

Abstract
Diabetes could directly induce cardiac injury, leading to cardiomyopathy. However, treatment strategies for diabetic cardiomyopathy remain limited. ZNF593-AS knockout and cardiomyocyte-specific transgenic mice were constructed. In addition, high-fat diet (HFD)-induced diabetic mouse model and db/db mice, another classic diabetic mouse model, were employed. ZNF593-AS was silenced using GapmeR, a modified antisense oligonucleotide, while overexpressed using a recombinant adeno-associated virus serotype 9-mediated gene delivery system. Transcriptome sequencing, RNA pull-down assays, and RNA immunoprecipitation assays were also performed to investigate the underlying mechanisms. ZNF593-AS expression was decreased in diabetic hearts. ZNF593-AS attenuated the palmitic acid-induced apoptosis of cardiomyocytes in vitro. In HFD-induced diabetic mice, ZNF593-AS deletion aggravated cardiac dysfunction and enhanced cardiac apoptosis and inflammation. In contrast, HFD-induced cardiac dysfunction was improved in ZNF593-AS transgenic mice. Consistently, ZNF593-AS exerted the same cardioprotective effects in db/db mice. Mechanistically, ZNF593-AS directly interacted with the functional domain of interferon regulatory factor 3 (IRF3), and suppressed fatty acid-induced phosphorylation and activation of IRF3, contributing to the amelioration of cardiac cell death and inflammation. In conclusion, our results identified the protective role of ZNF593-AS in diabetic cardiomyopathy, suggesting a novel potential therapeutic target.
Keywords: MT: Non-coding RNAs, diabetic cardiomyopathy, ZNF593-AS, IRF3, lncRNA, diabetes, cardiomyocytes
Graphical abstract
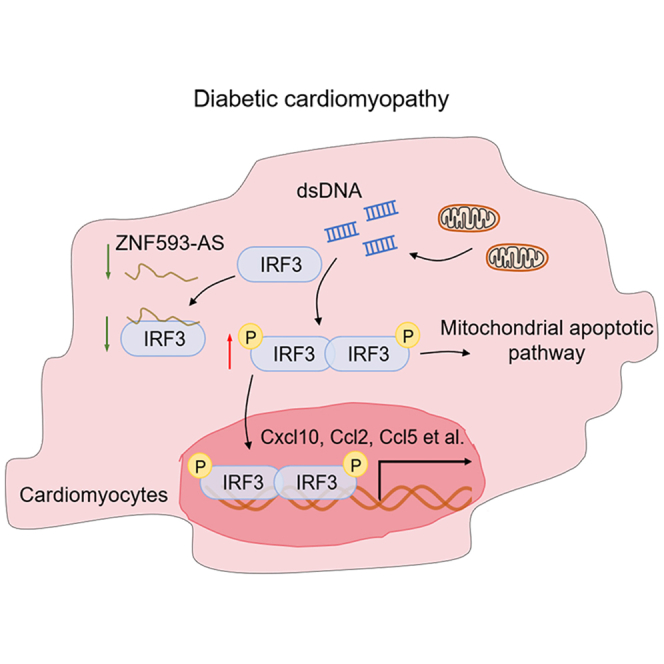
Wang and colleagues demonstrate that a conserved lncRNA, ZNF593-AS, protects against diabetes-induced cardiac injury via suppressing IRF3 phosphorylation, providing a new target for the treatment of diabetic cardiomyopathy.
Introduction
Diabetes mellitus is a widespread metabolic disorder with a substantially increasing prevalence.1 Among patients with diabetes, cardiovascular disease, as a common comorbidity, is the major cause of death.2 Coronary artery disease is known to increase the prevalence of heart failure in patients with diabetes. Metabolic abnormalities can also directly damage cardiomyocytes, leading to diabetic cardiomyopathy. Diabetic cardiomyopathy refers to a clinical condition in patients with diabetes combined with ventricular dysfunction in the absence of coronary artery disease and hypertension.3 The pathophysiological abnormalities contributing to cardiac dysfunction in diabetic hearts involve impaired mitochondrial function, increased oxidative stress, exacerbated insulin resistance, abnormal calcium handling, and enhanced apoptosis.4 In addition to the pivotal roles of cardiomyocytes, the immune system also contributes greatly. Some bio-mediators, such as cytokines and chemokines, released by diabetes-damaged cardiomyocytes, can trigger the maladaptive pro-inflammatory response, which in turn aggravates metabolic disorders.5 For instance, high glucose and elevated fatty acid-induced reactive oxygen species (ROS) overproduction could promote the activation of the NLRP3 inflammasome in cardiomyocytes and trigger the subsequent processing and secretion of mature interleukin (IL)-18 and IL-1β, which contributed to the influx of immune cells, apoptosis of cardiomyocytes, and activation of fibroblasts in diabetic hearts.6,7 Thus, blocking the process of inflammation may be important to treat diabetic cardiomyopathy.
Long non-coding RNAs (lncRNAs) are a class of functional transcripts longer than 200 nucleotides without protein-coding capacity. In recent years, various lncRNAs have been reported to regulate gene expression at both transcriptional and post-transcriptional levels in cardiovascular diseases via multiple molecular mechanisms, which suggests the possibility for lncRNAs to serve as potential therapeutic targets.8,9 For instance, lncRNA Mhrt could sequester Brg1, a chromatin-remodeling factor from its chromatin targets, such as MYH6 and MYH7, to inhibit MYH6/MYH7 isoform switch in pressure-overloaded hearts, thereby preventing stress-induced cardiac remodeling.10 Recently, we have identified a novel conserved cardiomyocyte-enriched lncRNA, ZNF593-AS, which improves calcium handling and contractile function of cardiomyocytes by stabilizing RYR2 mRNA in pressure-overload-induced heart failure.11 However, positive inotropic therapies have failed to improve long-term clinical outcomes of patients with chronic heart failure in multiple clinical trials.12,13 Diabetes is particularly prevalent in chronic heart failure patients and associated with a poor prognosis.14 Considering the direct myocardial damage caused by diabetes, we wonder whether ZNF593-AS can also participate in diabetes-induced cardiac injury.
Interferon regulatory factor 3 (IRF3) is a transcription factor that acts as an important mediator of the cGAS-STING signaling pathway. Generally, cGAS senses cytosolic pathogen DNA during viral or bacterial infection and activates its adaptor STING. Subsequently, IRF3 dimerizes and translocates to the nucleus after being phosphorylated by TBK1, which is activated by interaction with STING. Functioning as a transcription factor, activated nuclear IRF3 triggers the expression of type I interferon and chemokines to defend against infections.15 It is worth noting that in addition to these classical pathogen-associated molecular patterns, damage-associated molecular patterns such as cytosolic double-stranded DNA released from damaged mitochondria can activate IRF3 via the cGAS-STING signaling pathway. Under such non-infectious conditions, an overactivated IRF3 pathway can cause sterile autoinflammation, contributing to tissue injury.16,17,18
In the present study, we found that ZNF593-AS expression was decreased in diabetic hearts, and ZNF593-AS overexpression attenuated diabetes-induced cardiac dysfunction in two diabetic mouse models. Mechanistically, we demonstrated that cytosolic ZNF593-AS sequestered IRF3 in the cytoplasm by inhibiting IRF3 phosphorylation, thereby preventing the release of the downstream pro-inflammatory factors. Taken together, our results suggest that ZNF593-AS is a potential therapeutic target in diabetic cardiomyopathy.
