

Abstract
A variety of inflammatory conditions may directly involve the endocrine glands, leading to endocrine dysfunction that can cause severe consequences on patients’ health, if left untreated. Inflammation of the endocrine system may be caused by either infectious agents or other mechanisms, including autoimmune and other immune-mediated processes. Not infrequently, inflammatory and infectious diseases may appear as tumor-like lesions of endocrine organs and simulate neoplastic processes. These diseases may be clinically under-recognized and not infrequently the diagnosis is suggested on pathological samples. Thus, the pathologist should be aware of the basic principles of their pathogenesis, as well as of their morphological features, clinicopathological correlates, and differential diagnosis. Interestingly, several systemic inflammatory conditions show a peculiar tropism to the endocrine system as a whole. In turn, organ-specific inflammatory disorders are observed in endocrine glands. This review will focus on the morphological aspects and clinicopathological features of infectious diseases, autoimmune disorders, drug-induced inflammatory reactions, IgG4-related disease, and other inflammatory disorders involving the endocrine system. A mixed entity-based and organ-based approach will be used, with the aim to provide the practicing pathologist with a comprehensive and practical guide to the diagnosis of infectious and inflammatory disorders of the endocrine system.
Keywords: Inflammatory and infectious disorders, Endocrine pathology, Endocrine glands, Immune check-point inhibitors, IgG4-related disease, Autoimmune polyendocrine syndrome, Autoimmune disorders
Introduction
The complex interplay between the endocrine and immune systems causes the insurgence of endocrinopathies following severe inflammatory conditions and, in turn, the propensity to develop inflammation as a complication of endocrine diseases [1]. However, the endocrine glands may also be directly involved by infectious and inflammatory processes, both in the course of systemic disorders and in the form of organ-specific, or even cell-specific, diseases. Almost invariably, infectious and inflammatory disorders of endocrine tissues lead to a decrease in hormone production and activity and may ultimately result in severe and even life-threatening conditions if left untreated. As these disorders may be clinically under-recognized, the pathologist should be aware of the basic principles of their pathogenesis and be prepared to recognize their morphological features and clinicopathological correlations when faced with histopathological samples of the affected endocrine organs. Not infrequently, inflammatory and infectious diseases may appear as tumor-like lesions of endocrine organs and simulate neoplastic processes. Indeed, the careful observation and identification of the inflammatory and stromal cell populations, supported, when necessary, by the wise employment of a selected panel of immunohistochemical markers, may not only help to recognize the non-neoplastic nature of a pathological process [2] but can also provide important clues to the pathogenic mechanism and, in some cases, the etiological agent involved in the disease.
Infectious disease of endocrine organs may be caused by a variety of microbial agents, including viruses, bacteria, fungi, and even parasites. The clinical correlates and the histopathological aspects of these diseases vary according to the type of microorganism, the immune status of the host, and the organ involved. Generally speaking, bacterial infections are very rare and are more frequently observed in the adrenal glands, whereas viral infections tend to be more frequent and may involve all endocrine organs. Intriguingly, viral infections of endocrine organs have also been suggested to have a role in the development of autoimmune endocrinopathies [3]. Fungal infections and parasitic infestations are exceedingly rare. Although the exact identification of the microbial agent responsible for the infection should be performed in the microbiology laboratory, the pathologist may rely on several tools to suspect or even confirm the etiology of an infectious disease. A well-performed hematoxylin–eosin stain is frequently enough to determine the type of inflammatory cell populations (e.g., neutrophilic granulocytes in pyogenic bacterial infection; epithelioid macrophages, clustered or not in granulomas during mycobacterial, fungal infections, or parasitic infestations) and the cytological changes (nuclear or cytoplasmic) in viral infections. Large colonies of bacteria or fungi are easily identifiable on routinely stained histopathological sections. In some situations, traditional histochemical stains (e.g., Gram stain for bacteria, PAS stain for fungi, argentic stains for bacteria and fungi, Ziehl–Neelsen stain for mycobacteria) and electron microscopy have been replaced by more specific immunohistochemical stains or in situ hybridization studies. The new gold standard for the identification and typing of microbes, however, is molecular techniques that can now be performed on formalin-fixed and paraffin embedded samples, providing pathologists with sensitive and specific methods to make an accurate microbiological diagnosis [4].
Non-infectious inflammatory diseases of the endocrine organs encompass a variety of autoimmune and, more generally, immune-mediated conditions that can selectively affect one gland or involve multiple sites of the endocrine system (autoimmune polyendocrine syndromes). The propensity of some endocrine organs to develop immune-mediated diseases has driven specific attention of the scientific community, also following the observation that the endocrine organs are among the sites of immune check point inhibitors adverse effects [5, 6]. The immunological and genetic bases of the complex mechanisms disrupting innate and specific immunity in endocrinopathies are beyond the scope of this article. However, in diagnostic practice, it is easy to find that autoimmune disorders are more frequently observed in some endocrine organs, such as the thyroid and adrenal, than in others, like the pituitary or parathyroid. It has been recently proposed that thyroid, adrenals, and pancreatic beta cells hypersecreting mutants, which may generate life-threatening conditions (hyperthyroidism, hypercortisolism, and hypoglycemia) when proliferating without control, elicit a stronger immune surveillance than, for example, parathyroid, pancreatic alpha cells, or non-ACTH pituitary cells hypersecreting mutants that would induce milder clinical abnormalities [5]. In a finalistic view, the higher frequency of autoimmune diseases of the thyroid, adrenals, and islet beta-cells would be attributable to the cost of a highly efficient immune surveillance, against the greater prevalence of proliferative diseases of the pituitary, parathyroid, and islet alpha cells, where the immune surveillance seems to be less efficient [5].
Regarding the adverse effects of immune therapy, which represent the main drug-induced inflammatory reactions in endocrine organs, the responsible mechanism for the propensity of the endocrine system to develop such complications has not been clarified, yet. Although the pathogenic mechanisms underlying these conditions have been reported to be similar to those of autoimmune diseases, endocrinopathies induced by immune therapy differ from classical autoimmunity from many points of view and deserve a separate discussion [7, 8].
A peculiar inflammatory condition involving endocrine glands is IgG4-related disease (IgG4-RD), an immune-mediated condition causing fibrosis and inflammation that may involve virtually any anatomical location, presenting at single or multiple sites. IgG4-RD typically presents as a mass lesion and may mimic other mass-forming disorders, from both a clinical and a morphological point of views. The pathologic diagnostic criteria for IgG4-RD were published in 2012, acknowledging organ-specific morphological manifestations of the disease [9]. Since then, the concept of IgG4-RD has evolved, and its intersections with other inflammatory, autoimmune, and neoplastic conditions have been explored [10, 11]. In the endocrine system, the thyroid, the pituitary, and, more rarely, the adrenals and the endocrine pancreas may be involved in this disease [12]. The correct recognition of this entity is of paramount importance as, in most cases, a proper therapy may completely resolve the endocrinopathy and avoid undue surgery [12].
This review will recapitulate the spectrum of infectious diseases and inflammatory conditions directly involving endocrine organs, focusing on the clincopathological correlations and diagnostic criteria of each entity and giving insight into the organ-specific manifestations of these disorders.
Infectious Diseases in Endocrine Organs: A Pathological Point of View
Infections of endocrine organs are uncommon and may be caused by a variety of microorganisms. While rare, infections can lead to significant complications and clinical manifestations of endocrine abnormalities.
Thyroid Infections
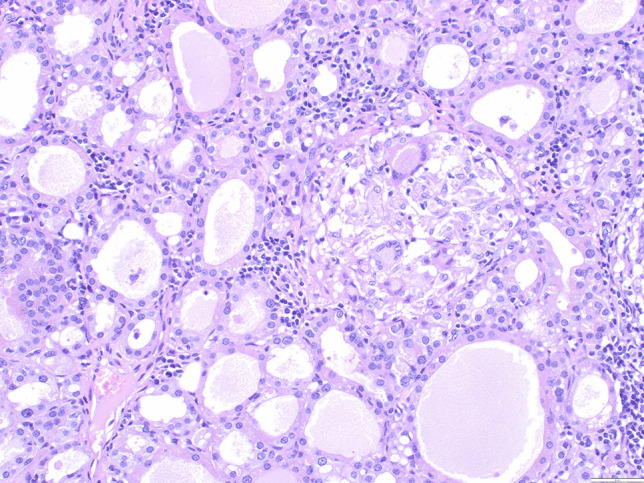
One of the most common manifestations of thyroid infection is acute (suppurative) thyroiditis. Acute thyroiditis is a rare condition, clinically characterized by fever, chills, neck pain, and signs and symptoms of neck mass related to increased thyroid volume [13]. Thyroid function is usually normal, but thyrotoxicosis may intervene due to thyroid cell destruction. The most common causes of acute thyroiditis are bacterial infections, most commonly sustained by Streptococcus pyogenes, Streptococcus pneumonia, or Staphylococcus aureus [14]. Primary thyroid infections are exceedingly rare and acute thyroiditis, usually represents secondary localizations of systemic infections or adjacent structures involvement by pyogenic bacterial infections. Congenital abnormalities, such as pyriform sinus fistula or, more rarely, branchial pouch remnants, are the most frequent causes of acute thyroiditis [15]; thus, this condition is more frequent in children than in adults [14]. Other conditions at risk, mainly responsible for adult cases, are immunodeficiencies of various types, trauma, or, rarely, medical procedures such as fine needle aspiration, or thyroid and parathyroid surgery [14]. Thyroid carcinoma may be another predisposing cause [16]. The morphological aspects of acute thyroiditis are macroscopically characterized by mild thyroid enlargement, that can be focal or diffuse, depending on the extent of the infection. Suppurative foci may be evident on the cut surface. The histopathological picture is dominated by neutrophilic granulocyte infiltration, which can be associated with suppurative necrosis (Fig. 1A). The diagnosis is usually confirmed by fine needle aspiration, which allows samples to be obtained for both cultures and cytological examination. In cytological samples, acute thyroiditis is characterized by numerous neutrophilic granulocytes, necrosis, and fibrin, depending on the severity of inflammation [17]. Microorganisms may be identified on hematoxylin–eosin or using ancillary stains (tissue gram stains, silver stains, PAS stain, and mycobacterial stains), organisms may be identified (Fig. 1B; Fig. 2), but culture continues to be the gold standard for diagnosis. The prognosis of acute bacterial thyroiditis is good if the correct diagnosis is made and effective antimicrobic therapy is administered [13]. Surgical drainage may be useful in case of abscess formation and resistance to antibiotic treatment [13]. Large abscesses may require surgical intervention with drainage [18, 19]. In rare refractory cases, surgery with hemithyroidectomy or total thyroidectomy may be needed [14].
Fig. 1.
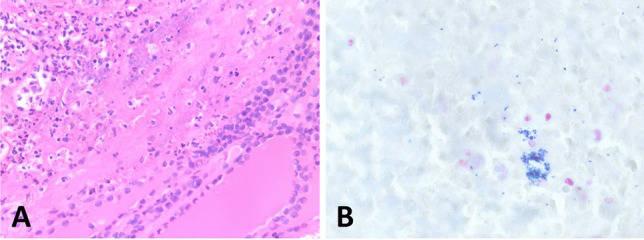
Acute suppurative thyroiditis. A dense infiltrate of neutrophils effaces thyroid parenchyma (A). Gram stain highlights the presence of bacteria (B)
Fig. 2.
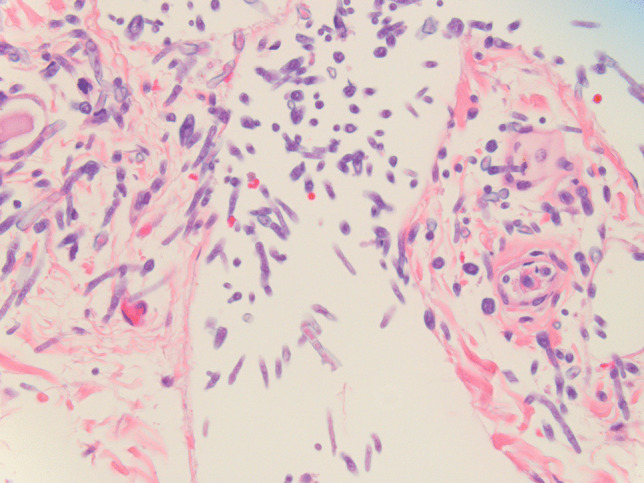
Colonization of thyroid parenchyma by Aspergilli. Mycotic structures are easily identifiable on hematoxylin–eosin stained slides
Systemic viral infections may cause granulomatous thyroiditis similar to De Quervain’s thyroiditis (see later in the text), which, however, is more likely caused by an autoimmune reaction within the gland. Interestingly, subacute thyroiditis has been seen as a complication of SARS-COV-2 infection and following SARS-COV-2 vaccination [20–23]. In immunocompromised hosts, fungal thyroiditis may arise, mostly in the context of an Aspergillus systemic infection [24]. In these cases, symptoms related to thyroid function are frequently neglected and the diagnosis is made at autopsy [24]. Cases of thyroid infections by Pneumocystis carinii have been reported in patients with acquired immunodeficiency syndrome (AIDS) [25], but in the era of anti-retroviral drugs, they have disappeared.
Adrenal Infections
Infections of the adrenal glands are uncommon and are mainly the result of disseminated disease. Interestingly, infections are often asymptomatic unless they destroy a significant amount of adrenal cortical tissue, resulting in adrenal insufficiency (Addison’s disease) [26]. Worldwide, mycobacterial infections, particularly, M. tuberculosis causes a significant number of cases of Addison’s disease in developing countries [27, 28]. Tuberculosis is the second most common cause (after autoimmune adrenalitis) of primary adrenal insufficiency, particularly in the non-Western world where tuberculosis remains endemic [29]. Systemic infections, particularly those associated with bacterial sepsis resulting in vascular collapse and disseminated intravascular coagulation (DIC) can result in acute adrenal insufficiency due to adrenal hemorrhage (Waterhouse-Friderichsen syndrome, WFS) [30]. WFS is most commonly associated with meningococcal septicemia but may be seen with other types of bacterial sepsis and rarely with viral infections [31, 32]. The adrenal gland is reported to be the most common endocrine organ affected by fungal infections, which can be either asymptomatic or associated with adrenal insufficiency [33]. Individuals who are immunocompromised have increased susceptibility to infection; however, immunocompetent individuals can have isolated adrenal gland involvement from by a variety of fungal organisms, including histoplasmosis, blastomycosis, and paracoccidioides [33–35]. A recent study from India found male predominance and non-compromised immune status to be particular characteristics of adrenal histoplasmosis [35]. Histologically, the adrenal gland may show acute and chronic inflammation, granulomatous inflammation, and necrosis. Fungal involvement of the adrenal gland may be incidental and asymptomatic or can be associated with adrenal insufficiency. Viral infections may cause adrenal necrosis and vasculitis either by a direct cytopathic effect or as secondary effect of the inflammatory process generated by their presence (Fig. 3) [36]. Viruses such as the human immunodeficiency virus (HIV) may be associated with adrenal insufficiency. Histologically, the adrenal gland was found to be compromised in more than 99% of 128 autopsy patients with acquired immunodeficiency syndrome (AIDS) by necrosis, fibrosis, hemorrhage, or neoplasms [37]. Additionally, HIV predisposes individuals to infections, including those due to other viruses such as cytomegalovirus, and can be associated with adrenal dysfunction [38]. In historic autopsy studies, in the setting of AIDS, the adrenal gland was involved in CMV adrenalitis in approximately 50% of individuals [37, 39, 40]. Among individuals with AIDS and CMV, the adrenal glands were found to be affected by CMV adrenalitis in 84% [41]. Although adrenal insufficiency can occur, destruction of adrenal tissue is generally not widespread enough in these cases of CMV adrenalitis in the setting of AIDS to result in adrenal cortical deficiency [40].
Fig. 3.
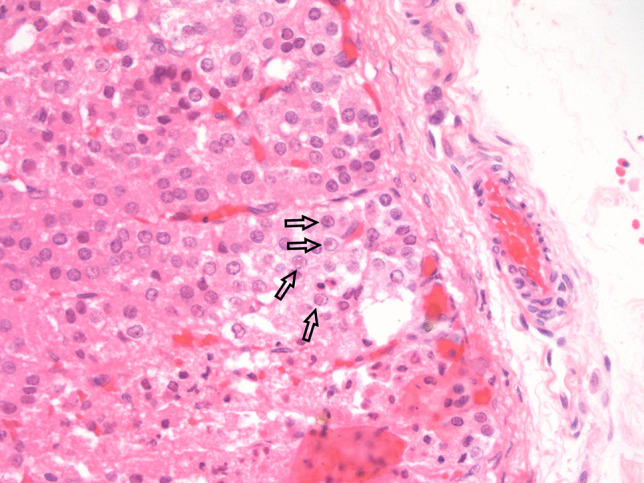
Fatal congenital HSV infection in a newborn: cortical adrenal cells show characteristic nuclear viral inclusions (arrows)
Most recently, the impact of SARS-CoV-2 virus infection and vaccines on the endocrine system, including the adrenal gland, has come to the forefront of the literature involving infections and the adrenal gland [36, 42–45]. Angiotensin-converting enzyme 2 (ACE2) and transmembrane protease serine 2 (TMPRSS2) are viral receptors using which SARS-CoV-2 enters host cells [46]. As both ACE2 and TMPRSS2 are expressed in adrenal tissue, it is possible that SARS-CoV-2 could infect the endocrine system [42, 47]. ACE2 and TMPRSS are co-localized in adrenal cortical cells [48]. Individuals with COVID-19 who are critically ill have been found to have lower cortisol levels compared to critically ill patients without COVID-19, and adrenal insufficiency has been reported in the setting of COVID-19. However, it remains unclear whether the hypocortisolism is due to primary infection of the adrenal gland with SARS-CoV-2 or to dysfunction of the hypothalamic pituitary axis [43, 48–50]. COVID-19 vaccines have been associated rarely with endocrine side effects, particularly in thyroid disorders with involvement of the adrenal gland being anecdotal [51].
Pituitary Infections
Infections of the pituitary are rare and often difficult to diagnose since they can mimic neoplasms clinically and by imaging. Infections can result from disseminated spread, trauma/surgery in the area, or locoregional spread (sinonasal tract most commonly).
Pituitary Abscess
Abscesses of the pituitary gland are a result of either hematogenous spread or direct extension from infected surrounding structures [52]. While most abscesses present with mass symptoms similar to pituitary adenomas, panhypopituitarism, involving both the anterior and posterior pituitary function, may arise in some cases [52]. The most common surgical finding is pus in the sella; on pathologic examination, acute and chronic inflammation are seen. Special stains may be helpful in elucidating a causative organism; however, an etiologic agent is identified in < 20% of cases, with Streptococcus and Staphylococcus being the most common ones [52].
Other Infections of the Pituitary Gland
The pituitary may rarely be infected by other bacteria such as Treponema pallidum (these days mainly in immunocompromised patients) and M. tuberculosis, particularly in developing countries [53]. Fungi may also rarely infect the pituitary, usually by direct extension from surrounding structures or disseminated disease [53]. A variety of viral diseases may also affect the pituitary, including herpes viruses (Fig. 4), tick-borne viruses, and hemorrhagic viruses. These viruses often cause meningitis and involve the pituitary in a secondary fashion. More recently, infection by SARS-COV-2 has been associated with pituitary hemorrhage, infarction, and hypofunction [54, 55].
Fig. 4.
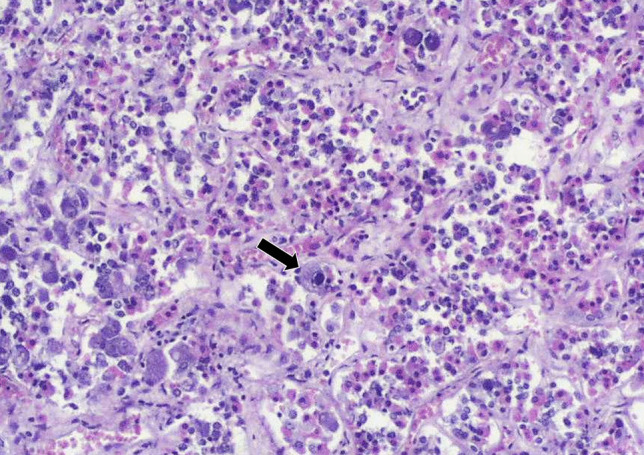
Cytomegalovirus infection in the pituitary of an immune suppressed patient. Viral inclusion is visible in pituitary cells (arrow). Courtesy of Prof. Stefano La Rosa, University of Insubria, Varese, Italy
Parathyroid Infections
Infections of the parathyroid are rarely of clinical significance and are mainly due to systemic infections identified at postmortem examination [56].
Non-infectious Inflammatory Diseases Involving the Endocrine System
Autoimmune Polyendocrine Syndromes
Autoimmune polyendocrine syndromes include a variety of clinical conditions associated with a loss of immune tolerance affecting multiple endocrine glands. There are two broad types of autoimmune polyendocrine syndromes — type 1 (APS1) and type 2 (APS2). Although both affect endocrine and often non-endocrine organs, they are genetically different (one is rare and monogenic, and one is more common and polygenic), and they affect different ages, different sexes, and different populations with different frequencies. These syndromes can occur over the lifetime, and there is significant variation in the frequencies and patterns of autoimmunity and the various organ specific autoimmune diseases that occur [57]
Autoimmune polyendocrine syndrome (APS1) is an autosomal recessive disorder caused by a single-gene abnormality involving AIRE, an autoimmune regulator gene on 21q22.3, [58, 59]. AIRE encodes an autoimmune regulator protein, AIRE, a transcription factor expressed predominantly in the medullary thymus, is involved in removing self-reactive T cells that disturb an individual’s immunological tolerance and cause autoimmune disease [60–64]. As a transcriptional and post-transcriptional regulator of peripheral tissue antigens in medullary thymic epithelial cells, AIRE is associated with negative selection of auto-reactive thymus cells regulating central tolerance and preventing autoimmune diseases [64]. APS1 is also known as autoimmune polyendocrinopathy, candidiasis, and ectodermal dysplasia (APECED). This is a rare disease, with an estimated prevalence of 1:200,000 in northern Italy, 1:90,000 in Norway [65], 1:25,000 in Finland, and 1:6500 to 1:9000 Iranian Jews [66], with an overall prevalence of approximately 1:100,000 [67]. There is a predominance of certain aberrations associated with particular locals. For example, the R257X allele is predominant in Finnish (“Finnish major mutation”) and northern Italians as well as Russians and Polish [68–70], p.Y85C is predominant in Persian Jews [66], R139X in Sardinians [71], and a 13 base-pair deletion (p.C322del13) involving plant homeodomain 1 being common in North Americans [72], British kindreds [73], and Norwegians [74].
