

Abstract
Objective:
Diffuse alveolar hemorrhage in children is a rare condition resulting from different underlying diseases. This study aimed at describing characteristics and diagnostic measures in children with ILD (children’s interstitial lung disease, chILD) and diffuse alveolar hemorrhage (DAH) to improve the diagnostic approach by increasing clinician’s awareness of diagnostic shortcomings.
Patients and Methods:
A retrospective data analysis of patients with ILD and DAH treated in our own or collaborating centers between 01/07/1997 and 31/12/2020 was performed. Data on clinical courses and diagnostic measures were systematically retrieved as case-vignettes and investigated. To assess suitability of diagnostic software-algorithms, the Human Phenotype Ontology (HPO) was revised and expanded to optimize conditions of its associated tool the “Phenomizer”.
Results:
For 97 (74%) of 131 patients, etiology of pulmonary hemorrhage was clarified. For 34 patients (26%), no underlying condition was found (termed as idiopathic pulmonary hemorrhage, IPH). Based on laboratory findings or clinical phenotype/ comorbidities, 20 of these patients were assigned to descriptive clusters: IPH associated with autoimmune features (9), eosinophilia (5), renal disease (3) or multi-organ involvement (3). For 14 patients, no further differentiation was possible.
Conclusion:
Complete and sometimes repeated diagnostics are essential for establishing the correct diagnosis in children with DAH. We suggest assignment of patients with IPH to descriptive clusters, which may also guide further research. Digital tools such as the Phenomizer/ HPO are promising, but need to be extended to increase diagnostic accuracy.
Keywords: Diffuse pulmonary hemorrhage, pediatric, children’s interstitial lung disease, human phenotype ontology, idiopathic pulmonary hemosiderosis
INTRODUCTION
Diffuse alveolar hemorrhage (DAH) in children is rare and may be the result of a variety of underlying diseases and conditions, leading to single or recurrent bleeding events (1, 2). The causes for pulmonary hemorrhage and DAH differ widely from diseases that cause pulmonary vasculitis/ capillaritis (e.g. microscopic polyangiitis, anti-glomerular basement membrane disease/ Goodpasture’s syndrome, systemic lupus erythematosus) to coagulation disorders, drug toxicity, immune dysregulation (e.g. COPA syndrome), transplantation/ rejection and cardiovascular diseases (e.g. pulmonary hypertension, arteriovenous malformations, pulmonary veno-occlusive disease) (3–5). The condition of recurrent pulmonary hemorrhages that cannot be assigned to a certain diagnosis is generally referred to as “idiopathic pulmonary hemosiderosis” (IPH) (6, 7). The classical triad of symptoms of DAH includes hemoptysis, diffuse parenchymal infiltrates on imaging and iron deficiency anaemia. However, the disease may also present with less specific symptoms such as tachypnea, dyspnea, cough and failure to thrive (1, 2, 7–9). Exacerbations may lead to severe clinical deterioration due to hypoxemia with need for ventilation and blood transfusion, and prognosis has been considered to be poor due to a mortality of up to 60% (2, 10). As treatment options for children with pulmonary hemorrhage differ depending on the underlying cause, precise diagnoses are essential: Immunosuppressive treatment, which is frequently used if an immunological mechanism is suspected as it may be the case for patients with systemic lupus erythematosus or vasculitis, may not be the best long-term treatment option for a patient with Lane-Hamilton-syndrome (long-term treatment includes gluten-free diet) or a patient with hereditary hemorrhagic telangiectasia (HHT; treatment includes embolization of pulmonary arteriovenous malformations) (11–13).
However, up to date data are scarce and there is no conclusive classification of these conditions, as previous studies focussed mainly on descriptions of clinical phenotype and course, treatment options and prognosis (6–9, 14–16). Based on an existing large cohort of real-life cases of diffuse lung disease, our goal was to find a straightforward approach for diagnosing and classifying DAH that is useful to practicing subspecialty clinicians.
To find an approach for diagnosing DAH, we quantitatively analysed the diagnostic procedures taken in a large group of patients to identify diagnostic shortcomings. Besides analysing established diagnostic measures, we were interested if computer-based algorithms might contribute to making an exact diagnosis, and assessed the usefulness of the “Phenomizer”, a web-based tool for the diagnosis of rare genetic conditions, for children with DAH. The resulting simple empirical algorithm provides a practical etiology-based approach to the child with unclear DAH.
To provide an approach to classification of DAH in children, we evaluated the grouping of children with DAH in our inhouse database and in the chILD-EU-register (international management platform for children’s interstitial lung disease) (17). We aimed to classify children into those where a plausible cause or underlying condition for pulmonary hemorrhage was identified and those where none was identified (“idiopathic” pulmonary hemorrhage). For the latter group, we formed descriptive clusters to obtain hints for etiology.
2. METHODS
2.1. Study Design and Subjects
For this retrospective study focusing on DAH, we retrieved patients with DAH from our inhouse database and the chILD-EU-register, covering subjects with ILD between 01/07/1997 and 31/12/2020, and analyzed them (Figure 1). We examined the following disease categories, as predefined in the chILD-EU-register and the inhouse database, manually case by case to identify patients with DAH: ILD-related to systemic disease processes (B1) and ILD- related to lung vessels structural processes (B4) including mature/ almost mature neonates with pulmonary hypertension (Ax, Ay) (18, 19). We limited the study to these categories (inclusion criteria) as most of the well-known causes for DAH described in previous studies can be found there (3–5). As nomenclature regarding pulmonary bleedings may be confusing (for example IPH is actually a histological diagnosis (20, 21), definitions of various terms related to pulmonary hemorrhage and DAH are listed in Supplemental Table 2.
Figure 1.
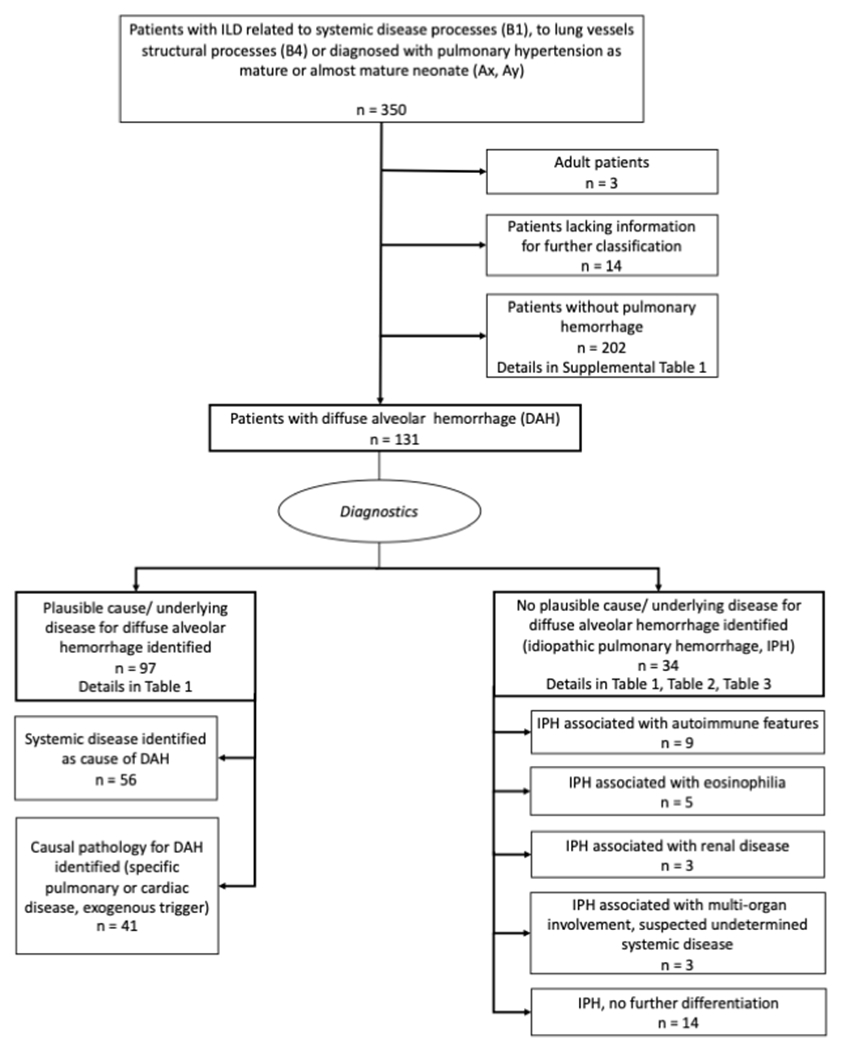
Study enrollment and distribution of patients with diffuse alveolar hemorrhage (DAH).
