engaged by this compound (Figures 2A, B, and S2A and Dataset S1). Among the cysteines stereoselectively liganded by MY-1B was PSME1_C22 (Figure 2A), which resides near the PSME1-PSME2 interface (Figure 2C). One additional cysteine was quantified in PSME1 (C106), as well as one cysteine in PSME2 (C91), and these cysteines both exhibited stereoselective increases in IA-DTB reactivity in MY-1B-treated cells (Figure 2D). PSME1_C106 and PSME2_C91 are also located near the PSME1-PSME2 interface (Figure 2C), suggesting that these residues may become more solvent accessible and IA-DTB-reactive following MY-1B engagement of PSME1_C22. MY-1B produced a similar profile of stereoselective PSME1/2 cysteine reactivity changes in another human cancer cell line (Ramos cells; Dataset S1), and we further observed robust stereoselective enrichment of PSME1 by an alkyne analogue of MY-1B (MY-11B) compared to the enantiomer (MY-11A) (Figure 2E, F and Dataset S1). Finally, we also performed cysteine-directed ABPP experiments with the tryptoline acrylamides, but we did not detect EV-96-sensitive cysteines in DDX42 (Figure S2B and Dataset S1). We will return to the mechanistic characterization of EV-96 below.
Figure 2.
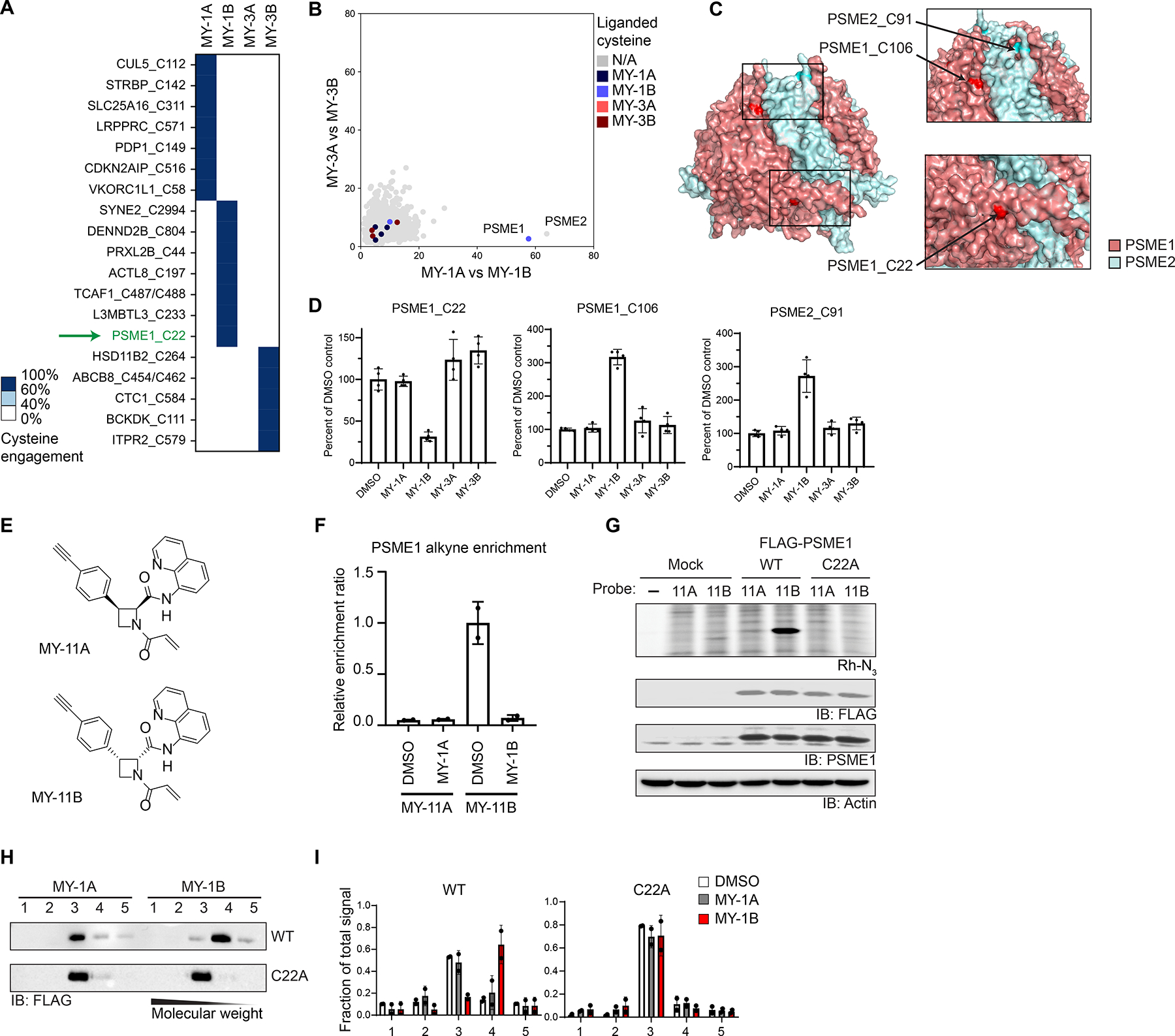
Electrophilic compounds disrupt the PA28 complex by engaging C22 of PSME1. A. Heatmap showing cysteines stereoselectively liganded by azetidine acrylamides in 22Rv1 cells (20 μM compound, 1 h) as determined by cysteine-directed ABPP. Cysteines were considered stereoselectively liganded if they showed > 60% reduction in IA-DTB labeling by one stereoisomeric compound and < 40% reduction for the other three stereoisomeric compounds. Data are average values from n = 4 independent experiments.
B. Graph showing size shifts of proteins in pairwise comparisons of SEC profiles for 22Rv1 cells treated with azetidine acrylamide enantiomers (MY-1A vs MY-1B, x-axis; MY-3A vs MY-3B; y-axis; reanalysis of data from Figure 1E), where proteins with stereoselectively liganded cysteines are color-coded.
C. Crystal structure of PSME1 and PSME2 complex (PDB: 7DRW).
D. Reactivity of cysteines in PSME1 and PSME2 quantified in cysteine-directed ABPP experiments.
E. Structures of alkynylated azetidine acrylamide probes MY-11A (inactive) and MY-11B (active).
F. Quantification of stereoselective enrichment of PSME1 by MY-11B (5 μM, 1 h) compared to MY-11A (5 μM, 1 h) and blockade of enrichment by MY-1A and MY-1B (20 μM, 2 h pretreatment) in Ramos cells. Data are average values ± SD normalized to MY-11B treatment group, n = 2 independent experiments.
G. MY-11B, but not MY-11A (2. 5 μM 30 min), reacts with recombinantly expressed WT-PSME1, but not a C22A-PSME1 mutant expressed in HEK293T cells as determined by gel-ABPP. Top image, ABPP data (top image); bottom images, western blots. Results are from a single experiment representative of two independent experiments.
H. Western blot analysis of SEC profiles for recombinant WT and C22A-PSME1 expressed in 22Rv1 cells treated with MY-1A or MY-1B (10 μM, 3 h) prior to analysis by SEC.
I. Quantification of data shown in panel H. Data are average values ± SD from n = 2 independent experiments.
We confirmed by gel-ABPP that MY-11B stereoselectively reacted with recombinantly expressed WT-PSME1, but not a C22A-PSME1 mutant (Figures 2G and S2C). Recombinant PSME1 has been shown to form homooligomeric structures in the absence of PSME2,38 and we accordingly found that recombinant PSME1 exhibited a similar SEC elution profile to endogenous PSME1 (Figure S2D). Treatment of cells with MY-1B, but not MY-1A, caused a clear size shift for recombinant WT-PSME1 to fraction 4, while the C22A-PSME1 mutant was unaffected (Figures 2H, I). These data thus support that MY-1B disrupts PSME1-mediated PPIs by specifically engaging C22.
Previous studies have shown that substituting an acrylamide with a less reactive butynamide electrophile can improve the selectivity of covalent ligands.43 We found here that the butynamide analogue of MY-1B – MY-45B (Figure 3A) – but not its enantiomer MY-45A (Figure 3A), engaged recombinant and endogenous PSME1_C22 with greater potency (IC50 value of ~0.4 μM) and selectivity compared to MY-1B (Figures 3B–E and S2E, F and Dataset S1). Across > 10,000 quantified cysteines in human 22Rv1 cells, MY-45B (5 μM, 3 h) substantially engaged seven cysteines, only two of which stereoselectively reacted with MY-45B compared to MY-45A – C22 of PSME1 and C258 of the helicase DDX49 (Figure 3D, E), a protein that has not been implicated in proteasome regulation. MY-45B, but not MY-45A, caused the expected size shifts for endogenous PSME1 and PSME2 (Figures 3F and S2G), as well as recombinant WT-PSME1 (Figure S2H).
Figure 3.
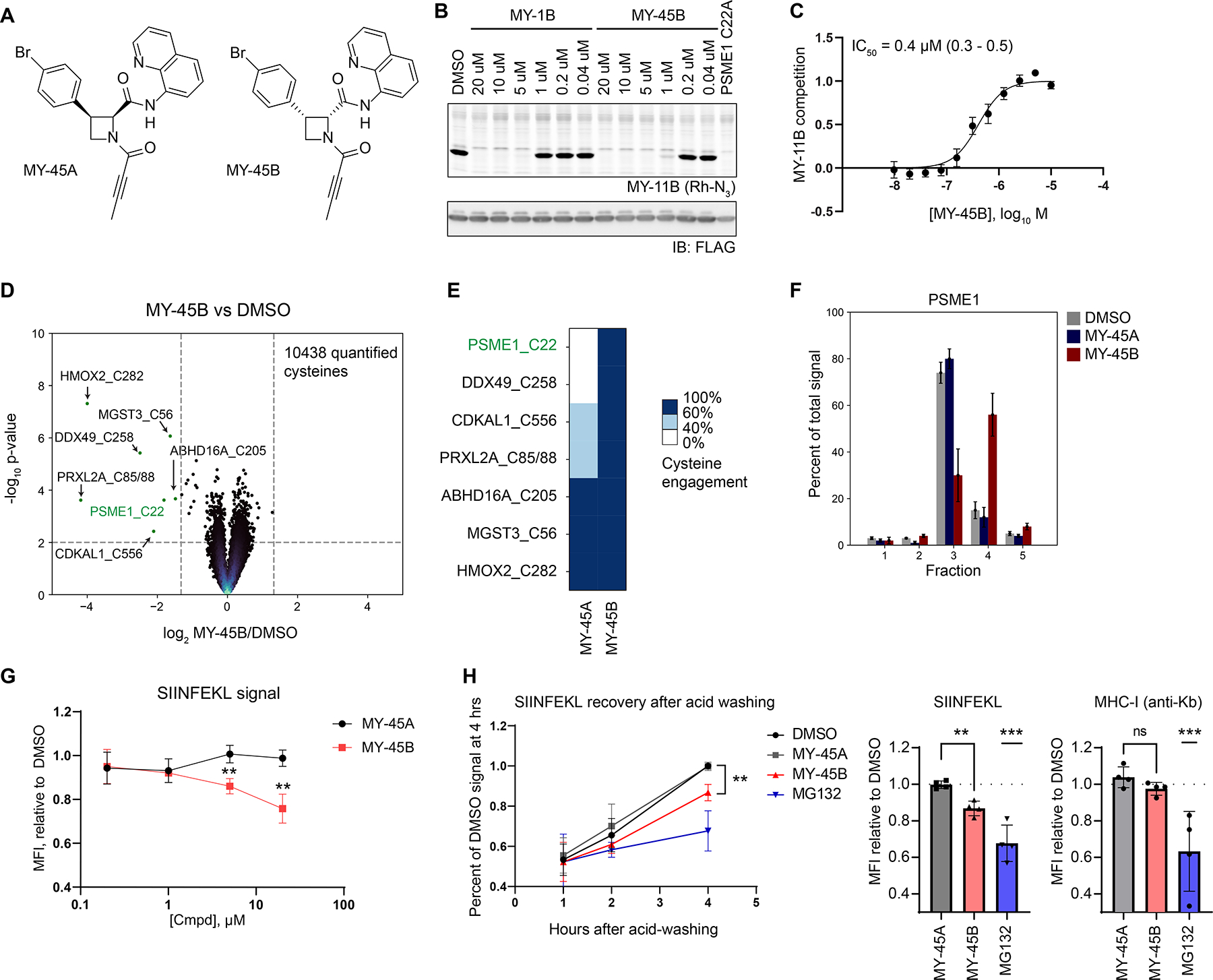
Covalent ligands targeting PSME1_C22 functionally impair MHC-I antigenic peptide presentation. A. Structures for azetidine butynamides MY-45A (inactive), and MY-45B (active).
B. Comparison of potency of engagement of PSME1 by MY-1B and MY-45B (2 h, pretreatment before addition of MY-11B (2.5 μM, 30 min) as determined by gel-ABPP. Results are from a single experiment representative of two independent experiments.
C. Concentration-dependent engagement of PSME1 by MY-45B as determined by gel-ABPP (see panel B). Data are average values ± SEM from n = 6 independent experiments, IC50 and 95% CI (confidence intervals) are listed.
D. Volcano plot showing cysteines substantially (> 60% reduction in IA-DTB labeling) and significantly (p value < 0.01) liganded by MY-45B (5 μM, 3 h) in 22Rv1 cells as determined by cysteine-directed ABPP. Data are average values from n = 4 independent experiments.
E. Heatmap displaying MY-45B-liganded cysteines (from panel D) and their reactivity with enantiomer MY-45A.
F. SEC-MS elution profile for endogenous PSME1 in 22Rv1 cells treated with DMSO, MY-45A, or MY-45B (20 μM, 3 h). Data are average values ± SEM from n = 2–11 independent experiments.
G. Concentration-dependent effects of MY-45B on MHC-I antigen presentation. Mouse T lymphoma cells expressing chicken ovalbumin (E.G7-Ova) were treated with DMSO or MY-45A or MY-45B for 4 h, subject to mild acid elution of MHC I-bound peptides, recovered for 4 h, and analyzed for SIINFEKL peptide presentation by FACS (MFI, mean fluorescence intensity). Data are average values ± SD from n = 3 independent experiments. **, p < 0.01 compared to MY-45A treatment.
H. Time-dependent effects of MY-45B on MHC-I antigen presentation. Experiments performed as described in panel G; MG132 (10 μM). MFI for SIINFEKL peptide (left panel) and overall MHC-I (right panel, measured at 4 h post-acid wash). Data are average values ± SD from n = 4 independent experiments. **, p < 0.01, *** p < 0.001 compared to MY-45A treatment.
The PA28 complex has been found to impact MHC class I (MHC-I) presentation of a select, but incompletely determined, set of peptides,41,44 including the chicken ovalbumin peptide SIINFEKL.41 Using a kinetic assay that measures the rate of recovery of MHC-I peptide presentation following acid washing45,46 (Figures S3A, B), we found that mouse T lymphoma cells constitutively expressing chicken ovalbumin (E.G7-Ova) showed a time- and concentration-dependent reduction in SIINFEKL presentation – but not overall MHC-I surface expression – following treatment with MY-45B, but not MY-45A (Figures 3G, H), and these data correlated with stereoselective engagement of mouse PSME1_C22 by MY-45B (Figure S3C and Dataset S1). In contrast, the direct proteasome inhibitor MG132 suppressed both SIINFEKL peptide and MHC-I presentation (Figure 3H).
These results, taken together, indicated that azetidine acrylamides and butynamides stereoselectively disrupt the structure and function of the PA28 complex by engaging PSME1_C22. We next asked how these covalent PSME1 ligands more globally impact MHC-I peptide presentation.
PSME1 disruption modulates MHC-I-peptide interactions in human leukemia cells
We first generated cell models genetically disrupted for PSME1 or PSME2 using CRISPR/Cas9 in the human leukemia cell line KBM7 (Figure 4A). Genetic disruption of either PSME1 or PSME2 led to loss of both proteins (Figure 4A), suggesting that the proteins depended on one another for stability. MS-based proteomics of sgControl versus sgPSME1 cells, as well as cells treated with MY-45B or MY-45A, revealed few changes in protein abundance (Figure S3D), indicating that genetic or chemical disruption of the PA28 complex did not perturb the general protein-degrading function of the proteasome. A dramatic decrease (> 90%) in signals for the tryptic peptide containing PSME1_C22 (R.EDLCTK) was observed, however, in this proteomic experiment for MY-45B-, but not MY-45A-treated cells (Figure S3E), thus confirming site-specific and stereoselective engagement of PSME1_C22 by MY-45B.
Figure 4.
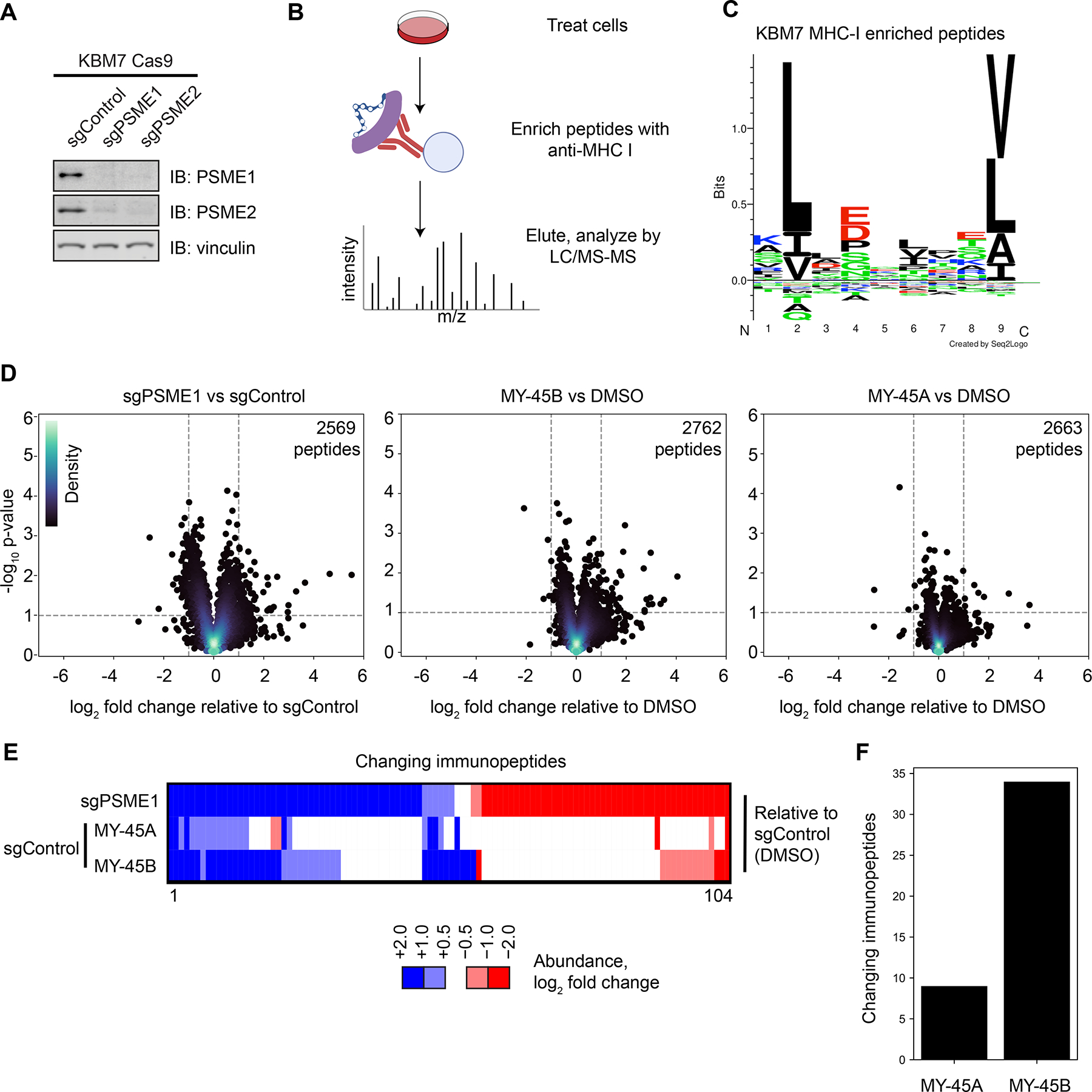
Chemical or genetic perturbation of PSME1 modulates MHC-I-immunopeptide interactions in human leukemia cells. A. Western blots of PSME1 and PSME2 in KBM7 CRISPR/Cas9 control (sgAAVS1), PSME1 (sgPSME1), or PSME2 (sgPSME2) cell lines. Results are from a single experiment representative of two independent experiments.