Recently, autosomal dominant pathogenic variants in AIRE (involving the first plant homeodomain (PHD1 zinc finger of AIRE) have been reported in common-organ-specific autoimmune diseases characterized by milder disease, reduced penetrance, and later onset [74]. These mutations suppress wild-type AIRE in a dominant negative manner and are found at relatively high frequencies in the population [74]. With sequencing of AIRE, individuals with two mutations are thought to have a APS1 while those with dominant negative mutations are characterized as having non-classic APS 1 [56]. Before genetic testing is performed, individuals suspected of having APS1 may be screened for interferon autoantibodies if the testing is available before AIRE sequencing is performed [57].
In a systemic literature review of the clinical, immunologic, and genetic features of 938 individuals with APS1, children are most often affected, but 5.2% of individuals were older than 18 years at disease onset, and males and females are affected with a ratio of 3:4 [75]. There is a long diagnostic delay in testing for the AIRE pathogenic variant in about half of the patients [75]. APS1 is characterized by a classic triad of mucocutaneous candidiasis (82.5%), which is usually the first manifestation, followed by hypoparathyroidism (84.2%) which is reportedly the most common, and finally primary adrenal insufficiency (Addison disease) (72.2%) [75]. Additional endocrine manifestations may include type 1 diabetes (2–33%), hypogonadism (12%), and autoimmune thyroid disease (10%) [76]. Additional non-endocrine manifestations include ectodermal dystrophy, infections, enamel hypoplasia, hyposplenism, nephritis, pneumonitis, exocrine pancreatitis, alopecia, vitiligo, gastrointestinal disorders (atrophic gastritis), gonadal failure, neurologic involvement (cerebellar ataxia with pseudotumor), and ocular manifestations (keratitis, keratoconjunctivitis, retinitis pigmentosa, ptosis), among others [69, 75, 76]. Histologically, the endocrine organs are involved by a lymphocytic infiltrate with loss of epithelial cells and may be associated with fibrosis.
Autoimmune polyendocrine syndrome type 2 (APS2), with a prevalence of 1:20,000, is more common than APS1 which has an overall prevalence of 1:100,000) [76]. Unlike APS1 which generally begins in childhood, APS2 usually has an onset in young adulthood. APS2 affects women more than men (3:1). Individuals with APS2 have at least one of the following: autoimmune thyroid disease (70–75%), type 1 diabetes (40–60%), or primary adrenal insufficiency (40–50%) [76]. Other endocrine organs can occasionally be involved, such as parathyroid (hypoparathyroidism), gonads (hypogonadism), and pituitary (hypopituitarism). Non-endocrine organs may also be involved including the gastrointestinal tract (autoimmune gastritis, celiac disease, and autoimmune hepatitis) and the skin (vitiligo, alopecia, psoriasis, urticaria, neurodermatitis, pemphigus, and atopic eczema). Other associated disorders include rheumatoid arthritis, systemic lupus erythematosus, Sjogren syndrome, and pernicious anemia [76].
A variety of genes are involved in APS2, often the same genes, and single nucleotide polymorphisms are associated with several autoimmune diseases [57]. Individuals with variants in DR3-DQ2 and DR4-DQ8 have increased risk for celiac disease, type 1 diabetes, autoimmune thyroid disease, and primary adrenal insufficiency [57]. The susceptibility genes include PTPN22 (protein tyrosine phosphatase non receptor type 22), CTLA4 (cytotoxic T lymphocyte antigen 4), and sex-dependent alteration of HLA [77]. Testing for APS2 genetically is complicated. Often, organ-specific antibodies (glutamic acid decarboxylase, thyroid peroxidase, and 21-hydroxylase) are evaluated, as they may occur very early in disease.
Recently, APS2 has been reported in some individuals following treatment with immune checkpoint inhibitors [78]. In a literature review searching for individuals with two or more endocrine disorders after treatment with immune checkpoint inhibitor therapy, 23 patients were identified, approximately 61% received anti programmed cell death 1 therapy, 17% received anti programmed cell death ligand therapy, and 4.3% received anti-cytotoxic T lymphocyte antigen 4 therapy [78]. The thyroid gland was the endocrine organ most frequently affected (78%) with the pancreatic islets affected next most commonly (74%), followed by the pituitary (48%), and adrenal (9%) [78].
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a very serious multi-system autoimmune disorder due to pathogenic variants, involving FOXP3 (forkhead box P3) transcription factor which is critical for CD4 T regulatory cells [79]. This disorder is extremely rare and has as an onset in infancy. In recent review, the disorder is characterized by individuals having early onset type 1 diabetes, autoimmune enteropathy with intractable diarrhea and malabsorption, dermatitis, and frequent eosinophilia with elevated IgE levels, with kidney disease in some patients and late manifestations of autoimmune thyroid disease, alopecia, cytopenias, hepatitis, and pancreatitis [57]. With an X-linked recessive inheritance, the individuals affected are males [79]. In a systemic review of the literature, which included 195 patients with IPEX, approximately 98% had enteropathy, 62% skin disorders, 53% endocrinopathy, 39% hematologic abnormalities, 40% infections, 22% other immune complications, and 16% had kidney involvement [79]. Alterations of FOXP3 affecting the forkhead domain were most common, followed by, the repressor domain, and the leucine zipper domain. Genotype–phenotype correlations were found, associating the forkhead domain alterations with autoimmune hemolytic anemia, and the leucine zipper domain with autoimmune hemolytic anemia, protein-losing enteropathy, and other hematologic presentations as well as hepatomegaly, and splenomegaly. In addition, zinc finger domain abnormalities often presented with autoimmune cytopenias and diarrhea [79]. Interestingly, in this literature survey, 65% of individuals had only one endocrine disorder rather than multiple endocrinopathies, with patients’ clinical presentations consisting mostly of single symptoms rather than multiple symptoms [79].
IgG4-related Disease
The IgG class of immunoglobulins includes four subclasses, named IgG1, IgG2, IgG3, and IgG4, in descending order of their serum concentration. The IgG4 subclass is functionally characterized by reduced effector binding to both C1q protein complex and to FC receptors compared to the other IgG subclasses [80]. These properties have been related to the fact that IgG4 are dynamic molecules, able to spontaneously exchange one of the two Fab fragments with each other (“half-antibody exchange reaction”), giving rise to bi-specific antibodies, which may bind with two different antigens, but are monovalent for each of them, thus unable to form circulating immune complexes [81]. Overall, the reduced effector binding of IgG4 has been linked to a “down regulatory” effect on the immune system, interpreted at times as “anti-inflammatory” or “tolerogenic,” that may help in tuning the immune response against recurrent and/or chronic antigenic stimulations, although their functions are still to be clarified [82]. IgG4 are characterized by lower mean serum concentration levels than the other IgG subclasses and show a unique inter-individual variability of their concentration levels in healthy subjects [83]. The highest levels are observed in elderly males [83].
The involvement of IgG4 in human pathology was first highlighted at the beginning of our century, when Hamano and colleagues demonstrated that patients with sclerosing (autoimmune) pancreatitis had high serum levels of this immunoglobulin, decreasing after glucocorticoid therapy, along with clinical resolution of symptoms [84]. After that first report, an increasing number of immune-mediated diseases in different anatomical locations were identified, featuring high IgG4 serum levels, high numbers of intralesional IgG4-producing plasma cells, and prominent mass-forming fibrosis. All of these diseases, most of which were formerly known by other names (e.g., Mikulicz disease, retroperitoneal fibromatosis, angiolymphoid hyperplasia with eosinophilia), were finally grouped under the heading of IgG4-related disease (IgG4-RD) [85]. Importantly, the recognition of IgG4-RD relies heavily on the histopathological features of the lesions, which include lymphoplasmacytic infiltrate, storiform fibrosis, and obliterative phlebitis, along with high numbers of IgG4 positive (IgG4 +) plasma cells and a high IgG4 + /IgG + plasma cell ratio. The relevance of histopathological features and the criteria for the diagnosis of IgG4-RD were stated in a consensus paper [9]. Although in some cases a biopsy of the diseased organ may be difficult to obtain and clinical, radiological, and serological features may point to a possible or probable IgG4-RD, histopathological analysis is still required for establishing a definite diagnosis [11]. Moreover, it has been clarified that the microscopic appearance of the lesion, in terms of lymphoplasmacytic infiltrate, storiform fibrosis, and, but not mandatory, obliterative phlebitis, is requested for the diagnosis, that cannot rely only on the finding of high numbers of IgG4 + plasma cells and IgG4 + /IgG + plasma cell ratio [9]. In establishing the histopathological diagnosis of IgG4-RD, some exclusion criteria must be considered, such as the presence of granulomas, prominent neutrophilic infiltration, prominent necrosis, cell atypia suspicious for malignancy and not fully investigated, or features of other mass forming lesions, such as inflammatory myofibroblastic tumor and Rosai-Dorfman disease [10]. Importantly, histopathological features of IgG4-RD may vary in different organs, and site-specific criteria have been acknowledged since the beginning [9, 11]. In fact, IgG4-RD may arise in virtually every anatomical location, and not infrequently multiple sites are involved in the same patient.
The most affected sites by IgG4-RD are the pancreas, major salivary glands, lacrimal glands, and retroperitoneum, followed by bile ducts, lungs, kidneys, orbits, and, less frequently, the thyroid, pachymeninges, and aorta [86]. In fact, two of the 11 typically involved organs, i.e., the pancreas and the thyroid, belong to the endocrine system. In addition, the pituitary is a rare, but still significant, target organ of this disease. The adrenals are very rare sites of disease, and a direct involvement of the parathyroid glands has not been reported, to date. In general, IgG4-RD in endocrine glands represents a challenge from both clinical and pathological points of view. Establishing the correct diagnosis is crucial for the correct management of the patient, as effective, though not yet standardized, treatment strategies, including corticosteroid drugs, immunosuppressants, and B cell targeted therapies, are available [86]. This is why a careful radiological study of the lesion is needed in organs like the pancreas or the pituitary, in which IgG4-RD may simulate a neoplasm, and a preliminary biopsy approach is needed to avoid unnecessary surgery [87]. On the other hand, the diagnosis of IgG4-RD on small samples may not be straightforward [9] not only due to the scarce material and frequent crush artifacts but also due to the variability of the morphological features of the disease in its various stages. In fact, in the early phases of IgG4-RD, the lymphoplasmacytic infiltrate may dominate the pathological picture, making it easy to highlight IgG4 + plasma cells. In the later stages, dense hypocellular fibrosis may prevail, obscuring the diagnostic features of the disease. In addition, in the endocrinological practice, the replacement therapy for endocrine gland hypofunction should be carefully tuned according to the stage of the disease and to its response to the pathogenesis-based treatment. The detailed clinicopathological features of IgG4-RD in the pituitary gland, thyroid, pancreas, and adrenals will be discussed in specific paragraphs of this review. Figure 5 summarizes the crucial data.
Fig. 5.

IgG4-related disease (IgG4-RD) manifestations in endocrine organs
Drug-induced Inflammatory Reactions in Endocrine Organs
Although a wide variety of therapeutic drugs may have morphologically evident consequences on endocrine organs (e.g., “black thyroid” induced by tetracyclines, thyroid hyperplasia induced by amiodarone, adrenal hyperplasia induced by exogenous corticosteroids), drug-related inflammatory lesions in the endocrine system are typically induced by immune check point inhibitors (ICIs). These are a group of monoclonal antibodies that specifically block proteins of the immune check point, such as CTLA-4, PD-1, and PD-L1, which are inhibitory molecules that attenuate T cell activity. These molecules have the physiological role of downregulating exaggerated immune reactions against external antigens avoiding auto-immune disorders [88]. However, their action may be exploited by cancer cells and used as an escape mechanism to elude the attack by specific immunity [88]. In fact, the blockage of these molecules by ICIs removes the acquired peripheral immune tolerance towards cancer cells and allows the immune response to be fully effective [89]. In other words, the antitumor activity of ICIs is due to their ability to restore an effective immune response to cancer antigens, which contributes to the destruction of tumor cells [90].
Since 2011, when the first ICI inducing CTLA-4 blockage (ipilumab) was approved, the introduction of anti-CTLA-4, PD-1, and PD-L1 antibodies in clinical practice has dramatically changed the therapeutic approach to a wide spectrum of cancers, improving patients’ outcome [91]. However, the activation of the immune system induced by ICIs is not a tumor-specific event and involves immune cells in the whole organism, implying adverse effects that, in many ways, recall autoimmune disorders [92]. The systemic consequences of ICIs may be seen in every anatomical location; however, the skin, the digestive tract, the liver, and the endocrine system present the most frequent and impactful adverse events [92, 93]. It has been reasoned that the propensity of endocrine organs to develop adverse effects of ICI therapy parallels that seen for autoimmune disorders. This may be due to a peculiar tolerogenic profile of endocrine tissues [7, 8]. Nevertheless, ICI-induced adverse effects in the endocrine system differ from classical autoimmune conditions in at least three ways [7]. (1) The organs involved: although the thyroid and pancreatic beta cells are still among the most targeted organs, the pituitary, in which autoimmune conditions are rare, is one of the most targeted organs, and the parathyroid may be involved as well. (2) The autoantibodies repertoire: there is evidence that the classical autoantibodies involved in autoimmunity are not constantly present in patients who develop endocrinopathies during immune checkpoint inhibition, suggesting the existence of alternative autoantigens. (3) The severity of the disease: the inflammatory process developing in patients treated with immune therapy causes rapid and irreversible disruption in the majority of cases, differently from classical autoimmunity that may show a spectrum of severity. This is possibly related to the massive recruitment of cytotoxic T cells induced by the immune checkpoint blockage, which is not observed in classical autoimmunity.
The involvement of endocrine organs as ICI adverse effect is, globally, a frequent event that may affect up to 40% of patients treated with these drugs [94]. However, the frequency of endocrinopathies, the site of involvement, and the time of insurgence vary according to the specific drug (or drugs) administered [92, 93] (Fig. 6). The exhaustive discussion of the clinical features and the pathogenetic mechanisms of ICI-related endocrinopathies is beyond the aim of this review. In general, combined therapy with both anti-CTLA-4 and anti-PD-1/PD-L1 antibodies cause more endocrine adverse effects than the use of a single drug [92, 93]. Nevertheless, it has been observed that pituitary inflammation is more associated with CTLA-4 blockade than with PD-1/PD-L1 inhibition, whereas the reverse is true for thyroid disorders and type 1 diabetes mellitus following endocrine pancreas involvement. Adrenalitis is equally seen with either therapy [8]. Interestingly, several cases of autoimmune polyendocrine syndrome type 2 following ICI-treatment have been described, again highlighting the propensity of the endocrine system as a whole to be a target of side effects of these drugs [78].
Fig. 6.

Immune checkpoint inhibitor-related endocrine adverse effects
The microscopic features of adverse effects due to ICIs in endocrine organs are not fully established, as histopathological examination is not routinely performed in these cases and the diagnosis mainly relies on clinical observations and laboratory data, whereas the therapy is mainly based on medical therapy with hormone replacement. ICI-related thyroid disorders may be associated either with hyperthyroidism or with hypothyroidism. Usually, an hyperfunctional early phase is followed by a decrease in thyroid hormone production. Secondary hypothyroidism due to ICI-related pituitary inflammation has been described [92, 93]. Thyroid surgery is reserved to anecdotal cases of patients presenting with thyrotoxicosis refractory to medical treatment [95–97], whereas a fine needle biopsy may be useful, independently of the endocrinological picture, when the thyroid shows nodular lesions during restaging of the neoplastic disease treated with ICIs [98]. The shared features of inflammatory lesions induced by nivolumab and pembrolizumab in the thyroid are different from those of autoimmune thyroid disorders (Graves’ disease and Hashimoto thyroiditis) and are represented by dense lymphocytic and histiocytic infiltrates that efface the normal parenchyma and destroy epithelial cells [95–97]. T cell infiltrates have been reported to be predominant, with a CD8 + cytotoxic profile, and perifollicular histiocytes express PD-L1 [95, 97]. Hypophysitis is a rarer adverse effect of ICI therapy than thyroiditis, although it is clinically well-characterized [99]. Pituitary inflammation related to anti-CTLA-4 therapy is distinct from that induced by anti-PD-1/PD-L1: the former is characterized by sellar expansion at MRI with panhypopituitarism and diabetes insipidus due to mass-effect, whereas the latter features isolated ACTH insufficiency and consequent secondary hypocortisolism [99–101]. The histological pictures have only been described in two autopsy cases, one treated with tremelimumab (an anti-CTLA-4 antibody) [100] and one treated with nivolumab (an anti-PD-1 antibody) [101]. Interestingly, the microscopic aspects of the two cases were distinct: a necrotizing hypophysitis with dense mononucelar infiltrate and fibrosis in vital areas was observed in the patient treated with tremelimumab [100], whereas a lymphocytic hypophysitis without necrosis or fibrosis was seen in the nivolumab-treated patient [101]. In the latter case, a significant reduction of ACTH-producing cells was seen, in the presence of comparatively normal numbers of GH-, PRL-, TSH-, FSH-, and LH-producing cells, and it was associated with isolated ACTH deficiency [101]. The lymphoid infiltrate, in the nivolumab-treated patient, was predominantly composed of cytotoxic CD8 + T cells [101], whereas a predominance of CD4 + T cells and CD20 + B cells was observed in the tremelimumab-related pituitary inflammation [100]. IC-related adrenal insufficiency is secondary to pituitary ACTH deficiency in more than 90% of cases. However, a few cases of ICI-induced adrenalitis have been reported, although no histopathological report exists about this entity [102, 103]. ICI-associated hypoparathyroidism is restricted to patients treated with anti-PD-1/PD-L1 antibodies and is associated with irreversible hypocalcemia [104], and the morphological features of involved parathyroid gland have not been reported. ICI-induced type I diabetes mellitus is the consequence of inflammatory damage to pancreatic beta cells and arises during anti-PD-1/PD-L1 treatment, whereas it is much rarer following anti-CTLA-4 therapy. Affected patients show rapidly increasing hyperglycemia with low levels of glycated hemoglobin and, if untreated, may rapidly evolve to insulin deficiency and diabetic ketoacidosis [105]. A systematic histopathological documentation of Langerhans islets by the ICI-related inflammatory process is lacking. Increased infiltration of CD8 + T lymphocytes, similar to what is observed in classical type I diabetes mellitus, is possibly the histopathological correlate of the disease [8]
Organ-specific Inflammatory Diseases
Pituitary Gland
Hypophysitis is a broad collective term for various forms of pituitary gland inflammation, encountered in approximately 2% of pituitary surgery specimens [106]. The spectrum of diseases associated with pituitary inflammation is wide. General approaches to classifying hypophysitis are based on anatomic localization, histopathology of immune cell infiltrate, and etiology. Histopathology of pituitary inflammation is commonly distinguished into lymphocytic, granulomatous, xanthomatous, necrotizing, and plasmacytic/IgG4-related types or mixtures thereof [107–110]. The etiological classification generally separates primary and secondary hypophysitis forms. In primary hypophysitis, inflammation originates in the pituitary gland, presumably due to a poorly understood autoimmune mechanism. In secondary hypophysitis, pituitary inflammation occurs as part of or as a result of a distinct disease of known etiology and/or pathogenesis. Hypophysitis may result in pituitary hormone insufficiency with potentially lethal complications. Management and prognosis are dictated by a timely diagnosis, which is facilitated via multi-disciplinary work-up and supported by surgical interventions and pathological investigations [109, 111].
Current Status and Challenges of Hypophysitis Classification
Primary hypophysitis is considered an autoimmune disease that is limited to the pituitary gland. The pathogenesis is currently poorly understood. Pituitary autoimmunity has been linked to the occurrence of circulating serum autoantibodies directed against various cellular components of pituitary cells [112–117]. However, circulating anti-pituitary antibodies are not regarded as being disease-specific, as they are also detected in paraneoplastic syndromes, congenital anomalies like cryptorchism, or non-pituitary autoimmune conditions [114, 118, 119]. Thus, pituitary autoantibodies are commonly recognized as surrogate markers of pituitary autoimmunity, but their causal role in disease development remains a matter of debate [113, 114, 119].
The most commonly named prevalence of autoimmune hypophysitis is 1 in 9 million [110, 111, 120–124], derived from a study in 2001, reporting 5 cases of hypophysitis that occurred over a 15-year time span in a community of 3 million people [125]. However, actual numbers may differ as not all patients with hypophysitis are subjected to surgery, and therefore, diagnosis is not histologically substantiated in many cases [126]. Moreover, new pathological insights of the last decades vindicate reclassification of former hypophysitis diagnoses and, thus, reevaluation of many previously reported statistics may be appropriate [106, 107, 127, 128].
Secondary hypophysitis generally refers to pituitary inflammation occurring as a result of a pre-existing condition. Possible causes are adjacent pituitary lesions, infectious diseases, systemic inflammation, paraneoplastic syndromes, or medical treatments (iatrogenic). The latter has gained increasing attention over the last decades, particularly with reference to hypophysitis as an immune-related adverse event in patients treated with immune checkpoint inhibitors in cancer therapy [99, 129].