Informed consent for review of diagnostics was obtained from patients and/or caregivers. The retrospective analysis of cases, the register study, data evaluation and reporting were approved by the Ethics committee of LMU Munich (EK 23-5-2011, EK 111-13, 20-329).
The cases were investigated for the presence of DAH. DAH was defined to be present if at least one of the following criteria was demonstrated in a patient:
Clinical signs for pulmonary hemorrhage: Hemoptysis or bronchoscopic detection of acute or former airway bleeding (e.g. diffuse blood residuals/ crusts or thrombi in the tracheobronchial system). Patients whose hemoptysis or airway bleeding turned out to be due to airway diseases or airway injuries were excluded from this study.
Bronchoalveolar lavage (BAL) finding on bronchoscopy: Increasing bloody appearance of lavage fluid fractions while obtaining lavage, a specific sign for alveolar hemorrhage (22).
Histological examination of lung tissue (collected by biopsy or autopsy): Hemosiderin-laden macrophages/ siderophages (in Prussian blue reaction) or description of bleedings in pathologist’s report except for bleedings that were interpreted as procedure-related.
Hemosiderin-laden macrophages in bronchoalveolar lavage fluid: Hemosiderin-laden macrophages are usually elevated in BAL fluid of patients with pulmonary hemorrhage (normal range is 7.5 ± 10.7 % (mean ± SD) of all macrophages (23)). Patients with 10% or more hemosiderin-laden macrophages were included in the analysis. Patients after lung transplantation with repeated transbronchial biopsies were not taken into account. The percentage of hemosiderin-laden macrophages of patients in this study was calculated by local laboratories quantifying the BAL cytology.
Combination of respiratory symptoms with strong contemporaneous hints for bleeding on imaging (chest CT scan) and in laboratory findings (anaemia).
If patients were included in the chILD-EU-register or inhouse ILD-database but over time were diagnosed with other conditions besides ILD that may cause tracheobronchial hemorrhage, they were excluded from this analysis (infections, airway diseases including cystic fibrosis, patent ductus arteriosus in neonates, extreme premature infants (<28 weeks of gestational age) if the pulmonary hemorrhage was occurring within the neonatal period on mechanical ventilation, focal airway injury, tumor; exclusion criteria).
Clinical bleedings signs were considered most useful indicators for pulmonary hemorrhage as well as diffuse alveolar hemorrhage on bronchoscopy and in histological evaluation of lung tissue. Less certain indicators for a former pulmonary hemorrhage, especially when isolated, were mild or moderate elevation of hemosiderin-laden macrophages in BAL. Patients included in the cohort only because of positive staining for hemosiderin-laden macrophages in BAL are therefore marked separately in Table 1.
Table 1.
Children with DAH. Overview of diagnostic procedures.
| Diagnosis of pulmonary hemorrhage by: | |||||||
|---|---|---|---|---|---|---|---|
| Disease category/ subcategory | Number of patients in subcategory | Patients with DAH (n (percent of total) ) | Clinical diagnosis* | Bronchoscopic diagnosis** | Histopathologic diagnosis | BAL cytology additionally to other criteria (n (% hem.-laden macroph.)) | BAL cytology only*** (n (%hem.-laden macroph.)) |
| Systemic disease with DAH (n = 56; B1) | |||||||
| Sarcoidosis | 17 | 3 | 1 | 0 | 1 | 2 (99%, 25%) | 1 (11%) |
| Granulomatosis with polyangiitis (GPA) | 14 | 11 | 8 | 2 | 3 | 4 (39%, 60%, 19%, 81%) | 1 (40%) |
| Eosinophilic granulomatosis with polyangiitis (EGPA) | 6 | 3 | 1 | 0 | 2 | 0 | 0 |
| Langerhans cell histiocytosis | 11 | 2 | 0 | 0 | 2 | 0 | 0 |
| Hereditary hemorrhagic telangiectasia (HHT) | 9 | 3 | 3 | 1 | 1 | 0 | 0 |
| Ehlers Danlos syndrome | 2 | 2 | 1 | 1 | 0 | 1 (40%) | 0 |
| Filamin A deficiency | 9 | 3 | 1 | 0 | 1 | 1 (45%) | 1 (57%) |
| Aminoacyl-tRNA synthetase deficiency (-ARS) | 6 | 2 | 0 | 0 | 1 | 0 | 1 (12%) |
| Storage diseases | 8 | 1 | 0 | 0 | 0 | 0 | 1 (25%) |
| Cantu syndrome | 3 | 1 | 0 | 0 | 1 | 0 | 0 |
| Familial dysautonomia | 2 | 1 | 0 | 0 | 0 | 0 | 1 (% not given) |
| Systemic lymphatic disorder | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| Immune-mediated/ collagen vascular disorders | 20 | 4 | 3 | 1 | 3 | 3 (58%, 2 not given) | 0 |
| Lane-Hamilton syndrome | 7 | 7 | 5 | 1 | 3 | 4 (100%, 99%, 90%, 1 not given) | 0 |
| Systemic lupus erythematosus (SLE) | 5 | 1 | 0 | 0 | 1 | 0 | 0 |
| Anti-glomerular basement membrane disease (Goodpasture’s syndrome) | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| Diffuse alveolar hemorrhage due to vasculitis disorders | 2 | 2 | 1 | 1 | 0 | 1 (13%) | 0 |
| Microscopic polyangiitis | 1 | 1 | 1 | 1 | 0 | 1 (85%) | 0 |
| Pulmonary thrombotic microangiopathy due to atypical hemolytic uremic syndrome | 1 | 1 | 0 | 1 | 1 | 0 | 0 |
| Autoimmune interstitial lung, joint and kidney disease (COPA-syndrome) | 6 | 5 | 2 | 0 | 2 | 3 (53%, 25%, 31%) | 0 |
| Hyper-IgG4-syndrome | 1 | 1 | 0 | 0 | 0 | 0 | 1 (15%) |
| Cardiac disease with DAH (n = 4; B4) | |||||||
| Congestion due to congenital heart defect/cardiac dysfunction | 7 | 4 | 1 | 1 | 1 | 1 (70%) | 2 (20%, 60%) |
| Abnormalities of pulmonary vessels with DAH (blood, lymphatic; n = 33; B4, Ax, Ay) | |||||||
| Primary pulmonary hypertension (28 patients Ax/Ay, 2 of them with bleedings; 50 patients B4) | 78 | 15 | 1 | 1 | 9 | 2 (68%, 50%) | 3 (32%, 32%, 50%) |
| Pulmonary capillary hemangiomatosis/pulmonary veno-occlusive disease | 21 | 9 | 1 | 0 | 8 | 1 (% not given) | 0 |
| Pulmonary hemorrhage due to pulmonary vascular malformations | 3 | 3 | 2 | 1 | 2 | 2 (88%, 66%) | 0 |
| Pulmonary capillaritis | 3 | 3 | 3 | 0 | 2 | 2 (% not given) | 0 |
| Abnormalities of pulmonary lymphatic vessels | 9 | 3 | 0 | 1 | 2 | 0 | 1 (17%) |
| Exogenous trigger for DAH (n = 4; B4) | |||||||
| Pulmonary hemorrhage due to medication | 2 | 2 | 2 | 0 | 0 | 0 | 0 |
| Fictitious pulmonary hemorrhage | 2 | 2 | 2 | 0 | 0 | 0 | 0 |
| All patients from above categories (n = 97, plausible cause/ underlying condition for DAH identified) | 257 | 97 (38%) | 41 | 13 | 46 | 28 | 13 |
| No plausible cause/ underlying condition for DAH identified | 34 | 34 (100%) | 27 | 7 | 13 | 25 (37-99%; 13 not given) | 0 |
| All patients | 291 # | 131 (45%) | 68 | 20 | 59 | 53 | 13 |
Some patients fulfilled more than one criterion for diagnosis of DAH.