Results
LncRNA ZNF593-AS expression is decreased in diabetic hearts
To determine whether ZNF593-AS participates in diabetic cardiomyopathy, we first examined ZNF593-AS expression levels in two mouse models of diabetes: leptin receptor-deficient db/db mice and high-fat diet (HFD)-induced obese mice. The expression level of ZNF593-AS was decreased by half in the hearts of the two diabetic models (Figure 1A). Previously, we identified ZNF593-AS as a cardiomyocyte-enriched lncRNA mainly localized in the cytoplasm.11 To further confirm the expression pattern of cardiac ZNF593-AS in diabetes, we conducted a fluorescence in situ hybridization (FISH) assay in the heart, and found that the intensity of ZNF593-AS signal that co-localized with the cardiomyocyte-specific marker, cTnT, was substantially decreased in the diabetic state (Figure 1B).
Figure 1.

LncRNA ZNF593-AS expression is decreased in diabetic hearts
(A) ZNF593-AS expression in the hearts of db/db mice and high-fat-diet mice using RT-qPCR. (B) Co-localization of ZNF593-AS with cardiomyocyte-specific marker using FISH assay. (C) RT-qPCR analysis of ZNF593-AS in AC16 and HL-1 cells under the stimulation of PA. (D) FISH of ZNF593-AS in AC16 and HL-1 cells under the stimulation of PA. Comparisons were performed by Student’s t test for (A) and (C).
In diabetic hearts, due to insulin resistance and/or insulin deficiency, the utilization of glucose decreases with fatty acids becoming almost the complete energy source.19 As a result, excessive fatty acids infusion induces intramyocardial lipid accumulation and lipotoxic injury, contributing to cardiomyocyte apoptosis and cardiac dysfunction.20 Therefore, in the in vitro cell model, we used palmitic acid (PA) to mimic such fatty acids overload condition, a feature closely related to diabetic cardiomyopathy.21,22,23,24 As expected, PA reduced the transcript levels of ZNF593-AS in human AC16 and murine HL-1 cells (Figure 1C). Consistently, the expression of ZNF593-AS was decreased in highly purified neonatal murine ventricular myocytes (NMVMs) under PA treatment, as well (Figures S1A and S1B). In addition, FISH assay further confirmed that ZNF593-AS was predominantly localized in the cytoplasm and decreased by PA treatment in cultured AC16 and HL-1 cells (Figure 1D).
Collectively, the decreased abundance of ZNF593-AS in the hearts of mice with diabetes indicated a potential role of ZNF593-AS in diabetic cardiomyopathy.
LncRNA ZNF593-AS attenuates diabetes-induced cardiac dysfunction
To investigate the potential function of ZNF593-AS in vivo, we generated ZNF593-AS knockout (KO) mice using the CRISPR-Cas9-mediated genome editing system (Figure 2A). The PCR assay confirmed the deletion of exons 1–3 region in the genome of ZNF593-AS KO mice (Figure S2A). We further validated ZNF593-AS expression in the hearts of ZNF593-AS KO mice using RT-qPCR (Figure S2B) and did not observe any change in the expression of the Zfp593 gene, which encodes ZNF593 on the opposite strand of ZNF593-AS (Figure S2C).
Figure 2.

ZNF593-AS attenuates diabetes-induced cardiac dysfunction
(A) Schematic of the strategy used for ZNF593-AS deletion. (B, C, and D) Echocardiographic analysis and hemodynamic parameters of ZNF593-AS KO and WT mice fed with high-fat or normal diet: EF value, FS value (B), E/A ratio (C), dp/dtmax and dp/dtmin (D). (E and F) Immunostaining of TUNEL staining in the hearts of ZNF593-AS KO and WT mice fed with high-fat or normal diet. (G) Schematic of the strategy used for ZNF593-AS cardiomyocyte-specific Tg mice construction. (H, I, and J) Echocardiographic analysis and hemodynamic parameters of ZNF593-AS Tg and WT mice fed with high-fat or normal diet: EF value, FS value (H), E/A ratio (I), dp/dtmax, and dp/dtmin (J). (K and L) Immunostaining of TUNEL staining in the hearts of ZNF593-AS Tg and WT mice fed with high-fat or normal diet. One-way ANOVA with the Tukey post-test was used for comparisons.
A diabetic mouse model was established through HFD feeding. Echocardiography was performed 6 months after HFD feeding. HFD remarkably induced systolic and diastolic dysfunction with reduced ejection fraction, fractional shortening, and mitral E/A ratio (Figures 2B, 2C, S3A, and S3B). Consistently, hemodynamic parameters, the maximum rate of rise of left ventricular pressure during contraction (dp/dtmax), and the maximum rate of rise of left ventricular pressure during relaxation (dp/dtmin) measured by Millar pressure catheter indicated that ZNF593-AS KO mice developed aggravated systolic and diastolic dysfunction compared with littermate wild-type mice when fed with HFD (Figure 2D). A considerable increase in the percentage of TUNEL-positive nuclei was observed in the hearts of ZNF593-AS KO mice fed with HFD, suggesting enhanced apoptosis (Figures 2E and 2F). However, the HFD-induced increase in body weight and fasting blood glucose were unaffected by ZNF593-AS deletion (Figures S4A and S4B).
To further confirm the function of ZNF593-AS under diabetic conditions, we used another two classic diabetic models, db/db mice and streptozotocin (STZ)-induced diabetes, in which ZNF593-AS was silenced with GapmeR, a modified antisense oligonucleotide. At the end of the treatment period, we observed elevated blood glucose in db/db and STZ-treated mice (Figures S5A and S5B). As expected, the GapmeR-mediated knockdown of ZNF593-AS exacerbated cardiac dysfunction in db/db and STZ-treated mice (Figures S5C–S5H). Silencing efficiency was validated using RT-qPCR (Figures S5I and S5J). In addition, we did not observe any obvious effects of ZNF593-AS on cardiac function in non-diabetic mice, suggesting that ZNF593-AS functions selectively in the diabetic state.
Indeed, ZNF593-AS KO led to global deletion of ZNF593-AS, and the same case for GapmeR. However, we observed that kidney and liver function, and serum levels of blood glucose, as well as hepatic metabolic parameters remained unchanged in ZNF593-AS KO mice (Figures S6A–S6H), suggesting that the deteriorated cardiac function mediated by ZNF593-AS KO might be directly contributed by the heart rather than the indirect effects from other metabolic organs such as the liver.