B. Cartoon schematic for anti-MHC class I immunopeptidomics protocol. After compound treatment, cells are lysed, and MHC-I bound peptides immunoprecipitated with anti-MHC-I antibody, eluted, and analyzed by LC-MS.
C. Motif analysis of peptides enriched by MHC-I co-immunoprecipitation from KBM7 cells.
D. Volcano plots showing substantially (> two-fold increase or decrease) and significantly (p value < 0.05) changing MHC-I bound immunopeptides in sgPSME1 vs sgControl (sgAAVS1) cells (left), DMSO- vs MY-45B-treated sgControl cells (middle), or DMSO- vs MY-45A-treated sgControl cells (right) (10 μM compound, 8 h). Data are average values from n= 3–4 independent experiments.
E. Heatmap of MHC-I-bound immunopeptides that are substantially and significantly changing in at least one comparison group (sgPSME1 vs sgControl; MY-45A- vs DMSO-treated sgControl; or MY-45B- vs DMSO-treated sgControl) as defined in panel D.
F. Bar graph showing the number of MHC-I-bound immunopeptides that are substantially and significantly changing in MY-45A- vs DMSO-treated sgControl or MY-45B- vs DMSO-treated sgControl cells as defined in panel D.
We next evaluated the antigenic peptide profiles of sgPSME1 cells compared to sgControl cells, as well as sgControl cells treated with MY-45B (10 μM, 8 h) compared to MY-45A- or DMSO-treated cells, following an established protocol for the affinity enrichment and LC-MS analysis of MHC-I-bound peptides47 (Figure 4B). Motif analysis of the eluted immunopeptides verified a consensus motif for the KBM7 HLA haplotype HLA-A*02:0148 (Figure 4C). sgPSME1 cells exhibited substantial (log2 fold change > 1) and significant (p < 0.05) changes in ~100 of > 2,500 quantified immunopeptides, which reflected both decreases and increases in co-enrichment with MHC-I (Figure 4D, E). A smaller subset of MHC-I-associated peptides were altered by MY-45B (34 peptides) in sgControl cells (Figure 4D–F), and these peptides showed a striking (~70%) overlap with the peptides altered in sgPSME1 cells (Figure 4D–F). In contrast, the inactive enantiomer MY-45A caused far fewer changes in the antigenic peptide profile of KBM7 cells (< 10 peptide changes; Figure 4D–F). These findings, taken together, indicate that the acute, pharmacological disruption of the PA28 by covalent ligands engaging C22 of PSME1 produces discrete changes in MHC-I-associated peptides that are largely consistent with those caused by chronic genetic loss of PSME1/PSME2.
Tryptoline acrylamides stereoselectively alter spliceosome composition and function
Returning to our finding that EV-96 promotes a stereoselective shift of DDX42 to a higher MW form despite not apparently engaging any cysteines in this protein (Figure 1F, H), we gathered additional clues pertaining to mechanism of action by quantitative proteomics, which revealed a striking, stereoselective reduction in several proteins in EV-96-treated 22Rv1 cells (Figure 5A). These changing proteins were enriched in cell division and cell cycle functions (Figure 5B), suggesting that EV-96 may affect cancer cell proliferation. Consistent with this hypothesis, we found that EV-96 caused a stereoselective blockade in the growth of 22Rv1 cells (Figure 5C) and other human cancer cell lines (Figure S4A). The EV-96-induced protein changes minimally overlapped with a “frequent responder” group of proteins previously shown to represent common changes caused by diverse cytotoxic compounds12 (Figure S4B), suggesting a discrete mechanism of anti-proliferative action for EV-96.
Figure 5.
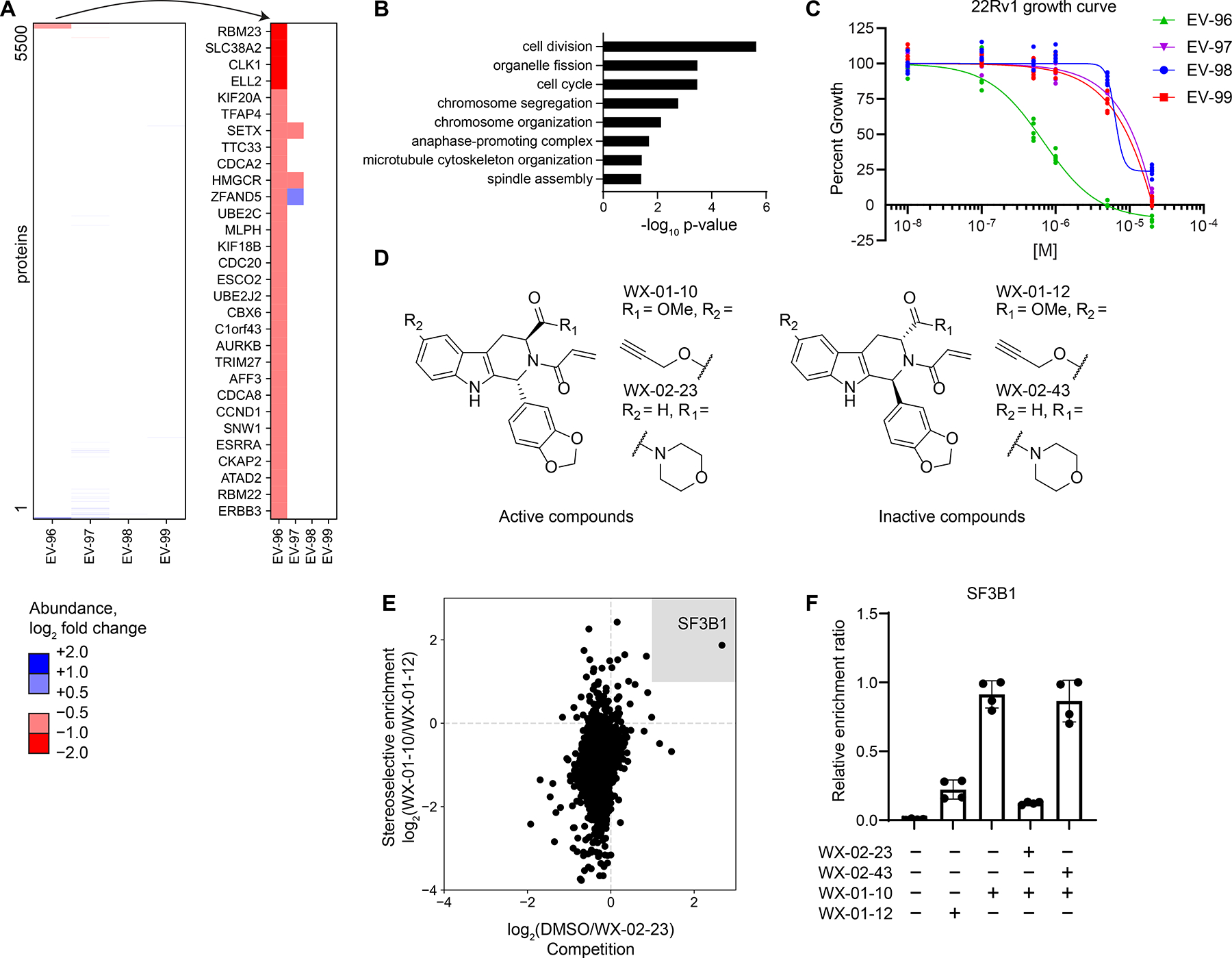
Tryptoline acrylamides that stereoselectively engage SF3B1 alter protein abundances and block the proliferation of cancer cells. A. Left panel, Heatmap of protein abundance changes in 22Rv1 cells treated with tryptoline acrylamides (20μM, 8 h). Right panel, Blow up of heatmap showing proteins with > 33% decreases in abundance in 22Rv1 cells treated with EV-96. Data are average values from n = 4–6 independent experiments.
B. Gene ontology enrichment for proteins stereoselectively decreased in abundance by EV-96 (panel A).
C. Cell growth effects of tryptoline acrylamides. Cells were treated with compounds for 72 h prior to CellTiter-Glo measurement. Data are relative to DMSO control from n = 6 independent experiments.
D. Structures of alkyne (WX-01-10 and WX-01-12) and morpholino amide (WX-02-23 and WX-02-43) analogues of EV-96 and EV-97.
E. Chemical proteomic identification of SF3B1 as a protein that is stereoselectively enriched by WX-01-10 and stereoselectively competed in enrichment by WX-02-23. X-axis: log2 competition ratio values for proteins enriched by alkyne probe WX-01-10 (10 μM, 1 h) in 22Rv1 cells pretreated with DMSO or WX-02-23 (5 μM, 2 h pretreatment) as a competitor. Y-axis: log2 enrichment ratio values for proteins treated with active alkyne probe WX-01-10 vs inactive probe WX-01-12 (10 μM, 1 h). Data are average values from n = 4 independent experiments (See also Figure S4I for schematic of this experiment).
F. Quantification of stereoselective enrichment and competition of SF3B1 by active alkyne probe (WX-01-10) and competitor (WX-02-23) vs inactive enantiomer alkyne probe (WX-01-12) and inactive enantiomer competitor (WX-02-43). Data are average values ± SD normalized to WX-01-10 treatment group for n = 4 independent experiments.
We found that a non-electrophilic propanamide analogue of EV-96 did not impair cancer cell growth (Figures S4C and S4D), supporting a covalent mode of action for the compound. Recognizing that some electrophilic compound-sensitive cysteines may evade detection by cysteine-directed ABPP if, for instance, they reside on non-proteotypic peptides,49 we adopted a complementary chemical proteomic strategy to identify proteins stereoselectively engaged by EV-96 using an alkyne analogue WX-01-10, which maintained stereoselective cell growth inhibition compared to its enantiomer WX-01-12 (Figures 5D and S4E). We concurrently discovered that a morpholine amide analogue of EV-96 – WX-02-23 – exhibited ~4-fold greater antiproliferative activity (IC50 of 170 nM) with preserved stereoselectivity compared to its enantiomer WX-02-43 (Figures 5D and S4F–H). We then performed MS-based proteomic experiments involving the pre-treatment of cancer cells with WX-02-23 or WX-02-43 (or DMSO), followed by treatment with WX-01-10 or WX-01-12, where a protein target responsible for the observed anti-proliferative effect should display: i) stereoselective enrichment by WX-01-10 (in comparison to WX-01-12); and ii) competition in its enrichment by WX-02-23, but not WX-02-43 (Figure S4I). Only a single protein – the spliceosome factor SF3B1 – was found to meet these criteria (Figures 5E, F and Dataset S1). We also found that SF3B1 enrichment by WX-01-10 was blocked by pretreatment with EV-96, but not EV-97 (Figure S4J and Dataset S1).
SF3B1 is an ~150 kDa component of the spliceosome involved in stabilizing the branch point adenosine prior to intron removal.50 Consistent with SF3B1 being a direct, stereoselective target of tryptoline acrylamides, gel-ABPP identified an ~150 kDa protein in 22Rv1 cells that reacted with WX-01-10, but not WX-01-12, and this interaction was blocked by WX-02-23, but not WX-02-43 (Figures 6A and S5A). WX-01-10 labeling of the 150 kDa protein was also blocked by pre-treatment with pladienolide B (Figure 6A and S5A), a natural product modulator of SF3B1.51, 52 WX-02-23 and pladienolide B both induced expression of p27 (Figure 6A), a previously described feature of spliceosome modulators.53
Figure 6.
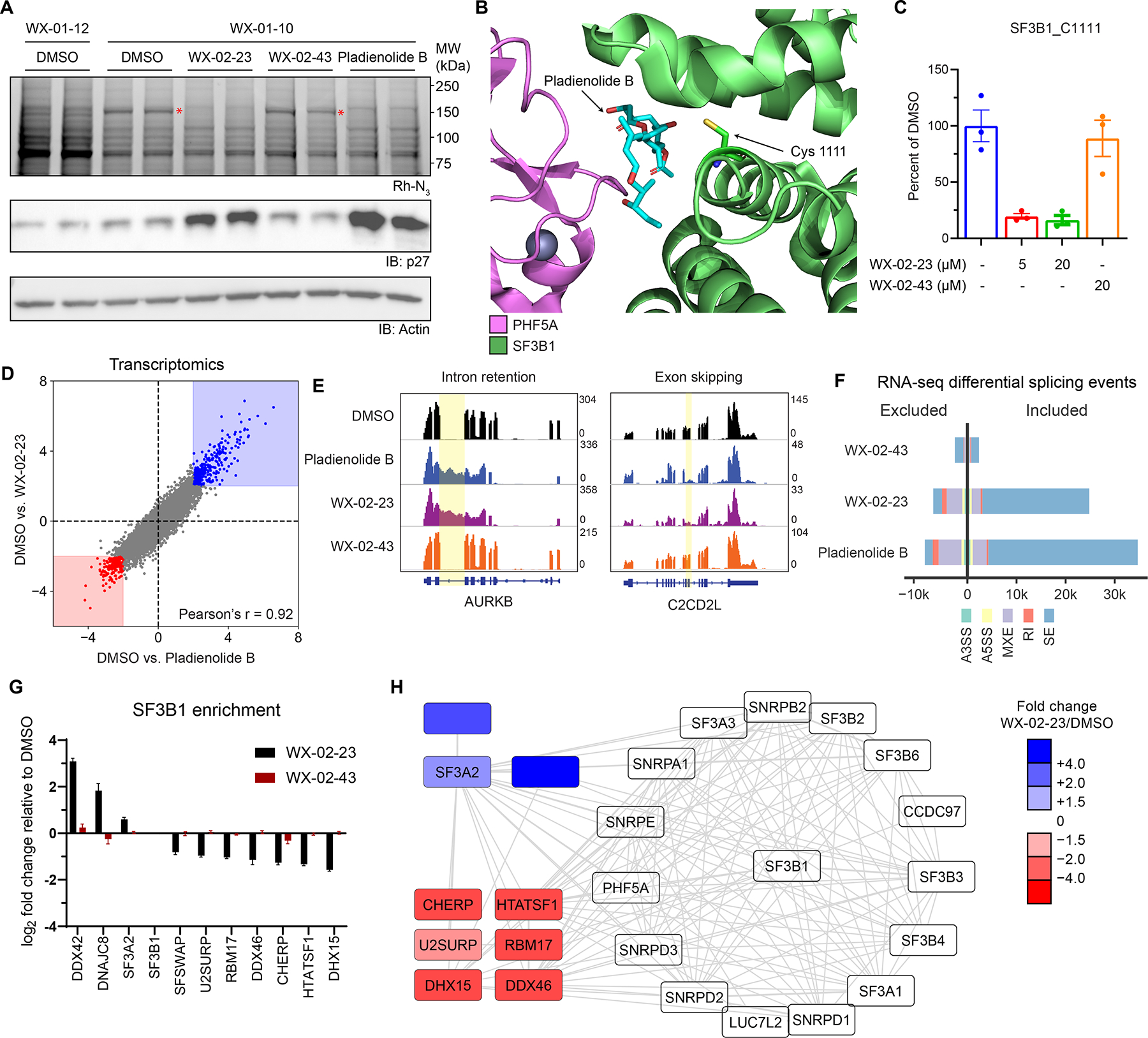
Tryptoline acrylamides engage C1111 of SF3B1 and stereoselectively modulate spliceosome structure and function. A. Stereoselective labeling of a 150 kDa protein (red asterisk) in 22Rv1 cells as determined by gel-ABPP. Top panel, Gel-ABPP, where cells were pre-treated with DMSO, WX-02-23 (1 μM), WX-02-43 (1 μM) or pladienolide B (10 nM) for 24 h followed by treatment with WX-01-10 or WX-01-12 (1 μM, 1 h). Lower panels, western blots. Results are from a single experiment representative of two independent experiments. B. Crystal structure of SF3B1-PHF5A complex bound to pladienolide B, highlighting the location of C1111 (PDB: 6EN4).
C. Quantification of stereoselective engagement of SF3B1_C1111 by WX-02-23 as measured by targeted cysteine-directed ABPP of 22Rv1 cells treated with 5 or 20 μM compound (3 h). Data are average values ± SD from n = 2–3 independent experiments.
D. Scatter plot of mRNA abundance changes in 22Rv1 cells treated with WX-02-23 (5 μM), pladienolide B (10 nM), or DMSO for 8 h. RNA-seq data are average values shown as log2 fold change relative to DMSO for n = 3 independent experiments.
E. Examples of intron retention in AURKB and exon skipping in C2CD2L caused by pladienolide B and WX-02-23 in 22Rv1 cells.
F. Summary of alternative splicing events caused by pladienolide B (10 nM), WX-02-23 (5 μM), and inactive enantiomer WX-02-43 (5 μM) in 22Rv1 cells (8 h) compared to DMSO treatment, as identified with rMATS by threshold of |PSI| > 0.1 and FDR < 0.05. Data represent values from three independent experiments.
G. Differential co-immunoprecipitation of proteins with SF3B1 (> 1.5 fold increase or decrease) in HEK293T cells treated with DMSO, WX-02-23, or WX-02-43 (5 μM, 3 h). Co-immunoprecipitated performed with anti-SF3B1 antibody (CST #14434). Data are average log2 fold changes ± SD from n = 4–7 independent experiments. See Dataset S1 for co-immunoprecipitation data from cells treated with pladienolide B (10 nM, 3 h).
H. Interactome map from STRING database filtered for proteins identified as SF3B1 interactors in the co-immunoprecipitation experiments from panel I. Data are average log2 fold changes from n = 4-7 independent experiments.
The co-crystal structure of a pladienolide B-SF3B1 complex has confirmed that this interaction is reversible involving contacts with both SF3B1 and PHF5A.54 Review of this structure identified a single SF3B1 cysteine (C1111) in the pladienolide B binding pocket as a candidate for covalent modification by WX-02-23 (Figure 6B). SF3B1_C1111 has rarely been quantified by cysteine-directed ABPP,6,27,32 suggesting this cysteine may reside on a non-proteotypic tryptic peptide. Consistent with this hypothesis, we found that the IA-DTB adduct of a tryptic peptide containing C1111 eluted at the tail end of our standard liquid chromatography gradient (~38% acetonitrile) (Figure S5B). Using a targeted proteomic assay, we found that WX-02-23 stereoselectively blocked IA-DTB reaction with C1111 (Figures 6C and S5C), but did not affect the IA-DTB reactivity of other cysteines in SF3B1 (Figure S5D–F). Finally, our cysteine-directed ABPP data also confirmed that EV-96 and WX-02-23 did not engage PHF5A_C26 (Dataset S1), which was recently found to be targeted by spliceostatin A, another natural product modulator of the spliceosome.53,55
WX-02-23 and pladienolide B caused similar changes to the transcriptomes (Figure 6D) and proteomes (Figure S6A) of cancer cells, and these changes were not observed with WX-02-43 (Figure S6B). WX-02-23 and pladienolide B also showed modestly greater anti-proliferative effects in Panc 05.04 cells expressing the cancer-associated K700E-SF3B1 mutant compared to Panc 04.03 cells expressing WT-SF3B156 (Figure S6C), which is consistent with previous findings with pladienolide B analogues.57 WX-02-23 and pladienolide B further altered mRNA splicing in similar ways, including the induction of both exon skipping and, to a lesser extent, intron retention events (Figures 6E, F and S6D). No such splicing effects were observed with WX-02-43 (Figure 6E, F and S6D). We finally found that 22Rv1 cells stably overexpressing Y36C-PHF5A, a point mutant that has been found to confer resistance to the anti-proliferative activity of pladienolide B,58 also protected against the growth inhibitory effects of WX-02-23 (Figure S6E). SF3B1 reactivity with the alkyne probe WX-01-10 was additionally disrupted in 22Rv1 cells expressing Y36C-PHF5A (Figure S6F), and modeling studies also supported the importance of PHF5A_Y36 in promoting interactions with WX-02-23 (Figure S6G).