hemoptysis or combination of respiratory symptoms with strong hints for bleeding in chest CT and laboratory (anaemia),
macroscopic signs for bleeding in bronchoscopy and/or alveolar hemorrhage in procedure (bloody BAL),
hem-laden macroph. (hemosiderin-laden macrophages) > 10%.
B1: ILD related to systemic disease processes; B4: ILD related to lung vessels structural processes; Ax: ILD-unclear RDS in the mature neonate; Ay: ILD-unclear RDS in the almost mature neonate. Disease categories/ subcategories were adapted from Griese et al. (20, 21).
These subjects plus those in a category without cases with DAH (Supplemental Table 1) plus the 3 adult patients plus 14 patients lacking information add up to the 350 patients depicted in Figure 1.
Please note: In our databases, four more patients that were assigned to other disease categories as B1, B4, Ax/ pulmonary hypertension, Ay/ pulmonary hypertension showed pulmonary hemorrhage: a patient with an extralobar sequester (disease category C1: Locally, gross structural abnormalities of the lung), a patient with congenital alveolar dysplasia (disease category A1: Diffuse developmental disorders), a patient with an unclear diffuse ILD (disease category By: Unclear diffuse parenchymal lung disease in the non-neonate) and a patient with pulmonary graft-versus-host-disease after stem cell transplantation (disease category B3: Diffuse parenchymal lung diseases in the immunocompromised or transplanted host).
Patients were then classified either as having an identified plausible cause of pulmonary hemorrhage or as lacking an identifiable cause/ underlying etiology; this group was subsequently analyzed separately and categorized further by creating clusters, which was done by qualitative analysis of the cases (Figure 1). A plausible cause was defined as condition previously described as associated with pulmonary hemorrhage or a diagnosis considered being very likely to etiologically cause pulmonary hemorrhage.
2.2. Evaluation of diagnostic work-up for patients with DAH
Data at baseline, on clinical course and diagnostic measures were systematically retrieved and collected as case-vignettes, reviewed and adjudicated by the first author and the principal investigator. During this process, each patient’s record was assessed on basis of available clinical information and diagnostic tests to identify an etiological cause for patient’s diffuse alveolar hemorrhage.
Diagnostic algorithms for evaluation of patients with (suspected) pulmonary hemorrhage have been described before (5, 6). A schematic algorithm was used for systematical investigation of the performed diagnostic procedures in our cohort (Figure 2A). For dealing with children with DAH in clinical practice, we provide a check list in the supplement (Supplemental Table 3).
Figure 2.
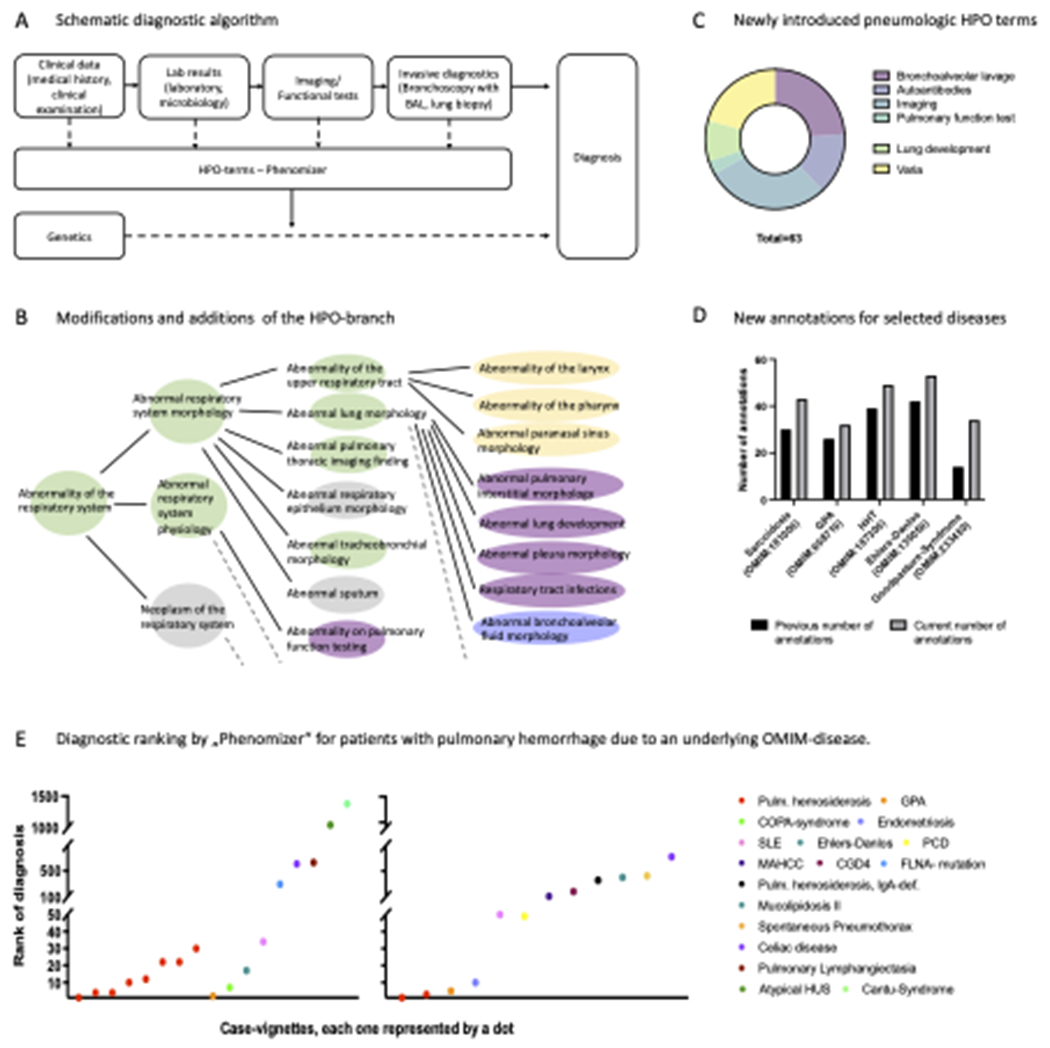
Pulmonary hemorrhage as an example for rare pneumological conditions and Human Phenotype Ontology (HPO). A: Schematic diagnostic algorithm for patients with pulmonary hemorrhage. B: Modifications and additions of the the HPO-branch „Abnormality of the respiratory system“. Green, changes in organisation of HPO-branches. Yellow, introduction of new parent terms. Purple, reassignment and restructuring of terms. Blue, entirely new branch. Broken lines symbolize that the HPO contains additional branches/ terms that are not listed in the figure. Figure 2B is inspired by Haimel et al. (50). C: Creation of new HPO-terms within the field of pneumology. D: Number of annotated terms to selected diseases causing pulmonary hemorrhage before/ after creating new annotation. E: Diagnostic ranking by the „Phenomizer“ for patients with pulmonary hemorrhage due to an underlying OMIM®-disease. Each dot represents a case-vignette with a specific diagnosis. Left graph: cases from our cohort (n = 17). Right graph: case reports from specialized literature (n = 12). Patients with pulmonary hemosiderosis (n=10) are marked in red.