To further confirm the cardioprotective role of ZNF593-AS in cardiomyocytes, cardiomyocyte-specific transgenic (Tg) mice with ZNF593-AS overexpression driven by the myosin heavy chain 6 (Myh6) promoter were constructed (Figures 2G, S7A, and S7B). After 6 months of HFD feeding, blood glucose level was increased in HFD-fed mice (Figure S8). Cardiac function was ameliorated in ZNF593-AS Tg mice compared with that of WT mice, characterized by increased ejection fraction, fractional shortening, E/A ratio, dp/dtmax, and dp/dtmin (Figures 2H–2J, S9A, and S9B), and we observed a decrease in the number of apoptotic cells in the hearts of ZNF593-AS Tg mice (Figures 2K and 2L). In line with this, we attempted to overexpress ZNF593-AS in diabetic db/db mice using a gene delivery system mediated by recombinant adeno-associated virus serotype 9 (rAAV9) with a cardiomyocyte-specific troponin T promoter (Figures S10A and S10B). We found that cardiac overexpression of ZNF593-AS attenuated left ventricular contractile dysfunction in diabetic db/db mice (Figures S10C and S10D).
Together, these in vivo data indicated that ZNF593-AS could alleviate diabetes-induced cardiac dysfunction.
LncRNA ZNF593-AS interrupts IRF3-dependent signaling via suppressing IRF3 phosphorylation
Next, we confirmed the function of ZNF593-AS following PA stimulation in vitro. In HL-1 cells, silencing of ZNF593-AS with GapmeR enhanced PA-induced apoptosis (Figures 3A and S11A). By contrast, PA-induced apoptosis was suppressed by ZNF593-AS overexpression (Figures 3B and S11B).
Figure 3.

ZNF593-AS interrupts IRF3-dependent signaling via suppressing IRF3 phosphorylation
(A) Flow cytometry analysis showed ZNF593-AS knockdown accelerated PA-induced apoptosis of HL-1 cells. (B) Flow cytometry analysis showed ZNF593-AS overexpression antagonized PA-induced apoptosis of HL-1 cells. (C) Scatterplot diagram of DEGs between control and ZNF593-AS overexpression groups in PA-treated HL-1 cells. Blue points represent downregulated genes, red points represent upregulated genes, gray points represent non-DEGs, and black points represent Irf3 (fold change >2 or <0.5, p < 0.05). (D) RT-qPCR validation of the expression of representative IRF3-dependent genes. (E) Western blotting analysis of phosphorylated and total IRF3 in HL-1 lysates of control and ZNF593-AS overexpression under PA treatment. Student’s t test was used for comparisons in (D) and (E). One-way ANOVA with the Tukey post-test was used in (A) and (B).
To further investigate the underlying molecular mechanism, we performed transcriptome analysis of HL-1 cells with or without ZNF593-AS overexpression under PA stimulation, which identified approximately 400 differentially expressed genes (fold change >2 or <0.5, p < 0.05). Among the downregulated genes in volcano plots, the majority were prototypical IRF3-dependent genes, such as Ifnb1, Cxcl10, Ifit1, Ccl2, Ccl5, Mx1, and Isg15 (Figure 3C). Consistently, a dramatic decrease in the expression of these IRF3-dependent genes was confirmed using RT-qPCR (Figure 3D); however, the mRNA levels of IRF3 were not affected by ZNF593-AS (Figure 3C, black point). Then, we investigated whether the protein level of IRF3 was regulated by ZNF593-AS. Results showed that IRF3 protein levels were not affected by ZNF593-AS overexpression following PA treatment, either (Figure 3E). Therefore, we further measured the levels of the activated form of IRF3, phosphorylated IRF3 (pIRF3). As expected, we observed that ZNF593-AS overexpression decreased the level of pIRF3, suggesting that ZNF593-AS inhibited the transcription of IRF3-related cytokines and chemokines by suppressing the phosphorylation of IRF3 (Figure 3E). In addition, overexpression of ZNF593-AS suppressed the phosphorylation level of IRF3 in NMVMs, as well as in HL-1 cells (Figure S12).
Taken together, these data suggested that ZNF593-AS could inhibit the IRF3 signaling pathway by suppressing IRF3 phosphorylation under PA treatment.
IRF3 signaling pathway is activated in diabetic hearts
PA could activate the STING-IRF3 pathway by triggering the leakage of mtDNA from damaged mitochondria into the cytosol in endothelial cells and cardiomyocytes.18,25 Consistently, we found that PA treatment substantially increased the amount of mtDNA in the cytosol of HL-1 cells (Figure 4A), which could lead to the phosphorylation of IRF3 by subsequent activation of the cGAS-STING pathway. As expected, we observed that pIRF3 was dramatically increased in the hearts of db/db and HFD-induced diabetic mice, suggesting the activation of IRF3 signaling (Figure 4B).
Figure 4.

IRF3 signaling pathway is activated in diabetic heart
(A) qPCR analysis of mtDNA in cytosolic fraction. Nuclear DNA,18S rDNA served as loading control. (B) Western blotting analysis of phosphorylated and total IRF3 in the hearts of HFD and db/db mice (mouse IRF3 band was observed in approximately 50 kDa). (C) Western blotting analysis of phosphorylated and total IRF3 in the hearts of heart failure patients with or without diabetes (human IRF3 band was observed in approximately 55 kDa). (D) RT-qPCR analysis of ZNF593-AS levels in the hearts of heart failure patients with or without diabetes. (E) Correlation analysis between ZNF593-AS expression and IRF3 phosphorylation level. Student’s t test was used for comparisons in (A), (B), (C), and (D). Pearson correlation analysis was used in (E).
To verify whether IRF3 was activated in human diabetic hearts, we obtained several heart samples from patients with heart failure combined with diabetes. Consistently, the ratio of pIRF3/IRF3 was remarkably increased in diabetic failing hearts compared with that in non-diabetic failing hearts (Figure 4C). Collectively, these data provide strong evidence for the activation of IRF3 in diabetic hearts. Meanwhile, we examined the expression of ZNF593-AS in the same human heart samples and found that hsa-ZNF593-AS expression was decreased in diabetic failing hearts compared with that in non-diabetic failing hearts (Figure 4D). Furthermore, correlation analysis revealed a negative correlation between ZNF593-AS expression and the IRF3 phosphorylation ratio, indicating that hsa-ZNF593-AS could potentially suppress the phosphorylation of IRF3 in diabetic cardiomyopathy (Figure 4E).
These data implied that phosphorylation of IRF3 was significantly enhanced in diabetic hearts, which might lead to the activation of downstream pathways.
LncRNA ZNF593-AS inhibits diabetes-induced activation of IRF3 signaling pathway
As a transcription factor, IRF3 translocates to the nucleus and triggers the transcription of pro-inflammatory factors when it is phosphorylated. Therefore, to further evaluate the function of ZNF593-AS, we isolated a highly purified nuclear fraction (Figure S13). In HL-1 and AC16 cells, ZNF593-AS overexpression substantially blocked the PA-induced-IRF3 nuclear translocation (Figures 5A and 5B).
Figure 5.