We interpret these data, taken together, as strong evidence that WX-02-23 and related tryptoline acrylamides produce their anti-proliferative effects through covalent modification of SF3B1, which in turn perturbs spliceosome function in a manner that resembles the effects of structurally unrelated natural product modulators of the spliceosome such as pladienolide B.
Covalent SF3B1 ligands stabilize a spliceosome state with enhanced binding to DDX42
Curious to understand whether and how covalent modification of SF3B1 might relate to the SEC migration change in DDX42 caused by tryptoline acrylamides, we noted some literature precedence for DDX42 (or SF3b125) physically associating with the spliceosome.59 Interestingly, we found that fraction 1, to which DDX42 shifted in EV-96 or WX-02-23-treated cells, also contained other spliceosome components, including SF3B1 (Figure S7A), suggesting that SF3B1 ligands may promote DDX42 binding to the spliceosome. Consistent with this hypothesis, immunoprecipitation (IP)-MS proteomics revealed that DDX42 associated with SF3B1 to a much greater extent in WX-02-23-treated (and pladienolide B-treated) cancer cells compared to DMSO- or WX-02-43-treated cancer cells (Figure 6G and Dataset S1). WX-02-23 also caused a broader remodeling of SF3B1 interactions, including enhanced association with splicing factor DNAJC8 and decreased interactions with other spliceosome components (Figures 6G, H).
DDX42 contributions to the spliceosome remain poorly understood, and the limited functional studies performed on this helicase to date have mainly focused on spliceosome-independent activities.60,61 To investigate DDX42’s role in splicing, we introduced an N-terminal degradation tag (dTAG)62 into the endogenous DDX42 locus of HCT-116 cells. The resulting DDX42-dTAG fusion protein could be degraded to near-completion within 1 h of treatment with the Von-Hippel-Lindau (VHL)-recruiting heterobifunctional small molecule dTAGv−163 (Figure 7A). Longer-term treatment with dTAGv-1 (72 h) caused noticeable growth impairment (Figure 7B).
Figure 7.
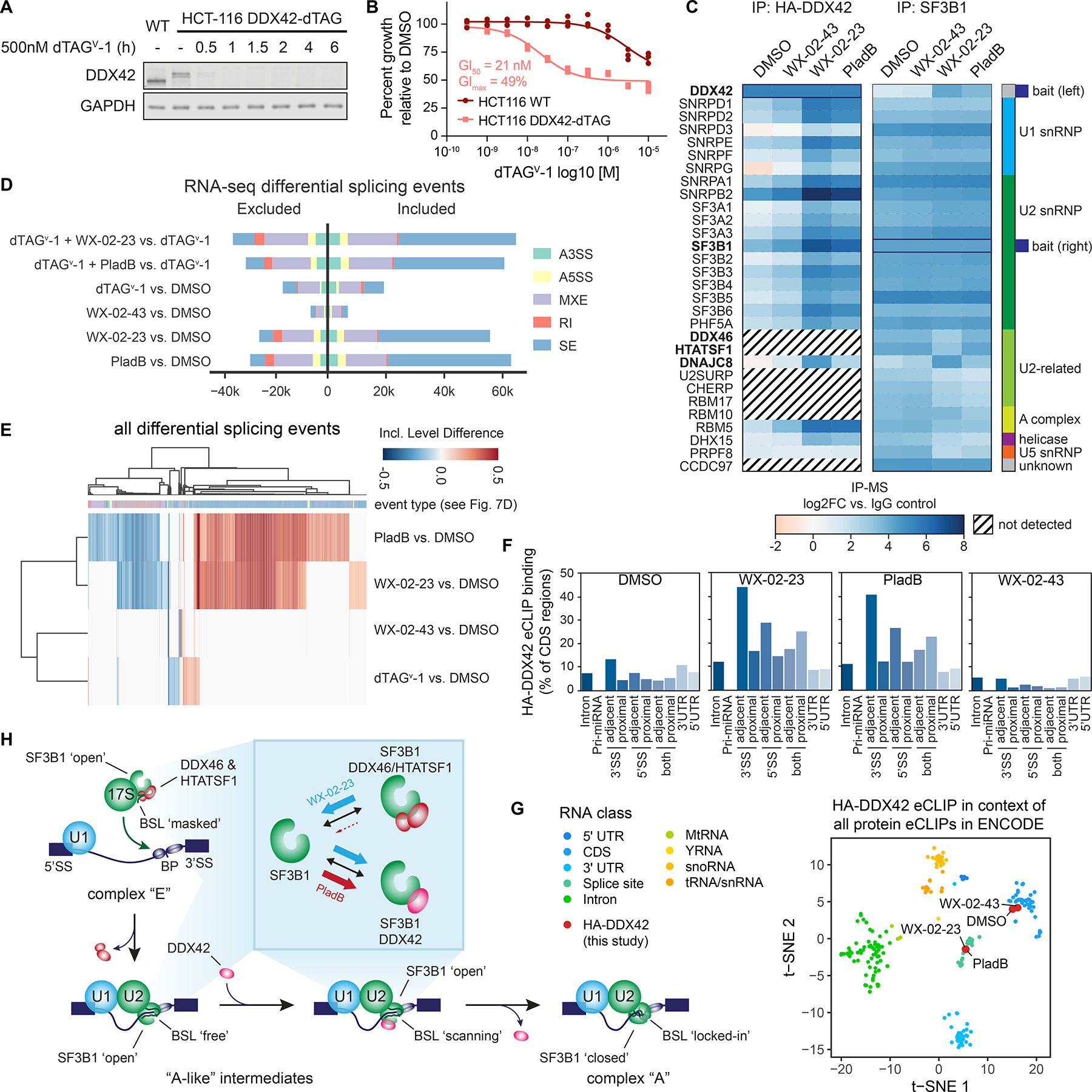
DDX42 facilitates spliceosome branch point selection. A. Western blot analysis of dTAGv-1 ligand-induced DDX42 degradation in DDX42-dTAG HCT-116 cells.
B. Cell growth curves for DDX42-dTAG or wild-type HCT-116 cells treated with dTAGv-1 for 72 h. Data are from n = 3 independent experiments.
C. Heatmap showing proteins enriched in HA-DDX42 (left) or SF3B1 (right) immunoprecipitation-MS experiments in DDX42-dTAG HCT-116 cells treated with WX-02-23 (5 μM), WX-02-43 (5 μM), pladienolide B (PladB, 10 nM), or DMSO for 3 h. Proteins were included if they were either enriched in DMSO vs. IgG (log2FC >1 for HA,
>2 for SF3B1) or in WX-02-23 vs. DMSO (log2FC >2). Results are average values from 2–4 independent experiments. Interacting proteins were input into the StringDB database and the largest connected component of 30 proteins form the basis of the heatmap. Data were normalized to corresponding bait and are shown as log2 fold-enrichment vs. IgG control. Diagonal black lines indicate proteins that were not detected in HA-DDX42 IP-MS experiments. Bold type mark proteins substantially affected in interactions with SF3B1 by WX-02-23 and/or pladienolide B.
D. Alternative splicing events triggered by indicated combinations of DDX42 degradation +/− treatment with pladienolide B (PladB, 10 nM), WX-02-23 (5 μM) or WX-02-43 (5 μM) in DDX42-dTAG HCT-116 cells. Cells were pre-treated for 1 h with either 500 nM dTAGv-1 or DMSO, followed by addition of either DMSO or compounds for 8 h. Splicing changes were identified with rMATS threshold of |PSI| > 0.1 and FDR < 0.05. Data represent values from three independent experiments.
E. Clustered heatmap of inclusion level differences between indicated compound treatments and DMSO control for alternative splicing events from panel D. Columns are annotated by the type of alternative splicing event in the color scheme of panel D. Data represent values from three independent experiments.
F. DDX42 RNA-binding profiles in DMSO- vs compound-treated DDX42-dTAG HCT-116 cells measured by eCLIP-seq using the HA-tag of the dTAG fusion. DDX42-dTAG HCT-116 cells were treated with WX-02-23 (5 μM), WX-02-43 (5 μM), or Pladienolide B (100 nM) for 3 h. Data are average values from two independent experiments. eCLIP enriched windows (FDR<0.2) w are depicted as percent binding relative to coding sequence (CDS). Proximal denotes within 500 bases and adjacent denotes within 100 bases from the annotated splice site (SS).
G. tSNE of HA-DDX42 eCLIP samples in the context of all available eCLIP datasets that were generated by the ENCODE consortium. DDX42-dTAG HCT-116 cells were treated with WX-02-23 (5 μM), WX-02-43 (5 μM), or Pladienolide B (100 nM) for 3 h. Data are average values from n = 2 independent experiments.
H. Proposed model for function of DDX42 in facilitating the branch point selection step of spliceosome function. Inset summarizes differential SF3B1 complexation effects caused by synthetic, covalent (WX-02-23) vs natural product, reversible (pladienolide B) SF3B1 ligands.
The dTAGv-1 system revealed that degradation of DDX42 did not substantially alter the stereoselective reactivity of alkyne probe WX-01-10 with SF3B1 (Figure S7B, C). Additionally, cells lacking DDX42 still displayed robust anti-proliferative responses to WX-02-23 and pladienolide B (Figure S7D). These data suggested that ligand binding to SF3B1 does not require DDX42, but instead stabilizes a spliceosome assembly state that displays enhanced affinity for DDX42. Consistent with this model, IP-MS studies using the HA-tag of the DDX42-dTAG fusion construct revealed that DDX42 principally bound to components of the U1, U2, and SF3b spliceosome subcomplexes, and these interactions were strongly and stereoselectively enhanced by WX-02-23 treatment (or by pladienolide B treatment) (Figure 7C and Dataset S1).
DDX42 facilitates spliceosome branch point selection
RNA-seq analysis of cells post-degradation of DDX42 (dTAGv-1, 9 h) revealed substantial splicing changes, including exon skipping and, to a lesser degree, intron retention events (Figure 7D). These splicing alterations were less dramatic than those caused by SF3B1 ligands, which also maintained their major splicing effects in cells lacking DDX42 (Figure 7D). Interestingly, the splicing changes promoted by DDX42 loss differed from those triggered by SF3B1 ligands (Figure 7E). We specifically observed that DDX42 loss increased the usage of stronger branch point sequences and decreased the usage of weaker branch points at differentially included exons (Figure S7E), suggesting that DDX42 may facilitate branch point selection. To test this hypothesis more directly, we evaluated DDX42-mRNA interactions by eCLIP-seq (enhanced crosslinking and immunoprecipitation followed by sequencing)64 using the hemagglutinin (HA) epitope tag component of the dTAG fusion. These eCLIP-seq experiments revealed that, in untreated cells, DDX42 mainly bound to the coding region of (pre-)mRNAs, but, following exposure of cells to SF3B1 ligands WX-02-23 or pladienolide B, the DDX42-RNA interaction preferences strongly shifted towards regions within or near splice sites (Figure 7F, G and Dataset S2).65,66 Our results, taken together, thus suggest that DDX42 dynamically binds SF3B1 as part of an A-like spliceosome complex that is in close contact with (pre-)mRNAs and splice sites to facilitate branch point selection. The distinct splicing effects caused by DDX42 degradation versus SF3B1 ligands may in turn reflect differences in the functional outcomes associated with physical loss of DDX42 versus ligand-induced trapping of the spliceosome in specific subcomplexes that include a state showing enhanced interactions with DDX42.
As part of the 17S U2 snRNP and before binding pre-mRNA, SF3B1 is found in an ‘open’ conformation with the U2 snRNA branchpoint-interacting stem-loop (BSL) masked by two proteins, HTATSF1 and the RNA helicase DDX46.55,67 Upon binding pre-mRNA and interacting with other spliceosome components, DDX46 releases HTATSF1 and itself, presumably to allow scanning by the BSL for pre-mRNA branchpoint sequences. Upon recognition of a branchpoint sequence, SF3B1 switches to a closed conformation to stabilize the newly formed BSL-branchpoint helix.68 Structural work has revealed that pladienolide B impairs this conformational switch to the closed state by acting as a wedge in an SF3B1 hinge region, leading to reduced fidelity of branchpoint selection.54 Intriguingly, DDX46 and HTATSF1 were not among the proteins that co-immunoprecipitated with DDX42 (Figure 7C and Dataset S1), and both DDX46 and HTATSF1 displayed reduced binding to SF3B1 in WX-02-23-treated cells (Figure 6G, H, Figure 7C, and Dataset S1). It thus appears that HTATSF1/DDX46 and DDX42 binding to SF3B1 are mutually exclusive, as supported by a recent cryo-EM structure of a reconstituted DDX42-SF3b complex that has revealed overlapping binding interfaces for the SF3B1-DDX42 and SF3B1-HTATSF1/DDX46 complexes (Figure S7F).69 Finally, we found that the WX-02-23-dependent disruption of DDX46 and HTATSF1 interactions with SF3B1 was preserved in cells lacking DDX42 (Figure S7H), providing further evidence that covalent SF3B1 ligands bind to an open state of the spliceosome lifecycle that shows enhanced affinity for DDX42 and reduced interactions with DDX46 and HTATSF1. Considering the prevailing model that DDX46 activity and HTATSF1 release prime the scanning of the U2 BSL for branch point sequences.55,67 as well as our finding that DDX42 preferentially binds near splice sites when induced to associate with SF3B1 following WX-02-23 treatment, we suggest that DDX42 functions after DDX46/HTATSF1 to facilitate the stable selection of branch points as part of the SF3B1 open complex (Figure 7H). Alternatively, we cannot exclude that DDX42 may also act at an earlier step in spliceosome assembly, as has recently been suggested.69
Before concluding, we call attention to a curious finding that WX-02-23 and pladienolide B, despite binding to the same SF3B1-PHF5A pocket, appear to differentially reshape SF3B1 interactions. While both compounds promoted SF3B1 binding to DDX42, only WX-02-23 impaired interactions with DDX46 and HTATSF1 (Figure 7C and Dataset S1). Additionally, while WX-02-23 and pladienolide B generally caused similar splicing changes, some differences were observed (Figure 7E). While we do not yet understand how such compound-specific effects on spliceosome composition and function may impact cellular biochemistry and physiology, we believe that the covalent SF3B1 ligands reported herein offer a mechanistically distinct and synthetically accessible class of compounds for further structural and functional exploration of this important topic.
Discussion
Chemical proteomics has emerged as an attractive strategy to discover small-molecule binders for proteins in native biological systems.8,70–72 Original ABPP strategies using active site-directed chemical probes produced data where small-molecule binding to a protein could be inferred to cause a functional effect.73–75 As the concepts of ABPP have been extended to enable assessment of small-molecule binding far beyond enzyme active sites to include virtually any protein in the human proteome,6,27,32 fundamental challenges emerge in relating binding events to functional outcomes. Establishing this connection with assays that assess the functional features of many proteins in parallel, preferably in physiologically relevant settings, could greatly accelerate chemical probe discovery versus a more traditional one-at-a-time investigation of individual small molecule-protein interactions. Here, we have introduced a “function-first” proteomic platform capable of sifting through a broad swath of small molecule-protein binding events to identify those that perturb protein complexes in cells.
To our knowledge, the azetidine acrylamides and butynamides described herein that engage C22 of PSME1 represent the first chemical probes targeting the PA28 proteasome regulatory complex. Consistent with the proposed role of the PA28 complex in regulating MHC class I antigenic peptide processing,41,42 we found that covalent PSME1 ligands disrupting this complex alter the presentation of a discrete set of MHC-I immunopeptides. It would be interesting, in the future, to determine the impact of PA28 disruption on the MHC-I antigenic peptide repertoire of disease-relevant systems, such as autoimmune syndromes or cancer cells that have developed resistance to proteasome inhibitors.76
The tryptoline acrylamides that engage C1111 of SF3B1 are distinct from other chemical probes described for this protein, which are reversible natural product-derived compounds of greater structural complexity. Considering further that individual pladienolide B analogues have been found to differentially modulate splicing outcomes57 and are in clinical development for cancers, including those with high-frequency mutations in spliceosome components like SF3B1,77,78 we wonder if covalent tryptoline acrylamide ligands can be optimized to preferentially impact the functions of common cancer-related SF3B1 mutants (e.g., K700E-SF3B1). Finally, our studies have also contributed to a more fundamental understanding of the function of DDX42 as a dynamic component of the spliceosome, where this protein appears to specifically contribute to branch site selection. The transient nature of DDX42 binding to the spliceosome may explain why a recent structure of the DDX42-associated SF3b complex was solved by reconstitution of recombinantly expressed proteins,69 and we are hopeful that the stabilization of endogenous DDX42 binding to the spliceosome by covalent SF3B1 ligands may offer a path to additional structures that include a greater proportion of spliceosome components, including additional dynamic subunits that are stabilized in binding to SF3B1 in the presence of WX-02-23 (e.g., DNAJC8; Figure 6G, H, Figure 7C, Figure S7H, and Dataset S1).
Reflecting on why our approach was successful at identifying selective, cell-active chemical probes that perturb PPIs from a small number of test compounds (two sets of four stereoisomeric acrylamides), we posit a few important factors. First, covalent ligands, by leveraging a combination of reactivity and recognition features, may provide an advantage for chemically targeting historically challenging protein classes such as adaptor proteins like PSME1. Second, we believe that the DOS principles underpinning our focused compound libraries (e.g., sp3-rich cores that are stereochemically defined, entropically constrained, and densely functionalized) may further improve the probability of engaging PPI sites, at least when compared to more fragment-like and/or sp2-based small-molecule libraries. An additional advantage of DOS-constructed libraries is that they can furnish chemical probes paired with physicochemically matched, inactive enantiomeric control compounds for cell biological studies. 32,79,80
Projecting forward, we are intrigued by the prospects of profiling larger electrophilic compound libraries. The SEC-MS method, while not high-throughput, should prove capable of assaying up to 30 compounds per week with a single FPLC/LC-MS system. And, if one elected to perform targeted (rather than untargeted) proteomic analyses81 focused on specific protein complexes of interest, the throughput of the method could likely be further improved. Additionally, many PPI interfaces may lack cysteine residues, and it is therefore provocative to consider whether incorporating alternative electrophiles targeting other nucleophilic amino acids71,82,83 into DOS designs, or replacing the electrophile with a photoreactive group to capture reversible binding interactions,7 might further enhance the chemical proteomic discovery of compounds that modulate protein complexes in cells.
We finally call attention to the numerous electrophilic compound-cysteine interactions discovered herein that did not apparently affect protein complexation state (e.g., Figures 2A, B, 3E, and S2A, and Dataset S1). These covalent liganding events, which were found on diverse protein types, may manifest functionality when analyzed by complementary methods to globally profile protein-protein84–86 or other biomolecular (e.g., protein-RNA64/DNA87) interactions, or that measure the turnover, post-translational modification state88,89 and localization90,91 of proteins. These findings, along with the continued discovery of allosteric inhibitors for diverse proteins79,92–95 and the structural mapping of hotspots for small-molecule binding at protein interfaces,96 suggest that many ligandable sites on proteins have functional potential. Fully unlocking this functionality appears poised to gain from integrating structurally diversified electrophilic chemistry with generalized assays that record the myriad ways compounds modulate protein activities in human cells.