There is considerable overlap in the histopathology of primary and secondary hypophysitis. Thus, the classification of pituitary inflammation is challenging, and cannot be exclusively based on histopathology. In practice, hypophysitis classification requires knowledge of pre-existing conditions and integration of data from endocrinology, radiology, and oncology. Due to these prevailing challenges in diagnostics and the poorly understood pathomechanisms associated with various forms of pituitary inflammation, the current literature is often vague or inconsistent in terms of hypophysitis classification terminology.
With the aim to harmonize the field of pituitary inflammation, we will review commonly described histological types of hypophysitis, and elaborate on their respective diagnostic significance, etiological context, and associated terminology.
With the aim of harmonizing the field of pituitary inflammation, we will review the commonly described histological types of hypophysitis, and elaborate on their respective diagnostic significance, etiological context, and associated terminology. Moreover, we propose a classification scheme that incorporates the consideration of multidisciplinary patient data and diagnostic certainty. Finally, we demonstrate a pathology-centered diagnostic algorithm, which may be helpful to guide interdisciplinary work-up of hypophysitis, and aid in the uniform implementation of classification terminology to facilitate improved comparability among future studies.
Lymphocytic Hypophysitis
Lymphocytic inflammation is the most common histological type of primary hypophysitis, accounting for roughly 70–90% of primary hypophysitis cases [106, 110, 122]. Primary lymphocytic hypophysitis has been described to occur more frequently in women and in association with pregnancy. Management of the disease encompasses conservative approaches, anti-inflammatory therapy, radiotherapy, and surgery [109, 130]. Although lethal complications due to irreversible adrenal insufficiency may occur, the overall prognosis is considered good if diagnosis is timely.
The histopathology of lymphocytic hypophysitis is characterized by diffuse destructive infiltration of polyclonal B- and T-lymphocytes into the pituitary gland, which may arrange into lymphoid follicles with germinal centers [111] (Fig. 7). Plasma cells, histiocytes, macrophages, and eosinophilic granulocytes are also encountered [120, 131]. Pituitary cells may be significantly reduced in number and may show oncocytic changes [131]. Fibrosis may be seen to a variable extent, especially in later disease stages [131, 132]. Rarely, necrosis may also be present [120].
Fig. 7.
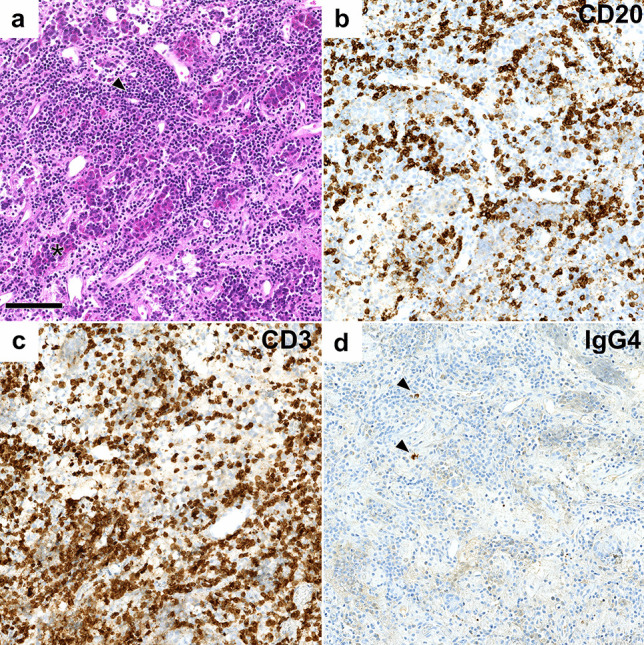
Lymphocytic hypophysitis: a anterior pituitary hormone-secreting cells (asterisk) are overrun by a dense lymphocytic infiltrate (arrowhead), comprised of CD20 + B cells (b) and CD3 + T cells (c). Very few IgG4-expressing plasma cells were found (arrowheads, d). Systemic infections or inflammatory disease were ruled out in this patient leading to the classification as certain primary lymphocytic hypophysitis
Hematopoietic neoplasms should be excluded as differential diagnoses of lymphocytic hypophysitis. Primary pituitary lymphomas histologically resemble systemic lymphomas, and are commonly B cell non-Hodgkin lymphomas. In contrast to lymphocytic hypophysitis, pituitary lymphomas may often present more aggressively and extend beyond the sellar or parasellar regions [2, 133–136].
Lymphocytic infiltration of the pituitary gland may be encountered as part of a secondary reaction to other focal pituitary lesions or systemic diseases. Lymphocytic inflammation of pituitary tissue may be found associated with non-neoplastic pituitary lesions (e.g., arachnoid cyst [137], or Rathke cleft cyst [138] (Fig. 8) or neoplastic pituitary lesions (e.g., germinoma) [139, 140], craniopharyngioma [106], or pituitary neuroendocrine tumors/adenomas [141] (Fig. 9). Coexistence of non-pituitary autoimmune conditions is reported in 25–50% of patients with lymphocytic hypophysitis [142, 143]. Among the encountered diseases are autoimmune thyroid disease, adrenalitis, and pernicious anemia. Autoimmune diseases may precede or follow the onset of lymphocytic hypophysitis [144].
Fig. 8.
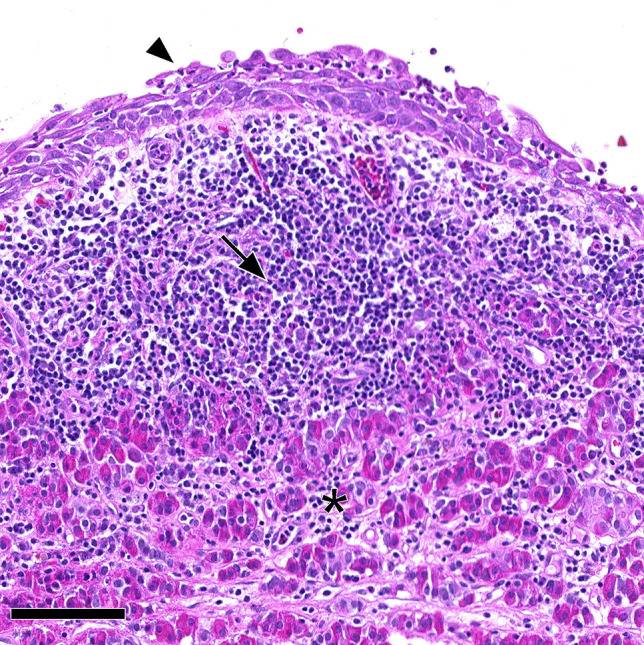
Rathke cleft cyst associated with lymphocytic hypophysitis. Histopathology showed dense lymphocytic infiltration (arrow) of residual pituitary gland (asterisk) in proximity of a Rathke cleft cyst with squamous metaplasia (arrowhead). This case was classified as Rathke cleft cyst with certain secondary lymphocytic hypophysitis
Fig. 9.
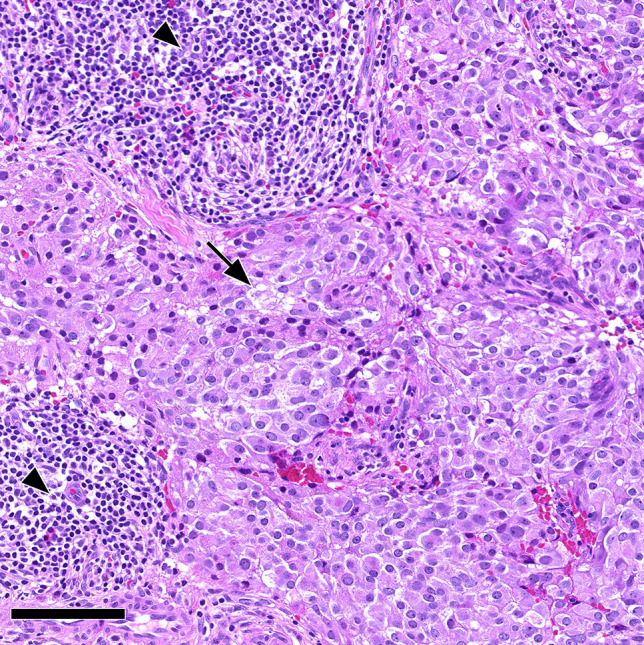
Acidophil stem cell PitNET/adenoma associated with lymphocytic inflammation. Histomorphology of an acidophil stem cell pituitary neuroendocrine tumor/adenoma composed of oncocytic cells with intracytoplasmic vacuoles reflecting giant mitochondria (arrow). This tumor showed well-marked accompanying lymphocytic inflammation as follicular aggregates (arrowheads). This case was classified as acidophil stem cell PitNET/adenoma with secondary lymphocytic hypophysitis
While co-occurrence of hypophysitis and non-pituitary autoimmune diseases supports the hypothesis of an autoimmune mechanism in primary hypophysitis, this causes some inconsistency in the classification terminology encountered in the literature. Some authors classify autoimmune hypophysitis, which occurs together with other autoimmune diseases as a form of secondary hypophysitis [111, 122, 123]. Other authors do not draw such distinctions [107, 120, 142]. Moreover, the stand-alone term “lymphocytic hypophysitis” is commonly used interchangeably with “primary lymphocytic hypophysitis” in the literature [109, 111, 131, 145, 146]. However, “lymphocytic hypophysitis” may also refer to lymphocytic variants of secondary hypophysitis [120, 138, 147]. Previous studies have recognized that these differing approaches to classification terminology may cause reader confusion [121].
Granulomatous Hypophysitis
Granulomatous inflammation is the second most common type of primary hypophysitis (often referred to as idiopathic granulomatous hypophysitis), and accounts for roughly 10–20% of primary hypophysitis cases [106, 110, 122, 148]. Compared to primary lymphocytic hypophysitis, this type is less associated with pregnancy, although a female predilection has also been reported [148, 149]. Clinically, primary granulomatous hypophysitis is often less responsive to steroid treatment, and clinical presentation is commonly more severe than in primary lymphocytic hypophysitis [122, 145, 149].
Histopathology of granulomatous hypophysitis is characterized by granulomas of epithelioid histiocytes with or without multinucleated giant cells and lymphocytic infiltrates (Fig. 10). Fibrosis and necrosis may be seen [148]. Previous studies have suggested that primary lymphocytic and granulomatous hypophysitis may represent an autoimmune spectrum, with the lymphocytic form predominantly constituting early lesions and the granulomatous form appearing later [148, 150]. This assumption is corroborated by the slightly higher age of patients presenting with granulomatous hypophysitis and the finding that hypophysitis mouse models exhibit both lymphocytic and granulomatous histopathology [148, 151]. Other studies have proposed that primary granulomatous hypophysitis is immunologically distinct from primary lymphocytic hypophysitis. This may be substantiated by the different clinical courses seen in the two forms and the distinct immune profile of primary granulomatous hypophysitis, which was reported to be compatible with a type IV hypersensitivity response [120, 146]. Thus, the etiopathogenesis of primary granulomatous hypophysitis is currently unknown, and the association with primary lymphocytic hypophysitis is unclear.
Fig. 10.
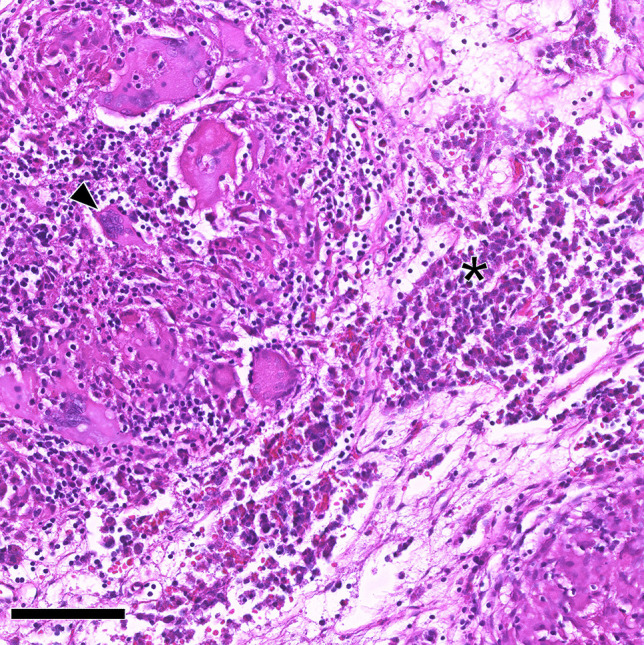
Granulomatous hypophysitis. Non-necrotizing granulomas with multinucleated giant cells (arrowhead) and interspersed residual anterior pituitary cells (asterisk). Systemic infectious/inflammatory diseases were ruled out in this patient, leading to the classification as certain primary granulomatous hypophysitis
An association with autoimmune diseases is less often encountered in granulomatous compared to lymphocytic hypophysitis [149]. Only a few reports have described patients with co-occurrence of conditions like Crohn’s disease [152], granulomatosis with polyangiitis (formerly called Wegener’s) [153], Takayasu’s arteritis [154], or Hashimoto’s thyroiditis [149]. Moreover, pituitary manifestations may rarely occur in sarcoidosis [155]. Granulomatous inflammation of the pituitary gland may be encountered secondary to systemic infectious diseases like tuberculosis or syphilis [156]. The diagnosis of primary granulomatous hypophysitis therefore needs the exclusion of secondary etiology by means of extensive diagnostic steps. These steps include Mantoux skin testing, M. tuberculosis-DNA PCR of cerebrospinal fluid and pituitary specimens, chest X-ray, serologic syphilis testing and measurement of ACE and ANCA serum levels [148, 157]. Previous studies have recognized the diagnostic challenges associated with the exclusion of secondary etiology in granulomatous hypophysitis [148, 158, 159].
Xanthomatous and Xanthogranulomatous Hypophysitis
Xanthomatous hypophysitis is commonly recognized as a rare type of primary hypophysitis encountered in about 3% of cases [107, 120, 123, 149, 160, 161]. Patients are usually younger than in primary lymphocytic and granulomatous hypophysitis. No association with pregnancy has been described [149]. Co-occurrence with other autoimmune diseases is rare [162]. While data on management and prognosis are limited, it is currently suggested that anti-inflammatory treatment is of low value compared to surgery. The prognosis is variable [160, 163, 164].
Histologically, xanthomatous hypophysitis is characterized by the infiltration of foamy macrophages and lymphocytes into the pituitary gland, with evidence of little or no hemosiderin pigment (Fig. 11). Recent reports have suggested that xanthomatous hypophysitis may represent a secondary response to ruptured cysts, most commonly Rathke cleft cysts [165–167]. Although not all cases of xanthomatous hypophysitis can definitely be linked to sellar cysts, it has been proposed that this type should be recognized as a form of secondary hypophysitis [167]. Xanthomatous hypophysitis is suggested to represent part of a histological spectrum with many cases transitioning into xanthogranulomatous inflammation types [166]. The histomorphology of xanthogranulomatous hypophysitis, often referred to as sellar xanthogranuloma, is characterized by cholesterol clefts, foamy and multinucleated giant cells, and prominent hemosiderin deposits (Fig. 12). It is widely recognized, that xanthogranulomatous hypophysitis shows features of a secondary reaction to chronic inflammatory processes, mostly associated with Rathke cleft cysts, craniopharyngiomas, PitNET/adenomas or other cystic lesions [168–170]. However, xanthogranulomatous hypophysitis has also been described as occurring as part of systemic conditions with an autoimmune etiology or without evidence of adjacent pituitary lesions [168, 171].
Fig. 11.
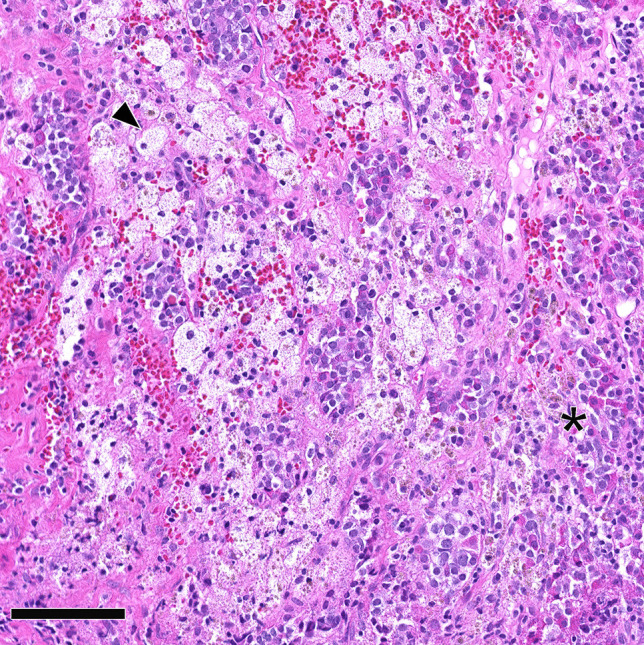
Xanthomatous hypophysitis. Clusters of histiocytes having abundant foamy cytoplasm and bland, round nuclei (arrowhead), with interspersed and scattered lymphocytes. The asterisk marks anterior residual pituitary cells. Histopathology provided evidence of a Rathke cleft cyst, leading to the classification as Rathke cleft cyst with certain secondary xanthomatous hypophysitis
Fig. 12.
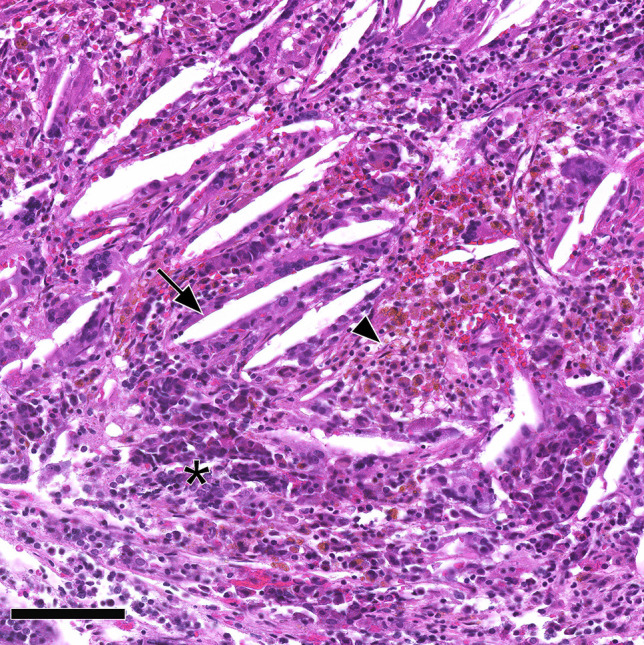
Xanthogranulomatous hypophysitis. Abundant cholesterol clefts (arrow), hemosiderin deposits, and macrophage clusters (arrowhead) among residual pituitary cells (asterisk). An MRI of the patient had provided evidence of a cystic sellar lesion prior to surgery. No cyst wall remnants were encountered upon histological examination, leading to the classification as probable secondary xantho-granulomatous hypophysitis
Necrotizing Hypophysitis
Necrotizing hypophysitis is often mentioned as an extremely rare type of primary hypophysitis [110, 122, 123, 164]. Only five histologically confirmed cases have been reported in the literature [172–175]. The histopathology of necrotizing hypophysitis was described as being characterized by significant non-hemorrhagic pituitary necrosis and marked lymphocytic pituitary inflammation. The affected patients demonstrated similar clinical courses comprising sudden-onset pituitary insufficiency, diabetes insipidus and radiologic features of ischemic pituitary apoplexy. Some authors have acknowledged that it is unclear whether necrotizing hypophysitis represents a distinct entity or a variant of known hypophysitis types [111, 120]. Due to the limited case numbers presented in the literature, sufficient data to understand the pathogenesis of necrotizing hypophysitis is not available. Moreover, necrosis is not uncommon in the pituitary gland and has been described in association with lymphocytic and granulomatous hypophysitis [148, 176], or in the context of apoplexy in pituitary tumors [177] or following significant post-partum bleeding [178]. Pituitary inflammation may also be triggered secondary to pituitary necrosis [172, 179]. Consequently, careful histological interpretation is warranted when pituitary necrosis is encountered together with pituitary inflammation (Fig. 13).
Fig. 13.
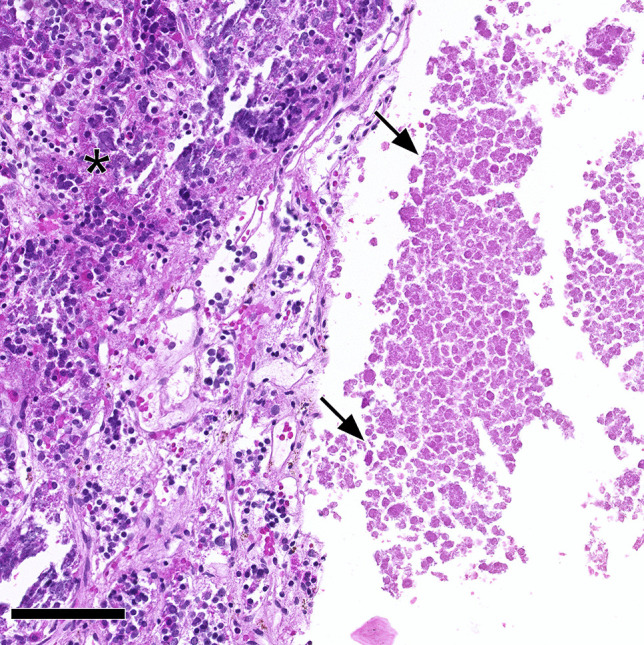
Pituitary necrosis and lymphocytic hypophysitis. Eosinophilic amorphous necrosis (arrows) next to pituitary gland with mild lymphocytic inflammation (asterisk). Pituitary apoplexy leading to necrosis of a PitNET/adenoma with secondary lymphocytic hypophysitis was suspected in this case. The case was classified as pituitary necrosis and probable secondary lymphocytic hypophysitis
Plasmacytic/IgG4-related Hypophysitis
The pituitary manifestation of IgG4-RD is referred to as IgG4-related hypophysitis (IgG4-RH; Fig. 8). IgG4-RH may occur as isolated IgG4-RH, with inflammation restricted to the pituitary gland or in the context of systemic IgG4-RD with involvement of further organs. Isolated IgG4-RH frequency among primary hypophysitis was previously reported as 4% in a study summarizing publications from 1917 to 2016 [100]. However, the incidence of IgG4-RD is increasing, presumably due to the raised awareness of the disease among physicians [106, 127, 180]. Recently, the frequency of isolated IgG4-RH among primary is reported to be up to 36–65% [106, 127, 181]. Pituitary involvement in patients with pre-existing systemic IgG4-RD is rare, with numbers ranging from 0–8% of IgG4-RD patients [182–185]. Recent studies have shown that there is a female predominance in isolated IgG4-RH as opposed to systemic IgG4-RD, which is more common in men [182, 184]. Furthermore, patients with isolated IgG4-RH are usually younger [127, 181]. Previous studies have suggested that the differing clinical characteristics of isolated IgG4-RH and systemic IgG4-RD with pituitary involvement might be due to distinct etiopathogenesis [127]. However, it is currently unclear if these two disease manifestations represent distinct pathological entities.