Abbreviations: BAL: bronchoalveolar lavage; CDG4: Chronic granulomatous disease, autosomal recessive, 4; FLNA-mutation: Filamin A-mutation; GPA: Granulomatosis with polyangiitis; HHT: hereditary hemorrhagic teleangiectasia; HPO: Human Phenotype Ontology; HUS: Hemolytic uremic syndrome; MAHCC: methylmalonic aciduria and homocystinuria type cblC; PCD: Primary ciliary dyskinesia; SLE: Systemic lupus erythematosus; Pulm. hemosiderosis, IgA-def.: Pulmonary hemosiderosis with IgA-deficiency.
2.3. Evaluation of diagnostic suitability of a software-tool (Human Phenotype Ontology)
We used the “Phenomizer”, an associated tool of the Human Phenotype Ontology (HPO) that links phenotype with genetics. As others have tried before, we aimed to evaluate its contribution to making a diagnosis (24–28). A detailed description of how to use this tool was provided by Kohler et al. (24). The Phenomizer is available under https://compbio.charite.de/phenomizer/.
Here, we first revised and expanded the HPO database from a pulmonological point of view by adding new terms and annotations and by reorganizing existing branches/ terms to adjust for the DAH conditions (Figure 2, B–D). Then, randomly 25 case-vignettes of our cohort were chosen. In addition, we searched Pediatric Pulmonology, a journal specialized in pediatric lung diseases, for another 25 case reports dealing with one of the diagnoses mentioned in Table 1. For this analysis, we explicitly decided to stick to case reports of one single journal in order to prevent the results to be biased by divergent journal guidelines (for example on text length). As analysis concerning accuracy of diagnosis with the Phenomizer is currently possible for rare diseases with an OMIM® (Online Mendelian Inheritance in Man®) number only, we selected those 29 out of the 50 descriptions which were describable by OMIM® (17 case-vignettes, 12 case reports from Pediatric Pulmonology). HPO-terms from each case-vignettes were then manually extracted and put in the Phenomizer to calculate probable underlying (genetic) diagnoses. The calculated diagnosis was then compared to the correct/ real diagnosis.
RESULTS
350 patients from the disease categories “ILD related to systemic diseases processes (B1)”, “ILD related to lung vessels structural processes (B4)” and “Suspected ILD due to unclear RDS caused by pulmonary hypertension in mature/ almost mature neonate” (Ax, Ay) were retrieved and further analyzed. Of these, 131 pediatric patients with DAH were identified (Figure 1). Diseases from disease categories B1 and B4 where no pulmonary hemorrhages were observed are listed in Supplemental Table 1.
Patients with a plausible cause/ underlying disease known to cause DAH
In 97 patients (74%), the etiology of DAH was clarified by the performed investigations. In roughly half of those (n = 56), a systemic disease known to cause lung bleedings was diagnosed, and in the remaining patients (n = 41), a specific pulmonary disorder, a cardiac disease or an exogenous trigger were identified (Table 1). Several of the patients underwent genetic testing because of a clinical suspicion, leading to identification of underlying monogenetic diseases (pathogenic variants in FLNA (filamin A deficiency), FARSA (aminoacyl-tRNA synthetases deficiency), COL3A1 (vascular Ehler’s Danlos Syndrome), COPA (autoimmune interstitial lung, joint and kidney disease), ABCC9 (Cantu syndrome), ALK1 (hereditary hemorrhagic teleangiectasia), FLT4 (Milroy disease), BMPR2 (primary pulmonary hypertension, with or without hereditary hemorrhagic teleangiectasia)). Mean age at first bleedings and at the end of follow-up in this group was 8.3 (range: 0-17.9)/ 12.3 years (range: 0.1-26.0) years, respectively (Supplemental Table 4). Median observation time was 4.7 years.
At the end of follow-up, 10% of the patients were asymptomatic, 38% improved (sick-better) compared to baseline-visit, 24% were stable (sick-same), 6% worsened (sick-worse), 15% were dead (no information about clinical condition at the end of follow-up: 6%; Supplemental Table 4).
Thirteen patients in this group did not show any other indicators for pulmonary bleeding than a positive staining for hemosiderin-laden macrophages in BAL fluid (11-60% hemosiderin-laden macrophages). About half of them were diagnosed with an underlying cardiac disease or primary pulmonary hypertension, suggesting pulmonary hypertension as an underlying condition for destructive changes in pulmonary arterial vessels with subsequent microscopic pulmonary hemorrhage (21).
Patients with no plausible cause/ underlying disease known to cause DAH: “Idiopathic” pulmonary hemorrhage (IPH)
In about 25% of our cohort (34 patients) no underlying cause for DAH was identified. 28 patients had recurrent, bilateral DAH; six patients had a single, bilateral DAH event.
The diagnostic measures were analyzed and revealed incomplete evaluation in some cases (Table 2). For two patients, pulmonary imaging lacked a chest CT scan, and eight patients did not undergo bronchoscopy. Only about one third (35%, 12 patients) had a lung biopsy. There were no patients who did not have any of these three examinations. Extensive laboratory diagnostics were done in most, but not in all patients, concerning testing for autoantibodies in particular (Table 2, Table 3). Underlying COPA-Syndrome was excluded in 25 patients by targeted genetic screening for a pathogenic/ likely-pathogenic variant in exons 8 and 9 of the COPA-gene. In four patients, genetic testing was extended by exome analysis; however, this did not reveal any relevant disease-causing variants.
Table 2.