ZNF593-AS inhibits diabetes-induced activation of IRF3 signaling pathway
(A and B) Western blotting analysis of IRF3 in cytoplasmic and nuclear extracts of HL-1 (A) and AC16 cells (B) transfected with ZNF593-AS under the stimulation of PA. GAPDH and Lamin B1 served as loading control. (C, D, and E) Western blotting analysis of phosphorylated and total IRF3 (C), western blotting analysis of cytoplasmic and nuclear IRF3 (D), and RT-qPCR analysis of the expression of Cxcl10, Ccl2, and Ccl5 mRNA levels (E) in ZNF593-AS KO and WT mice hearts. (F, G, and H) Western blotting analysis of phosphorylated and total IRF3 (F), western blotting analysis of cytoplasmic and nuclear IRF3 (G), and RT-qPCR analysis of the expression of Cxcl10, Ccl2, and Ccl5 mRNA levels (H) in ZNF593-AS Tg and WT mice hearts. One-way ANOVA with the Tukey post-test was used for comparisons.
Given that we demonstrated that ZNF593-AS could prevent PA-induced-IRF3 activation in vitro, we next investigated this in vivo. As predicted, we found that ZNF593-AS deletion significantly enhanced IRF3 phosphorylation and nuclear translocation in the cardiac tissues of HFD-fed mice (Figures 5C and 5D). The HFD-induced expression of typical IRF3-dependent chemokines, such as Cxcl10, Ccl5, and Ccl2, were further enhanced by ZNF593-AS deletion (Figure 5E).
HFD-induced cardiac IRF3 activation, manifested by phosphorylation and nuclear translocation, was inhibited in ZNF593-AS Tg mice (Figures 5F and 5G). In addition, we observed a modest reduction in the expression of IRF3-dependent chemokines in the hearts of ZNF593-AS Tg mice fed with HFD (Figure 5H).
To determine if nuclear translocation was increased in which cell type of IRF3, we stained IRF3 with different cardiac cell type markers such as troponin I (TNI), CD31, and vimentin. Interestingly, reduced nuclear IRF3 was observed in cardiomyocytes, fibroblasts, and endothelial cells in ZNF593-AS global KO mice heart (Figure S14A). However, in ZNF593-AS Tg mice heart, only cardiomyocyte localized nuclear IRF3 was decreased while nuclear IRF3 in fibroblasts and endothelial cells remained unchanged (Figure S14B). These results indicated that cardiac-specific ZNF593-AS overexpression-mediated IRF3 nuclear translocation was specifically localized in cardiomyocytes. However, global ZNF593-AS KO clearly regulated nuclear IRF3 translocation in non-cardiomyocytes, and therefore, it is possible that ZNF593-AS overexpression in cardiomyocytes and non-cardiomyocytes might synergistically protect against diabetic cardiomyopathy, which is an intriguing subject for future investigation.
Collectively, these in vitro and in vivo data confirmed that ZNF593-AS can interrupt diabetes-induced-IRF3 activation and chronic inflammation.
LncRNA ZNF593-AS directly binds IRF3
To investigate whether the effect of ZNF593-AS on diabetes was attributed to the inhibition of IRF3 phosphorylation, we used IRF3 small interfering RNA (siRNA) and BX795, a TBK1 inhibitor, to suppress the IRF3 signaling pathway (Figures S15A and S15B).26 Under the stimulation of PA, the protection of ZNF593-AS against apoptosis was diminished when treated with IRF3 siRNA or BX795, suggesting the function of ZNF593-AS was mediated via IRF3 signaling (Figures 6A, 6B, S16A, and S16B).
Figure 6.

ZNF593-AS directly binds IRF3
(A and B) Anti-apoptotic effect of ZNF593-AS overexpression was blocked by IRF3 knockdown (A) and BX795 treatment (4 μM) (B). (C) RNA pull-down assay followed by western blotting of IRF3. HL-1 and AC16 cell lysates were incubated with biotin-labeled sense or antisense (negative control) of ZNF593-AS. (D) RIP assay to determine the binding of IRF3 and ZNF593-AS by using IRF3 antibody or IgG (negative control) in HL-1 and AC16 cells. (E) Schematic diagram of IRF3 full-length and truncations deleting different domain with FLAG tag at C-terminal (upper panel). Western blotting analysis of HL-1 cells transfected with IRF3 full-length and different truncations (lower panel). (F) RIP assay with FLAG antibody in HL-1 cells transfected with IRF3 full-length and different truncations. (G) Schematic diagram of the function of ZNF593-AS in diabetic cardiomyopathy. Student’s t test was used for comparisons in (D). One-way ANOVA with the Tukey post-test was used (A) and (B).
We then investigated whether ZNF593-AS affected the phosphorylation state of IRF3 by directly binding to IRF3. First, we synthesized ZNF593-AS and its antisense strand via in vitro transcription, labeled the 3′ end of the RNA with biotin, and performed RNA pull-down assays using HL-1 and AC16 cell lysates. As expected, hsa-ZNF593-AS and mmu-ZNF593-AS strongly bound to IRF3 (Figure 6C). To further confirm the interaction between ZNF593-AS and IRF3 in vivo, an RNA immunoprecipitation (RIP) assay was conducted (Figures S17A and S17B). Consistent with the results of the RNA pull-down assay, ZNF593-AS was substantially enriched with IRF3 antibody (Figure 6D).
To further determine the domain of IRF3 that contributed to the binding with ZNF593-AS, we constructed a series of vectors, expressing full-length IRF3 and different domain deletion mutants with FLAG tag, respectively (Figure 6E). Next, we performed RIP assay using an FLAG antibody. The truncation-deleting signal response domain (SRD), also called serine-rich domain, which contained several phosphorylation sites and contributed to the activation of IRF3, showed less enrichment for ZNF593-AS, suggesting that the interaction between IRF3 and ZNF563-AS depends on the SRD region (Figure 6F).
Thus, we speculated that ZNF593-AS may directly bind to the SRD of IRF3, impairing IRF3 phosphorylation and activation.
Discussion
In the current study, we identified a diabetes-related lncRNA, ZNF593-AS, that could attenuate diabetes-induced cardiac dysfunction by preventing the activation of the IRF3 pathway, which provided a novel therapeutic strategy to target cardiac injury in diabetes (Figure 6G).
Glycemic control has long been the focus of diabetes treatment. However, considering the prevalence of cardiac dysfunction in diabetic patients, evidence from clinical studies has revealed that glucose management with insulin is associated with worse outcomes in diabetic patients with heart failure.27,28 Therefore, in addition to intensive glycemic control therapy, treatments targeting diabetes-induced cardiac injury need to be taken into consideration. Sodium-glucose cotransporter 2 (SGLT2) inhibitors, a novel class of hypoglycemic agents, reduce adverse cardiovascular outcomes in heart failure patients with diabetes; this cardiovascular benefit is independent of the hypoglycemic ability, given that cardioprotective effects also exist in the absence of diabetes.29 Numerous new potential treatment strategies targeting the pathophysiology of diabetic cardiomyopathy, including increased oxidative stress, overactivated inflammation, and metabolic disturbances, show favorable outcomes in animal models of diabetes for preclinical research.30,31,32 Over the past decades, lncRNAs have been identified to regulate the expression of various genes to exert biological functions, thus emerging as a new prospect for therapy.33,34 Many studies have demonstrated that lncRNAs play an important role in the development and progression of diabetic cardiomyopathy.35,36,37 ZNF593-AS is a cardiomyocyte-enriched lncRNA, which is conserved between humans and mice, and is known to exert protective effects in pressure-overload-induced heart failure. In this study, we found that the expression of ZNF593-AS was decreased in diabetic failing hearts of patients and mouse models. Using HFD and db/db mouse models, we demonstrated that ZNF593-AS knockdown exacerbated cardiac dysfunction, increased apoptosis, and aggravated inflammatory reactions, and ZNF593-AS overexpression showed the opposite effect. Although ZNF593-AS knockdown is global in vivo, global knockdown of ZNF593-AS had no effects on kidney and liver function, as well as hepatic metabolic parameters, which suggested that ZNF593-AS might exert its cardioprotective function directly on the heart. To specifically target the heart, our subsequent study using cardiomyocyte-specific promoter to drive ZNF593-AS overexpression further confirmed the protective role of ZNF593-AS directly in cardiomyocytes. However, we are indeed unable to rule out the effects of ZNF593-AS in other cardiac cells, such as cardiac fibroblasts or cardiac endothelial cells, which is an intriguing subject for future study.