The histopathological diagnosis of IgG4-RH is challenging and deserves closer attention. In general, the diagnosis of IgG4-RD is based on a combination of radiological, serological, and pathological features [10, 11]. According to the 2020 revised comprehensive diagnostic (RCD) criteria by the Japanese IgG4 team [11], IgG4-RD can be made when two of three histological criteria are fulfilled in an organ: (I) presence of dense lymphocyte and plasma cell infiltration with fibrosis (Fig. 14a); (II) ratio of IgG4-/IgG-positive plasma cells greater than 40% and more than 10 IgG4-positive plasma cells per high power field (HPF) (Fig. 14b); (III) presence of tissue fibrosis, particularly storiform fibrosis, or obliterative phlebitis [11]. The consensus statement additionally recognizes organ-specific histological criteria to strengthen diagnostic certainty obtained from pathological features. However, to date, no pituitary-specific histological criteria have been defined. The American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) also recently defined classification criteria for IgG4-RD [10]. This classification is based on complex sets of inclusion and exclusion criteria for IgG4-RD, with the purpose of increasing specificity and excluding patients with infections, malignancies, hematological or immunological diseases. The ACR/EULAR classification is focused on frequently affected organs. Due to its rather uncommon involvement in IgG4-RD, the pituitary gland is also not included in these diagnostic criteria. In 2011, Leporati et al. proposed the presence of mononuclear cell infiltrates within the pituitary gland, rich in plasma cells and lymphocytes, with more than 10 IgG4-positive cells per HPF, as the histopathological criteria for IgG4-RH diagnosis [186]. These criteria were widely accepted by many authors [111, 124, 181, 187]. For IgG4-RH diagnosis, some authors also employed the IgG4-RD consensus criteria and additionally required the IgG4/IgG ratio cut-off to be > 40%, as proposed by Umehara et al. [11]. A recent study has suggested that these commonly used diagnostic criteria lead to an overestimation of IgG4-RH and proposed the application of the ACR/EULAR exclusion criteria to increase diagnostic specificity in the pituitary gland [188]. The general need for improved specific histopathological IgG4-RH criteria is reflected by various findings. For example, IgG4-positive plasma cell infiltration may be seen in association with focal pituitary lesions (e.g., Rathke cleft cyst) and also non-IgG4-related systemic diseases (e.g., hematological neoplasms) [188–191]. Moreover, obliterative phlebitis and storiform fibrosis patterns, which resemble histopathological characteristics of IgG4-RD in other organs, may be of poor diagnostic value in the pituitary gland [127]. Finally, the assessment of IgG4-positive cells per HPF is vague, since field area sizes are highly variable between different high-power objective lenses [192].
Fig. 14.
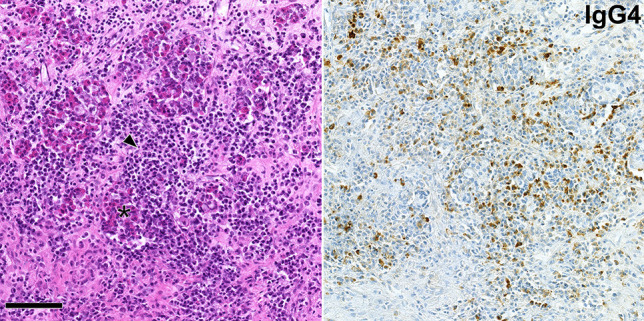
IgG4-related hypophysitis. Pituitary cells (asterisk) are overrun by a dense lymphocytic and plasma cell infiltrate (left image, arrowhead). IgG4-expressing plasma cells are numerous (right image). The illustrated tissue section is roughly 0.32 mm2 of size, corresponding to the field area of a high-power objective lens with a 0.64-mm field diameter. In this case, there was no evidence of IgG4-related disease in other organs of the patient, leading to the classification as certain primary IgG4-related hypophysitis. Scale bar is 100 µm
The literature presents inconsistent use of terminology referring to the etiological classification of IgG4-RH. Some authors exclusively refer to IgG4-RH as a form of primary hypophysitis, irrespective of further organ involvement [108, 121, 123, 193]. Others have also referred to pituitary involvement in systemic IgG4-RD as a form of secondary hypophysitis [107, 110, 122].
Refinement of Hypophysitis Classification
Hypophysitis classification resembles a continuously changing and evolving field with slow progress in the comprehension of the inflammatory pathomechanisms. Some changes in hypophysitis classification are attributable to newly emerging entities, such as IgG4-RD. Other changes come from novel iatrogenic causes, such as cancer treatment with immune checkpoint inhibitors. In further instances, diagnostic uncertainty prevails concerning the etiology of hypophysitis, especially with respect to xanthomatous or necrotizing forms. Thus, a combination of different factors provokes inconsistent classification approaches and variable terminology across studies.
In order to form more homogenous case groups, facilitate research comparability, and enable further progress in the currently poorly understood field of pituitary autoimmunity, the application of uniform terminology and classification is desirable. Table 1 summarizes diseases associated with pituitary inflammation and their etiology-based classification. Figure 15 illustrates our proposed algorithm of a pathology-centered approach to match pathological and etiological hypophysitis classification terminology in due consideration of multi-disciplinary findings. On the grounds of the preceding literature review, we highlight six bullet points that may improve consistency in hypophysitis classification.
-
Incorporation of diagnostic certainty into the classification of hypophysitis:
The matching of pathology and etiology requires multi-disciplinary information, which may not be readily available in routine diagnostics or retrospective studies. We suggest that diagnostic certainty be incorporated into the etiology-based hypophysitis classification. We propose that the terms “certain” or “probable” should standardly precede the terms “primary” or “secondary” in etiological hypophysitis classification. Hypophysitis of unclear origin should be referred to as “indeterminate”.
-
Combination of pathology- and etiology-based terminology:
The inconsistent use of hypophysitis terminology to refer to either entity-specific or general descriptive attributes can lead to irritation. We propose that classification terms based on etiology (“indeterminate” or “certain”/ “probable” preceding “primary”/ “secondary”) and pathology (“lymphocytic”/ “granulomatous”/ “xanthomatous” …), should be consistently combined to avoid confusion. Example 1: “Probable primary lymphocytic hypophysitis” refers to a lymphocytic hypophysitis in a patient without known pre-existing systemic diseases or adjacent pituitary lesion, where systemic inflammatory diseases have, however, not been ruled out yet. Example 2: “Certain secondary xanthomatous hypophysitis” refers to xanthomatous hypophysitis in a patient with histological evidence of a ruptured Rathke cleft cyst. When secondary hypophysitis is identified, the final diagnosis should also name the causative disease entity.
-
Diagnostic criteria of IgG4-RH:
Although commonly recognized by previous studies, the diagnostic criterion for IgG4-RH as defined by Leporati et al. [186] (> 10 IgG4-positive plasma cells/HPF) likely leads to an overdiagnosis of the disease. We propose that the additional requirement of an IgG4/IgG ratio cut-off > 40%, as proposed by Umehara et al. [11] along with typical histomorphology, should be met to make the diagnosis. Future studies will show if these criteria require further refinement.
-
Etiological classification of autoimmune hypophysitis co-occurring with other autoimmune diseases:
The classification of hypophysitis occurring in the context of pre-existing autoimmune diseases is a matter of debate. It is currently not known if isolated primary autoimmune hypophysitis, as opposed to hypophysitis in systemic autoimmune diseases, represents distinct entities. In order to facilitate distinguishability between these cases and avoid semantic overlap, we propose that the etiological classification term “primary” should be restricted to cases in which inflammation is limited to the pituitary gland. Hypophysitis occurring as part of a systemic autoimmune disease should be referred to as “secondary hypophysitis,” while the underlying pathomechanisms are unknown.
-
Secondary etiology of xanthomatous/xanthogranulomatous hypophysitis:
We support the recognition of xanthomatous and xanthogranulomatous hypophysitis as types of secondary hypophysitis, which most likely occur as a result of leakage, rupture, or bleeding of a pre-existing pituitary lesion. Therefore, we propose that xanthomatous/xanthogranulomatous hypophysitis with or without evidence of an adjacent or preceding pituitary lesion should be classified as certain or probable secondary xanthomatous/xanthogranulomatous hypophysitis, respectively.
-
Classification of necrotizing hypophysitis:
The recognition of necrotizing hypophysitis as a distinct type of primary hypophysitis is questionable due to the limited case numbers and significant clinical and pathological overlap with other entities. We propose that the etiology of this pathological variant and, consequently, its classification as primary or secondary should be left to speculation at present. Moreover, it is important to consider that pituitary necrosis may occur secondary to other causes, some of which are often left unresolved.
Table 1.
Etiology-based hypophysitis classification and associated disease entities
| Hypophysitis etiology | ||
|---|---|---|
| Cause | Primary | Secondary |
| Inflammatory | - Isolated hypophysitis (lymphocytic, plasmacytic, granulomatous, IgG4-related, or mixed forms) |
- Systemic IgG4-related disease - Type 1 diabetes mellitus - Polyglandular syndrome - Primary biliary cirrhosis - Autoimmune adrenalitis - Autoimmune thyroiditis |
| Neoplastic |
- Pituitary neuroendocrine tumour/adenoma - Craniopharyngioma - Germinoma - Astrocytoma - Meningioma - Metastasis |
|
| Infectious |
- Tuberculosis - Syphilis - other bacterial, mycotic, viral, parasitic pathogens |
|
| Vascular |
- Granulomatosis with polyangiitis (formerly Wegener ‘s) - Temporal arteritis - Takayasu arteritis - Apoplexy - Aneurysm |
|
| Iatrogenic |
- Immune checkpoint inhibitors - Immunosuppressants - Interferons - Surgery - Irradiation |
|
| Other |
- Rathke cleft cyst - Arachnoid cyst - Colloid cyst - (Epi-)Dermoid cyst - Paraneoplastic anti-PIT1-antibody syndrome |
|
Fig. 15.

Diagnostic algorithm for pathological and etiological classification of pituitary inflammation. Starting point of the algorithm is the histological evidence of pituitary inflammation. End points are the etiological hypophysitis classification combined with the degree of diagnostic certainty. The preceding term “probable” should prompt further multidisciplinary diagnostic work-up in order to clarify the diagnosis. If new relevant aspects arise from other disciplines, the algorithm returns to the starting point. The final diagnosis unifies the degree of diagnostic certainty (certain/probable/indeterminate), etiology (primary/secondary), and pathological classification (lymphocytic/plasmacytic/granulomatous/xanthomatous/…). *The recognition of necrotizing primary hypophysitis as a distinct entity is questionable. Detection of pituitary necrosis should prompt characterization of the inflammatory infiltrate found in the intact pituitary tissue. Vascular pathogenesis (pituitary apoplexy) or tumor necrosis should be considered
In conclusion, primary hypophysitis is a poorly understood disease. Our comprehension of the pathomechanisms is obstructed by restricted surgical accessibility of the pituitary gland, overlapping pathological and clinical features of various entities, and the challenges of interdisciplinary work-up for adequate classification. Harmonization of classification terminology is desirable in order to form homogeneous case groups. Our proposed diagnostic algorithm and classification terminology may be helpful to enhance multidisciplinary diagnostic interplay and facilitate comparability between different studies.
Thyroid Gland
Inflammatory processes of the thyroid gland cover a wide spectrum of pathologic conditions, characterized by different etiologies (infectious, autoimmune, immune-mediated), morphological pictures (according to the type/s of inflammatory cells and cytoarchitectural changes in the thyroid parenchyma), and clinical presentation (acute, subacute, or chronic; associated with hypo- or hyperthyroidism). Thus, these disorders have been variably classified according to these parameters (Figs. 16 and 17). However, it must be recognized that most of them represent definite clinicopathological entities. Acute thyroiditis is usually caused by infections and has already been discussed. In the following text, the most frequent and clinically significant non-acute inflammations of the thyroid gland will be reviewed.
Fig. 16.

Classification criteria for thyroiditis
Fig. 17.

Histopathology-based classification of thyroiditis with incorporation of clinical and etiopathogenetic information. Relevant clinicopathological entities are highlighted in bold
Subacute Granulomatous Thyroiditis (de Quervain Thyroiditis)
This type of thyroiditis is also known as painful subacute thyroiditis, giant cell thyroiditis, or post-viral thyroiditis. It is clinically characterized by low grade fever, anterior neck pain, an enlarged, palpable, and tender thyroid, transient hyperthyroidism, and elevation of serum inflammation indexes, in absence of anti-thyroid antibodies [194]. Women are significantly more affected than men and the age of occurrence is in early adulthood (25–35 years) [194]. Viral infections are thought to be the main cause of subacute thyroiditis, although a true causal relationship has not been demonstrated. Coxsackievirus and Echovirus, which are responsible for seasonal parainfluenza syndromes, have been implicated in the pathogenesis, as have other viruses causing mumps, measles, and influenza [195], and SARS-CoV-2 has been demonstrated to be associated with this disease, too [196]. Several drugs, including interferon and tumor necrosis factor alpha have also been claimed to be related to subacute thyroiditis [197, 198]
Although nowadays the diagnosis of subacute thyroiditis is mostly made on clinical grounds and with the aid of ultrasonography and fine needle aspiration, the pathological features of the disease have been well characterized in the past [199]. Macroscopically, the thyroid is asymmetrically enlarged and homogeneously firm, but nodules may be present. The histopathological picture varies according to the stage of the disease, being characterized by follicular cell disruption and neutrophilic inflammatory infiltrate in the early phases; mononuclear infiltrate with granuloma formation appears later. Granulomas are, at least initially, centered on follicles and contain epithelioid macrophages and giant multinucleated cells. Along time, reparative changes appear and fibrosis begins to dominate the picture. In fine needle aspiration specimens, isolated giant cells, macrophages, or even granulomas are seen, but neutrophils may appear in early stages of the disease. In later fibrotic stages, cytology samples may be non-diagnostic, due to fibrosis-related hypocellularity [200]. Rarely, reactive atypia of follicular cells may prompt a diagnosis of an atypical follicular lesion [201].
Subacute thyroiditis has a benign course in the majority of patients, with recovery of thyroid function in less than a year after transient phases of hyperthyroidism and, later, hypothyroidism. Persistent hypothyroidism has been reported in less than 10% of patients [202]. Anti-inflammatory drugs are used to relieve symptoms.
Other Granulomatous Thyroiditis
Granulomatous inflammation may be seen in various conditions, apart from de Quervain’s thyroiditis. So called palpation thyroiditis is a relatively frequent finding in surgical specimens and may be related either to medical physical examination or to self-touching in patients with large goiters. It is microscopically characterized by focal effacement of single or clustered follicles, which are ringed by multinucleated giant cells, lymphocytes, and plasma cells (Fig. 18). Palpation thyroiditis is distinguished from de Quervain’s thyroiditis thanks to the very limited damage to thyroid parenchyma [203].
Fig. 18.
Palpation thyroiditis. A well-formed granuloma, composed of epithelioid histiocytes and multinucleated giant cells, surrounded by lymphoplasmacytic infiltrate is seen. The lesion is focal, no fibrosis is observed, and the damage to follicular thyroid cells is very limited. Courtesy of Prof. Sefano La Rosa, University of Insubria, Varese, Italy
Tuberculosis may involve the thyroid gland with typical caseating granulomas, in which the mycobacterium may be detected either with Ziehl–Neelsen method or, more accurately, using a specific polymerase chain reaction for bacterial DNA.
Sarcoidosis rarely affects the thyroid gland and its recognition is not straightforward, as this is a diagnosis of exclusion after having checked other possible causes of thyroid inflammation. Anecdotal cases of sarcoidosis-related hyperthyroidism and hypothyroidism have been described [204, 205].
Autoimmune Thyroid Disorders
Autoimmune thyroid disorders (AITDs) are discussed here together, as they are morphologically characterized by lymphoid infiltration of the parenchyma and thus may be included, as a group, in the inflammatory diseases of the thyroid gland. Classically, AITDs include autoimmune thyroiditis (or Hashimoto thyroiditis, HT) and Graves’ disease (GD); however, other entities, such as postpartum thyroiditis and silent thyroiditis, have a demonstrated autoimmune pathogenesis. The diagnosis of AITDs relies on clinical and laboratory data. However, the pathologist is not infrequently faced with these disorders, mainly HT and GD, and should be aware of their morphological features and of the possible proliferative lesions that may arise in their context.
HT, also known as autoimmune thyroiditis or chronic lymphocytic thyroiditis, is clinically characterized by the progressive emergence of hypothyroidism (a transient hyperthyroidism may appear in the early phase of the disease), associated with circulating anti-thyroid peroxidase and anti-thyroglobulin. It is most commonly seen in middle aged women (M:F ratio 5–7:1), but children and elderly people may also be affected. A genetic predisposition to this disease is documented by studies on families and twins [206]. Although the management of HT mostly relies on medical treatment based on thyroid hormone replacement to reach euthyroidism, thyroid surgery may be indicated in various settings, such as the presence of a dominant nodule, or diffuse thyroid enlargement with compression of cervical structures, or as an option in case of medical treatment failure [207]. In these circumstances, a careful pathological examination is important to confirm the diagnosis and rule out neoplastic disease. Macroscopically, the thyroid with HT is diffusely enlarged, with a lobulated cut surface, which is generally paler than a normal thyroid. Nodules and cystic lesions may be present. Microscopically, the gland parenchyma is effaced by a dense mononuclear infiltrate, composed of lymphocytes and plasma cells often forming lymphoid follicles with activated germinal centers (Fig. 19). The lobular architecture of the thyroid is more evident than normal, due to the presence of augmented fibrous septa, and in advanced stages of the disease, fibrosis may become prominent, inducing atrophy of thyroid cells. Thyroid follicular cells typically undergo oncocytic change (Fig. 19, inset) and squamous metaplasia may also be observed. Several histological variants of HT have been described, including the fibrous, the fibrous atrophy, the juvenile, and the cystic variant [208]. An IgG4-related disease variant of HT has been described, but we believe it should be considered a separate entity and will be discussed later. Reactive atypia of follicular thyroid cells is not an unusual finding in HT: however, in a subset of cases, distinct clusters of thyrocytes with an irregular nuclear membrane and other features resembling thyroid papillary carcinoma nuclei may be seen. These have been called follicular epithelial dysplasia (FED) (Fig. 20) and are thought to be precursor lesions of papillary thyroid carcinoma (PTC) arising in HT [209, 210]. The differentiation of FED from small foci of PTC relies on the absence of infiltration and papillary architecture in the former lesions, which, on the other hand, lack nuclear pseudoinclusions and other overt features of PTC nuclei [209, 210]. The relationships of HT with thyroid neoplasms is complex and regards both epithelial and non-epithelial neoplasms. Papillary carcinoma is the most common neoplasm associated with HT and a recent meta-analysis reported the co-existence of PTC in 8% (only male population) to 36.4% (both male and female population) of surgically removed thyroid specimens with HT [211]. The prognostic correlates of PTC arising on HT are not yet well established: some studies report a good clinical outcome related to the small size, the reduced rate of extrathyroidal infiltration and lymph node metastases, and the lack of BRAF mutation [212, 213], but others did not find any significant better prognosis in this group of patients, compared to non-HT-associated PTC [214]. Although a definite association of HT with a specific PTC subtype has not been demonstrated, the rare Warthin-like subtype of PTC mostly arises in the context of HT [215]. Interestingly, a rare pathological entity, named multifocal fibrosing thyroiditis, which can overlap with HT, represents a morphological mimicker of PTC and must be distinguished from it [216]. Another HT-related malignancy is thyroid extranodal marginal zone lymphoma of MALT type (MALT lymphoma). This type of lymphoma may arise in both mucosal and glandular tissues and is generally associated with chronic inflammation [217]. In the thyroid, MALT lymphoma is strongly associated with HT and the differential diagnosis between the two entities may be challenging, as discussed elsewhere [2]. A destructive growth, with complete effacement of the thyroid follicles and extrathyroidal extension, is highly suggestive of lymphoma. The application of a proper immunohistochemical panel and molecular analysis for immunoglobulin gene rearrangement helps in the correct differential diagnosis between MALT lymphoma and HT or other types of lymphoma [2].
Fig. 19.

Hashimoto thyroiditis. The thyroid parenchyma is overrun by dense lymphocytic infiltration, with activated lymphoid follicles, inducing reactive changes in thyroid follicular cells, that frequently undergo oncocytic change (inset)
Fig. 20.