Patients with DAH, no plausible cause identified (“idiopathic pulmonary hemorrhage”, IPH; n = 34): Suggested disease entities, observation time, outcome and performed diagnostics.
| Number of performed diagnostic tests/ missing diagnostic tests/ tests with pathological results respectively positive findings for pulmonary hemorrhage | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Suggested disease entity | Observation time (years; mean (min/ max)) | Long-term course (healthy/ sick-better/ sick-same/ sick-worse/ dead) | Autoantibodies ANA/ ANCA (except tTG- autoantibodies) | tTG-auto-antibodies | Screening for immunodeficiency | Chest CT scan | Bronchoscopy and bronchoalveolar lavage | Lung biopsy | Targeted genetic testing for COPA-mutation (exon 8,9) |
| IPH associated with autoimmune features (n = 9) | 3.8 (0/ 10.7) | 3/ 2/ 1/ 2/ 1 (9y) | 9/ 0/ 9 | 5/ 4/ 1** | 8/ 1/ 1 | 9/ 0/ 9 | 5/ 4/ 5 | 4/ 5/ 4 | 6, +1 WES/ 2/ 0 |
| IPH associated with eosinophilia (n = 5) | 3.2 (0/ 6.7) | 3/ 0/ 1/ 1/ 0 | 5/ 0/ 0 | 3/ 2/ 0 | 5/ 0/ 0 | 5/ 0/ 5 | 4/ 1/ 4 | 1/ 4/ 1 | 4/ 1/ 0 |
| IPH associated with renal disease (n = 3) | 4.2 (2.1/ 7.2) | 0/ 2/ 0/ 1/ 0 | 3/ 0/ 1 | 1/ 2/ 0 | 3/ 0/ 0 | 3/ 0/ 3 | 3/ 0/ 2 | 1/ 2/ 1 | 2, +1 WES/ 0/ 0 |
| IPH associated with multi-organ involvement, suspected undetermined systemic disease (n = 3) | 2 (0.4/ 1.1) | 0/ 2/ 0/ 1/ 0 | 2/ 1/ 0 | 0/ 3/ 0 | 3/ 0/ 1 | 3/ 0/ 3 | 3/ 0/ 3 | 2/ 1/ 2 | 1/ 2/ 0 |
| IPH, no further classification (n = 14) | 4.4 (0.3/ 17.7) | 4/ 7/ 2/ 1/ 0 | 10/ 1, 3*/ 0 | 4/ 10/ 0 | 13/ 1/ 1 | 12/ 2/ 12 | 13/ 1/ 12 | 4/ 10/ 4 | 8, +2 WES/ 4/ 0 |
| All (n = 34) | 4.3 (0/ 17.7) | 10/ 13/ 4/ 6/ 1 | 29/ 2, 3*/ 10 | 13/ 21/ 1 | 32/ 2/ 3 | 32/ 2/ 34 | 28/ 6/ 26 | 12/ 22/ 12 | 25/ 9/ 0 |
| Missing diagnostics (%) | 15 % | 62 % | 6 % | 6 % | 18 % | 65 % | 26 % | ||
incomplete: either ANA or ANCA missing;
at this time, the patient had a normal gut biopsy under gluten-containing diet
WES: whole exome sequencing; tTG: tissues transglutaminase
“sick-better” is defined as signs or symptoms of the disease having weakened in intensity and/or number, ”sick-same” is defined as signs or symptoms with same intensity and number, “sick-worse” is defined as signs or symptom having increased in intensity and/ or number compared to baseline (52).
Note: Patients showed elevated ANCAs in only two cases in the cluster of IPH associated with autoimmune features (isolated elevation of ANCAs) and in one case in the cluster of IPH associated with renal disease (elevation of ANCAs as well as ANAs).
Table 3.
Patients with DAH, no plausible cause identified (“idiopathic pulmonary hemorrhage”, IPH, n = 34): Patient’s characteristics including relevant diagnostic findings.
| n=34 | Relevant extrapulmonary comorbidities/symptoms | Relevant laboratory findings | Need for transfusion | Evaluation for immunodeficiency | Evaluation for autoimmunity | Histologic pattern (lung biopsy) | Genetic analysis (all negative results) | |
|---|---|---|---|---|---|---|---|---|
| Recurrent pulmonary hemorrhage | IPH associated with autoimmune features (n=7) | fatigue (1) weight loss (1) adipositas/ iatrogenic Cushing syndrome (1) sinus vein thrombosis (1) CVID (IgG-/ IgA-deficiency) (1) |
anaemia (7) intermittent LDH elevation (2) |
yes (3) no (3) unknown (1) |
IgE elevated (1) insufficient antibody production (IgG, IgA) (1) |
ANCA positive (2) ANA elevated (4) (1:100, 1:100, 1:160, 1:240) tTG-autoantibodies positive (1; gut biopsy negative for celiac disease while being on gluten-containing diet) |
Major pattern: hemosiderosis (4) Minor pattern: chronic Bronchiolitis (1), diffuse lymphoid hyperplasia (1) |
COPA (6), ABCA3, SFTPC, SFTPC, NKX-2.1 (1), whole exome analysis (1), chromosome analysis (1), not done- no material (3) |
| IPH associated with eosinophilia (n=3) | hepatomegaly, hemolytic anaemia (1) cow milk protein allergy, ASD II (1) late prematurity (34 weeks of gestational age) |
anaemia (3) peripheral eosinophilia (3; max. 14%,) eosinophilia in bone marrow aspirate (1) |
yes (1) no (1) unknown (1) |
hypocomplementemia (1) | ANA max. 1:80 (1) | Major pattern: hemosiderosis (1) Minor patterns: follicular bronchiolitis (1), obliterative bronchiolitis (1) |
COPA (3), not done- no material (1) | |
| IPH associated with active renal disease (n=3) | failure to thrive, unilateral renal lithiasis and atrophic kidney, recurrent facial dermatitis (1) mesangioproliferative glomerulonephritis, failure to thrive, splenomegaly, pilomatrixomata, cystic skin lesions, chronic urticaria (1) perinatal asphyxia, glomerulonephritis/ HSP-nephritis (1) |
anaemia (3) transient hyperphosphatasaemia (1) |
yes (3) | IgM mildly diminished (1) | anti-GBM-antibodies negative (2) ANA elevated (max. 1:5120) (1) MPO-ANCAs intermittently elevated (1) |
Major pattern: hemosiderosis (1) Minor patterns: follicular bronchiolitis (1), diffuse lymphoid hyperplasia (1) |
COPA (3), whole exome analysis (1) | |
| IPH associated with multi-organ involvement, suspected undetermined systemic disease (n=3) | Pierre-Robin sequence with cleft soft palate, obstructive sleep apnea failure to thrive, dysphagia, developmental delay, muscular hypotension; GERD, state after cardiac arrest, mid-tracheal narrowing prematurity (27 weeks of gestational age) with long-term ventilation, failure to thrive, diabetes mellitus type 1, paresthesia (hands/feet), GERD, state after recurrent sepsis, chronic constipation, opiat dependency due to chronic chest pain |
anaemia (3) LDH elevation (1) |
yes (3) | IgM/IgA mildly diminished (1) | negative (2) | Major pattern: hemosiderosis (2) Minor pattern: alveolar hypoplasia (1) |
COPA (1), chromosome analysis (1), not done- no material (1) | |
| IPH, no further classification (n=12) | failure to thrive (3), hepatomegaly (1), pectus deformity (1), fatigue (2), iatrogenic Cushing syndrome (2), steroid-induced diabetes mellitus (1), ADHD (1), atopic dermatitis (2), pollinosis (1), nicotin- and cannabis-abusus (1), lactose/ fructose malabsorption (1), PDA (1), ASD II (1), adipositas (1), Gilbert’s syndrome (1), none (2), neonatal resuscitation due to meconium aspiration with bilateral pneumothoraces, hypoxic seizures, atopic dermatitis, food allergies (1) |
anaemia (11) LDH elevation (4) |
yes (2) no (2) unknown (8) |
IgG/ IgM/ IgA normal (10) IgE normal (6) IgG/IgA mildly diminished (1) intermittent IgE elevation (2) insufficient antibody production (IgG) (1) |
negative (11) | Major patterns: hemosiderosis (3), alveolar hypoplasia (1) Minor patterns: chronic bronchiolitis (1), hemosiderosis (1) |
COPA (6), whole exome analysis (2), not done- no material (4) | |
| Pulmonary hemorrhage, single event | IPH associated with autoimmune features (n=2) | developmental delay recurrent infections (parotitis, pyelonephritis, respiratory tract), state after CMV-infection |
anaemia (2) | yes (1) no (1) |
immunoglobulins normal (2) | ANA positive (1:100, 1:200) | no lung biopsies obtained | COPA (2) |
| IPH associated with eosinophilia (n=2) | failure to thrive, hepatomegaly unilateral atrophic kidney (no active disease), hypothyroidism, pollinosis |
anaemia (1) peripheral eosinophilia (2; max. 11%) eosinophilia in bone marrow aspirate (1) milk specific IgE positive (1) |
no (2) | immunoglobulins normal (2) | negative (2) | no lung biopsies obtained | COPA (2) | |
| IPH, no further classification (n=2) | none (2) | anaemia (2) | yes (1) no (1) |
immunoglobulins normal (1) | negative (2) | no lung biopsies obtained | COPA (2) |
In brackets: number of affected subjects.