Limitations of the study
Certain proteins showing electrophilic compound-induced size shifts by SEC may not possess cysteines engaged by these compounds. There are multiple potential reasons for this apparent disconnect. First, as we found for PSME2 and DDX42, electrophilic compounds may cause size shifts for proteins within a complex in addition to, or even other than, the direct target of the compounds. Fortunately, proteomic efforts to globally map protein complexes in human cells19 have provided a rich set of data to draw upon when relating covalent ligandability maps to protein size shifts measured by SEC, such that compound effects on both direct targets and indirect associated partners in a complex may be interpretable. An electrophilic compound-induced size shift may also prove difficult to connect to a specific liganding event using cysteine-directed ABPP if the relevant cysteine is on a non-proteotypic peptide (as we initially found for C1111 of SF3B1) or is engaged by the compound with low stoichiometry. Either of these challenges should be addressable by using alkyne variants of electrophilic compounds, which enable direct enrichment and analysis of covalent protein targets. It is also important to acknowledge that some protein size shifts caused by electrophilic compounds may be overlooked by our current SEC protocol, including those reflecting exchange between two large complexes or that are parts of highly dynamic or membrane-associated complexes. These technical gaps may be addressed by alternative approaches for mapping protein-protein interactions, such as in situ chemical crosslinking,84–86 cellular thermal shift profiling,97, or proximity labeling.19,98
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resource and reagents should be directed to and will be fulfilled by the lead contact, Benjamin F. Cravatt (cravatt@scripps.edu)
Materials availability
All compounds generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
All RNA-sequencing data, raw proteomic data and gel images have been deposited at PRIDE,99 GEO and Mendeley Data respectively, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PA28α Antibody | Cell Signaling Tech | Cat#: 2408S; RRID:AB_2170937 |
| PA28β Antibody | Cell Signaling Tech | Cat#: 2409S; RRID:AB_2171085 |
| Monoclonal ANTI-FLAG® M2 antibody produced in mouse | Sigma-Aldrich | Cat#: F1804-200UG; RRID:AB_262044 |
| HA-Tag (C29F4) Rabbit mAb | Cell Signaling Tech | Cat#: 3724S; RRID:AB_1549585 |
| HA-Tag (6E2) Mouse mAb | Cell Signaling Tech | Cat#: 2367S; RRID:AB_10691311 |
| Vinculin (E1E9V) XP® Rabbit mAb | Cell Signaling Tech | Cat#: 13901S; RRID:AB_2728768 |
| GAPDH (14C10) Rabbit mAb | Cell Signaling Tech | Cat#: 2118S; RRID:AB_561053 |
| DDX42 polyclonal antibody rabbit | Bethyl Laboratories | Cat#: 303-353A; RRID:AB_10951853 |
| Anti-β-Actin Antibody (C4) | Santa Cruz Biotechnology | Cat#: sc-47778; RRID:AB_626632 |
| β-Actin (13E5) Rabbit mAb | Cell Signaling Tech | Cat#: 4970S; RRID:AB_2223172 |
| SF3B1 (D7L5T) Rabbit mAb | Cell Signaling Tech | Cat#: 14434S; RRID:AB_2798479 |
| p27 Kip1 (D69C12) XP® Rabbit mAb | Cell Signaling Tech | Cat#: 3686S; RRID:AB_2077850 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Tech | Cat#: 7074S; RRID:AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Tech | Cat#: 7076S; RRID:AB_330924 |
| rabbit anti-goat IgG-HRP | Santa Cruz Biotechnology | Cat#: sc-2768; RRID:AB_656964 |
| InVivoMAb anti-human MHC Class I (HLA-A, HLA-B, HLA-C) Clone: W6/32 | BioXCell | Cat#: BE0079; RRID:AB_1107730 |
| IRDye® 680RD Goat anti-Rabbit IgG Secondary Antibody | VWR | Cat#: 926-68071; RRID:AB_10956166 |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | VWR | Cat#: 925-32211; RRID:AB_2651127 |
| IRDye® 680RD Goat anti-Mouse IgG Secondary Antibody | VWR | Cat#: 926-68070; RRID:AB_10956588 |
| IRDye® 800CW Goat anti-Mouse IgG Secondary Antibody | VWR | Cat#: 926-32210; RRID:AB_621842 |
| PE anti-mouse H-2Kb/H-2Db Antibody | BioLegend | Cat#: 114608; RRID:AB_313599 |
| PE Mouse IgG2a, κ Isotype Ctrl Antibody | BioLegend | Cat#: 400211; RRID:AB_326460 |
| APC anti-mouse H-2Kb bound to SIINFEKL Antibody | BioLegend | Cat#: 141606; RRID:AB_11219595 |
| APC Mouse IgG1, κ Isotype Ctrl Antibody | BioLegend | Cat#: 400120; RRID:AB_2888687 |
| APC/Cyanine7 Mouse IgG1, κ Isotype Ctrl Antibody | BioLegend | Cat#: 400128; RRID:AB_2892538 |
| Bacterial and virus strains | ||
| NEB 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | Cat#: C2987H |
| Chemicals, peptides, and recombinant proteins | ||
| MEM Non-Essential Amino Acids Solution (100X) | Thermo Scientific | Cat#: 11140050 |
| Lipofectamine™ 2000 Transfection Reagent | Thermo Scientific | Cat#: 11668019 |
| Polybrene | Santa Cruz Biotechnology | Cat#: sc-134220A |
| Geneticin™ Selective Antibiotic (G418 Sulfate) | Thermo Scientific | Cat#: 10131035 |
| Pladienolide B | Tocris Bioscience | Cat#: 60-705-00U |
| Desthiobiotin polyethyleneoxide iodoacetamide | Santa Cruz Biotechnology | Cat#: sc-300424 |
| Tetramethylrhodamine (TAMRA) azide | Synthesized in-house | N/A |
| Biotin-PEG4-azide | ChemPep | Cat#: 271606 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Scientific | Cat#: 34580 |
| Novex 4-20% Tris-Glycine Mini Gels | Invitrogen | Cat#: XP04205BOX |
| Nitrocellulose western blotting membrane, 0.45 μM | GE Healthcare Amersham | Cat#: 10600002 |
| Affi-Gel 10 Gel | Bio-rad | Cat#: 1536046 |
| DMSO | Corning | Cat#: 25-950-CQC |
| EDTA (0.5M, pH 8.0) | Invitrogen | Cat#: AM9260G |
| Urea | Fisher Scientific | Cat#: M1084871000 |
| Iodoacetamide | Sigma-Aldrich | Cat#: I1149-25G |
| Dithiothreitol (DTT) | Fisher Bioreagents | Cat#: BP172-25 |
| T ris(benzyltriazolylmethyl)amine (TBTA) | TCI | Cat#: T2993 |
| Copper(II) sulfate, anhydrous | Sigma-Aldrich | Cat#: 451657-10G |
| Tris(2-carboxyethyl)phosphine HCl (TCEP) | Sigma-Aldrich | Cat#: 75259 |
| Sequencing grade modified trypsin | Promega | Cat#: V5111 |
| Lysyl Endopeptidase, Mass Spectrometry Grade (Lys-C) | Fujifilm Wako | Cat#: 125-05061 |
| Streptavidin agarose resin | Fisher Scientific | Cat#: 20349 |
| Micro bio-spin column | Bio-rad | Cat#: 7326204 |
| Tween 20 | Fisher Bioreagents | Cat#: BP337-500 |
| Nonidet P40 substitute (Igepal CA-630) | USB Corporation | Cat#: 19628 |
| Triethylammonium bicarbonate (TEAB) buffer | Sigma-Aldrich | Cat#: T7408-500ML |
| TMT10plex Isobaric Label Reagent Set | Thermo Scientific | Cat#: 90406 |
| TMT16plex Isobaric Label Reagent Set | Thermo Scientific | Cat#: A44520 |
| Hydroxylamine solution | Sigma-Aldrich | Cat#: 467804-10ML |
| Formic acid, ~98%, for mass spectrometry | Honeywell Fluka | Cat#: 94318-250ML-F |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat#: G7573 |
| Caspase-Glo® 3/7 Assay System | Promega | Cat#: G8093 |
| NEBNext Ultra II RNA Library Prep Kit for Illumina | New England Biolabs | Cat#: E7770 |
| RNeasy Mini Plus Kits | QIAGEN | Cat#: 74034 |
| eBioscience™ Fixable Viability Dye eFluor™ 780 | Invitrogen | Cat#: 65-0865-18; N/A |
| Deposited data | ||
| All raw proteomic data have been uploaded to PRIDE | This study | PXD029655 |
| All RNA-sequencing data have been uploaded to GEO | This study | GSE185373, GSE220185, GSE220845 |
| All uncropped gels have been uploaded to Mendeley Data | This study | DOI: 10.17632/r6t9f3n9wr.1 |
| Experimental models: Cell lines | ||
| 22Rv1 | ATCC | CRL-2505; RRID:CVCL_1045 |
| MCF7 | ATCC | HTB-22; RRID:CVCL_0031 |
| Ramos | ATCC | CRL-1596; RRID:CVCL_0597 |
| THP1 | ATCC | TIB-202; RRID:CVCL_0006 |
| HEK293T | ATCC | CRL-3216; RRID:CVCL_0063 |
| HEK293FT | Thermo Scientific | R70007; RRID:CVCL_6911 |
| KBM7 | Georg Winter, CeMM, Vienna | RRID:CVCL_A426 |
| HCT-116 | ATCC | CCL-247; RRID:CVCL_0291 |
| Panc 04.03 | ATCC | CRL-2555; RRID:CVCL_1636 |
| Panc 05.04 | ATCC | CRL-2557; RRID:CVCL_1637 |
| E.G7-Ova | ATCC | CRL-2113; RRID:CVCL_3505 |
| Oligonucleotides | ||
| sgDDX42_N_sense 5’-CACCGattcctaacaggtcagtcat | This study | N/A |
| sgDDX42_N_anti 5’-AAACatgactgacctgttaggaatC | This study | N/A |
| RepDDX42N_1_P_F 5’-gcgttacatagcatcgtacgcgtacgtgtttggcttattcctaacaggtcagtatgaccgagtacaagcccacg | This study | N/A |
| RepDDX42N_1_P_R 5’-agcattctagagcatcgtaCGCGTACGTGTTTGGtccagttcatggtgccaatgGAagatccgccgccacc | This study | N/A |
| DDX42_N_seqF 5’-gcccttggggctatacacttt | This study | N/A |
| DDX42_N_seqR 5’-ccagcactgatggcaaaacc | This study | N/A |
| sgPSME1_sense 5’-CACCGccagcccgaggcccaagcca | This study | N/A |
| sgPSME2_sense 5’ CACCGaaatccagagacttacctcc | This study | N/A |
| sgControl-AAVS1_sense 5’-caccgGGGGCCACTAGGGACAGGAT | This study | N/A |
| Recombinant DNA | ||
| pLenti3.3/TR | Thermo Scientific | A11144 |
| psPAX2.0 | Addgene | 12260 |
| CMV VSV-G | Addgene | 98286 |
| pLenti6.3 | Thermo Scientific | A11144 |
| pLEX304 | Addgene | 25890 |
| Software and algorithms | ||
| PRISM Version 9.0.0 | GraphPad | https://www.graphpad.com/features |
| MaxQuant (v2.0.3.1) | N/A | https://www.maxquant.org/ |
| Integrated Proteomics Pipeline (IP2) | Integrated Proteomics Applications, Inc | http://proteomicswiki.com/wiki/index.php/IP2 |
| FlowJo (v10.0.7) | TreeStar Inc. | https://www.flowjo.com/ |
| edgeR (v3.32.1) | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| DESeq2 (v1.30.1) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| limma voom (v3.46.0) | Law et al., 2014 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| PyMOL (v2.5.2) | Schrödinger | https://pymol.org/2/ |
| Trim_galore (v0.6.4) | Martin et al., 2011 | https://github.com/FelixKrueger/TrimGalore |
| STAR (v2.7.5) | Dobin et al., 2013 | https://github.com/alexdobin/STAR/ |
| samtools (v1.9) | Danecek et al., 2021 | http://www.htslib.org/ |
| bamCoverage (part of the Deeptools package; v3.3.1) | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/index.html |
| featureCounts (part of the subread package; v1.5.0) | Liao et al., 2014 | https://subread.sourceforge.net/ |
| ChimeraX | Pettersen et al., 2021 | https://www.cgl.ucsf.edu/chimerax/ |
| Glide | Schrödinger | https://www.schrodinger.com/products/glide |
| cutadapt 3.4 | Martin et al., 2011 | https://cutadapt.readthedocs.io/en/v3.4/# |
| rMATS (v4.1.1) | Shen et al., 2014 | https://rnaseq-mats.sourceforge.net/rmats4.1.1/index.html |
| Branchpointer | Signal et al., 2018 | https://github.com/signalbash/branchpointer |
| Skipper | Boyle et al., 2022 | https://github.com/YeoLab/skipper |
| skewer | Jiang et al., 2014 | https://github.com/relipmoc/skewer |
| fastp 0.11.5 | Chen et al., 2018 | https://github.com/OpenGene/fastp |
| UMIcollaps | Liu et al., 2019 | https://github.com/Daniel-Liu-c0deb0t/UMICollapse |
| Other | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
22Rv1 (ATCC, CRL-2505), MCF7 (ATCC, HTB-22), Ramos (ATCC, CRL-1596), THP1 (ATCC, TIB-202), HEK293T (ATCC, CRL-3216), HEK293FT (Thermo, R70007), KBM7 (Georg Winter, CeMM, Vienna; RRID:CVCL_A426), HCT-116 (ATCC, CCL-247), Panc 04.03 (ATCC, CRL-2555), Panc 05.04 (ATCC, CRL-2557), and E.G7-Ova (ATCC, CRL-2113) were grown in in RPMI (22Rv1, Ramos, THP1, Panc 04.03, Panc 05.04, E.G7-Ova), DMEM (HEK293T, HEK293FT, HCT-116), IMDM (KBM7), or EMEM (MCF7), supplemented with 10% fetal bovine serum (FBS), 2 mM L-alanyl-L-glutamine (GlutaMAX), penicillin (100 U/ml), and streptomycin (100 μg/ml) and maintained at 37 °C with 5% CO2.
Generation of stably transduced clonal 22Rv1 cells expressing TetR:
HEK293FT cells (Thermo R70007, supplemented with MEM nonessential amino acids, 25-025-Cl Corning) (5×106) were plated in 10-cm plates and allowed to attach overnight. To 1mL of serum-free DMEM the following were added: 3 μg of pLenti3.3/TR, 2.25 μg psPAX2.0 plasmid (Addgene, catalog number: 12260) and 0.75 μg CMV VSV-G (Addgene, catalog number: 98286) and 36 μL lipofectamine 2000 transfection reagent (Invitrogen). Reagents were flicked to mix, and after 15 min incubation, the transfection mixture was added dropwise to plates containing cells. Medium was exchanged approximately 16 h post transfection, virus-containing supernatants were then collected 48 h post transfection and filtered, and then used to infect 22Rv1 cells in 6-well plates in the presence of 6 μg/ml Polybrene (Santa Cruz). The media was replaced 24 h later, cells were allowed to recover for an additional 24 h, and then geneticin (400 μg/mL) was added for selection. Clonal pLenti3.3/TR expressing cells were selected by cloning cylinder and subsequently transfected with constructs of interest.
Transfection of epitope tagged and mutant proteins of interest:
Clonal pLenti3.3/TR expressing 22Rv1 cells were seeded into 6cm dishes and 24 h later transfected with proteins of interest cloned into pLenti6.3. 1 μg of pLenti6.3 plasmid with gene of interest and 3 μL of PEI (1 μg/μL) were added to 200 μL serum-free RPMI and incubated for 15 minutes. Medium of cells was replaced to contain tetracycline (0.1–1 μg/mL final concentration) and then transfection mixture was added dropwise. Cells were assayed 24–48 h later.
Generation of DDX42-dTAG knock-in HCT-116 cells:
Knock-in of a dTAG cassette into the endogenous DDX42 locus was performed as previously described100. A pX330A_sgPITCh_sgDDX42_N sgRNA/Cas9 plasmid was cloned via standard oligo-annealing and ligation methods using two sense/antisense DNA oligos (sgDDX42_N_sense 5’-CACCGattcctaacaggtcagtcat; sgDDX42_N_anti 5’-AAACatgactgacctgttaggaatC). A pCRIS-PITChv2 repair template plasmid was cloned using pCRIS-PITChv2-Puro-dTAG (BRD4) (Addgene #91793) as a PCR template to introduce 20–22 bp microhomology arms corresponding to the genomic DNA sequences immediately 5’ and 3’ of the sgRNA cut site (primers: RepDDX42N_1_P_F 5’-gcgttacatagcatcgtacgcgtacgtgtttggcttattcctaacaggtcagtatgaccgagtacaagcccacg; RepDDX42N_1_P_R 5’-agcattctagagcatcgtaCGCGTACGTGTTTGGtccagttcatggtgccaatgGAagatccgccgccacc). The resulting fragment was gel-purified and re-introduced into an MluI-linearized pCRIS-PITChv2 backbone using NEBuilder 2xHiFi Gibson assembly master mix (New England Biolabs, USA). The sgRNA (6 μg) and repair (6 μg) plasmids were then co-electroporated into 2 million wild-type HCT-116 cells using Lonza Nucleofector II with reagent kit V and program D-032. After recovery for 3 days, selection for successfully tagged clones was initiated by DMEM containing 2 μg/mL puromycin. Surviving colonies appeared after 5 days of puromycin selection and cells were seeded in limiting dilution of 0.1–0.5 cells per 50 μL to four 384-well plates in puro-containing DMEM. After 14 days, wells with a single colony were expanded to 24-well plates and tested for positive dTAG integration via HA-tag and DDX42 antibody western blot. Clones with positive western blot signal were next genotyped by extracting genomic DNA and amplifying the region surrounding the sgRNA cut site (primers DDX42_N_seqF 5’-gcccttggggctatacacttt; DDX42_N_seqR 5’-ccagcactgatggcaaaacc). Clones showing a large band (integrated cassette), but no short band (no integration at the cut site) were selected for Sanger sequencing of the purified PCR reaction. One successful clone was selected and designated the HCT-116 dTAG-DDX42 working clone, which showed homozygous and seamless, indel-free integration of the dTAG cassette. Four more clones with varying integration outcomes (2x seamless, 2x in-frame insertions) were kept as backup clones but not used for any experiments.
Generation of sgPSME1, sgPSME2 and sgControl KBM7 cells:
Stable knock-out cell lines were generated by transduction of KBM7_Cas9-P2A-blast (Cas9 lentivirus produced from Addgene #52962) with lentiGuide-puro virus (sgRNAs cloned into Addgene #52963) using standard CRISPR/Cas9 and lentivirus protocols. Briefly, sgRNAs were cloned into lentiGuide-puro via BsmBI restriction and annealed oligo ligation cloning (sgPSME1_sense 5’-CACCGccagcccgaggcccaagcca; sgPSME2_sense 5’ CACCGaaatccagagacttacctcc; sgControl-AAVS1_sense 5’-caccgGGGGCCACTAGGGACAGGAT).
Lentivirus was produced using the polyethylenimine (PEI) protocol101. Briefly, 2 million Lenti-X 293T cells (Takara) were seeded in 5 mL DMEM to 6 cm dishes 20 h before transfection. The next day, a PEI master mix was prepared (61.8 μL serum-free DMEM, 1.7 μg pSPAX2, 0.85 μg pMD2.G, 1.29 μg expression plasmid DNA per transfection), mixed with 18.5 μL PEI (1 mg/mL in water, neutralized with NaOH to pH 7.0; 40,000 MW) and incubated for 10 minutes at room temperature. The Lenti-X supernatant was changed to 3 mL fresh full DMEM and PEI/plasmid mix was added to the medium dropwise. Viral supernatant was harvested once after 70 h, sterile filtered using .45 μm syringe filters and frozen in aliquots at −80 °C.
For transduction, 1 million KBM7 cells were mixed with 50 μL of freshly thawed sgPSME1, sgPSME2, or sgControl/AAVS1 viral supernatant in a total of 3mL full IMDM supplemented with 8 μg/mL polybrene in 12-well plates. Cells were spin-infected at 2000 rpm and 30 °C for 1 h and incubated for 24 h at 37 °C. Selection was initiated with 1 μg/mL puromycin and considered complete after 4 days, when the no-virus control cells were completely killed. Selected pools were characterized using gDNA PCR and western blot and used for knock-out cell line experiments.