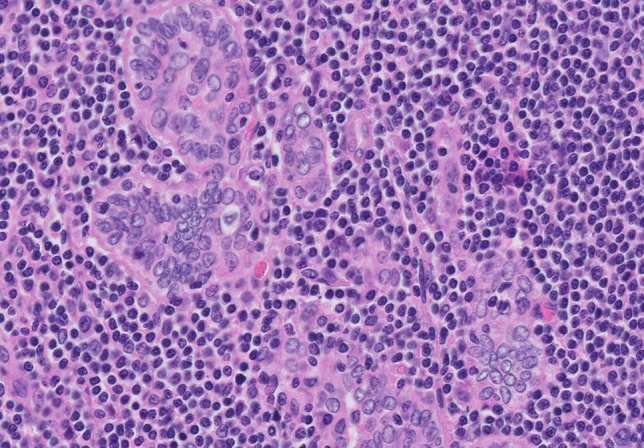
Follicular epithelial dysplasia in Hashimoto thyroiditis. These follicles, embedded in a dense lymphocytic infiltrate, show irregular shape, nuclear crowding, irregular nuclear membrane with occasional grooving, and nuclear clarification. There are no signs of invasion, papillary growth, or nuclear pseudoinclusions
GD, also known as Basedow disease or diffuse toxic hyperplasia, is the most common cause of persistent hyperthyroidism in iodine-replete areas and one of the most common autoimmune diseases worldwide [218]. The male-to-female ratio is 3–8:1, and the most frequent age of insurgence is in the 4th decade of life. Besides thyroid involvement, GD is clinically characterized by unique ophthalmopathy and dermatopathy [219]. The hyperthyroidism in GD is caused by circulating anti-TSH receptor antibodies that act as agonists of the receptor, stimulating thyroid hormone production. An association with specific HLA haplotypes (HLA-B8 and HLA-DR3) has been demonstrated, as well as a stronger familial predisposition than for HT [220]. Patients with GD are treated using antithyroid drugs, radioactive iodine, or thyroidectomy. According to the most recent guidelines, surgery is recommended for women planning a pregnancy and for patients with large compressive goiter or with suspicious thyroid nodule, or resistant to radioactive iodine therapy or with moderate/severe ophthalmopathy [221]. The morphological features of GD vary in previously treated and untreated cases. Macroscopically, untreated cases show a diffuse enlargement of the thyroid gland, with a homogenous, dark red, cut surface. In contrast, the gland may be smaller and paler in previously treated patients, due to the lower vascularization of thyroid parenchyma. Accordingly, the histopathological picture of untreated GD is dominated by diffuse hyperplasia of follicular thyroid cells that, in untreated cases, show a columnar shape and are arranged in papillae infolding into follicular lumina. The hyperfunctioning state of thyrocytes is documented by the low amounts and the scalloping of colloid, which is paler than in a normal thyroid (Fig. 21). Cases treated with anti-thyroid drugs may show greater amounts of colloid and less pronounced papillary hyperplasia; nuclear pleomorphism and hyperchromasia may be present (Fig. 22). GD treated with radioiodine therapy are characterized by fibrosis, reduction of hyperplastic features and radioiodine-induced nuclear atypia. Lymphocytic infiltration is also present in the form of lymphoid follicles but is far less prominent than in HT. Nodular follicular thyroid lesions may be present in GD, a fraction of which is represented by thyroid carcinoma. The frequency of thyroid cancer in GD is reported to be between 2.3 and 21.1%, with an incidence 2.5 times higher than in the general population, and the most common histotype was PTC [222]. In this respect, it should be noted that the differential diagnosis between GD and PTC may pose some difficulties to general pathologists, due to the papillary architecture of GD and the occasional presence of psammoma bodies and hyperchromatic nuclei. In such circumstances, the histological evidence of hyperfunction, the presence of fibrovascular axes in papillae, the absence of clear-cut nuclear features of papillary carcinoma, and the diffuse nature of cytoarchitectural changes favor GD (Fig. 22). In challenging situations, a panel of immunostains including, but not limited to HBME1 (positive in PTC and negative in non-neoplastic follicular cells) and CD56 (negative in PTC and positive in non-neoplastic follicular cells) may be taken in consideration to support diagnosis, but the role of immunohistochemistry for the differential diagnosis of thyroid lesions is still controversial [223, 224].
Fig. 21.
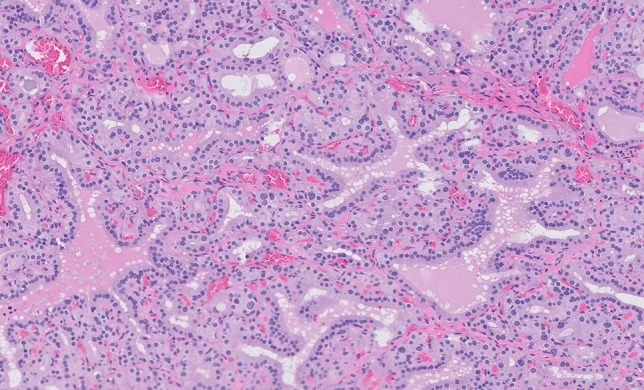
Untreated Graves’ disease. Diffuse hyperplasia of hyperfunctioning thyroid follicles lined by columnar follicular cells and containing pale colloid with scalloping due to active resorption by follicular cells
Fig. 22.
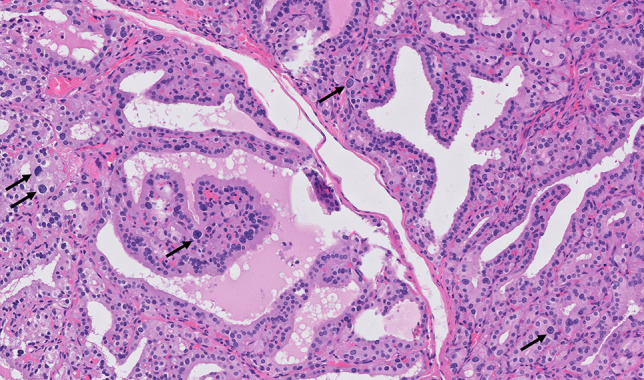
Treated Graves’ disease. In this case, a papillary overgrowth of follicular cells is observed, as well as focal nuclear enlargement and hyperchromasia (arrows). The presence of fibrovascular cores in papillary structures, the absence of nuclear clarification, grooves and pseudoinclusions, the hyperfunctioning morphology of thyroid cells, and the diffuse character of these features exclude the diagnosis of a papillary carcinomas. Nuclear pleomorphism was due to previous anti-thyroid therapy
IgG4-related Thyroid Disease
As already discussed earlier in this review, the diagnosis of IgG4-RD heavily relies on the recognition of a peculiar histological picture in the target organs, dominated by dense lymphoplasmacytic infiltrates and storiform fibrosis, with a variable presence of obliterative phlebitis and infiltration of eosinophils [9]. In fact, it should be acknowledged that these features may be present in varying combinations, giving rise to a spectrum of histopathological appearances. In the thyroid, this heterogeneity is reflected in morphological pictures that may fall into different clinicopathological entities, including HT, the fibrous variant of HT, particular cases of GD, and invasive fibrous thyroiditis (Riedel thyroiditis) [225]. However, despite the morphological similarities, IgG4-related thyroid diseases differ from their non-IgG4 counterpart in terms of clinical features and the composition of the inflammatory infiltrate which is, of course, dominated by IgG4 + plasma cells. IgG4-related HT, for example, is more common in males, more rapidly evolves into hypothyroidism, has higher serum levels of anti-thyroid antibodies, and frequently presents hypoechoic nodules than non-IgG4 HT. In addition, elevated IgG4 serum levels may be present, which decrease after thyroidectomy [226]. Although specific treatment guidelines for IgG4-related HT have not been established, yet the distinction between this entity and non-IgG4 HT may be useful for patient management. Specific cut-offs for IgG4 + plasma cell infiltration have been suggested for this purpose: IgG4 + plasma cells > 20/HPF, and IgG4 + /IgG + plasma cell ratio > 30% [227] (Fig. 23). Invasive fibrous thyroiditis (Riedel thyroiditis) is a rare disease, characterized by sudden enlargement of the thyroid gland, which is nontender and firm to palpation and causes progressive compression of cervical structures. It is now established that the fibro-inflammatory process underlying this disease is part of the IgG4-RD [228]. Macroscopically, the thyroid glands involved in Riedel thyroiditis are enlarged, harder than normal, and have a pale cut surface, on which the normal lobulation of the gland is not recognizable. Fibrosis often extends into perithyroidal tissues. Histologically, storiform and keloid-like fibrosis are present, along with a lymphoplasmacytic infiltrate with numerous plasma cells (Fig. 24). Oncocytic change in thyroid cells is not observed and, if present, should suggest a fibrous variant of HT.
Fig. 23.
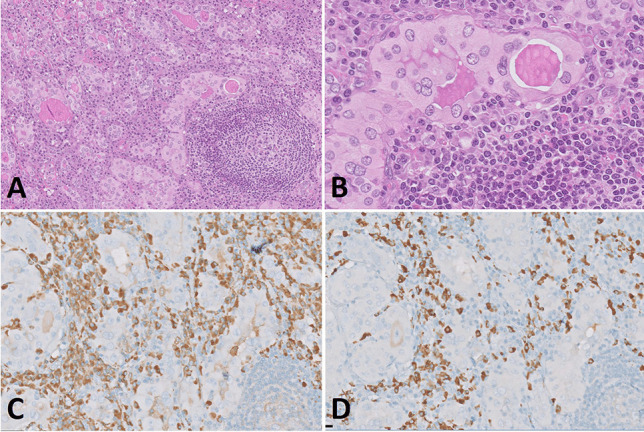
IgG4-related Hashimoto thyroiditis. An activated lymphoid follicle is evident, but the interstitial inflammatory infiltrate is mainly composed by mature plasma cells (A, B). Oncocytic changes of follicular thyroid cells is prominent (B). Interstitial plasma cells are IgG + (C), with numerous IgG4 + (D) plasma cells and an IgG4/IgG ratio > than 40%
Fig. 24.
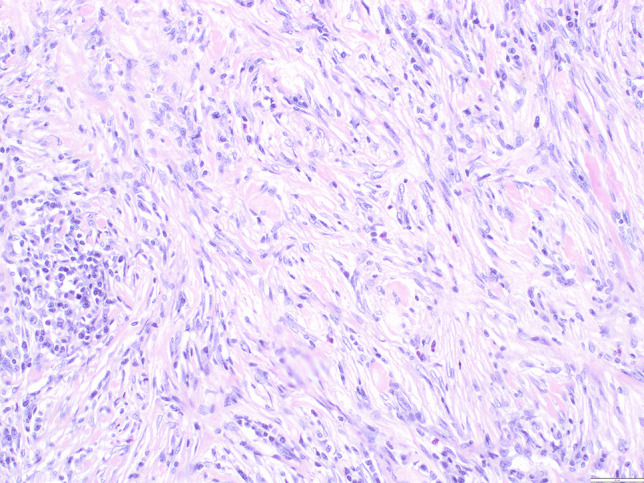
Riedel’s thyroiditis. Dense, storiform fibrosis and plasma cell infiltrate. Thyroid parenchyma is no more recognizable. Courtesy of Prof. Sefano La Rosa, University of Insubria, Varese, Italy
Adrenal Gland
Inflammatory and infectious disorders affect the adrenal gland encompass a variety of entities in disorders ranging from focal adrenalitis, to autoimmune involvement of the adrenal gland, and bacterial, fungal, or viral infections. Most recently, the effects of SARS-CoV-2 and associated vaccine have been described involving the adrenal gland. Focal adrenalitis is a common incidental finding seen in the adrenal glands of elderly (Fig. 25) [229]. Unlike autoimmune adrenalitis, focal adrenalitis lacks the more diffuse inflammation seen in autoimmune lymphocytic adrenalitis. Focal adrenalitis is not known to cause endocrine dysfunction of the adrenal gland unlike autoimmune lymphocytic adrenalitis or infections.
Fig. 25.

Focal lymphocytic adrenalitis in an elderly man. Courtesy of Prof. Stefano La Rosa, University of Insubria, Varese, Italy
Autoimmune lymphocytic adrenalitis is rare, but it is the most frequent cause of Addison disease. The lymphoplasmacytic infiltrate is associated with destruction of the adrenal cortex and becomes clinically apparent only when 90% of the cortex is destroyed [29, 230]. A recent genome wide association study of 1223 cases of autoimmune Addison disease (excluding individuals with autoimmune polyendocrine syndrome type-1) and 4097 healthy controls identified 9 independent risk foci including four foci seen in other autoimmune diseases (HLA, BACH2, PTPN22, and CTLA4) as well as LPP, SH2B3, SIGLEC5, UBASH3A, and two alterations in AIRE (Autoimmune Regulator gene) [231]. These associations explained 35–41% of the cases. Four of the 9 loci (LPP, SH2B3, SIGLEC5, and UBASH3A) had not previously been identified in association with autoimmune Addison disease [231]. The strongest risk locus in autoimmune Addison disease was found to be the HLA region and dominated by HLA-DQB1 [231].
IgG4-related disease of the adrenal glands is very uncommon, and only anecdotal cases have been reported, presenting as pseudotumor masses that, at histological analysis, showed the characteristic features of IgG4-RD (storiform fibrosis, dense plasma cell infiltrate, high IgG4/IgG ratio, and high numbers of IgG4 + plasma cells) [232, 233].
Medications can be associated with endocrine dysfunction, including the adrenal gland [234]. Immune checkpoint inhibitors may be associated with endocrine abnormalities in 25–50% of individuals on these medications [8]. Tyrosine kinase inhibitors targeting the vascular endothelial growth factor receptor have also been associated with adrenal insufficiency [235].
Conclusions
Infectious and inflammatory disorders of the endocrine system are relatively rare conditions that encompass a spectrum of etiologies, clinical manifestations, and prognoses. The pathologist should be aware of their morphological features, in order to guide appropriate patient management. Distinguishing among inflammatory conditions of different etiologies/pathogenesis or, in the case of mass-forming inflammation, excluding a neoplastic process is an important task in diagnostic daily practice.
Author Contribution
SU and MD designed and coordinated the review and drafted the manuscript; LE, JW, KM, and WG drafted the manuscript. All Authors approved the final version of the manuscript.
Funding
WS received funding for the German Pituitary Tumor Registry to WS from the Novartis Pharma GmbH (Nuremberg, Germany), Novo Nordisk Pharma GmbH (Mainz, Germany), Pfizer Pharma GmbH (Berlin, Germany), and Ipsen Pharma GmbH (Ettlingen, Germany).
Availability of Data and Materials
Not applicable.
Declarations
Ethics Approval
Not applicable.
Competing Interests
The authors declare no competing interests.
Footnotes
Silvia Uccella and Matthias Dottermusch are co-first authors
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Silvia Uccella, Email: silvia.uccella@gmail.com, Email: silvia.uccella@hunimed.eu.
Matthias Dottermusch, Email: m.dottermusch@uke.de.
Lori Erickson, Email: Erickson.Lori@mayo.edu.
Julia Warmbier, Email: julia.warmbier@web.de.
Kathleen Montone, Email: Kathleen.Montone@pennmedicine.upenn.edu.
Wolfgang Saeger, Email: w.saeger@uke.de.
References
- 1.Straub RH. Interaction of the endocrine system with inflammation: a function of energy and volume regulation. Arthritis Res Ther. 2014;16(1):203. doi: 10.1186/ar4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uccella S, Magnoli F, Amaglio C, Sessa F, La Rosa S. The spectrum of lymphoproliferative disorders in endocrine organs: from histology to molecular genetics. Diagn Histopathol. 2019;25(5):166–177. doi: 10.1016/j.mpdhp.2019.02.003. [DOI] [Google Scholar]
- 3.Kino T, Chrousos GP. Virus-mediated modulation of the host endocrine signaling systems: clinical implications. Trends Endocrinol Metab. 2007 May-Jun;18(4):159–66. [DOI] [PMC free article] [PubMed]
- 4.Morris GB, Ridgway EJ, Suvarna SK. Traditional stains and modern techniques for demonstrating microorganisms in histology. In: Suvarna SK, Layton C, Bancroft JD, editors. Bancroft’s Theory and Practice of Histological Techniques (Eighth Edition) Elsevier; 2019. pp. 254–279. [Google Scholar]
- 5.Korem Kohanim Y, Tendler A, Mayo A, Friedman N, Alon U. Endocrine Autoimmune Disease as a Fragility of Immune Surveillance against Hypersecreting Mutants. Immunity. 2020;52(5):872–884.e5. doi: 10.1016/j.immuni.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryder M, Callahan M, Postow MA, Wolchok J, Fagin JA. Endocrine-related adverse events following ipilimumab in patients with advanced melanoma: a comprehensive retrospective review from a single institution. Endocr Relat Cancer. 2014;21(2):371–381. doi: 10.1530/ERC-13-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Percik R, Shoenfeld Y. Check point inhibitors and autoimmunity: Why endocrinopathies and who is prone to? Best Pract Res Clin Endocrinol Metab. 2020;34(1):101411. doi: 10.1016/j.beem.2020.101411. [DOI] [PubMed] [Google Scholar]
- 8.Wright JJ, Powers AC, Johnson DB. Endocrine toxicities of immune checkpoint inhibitors. Nat Rev Endocrinol. 2021;17(7):389–399. doi: 10.1038/s41574-021-00484-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, Klöppel G, Heathcote JG, Khosroshahi A, Ferry JA, Aalberse RC, Bloch DB, Brugge WR, Bateman AC, Carruthers MN, Chari ST, Cheuk W, Cornell LD, Fernandez-Del Castillo C, Forcione DG, Hamilos DL, Kamisawa T, Kasashima S, Kawa S, Kawano M, Lauwers GY, Masaki Y, Nakanuma Y, Notohara K, Okazaki K, Ryu JK, Saeki T, Sahani DV, Smyrk TC, Stone JR, Takahira M, Webster GJ, Yamamoto M, Zamboni G, Umehara H, Stone JH. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–1192. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- 10.Wallace ZS, Naden RP, Chari S, Choi H, Della-Torre E, Dicaire JF, Hart PA, Inoue D, Kawano M, Khosroshahi A, Kubota K, Lanzillotta M, Okazaki K, Perugino CA, Sharma A, Saeki T, Sekiguchi H, Schleinitz N, Stone JR, Takahashi N, Umehara H, Webster G, Zen Y, Stone JH; American College of Rheumatology/European League Against Rheumatism IgG4-Related Disease Classification Criteria Working Group. The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-Related Disease. Arthritis Rheumatol. 2020 Jan;72(1):7–19. [DOI] [PubMed]
- 11.Umehara H, Okazaki K, Kawa S, Takahashi H, Goto H, Matsui S, Ishizaka N, Akamizu T, Sato Y, Kawano M; Research Program for Intractable Disease by the Ministry of Health, Labor and Welfare (MHLW) Japan. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol. 2021 May;31(3):529–533. [DOI] [PubMed]
- 12.Rzepecka A, Babińska A, Sworczak K. IgG4-related disease in endocrine practice. Arch Med Sci. 2019;15(1):55–64. doi: 10.5114/aoms.2017.70889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paes JE, Burman KD, Cohen J, Franklyn J, McHenry CR, Shoham S, Kloos RT. Acute bacterial suppurative thyroiditis: a clinical review and expert opinion. Thyroid. 2010;20(3):247–255. doi: 10.1089/thy.2008.0146. [DOI] [PubMed] [Google Scholar]
- 14.Falhammar H, Wallin G, Calissendorff J. Acute suppurative thyroiditis with thyroid abscess in adults: clinical presentation, treatment and outcomes. BMC Endocr Disord. 2019;19(1):130. doi: 10.1186/s12902-019-0458-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamada H, Fujita K, Tokuriki T, Ishida R. Nine cases of piriform sinus fistula with acute suppurative thyroiditis. Auris Nasus Larynx. 2002;29(4):361–365. doi: 10.1016/S0385-8146(02)00019-6. [DOI] [PubMed] [Google Scholar]
- 16.Kalladi Puthanpurayil S, Francis GL, Kraft AO, Prasad U, Petersson RS. Papillary thyroid carcinoma presenting as acute suppurative thyroiditis: a case report and review of the literature. Int J Pediatr Otorhinolaryngol. 2018;105:12–15. doi: 10.1016/j.ijporl.2017.11.032. [DOI] [PubMed] [Google Scholar]
- 17.Nishihara E, Miyauchi A, Matsuzuka F, Sasaki I, Ohye H, Kubota S, Fukata S, Amino N, Kuma K. Acute suppurative thyroiditis after fine-needle aspiration causing thyrotoxicosis. Thyroid. 2005;15(10):1183–1187. doi: 10.1089/thy.2005.15.1183. [DOI] [PubMed] [Google Scholar]
- 18.Lesh R, Hellums R, Pichardo P, Wong J, Pellitteri P, Purdy N, Stavrides K, Haugen TW. Thyroid Abscess: A Case Series and Literature Review. Ear Nose Throat J. 2023;5:1455613221150128. doi: 10.1177/01455613221150128. [DOI] [PubMed] [Google Scholar]
- 19.Yedla N, Pirela D, Manzano A, Tuda C, Lo Presti S. Thyroid abscess: Challenges in diagnosis and management. J Investig Med High Impact Case Rep. 2018;6:2324709618778709. doi: 10.1177/2324709618778709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossetti CL, Cazarin J, Hecht F, Beltrão FEL, Ferreira ACF, Fortunato RS, Ramos HE, de Carvalho DP. COVID-19 and thyroid function: What do we know so far? Front Endocrinol (Lausanne). 2022;19(13):1041676. doi: 10.3389/fendo.2022.1041676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reisi-Vanani V, Farzan M, Farzan M, Ataei-Goujani H, Keihani M, Taghipour-Boroujeni G. Role of the immune system and possible mechanisms in COVID-19 vaccine-induced thyroiditis: Case report and literature review. J Clin Transl Endocrinol Case Rep. 2022;26:100138. doi: 10.1016/j.jecr.2022.100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ando Y, Ono Y, Sano A, Fujita N, Ono S. Subacute Thyroiditis after COVID-19: A Literature Review. Am J Trop Med Hyg. 2022;107(5):1074–1082. doi: 10.4269/ajtmh.21-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Șandru F, Carsote M, Petca RC, Gheorghisan-Galateanu AA, Petca A, Valea A, Dumitrașcu MC. COVID-19-related thyroid conditions (Review) Exp Ther Med. 2021;22(1):756. doi: 10.3892/etm.2021.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldani LZ, Zavascki AP, Maia AL. Fungal thyroiditis: an overview. Mycopathologia. 2006;161(3):129–139. doi: 10.1007/s11046-005-0239-3. [DOI] [PubMed] [Google Scholar]
- 25.Danahey DG, Kelly DR, Forrest LA. HIV-related Pneumocystis carinii thyroiditis: a unique case and literature review. Otolaryngol Head Neck Surg. 1996;114(1):158–161. doi: 10.1016/S0194-59989670304-2. [DOI] [PubMed] [Google Scholar]
- 26.Paolo WF, Jr, Nosanchuk JD. Adrenal infections. Int J Infect Dis. 2006;10(5):343–353. doi: 10.1016/j.ijid.2005.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Upadhyay J, Sudhindra P, Abraham G, Trivedi N. Tuberculosis of the adrenal gland: a case report and review of the literature of infections of the adrenal gland. Int J Endocrinol. 2014;2014:876037. doi: 10.1155/2014/876037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kibirige D. Endocrine dysfunction among adult patients with tuberculosis: An African experience. Indian J Endocrinol Metab. 2014;18(3):288–294. doi: 10.4103/2230-8210.131136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saeger W. Adrenalitis [Adrenalitis]. Pathologe. 2016 May;37(3):238–44. German. [DOI] [PubMed]
- 30.Karki BR, Sedhai YR, Bokhari SRA. Waterhouse-Friderichsen Syndrome. 2022 Sep 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan–. [PubMed]
- 31.Shimizu S, Tahara Y, Atsumi T, Imai Y, Ueda H, Seo R, Higashibeppu N, Mima H, Yamazaki K. Waterhouse-friderichsen syndrome caused by invasive haemophilus influenzae type B infection in a previously healthy young man. Anaesth Intensive Care. 2010;38(1):214–215. [PubMed] [Google Scholar]
- 32.Hale AJ, LaSalvia M, Kirby JE, Kimball A, Baden R. Fatal purpura fulminans and Waterhouse-Friderichsen syndrome from fulminant Streptococcus pneumoniae sepsis in an asplenic young adult. IDCases. 2016;16(6):1–4. doi: 10.1016/j.idcr.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhattacharya S, Kubiha S, Tyagi P. Fungi and Endocrine Dysfunction. 2021 Jun 25. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000–. PMID: 34288619.