Next, IRF3 was identified as the target of ZNF593-AS in cardiomyocytes in the diabetic state. ZNF593-AS directly binds to IRF3 to block its phosphorylation, leading to prevention of apoptosis and inflammation.
The activated STING-IRF3 signaling pathway has been previously reported to participate in the lipotoxic damage of islet β cells in type 2 diabetes, and IRF3 knockdown can ameliorate inflammation and apoptosis of insulinoma cells induced by PA in vitro.38 Similarly, in high-fat-fed mice, IRF3 is involved in the inflammation and insulin resistance of adipose tissues.18,39 Here, we observed that IRF3 was phosphorylated and activated in the cardiac tissue of heart failure patients with diabetes and diabetic mice, which was consistent with the results of a previous study using the db/db mouse model.25 In addition, inhibition of the STING-IRF3 pathway by using an STING inhibitor has been reported to ameliorate cardiac remodeling and inflammation in an animal model of diabetic cardiomyopathy.25 The elevated level of non-esterified fatty acids, which could induce mitochondrial damage and leakage of mtDNA into the cytosol, might be the main contributor to the activation of IRF3 in diabetic hearts. Consistently, under PA treatment, IRF3 was phosphorylated and translocated into the nucleus in cultured cardiomyocytes. Furthermore, transcriptome analysis revealed that ZNF593-AS markedly inhibited the IRF3 signaling pathway in vitro. Next, we found that IRF3 phosphorylation and activation of downstream IRF3-regulated genes were blocked by ZNF593-AS overexpression in the hearts of HFD-fed mice. In our heart samples of heart failure patients with diabetes, the IRF3 phosphorylation ratio was negatively correlated with the expression of ZNF593-AS, providing additional supporting evidence. In addition, we demonstrated that the protective effect of ZNF593-AS was mediated by inhibition of IRF3 phosphorylation by knocking down IRF3 and using a TBK1 inhibitor in vitro. However, further in vivo studies are required to validate this hypothesis.
Considering that the downstream genes of IRF3 such as Ccl2/Cxcl10 are more macrophage-enriched compared with cardiomyocytes, we wondered whether ZNF593-AS and IRF3 also play roles in macrophages. In RAW 264.7 cells, a murine macrophage cell line, we found that ZNF593-AS indeed suppressed PA-induced IRF3 phosphorylation, similar to that observed in cardiomyocytes (Figure S18A). However, ZNF593-AS expression remained unchanged under PA stimulation in RAW 264.7 cells (Figure S18B), different from the downregulation of ZNF593-AS in cardiomyocytes under the same PA treatment. These findings suggested that even though exogenous ZNF593-AS overexpression in macrophages might have some regulatory effects, endogenous ZNF593-AS in macrophages was not the leading trigger for cardiomyopathy during diabetic condition.
Previously, we have identified RYR2 as a downstream target of ZNF593-AS in pressure-overload-induced heart failure. To further explore the relationship between RYR2 and the new target IRF3 in diabetic cardiomyopathy, we first examined the expression of RYR2 in ZNF593-AS KO and Tg mice hearts using RT-qPCR, and found that RYR2 mRNA level was decreased in ZNF593-AS KO hearts, and conversely, RYR2 mRNA level was increased in ZNF593-AS TG mice hearts. (Figures S19A and S19B). However, RYR2 was unable to affect IRF3 phosphorylation, and IRF3 was unable to regulate RYR2 expression, vice versa (Figures S19C and S19D). These results suggested that ZNF593-AS might regulate RYR2 expression and IRF3 phosphorylation simultaneously, but independently, in diabetic cardiomyopathy. Notably, we have demonstrated that ZNF593-AS inhibited PA-induced apoptosis via suppressing IRF3 phosphorylation. However, RYR2 knockdown failed to diminish such anti-apoptotic effect mediated by ZNF593-AS overexpression (Figures S19E and S19F). Therefore, even though we were unable to completely rule out the participation of the ZNF593-AS/RYR2 pathway in diabetic cardiomyopathy, our in vitro data showed that ZNF593-AS/IRF3 rather than the ZNF593-AS/RYR2 pathway alleviated lipotoxicity-induced cardiomyocyte apoptosis and inflammation, the characteristic pathophysiological abnormalities that contributed to cardiac dysfunction in diabetic hearts.
Several lncRNAs, such as lncRNA-ISIR, lncLrrc55-AS, and lncBST2-2, have been shown to enhance the phosphorylation of IRF3 via different regulatory mechanisms, which play important roles in the immune response to viral infection.40,41,42 For instance, lncLrrc55-AS can promote phosphatase methylesterase 1 (PME-1)-mediated demethylation and inactivation of phosphatase 2A, an inhibitor of the IRF3 signaling pathway, by binding to PME-1, thus strengthening IRF3 phosphorylation.42 In this study, we found an lncRNA exerting a suppressive effect on the phosphorylation of IRF3. By performing RNA pull-down and RIP assays, we demonstrated that ZNF593-AS could directly bind to the IRF3 protein. Specifically, the SRD region of IRF3, which possesses a phosphorylation site, interacts with ZNF593-AS. A previous study revealed that the cytosolic lncRNA NKILA can inhibit IκB phosphorylation by directly masking the phosphorylation motifs of IκB.43 Therefore, we speculated that the interaction between ZNF593-AS and IRF3 could mask the phosphorylation site of IRF3 in the SRD region, contributing to the suppression of IRF3 phosphorylation.
Our study revealed the cardioprotective role of a conserved lncRNA, ZNF593-AS, in diabetic cardiomyopathy. By suppressing IRF3 phosphorylation, ZNF593-AS ameliorated diabetes-induced cardiac injury, providing a new strategy for the treatment of diabetic cardiomyopathy.
Materials and methods
Cell culture and treatments
Human cardiomyocyte AC16 cells were cultured in DMEM (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS, Gibco) at 37°C in 5% CO2. Murine HL-1 cells were maintained in Claycomb medium (Sigma-Aldrich, St. Louis, MO, USA) with 10% FBS, supplemented with 100 μM norepinephrine and 4 mM L-glutamine. NMVMs were separated from 1- to 3-day-old C57BL/6 mice and cultured in DMEM medium containing 10% FBS, BrdU (0.1 mM), and penicillin/streptomycin, as described previously.44 RAW 264.7 cells were cultured in DMEM medium with 10% FBS.