Generation of 22Rv1 cells stably transduced with PHF5A_WT or Y36C:
Lentiviral expression plasmids pLEX304_PHF5A-WT-STOP and pLEX304_PHF5A-Y36C-STOP were cloned using the gateway recombinase cloning strategy (Invitrogen, USA) from commercially synthesized gene blocks (IDT, USA). Lentivirus production and transduction of 22Rv1 cells was performed as described above. 22Rv1 cells stably transduced with pLEX304_PHF5A were selected with 10 μg/mL blasticidin for 7 days, at which point non-transduced control cells were completely killed.
METHOD DETAILS
Proteomic platforms: SEC-MS (related to Figures 1, 2)
Sample preparation:
Cells (adherent: 15cm dishes, suspension: 2 million cells/mL) were treated with electrophilic probes in situ for indicated times and concentrations (20 μM, 3 h), washed with ice-cold PBS, and flash frozen in liquid nitrogen. Cell pellets were lysed by sonication (8 pulses, 40% power) in 600 μL ice-cold PBS supplemented with cOmplete protease and PhosSTOP phosphatase inhibitors (Roche; 1 tablet/10mL of PBS), ultracentrifuged for 20 min at 100,000 g, then normalized to a standardized concentration (typically 1.5 to 2.5 mg/mL) using a standard DC protein concentration assay (Bio-Rad). 500 μL of clarified lysate was injected into a Superdex 200 Increase 10/300 GL column attached to an ÄKTA pure FPLC system (Cytiva). Proteins were fractionated using an isocratic gradient (PBS) running at 0.5 mL/min into 5 fractions 2 mL wide, beginning at 8 mL and ending at 18 mL. Eluate was collected into 15 mL tubes containing 12 mL of acetone at 4 °C. After completion of one set of 5 fractions, the above protocol was repeated a second time for the remaining 5 fractions. Proteins were precipitated overnight at −20 °C, then centrifuged at 4500 g for 20 minutes, yielding a white protein pellet. Acetone/PBS mixture was decanted off the pellets, which were dried at room temperature and then resuspended in 125 μL of 8 M urea, 10 mM DTT in 100 mM EPPS for 15 minutes at 65 °C, followed by probe sonication to complete resuspension. 6.25 μL of 500 mM iodoacetamide was added to samples for alkylation (30 minutes, 37 °C), followed by dilution with 370 μL of 100 mM EPPS, and addition of 2 μg trypsin per fraction, and digested overnight at 37 °C. 70 μL of tryptic digest was aliquoted into a clean microcentrifuge tube, followed by 30 μL of dry acetonitrile and 4.5 μL (20 μg/μL) of TMT tag in dry acetonitrile. Samples were labeled at room temperature for 75 minutes, then quenched with 6 μL of 5% hydroxylamine for 15 minutes and acidified with 5 μL of formic acid. The set of 10 fractions were then combined into a single tube and evaporated by speed vac. Samples were desalted via Sep-Pak and then high pH fractionated as described below.
Data processing:
Fractional distributions for each peptide-spectra match with a total TMT reporter ion intensity were calculated by dividing the reporter ion intensity for each TMT channel by the summed intensity across the 5 TMT channels corresponding to a single treatment condition (Equation 1). Protein-level SEC elution profiles were then generated by averaging together peptide-level elution profiles from unique peptides with summed reporter ion intensities >5,000. Two unique peptide sequences were required per-protein. Bar graph elution profiles are represented as the mean ± standard error of the mean across all replicates. Euclidean distances (SEC shift scores, Equation 2) were calculated using the average elution profile for each protein across replicates, combined by treatment condition and cell line. Figures reporting mean elution times used Equation 3 for calculation. Figures 1E, 1F use Equation 4 for calculating the delta-SEC shift score for an enantiomeric pair.
Filtering of SEC-MS data:
Replicates were combined based on cell line and treatment condition (probe, concentration, duration) by averaging SEC elution profiles for each protein across all experiments. A coefficient of variation (CV) filter was applied by taking the average the per-fraction CV across all 5 fractions. Proteins with a CV > 0.5 were removed from analysis.
| Equation 1. |
| Equation 2. |
| Equation 3. |
| Equation 4. |
where , represent protein-level fractional distributions for individual treatment conditions.
SEC analysis by Western blot (related to Figure 2)
For experiments using transiently expressed constructs (FLAG-PSME1 WT/C22A), stable clonal 22Rv1 pLenti3.3/TR were transfected using polyethylenimine and treated with tetracycline (0.1 μg/mL) for 48 h prior to treatment with electrophilic probes.
Cells were lysed and fractioned by SEC as described above. After acetone precipitation and resuspension, 4x gel loading buffer was added to eluates, and proteins were resolved using SDS-PAGE (4–20%, Tris-glycine gel), and transferred to a nitrocellulose membrane (350 mA for 90 min). The membrane was blocked with 5% milk in Tris-buffered saline (20 mM Tris-HCl 7.6, 150 mM NaCl) with 0.1% tween (TBST) and incubated with primary antibody overnight at 4 C. After TBST wash (3 times), the membrane was incubated with secondary antibody for 1 h at room temperature, washed with TBST again, developed with ECL western blotting detection reagents, and imaged on a Bio-Rad ChemiDoc MP. Relative band intensities were analyzed using ImageJ.
Gel-ABPP studies of PSME1 (related to Figure 3)
C-terminally FLAG-tagged PSME1 WT or C22A were transiently expressed in HEK293T cells, harvested 48 h later, then cells were divided into aliquots, flash frozen and stored at −80 °C. On the day of the experiment, an aliquot of cells were thawed, lysed by sonication in ice-cold PBS supplemented with cOmplete protease inhibitors (1 tablet/10mL of PBS). normalized to 2.0 mg/mL using a standard DC protein assay (Bio-Rad) and divided into 25 μL aliquots. 1 μL of DMSO or compound (25x stock) was incubated with lysates for 2 h at RT, followed by incubation with 1 μL of 62.5 μM MY-11B alkyne probe (final concentration 2.4 μM) for an additional 30 minutes. Reagents for the copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction were pre-mixed prior to addition to the samples, as described previously.6 After 1 h of labeling, 4x SDS loading buffer was added to samples, which were then analyzed by SDS-PAGE. ImageJ was used to quantify rhodamine band intensities, and GraphPad PRISM Version 9.0.0 was used to generate IC50 curves (four-parameter variable slope least squares regression)
FACS analysis with citric acid wash (related to Figure 3)
E.G7-Ova cells (2 million/mL) were treated with compounds for 4 h, washed with PBS, and then washed with 1 mL of mild acid elution buffer (131 mM citric acid, 66 mM NaH2PO4; pH 3.0) for 2 minutes, and then washed with media three times. Cells were then resuspended in warm RPMI and allowed to recover for the indicated period, after which cells were washed with PBS and transferred to 96 well plates for staining. Each well was washed with 200 μL PBS, and then stained for 20 minutes with 50 μL of staining solution: 1:1000 fixable viability dye stain (Invitrogen) and 1:200 dilution of anti-SIINFEKL (BioLegend) or anti-mouse H-2Kb (BioLegend) antibody in FACS buffer (2% FBS in PBS supplemented with PenStrep and sterile filtered). Additional wells were used as unstained or singly-stained controls. After staining, cells were washed with PBS, then fixed in 200 μL 4% paraformaldehyde in PBS for 15 minutes. Cells were resuspended in 75 μL FACS buffer and analyzed on a Novocyte flow cytometer and analyzed using FlowJo (v10.0.7). The corresponding data in Figure 3G are presented as the average percentage of median fluorescence intensity (MFI) relative to DMSO (4 h timepoint) treated control ± SD from n = 4 replicates. The corresponding data in Figure 3H are presented as the average percentage of MFI relative to DMSO treated control ± SD from n = 3 replicates. Statistical analysis was performed using GraphPad PRISM Version 9.0.0.
Cysteine-directed ABPP (Related to Figures 2, 3)
Sample preparation:
Cells (adherent: 15 cm dishes, suspension: 2 million cells/mL) were treated with electrophilic probes in situ for indicated times and concentrations, washed with ice-cold PBS, and flash frozen in liquid nitrogen. Cell pellets were lysed by sonication (8 pulses, 40% power) in 500 μL ice-cold PBS supplemented with cOmplete protease inhibitors (1 tablet/10 mL of PBS). Whole cell lysates protein content was measured using a standard DC protein assay (Bio-Rad) and 500 μL (2 mg/mL protein content) were treated with 5 μL of 10 mM IA-DTB (in DMSO) for 1 h at ambient temperature with occasional vortexing. Samples were methanol-chloroform precipitated with the addition of 500 μL ice-cold MeOH and 200 μL CHCl3, vortexed, and centrifuged (10 min, 10,000 g). Without disrupting the protein disk, both top and bottom layers were aspirated, and the protein disk was washed with 1 mL ice-cold MeOH and centrifuged (10 min, 10,000 g). The pellets were allowed to air dry, and then re-suspended in 90 μL buffer (9 M urea, 10 mM DTT, 50 mM TEAB pH 8.5). Samples were reduced by heating at 65 °C for 20 minutes and water bath sonicated as needed to resuspend the protein pellets, followed by alkylation via addition of 10 μL (500 mM) iodoacetamide and incubated at 37 °C for 30 min with shaking. Samples were diluted with 300 μL buffer (50 mM TEAB pH 8.5) to reach final concentration of 2 M urea, briefly centrifuged, and probe sonicated once more to ensure complete resuspension. Trypsin (4 μL of 0.25 μg/μL in trypsin resuspension buffer with 25 mM CaCl2) was added to each sample and digested at 37 °C for 2 h to overnight. Digested samples were then diluted with 300 μL wash buffer (50 mM TEAB pH 8.5, 150 mM NaCl, 0.2% NP-40) containing streptavidin-agarose beads (50 μL of 50% slurry in wash buffer) and were rotated at room temperature for 2 h. Enriched samples were pelleted by centrifugation (2000 g, 2 min) and transferred to BioSpin columns and washed (3×1 mL wash buffer, 3×1 mL PBS, 3×1mL water). Enriched peptides were eluted with 300 μL of 50% acetonitrile with 0.1% formic acid and eluate evaporated to dryness via speedvac. IA-DTB labeled and enriched peptides were resuspended in 100 μL EPPS buffer (200 mM, pH 8.0) with 30% acetonitrile, vortexed, and water bath sonicated. Samples were TMT labeled with 3 μL of corresponding TMT tag (20 μg/μL), vortexed, and incubated at room temperature for 1 h. TMT labeling was quenched with the addition of hydroxylamine (5 μL 5% solution in H2O) and incubated for 15 minutes at room temperature. Samples were then acidified with 5 μL formic acid, combined and dried using a SpeedVac. Samples were desalted via Sep-Pak and then high pH fractionated as described below.
Data processing:
Cysteine engagement ratios (probe vs DMSO) were calculated for each peptide-spectra match by dividing each TMT reporter ion intensity by the average intensity for the channels corresponding to DMSO treatment. Peptide-spectra matches were then grouped based on protein ID and residue number (e.g., PSME1 C22), excluding peptides with summed reporter ion intensities for the DMSO channels < 10,000, coefficient of variation for DMSO channels > 0.5, and non-unique or non-tryptic peptide sequences. TMT reporter ion intensities were normalized to the median summed signal intensity across channels.
Filtering of cysteine data:
Replicates were combined based on cell line and treatment condition (probe, concentration, duration) by averaging cysteine engagement ratios across all experiments.
Immunopeptidomics (related to Figure 4)
Sample preparation:
KBM7 cells (10 million cells/mL, 500 million cells per sample) were treated with electrophilic probes (from 50 mM stocks) as indicated for 8 h, washed with ice-cold PBS, and flash frozen in liquid nitrogen. MHC enrichments were processed as previously described (Purcell et al., 2019). Briefly, cells were lysed with 3 mL of lysis buffer (0.5% (vol/vol) NP-40 (Igepal CA-630), 50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.2 mM Iodoacetamide, and protease inhibitor cocktail (Roche)), rotating at 4 C for 30 minutes in a 5 mL low-bind Eppendorf. Samples were centrifuged at 18,000 g for 10 minutes and supernatant was taken for enrichment by anti-MHC antibody (BioXCell, BE0079) conjugated to Affi-gel 10 matrix (~2 mg of antibody per sample). Enrichments rotated at 4 °C for 2 h and transferred to Bio-spin filter column for washing with 3×1 mL lysis buffer, wash buffer 1 (50 mM Tris pH 8, 150 mM NaCl), wash buffer 2 (50 mM Tris pH 8, 400 mM NaCl), and wash buffer 3 (50 mM Tris pH 8). MHC-conjugated peptides were eluted with 1 mL of 10% acetic acid in HPLC grade water. Samples were immediately desalted using Sep-Pak, dried via speedavac, and then ready for injection into the mass spectrometer.
Mass spectrometry:
Samples were analyzed by liquid chromatography tandem mass-spectrometry using an Orbitrap Eclipse Tribrid Mass Spectrometer (Thermo Scientific) coupled to an UltiMate 3000 Series Rapid Separation LC system and autosampler (Thermo Scientific Dionex). The peptides were eluted onto an EASY-Spray HPLC column (Thermo ES902, ES903) using an Acclaim PepMap 100 (Thermo 164535) loading column, and separated at a flow rate of 0.25 μL/min. Peptides were separated across a 90 minute gradient of 2–15%, 5 minutes 15–20%, and then 5 minutes 20–95% acetonitrile (0.1% formic acid) followed by column equilibration. Data was acquired using a data-dependent acquisition in profile mode. Briefly, the scan sequence began with an MS1 master scan (Orbitrap analysis, resolution 120,000, 350–1400 m/z, RF lens 30%, automatic gain control [AGC] target 250%, maximum injection time 50 ms, profile mode) with dynamic exclusion enabled (repeat count 1, duration 15 s). The top precursors were then selected for MS2 analysis within a 2.5 duty cycle. MS2 analysis consisted of quadrupole isolation (isolation window 0.7) of precursor ion followed by collision-induced dissociation (CID) in the orbitrap (AGC 300%, normalized collision energy 35%, maximum injection time 250 ms). Raw files were searched using MaxQuant (v2.0.3.1) with 5% PSM FDR again the Human UniProt database (release 2016), unspecific digestion, 8–12 amino acids in length, 20 ppm peptide tolerance, and match-between-runs enabled (1 minute match time window, 20 minute alignment time window). Subsequent results were further filtered to require an identification posterior error probability < 0.01.
General protein abundance proteomics (related to Figures 5, 6)
Sample preparation:
Cells (adherent: 15cm dishes, suspension: 2 million cells/mL) were treated with electrophilic probes in situ for indicated times and concentrations (1 h or 3 h as noted), washed with ice-cold PBS, and flash frozen in liquid nitrogen. Cell pellets were lysed by sonication (8 pulses, 40% power) in 200 μL ice-cold PBS supplemented with cOmplete protease inhibitors (1 tablet/10 mL of PBS) and 1 mM PMSF (100 mM stock in ethanol). Whole cell lysates protein content was measured using a standard DC protein assay (Bio-Rad) and a volume corresponding to 200 μg was transferred to a new low-bind Eppendorf tube (containing 48 mg urea) and brought to 100 μL total volume. Samples were reduced with DTT (5 μL 200 mM stock in H2O, 10 mM final concentration) and incubated at 65 °C for 15 minutes, then alkylated with iodoacetamide (5 μL 400 mM stock in H2O, 20 mM final concentration) incubated at 37 °C for 30 minutes in the dark. Samples were precipitated with the addition of ice-cold MeOH (600 μL), CHCl3 (180 μL), and H20 (500 μL), and vortexed and centrifuged (16,000 g, 10 minutes, 4 °C). The top layer above the protein disc was aspirated and an additional 1mL of icecold MeOH was added. The samples were again vortexed and centrifuged (16,000 g, 10 minutes, 4 °C), the supernatant aspirated and the protein pellets were allowed to air dry and be stored at −80 °C or proceeded to resuspension and digestion. Samples were resuspended in 160 μL EPPS buffer (200 mM pH 8.0) using a probe sonicator (10–15 pulses). Proteomes were first digested with Lys-C (0.5 μg/μL in HPLC grade water, 4 μL per sample) for 2 h at 37 °C. Then trypsin (8 μL per sample, 0.5 μg/μL in trypsin resuspension buffer with 20 mM CaCl2) was added and samples were incubated at 37 °C overnight. Peptide concentrations were estimated using Micro BCA™ assay (Thermo Scientific), and a volume corresponding to 25 μg was transferred to a new low-bind Eppendorf tube and brought to 35 μL with EPPS buffer. Samples were diluted with 9 μL acetonitrile and then TMT labeled with 5 μL of corresponding TMT tag (20 μg/μL), vortexed, and incubated at room temperature for 1 h. TMT labeling was quenched with the addition of hydroxylamine (5 μL 5% solution in H2O) and incubated for 15 minutes at room temperature. Samples were then acidified with 2.5 μL formic acid and an equal volume (20 μL corresponding to ~8.85 μg per channel) was combined and dried using a SpeedVac. Samples were desalted via Sep-Pak and then high pH fractionated as described below.
Data processing:
Protein abundance ratios (probe vs DMSO) were calculated for each peptide-spectra match by dividing each TMT reporter ion intensity by the average intensity for the channels corresponding to DMSO treatment. Peptide-spectra matches were then grouped based on protein ID, excluding peptides with summed reporter ion intensities for the DMSO channels < 10,000, coefficient of variation for DMSO channels > 0.5, non-unique or non-tryptic peptide sequences. TMT reporter ion intensities were normalized to the median summed signal intensity across channels.
Filtering of whole proteome data:
Whole proteome data was filtered at peptide-level by removing any peptide-spectra matches with a standard deviation of abundance ratio > 100%. At a replicate-level, proteins with at least 2 distinct peptide-spectra matches were retained for analysis. Replicates were then combined based on cell line and treatment condition (probe, concentration, duration) by averaging protein abundance ratios across all experiments.
Offline fractionation
High pH offline fractionation was performed as previously described32,102. Briefly, samples fractionated via Peptide Desalting Spin Columns (Thermo 89852) were resuspended in buffer A (5% acetonitrile, 0.1% formic acid) and bound to the spin columns. Bound peptides were then washed in water, 10 mM NH4HCO3 containing 5% acetonitrile, and eluted in fractions of increasing acetonitrile, concatenated, and then dried using a SpeedVac vacuum concentrator. Resulting fractions were resuspended in buffer A (5% acetonitrile, 0.1% formic acid) and analyzed by mass spectrometry.
Samples fractionated by HPLC were resuspended in buffer A (500 μL) and fractionated into a 96 deep-well plate using HPLC (Agilent). The peptides were eluted onto a capillary column (ZORBAX 300Extend-C18, 3.5 μm) and separated at a flow rate of 0.5 mL/min using the following gradient: 100% buffer A from 0–2 min, 0%–13% buffer B from 2–3 min, 13%–42% buffer B from 3–60 min, 42%–100% buffer B from 60–61 min, 100% buffer B from 61–65 min, 100%–0% buffer B from 65–66 min, 100% buffer A from 66–75 min, 0%–13% buffer B from 75–78 min, 13%–80% buffer B from 78–80 min, 80% buffer B from 80–85 min, 100% buffer A from 86–91 min, 0%–13% buffer B from 91–94 min, 13%–80% buffer B from 94–96 min, 80% buffer B from 96–101 min, and 80%–0% buffer B from 101–102 min (buffer A: 10 mM aqueous NH4HCO3; buffer B: acetonitrile). Each well in the 96-well plate contained 20 μL of 20% formic acid to acidify the eluting peptides. The eluent was evaporated to dryness in the plate using SpeedVac vacuum concentrator. The peptides were resuspended in 80% acetonitrile, 0.1% formic acid buffer (125 μL/column) and every 12th fraction was combined. Samples were dried using SpeedVac vacuum concentrator, the resulting 12 combined fractions were re-suspended in buffer A (5% acetonitrile, 0.1% formic acid) and analyzed by mass spectrometry.