- 34.Peçanha PM, Batista Ferreira ME, Massaroni Peçanha MA, Schmidt EB, Lamas de Araújo M, Zanotti RL, Potratz FF, Delboni Nunes NE, Gonçalves Ferreira CU, Delmaestro D, Falqueto A. Paracoccidioidomycosis: Epidemiological and Clinical Aspects in 546 Cases Studied in the State of Espírito Santo, Brazil. Am J Trop Med Hyg. 2017 Sep;97(3):836–844. [DOI] [PMC free article] [PubMed]
- 35.Pal N, Banu HN, Chakraborty M, Jain N, Maiti PK. Current perspective of adrenal histoplasmosis in India: A prospective study in a tertiary care hospital. Eastern India. Indian J Med Microbiol. 2022;S0255–0857(22):00190–196. doi: 10.1016/j.ijmmb.2022.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Somasundaram NP, Gunatilake SSC. Infections in Endocrinology: Viruses. 2021 Mar 14. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000–.
- 37.Rodrigues D, Reis M, Teixeira V, Silva-Vergara M, Filho DC, Adad S, Lazo J. Pathologic findings in the adrenal glands of autopsied patients with acquired immunodeficiency syndrome. Pathol Res Pract. 2002;198(1):25–30. doi: 10.1078/0344-0338-00180. [DOI] [PubMed] [Google Scholar]
- 38.Mifsud S, Gauci Z, Gruppetta M, Mallia Azzopardi C, Fava S. Adrenal insufficiency in HIV/AIDS: a review. Expert Rev Endocrinol Metab. 2021;16(6):351–362. doi: 10.1080/17446651.2021.1979393. [DOI] [PubMed] [Google Scholar]
- 39.Glasgow BJ, Steinsapir KD, Anders K, Layfield LJ. Adrenal pathology in the acquired immune deficiency syndrome. Am J Clin Pathol. 1985;84(5):594–597. doi: 10.1093/ajcp/84.5.594. [DOI] [PubMed] [Google Scholar]
- 40.Chiu B, Butany J, Kovacs K, Cheng Z. Cytomegalovirus adrenalitis in acquired immunodeficiency syndrome. Endocr Pathol. 1994;5(4):218–222. doi: 10.1007/BF02921489. [DOI] [PubMed] [Google Scholar]
- 41.Pulakhandam U, Dincsoy HP. Cytomegaloviral adrenalitis and adrenal insufficiency in AIDS. Am J Clin Pathol. 1990;93(5):651–656. doi: 10.1093/ajcp/93.5.651. [DOI] [PubMed] [Google Scholar]
- 42.Lisco G, De Tullio A, Stragapede A, Solimando AG, Albanese F, Capobianco M, Giagulli VA, Guastamacchia E, De Pergola G, Vacca A, Racanelli V, Triggiani V. COVID-19 and the Endocrine System: A Comprehensive Review on the Theme. J Clin Med. 2021;10(13):2920. doi: 10.3390/jcm10132920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mirza SA, Sheikh AAE, Barbera M, Ijaz Z, Javaid MA, Shekhar R, Pal S, Sheikh AB. COVID-19 and the Endocrine System: A Review of the Current Information and Misinformation. Infect Dis Rep. 2022;14(2):184–197. doi: 10.3390/idr14020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanczkowski W, Gaba WH, Krone N, Varga Z, Beuschlein F, Hantel C, Andoniadou C, Bornstein SR. Adrenal Gland Function and Dysfunction During COVID-19. Horm Metab Res. 2022;54(8):532–539. doi: 10.1055/a-1873-2150. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, Wu X. Influence of COVID-19 vaccines on endocrine system. Endocrine. 2022;78(2):241–246. doi: 10.1007/s12020-022-03119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA, Drosten C, Pöhlmann S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020 Apr 16;181(2):271–280.e8. 10.1016/j.cell.2020.02.052. Epub 2020 Mar 5. PMID: 32142651; PMCID: PMC7102627. [DOI] [PMC free article] [PubMed]
- 47.Lazartigues E, Qadir MMF, Mauvais-Jarvis F. Endocrine Significance of SARS-CoV-2’s Reliance on ACE2. Endocrinology. 2020 Sep 1;161(9):bqaa108. [DOI] [PMC free article] [PubMed]
- 48.Mao Y, Xu B, Guan W, Xu D, Li F, Ren R, Zhu X, Gao Y, Jiang L. The Adrenal Cortex, an Underestimated Site of SARS-CoV-2 Infection. Front Endocrinol (Lausanne). 2021;8(11):593179. doi: 10.3389/fendo.2020.593179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beshay L, Wei K, Yang Q. Newly diagnosed autoimmune Addison’s disease in a patient with COVID-19 with autoimmune disseminated encephalomyelitis. BMJ Case Rep. 2022 Dec 5;15(12):e250749. 10.1136/bcr-2022-250749. PMID: 36593594; PMCID: PMC9723877. [DOI] [PMC free article] [PubMed]
- 50.Eskandari D, Ziaee A, Amirfarhangi Anbardan A, Zeinali E, Tirkan A. Primary adrenal insufficiency and myocarditis in COVID-19 disease: a case report. BMC Endocr Disord. 2022;22(1):336. doi: 10.1186/s12902-022-01257-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pezzaioli LC, Gatta E, Bambini F, Facondo P, Gava M, Cavadini M, Buoso C, Di Lodovico E, Rotondi M, Ferlin A, Cappelli C. Endocrine system after 2 years of COVID-19 vaccines: A narrative review of the literature. Front Endocrinol (Lausanne). 2022;10(13):1027047. doi: 10.3389/fendo.2022.1027047.PMID:36440218;PMCID:PMC9685624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao L, Guo X, Tian R, Wang Q, Feng M, Bao X, Deng K, Yao Y, Lian W, Wang R, Xing B. Pituitary abscess: clinical manifestations, diagnosis and treatment of 66 cases from a large pituitary center over 23 years. Pituitary. 2017;20:189–194. doi: 10.1007/s11102-016-0757-7. [DOI] [PubMed] [Google Scholar]
- 53.Pekic S, Miljic D, Popovic V. Infections of the Hypothalamic-Pituitary Region. 2021 Aug 9. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000–.
- 54.Martinez-Perez R, Kortz MW, Carroll BW, Duran D, Neill JS, Luzardo GD, Zachariah MA. Coronavirus disease 2019 and pituitary apoplexy: a single-center case series and review of the literature. World Neurosurg. 2021;S1878–8750(21):00829–839. doi: 10.1016/j.wneu.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bordes SJ, Phang-Lyn S, Najera E, Borghei-Razavi H, Adada B. Pituitary apoplexy attributed to COVID-19 infection in the absence of an underlying macroadenoma or other identifiable cause. Cureus. 2021;13:e13315. doi: 10.7759/cureus.13315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Talat N, Diaz-Cano S, Schulte KM. Inflammatory diseases of the parathyroid gland. Histopathology. 2011;59(5):897–908. doi: 10.1111/j.1365-2559.2011.04001.x. [DOI] [PubMed] [Google Scholar]
- 57.Husebye ES, Anderson MS, Kämpe O. Autoimmune Polyendocrine Syndromes. N Engl J Med. 2018;378(12):1132–1141. doi: 10.1056/NEJMra1713301.PMID:29562162;PMCID:PMC6007870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Finnish-German APECED Consortium An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet. 1997;17(4):399–403. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- 59.Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE, Kawasaki K, Asakawa S, Ito F, Shimizu N. Positional cloning of the APECED gene. Nat Genet. 1997;17(4):393–398. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- 60.Halonen M, Kangas H, Rüppell T, Ilmarinen T, Ollila J, Kolmer M, Vihinen M, Palvimo J, Saarela J, Ulmanen I, Eskelin P. APECED-causing mutations in AIRE reveal the functional domains of the protein. Hum Mutat. 2004;23(3):245–257. doi: 10.1002/humu.20003. [DOI] [PubMed] [Google Scholar]
- 61.Pitkänen J, Peterson P. Autoimmune regulator: from loss of function to autoimmunity. Genes Immun. 2003;4(1):12–21. doi: 10.1038/sj.gene.6363929. [DOI] [PubMed] [Google Scholar]
- 62.Rizzi M, Ferrera F, Filaci G, Indiveri F. Disruption of immunological tolerance: role of AIRE gene in autoimmunity. Autoimmun Rev. 2006;5(2):145–147. doi: 10.1016/j.autrev.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 63.Fierabracci A. Recent insights into the role and molecular mechanisms of the autoimmune regulator (AIRE) gene in autoimmunity. Autoimmun Rev. 2011;10(3):137–143. doi: 10.1016/j.autrev.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 64.Passos GA, Speck-Hernandez CA, Assis AF, Mendes-da-Cruz DA. Update on Aire and thymic negative selection. Immunology. 2018;153(1):10–20. doi: 10.1111/imm.12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wolff AS, Erichsen MM, Meager A, Magitta NF, Myhre AG, Bollerslev J, Fougner KJ, Lima K, Knappskog PM, Husebye ES. Autoimmune polyendocrine syndrome type 1 in Norway: phenotypic variation, autoantibodies, and novel mutations in the autoimmune regulator gene. J Clin Endocrinol Metab. 2007;92(2):595–603. doi: 10.1210/jc.2006-1873. [DOI] [PubMed] [Google Scholar]
- 66.Zlotogora J, Shapiro MS. Polyglandular autoimmune syndrome type I among Iranian Jews. J Med Genet. 1992;29(11):824–826. doi: 10.1136/jmg.29.11.824.PMID:1453436;PMCID:PMC1016181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ricotta EE, Ferré EMN, Schmitt MM, DiMaggio T, Lionakis MS. Prevalence of APECED-Like Clinical Disease in an Electronic Health Record Database, USA. J Clin Immunol. 2022;42(4):904–906. doi: 10.1007/s10875-022-01254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scott HS, Heino M, Peterson P, Mittaz L, Lalioti MD, Betterle C, Cohen A, Seri M, Lerone M, Romeo G, Collin P, Salo M, Metcalfe R, Weetman A, Papasavvas MP, Rossier C, Nagamine K, Kudoh J, Shimizu N, Krohn KJ, Antonarakis SE. Common mutations in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients of different origins. Mol Endocrinol. 1998;12(8):1112–1119. doi: 10.1210/mend.12.8.0143. [DOI] [PubMed] [Google Scholar]
- 69.Orlova EM, Sozaeva LS, Kareva MA, Oftedal BE, Wolff ASB, Breivik L, Zakharova EY, Ivanova ON, Kämpe O, Dedov II, Knappskog PM, Peterkova VA, Husebye ES. Expanding the Phenotypic and Genotypic Landscape of Autoimmune Polyendocrine Syndrome Type 1. J Clin Endocrinol Metab. 2017;102(9):3546–3556. doi: 10.1210/jc.2017-00139. [DOI] [PubMed] [Google Scholar]
- 70.Stolarski B, Pronicka E, Korniszewski L, Pollak A, Kostrzewa G, Rowińska E, Włodarski P, Skórka A, Gremida M, Krajewski P, Ploski R. Molecular background of polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome in a Polish population: novel AIRE mutations and an estimate of disease prevalence. Clin Genet. 2006;70(4):348–354. doi: 10.1111/j.1399-0004.2006.00690.x. [DOI] [PubMed] [Google Scholar]
- 71.Rosatelli MC, Meloni A, Meloni A, Devoto M, Cao A, Scott HS, Peterson P, Heino M, Krohn KJ, Nagamine K, Kudoh J, Shimizu N, Antonarakis SE. A common mutation in Sardinian autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. Hum Genet. 1998;103(4):428–434. doi: 10.1007/s004390050846. [DOI] [PubMed] [Google Scholar]
- 72.Heino M, Scott HS, Chen Q, Peterson P, Mäebpää U, Papasavvas MP, Mittaz L, Barras C, Rossier C, Chrousos GP, Stratakis CA, Nagamine K, Kudoh J, Shimizu N, Maclaren N, Antonarakis SE, Krohn K. Mutation analyses of North American APS-1 patients. Hum Mutat. 1999;13(1):69–74. doi: 10.1002/(SICI)1098-1004(1999)13:1<69::AID-HUMU8>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 73.Pearce SH, Cheetham T, Imrie H, Vaidya B, Barnes ND, Bilous RW, Carr D, Meeran K, Shaw NJ, Smith CS, Toft AD, Williams G, Kendall-Taylor P. A common and recurrent 13-bp deletion in the autoimmune regulator gene in British kindreds with autoimmune polyendocrinopathy type 1. Am J Hum Genet. 1998;63(6):1675–1684. doi: 10.1086/302145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bruserud Ø, Oftedal BE, Landegren N, Erichsen MM, Bratland E, Lima K, Jørgensen AP, Myhre AG, Svartberg J, Fougner KJ, Bakke Å, Nedrebø BG, Mella B, Breivik L, Viken MK, Knappskog PM, Marthinussen MC, Løvås K, Kämpe O, Wolff AB, Husebye ES. A Longitudinal Follow-up of Autoimmune Polyendocrine Syndrome Type 1. J Clin Endocrinol Metab. 2016;101(8):2975–2983. doi: 10.1210/jc.2016-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sharifinejad N, Zaki-Dizaji M, Tebyanian S, Zainaldain H, Jamee M, Rizvi FS, Hosseinzadeh S, Fayyaz F, Hamedifar H, Sabzevari A, Matloubi M, Heropolitańska-Pliszka E, Aghamahdi F, Abolhassani H, Azizi G. Clinical, immunological, and genetic features in 938 patients with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED): a systematic review. Expert Rev Clin Immunol. 2021;17(8):807–817. doi: 10.1080/1744666X.2021.1925543. [DOI] [PubMed] [Google Scholar]
- 76.Kahaly GJ, Frommer L. Autoimmune polyglandular diseases. Best Pract Res Clin Endocrinol Metab. 2019;33(6):101344. doi: 10.1016/j.beem.2019.101344. [DOI] [PubMed] [Google Scholar]
- 77.Frommer L, Kahaly GJ. Autoimmune Polyendocrinopathy. J Clin Endocrinol Metab. 2019;104(10):4769–4782. doi: 10.1210/jc.2019-00602. [DOI] [PubMed] [Google Scholar]
- 78.Zhao Z, Wang X, Bao XQ, Ning J, Shang M, Zhang D. Autoimmune polyendocrine syndrome induced by immune checkpoint inhibitors: a systematic review. Cancer Immunol Immunother. 2021;70(6):1527–1540. doi: 10.1007/s00262-020-02699-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park JH, Lee KH, Jeon B, Ochs HD, Lee JS, Gee HY, Seo S, Geum D, Piccirillo CA, Eisenhut M, van der Vliet HJ, Lee JM, Kronbichler A, Ko Y, Shin JI. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: A systematic review. Autoimmun Rev. 2020;19(6):102526. doi: 10.1016/j.autrev.2020.102526. [DOI] [PubMed] [Google Scholar]
- 80.Nirula A, Glaser SM, Kalled SL, Taylor FR. What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol. 2011;23(1):119–124. doi: 10.1097/BOR.0b013e3283412fd4. [DOI] [PubMed] [Google Scholar]
- 81.Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy. 2009;39(4):469–477. doi: 10.1111/j.1365-2222.2009.03207.x. [DOI] [PubMed] [Google Scholar]
- 82.Collins AM, Jackson KJ. A Temporal Model of Human IgE and IgG Antibody Function. Front Immunol. 2013;9(4):235. doi: 10.3389/fimmu.2013.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aucouturier P, Danon F, Daveau M, Guillou B, Sabbah A, Besson J, Preud’homme JL. Measurement of serum IgG4 levels by a competitive immunoenzymatic assay with monoclonal antibodies. J Immunol Methods. 1984 Nov 16;74(1):151–62. [DOI] [PubMed]
- 84.Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N, Kiyosawa K. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344(10):732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 85.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–551. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 86.Perugino CA, Stone JH. IgG4-related disease: an update on pathophysiology and implications for clinical care. Nat Rev Rheumatol. 2020;16(12):702–714. doi: 10.1038/s41584-020-0500-7. [DOI] [PubMed] [Google Scholar]
- 87.Kamisawa T, Okazaki K. Diagnosis and Treatment of IgG4-Related Disease. Curr Top Microbiol Immunol. 2017;401:19–33. doi: 10.1007/82_2016_36. [DOI] [PubMed] [Google Scholar]
- 88.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pardoll D. Releasing the brakes on antitumor immune response. Science. 1996;271(5256):1691. doi: 10.1126/science.271.5256.1691. [DOI] [PubMed] [Google Scholar]
- 90.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discov. 2022;21(7):509–528. doi: 10.1038/s41573-021-00345-8. [DOI] [PubMed] [Google Scholar]
- 92.Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, Shabafrouz K, Ribi C, Cairoli A, Guex-Crosier Y, Kuntzer T, Michielin O, Peters S, Coukos G, Spertini F, Thompson JA, Obeid M. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16(9):563–580. doi: 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
- 93.de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A Systematic Review and Meta-Analysis of Endocrine-Related Adverse Events Associated with Immune Checkpoint Inhibitors. Horm Metab Res. 2019;51(3):145–156. doi: 10.1055/a-0843-3366. [DOI] [PubMed] [Google Scholar]
- 94.Wright JJ, Johnson DB. Approach to the Patient wiht Immune Checkpoint Inhibitor-Associated Endocrine Dysfunction. J Clin Endocrinol Metab. 2022 Dec 9:dgac689. 10.1210/clinem/dgac689. Epub ahead of print. PMID: 36481794. [DOI] [PMC free article] [PubMed]
- 95.Neppl C, Kaderli RM, Trepp R, Schmitt AM, Berger MD, Wehrli M, Seiler CA, Langer R. Histology of Nivolumab-Induced Thyroiditis. Thyroid. 2018;28(12):1727–1728. doi: 10.1089/thy.2018.0418. [DOI] [PubMed] [Google Scholar]
- 96.Imblum BA, Baloch ZW, Fraker D, LiVolsi VA. Pembrolizumab-Induced Thyroiditis. Endocr Pathol. 2019;30(2):163–167. doi: 10.1007/s12022-019-9579-2. [DOI] [PubMed] [Google Scholar]
- 97.Jabkowski J, Loidl A, Auinger B, Kehrer H, Sepp N, Pichler R. Pembrolizumab-Induced Thyroiditis Shows PD-L1Expressing Histiocytes and Infiltrating T Cells in Thyroid Tissue - A Case Report. Front Immunol. 2021;18(12):606056. doi: 10.3389/fimmu.2021.606056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Angell TE, Min L, Wieczorek TJ, Hodi FS. Unique Cytologic Features of Thyroiditis Caused by Immune Checkpoint Inhibitor Therapy for Malignant Melanoma. Genes Dis. 2018 Mar;5(1):46–48. 10.1016/j.gendis.2017.11.002. Epub 2017 Nov 21. PMID: 29619406; PMCID: PMC5879785. [DOI] [PMC free article] [PubMed]
- 99.Di Dalmazi G, Ippolito S, Lupi I, Caturegli P. Hypophysitis induced by immune checkpoint inhibitors: A 10-year assessment. Expert Review of Endocrinology and Metabolism. 2019;14:381–398. doi: 10.1080/17446651.2019.1701434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Caturegli P, Di Dalmazi G, Lombardi M, Grosso F, Larman HB, Larman T, Taverna G, Cosottini M, Lupi I. Hypophysitis Secondary to Cytotoxic T-Lymphocyte–Associated Protein 4 Blockade: Insights into Pathogenesis from an Autopsy Series. Am J Pathol. 2016;186:3225. doi: 10.1016/J.AJPATH.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Okabe N, Kobayashi T, Furuse J, Fujiwara M, Kamma H. An autopsy case study of lymphocytic hypophysitis induced by nivolumab treatment for esophageal malignant melanoma. Pathol Int. 2021;71(12):831–836. doi: 10.1111/pin.13161. [DOI] [PubMed] [Google Scholar]
- 102.Cui K, Wang Z, Zhang Q, Zhang X. Immune checkpoint inhibitors and adrenal insufficiency: a large-sample case series study. Ann Transl Med. 2022;10(5):251. doi: 10.21037/atm-21-7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grouthier V, Lebrun-Vignes B, Moey M, Johnson DB, Moslehi JJ, Salem JE, Bachelot A. Immune Checkpoint Inhibitor-Associated Primary Adrenal Insufficiency: WHO VigiBase Report Analysis. Oncologist. 2020;25(8):696–701. doi: 10.1634/theoncologist.2019-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Trinh B, Sanchez GO, Herzig P, et al. Inflammation-induced hypoparathyroidism triggered by combination immune checkpoint blockade for melanoma. J Immunother Cancer. 2019;7:52. doi: 10.1186/s40425-019-0528-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chang LS, Barroso-Sousa R, Tolaney SM, Hodi FS, Kaiser UB, Min L. Endocrine Toxicity of Cancer Immunotherapy Targeting Immune Checkpoints. Endocr Rev. 2019;40(1):17–65. doi: 10.1210/er.2018-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Warmbier J, Lüdecke DK, Flitsch J, Buchfelder M, Fahlbusch R, Knappe UJ, Kreutzer J, Buslei R, Bergmann M, Heppner F, Glatzel M, Saeger W. Typing of inflammatory lesions of the pituitary. Pituitary. 2022;25:131. doi: 10.1007/S11102-021-01180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yuen KCJ, Popovic V, Trainer PJ (2019) New causes of hypophysitis. Best Pract Res Clin Endocrinol Metab 33:101276. 10.1016/J.BEEM.2019.04.010 [DOI] [PubMed]
- 108.Sugihara H. Review on Recent Topics in Hypophysitis. J Nippon Med Sch. 2017;84:201–208. doi: 10.1272/JNMS.84.201. [DOI] [PubMed] [Google Scholar]
- 109.Joshi MN, Whitelaw BC, Carroll PV. MECHANISMS IN ENDOCRINOLOGY: Hypophysitis: diagnosis and treatment. Eur J Endocrinol. 2018;179:R151–R163. doi: 10.1530/EJE-17-0009. [DOI] [PubMed] [Google Scholar]
- 110.Prete A, Salvatori R. Hypophysitis. 2021 Oct 15. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Hofland J, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000–. PMID: 30160871.