ADHS: attention deficit hyperactivity disorder; ANA: antinuclear antibodies; ANCA: anti-neutrophil cytoplasmatic antibody; anti-GBM: anti-glomerular basement membrane; ASD: atrial septal defect; CMV: cytomegalovirus; GERD: gastro-esophageal reflux disease; HSP: Henoch Schönlein purpura; IgA/ IgG/ IgM/ IgE: immunglobuline A/ G/ M/ E; LDH: lactate dehydrogenase; MPO: myeloperoxidase; PDA: persistent ductus arteriosus; tTG: tissues transglutaminase.
Even when some diagnostics were missing, these patients were diagnosed as IPH. The referring centers considered their cases as sufficiently deep investigated by excluding known underlying causes or conditions for pulmonary hemorrhage.
In order to further differentiate the group of our patients with DAH without plausible etiology (IPH) we formed descriptive clusters among the different cases. These included patients with IPH and autoimmune features, eosinophilia, renal diseases, suspected undetermined systemic disease/ multi-organ involvement or no association (Table 2). Detailed characteristics of these patients, including relevant diagnostic findings, are depicted in Table 3.
IPH associated with autoimmune features.
Nine patients had autoimmune features with elevation of autoantibodies without meeting criteria for a specific autoimmune disease. As pediatric cut-off values for ANAs are frequently set from 1:80-1:100, we defined elevated ANAs as an ANA-titer exceeding 1:100 for this study (29, 30). For adults, ANA-cut-off values of 1:160 are more common (31).
IPH associated with eosinophilia.
Five patients had episodic, unexplained eosinophilia at the same time of pulmonary hemorrhage, defined as equal or more than 10% eosinophils in peripheral blood count. Two patients additionally showed eosinophilia in bone marrow aspirate.
IPH associated with renal disease.
Three patients had contemporaneously a chronic renal disease. Two had a glomerulonephritis, one showed a unilateral atrophic kidney of undefined cause and renal lithiasis. For one of these patients, evaluation for anti-GBM-autoantibodies was missing.
IPH associated with multi-organ involvement, suspected undetermined systemic disease.
An unknown or undefined multi-organ disease was present in three patients.
IPH, no further differentiation.
Overall, 20 of the patients without plausible cause for DAH (59%) could be assigned to one of the above phenotypic clusters. For 14 patients, a further differentiation was not possible (Table 2).
Evaluation of diagnostic suitability of the “Phenomizer” for DAH
After adding new terms and annotations and reorganizing pre-existing terms and branches (Figure 2, B–D), we tested if the program was able to suggest probable diagnoses if supplied with HPO-terms from DAH case-vignettes. We therefore extracted as many HPO-terms as possible manually from 29 case-vignettes (17 patients from our cohorts, 12 reports from specialized literature) and had the program calculate the rank of the correct diagnosis among all suggested diagnoses (Figure 2E). For patients with the diagnosis “Pulmonary hemosiderosis” (OMIM 178550, n=10), the correct diagnosis was always indicated within rank 1-30. For other diseases, ranking was very variable depending on the condition. There was no relevant difference in the ranking of the cases from our cohort and the ones retrieved from publications.
DISCUSSION
We presented a large cohort of 131 children with interstitial lung disease and DAH. In about 75% of the patients, an underlying plausible cause for DAH was identified. Finding a “plausible” cause refers to naming an underlying disease for DAH. Previously, cases that have not been assigned to a certain disease have often been described as simply “idiopathic”. By introducing the distinction between cases with a plausible cause and such ones where no plausible cause was found, our goal was to emphasize that “idiopathic” does not mean the patient already has a diagnosis; on the contrary, it is important to stress that these patients need continuous and repeated reevaluation to find the underlying cause of their symptoms.
We observed an overall high rate of mostly systemic conditions causing DAH within their common clinical spectrum. Obviously, some very high bleeding rates were due to low patient numbers of some diseases and selection of cases with pulmonary complications in the register. For almost all of these conditions, the occurrence of pulmonary hemorrhage was described in the literature before, often as case reports (1, 32–42). These results clearly suggest that in the diagnostic approach for DAH the broad range of specific conditions with ILD must be considered. That way, etiologic diagnosis may be identified in a large fraction of patients. Interestingly, compared with the French RespiRare® cohort, we did not observe cases with pulmonary hemorrhage and trisomy 21 (15, 43).
It is important to consider that these results may be biased to those with a likely underlying cause as we limited the analysis to disease categories B1, B4, Ax, Ay, as described in detail under “Methods” (18, 19). However, when electronically searching the remaining, excluded disease categories for DAH, we identified only a limited amount of cases in other categories (n = 4; Table 1, Supplemental Table 4).
We focused on the 34 patients (26%) with no plausible cause or underlying condition identified for their DAH. In this group, it is likely that there are at least three reasons for failure to identify an etiology of DAH: (1) the underlying disease entities are not yet fully understood or molecularly characterized, (2) the full expression of the phenotype is not yet established in the growing child, as the disease is developing or deteriorating over time, (3) the panel of diagnostic tests has not yet been completed. Standardized work-ups are essential for establishing a diagnosis in patients with DAH. Incomplete diagnostics may be due to not readily available check lists for a relatively rare condition (Supplemental Table 3), insufficient clinical knowledge on these rare to ultra-rare conditions among pediatricians or technical or organizational shortcomings, including the availability of lung biopsy options in small children or advanced genetic testing.
In the majority of the cases with no etiologic diagnosis identified, no lung biopsy was performed. Low numbers of lung biopsies were described earlier for cohorts of patients with IPH from France, Turkey or India (8, 9, 15). As lung biopsies are invasive, the procedure might not have been done in patients who are stable and thriving, if there was only a single bleeding event or if patients were already under treatment with steroids (44). However, as shown in Table 1, biopsy results contributed to clarification of an underlying disease in many cases (n = 46). We consider it reasonable to complete less invasive investigations which can be found in Supplemental Table 3 before obtaining a lung biopsy following a minimal invasive approach (as little as possible, as much as necessary). However, our data emphasize the yield of lung biopsies in the diagnostic process.
Interestingly, compared to the results of Fullmer et al. who found pulmonary capillaritis in eight of 23 lung biopsies of patients with pulmonary hemorrhage, pathologists less frequently identified pulmonary capillaritis in our population (3 out of 59 patients with lung biopsies; whenever biopsy material was available, this was confirmed by our reference pathologist) (32).