Parallel reaction monitoring (Related to Figure 6)
Dry peptide samples were reconstituted in a water/acetonitrile (85:15) mixture containing 0.1% formic acid (100 μL) and 15 μL was injected on to an EASY-Spray C18 loading column (5 μm particle size, 100 μm × 2 cm; Fisher Scientific, DX164564) and resolved on a custom analytical column (2 μM particle size, 75 μm × 15 cm) using a Dionex Ultimate 3000 nano-LC (Thermo Fisher Scientific). Peptides were separated over a 60-min gradient of 15 to 33% acetonitrile (0.1% formic acid) and analyzed on a Q-Exactive instrument (Thermo Fisher Scientific) using a parallel reaction monitoring method targeting the SF3B1 peptide containing C1111 (amino acids 1110 – 1149, missed tryptic, +3 charge state). Selected precursor ions were isolated and fragmented by high-energy collision dissociation and fragments were detected in the Orbitrap at 17,500 resolution.
Raw data files were uploaded analyzed in Skyline (v.21.1.0.278) to determine the abundance of each peptide in inhibitor-treated samples relative to vehicle-treated samples. Peptide quantification was performed by calculating the sum of the peak areas corresponding to six fragment ions from each peptide. The peptides and fragment ions were preselected from in-house reference spectral libraries acquired in data-dependent acquisition mode to identify authentic spectra for each peptide and normalized to internal retention time standards.
TMT liquid chromatography-mass-spectrometry (LC-MS) analysis
Samples were analyzed by liquid chromatography tandem mass-spectrometry using an Orbitrap Fusion Tribrid Mass Spectrometer (Thermo Scientific) or an Orbitrap Eclipse Tribrid Mass Spectrometer coupled to an UltiMate 3000 Series Rapid Separation LC system and autosampler (Thermo Scientific Dionex). The peptides were eluted onto a capillary column (75 μm inner diameter fused silica, packed with C18 (Waters, Acquity BEH C18, 1.7 μm, 25 cm)) or an EASY-Spray HPLC column (Thermo ES902, ES903) using an Acclaim PepMap 100 (Thermo 164535) loading column, and separated at a flow rate of 0.25 μL/min. Data was acquired using an MS3-based TMT method on Orbitrap Fusion or Orbitrap Eclipse Tribrid Mass Spectrometers. Briefly, for Fusion acquisition, the scan sequence began with an MS1 master scan (Orbitrap analysis, resolution 120,000, 400–1700 m/z, RF lens 60%, automatic gain control [AGC] target 2E5, maximum injection time 50 ms, centroid mode) with dynamic exclusion enabled (repeat count 1, duration 15 s). The top ten precursors were then selected for MS2/MS3 analysis. MS2 analysis consisted of quadrupole isolation (isolation window 0.7) of precursor ion followed by collision-induced dissociation (CID) in the ion trap (AGC 1.8E4, normalized collision energy 35%, maximum injection time 120 ms). Following the acquisition of each MS2 spectrum, synchronous precursor selection (SPS) enabled the selection of up to 10 MS2 fragment ions for MS3 analysis. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap (collision energy 55%, AGC 1.5E5, maximum injection time 120 ms, resolution was 50,000). For MS3 analysis, we used charge state–dependent isolation windows. For charge state z = 2, the MS isolation window was set at 1.2; for z = 3–6, the MS isolation window was set at 0.7. Scan parameters on Eclipse Tribrid Mass Spectrometers were the same with the following exceptions; full MS scan used RF lens 30%, standard AGC target and auto maximum injection time. MS2 scans in the Ion Trap used normalized collision energy 36%, standard AGC target and auto maximum injection time. MS3 SPS analysis used 30,000 resolution and 500% normalized AGC target and auto maximum injection time. The MS2 and MS3 files were extracted from the raw files using RAW Converter (version 1.1.0.22; available at http://fields.scripps.edu/rawconv/), uploaded to Integrated Proteomics Pipeline (IP2), and searched using the ProLuCID algorithm (publicly available at http://fields.scripps.edu/downloads.php) using a reverse concatenated, non-redundant variant of the Human UniProt database (release 2016). Cysteine residues were searched with a static modification for carboxyamidomethylation (+57.02146 Da). For cysteine profiling experiments, a dynamic modification for IA-DTB labeling (+398.25292 Da) was included with a maximum number of 2 differential modifications. N-termini and lysine residues were also searched with a static modification corresponding to the TMT tag (+229.1629 Da). Peptides were required to be at least 6 amino acids long, and to be fully tryptic, except for the GluC digested cysteine profiling samples which included K, R, E, and D cleavage sites. ProLuCID data was filtered through DTASelect (version 2.0) to achieve a peptide false-positive rate below 1%. The MS3-based peptide quantification was performed with reporter ion mass tolerance set to 20 ppm with Integrated Proteomics Pipeline (IP2).
Gene Ontology analysis (related to Figure 5)
A list of stereoselectively affected proteins was analyzed using GOnet tools103 (ontology version 2019-07-01) for GO term enrichment of biological processes using hierarchical output.
Proliferation and apoptosis assays (related to Figures 5–7)
Cells were seeded at 5000 cells per well (50 μL medium) in 96-well flat bottom white wall plates. After 24 h, 50 μL of medium containing DMSO or compound dilutions (2x final concentration from 1000x stocks) were added to the wells. At the time of compound addition, a reference plate to determine the cell population density at time 0 (T0) was assayed using CellTiter-Glo (Promega) reagent (50 μL added to each well). After 72 h in culture, the remaining plates were assayed using CellTiter-Glo. After 30 min of shaking at room temperature, luminescence was analyzed using a CLARIOstar (BMG Labtech) plate reader. Apoptosis assays were performed similarly as described above using Caspase-Glo 3/7 (Promega) following 24 h compound treatments. Raw values were normalized using GraphPad PRISM software version 9.0.0. Lines of best fit were generated using a four-parameter variable slope least squares regression. Proliferation assays linked to HCT-116 DDX42-dTAG cells were performed similarly, except that compound was prepared in a new 96-well plate as a 1:sqrt(10) serial dilution with 9 dose points (plus 1 untreated well) and adding 50 μL of this drug/medium solution to the 50 μL cells.
Chemical proteomic experiments with alkyne compounds (related to Figure 5)
22Rv1 cells were treated with indicated tryptoline acrylamides for 2 h, followed by incubation with corresponding alkyne analogues for 1 h, harvested and lysed in PBS with protease inhibitors (cOmplete), and then normalized to 2.0 mg/mL. Reagents for CuAAC conjugation to a biotin-PEG4-azide tag were pre-mixed prior to addition to the samples (55 μL of CuAAC reaction mix for 500 μL of lysate), as described previously described104. After 1 h, samples were precipitated with the addition of ice-cold MeOH (600 μL), CHCl3 (180 μL), and H20 (500 μL), and vortexed and centrifuged (16,000 g, 10 minutes, 4 °C). The top layer above the protein disc was aspirated and an additional 1mL of ice-cold MeOH was added. The samples were again vortexed and centrifuged (16,000 g, 10 minutes, 4 °C), the supernatant aspirated and the protein pellets were allowed to air dry and be stored at −80 °C or proceeded to resuspension and enrichment. Pellets were resuspended in 500 μL 8 M urea (in DPBS) with 10 μL 10% SDS and sonicated to ensure no large precipitate remained. Samples were reduced with 25 μL of 200 mM DTT and incubated at 65 °C for 15 minutes, then reduced with addition of 25 μL of 400 mM iodoacetamide and incubated in the dark at 37 °C for 30 minutes. For enrichment, 130 μL 10% SDS was added and then samples were diluted to 5.5 mL DPBS. Samples were enrichment with streptavidin beads (Thermo cat # 20353; 100 μL/sample) and incubated at room temperature for 1.5 h while rotating. After incubation, samples were pelleted by centrifugation (2,000 g, 2 min) and beads were washed with 0.2% SDS in DPBS (2 × 10 mL), DPBS (1 × 5 mL), then transferred to low-bind Eppendorf (cat # 0030108442), washed with water (2 × 1 mL), and 200 mM EPPS (1mL). Pelleted beads were resuspended in 200 μL 2 M urea (in 200 mM EPPS) and digested with 2 μg Trypsin with 1 mM CaCl2 (final concentration) overnight at 37 °C. Digested peptides were transferred to new low-bind Eppendorf and acetonitrile added (to 30% final) and TMT labeled with 6 μL of corresponding TMT tag (20 μg/μL), vortexed, and incubated at room temperature for 1 h. TMT labeling was quenched with the addition of hydroxylamine (6 μL 5% solution in H2O) and incubated for 15 minutes at room temperature. Samples were then acidified with 15 μL formic acid, combined and dried using a SpeedVac. Samples were desalted and high pH fractionated using Peptide Desalting Spin Columns (Thermo 89852) to a final of 3 fractions and analyzed by mass spectrometry.
Gel-ABPP for SF3B1 cysteine engagement (related to Figure 6)
22Rv1 or HCT-116 cells were treated with indicated compounds for 2–24 h, chased with alkyne probe for 1 h in situ, harvested and lysed in PBS with protease inhibitors (cOmplete), and then normalized to 2.0 mg/mL. Reagents for the CuAAC reaction were pre-mixed prior to addition to the samples (3 μL of CuAAC reaction mix for 25 μL of lysate), as previously reported6. After 1 h of labeling, 4x SDS loading buffer was added to samples, which were then analyzed by SDS-PAGE.
IP-ABPP:
HCT-116 DDX42-dTAG cells were treated in situ with DMSO or dTAGv-1 (500 nM) for 1 h. The samples were then treated with WX-01-10 or WX-01-12 (2.5 μM) for 1 h. The cells were washed with ice-cold DPBS and then collected via scraping. The pellets were flash frozen in liquid nitrogen and stored at −80 °C until analysis. The pellets were suspended in 0.5 mL DPBS containing 1.5 mM MgCl2, 1% NP40, and 1 mM PMSF (1% DMSO v/v final). The cells were lysed via sonication (1 × 10, 10%) on ice and then treated with Pierce Universal nuclease (1 μL). The samples were incubated for 1.5 h at 4 °C with rotation. The samples were then re-sonicated (1 × 10, 10%) and centrifuged at 16,000 RPM for 10 min. The supernatant was quantified via DC assay (BioRad) and normalized to 2 mg/mL total (550 μL each). 50 μL input was collected and treated with CuAAC mixture for 1 h at room temperature and then it was quenched with 4x loading buffer (store at −20 °C prior to analysis). The remaining sample was treated with 1 μg rabbit anti-SF3B1 overnight at 4 °C with rotation. Then 25 μL Pierce Protein A plus resin (pre-washed in DPBS supplemented with 0.2% NP40, suspended in 75 μL final) was added to each sample and allowed to incubate at 4 °C for 4 h. The beads were washed 3 × 1 mL with ice-cold DPBS containing 0.2% NP40 (pellet at 2000 rpm for 2 min to pellet beads). The beads were suspended in 50 μL DPBS and treated with CuAAC mixture for 1 h at room temperature. The SF3B1 was eluted by addition of 4x loading buffer and then heated for 10 min at 95 °C. The supernatant was collected and analyzed by SDS-PAGE. proteins were resolved using SDS-PAGE (10% Tris-glycine gel) and transferred to a methanol pre-activated PVDF membrane (60 V for 2 h). The membrane was blocked with 5% milk in Tris-buffered saline (20 mM Tris-HCl 7.6, 150 mM NaCl) with 0.1% tween (TBST) and incubated with primary antibody overnight. After TBST wash (3 times), the membrane was incubated with secondary antibody (mouse anti-rabbit, sc-2357), washed with TBST again, developed with ECL western blotting detection reagents (SuperSignal™ West Pico PLUS Chemiluminescent Substrate), and recorded on a Bio-Rad ChemiDoc MP.
RNA sequencing and analysis of differential gene expression (related to Figures 6 and 7)
Total RNA from compound or DMSO treated 22Rv1 or HCT-116 DDX42-dTAG cells was isolated using QIAshredder and RNeasy Mini Plus Kits (QIAGEN) according to manufacturer’s protocol and stored at −80 °C until further analysis. RNA sequencing libraries were prepared using the NEBNext Ultra II RNA Library Prep Kit for Illumina following manufacturer’s instructions (NEB, Ipswich, MA, USA). Briefly, mRNAs were first enriched with Oligo(dT) beads. Enriched mRNAs were fragmented for 15 minutes at 94 °C. First strand and second strand cDNAs were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3’ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by limited-cycle PCR. The sequencing libraries were validated on the Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA), and quantified by using Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA, USA). FASTQ files were first trimmed using Trim_galore (v0.6.4)105 to remove sequencing adapters and low quality (Q<15) reads. Trimmed sequencing reads were aligned to the human Hg19 reference genome (GENCODE, GRCh37.p13) using STAR (v2.7.5)106. SAM files were subsequently converted to BAM files, sorted, and indexed using samtools (v1.9)107. BAM files were used to generate bigwig files using bamCoverage (part of the Deeptools package; v3.3.1)108. Read counting across genomic features was performed using featureCounts (part of the subread package; v1.5.0)109 using the following parameters: -p -T 20 -O -F GTF -t exon. Differential gene expression analysis was performed using the edgeR (v3.32.1)110, DESeq2 (v1.30.1)111, and limma voom (v3.46.0)112 R packages. Data visualization and figure generation was performed in Rstudio (v1.3.1073) using the following packages: ggplot2 (v3.3.5), ggpubr (v0.4.0), complexHeatmap (v2.6.2), and VennDiagram (v1.6.0).
For quantification of alternative RNA splicing, fastq files are adapter-trimmed by cutadapt 3.4105 and aligned to GRCh38 by STAR 2.7.6106 and analyzed using rMATS (v4.1.1)113 using the GENCODE (v35) GTF annotation for GRCh38 and the following parameters: -t paired --libType fr-unstranded –readLength 150 --novelSS. Enumeration of isoform counts was performed using only reads that span the splice junction directly. To identify high confidence AS events, events were considered significant if (i) the inclusion level difference was greater than 10% compared to DMSO, (ii) the False Discovery Rate (FDR) was smaller than 0.05. For comparison of splicing events between conditions, we further limited to highly covered events where at least a total of 100 junctional reads cover the isoform for all conditions. The Inclusion level difference for each AS event were used to cluster the conditions. Data analysis and visualization was performed using custom scripts in python (3.7.12), pandas, scikit-learn, seaborn, Rstudio (v1.3.1073) using the following packages: ggplot2 (v3.3.5), ggrepel (v0.9.1), maser (v1.8.0), and VennDiagram (v1.6.0).
Branchpoint analysis:
Branchpoint analysis was performed using Branchpointer (https://github.com/signalbash/branchpointer)114, which is a machine learning model trained using sequence feature surrounding the 3′ splice site. To identify branchpoint associated with each isoform, we found −18 to −44 nucleotide upstream of 3′ splice site and fed into the model. We filtered with the suggested scoring threshold (prediction probability > 0.52) from the software for high confidence branchpoints. U2 binding energy predicted by branchpointer was then used to compare the difference between isoforms.
SF3B1 Co-immunoprecipitation studies (related to Figure 6 and 7)
HEK293T or HCT-116 DDX42-dTAG cells were treated in situ with DMSO, 5 μM of WX-02-23/WX-02-43, or 10 nM pladienolide B for 3 h. Cells were harvested, washed with ice-cold PBS, and lysed in CoIP lysis buffer (0.5% NP-40, 100 mM EPPS, 150 mM NaCl, cOmplete protease inhibitor). Lysates were clarified at 16,000 g for 3 minutes, normalized to 2.0 mg/mL and 500 μL of each lysate was aliquoted to a clean eppendorf tube.
Lysates were incubated with 5 μL of anti-IgG or anti-SF3B1 (CST) for 2 h at 4 °C, followed by 1 h with 25 μL of protein-A agarose beads. Beads were washed twice with CoIP lysis buffer, and then twice with 100 mM EPPS, followed by elution with 8 M urea in EPPS at 65 °C for 10 min. Eluates were reduced with DTT at 65 °C for 15 min (2.5 μL of 200 mM = 12.5 mM final), alkylated with iodoacetamide at 37 °C for 30 min (2.5 μL of 400 mM = 25 mM final), and diluted to 2 M urea by addition of 115 μL of EPPS. Samples were then trypsinized at 37 °C overnight (2 μg of trypsin per sample), then TMT labeled and desalted as described above.
A similar experiment was performed, where HCT-116 DDX42-dTAG cells were treated with 100 nM pladienolide B and the compound was included in the lysis buffer during lysis and immunoprecipitation to assess if any observed interactome differences between WX-02-23 and pladienolide B were due to washout of the reversibly binding pladienolide B during cell lysis/IP.
Enhanced CLIP-seq (related to Figure 7)
Library preparation:
5 million cells were seeded in 10 mL media in a 10 cm plate and grown for 20h. Media was then aspirated and replaced with 7 mL media containing the indicated compounds or 0.1% DMSO for 3h. Following this incubation period, media was aspirated, cells were washed with PBS and protein–RNA interactions were stabilized with UV crosslinking (254 nm, 400 mJ/cm2) in cold DPBS. Cells were scraped, harvested and pelleted in 1.5 mL tubes (2000 g, 3 min, 4°C). Cell pellets were flash-frozen in liquid N2 and stored at −80°C until further analysis. Thawed pellets were then lysed in 1 mL of iCLIP lysis buffer and digested with RNase I (Ambion). Immunoprecipitation of HA-DDX42–RNA complexes with HA-Tag (C29F4) Rabbit mAb (10 μg per mL of lysate) was performed using magnetic beads with pre-coupled secondary antibody (M-280 Sheep Anti-Rabbit IgG Dynabeads, ThermoFisher Scientific 11204D) and beads were stringently washed. After dephosphorylation with FastAP (ThermoFisher) and T4 PNK (NEB), a barcoded RNA adapter was ligated to the 3′ end (T4 RNA Ligase, NEB). Ligations were performed on-bead (to allow washing away unincorporated adapter) in high concentration of PEG8000. Samples were then run on standard protein gels and transferred to nitrocellulose membranes, and a region 75 kDa (~220 nt of RNA) above the protein size was isolated and proteinase K (NEB) treated to isolate RNA. RNA was reverse transcribed with Superscript III (Invitrogen), and treated with ExoSAP-IT (Affymetrix) to remove excess oligonucleotides. A second DNA adapter (containing a random-mer of 10 (N10) random bases at the 5′ end) was then ligated to the cDNA fragment 3′ end (T4 RNA Ligase, NEB), performed with high concentration of PEG8000 (to improve ligation efficiency) and DMSO (to decrease inhibition of ligation due to secondary structure). After cleanup (Dynabeads MyOne Silane, ThermoFisher), an aliquot of each sample was first subjected to qPCR (to identify the proper number of PCR cycles), and then the remainder was PCR amplified (Q5, NEB) and size selected via agarose gel electrophoresis. Samples were sequenced on the Illumina NovaSeq 6000 platform as two single-end 75bp reads.
Data analysis:
After standard NovaSeq demultiplexing, eCLIP libraries processed with Skipper (https://github.com/YeoLab/skipper)65. Briefly, reads were adapter trimmed with skewer (https://github.com/relipmoc/skewer)115 and reads less than 20 bp were discarded. UMI were extracted using fastp 0.11.5 (https://github.com/OpenGene/fastp)116. Mapping was then performed against the full human genome (hg38) including a database of splice junctions with STAR (v 2.7.6)106, allowing up to 100 multimapped regions. When multimapping occurred, reads are randomly assigned to any of the matching position. Reads were then PCR duplicated using UMIcollaps (https://github.com/Daniel-Liu-c0deb0t/UMICollapse)117. Enriched “windows” (IP versus SM-Input) was called on deduplicated reads using a GC-bias aware beta-binomial model. Each window (~100 b.p.) were partitioned from Gencode v38 and associated with a specific type of genomic region (CDS, UTR, proximal introns near splice site… etc). Windows are filtered using FDR < 0.2 and only reproducible windows between two replicates are used.
Differential binding analysis:
To analyze how WX-02-23, WX-02-43 and PladB change DDX42 binding, we ran skipper with the same method as described above but replaced the SM-Input library with the DMSO IP library. This way, the output from skipper represents windows that contain enriched binding after treatment, compared to DMSO.