- 111.Gubbi S, Hannah-Shmouni F, Verbalis JG, Koch CA (2019) Hypophysitis: An update on the novel forms, diagnosis and management of disorders of pituitary inflammation. Best Pract Res Clin Endocrinol Metab 33:. 10.1016/J.BEEM.2019.101371 [DOI] [PMC free article] [PubMed]
- 112.Takao T, Nanamiya W, Matsumoto R, Asaba K, Okabayashi T, Hashimoto K. Antipituitary Antibodies in Patients with Lymphocytic Hypophysitis. Horm Res Paediatr. 2001;55:288–292. doi: 10.1159/000050015. [DOI] [PubMed] [Google Scholar]
- 113.De Bellis A, Pane E, Bellastella G, Sinisi AA, Colella C, Giordano R, Giavoli C, Lania A, Ambrosio MR, Di Somma C, et al. Detection of antipituitary and antihypothalamus antibodies to investigate the role of pituitary or hypothalamic autoimmunity in patients with selective idiopathic hypopituitarism. Clin Endocrinol (Oxf) 2011;75:361–366. doi: 10.1111/J.1365-2265.2011.04056.X. [DOI] [PubMed] [Google Scholar]
- 114.Yamamoto M, Iguchi G, Bando H, Kanie K, Hidaka-Takeno R, Fukuoka H, Takahashi Y. Autoimmune Pituitary Disease: New Concepts With Clinical Implications. Endocr Rev. 2020;41:261–272. doi: 10.1210/ENDREV/BNZ003. [DOI] [PubMed] [Google Scholar]
- 115.Cocco C, Meloni A, Mariotti S, Cossu E, D’amato F, Zulian S, Tongiorgi E, Ferri GL (2012) Novel neuronal and endocrine autoantibody targets in autoimmune polyendocrine syndrome type 1. 45:485–494. 10.3109/08916934.2012.680632 [DOI] [PubMed]
- 116.Cocco C, Brancia C, D’Amato F, Noli B. Pituitary gonadotropins and autoimmunity. Mol Cell Endocrinol. 2014;385:97–104. doi: 10.1016/J.MCE.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 117.Cocco C, Brancia C, Corda G, Ferri GL (2017) The Hypothalamic–Pituitary Axis and Autoantibody Related Disorders. Int J Mol Sci 18. 10.3390/IJMS18112322 [DOI] [PMC free article] [PubMed]
- 118.Ricciuti A, De Remigis A, Landek-Salgado MA, De Vincentiis L, Guaraldi F, Lupi I, Iwama S, Wand GS, Salvatori R, Caturegli P. Detection of Pituitary Antibodies by Immunofluorescence: Approach and Results in Patients With Pituitary Diseases. J Clin Endocrinol Metab. 2014;99:1758–1766. doi: 10.1210/JC.2014-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Iwama S, Arima H. Anti-pituitary antibodies as a marker of autoimmunity in pituitary glands. Endocr J. 2020;67:1077–1083. doi: 10.1507/ENDOCRJ.EJ20-0436. [DOI] [PubMed] [Google Scholar]
- 120.Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune Hypophysitis. Endocr Rev. 2005;26:599–614. doi: 10.1210/ER.2004-0011. [DOI] [PubMed] [Google Scholar]
- 121.Hypophysitis Faje A. Evaluation and Management. Clin Diabetes Endocrinol. 2016;6(2):15. doi: 10.1186/s40842-016-0034-8.PMID:28702249;PMCID:PMC5471685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Langlois F, Varlamov EV, Fleseriu M. Hypophysitis, the Growing Spectrum of a Rare Pituitary Disease. J Clin Endocrinol Metab. 2022;107:10. doi: 10.1210/CLINEM/DGAB672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bellastella G, Maiorino MI, Bizzarro A, Giugliano D, Esposito K, Bellastella A, De Bellis A. Revisitation of autoimmune hypophysitis: knowledge and uncertainties on pathophysiological and clinical aspects. Pituitary. 2016;19:625. doi: 10.1007/S11102-016-0736-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bhargava R, Hussein Z, Dorward NL, Grieve JP, Jaunmuktane Z, Marcus HJ, Proctor I, Baldeweg SE. IgG4-related hypophysitis: a retrospective cohort study. Acta Neurochir (Wien) 2022;164:2095–2103. doi: 10.1007/S00701-022-05231-9/FIGURES/2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Buxton N, Robertson I, Powell M, Chatterjee K (2009) Lymphocytic and granulocytic hypophysitis: a single centre experience. 15:242–246. 10.1080/02688690120057664 [DOI] [PubMed]
- 126.Kyriacou A, Gnanalingham K, Kearney T. Lymphocytic hypophysitis: modern day management with limited role for surgery. Pituitary. 2017;20:241–250. doi: 10.1007/S11102-016-0769-3/FIGURES/3. [DOI] [PubMed] [Google Scholar]
- 127.Bernreuther C, Illies C, Flitsch J, Buchfelder M, Buslei R, Glatzel M, Saeger W. IgG4-related hypophysitis is highly prevalent among cases of histologically confirmed hypophysitis. Brain Pathol. 2017;27:839–845. doi: 10.1111/BPA.12459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Angelousi A, Alexandraki K, Tsoli M, Kaltsas G, Kassi E. Hypophysitis (Including IgG4 and Immunotherapy) Neuroendocrinology. 2020;110:822–835. doi: 10.1159/000506903. [DOI] [PubMed] [Google Scholar]
- 129.Jessel S, Weiss SA, Austin M, Mahajan A, Etts K, Zhang L, Aizenbud L, Perdigoto AL, Hurwitz M, Sznol M, Herold KC, Kluger HM. Immune Checkpoint Inhibitor-Induced Hypophysitis and Patterns of Loss of Pituitary Function. Front Oncol. 2022;12:784. doi: 10.3389/FONC.2022.836859/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Honegger J, Buchfelder M, Schlaffer S, Droste M, Werner S, Strasburger C, Störmann S, Schopohl J, Kacheva S, Deutschbein T, Elbelt U (2015) Treatment of Primary Hypophysitis in Germany. J Clin Endocrinol Metab 100:3460–3469. 10.1210/JC.2015-2146 [DOI] [PubMed]
- 131.Naran J, Can AS. Lymphocytic Hypophysitis. 2022 Aug 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan–. PMID: 32965926.
- 132.Fehn M, Sommer C, Lüdecke DK, Plöckinger U, Saeger W. Lymphocytic Hypophysitis: Light and Electron Microscopic Findings and Correlation to Clinical Appearance. Endocr Pathol. 1998;9:71–78. doi: 10.1007/BF02739954. [DOI] [PubMed] [Google Scholar]
- 133.Duan L, Liu J, Zhang Y, Cui L, Zhai X, Pan B, Lu L, Pan H, Yao Y, Zhu H. Primary Pituitary Lymphoma in Immunocompetent Patients: A Report on Two Case Studies and the Review of Literature. Front Endocrinol (Lausanne) 2021;11:1143. doi: 10.3389/FENDO.2020.562850/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Huang YY, Lin SF, Dunn P, Wai YY, Hsueh C, Tsai JS. Primary pituitary lymphoma presenting as hypophysitis. Endocr J. 2005;52:543–549. doi: 10.1507/ENDOCRJ.52.543. [DOI] [PubMed] [Google Scholar]
- 135.Giustina A, Gola M, Doga M, Rosei EA. Primary Lymphoma of the Pituitary: An Emerging Clinical Entity. J Clin Endocrinol Metab. 2001;86:4567–4575. doi: 10.1210/JCEM.86.10.7909. [DOI] [PubMed] [Google Scholar]
- 136.Uccella S, Amaglio C, Brouland JP, Bianconi E, Ippolito S, Messerer M, Rouiller N, Tanda ML, Sessa F, La Rosa S. Disease heterogeneity in IgG4-related hypophysitis: report of two histopathologically proven cases and review of the literature. Virchows Arch. 2019;475(3):373–381. doi: 10.1007/s00428-019-02564-2. [DOI] [PubMed] [Google Scholar]
- 137.Park KH, Gwak H-S, Hong EK, Lee SH (2013) Inflamed Symptomatic Sellar Arachnoid Cyst: Case Report. Brain Tumor Res Treat 1:28. 10.14791/BTRT.2013.1.1.28 [DOI] [PMC free article] [PubMed]
- 138.Yang C, Wu H, Bao X, Wang R. Lymphocytic Hypophysitis Secondary to Ruptured Rathke Cleft Cyst: Case Report and Literature Review. World Neurosurg. 2018;114:172–177. doi: 10.1016/J.WNEU.2018.03.086. [DOI] [PubMed] [Google Scholar]
- 139.Fehn M, Bettendorf M, Lüdecke DK, Sommer C, Saeger W. Lymphocytic Hypophysitis Masking a Suprasellar Germinoma in a 12-year-old Girl - A Case Report. Pituitary. 1999;1:303–307. doi: 10.1023/a:1009923029942. [DOI] [PubMed] [Google Scholar]
- 140.Houdouin L, Polivka M, Henegar C, Blanquet A, Delalande O, Mikol J. Pituitary germinoma and lymphocytic hypophysitis: a pitfall. Report of two cases. Ann Pathol. 2003;23:349–354. [PubMed] [Google Scholar]
- 141.Heshmati HM, Kujas M, Casanova S, Wollan PC, Racadot J, Van Effenterre R, Derome PJ, Turpin G. Prevalence of lymphocytic infiltrate in 1400 pituitary adenomas. Endocr J. 1998;45:357–361. doi: 10.1507/ENDOCRJ.45.357. [DOI] [PubMed] [Google Scholar]
- 142.Rivera J-A. (2006) Lymphocytic hypophysitis: Disease spectrum and approach to diagnosis and therapy. Pituit. 2006;91(9):35–45. doi: 10.1007/S11102-006-6598-Z. [DOI] [PubMed] [Google Scholar]
- 143.Hashimoto K, Takao T, Makino S. Lymphocytic adenohypophysitis and lymphocytic infundibuloneurohypophysitis. Endocr J. 1997;44:1–10. doi: 10.1507/ENDOCRJ.44.1. [DOI] [PubMed] [Google Scholar]
- 144.Beressi N, Beressi JP, Cohen R, Modigliani E. Lymphocytic hypophysitis. A review of 145 cases. Ann Med Interne (Paris) 1999;150:327–341. [PubMed] [Google Scholar]
- 145.Honegger J, Schlaffer S, Menzel C, Droste M, Werner S, Elbelt U, Strasburger C, Störmann S, Küppers A, Streetz-Van Der Werf C, Petersenn S (2015) Diagnosis of Primary Hypophysitis in Germany. J Clin Endocrinol Metab 100:3841–3849. 10.1210/JC.2015-2152 [DOI] [PubMed]
- 146.Rao S, Mahadevan A, Maiti T, Ranjan M, Shwetha SD, Arivazhagan A, Saini J. Granulomatous and lymphocytic hypophysitis - are they immunologically distinct? APMIS. 2016;124:1072–1077. doi: 10.1111/APM.12603. [DOI] [PubMed] [Google Scholar]
- 147.Deguchi-Horiuchi H, Koide H, Sakuma I, Gao Y, Higuchi S, Nagano H, Hashimoto N, Horiguchi K, Iwadate Y, Inoshita N, Yokote K, Tanaka T. Two cases of symptomatic secondary hypophysitis due to Rathke’s cleft cysts treated with glucocorticoids: long-term follow-up. Endocr J. 2021;68:269–279. doi: 10.1507/ENDOCRJ.EJ20-0361. [DOI] [PubMed] [Google Scholar]
- 148.Hunn BHM, Martin WG, Simpson S, Mclean CA. Idiopathic granulomatous hypophysitis: a systematic review of 82 cases in the literature. Pituitary. 2014;17:357–365. doi: 10.1007/S11102-013-0510-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gutenberg A, Hans V, Puchner MJA, Kreutzer J, Brück W, Caturegli P, Buchfelder M. Primary hypophysitis: clinical-pathological correlations. Eur J Endocrinol. 2006;155:101–107. doi: 10.1530/EJE.1.02183. [DOI] [PubMed] [Google Scholar]
- 150.Ludmerer K, Kissane J (1984) Primary hypothyroidism and hypopituitarism in a young woman. In: The American journal of medicine. pp 319–30 [DOI] [PubMed]
- 151.Tzou SC, Lupi I, Landek M, Gutenberg A, Tzou YM, Kimura H, Pinna G, Rose NR, Caturegli P. Autoimmune hypophysitis of SJL mice: clinical insights from a new animal model. Endocrinology. 2008;149:3461–3469. doi: 10.1210/EN.2007-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Force BK, Vogel TP, Nguyen DM, Heck KA, Sebastian S, Takashima M, Yoshor D, Samson SL. A Remarkable Response of Granulomatous Hypophysitis to Infliximab in a Patient With a Background of Crohn’s Disease—A Case Report. Front Endocrinol (Lausanne) 2020;11:350. doi: 10.3389/FENDO.2020.00350/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Goyal M, Kucharczyk W, Keystone E. Granulomatous Hypophysitis due to Wegener’s Granulomatosis. AJNR Am J Neuroradiol. 2000;21:1466. [PMC free article] [PubMed] [Google Scholar]
- 154.Tóth M, Szabó P, Rácz K, Szende B, Balogh I, Czirják S, Slowik F, Gláz E. Granulomatous hypophysitis associated with Takayasu’s disease. Clin Endocrinol (Oxf) 1996;45:499–503. doi: 10.1046/J.1365-2265.1996.8110821.X. [DOI] [PubMed] [Google Scholar]
- 155.Langrand C, Bihan H, Raverot G, Varron L, Androdias G, Borson-Chazot F, Brue T, Cathebras P, Pinede L, Muller G, Broussolle C, Cotton F, Valeyre D, Seve P. Hypothalamo-pituitary sarcoidosis: a multicenter study of 24 patients. QJM An Int J Med. 2012;105:981–995. doi: 10.1093/QJMED/HCS121. [DOI] [PubMed] [Google Scholar]
- 156.Vega-Beyhart A, Medina-Rangel IR, Hinojosa-Azaola A, Fernández-Barrio M, Vargas-Castro AS, García-Inciarte L, Guzmán-Pérez A, Torres-Victoria TR, Martínez-Sánchez FD, Pérez-Guzmán MC, Cuevas-Ramos D (2020) Pituitary dysfunction in granulomatosis with polyangiitis. Clin Rheumatol 39:595–606. 10.1007/S10067-019-04735-7 [DOI] [PubMed]
- 157.Sharifi G, Mohajeri-Tehrani MR, Navabakhsh B, Larijani B, Valeh T. Idiopathic granulomatous hypophysitis presenting with galactorrhea, headache, and nausea in a woman: A case report and review of the literature. J Med Case Rep. 2019;13:1–4. doi: 10.1186/S13256-019-2276-4/FIGURES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Agale S, Binayke R, Kumari G, D’Costa G. Idiopathic granulomatous hypophysitis: A report of an uncommon disorder. Indian J Pathol Microbiol. 2018;61:389. doi: 10.4103/IJPM.IJPM_77_17. [DOI] [PubMed] [Google Scholar]
- 159.Xu Y, Lou L, Wang TH, Zhao YY, Cai XX, Ma J, Lu G. Granulomatous hypophysitis: experience with eight surgical cases of a single center. Chinese Neurosurg J. 2016;2:1–10. doi: 10.1186/S41016-015-0019-Y/FIGURES/10. [DOI] [Google Scholar]
- 160.Zhu J, Wang Z, Wang W, Fan J, Zhang Y, Li X, Liu J, Jiang S, Deng K, Duan L, Yao Y, Zhu H. Xanthomatous Hypophysitis: A Case Report and Comprehensive Literature Review. Front Endocrinol (Lausanne) 2021;12:1258. doi: 10.3389/FENDO.2021.735655/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cheung CC, Ezzat S, Smyth HS, Asa SL. The spectrum and significance of primary hypophysitis. J Clin Endocrinol Metab. 2001;86:1048–1053. doi: 10.1210/JCEM.86.3.7265. [DOI] [PubMed] [Google Scholar]
- 162.Kini H, Rao R, Pai M. Xanthomatous hypophysitis: A rare case report with review of literature. Indian J Pathol Microbiol. 2019;62:448. doi: 10.4103/IJPM.IJPM_319_18. [DOI] [PubMed] [Google Scholar]
- 163.Hanna B, Li YM, Beutler T, Goyal P, Hall WA. Xanthomatous hypophysitis. J Clin Neurosci. 2015;22:1091–1097. doi: 10.1016/J.JOCN.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 164.Yaghoubi MA, Zabihyan S, Saeidinia A, Gharib M, Moghaddam RG (2022) Xanthogranulomatous hypophysitis: A rare presentation in a young female patient. Clin Case Reports 10:e6337. 10.1002/CCR3.6337 [DOI] [PMC free article] [PubMed]
- 165.Gezer E, Çabuk B, Bayrak BY, Cantürk Z, Çetinarslan B, Selek A, Sözen M, Köksalan D, Ceylan S (2022) Xanthomatous Hypophysitis Secondary to a Ruptured Rathke’s Cleft Cyst: A Case Report. Brain Tumor Res Treat 10:48. 10.14791/BTRT.2022.10.E24 [DOI] [PMC free article] [PubMed]
- 166.Kleinschmidt-DeMasters BK, Lillehei KO, Hankinson TC. Review of xanthomatous lesions of the sella. Brain Pathol. 2017;27:377–395. doi: 10.1111/BPA.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Duan K, Asa SL, Winer D, Gelareh Z, Gentili F, Mete O. Xanthomatous Hypophysitis Is Associated with Ruptured Rathke’s Cleft Cyst. Endocr Pathol. 2017;28:83–90. doi: 10.1007/S12022-017-9471-X/FIGURES/3. [DOI] [PubMed] [Google Scholar]
- 168.Ved R, Logier N, Leach P, Davies JS, Hayhurst C. Pituitary xanthogranulomas: clinical features, radiological appearances and post-operative outcomes. Pituitary. 2018;21:256. doi: 10.1007/S11102-017-0859-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Alugolu R, Chandrasekhar YBVK, Shukla D, Sahu BP, Srinivas BH. Xanthogranulomatous colloid cyst of the third ventricle. J Neurosci Rural Pract. 2013;4:183. doi: 10.4103/0976-3147.112761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nishiuchi T, Murao K, Imachi H, Kushida Y, Haba R, Kawai N, Tamiya T, Ishida T. Xanthogranuloma of the intrasellar region presenting in pituitary dysfunction: a case report. J Med Case Rep. 2012;6:119. doi: 10.1186/1752-1947-6-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gopal-Kothandapani JS, Bagga V, Wharton SB, Connolly DJ, Sinha S, Dimitri PJ (2015) Xanthogranulomatous hypophysitis: a rare and often mistaken pituitary lesion. Endocrinol diabetes Metab case reports 2015:140089. 10.1530/EDM-14-0089 [DOI] [PMC free article] [PubMed]
- 172.Gutenberg A, Caturegli P, Metz I, Martinez R, Mohr A, Brück W, Rohde V. Necrotizing infundibulo-hypophysitis: an entity too rare to be true? Pituitary. 2012;15:202. doi: 10.1007/S11102-011-0307-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ahmed SR, Aiello DP, Page R, Hopper K, Towfighi J, Santen RJ. Necrotizing infundibulo-hypophysitis: a unique syndrome of diabetes insipidus and hypopituitarism. J Clin Endocrinol Metab. 1993;76:1499–1504. doi: 10.1210/jcem.76.6.8501157. [DOI] [PubMed] [Google Scholar]
- 174.Magalhães-Ribeiro C, Furtado A, Baggen Santos R, Mascarenhas L, Costa Correia S, Rocha G, Resende M (2021) Necrotizing infundibulo-hypophysitis: case-report and literature review. Br J Neurosurg 1–4. 10.1080/02688697.2021.1940857 [DOI] [PubMed]
- 175.Ćaćić M, Marinković J, Kruljac I, Perić B, Čerina V, Stipić D, Pažanin L, Pećina HI, Vrkljan M (2018) Ischemic Pituitary Apoplexy, Hypopituitarism and Diabetes Insipidus: A Triad Unique to Necrotizing Hypophysitis. Acta Clin Croat 57:768. 10.20471/ACC.2018.57.04.20 [DOI] [PMC free article] [PubMed]