Repeated diagnostic evaluation of cases with DAH is an important task, in particular regarding the determination of autoantibodies. We have observed several patients in our cohort where the clinical symptoms preceded positive results for autoantibodies by several years (Supplemental Figure 1).
The remaining group of patients with suspected IPH was not homogeneous. We therefore further differentiated these patients by assigning them to descriptive clusters, which was possible for more than half of them. These clusters were meant as lead structures to help organizing possibly associated signs and symptoms and to foster further or repeated investigations.
The biggest specific cluster (9 of 34 patients) showed an association of IPH with autoimmune features respectively elevated autoimmune antibodies. The important role of autoimmunity in IPH was as well described in the French RespiRare® cohort, investigating 25 cases of children with IPH (15). The well-known and important interplay of immunological mechanisms in the development of DAH is additionally supported by the relevant number of autoimmune diseases in our study (6, 14).
The second biggest subset of patients (5 of 34 patients) showed IPH with contemporaneous eosinophilia. Eosinophils are already known to contribute to many chronic airway diseases via eosinophil-mediated inflammation, such as asthma, eosinophilic pneumonia or eosinophilic granulomatosis with polyangiitis (EGPA) (45, 46); eosinophils may thus be a clue for future etiologic research.
The well-known association between pulmonary and renal disease, also named pulmonary-renal syndrome, usually refers to the combination of diffuse alveolar hemorrhage and rapidly progressive glomerulonephritis caused mainly by small-vessel vasculitis (47, 48). It has mostly been described in adults, data in children is rare (48). In our cohort, three IPH-patients were clustered as IPH associated with renal disease. Interestingly, vasculitis was not observed in one patient’s lung biopsy. However, this patient and another patient intermittently had positive autoantibodies (ANAs, MPO-ANCAs), indicating the possibility of a vasculitic component of the disease.
A weakness of this study is that due to the multi-center patient collective and the retrospective character, duration of follow-up varied strongly and follow-up visits were not standardized in number and interval, and examinations were carried out following local standards which might not be uniform for all collaborating centers. Although this may mirror the typical clinical situation of children with DAH, further studies with sufficient follow-up and continuous update of investigations are necessary to refine the suggested classifications of children with DAH. There might be a bias for the study population as children were included in the databases only if clinicians suspected an interstitial lung disease and noticed DAH. Some patients/ disease entities might not have been recorded and thus were missed in this study which means that this analysis might not capture the entire spectrum of DAH in childhood.
The diagnostic software-tool Phenomizer was used on samples of case-vignettes from our cohort and for comparison also on published reports on DAH to assess applicability and benefit in clinical practice. The Phenomizer/ HPO are promising tools in digital medicine which are constantly expanded and optimized (49, 50). Here, we contributed to the field of pneumology with a focus on DAH-related issues. Given the relatively high occurence of DAH in defined systemic diseases (Table 1), non-pneumological annotations need to be improved in particular for these complex conditions for a satisfactory use of the program.
Here, were investigated a relatively large cohort of pediatric patients with ILD and DAH highlighting the broad disease spectrum that should be considered when managing this condition (7–9, 14–16). As COPA syndrome was described as one of the first monogenetically caused diseases strongly associated with pulmonary hemorrhage, we tested all patients with no identified underlying disease/ condition for disease-causing variants in COPA if consent and biomaterial were available. Despite of a high rate of genetic testing (73%), no variants were identified in these subjects suggesting that COPA syndrome accounts for a relevant, but small proportion of patients with DAH (n = 5; Table 1). Depending on concomitant symptoms and laboratory findings, genetics for other autoinflammatory/ interferonopathy disorders such as SAVI (stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy) should be considered as well (Supplemental Table 3) (51).
Overall, efforts to identify the underlying cause of DAH are of great relevance, as definite diagnosis enables clinicians not only to treat these patients more precisely using disease-specific therapeutic approaches but may also support estimation on patient’s prognosis. It enables disease-specific follow-up with description of response to standard treatment and/ or inclusion of patients in disease-specific clinical trials.
CONCLUSION
This study shows that the etiology behind pediatric DAH in children with interstitial lung disease is highly heterogeneous. Children presenting with pulmonary hemorrhage need precise and sometimes repeated diagnostics to reveal an underlying cause or condition; to plan complete diagnostic work-up, a check list (Supplemental Table 3) may be helpful. For patients with IPH, we suggest descriptive clusters (IPH associated with autoimmune features/ eosinophilia/ renal disease/ multi-organ involvement/ without further differentiation) which could guide future subgroup-specific research. Diagnostic software-tools like the Phenomizer (HPO) are promising, but need further development and especially input from all medical disciplines to increase diagnostic accuracy.
Supplementary Material
ACKNOWLEDGMENTS
We thank all patients and their families as well as all chILD EU collaborating physicians and centers. The chILD collaborators for this study were: Ahrens, Hamburg; Barker, Berlin; Bhatt, Nottingham; Brasch, Bielefeld; Bush, London; Chinedu, London; Cunningham, Edinburgh; Egermann, Munich; Firnhaber, Hamburg; Gibson, Glasgow; Grosse-Onnebrink, Essen; Hartmann, Erlangen; Hermon, Vienna; Herrmann, Sankt Augustin; Kaiser-Labusch, Bremen; Körner-Rettberg, Bochum; Kriebel, Göttingen; Krikovsky, Budapest; Kumpf, Tübingen; Lehmann, Aachen; Lange, Warsaw; López Andreu, Valencia; Marczak, Warsaw; Mayell, Liverpool; Mehl, Berlin; Mildenberger, Mainz; Nährlich, Giessen; Pawlita, Munich; Poplawska, Mainz; Prenzel, Leipzig; Renner, Neuburg/ Danube; Saianda, Lisbon; Schäfer, Hamburg; Schulze, Frankfurt; Seidenberg, Oldenburg; Stehling, Essen; Szepfalusi, Vienna; Tessmer, Worms; Teubner, Neuburg/ Danube; Zimmer, Giessen.
FINANCIAL SUPPORT
This study was supported by funding of German Research Foundation (DFG, Deutsche Forschungsgemeinschaft; DFG-Gr 970/9-1).
Footnotes
ETHICAL STATEMENT
The retrospective analysis of cases, the register study, data evaluation and reporting were approved by the Ethics committee of LMU Munich (EK 23-5-2011, EK 111-13, 20-329).