Metagene analysis:
To visualize differential eCLIP densities at near-nucleotide resolution, we used Metadensity118 to calculate relative information content (RI). RI values here represents differential binding of WX-02-23/WX-02-43/PladB compared to DMSO as the background. Specifically, for each nucleotide position in a transcript, , where and are the fraction of total reads in IP from either WX-02-23/WX-02-43/PladB and DMSO-treated IP libraries respectively that map to nucleotide i. We averaged the relative information over all transcripts with at least one differential enriched window detected by skipper (see section “Differential binding analysis”).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise stated, quantitative data are expressed in bar and line graphs as mean ± SD (error bar) shown. SEC elution profiles are expressed as mean ± SEM (error bar). Unless otherwise stated, differences between two groups were examined using an unpaired two-tailed Student’s t-test with equal or unequal variance as noted. Significant P values are indicated (*P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001).
Supplementary Material
Dataset S1. Proteomic and RNA-seq data and Reference Tables, Related to Figures 1, 2, 3, 4, 5, 6, and 7.
Dataset S2. eCLIP data, Related to Figure 7
Highlights.
Co-fractionation proteomics identifies covalent ligands altering protein complexes
Activity-based protein profiling identifies cysteines targeted by covalent ligands
Covalent probes are discovered that disrupt the PA28 proteasomal regulatory complex
Covalent probes are discovered that functionally remodel the spliceosome
ACKNOWLEDGEMENTS
We thank M. Hayward, S. Niessen, H. Murrey, B. Martin, R. Park, and A. Cognetta for helpful discussions, and B. Chen and X. Lieu (WuXi AppTec) for compound synthesis. This work was supported by the NIH (R35CA231991 (B.F.C.) F32CA265211 (C.J.R.)), American Cancer Society (J.R.R., PF-18-217-01-CDD), Vividion Therapeutics, Janssen Pharmaceuticals, Pfizer, an EMBO long-term fellowship (M.G.J., EMBO ALTF 255-2021), a Jane Coffin Childs Memorial Fellowship (K.E.D.), an HHMI Hanna H Gray Fellowship (E.N., GT15176), and a JSPS overseas research fellowship (D.O.). G.W.Y. is supported by NIH R01 HG004659, U24 HG009889 and an Allen Distinguished Investigator Award, a Paul G. Allen Frontiers Group advised grant of the Paul G. Allen Foundation.
Footnotes
DECLARATION OF INTERESTS
G.M. Simon, V.F. Vartabedian, and L.R. Whitby are employees of Vividion Therapeutics, and B.F. Cravatt is a founder and member of the Board of Directors of Vividion Therapeutics. G.W.Y. is a co-founder, member of the Board of Directors, on the SAB, equity holder, and paid consultant for Locanabio and Eclipse BioInnovations. G.W.Y.’s interests have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies. A US provisional patent has been filed related to the work disclosed in this manuscript. The authors declare no other competing financial interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as a gender minority in their field of research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Schreiber SL (2019). A Chemical Biology View of Bioactive Small Molecules and a Binder-Based Approach to Connect Biology to Precision Medicines. Israel Journal of Chemistry 59, 52–59. 10.1002/ijch.201800113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu W, Kostic M, Zhang T, Che J, Patricelli MP, Jones LH, Chouchani ET, and Gray NS (2021). Fragment-based covalent ligand discovery. RSC Chem Biol 2, 354–367. 10.1039/d0cb00222d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott DE, Coyne AG, Hudson SA, and Abell C (2012). Fragment-based approaches in drug discovery and chemical biology. Biochemistry 51, 4990–5003. 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- 4.Brenner S, and Lerner RA (1992). Encoded combinatorial chemistry. Proc Natl Acad Sci U S A 89, 5381–5383. 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gironda-Martinez A, Donckele EJ, Samain F, and Neri D (2021). DNA-Encoded Chemical Libraries: A Comprehensive Review with Succesful Stories and Future Challenges. ACS Pharmacol Transl Sci 4, 1265–1279. 10.1021/acsptsci.1c00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González-Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. (2016). Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parker CG, Galmozzi A, Wang Y, Correia BE, Sasaki K, Joslyn CM, Kim AS, Cavallaro CL, Lawrence RM, Johnson SR, et al. (2017). Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 168, 527–541 e529. 10.1016/j.cell.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spradlin JN, Zhang E, and Nomura DK (2021). Reimagining Druggability Using Chemoproteomic Platforms. Acc Chem Res 54, 1801–1813. 10.1021/acs.accounts.1c00065. [DOI] [PubMed] [Google Scholar]
- 9.Maurais AJ, and Weerapana E (2019). Reactive-cysteine profiling for drug discovery. Curr Opin Chem Biol 50, 29–36. 10.1016/j.cbpa.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J-P, Subramanian A, Ross KN, et al. (2006). The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 313, 1929–1935. 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437–1452 e1417. 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruprecht B, Di Bernardo J, Wang Z, Mo X, Ursu O, Christopher M, Fernandez RB, Zheng L, Dill BD, Wang H, et al. (2020). A mass spectrometry-based proteome map of drug action in lung cancer cell lines. Nat Chem Biol 16, 1111–1119. 10.1038/s41589-020-0572-3. [DOI] [PubMed] [Google Scholar]
- 13.Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Ruepp A (2019). CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res 47, D559–d563. 10.1093/nar/gky973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.UniProt Consortium, T. (2018). UniProt: the universal protein knowledgebase. Nucleic Acids Res 46, 2699. 10.1093/nar/gky092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arkin MR, Tang Y, and Wells JA (2014). Small-molecule inhibitors of protein-protein interactions: progressing towards the reality. Chemistry & biology 21, 1102–1114. 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin L, Wang W, and Fang G (2014). Targeting protein-protein interaction by small molecules. Annu Rev Pharmacol Toxicol 54, 435–456. 10.1146/annurev-pharmtox-011613-140028. [DOI] [PubMed] [Google Scholar]
- 17.Schreiber SL (2021). The Rise of Molecular Glues. Cell 184, 3–9. 10.1016/j.cell.2020.12.020. [DOI] [PubMed] [Google Scholar]
- 18.Fields S, and Song O. k. (1989). A novel genetic system to detect protein–protein interactions. Nature 340, 245–246. 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 19.Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, et al. (2021). Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040.e3028. 10.1016/j.cell.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirkwood KJ, Ahmad Y, Larance M, and Lamond AI (2013). Characterization of Native Protein Complexes and Protein Isoform Variation Using Size-fractionation-based Quantitative Proteomics. Molecular & Cellular Proteomics : MCP 12, 3851–3873. 10.1074/mcp.M113.032367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kristensen AR, Gsponer J, and Foster LJ (2012). A high-throughput approach for measuring temporal changes in the interactome. Nat Meth 9, 907–909. http://www.nature.com/nmeth/journal/v9/n9/abs/nmeth.2131.html#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heusel M, Bludau I, Rosenberger G, Hafen R, Frank M, Banaei-Esfahani A, van Drogen A, Collins BC, Gstaiger M, and Aebersold R (2019). Complex-centric proteome profiling by SEC-SWATH-MS. Mol Syst Biol 15, e8438. 10.15252/msb.20188438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mallam AL, Sae-Lee W, Schaub JM, Tu F, Battenhouse A, Jang YJ, Kim J, Wallingford JB, Finkelstein IJ, Marcotte EM, and Drew K (2019). Systematic Discovery of Endogenous Human Ribonucleoprotein Complexes. Cell reports 29, 1351–1368.e1355. 10.1016/j.celrep.2019.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skinnider MA, and Foster LJ (2021). Meta-analysis defines principles for the design and analysis of co-fractionation mass spectrometry experiments. Nature Methods 18, 806–815. 10.1038/s41592-021-01194-4. [DOI] [PubMed] [Google Scholar]
- 25.Smith MC, and Gestwicki JE (2012). Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev Mol Med 14, e16. 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashkenazi A, Fairbrother WJ, Leverson JD, and Souers AJ (2017). From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nature reviews. Drug discovery 16, 273–284. 10.1038/nrd.2016.253. [DOI] [PubMed] [Google Scholar]
- 27.Bar-Peled L, Kemper EK, Suciu RM, Vinogradova EV, Backus KM, Horning BD, Paul TA, Ichu T-A, Svensson RU, Olucha J, et al. (2017). Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 171, 696–709.e623. 10.1016/j.cell.2017.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hacker SM, Backus KM, Lazear MR, Forli S, Correia BE, and Cravatt BF (2017). Global profiling of lysine reactivity and ligandability in the human proteome. Nature Chemistry 9, 1181–1190. 10.1038/nchem.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harvey EP, Hauseman ZJ, Cohen DT, Rettenmaier TJ, Lee S, Huhn AJ, Wales TE, Seo HS, Luccarelli J, Newman CE, et al. (2020). Identification of a Covalent Molecular Inhibitor of Anti-apoptotic BFL-1 by Disulfide Tethering. Cell Chem Biol 27, 647–656.e646. 10.1016/j.chembiol.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akçay G, Belmonte MA, Aquila B, Chuaqui C, Hird AW, Lamb ML, Rawlins PB, Su N, Tentarelli S, Grimster NP, and Su Q (2016). Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat Chem Biol 12, 931–936. 10.1038/nchembio.2174. [DOI] [PubMed] [Google Scholar]
- 31.Zhang M, Aguilar A, Xu S, Huang L, Chinnaswamy K, Sleger T, Wang B, Gross S, Nicolay BN, Ronseaux S, et al. (2021). Discovery of M-1121 as an Orally Active Covalent Inhibitor of Menin-MLL Interaction Capable of Achieving Complete and Long-Lasting Tumor Regression. J Med Chem 64, 10333–10349. 10.1021/acs.jmedchem.1c00789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vinogradova EV, Zhang X, Remillard D, Lazar DC, Suciu RM, Wang Y, Bianco G, Yamashita Y, Crowley VM, Schafroth MA, et al. (2020). An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 182, 1009–1026.e1029. 10.1016/j.cell.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuljanin M, Mitchell DC, Schweppe DK, Gikandi AS, Nusinow DP, Bulloch NJ, Vinogradova EV, Wilson DL, Kool ET, Mancias JD, et al. (2021). Reimagining high-throughput profiling of reactive cysteines for cell-based screening of large electrophile libraries. Nat Biotechnol 39, 630–641. 10.1038/s41587-020-00778-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grossman EA, Ward CC, Spradlin JN, Bateman LA, Huffman TR, Miyamoto DK, Kleinman JI, and Nomura DK (2017). Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-cancer Natural Products. Cell Chem Biol 24, 1368–1376.e1364. 10.1016/j.chembiol.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, and Hamon C (2003). Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75, 1895–1904. 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 36.Tao Y, Remillard D, Vinogradova EV, Yokoyama M, Banchenko S, Schwefel D, Melillo B, Schreiber SL, Zhang X, and Cravatt BF (2022). Targeted Protein Degradation by Electrophilic PROTACs that Stereoselectively and Site-Specifically Engage DCAF1. J Am Chem Soc 144, 18688–18699. 10.1021/jacs.2c08964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schreiber Stuart L (2000). Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 287, 1964–1969. 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 38.Huber EM, and Groll M (2017). The Mammalian Proteasome Activator PA28 Forms an Asymmetric α(4)β(3) Complex. Structure 25, 1473–1480.e1473. 10.1016/j.str.2017.07.013. [DOI] [PubMed] [Google Scholar]
- 39.Chen J, Wang Y, Xu C, Chen K, Zhao Q, Wang S, Yin Y, Peng C, Ding Z, and Cong Y (2021). Cryo-EM of mammalian PA28αβ-iCP immunoproteasome reveals a distinct mechanism of proteasome activation by PA28αβ. Nat Commun 12, 739–739. 10.1038/s41467-021-21028-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Groettrup M, Ruppert T, Kuehn L, Seeger M, Standera S, Koszinowski U, and Kloetzel PM (1995). The Interferon-inducible 11 S Regulator (PA28) and the LMP2/LMP7 Subunits Govern the Peptide Production by the 20 S Proteasome in vitro. Journal of Biological Chemistry 270, 23808–23815. 10.1074/jbc.270.40.23808. [DOI] [PubMed] [Google Scholar]
- 41.Yamano T, Murata S, Shimbara N, Tanaka N, Chiba T, Tanaka K, Yui K, and Udono H (2002). Two distinct pathways mediated by PA28 and hsp90 in major histocompatibility complex class I antigen processing. J Exp Med 196, 185–196. 10.1084/jem.20011922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raule M, Cerruti F, Benaroudj N, Migotti R, Kikuchi J, Bachi A, Navon A, Dittmar G, and Cascio P (2014). PA28αβ reduces size and increases hydrophilicity of 20S immunoproteasome peptide products. Chem Biol 21, 470–480. 10.1016/j.chembiol.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Barf T, Covey T, Izumi R, van de Kar B, Gulrajani M, van Lith B, van Hoek M, de Zwart E, Mittag D, Demont D, et al. (2017). Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. The Journal of pharmacology and experimental therapeutics 363, 240–252. 10.1124/jpet.117.242909. [DOI] [PubMed] [Google Scholar]
- 44.Keller M, Ebstein F, Bürger E, Textoris-Taube K, Gorny X, Urban S, Zhao F, Dannenberg T, Sucker A, Keller C, et al. (2015). The proteasome immunosubunits, PA28 and ER-aminopeptidase 1 protect melanoma cells from efficient MART-126-35-specific T-cell recognition. European journal of immunology 45, 3257–3268. 10.1002/eji.201445243. [DOI] [PubMed] [Google Scholar]
- 45.Shunji S, Toru A, and Katsuo K (1987). A simple method to eliminate the antigenicity of surface class I MHC molecules from the membrane of viable cells by acid treatment at pH 3. Journal of Immunological Methods 100, 83–90. 10.1016/0022-1759(87)90175-X. [DOI] [PubMed] [Google Scholar]
- 46.Sturm T, Sautter B, Wörner TP, Stevanović S, Rammensee H-G, Planz O, Heck AJR, and Aebersold R (2021). Mild Acid Elution and MHC Immunoaffinity Chromatography Reveal Similar Albeit Not Identical Profiles of the HLA Class I Immunopeptidome. Journal of Proteome Research 20, 289–304. 10.1021/acs.jproteome.0c00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Purcell AW, Ramarathinam SH, and Ternette N (2019). Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat Protoc 14, 1687–1707. 10.1038/s41596-019-0133-y. [DOI] [PubMed] [Google Scholar]
- 48.Duncan LM, Timms RT, Zavodszky E, Cano F, Dougan G, Randow F, and Lehner PJ (2012). Fluorescence-based phenotypic selection allows forward genetic screens in haploid human cells. PLoS One 7, e39651. 10.1371/journal.pone.0039651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mallick P, Schirle M, Chen SS, Flory MR, Lee H, Martin D, Ranish J, Raught B, Schmitt R, Werner T, et al. (2007). Computational prediction of proteotypic peptides for quantitative proteomics. Nature Biotechnology 25, 125–131. 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 50.Larsen NA (2021). The SF3b Complex is an Integral Component of the Spliceosome and Targeted by Natural Product-Based Inhibitors. Sub-cellular biochemistry 96, 409–432. 10.1007/978-3-030-58971-4_12. [DOI] [PubMed] [Google Scholar]
- 51.Lee SC, and Abdel-Wahab O (2016). Therapeutic targeting of splicing in cancer. Nature medicine 22, 976–986. 10.1038/nm.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, and Mizui Y (2007). Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nature Chemical Biology 3, 570–575. 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- 53.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, et al. (2007). Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol 3, 576–583. 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 54.Cretu C, Agrawal AA, Cook A, Will CL, Fekkes P, Smith PG, Lührmann R, Larsen N, Buonamici S, and Pena V (2018). Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Molecular Cell 70, 265–273.e268. 10.1016/j.molcel.2018.03.011. [DOI] [PubMed] [Google Scholar]
- 55.Cretu C, Gee P, Liu X, Agrawal A, Nguyen T-V, Ghosh AK, Cook A, Jurica M, Larsen NA, and Pena V (2021). Structural basis of intron selection by U2 snRNP in the presence of covalent inhibitors. Nat Commun 12, 4491. 10.1038/s41467-021-24741-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Foy A, and McMullin MF (2019). Somatic SF3B1 mutations in myelodysplastic syndrome with ring sideroblasts and chronic lymphocytic leukaemia. Journal of Clinical Pathology 72, 778–782. [DOI] [PubMed] [Google Scholar]
- 57.Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. (2018). H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nature medicine 24, 497–504. 10.1038/nm.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teng T, Tsai JH, Puyang X, Seiler M, Peng S, Prajapati S, Aird D, Buonamici S, Caleb B, Chan B, et al. (2017). Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat Commun 8, 15522. 10.1038/ncomms15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Will CL, Urlaub H, Achsel T, Gentzel M, Wilm M, and Luhrmann R (2002). Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J 21, 4978–4988. 10.1093/emboj/cdf480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonaventure B, Rebendenne A, Chaves Valadao AL, Arnaud-Arnould M, Gracias S, Garcia de Gracia F, McKellar J, Labaronne E, Tauziet M, Vivet-Boudou V, et al. (2022). The DEAD box RNA helicase DDX42 is an intrinsic inhibitor of positive-strand RNA viruses. EMBO Rep, e54061. 10.15252/embr.202154061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uhlmann-Schiffler H, Jalal C, and Stahl H (2006). Ddx42p--a human DEAD box protein with RNA chaperone activities. Nucleic Acids Res 34, 10–22. 10.1093/nar/gkj403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. (2017). Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274. 10.1038/nature21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nabet B, Ferguson FM, Seong BKA, Kuljanin M, Leggett AL, Mohardt ML, Robichaud A, Conway AS, Buckley DL, Mancias JD, et al. (2020). Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat Commun 11, 4687. 10.1038/s41467-020-18377-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13, 508–514. 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boyle EA, Her H-L, Mueller JR, Nguyen GG, and Yeo GW (2022). Skipper analysis of RNA-protein interactions highlights depletion of genetic variation in translation factor binding sites. bioRxiv, 2022.2010.2008.511447. 10.1101/2022.10.08.511447. [DOI] [Google Scholar]