HL-1 and AC16 cells were treated with 0.3 mM PA conjugated with 10% fatty acid-free bovine serum albumin (BSA) for 24 h, and cells in the control group were treated with equal concentrations of BSA solution. The plasmid, siRNA, and GapmeR were transfected into cells using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA), according to the manufacturer’s instructions.
Animals
All experiments were approved by the Animal Care and Use Committee of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China). The animals were maintained under a 12-h light/12-h dark cycle at constant temperature (22 °C).
ZNF593-AS KO mice were generated by GemPharmatech Co., Ltd. (Nanjing, China) using the CRISPR-Cas9 system in a C57BL/6 background. In brief, four guide RNAs (gRNAs) were designed and synthesized to target the exon 1–3 region of the ZNF593-AS gene. Cas9 mRNA and gRNA were injected into zygotes that were subsequently transplanted into pseudo-pregnant female mice. Founder mice were backcrossed with wild-type mice for five generations to remove potential off-target sites. PCR assays were performed to confirm the genotypes of the mice. The sequences of the primers used are as follows: KO-F: 5′-CCTTTGTCACCCTCAAGCCC-3′ and KO-R: 5′-CGTGCCCACAGAAGTGTCCA-3′.
ZNF593-AS cardiomyocyte-specific transgenic (CM-Tg) mice were generated according to standard procedures under control of Myh6 promoter. The presence of the ZNF593-AS transgene was identified via PCR by using the following primer pair: F: 5′-CTACATTGTTGGCTGTCCTCG-3′ and R: 5′-CAGGAGGTACTTCATCGATTCAG-3′.
The KO and CM-Tg mice used in the experiments were males. At the age of 8 weeks, the mice were randomly divided into chow diet and HFD groups, followed by continuous feeding of the corresponding diets for 6 months. The HFD (containing 60% fat, Cat# 12033) and normal control diet (containing 10% fat, Cat# 12031) were purchased from MediScience Diets Co. Ltd. (Yangzhou, China).
Male db/db (BKS-Leprem2Cd479/Gpt) mice and their wild-type littermates were purchased from GemPharmatech Co., Ltd. (Nanjing, China). GapmeR specific to ZNF593-AS and its negative control (20 mg/kg), designed and synthesized by Qiagen (Valencia, CA, USA), were administered weekly via intraperitoneal injection. At 10 weeks, db/db and control mice received via tail vein injection 5 × 1011 copies of the rAAV9 harboring either the ZNF593-AS or GFP sequence under the control of the troponin T (tnt) promoter. After 16 weeks, the cardiac function of db/db mice and their wild-type littermates was assessed using echocardiography and the Millar catheter system.
As for STZ (Sigma-Aldrich) administration, 8-week-old male C57BL/6 mice were intraperitoneally injected with 40 mg/kg STZ (dissolved in sodium citrate buffer) for 5 consecutive days, and the control mice were injected with an equivalent volume of sodium citrate buffer. GapmeR (20 mg/kg) was administered weekly via intraperitoneal injection. Eight weeks after STZ administration, cardiac function was assessed using echocardiography and the Millar catheter system.
Echocardiography was performed using the Vero 1100 system (FUJIFILM VisualSonics, ON, Canada), and hemodynamic parameters were measured using the Millar catheter system, as described previously.11
Human donors
Human heart tissues were collected according to a protocol approved by the Clinical Research Committee of Tongji Medical College (Wuhan, China). The study also conformed to the principles outlined in the Declaration of Helsinki. The participants recruited in the study provided informed consent from individual participants or their immediate family members. Heart tissues were collected from patients with heart failure during heart transplantation. The clinical characteristics of patients are presented in Table S1.
RNA sequencing
RNA sequencing and data analysis were performed by Seqhealth Technology Co., LTD (Wuhan, China). Briefly, total RNA was extracted from HL-1 cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The KCTM Stranded mRNA Library Prep Kit was used to prepare the RNA sequencing library (Seqhealth), by following the manufacturer’s protocol. The libraries were then submitted to the Novaseq 6000 platform for high-throughput sequencing, and differentially expressed genes (DEGs) were analyzed using the edgeR software package. The raw sequencing data have been uploaded to the GEO database (GEO: GSE230434).
RNA FISH
RNA FISH of ZNF593-AS was performed as described previously.11
RNA pull-down
ZNF593-AS was synthesized by in vitro transcription using the TranscriptAid T7 High-Yield Transcription Kit (Thermo Fisher Scientific, Waltham, MA, USA), and then the Pierce RNA 3′ End Desthiobiotinylation Kit (Thermo Fisher Scientific) was used to label ZNF593-AS with biotin. Next, the biotin-labeled RNA-protein complex was pulled down using the Pierce Magnetic RNA-Protein Pull-Down Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The captured proteins were eluted for immunoblot analysis.
RNA immunoprecipitation
In brief, lysates from ultraviolet light cross-linked cells were incubated with IRF3 antibody or normal rabbit immunoglobulin G (IgG) (Proteintech, Chicago, IL, USA) at 4°C overnight. Next, the protein-RNA complex was enriched with protein A/G agarose beads. The RNA in the precipitate was extracted using TRIzol and analyzed using real-time quantitative PCR (qPCR).
Cytosolic mitochondrial DNA detection
The cytosolic fraction was isolated without mitochondrial contamination using the Mitochondria Isolation Kit (Cat# 89874, Thermo Fisher Scientific) according to the manufacturer’s instructions, and then DNA was extracted from the cytosolic fraction using the QIAquick Nucleotide Removal Kit (Qiagen). The quantity of mitochondrial DNA (mtDNA) was measured using qPCR and normalized to the copy number of nuclear DNA. The primer sequences used are as follows: mouse-ND1-F: 5′-TCCGAGCATCTTATCCACGC-3′, coupled with mouse-ND1-R: 5′-GTATGGTGGTACTCCCGCTG-3′, 18S rDNA-F: 5′-TAGAGGGACAAGTGGCGTTC-3′, coupled with 18S rDNA-R: 5′-CGCTGAGCCAGTCAGTGT-3′.
Subcellular fractionation
Nuclear and cytosolic fractions were separated using a Cell Fractionation Kit (Cell Signaling Technology, Danvers, MA, USA), according to the manufacturer’s instructions.
Western blotting
Western blotting was performed as previously described.11 The specific primary antibodies used in this study included IRF3 polyclonal antibody (Cat# 11312-1-AP, Proteintech), phospho-IRF3 (Ser396) polyclonal antibody (Cat# 4947, Cell Signaling Technology), lamin B1 polyclonal antibody (Cat# 12987-1-AP, Proteintech), and GAPDH monoclonal antibody (Cat# 60004-1-lg, Proteintech).
Quantitative reverse-transcription PCR
Total RNA was extracted with TRIzol and reverse-transcribed into cDNA using HiScript III First Strand cDNA Synthesis Kit (+gDNA wiper) (Vazyme Biotech Co., Ltd, China). RT-qPCR was performed using SYBR Green Mix (KAPA Biosystems, Wilmington, MA, USA) on a 7900HT FAST real-time PCR system (Life Technologies). GAPDH or 18S rRNA expression was used as a normalization control. The primer sequences used are listed in Table S2.