- 176.Dan NG, Feiner RID, Houang MTW, Turner JJ. Pituitary apoplexy in association with lymphocytic hypophysitis. J Clin Neurosci. 2002;9:577–580. doi: 10.1054/JOCN.2001.0975. [DOI] [PubMed] [Google Scholar]
- 177.Nishioka H, Haraoka JO, Miki T. Spontaneous Remission of Functioning Pituitary Adenomas without Hypopituitarism following Infarctive Apoplexy: Two Case Reports. Endocr J. 2005;52:117–123. doi: 10.1507/endocrj.52.117. [DOI] [PubMed] [Google Scholar]
- 178.Schury MP, Adigun R. Sheehan Syndrome. 2022 Sep 5. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan–. PMID: 29083621. [PubMed]
- 179.Goswami R, Kochupillai N, Crock PA, Jaleel A, Gupta N. Pituitary autoimmunity in patients with Sheehan’s syndrome. J Clin Endocrinol Metab. 2002;87:4137–4141. doi: 10.1210/JC.2001-020242. [DOI] [PubMed] [Google Scholar]
- 180.Bando H, Iguchi G, Fukuoka H, Taniguchi M, Yamamoto M, Matsumoto R, Suda K, Nishizawa H, Takahashi M, Kohmura E, Takahashi Y. The prevalence of IgG4-related hypophysitis in 170 consecutive patients with hypopituitarism and/or central diabetes insipidus and review of the literature. Eur J Endocrinol. 2013;170:161–172. doi: 10.1530/EJE-13-0642. [DOI] [PubMed] [Google Scholar]
- 181.Amirbaigloo A, Esfahanian F, Mouodi M, Rakhshani N, Zeinalizadeh M. IgG4-related hypophysitis. Endocrine. 2021;73:270–291. doi: 10.1007/S12020-021-02714-0. [DOI] [PubMed] [Google Scholar]
- 182.Ebbo M, Daniel L, Pavic M, Sève P, Hamidou M, Andres E, Burtey S, Chiche L, Serratrice J, Longy-Boursier M, Ruivard M, Haroche J, Godeau B, Beucher AB, Berthelot JM, Papo T, Pennaforte JL, Benyamine A, Jourde N, Landron C, Roblot P, Moranne O, Silvain C, Granel B, Bernard F, Veit V, Mazodier K, Bernit E, Rousset H, Boucraut J, Boffa JJ, Weiller PJ, Kaplanski G, Aucouturier P, Harlé JR, Schleinitz N. IgG4-related systemic disease: features and treatment response in a French cohort: results of a multicenter registry. Medicine (Baltimore). 2012;91(1):49–56. doi: 10.1097/MD.0b013e3182433d77. [DOI] [PubMed] [Google Scholar]
- 183.Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S, Stone JH. IgG4-Related Disease: Baseline clinical and laboratory features in 125 patients with biopsy-proven disease. Arthritis Rheumatol (Hoboken, NJ) 2015;67:2466. doi: 10.1002/ART.39205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lin W, Lu S, Chen H, Wu Q, Fei Y, Li M, Zhang X, Tian X, Zheng W, Leng X, … Lipsky PE (2015) Clinical characteristics of immunoglobulin G4-related disease: a prospective study of 118 Chinese patients. Rheumatology (Oxford) 54:1982–1990. 10.1093/RHEUMATOLOGY/KEV203 [DOI] [PubMed]
- 185.Liu Y, Wang L, Zhang W, Pan H, Yang H, Deng K, Lu L, Yao Y, Chen S, Chai X, Feng F, You H, Jin Z, Zhu H (2018) Hypophyseal Involvement in Immunoglobulin G4-Related Disease: A Retrospective Study from a Single Tertiary Center. Int J Endocrinol 2018. 10.1155/2018/7637435 [DOI] [PMC free article] [PubMed]
- 186.Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab. 2011;96:1971–1980. doi: 10.1210/JC.2010-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sosa GA, Bell S, Christiansen SB, Pietrani M, Glerean M, Loto M, Lovazzano S, Carrizo A, Ajler P, Day PF (2014) Histologically confirmed isolated IgG4-related hypophysitis: two case reports in young women. Endocrinol Diabetes Metab Case Reports 2014. 10.1530/EDM-14-0062 [DOI] [PMC free article] [PubMed]
- 188.Vasaitis L, Wikström J, Ahlström S, Gudjonsson O, Kumlien E, Edén Engström B, Casar-Borota O (2021) Histopathological findings in the landscape of IgG4-related pathology in patients with pituitary dysfunction: Review of six cases. J Neuroendocrinol 33:e12942. 10.1111/JNE.12942 [DOI] [PubMed]
- 189.Pal R, Chatterjee D, Singla R, Jain N, Bhansali A, Dutta P. Co-Occurrence of Craniopharyngioma and IgG4-Related Hypophysitis: An Epiphenomenon or a Mere Coincidence? World Neurosurg. 2020;136:193–197. doi: 10.1016/J.WNEU.2019.12.181. [DOI] [PubMed] [Google Scholar]
- 190.Liu L, Perry AM, Cao W, Smith LM, Hsi ED, Liu X, Mo JQ, Dotlic S, Mosunjac M, Talmon G, Weisenburger DD, Fu K. Relationship between Rosai-Dorfman disease and IgG4-related disease: study of 32 cases. Am J Clin Pathol. 2013;140:395–402. doi: 10.1309/AJCPFH0SJ6YILXJU. [DOI] [PubMed] [Google Scholar]
- 191.Deshpande V. The pathology of IgG4-related disease: critical issues and challenges. Semin Diagn Pathol. 2012;29:191–196. doi: 10.1053/J.SEMDP.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 192.Cree IA, Tan PH, Travis WD, Wesseling P, Yagi Y, White VA, Lokuhetty D, Scolyer RA. Counting mitoses: SI(ze) matters! Mod Pathol. 2021;34:1651–1657. doi: 10.1038/S41379-021-00825-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ngaosuwan K, Trongwongsa T, Shuangshoti S. Clinical course of IgG4-related hypophysitis presenting with focal seizure and relapsing lymphocytic hypophysitis. BMC Endocr Disord. 2015;15:1–8. doi: 10.1186/S12902-015-0062-X/TABLES/2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nishihara E, Ohye H, Amino N, Takata K, Arishima T, Kudo T, Ito M, Kubota S, Fukata S, Miyauchi A. Clinical characteristics of 852 patients with subacute thyroiditis before treatment. Intern Med. 2008;47(8):725–729. doi: 10.2169/internalmedicine.47.0740. [DOI] [PubMed] [Google Scholar]
- 195.Martino E, Buratti L, Bartalena L, Mariotti S, Cupini C, Aghini-Lombardi F, Pinchera A. High prevalence of subacute thyroiditis during summer season in Italy. J Endocrinol Invest. 1987;10(3):321–323. doi: 10.1007/BF03348138. [DOI] [PubMed] [Google Scholar]
- 196.Caron P. Thyroiditis and SARS-CoV-2 pandemic: a review. Endocrine. 2021;72(2):326–331. doi: 10.1007/s12020-021-02689-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Falaschi P, Martocchia A, D’Urso R, Proietti A. Subacute thyroiditis during interferon-alpha therapy for chronic hepatitis C. J Endocrinol Invest. 1997;20(1):24–28. doi: 10.1007/BF03347968. [DOI] [PubMed] [Google Scholar]
- 198.Yasuji I. Subacute thyroiditis in a patient with juvenile idiopathic arthritis undergoing etanercept treatment: a case report and review of the literature. Mod Rheumatol. 2013;23(2):397–400. doi: 10.3109/s10165-012-0670-5. [DOI] [PubMed] [Google Scholar]
- 199.MEACHIM G, YOUNG MH. De Quervain’s subacute granulomatous thyroiditis: histological identification and incidence. J Clin Pathol. 1963 May;16(3):189–99. [DOI] [PMC free article] [PubMed]
- 200.Ranganath R, Shaha MA, Xu B, Migliacci J, Ghossein R, Shaha AR. de Quervain’s thyroiditis: A review of experience with surgery. Am J Otolaryngol. 2016 Nov-Dec;37(6):534–537. 10.1016/j.amjoto.2016.08.006. Epub 2016 Aug 18. PMID: 27686396; PMCID: PMC5574176199. [DOI] [PMC free article] [PubMed]
- 201.Yu R, Yang SE, Rao J. Atypical de Quervain’s thyroiditis diagnosed as atypia of undetermined significance by cytology and suspicious for cancer by Afirma Genomic Sequencing Classifier. Diagn Cytopathol. 2021;49(8):E312–E315. doi: 10.1002/dc.24733. [DOI] [PubMed] [Google Scholar]
- 202.Slatosky J, Shipton B, Wahba H. Thyroiditis: differential diagnosis and management. Am Fam Physician. 2000 Feb 15;61(4):1047–52, 1054. [PubMed]
- 203.Carney JA, Moore SB, Northcutt RC, Woolner LB, Stillwell GK. Palpation thyroiditis (multifocal granulomatour folliculitis) Am J Clin Pathol. 1975;64(5):639–647. doi: 10.1093/ajcp/64.5.639. [DOI] [PubMed] [Google Scholar]
- 204.Papi G, Briganti F, Artioli F, Cavazza A, Carapezzi C, Roggeri A, Baldoni C, Carani C, Chiarini V, Roti E. Sarcoidosis of the thyroid gland associated with hyperthyroidism: review of the literature and report of two peculiar cases. J Endocrinol Invest. 2006;29(9):834–839. doi: 10.1007/BF03347380. [DOI] [PubMed] [Google Scholar]
- 205.Katsamakas M, Tzitzili E, Boudina M, Kiziridou A, Valeri R, Zafeiriou G, Chrisoulidou A. Thyroid sarcoidosis: a rare entity in the differential diagnosis of thyroid cancer. Endocrinol Diabetes Metab Case Rep. 2021 Sep 1;2021:21–0095. 10.1530/EDM-21-0095. Epub ahead of print. [DOI] [PMC free article] [PubMed]
- 206.Tomer Y. Genetic susceptibility to autoimmune thyroid disease: past, present, and future. Thyroid. 2010;20(7):715–725. doi: 10.1089/thy.2010.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ragusa F, Fallahi P, Elia G, Gonnella D, Paparo SR, Giusti C, Churilov LP, Ferrari SM, Antonelli A. Hashimotos’ thyroiditis: Epidemiology, pathogenesis, clinic and therapy. Best Pract Res Clin Endocrinol Metab. 2019 Dec;33(6):101367. 10.1016/j.beem.2019.101367. Epub 2019 Nov 26. PMID: 31812326. [DOI] [PubMed]
- 208.Basolo F, Proietti A, Ugolini C. (2022). Hashimoto Thyroiditis. In: La Rosa, S., Uccella, S. (eds) Endocrine Pathology. Encyclopedia of Pathology. Springer, Cham. 10.1007/978-3-030-62345-6_5062
- 209.Chui MH, Cassol CA, Asa SL, Mete O. Follicular epithelial dysplasia of the thyroid: morphological and immunohistochemical characterization of a putative preneoplastic lesion to papillary thyroid carcinoma in chronic lymphocytic thyroiditis. Virchows Arch. 2013;462(5):557–563. doi: 10.1007/s00428-013-1397-1. [DOI] [PubMed] [Google Scholar]
- 210.Kholová I, Kalfert D, Lintusaari J, Rajakorpi E, Ludvíková M. Follicular Epithelial Dysplasia as Hashimoto Thyroiditis-Related Atypia: a Series of 91 Specimens. Endocr Pathol. 2021 Sep;32(3):368–374. 10.1007/s12022-021-09679-w. Epub 2021 May 15. PMID: 33991306; PMCID: PMC8370905. [DOI] [PMC free article] [PubMed]
- 211.Feldt-Rasmussen U. Hashimoto’s thyroiditis as a risk factor for thyroid cancer. Curr Opin Endocrinol Diabetes Obes. 2020;27(5):364–371. doi: 10.1097/MED.0000000000000570. [DOI] [PubMed] [Google Scholar]
- 212.Huang BY, Hseuh C, Chao TC, Lin KJ, Lin JD. Well-differentiated thyroid carcinoma with concomitant Hashimoto’s thyroiditis present with less aggressive clinical stage and low recurrence. Endocr Pathol. 2011;22(3):144–149. doi: 10.1007/s12022-011-9164-9. [DOI] [PubMed] [Google Scholar]
- 213.Tang Q, Pan W, Peng L. Association between Hashimoto thyroiditis and clinical outcomes of papillary thyroid carcinoma: A meta-analysis. PLoS One. 2022 Jun 16;17(6):e0269995. 10.1371/journal.pone.0269995. PMID: 35709179; PMCID: PMC9202927. [DOI] [PMC free article] [PubMed]
- 214.Sakiz D, Sencar ME, Calapkulu M, Ozturk Unsal I, Aktas L, Ucan B, Ozbek M, Cakal E. The Effects of Chronic Lymphocytic Thyroiditis on Clinicopathologic Factors in Papillary Thyroid Cancer. Endocr Pract. 2021;27(12):1199–1204. doi: 10.1016/j.eprac.2021.07.011. [DOI] [PubMed] [Google Scholar]
- 215.Jun HH, Kim SM, Hong SW, Lee YS, Chang HS, Park CS. Warthin-like variant of papillary thyroid carcinoma: single institution experience. ANZ J Surg. 2016;86(6):492–494. doi: 10.1111/ans.12725. [DOI] [PubMed] [Google Scholar]
- 216.Orsatti A, De Leo A, Chiarucci F, Simoncini G, Cremonini N, Fornelli A, Amorosa L, Maloberti T, de Biase D, Tallini G. Multifocal Fibrosing Thyroiditis: an Under-recognized Mimicker of Papillary Thyroid Carcinoma. Endocr Pathol. 2022 Sep;33(3):335–345. 10.1007/s12022-022-09726-0. Epub 2022 Jul 11. PMID: 35819567; PMCID: PMC9420094. [DOI] [PMC free article] [PubMed]
- 217.Zucca E, Bertoni F. The spectrum of MALT lymphoma at different sites: biological and therapeutic relevance. Blood. 2016;127(17):2082–2092. doi: 10.1182/blood-2015-12-624304. [DOI] [PubMed] [Google Scholar]
- 218.Bartalena L. Diagnosis and management of Graves disease: a global overview. Nat Rev Endocrinol. 2013;9(12):724–734. doi: 10.1038/nrendo.2013.193. [DOI] [PubMed] [Google Scholar]
- 219.Tanda ML. (2022). Hyperthyroidism. In: La Rosa, S., Uccella, S. (eds) Endocrine Pathology. Encyclopedia of Pathology. Springer, Cham. 10.1007/978-3-030-62345-6_5287
- 220.Smith TJ, Hegedüs L. Graves’ Disease. N Engl J Med. 2016;375(16):1552–1565. doi: 10.1056/NEJMra1510030. [DOI] [PubMed] [Google Scholar]
- 221.Cohen O, Ronen O, Khafif A, Rodrigo JP, Simo R, Pace-Asciak P, Randolph G, Mikkelsen LH, Kowalski LP, Olsen KD, Sanabria A, Tufano RP, Babighian S, Shaha AR, Zafereo M, Ferlito A. Revisiting the role of surgery in the treatment of Graves’ disease. Clin Endocrinol (Oxf). 2022;96(6):747–757. doi: 10.1111/cen.14653. [DOI] [PubMed] [Google Scholar]
- 222.Keskin C, Sahin M, Hasanov R, Aydogan BI, Demir O, Emral R, et al. Frequency of thyroid nodules and thyroid cancer in thyroidectomized patients with Graves’ disease. Arch Med Sci. 2020;16:302–307. doi: 10.5114/aoms.2018.81136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Rossi ED, Raffaelli M, Mule’ A, Miraglia A, Lombardi CP, Vecchio FM, Fadda G. Simultaneous immunohistochemical expression of HBME-1 and galectin-3 differentiates papillary carcinomas from hyperfunctioning lesions of the thyroid. Histopathology. 2006 Jun;48(7):795–800. 10.1111/j.1365-2559.2006.02428.x. PMID: 16722927. [DOI] [PubMed]
- 224.Zargari N, Mokhtari M. Evaluation of Diagnostic Utility of Immunohistochemistry Markers of TROP-2 and HBME-1 in the Diagnosis of Thyroid Carcinoma. Eur Thyroid J. 2019 Jan;8(1):1–6. 10.1159/000494430. Epub 2018 Nov 22. PMID: 30800635; PMCID: PMC6381885. [DOI] [PMC free article] [PubMed]
- 225.Rotondi M, Carbone A, Coperchini F, Fonte R, Chiovato L. DIAGNOSIS OF ENDOCRINE DISEASE: IgG4-related thyroid autoimmune disease. Eur J Endocrinol. 2019;180(5):R175–R183. doi: 10.1530/EJE-18-1024. [DOI] [PubMed] [Google Scholar]
- 226.Li Y, Nishihara E, Hirokawa M, Taniguchi E, Miyauchi A, et al. Distinct clinical, serological, and sonographic characteristics of Hashimoto’s thyroiditis based with and without IgG4-positive plasma cells. J Clin Endocrinol Metab. 2010;95:1309–1317. doi: 10.1210/jc.2009-1794. [DOI] [PubMed] [Google Scholar]
- 227.Li Y, Wang X, Liu Z, Ma J, Lin X, Qin Y, Nishihara E, Miyauchi A, Kakudo K. Hashimoto’s Thyroiditis with Increased IgG4-Positive Plasma Cells: Using Thyroid-Specific Diagnostic Criteria May Identify Early Phase IgG4 Thyroiditis. Thyroid. 2020;30(2):251–261. doi: 10.1089/thy.2019.0063. [DOI] [PubMed] [Google Scholar]
- 228.Dahlgren M, Khosroshahi A, Nielsen GP, Deshpande V, Stone JH. Riedel’s thyroiditis and multifocal fibrosclerosis are part of the IgG4-related systemic disease spectrum. Arthritis Care Res (Hoboken) 2010;62:1312–1318. doi: 10.1002/acr.20215. [DOI] [PubMed] [Google Scholar]
- 229.Griffel B. Focal adrenalitis. Its frequency and correlation with similar lesions in the thyroid and kidney. Virchows Arch A Pathol Anat Histol. 1974;364(2):191–8. [DOI] [PubMed]
- 230.Huebener KH, Treugut H. Adrenal cortex dysfunction: CT findings. Radiology. 1984;150(1):195–199. doi: 10.1148/radiology.150.1.6689760. [DOI] [PubMed] [Google Scholar]
- 231.Eriksson D, Røyrvik EC, Aranda-Guillén M, Berger AH, Landegren N, Artaza H, Hallgren Å, Grytaas MA, Ström S, Bratland E, Botusan IR, Oftedal BE, Breivik L, Vaudel M, Helgeland Ø, Falorni A, Jørgensen AP, Hulting AL, Svartberg J, Ekwall O, Fougner KJ, Wahlberg J, Nedrebø BG, Dahlqvist P; Norwegian Addison Registry Study Group; Swedish Addison Registry Study Group; Knappskog PM, Wolff ASB, Bensing S, Johansson S, Kämpe O, Husebye ES. GWAS for autoimmune Addison’s disease identifies multiple risk loci and highlights AIRE in disease susceptibility. Nat Commun. 2021 Feb 11;12(1):959. [DOI] [PMC free article] [PubMed]
- 232.Saeger W, Lohe B, Engels CL, Werner U. IgG4-Associated Adrenalitis-a Case Report. Endocr Pathol. 2018;29(3):294–298. doi: 10.1007/s12022-018-9531-x. [DOI] [PubMed] [Google Scholar]
- 233.Lynnhtun K, Achan A, Lam V. IgG4 related pseudotumour (calcifying fibrous tumour) of adrenal gland. Pathology. 2013;45:519–521. doi: 10.1097/PAT.0b013e32836359a9. [DOI] [PubMed] [Google Scholar]
- 234.Kurokawa K, Mitsuishi Y, Shimada N, Ito N, Ogiwara M, Miura K, Asao T, Ko R, Shukuya T, Shibayama R, Goto H, Takahashi K. Clinical characteristics of adrenal insufficiency induced by pembrolizumab in non-small-cell lung cancer. Thorac Cancer. 2022 Dec 15. 10.1111/1759-7714.14761. Epub ahead of print. PMID: 36523162. [DOI] [PMC free article] [PubMed]
- 235.Raschi E, Fusaroli M, Giunchi V, Repaci A, Pelusi C, Mollica V, Massari F, Ardizzoni A, Poluzzi E, Pagotto U, Di Dalmazi G. Adrenal Insufficiency with Anticancer Tyrosine Kinase Inhibitors Targeting Vascular Endothelial Growth Factor Receptor: Analysis of the FDA Adverse Event Reporting System. Cancers (Basel). 2022;14(19):4610. doi: 10.3390/cancers14194610.PMID:36230533;PMCID:PMC9559636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.