- 66.Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen JY, Cody NAL, Dominguez D, et al. (2020). A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719. 10.1038/s41586-020-2077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Z, Will CL, Bertram K, Dybkov O, Hartmuth K, Agafonov DE, Hofele R, Urlaub H, Kastner B, Luhrmann R, and Stark H (2020). Molecular architecture of the human 17S U2 snRNP. Nature 583, 310–313. 10.1038/s41586-020-2344-3. [DOI] [PubMed] [Google Scholar]
- 68.Haselbach D, Komarov I, Agafonov DE, Hartmuth K, Graf B, Dybkov O, Urlaub H, Kastner B, Luhrmann R, and Stark H (2018). Structure and Conformational Dynamics of the Human Spliceosomal B(act) Complex. Cell 172, 454–464 e411. 10.1016/j.cell.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 69.Yang F, Bian T, Zhan X, Chen Z, Xing Z, Larsen NA, Zhang X, and Shi Y (2023). Mechanisms of the RNA helicases DDX42 and DDX46 in human U2 snRNP assembly. Nat Commun 14, 897. 10.1038/s41467-023-36489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jones LH (2019). Expanding chemogenomic space using chemoproteomics. Bioorg Med Chem 27, 3451–3453. 10.1016/j.bmc.2019.06.022. [DOI] [PubMed] [Google Scholar]
- 71.Cuesta A, and Taunton J (2019). Lysine-Targeted Inhibitors and Chemoproteomic Probes. Annu Rev Biochem 88, 365–381. 10.1146/annurev-biochem-061516-044805. [DOI] [PubMed] [Google Scholar]
- 72.Backus KM (2019). Applications of Reactive Cysteine Profiling. Curr Top Microbiol Immunol 420, 375–417. 10.1007/82_2018_120. [DOI] [PubMed] [Google Scholar]
- 73.Liu Y, Patricelli MP, and Cravatt BF (1999). Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A 96, 14694–14699. 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Greenbaum D, Medzihradszky KF, Burlingame A, and Bogyo M (2000). Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem Biol 7, 569–581. 10.1016/s1074-5521(00)00014-4. [DOI] [PubMed] [Google Scholar]
- 75.Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, and Kozarich JW (2007). Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 46, 350–358. 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- 76.Dytfeld D, Luczak M, Wrobel T, Usnarska-Zubkiewicz L, Brzezniakiewicz K, Jamroziak K, Giannopoulos K, Przybylowicz-Chalecka A, Ratajczak B, Czerwinska-Rybak J, et al. (2016). Comparative proteomic profiling of refractory/relapsed multiple myeloma reveals biomarkers involved in resistance to bortezomib-based therapy. Oncotarget 7, 56726–56736. 10.18632/oncotarget.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brunner AM, and Steensma DP (2020). Targeting Aberrant Splicing in Myelodysplastic Syndromes: Biologic Rationale and Clinical Opportunity. Hematol Oncol Clin North Am 34, 379–391. 10.1016/j.hoc.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.DeNicola AB, and Tang Y (2019). Therapeutic approaches to treat human spliceosomal diseases. Curr Opin Biotechnol 60, 72–81. 10.1016/j.copbio.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Feldman HC, Merlini E, Guijas C, DeMeester KE, Njomen E, Kozina EM, Yokoyama M, Vinogradova E, Reardon HT, Melillo B, et al. (2022). Selective inhibitors of SARM1 targeting an allosteric cysteine in the autoregulatory ARM domain. Proc Natl Acad Sci U S A 119, e2208457119. 10.1073/pnas.2208457119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim Y. k., Arai MA, Arai T, Lamenzo JO, Dean EF, Patterson N, Clemons PA, and Schreiber SL (2004). Relationship of Stereochemical and Skeletal Diversity of Small Molecules to Cellular Measurement Space. Journal of the American Chemical Society 126, 14740–14745. 10.1021/ja048170p. [DOI] [PubMed] [Google Scholar]
- 81.Uzozie AC, and Aebersold R (2018). Advancing translational research and precision medicine with targeted proteomics. Journal of Proteomics 189, 1–10. 10.1016/j.jprot.2018.02.021. [DOI] [PubMed] [Google Scholar]
- 82.Brulet JW, Borne AL, Yuan K, Libby AH, and Hsu KL (2020). Liganding Functional Tyrosine Sites on Proteins Using Sulfur-Triazole Exchange Chemistry. J Am Chem Soc 142, 8270–8280. 10.1021/jacs.0c00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abbasov ME, Kavanagh ME, Ichu TA, Lazear MR, Tao Y, Crowley VM, Am Ende CW, Hacker SM, Ho J, Dix MM, et al. (2021). A proteome-wide atlas of lysine-reactive chemistry. Nat Chem. 10.1038/s41557-021-00765-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wheat A, Yu C, Wang X, Burke AM, Chemmama IE, Kaake RM, Baker P, Rychnovsky SD, Yang J, and Huang L (2021). Protein interaction landscapes revealed by advanced in vivo cross-linking-mass spectrometry. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2023360118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kleiner RE, Hang LE, Molloy KR, Chait BT, and Kapoor TM (2018). A Chemical Proteomics Approach to Reveal Direct Protein-Protein Interactions in Living Cells. Cell Chem Biol 25, 110–120 e113. 10.1016/j.chembiol.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Larance M, Kirkwood KJ, Tinti M, Brenes Murillo A, Ferguson MA, and Lamond AI (2016). Global Membrane Protein Interactome Analysis using In vivo Crosslinking and Mass Spectrometry-based Protein Correlation Profiling. Mol Cell Proteomics 15, 2476–2490. 10.1074/mcp.O115.055467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruprecht B, Wei L, Zheng L, Bodea S, Mo X, Maschberger M, Stoehr G, Hahne H, Cornella-Taracido I, and Chi A (2022). Chemoproteomic profiling to identify activity changes and functional inhibitors of DNA-binding proteins. Cell Chem Biol 29, 1639–1648 e1634. 10.1016/j.chembiol.2022.10.008. [DOI] [PubMed] [Google Scholar]
- 88.Huang JX, Lee G, Cavanaugh KE, Chang JW, Gardel ML, and Moellering RE (2019). High throughput discovery of functional protein modifications by Hotspot Thermal Profiling. Nature Methods 16, 894–901. 10.1038/s41592-019-0499-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Navarrete-Perea J, Yu Q, Gygi SP, and Paulo JA (2018). Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of Proteome Research 17, 2226–2236. 10.1021/acs.jproteome.8b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, and O’Shea EK (2003). Global analysis of protein localization in budding yeast. Nature 425, 686–691. 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 91.Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels Lisa M, et al. (2017). A subcellular map of the human proteome. Science 356, eaal3321. 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 92.Nussinov R, and Tsai C-J (2015). The Design of Covalent Allosteric Drugs. Annual Review of Pharmacology and Toxicology 55, 249–267. 10.1146/annurev-pharmtox-010814-124401. [DOI] [PubMed] [Google Scholar]
- 93.Kavanagh ME, Horning BD, Khattri R, Roy N, Lu JP, Whitby LR, Ye E, Brannon JC, Parker A, Chick JM, et al. (2022). Selective inhibitors of JAK1 targeting an isoform-restricted allosteric cysteine. Nat Chem Biol 18, 1388–1398. 10.1038/s41589-022-01098-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen Y-NP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH-T, Chen Z, Cooke VG, et al. (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152. 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- 96.Wells JA, and McClendon CL (2007). Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450, 1001–1009. 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 97.Tan CSH, Go KD, Bisteau X, Dai L, Yong CH, Prabhu N, Ozturk MB, Lim YT, Sreekumar L, Lengqvist J, et al. (2018). Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 359, 1170–1177. 10.1126/science.aan0346. [DOI] [PubMed] [Google Scholar]
- 98.Cho KF, Branon TC, Udeshi ND, Myers SA, Carr SA, and Ting AY (2020). Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat Protoc 15, 3971–3999. 10.1038/s41596-020-0399-0. [DOI] [PubMed] [Google Scholar]
- 99.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–d450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jaeger MG, Schwalb B, Mackowiak SD, Velychko T, Hanzl A, Imrichova H, Brand M, Agerer B, Chorn S, Nabet B, et al. (2020). Selective Mediator dependence of cell-type-specifying transcription. Nat Genet 52, 719–727. 10.1038/s41588-020-0635-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, and Zhang F (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 12, 828–863. 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Remsberg JR, Suciu RM, Zambetti NA, Hanigan TW, Firestone AJ, Inguva A, Long A, Ngo N, Lum KM, Henry CL, et al. (2021). ABHD17 regulation of plasma membrane palmitoylation and N-Ras-dependent cancer growth. Nat Chem Biol 17, 856–864. 10.1038/s41589-021-00785-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pomaznoy M, Ha B, and Peters B (2018). GOnet: a tool for interactive Gene Ontology analysis. BMC Bioinformatics 19, 470. 10.1186/s12859-018-2533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Y, Dix MM, Bianco G, Remsberg JR, Lee HY, Kalocsay M, Gygi SP, Forli S, Vite G, Lawrence RM, et al. (2019). Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat Chem 11, 1113–1123. 10.1038/s41557-019-0351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17, 10–12. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 106.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, and Li H (2021). Twelve years of SAMtools and BCFtools. GigaScience 10. 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 110.Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology 11, R25. 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome biology 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A 111, E5593–5601. 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Signal B, Gloss BS, Dinger ME, and Mercer TR (2018). Machine learning annotation of human branchpoints. Bioinformatics 34, 920–927. 10.1093/bioinformatics/btx688. [DOI] [PubMed] [Google Scholar]
- 115.Jiang H, Lei R, Ding S-W, and Zhu S (2014). Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15, 182. 10.1186/1471-2105-15-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu D (2019). Algorithms for efficiently collapsing reads with Unique Molecular Identifiers. PeerJ 7, e8275. 10.7717/peerj.8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Her HL, Boyle E, and Yeo GW (2022). Metadensity: a background-aware python pipeline for summarizing CLIP signals on various transcriptomic sites. Bioinform Adv 2, vbac083. 10.1093/bioadv/vbac083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset S1. Proteomic and RNA-seq data and Reference Tables, Related to Figures 1, 2, 3, 4, 5, 6, and 7.
Dataset S2. eCLIP data, Related to Figure 7
Data Availability Statement
All RNA-sequencing data, raw proteomic data and gel images have been deposited at PRIDE,99 GEO and Mendeley Data respectively, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PA28α Antibody | Cell Signaling Tech | Cat#: 2408S; RRID:AB_2170937 |
| PA28β Antibody | Cell Signaling Tech | Cat#: 2409S; RRID:AB_2171085 |
| Monoclonal ANTI-FLAG® M2 antibody produced in mouse | Sigma-Aldrich | Cat#: F1804-200UG; RRID:AB_262044 |
| HA-Tag (C29F4) Rabbit mAb | Cell Signaling Tech | Cat#: 3724S; RRID:AB_1549585 |
| HA-Tag (6E2) Mouse mAb | Cell Signaling Tech | Cat#: 2367S; RRID:AB_10691311 |
| Vinculin (E1E9V) XP® Rabbit mAb | Cell Signaling Tech | Cat#: 13901S; RRID:AB_2728768 |
| GAPDH (14C10) Rabbit mAb | Cell Signaling Tech | Cat#: 2118S; RRID:AB_561053 |
| DDX42 polyclonal antibody rabbit | Bethyl Laboratories | Cat#: 303-353A; RRID:AB_10951853 |
| Anti-β-Actin Antibody (C4) | Santa Cruz Biotechnology | Cat#: sc-47778; RRID:AB_626632 |
| β-Actin (13E5) Rabbit mAb | Cell Signaling Tech | Cat#: 4970S; RRID:AB_2223172 |
| SF3B1 (D7L5T) Rabbit mAb | Cell Signaling Tech | Cat#: 14434S; RRID:AB_2798479 |
| p27 Kip1 (D69C12) XP® Rabbit mAb | Cell Signaling Tech | Cat#: 3686S; RRID:AB_2077850 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Tech | Cat#: 7074S; RRID:AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Tech | Cat#: 7076S; RRID:AB_330924 |
| rabbit anti-goat IgG-HRP | Santa Cruz Biotechnology | Cat#: sc-2768; RRID:AB_656964 |
| InVivoMAb anti-human MHC Class I (HLA-A, HLA-B, HLA-C) Clone: W6/32 | BioXCell | Cat#: BE0079; RRID:AB_1107730 |
| IRDye® 680RD Goat anti-Rabbit IgG Secondary Antibody | VWR | Cat#: 926-68071; RRID:AB_10956166 |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | VWR | Cat#: 925-32211; RRID:AB_2651127 |
| IRDye® 680RD Goat anti-Mouse IgG Secondary Antibody | VWR | Cat#: 926-68070; RRID:AB_10956588 |
| IRDye® 800CW Goat anti-Mouse IgG Secondary Antibody | VWR | Cat#: 926-32210; RRID:AB_621842 |
| PE anti-mouse H-2Kb/H-2Db Antibody | BioLegend | Cat#: 114608; RRID:AB_313599 |
| PE Mouse IgG2a, κ Isotype Ctrl Antibody | BioLegend | Cat#: 400211; RRID:AB_326460 |
| APC anti-mouse H-2Kb bound to SIINFEKL Antibody | BioLegend | Cat#: 141606; RRID:AB_11219595 |
| APC Mouse IgG1, κ Isotype Ctrl Antibody | BioLegend | Cat#: 400120; RRID:AB_2888687 |
| APC/Cyanine7 Mouse IgG1, κ Isotype Ctrl Antibody | BioLegend | Cat#: 400128; RRID:AB_2892538 |
| Bacterial and virus strains | ||
| NEB 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | Cat#: C2987H |
| Chemicals, peptides, and recombinant proteins | ||
| MEM Non-Essential Amino Acids Solution (100X) | Thermo Scientific | Cat#: 11140050 |
| Lipofectamine™ 2000 Transfection Reagent | Thermo Scientific | Cat#: 11668019 |
| Polybrene | Santa Cruz Biotechnology | Cat#: sc-134220A |
| Geneticin™ Selective Antibiotic (G418 Sulfate) | Thermo Scientific | Cat#: 10131035 |
| Pladienolide B | Tocris Bioscience | Cat#: 60-705-00U |
| Desthiobiotin polyethyleneoxide iodoacetamide | Santa Cruz Biotechnology | Cat#: sc-300424 |
| Tetramethylrhodamine (TAMRA) azide | Synthesized in-house | N/A |
| Biotin-PEG4-azide | ChemPep | Cat#: 271606 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Scientific | Cat#: 34580 |
| Novex 4-20% Tris-Glycine Mini Gels | Invitrogen | Cat#: XP04205BOX |
| Nitrocellulose western blotting membrane, 0.45 μM | GE Healthcare Amersham | Cat#: 10600002 |
| Affi-Gel 10 Gel | Bio-rad | Cat#: 1536046 |
| DMSO | Corning | Cat#: 25-950-CQC |
| EDTA (0.5M, pH 8.0) | Invitrogen | Cat#: AM9260G |
| Urea | Fisher Scientific | Cat#: M1084871000 |
| Iodoacetamide | Sigma-Aldrich | Cat#: I1149-25G |
| Dithiothreitol (DTT) | Fisher Bioreagents | Cat#: BP172-25 |
| T ris(benzyltriazolylmethyl)amine (TBTA) | TCI | Cat#: T2993 |
| Copper(II) sulfate, anhydrous | Sigma-Aldrich | Cat#: 451657-10G |
| Tris(2-carboxyethyl)phosphine HCl (TCEP) | Sigma-Aldrich | Cat#: 75259 |
| Sequencing grade modified trypsin | Promega | Cat#: V5111 |
| Lysyl Endopeptidase, Mass Spectrometry Grade (Lys-C) | Fujifilm Wako | Cat#: 125-05061 |
| Streptavidin agarose resin | Fisher Scientific | Cat#: 20349 |
| Micro bio-spin column | Bio-rad | Cat#: 7326204 |
| Tween 20 | Fisher Bioreagents | Cat#: BP337-500 |
| Nonidet P40 substitute (Igepal CA-630) | USB Corporation | Cat#: 19628 |
| Triethylammonium bicarbonate (TEAB) buffer | Sigma-Aldrich | Cat#: T7408-500ML |
| TMT10plex Isobaric Label Reagent Set | Thermo Scientific | Cat#: 90406 |
| TMT16plex Isobaric Label Reagent Set | Thermo Scientific | Cat#: A44520 |
| Hydroxylamine solution | Sigma-Aldrich | Cat#: 467804-10ML |
| Formic acid, ~98%, for mass spectrometry | Honeywell Fluka | Cat#: 94318-250ML-F |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat#: G7573 |
| Caspase-Glo® 3/7 Assay System | Promega | Cat#: G8093 |
| NEBNext Ultra II RNA Library Prep Kit for Illumina | New England Biolabs | Cat#: E7770 |
| RNeasy Mini Plus Kits | QIAGEN | Cat#: 74034 |
| eBioscience™ Fixable Viability Dye eFluor™ 780 | Invitrogen | Cat#: 65-0865-18; N/A |
| Deposited data | ||
| All raw proteomic data have been uploaded to PRIDE | This study | PXD029655 |
| All RNA-sequencing data have been uploaded to GEO | This study | GSE185373, GSE220185, GSE220845 |
| All uncropped gels have been uploaded to Mendeley Data | This study | DOI: 10.17632/r6t9f3n9wr.1 |
| Experimental models: Cell lines | ||
| 22Rv1 | ATCC | CRL-2505; RRID:CVCL_1045 |
| MCF7 | ATCC | HTB-22; RRID:CVCL_0031 |
| Ramos | ATCC | CRL-1596; RRID:CVCL_0597 |
| THP1 | ATCC | TIB-202; RRID:CVCL_0006 |
| HEK293T | ATCC | CRL-3216; RRID:CVCL_0063 |
| HEK293FT | Thermo Scientific | R70007; RRID:CVCL_6911 |
| KBM7 | Georg Winter, CeMM, Vienna | RRID:CVCL_A426 |
| HCT-116 | ATCC | CCL-247; RRID:CVCL_0291 |
| Panc 04.03 | ATCC | CRL-2555; RRID:CVCL_1636 |
| Panc 05.04 | ATCC | CRL-2557; RRID:CVCL_1637 |
| E.G7-Ova | ATCC | CRL-2113; RRID:CVCL_3505 |
| Oligonucleotides | ||
| sgDDX42_N_sense 5’-CACCGattcctaacaggtcagtcat | This study | N/A |
| sgDDX42_N_anti 5’-AAACatgactgacctgttaggaatC | This study | N/A |
| RepDDX42N_1_P_F 5’-gcgttacatagcatcgtacgcgtacgtgtttggcttattcctaacaggtcagtatgaccgagtacaagcccacg | This study | N/A |
| RepDDX42N_1_P_R 5’-agcattctagagcatcgtaCGCGTACGTGTTTGGtccagttcatggtgccaatgGAagatccgccgccacc | This study | N/A |
| DDX42_N_seqF 5’-gcccttggggctatacacttt | This study | N/A |
| DDX42_N_seqR 5’-ccagcactgatggcaaaacc | This study | N/A |
| sgPSME1_sense 5’-CACCGccagcccgaggcccaagcca | This study | N/A |
| sgPSME2_sense 5’ CACCGaaatccagagacttacctcc | This study | N/A |
| sgControl-AAVS1_sense 5’-caccgGGGGCCACTAGGGACAGGAT | This study | N/A |
| Recombinant DNA | ||
| pLenti3.3/TR | Thermo Scientific | A11144 |
| psPAX2.0 | Addgene | 12260 |
| CMV VSV-G | Addgene | 98286 |
| pLenti6.3 | Thermo Scientific | A11144 |
| pLEX304 | Addgene | 25890 |
| Software and algorithms | ||
| PRISM Version 9.0.0 | GraphPad | https://www.graphpad.com/features |
| MaxQuant (v2.0.3.1) | N/A | https://www.maxquant.org/ |
| Integrated Proteomics Pipeline (IP2) | Integrated Proteomics Applications, Inc | http://proteomicswiki.com/wiki/index.php/IP2 |
| FlowJo (v10.0.7) | TreeStar Inc. | https://www.flowjo.com/ |
| edgeR (v3.32.1) | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| DESeq2 (v1.30.1) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| limma voom (v3.46.0) | Law et al., 2014 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| PyMOL (v2.5.2) | Schrödinger | https://pymol.org/2/ |
| Trim_galore (v0.6.4) | Martin et al., 2011 | https://github.com/FelixKrueger/TrimGalore |
| STAR (v2.7.5) | Dobin et al., 2013 | https://github.com/alexdobin/STAR/ |
| samtools (v1.9) | Danecek et al., 2021 | http://www.htslib.org/ |
| bamCoverage (part of the Deeptools package; v3.3.1) | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/index.html |
| featureCounts (part of the subread package; v1.5.0) | Liao et al., 2014 | https://subread.sourceforge.net/ |
| ChimeraX | Pettersen et al., 2021 | https://www.cgl.ucsf.edu/chimerax/ |
| Glide | Schrödinger | https://www.schrodinger.com/products/glide |
| cutadapt 3.4 | Martin et al., 2011 | https://cutadapt.readthedocs.io/en/v3.4/# |
| rMATS (v4.1.1) | Shen et al., 2014 | https://rnaseq-mats.sourceforge.net/rmats4.1.1/index.html |
| Branchpointer | Signal et al., 2018 | https://github.com/signalbash/branchpointer |
| Skipper | Boyle et al., 2022 | https://github.com/YeoLab/skipper |
| skewer | Jiang et al., 2014 | https://github.com/relipmoc/skewer |
| fastp 0.11.5 | Chen et al., 2018 | https://github.com/OpenGene/fastp |
| UMIcollaps | Liu et al., 2019 | https://github.com/Daniel-Liu-c0deb0t/UMICollapse |
| Other | ||