Statistical analysis
Data are presented as mean ± SEM, and statistical analyses were performed using GraphPad Prism (v8.0) (San Diego, CA, USA). Statistical differences in multiple comparisons were analyzed using one-way ANOVA, and Student’s t test was used to compare the statistical significance between two groups. Statistical significance was set at p < 0.05.
Acknowledgments
We thank our colleagues in Dr. Wang’s group for their technical help and stimulating discussions during the course of this investigation. Murine cardiac muscle cells (HL-1) were a kind gift from Professor Claycomb. The recombinant adeno-associated virus serotype 9 (rAAV9)-mediated gene delivery system was a kind gift from Dr. Xiao Xiao (East China University of Science and Technology). This work was supported by a grant from the National Natural Science Foundation of China (Nos. U22A20266, 91839302, 82270363, and 82100400). The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.
Author contributions
R.X. and J.F. designed and conducted the experiments and wrote the paper; J.W., K.J., J.Z., S.Y., Y.T., X.N., Z.W., and H.L. participated in designing the experiments; C.C. and D.W.W. designed the experiments and wrote the paper.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.04.025.
Contributor Information
Chen Chen, Email: chenchen@tjh.tjmu.edu.cn.
Dao Wen Wang, Email: dwwang@tjh.tjmu.edu.cn.
Supplemental information
Data availability
The authors confirm that the data supporting the findings of this study are available within the article or its supplemental materials.
References
- 1.Domingo-Lopez D.A., Lattanzi G., H J Schreiber L., Wallace E.J., Wylie R., O'Sullivan J., Dolan E.B., Duffy G.P. Medical devices, smart drug delivery, wearables and technology for the treatment of Diabetes Mellitus. Adv. Drug Deliv. Rev. 2022;185 doi: 10.1016/j.addr.2022.114280. [DOI] [PubMed] [Google Scholar]
- 2.Dillmann W.H. Diabetic cardiomyopathy. Circ. Res. 2019;124:1160–1162. doi: 10.1161/CIRCRESAHA.118.314665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yancy C.W., Jessup M., Bozkurt B., Butler J., Casey D.E., Drazner M.H., Fonarow G.C., Geraci S.A., Horwich T., Januzzi J.L., et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of cardiology foundation/American heart association task force on practice guidelines. J. Am. Coll. Cardiol. 2013;62:e147–e239. doi: 10.1016/j.jacc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 4.Jia G., Hill M.A., Sowers J.R. Diabetic cardiomyopathy. Circ. Res. 2018;122:624–638. doi: 10.1161/CIRCRESAHA.117.311586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filardi T., Ghinassi B., Di Baldassarre A., Tanzilli G., Morano S., Lenzi A., Basili S., Crescioli C. Cardiomyopathy associated with diabetes: the central role of the cardiomyocyte. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20133299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X., Li Z., Li B., Zhu X., Lai X. Klotho improves diabetic cardiomyopathy by suppressing the NLRP3 inflammasome pathway. Life Sci. 2019;234 doi: 10.1016/j.lfs.2019.116773. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H., Chen X., Zong B., Yuan H., Wang Z., Wei Y., Wang X., Liu G., Zhang J., Li S., et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J. Cell Mol. Med. 2018;22:4437–4448. doi: 10.1111/jcmm.13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boon R.A., Jaé N., Holdt L., Dimmeler S. Long noncoding RNAs: from clinical genetics to therapeutic targets? J. Am. Coll. Cardiol. 2016;67:1214–1226. doi: 10.1016/j.jacc.2015.12.051. [DOI] [PubMed] [Google Scholar]
- 9.Devaux Y., Zangrando J., Schroen B., Creemers E.E., Pedrazzini T., Chang C.-P., Dorn G.W., Thum T., Heymans S., Cardiolinc network Long noncoding RNAs in cardiac development and ageing. Nat. Rev. Cardiol. 2015;12:415–425. doi: 10.1038/nrcardio.2015.55. [DOI] [PubMed] [Google Scholar]
- 10.Han P., Li W., Lin C.H., Yang J., Shang C., Nuernberg S.T., Jin K.K., Xu W., Lin C.-Y., Lin C.J., et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan J., Li H., Xie R., Zhang X., Nie X., Shi X., Zhan J., Yin Z., Zhao Y., Dai B., et al. LncRNA ZNF593-AS alleviates contractile dysfunction in dilated cardiomyopathy. Circ. Res. 2021;128:1708–1723. doi: 10.1161/CIRCRESAHA.120.318437. [DOI] [PubMed] [Google Scholar]
- 12.Cuffe M.S., Califf R.M., Adams K.F., Jr., Benza R., Bourge R., Colucci W.S., Massie B.M., O'Connor C.M., Pina I., Quigg R., et al. Short-term intravenous milrinone for acute exacerbation of chronic heart FailureA randomized controlled trial. JAMA. 2002;287:1541–1547. doi: 10.1001/jama.287.12.1541. [DOI] [PubMed] [Google Scholar]
- 13.Greenberg B., Butler J., Felker G.M., Ponikowski P., Voors A.A., Desai A.S., Barnard D., Bouchard A., Jaski B., Lyon A.R., et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387:1178–1186. doi: 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- 14.From A.M., Leibson C.L., Bursi F., Redfield M.M., Weston S.A., Jacobsen S.J., Rodeheffer R.J., Roger V.L. Diabetes in heart failure: prevalence and impact on outcome in the population. Am. J. Med. 2006;119:591–599. doi: 10.1016/j.amjmed.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 15.Galluzzi L., Vanpouille-Box C., Bakhoum S.F., Demaria S. SnapShot: CGAS-STING signaling. Cell. 2018;173:276–276.e1. doi: 10.1016/j.cell.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 16.King K.R., Aguirre A.D., Ye Y.X., Sun Y., Roh J.D., Ng R.P., Kohler R.H., Arlauckas S.P., Iwamoto Y., Savol A., et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017;23:1481–1487. doi: 10.1038/nm.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y., Jesus A.A., Marrero B., Yang D., Ramsey S.E., Sanchez G.A.M., Tenbrock K., Wittkowski H., Jones O.Y., Kuehn H.S., et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mao Y., Luo W., Zhang L., Wu W., Yuan L., Xu H., Song J., Fujiwara K., Abe J.I., LeMaire S.A., et al. STING–IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler. Thromb. Vasc. Biol. 2017;37:920–929. doi: 10.1161/ATVBAHA.117.309017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bayeva M., Sawicki K.T., Ardehali H. Taking diabetes to heart—deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J. Am. Heart Assoc. 2013;2 doi: 10.1161/JAHA.113.000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van de Weijer T., Schrauwen-Hinderling V.B., Schrauwen P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011;92:10–18. doi: 10.1093/cvr/cvr212. [DOI] [PubMed] [Google Scholar]
- 21.Tsushima K., Bugger H., Wende A.R., Soto J., Jenson G.A., Tor A.R., McGlauflin R., Kenny H.C., Zhang Y., Souvenir R., et al. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ. Res. 2018;122:58–73. doi: 10.1161/CIRCRESAHA.117.311307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu L., Wang K., Wang W., Wen Z., Wang P., Liu L., Wang D.W. Glucagon-like peptide-1 ameliorates cardiac lipotoxicity in diabetic cardiomyopathy via the PPARα pathway. Aging Cell. 2018;17 doi: 10.1111/acel.12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin L., Geng L., Ying L., Shu L., Ye K., Yang R., Liu Y., Wang Y., Cai Y., Jiang X., et al. FGF21–Sirtuin 3 Axis confers the protective effects of exercise against diabetic cardiomyopathy by governing mitochondrial integrity. Circulation. 2022;146:1537–1557. doi: 10.1161/CIRCULATIONAHA.122.059631. [DOI] [PubMed] [Google Scholar]
- 24.Kuwabara Y., Horie T., Baba O., Watanabe S., Nishiga M., Usami S., Izuhara M., Nakao T., Nishino T., Otsu K., et al. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ. Res. 2015;116:279–288. doi: 10.1161/CIRCRESAHA.116.304707. [DOI] [PubMed] [Google Scholar]
- 25.Ma X.M., Geng K., Law B.Y.K., Wang P., Pu Y.L., Chen Q., Xu H.W., Tan X.Z., Jiang Z.Z., Xu Y. Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell Biol. Toxicol. 2023;39:277–299. doi: 10.1007/s10565-021-09692-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark K., Plater L., Peggie M., Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IκB kinase ε: a distinct upstream kinase mediates SER-172 phosphorylation and activation. J. Biol. Chem. 2009;284:14136–14146. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cosmi F., Shen L., Magnoli M., Abraham W.T., Anand I.S., Cleland J.G., Cohn J.N., Cosmi D., De Berardis G., Dickstein K., et al. Treatment with insulin is associated with worse outcome in patients with chronic heart failure and diabetes. Eur. J. Heart Fail. 2018;20:888–895. doi: 10.1002/ejhf.1146. [DOI] [PubMed] [Google Scholar]
- 28.Shen L., Rørth R., Cosmi D., Kristensen S.L., Petrie M.C., Cosmi F., Latini R., Køber L., Anand I.S., Carson P.E., et al. Insulin treatment and clinical outcomes in patients with diabetes and heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2019;21:974–984. doi: 10.1002/ejhf.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMurray J.J.V., Solomon S.D., Inzucchi S.E., Køber L., Kosiborod M.N., Martinez F.A., Ponikowski P., Sabatine M.S., Anand I.S., Bělohlávek J., et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019;381:1995–2008. doi: 10.1056/NEJMoa1911303. [DOI] [PubMed] [Google Scholar]
- 30.Kim S.K., Zhao Z.S., Lee Y.J., Lee K.E., Kang S.M., Choi D., Lim S.-K., Chung N., Lee H.C., Cha B.S. Left-ventricular diastolic dysfunction may be prevented by chronic treatment with PPAR-α or -γ agonists in a type 2 diabetic animal model. Diabetes. Metab. Res. Rev. 2003;19:487–493. doi: 10.1002/dmrr.410. [DOI] [PubMed] [Google Scholar]
- 31.Tan X., Hu L., Shu Z., Chen L., Li X., Du M., Sun D., Mao X., Deng S., Huang K., Zhang F. Role of CCR2 in the development of streptozotocin-treated diabetic cardiomyopathy. Diabetes. 2019;68:2063–2073. doi: 10.2337/db18-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia Z., Kuo K.H., Nagareddy P.R., Wang F., Guo Z., Guo T., Jiang J., McNeill J.H. N-acetylcysteine attenuates PKCβ2 overexpression and myocardial hypertrophy in streptozotocin-induced diabetic rats. Cardiovasc. Res. 2007;73:770–782. doi: 10.1016/j.cardiores.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 33.Khorkova O., Hsiao J., Wahlestedt C. Basic biology and therapeutic implications of lncRNA. Adv. Drug Deliv. Rev. 2015;87:15–24. doi: 10.1016/j.addr.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winkle M., El-Daly S.M., Fabbri M., Calin G.A. Noncoding RNA therapeutics — challenges and potential solutions. Nat. Rev. Drug Discov. 2021;20:629–651. doi: 10.1038/s41573-021-00219-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng Y., Xu W., Zhang W., Wang W., Liu T., Zhou X. LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy. Theranostics. 2019;9:4558–4566. doi: 10.7150/thno.31052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meng L., Lin H., Huang X., Weng J., Peng F., Wu S. METTL14 suppresses pyroptosis and diabetic cardiomyopathy by downregulating TINCR lncRNA. Cell Death Dis. 2022;13:38. doi: 10.1038/s41419-021-04484-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou X., Zhang W., Jin M., Chen J., Xu W., Kong X. lncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging miR-22-3p in diabetic cardiomyopathy. Cell Death Dis. 2017;8:e2929. doi: 10.1038/cddis.2017.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu H.Q., Qiao J.T., Liu F.Q., Wang J.B., Sha S., He Q., Cui C., Song J., Zang N., Wang L.S., et al. The STING-IRF3 pathway is involved in lipotoxic injury of pancreatic β cells in type 2 diabetes. Mol. Cell. Endocrinol. 2020;518 doi: 10.1016/j.mce.2020.110890. [DOI] [PubMed] [Google Scholar]
- 39.Kumari M., Wang X., Lantier L., Lyubetskaya A., Eguchi J., Kang S., Tenen D., Roh H.C., Kong X., Kazak L., et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Invest. 2016;126:2839–2854. doi: 10.1172/JCI86080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen S., Huang X., Xie Q., Liu Q., Zhu H., James Ou J.H. The role of long noncoding RNA BST2-2 in the innate immune response to viral infection. J. Virol. 2022;96 doi: 10.1128/jvi.00207-22. e00207–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J., Wang P., Li Z., Li Z., Han D., Wen M., Zhao Q., Zhang L., Ma Y., Liu W., et al. IRF3-binding lncRNA-ISIR strengthens interferon production in viral infection and autoinflammation. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.109926. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Y., Li M., Xue Y., Li Z., Wen W., Liu X., Ma Y., Zhang L., Shen Z., Cao X. Interferon-inducible cytoplasmic lncLrrc55-AS promotes antiviral innate responses by strengthening IRF3 phosphorylation. Cell Res. 2019;29:641–654. doi: 10.1038/s41422-019-0193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu B., Sun L., Liu Q., Gong C., Yao Y., Lv X., Lin L., Yao H., Su F., Li D., et al. A cytoplasmic NF-κB interacting long noncoding RNA blocks IκB phosphorylation and suppresses breast cancer metastasis. Cancer Cell. 2015;27:370–381. doi: 10.1016/j.ccell.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 44.Ravi V., Jain A., Taneja A., Chatterjee K., Sundaresan N.R. Isolation and culture of neonatal murine primary cardiomyocytes. Curr. Protoc. 2021;1:e196. doi: 10.1002/cpz1.196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article or its supplemental materials.