DECLARATION OF INTERESTS
There are no financial or personal relationships with other people or organizations that have influenced this work.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1.States LJ, Fields JM. Pulmonary hemorrhage in children. Semin Roentgenol. 1998;33(2):174–86. [DOI] [PubMed] [Google Scholar]
- 2.Godfrey S. Pulmonary hemorrhage/hemoptysis in children. Pediatr Pulmonol. 2004;37(6):476–84. [DOI] [PubMed] [Google Scholar]
- 3.Lara AR, Schwarz MI. Diffuse alveolar hemorrhage. Chest. 2010;137(5):1164–71. [DOI] [PubMed] [Google Scholar]
- 4.Susarla SC, Fan LL. Diffuse alveolar hemorrhage syndromes in children. Curr Opin Pediatr. 2007;19(3):314–20. [DOI] [PubMed] [Google Scholar]
- 5.Eber E, Midula F. ERS Handbook of Paediatric Respiratory Medicine: EUROPEAN RESPIRATORY SOCIETY; 2021. [Google Scholar]
- 6.Saha BK. Idiopathic pulmonary hemosiderosis: A state of the art review. Respir Med. 2021;176:106234. [DOI] [PubMed] [Google Scholar]
- 7.Saeed MM, Woo MS, MacLaughlin EF, Margetis MF, Keens TG. Prognosis in pediatric idiopathic pulmonary hemosiderosis. Chest. 1999;116(3):721–5. [DOI] [PubMed] [Google Scholar]
- 8.Kabra SK, Bhargava S, Lodha R, Satyavani A, Walia M. Idiopathic pulmonary hemosiderosis: clinical profile and follow up of 26 children. Indian Pediatr. 2007;44(5):333–8. [PubMed] [Google Scholar]
- 9.Kiper N, Gocmen A, Ozcelik U, Dilber E, Anadol D. Long-term clinical course of patients with idiopathic pulmonary hemosiderosis (1979-1994): prolonged survival with low-dose corticosteroid therapy. Pediatr Pulmonol. 1999;27(3):180–4. [DOI] [PubMed] [Google Scholar]
- 10.Chryssanthopoulos C, Cassimos C, Panagiotidou C. Prognostic criteria in idiopathic pulmonary hemosiderosis in children. Eur J Pediatr. 1983;140(2):123–5. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Martinez MU, Oostdam DAH, Abud-Mendoza C. Diffuse Alveolar Hemorrhage in Autoimmune Diseases. Curr Rheumatol Rep. 2017;19(5):27. [DOI] [PubMed] [Google Scholar]
- 12.Tryfon S, Papadopoulou E, Psarros G, Agrafiotis M, Saroglou M. Celiac disease and idiopathic pulmonary hemosiderosis: A literature review of the Lane-Hamilton Syndrome. Postgrad Med. 2022. [DOI] [PubMed] [Google Scholar]
- 13.Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48(2):73–87. [DOI] [PubMed] [Google Scholar]
- 14.Le Clainche L, Le Bourgeois M, Fauroux B, Forenza N, Dommergues JP, Desbois JC, et al. Long-term outcome of idiopathic pulmonary hemosiderosis in children. Medicine (Baltimore). 2000;79(5):318–26. [DOI] [PubMed] [Google Scholar]
- 15.Taytard J, Nathan N, de Blic J, Fayon M, Epaud R, Deschildre A, et al. New insights into pediatric idiopathic pulmonary hemosiderosis: the French RespiRare((R)) cohort. Orphanet J Rare Dis. 2013;8:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Luo F, Wang N, Song Y, Tao Y. Clinical characteristics and prognosis of idiopathic pulmonary hemosiderosis in pediatric patients. J Int Med Res. 2019;47(1):293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griese M, Seidl E, Hengst M, Reu S, Rock H, Anthony G, et al. International management platform for children’s interstitial lung disease (chILD-EU). Thorax. 2018;73(3):231–9. [DOI] [PubMed] [Google Scholar]
- 18.Griese M. Etiologic Classification of Diffuse Parenchymal (Interstitial) Lung Diseases. J Clin Med. 2022;11(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griese M, Irnstetter A, Hengst M, Burmester H, Nagel F, Ripper J, et al. Categorizing diffuse parenchymal lung disease in children. Orphanet J Rare Dis. 2015;10:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zander DS, Farver CF. Pulmonary Pathology: Elsevier; 2017. [Google Scholar]
- 21.Houser S, Mark EJ, Balis UJ. Lung Pathology: Humana Press; 2007. [Google Scholar]
- 22.Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185(9):1004–14. [DOI] [PubMed] [Google Scholar]
- 23.Salih ZN, Akhter A, Akhter J. Specificity and sensitivity of hemosiderin-laden macrophages in routine bronchoalveolar lavage in children. Arch Pathol Lab Med. 2006;130(11):1684–6. [DOI] [PubMed] [Google Scholar]
- 24.Kohler S, Oien NC, Buske OJ, Groza T, Jacobsen JOB, McNamara C, et al. Encoding Clinical Data with the Human Phenotype Ontology for Computational Differential Diagnostics. Curr Protoc Hum Genet. 2019;103(1):e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbosa-Gouveia S, Vazquez-Mosquera ME, Gonzalez-Vioque E, Hermida-Ameijeiras A, Sanchez-Pintos P, de Castro MJ, et al. Rapid Molecular Diagnosis of Genetically Inherited Neuromuscular Disorders Using Next-Generation Sequencing Technologies. J Clin Med. 2022;11(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baas M, Stubbs AP, van Zessen DB, Galjaard RH, van der Spek PJ, Hovius SER, et al. Identification of Associated Genes and Diseases in Patients With Congenital Upper-Limb Anomalies: A Novel Application of the OMT Classification. J Hand Surg Am. 2017;42(7):533–45 e4. [DOI] [PubMed] [Google Scholar]
- 27.Rae W, Ward D, Mattocks C, Pengelly RJ, Eren E, Patel SV, et al. Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin Genet. 2018;93(3):647–55. [DOI] [PubMed] [Google Scholar]
- 28.Muller T, Jerrentrup A, Schafer JR. [Computer-assisted diagnosis of rare diseases]. Internist (Berl). 2018;59(4):391–400. [DOI] [PubMed] [Google Scholar]
- 29.Aksu G, Gulez N, Azarsiz E, Karaca N, Kutukculer N. Determination of cut-off titers and agreement between immunofluorescence and immunoblotting methods for detecting antinuclear antibodies in children. J Clin Lab Anal. 2010;24(4):230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haslak F, Yildiz M, Altun I, Yilmaz G, Adrovic A, Sahin S, et al. Anti-nuclear antibody testing in children: How much is really necessary? Pediatr Int. 2021;63(9):1020–5. [DOI] [PubMed] [Google Scholar]
- 31.Kang I, Siperstein R, Quan T, Breitenstein ML. Utility of age, gender, ANA titer and pattern as predictors of anti-ENA and -dsDNA antibodies. Clin Rheumatol. 2004;23(6):509–15. [DOI] [PubMed] [Google Scholar]
- 32.Fullmer JJ, Langston C, Dishop MK, Fan LL. Pulmonary capillaritis in children: a review of eight cases with comparison to other alveolar hemorrhage syndromes. J Pediatr. 2005;146(3):376–81. [DOI] [PubMed] [Google Scholar]
- 33.Alexandre AT, Vale A, Gomes T. Diffuse alveolar hemorrhage: how relevant is etiology? Sarcoidosis Vasc Diffuse Lung Dis. 2019;36(1):47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Makhzoum JP, Grayson PC, Ponte C, Robson J, Suppiah R, Watts RA, et al. Pulmonary Involvement in Primary Systemic Vasculitides. Rheumatology (Oxford). 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang JC, Driver AG, Townsend CA, Kataria YP. Hemoptysis in sarcoidosis. Sarcoidosis. 1987;4(1):49–54. [PubMed] [Google Scholar]
- 36.Vece TJ, Watkin LB, Nicholas S, Canter D, Braun MC, Guillerman RP, et al. Copa Syndrome: a Novel Autosomal Dominant Immune Dysregulatory Disease. J Clin Immunol. 2016;36(4):377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong CY, Law GT, Wong KY, Shum TT, Yip HL. A case of Langerhans’ cell histiocytosis of the lung presenting with haemoptysis. Hong Kong Med J. 2004;10(1):49–51. [PubMed] [Google Scholar]
- 38.O’Keefe MC, Post MD. Pulmonary capillary hemangiomatosis: a rare cause of pulmonary hypertension. Arch Pathol Lab Med. 2015;139(2):274–7. [DOI] [PubMed] [Google Scholar]
- 39.Panda PK, Sriranga R, Kaur K, Sood R. Lane Hamilton Syndrome. Indian J Pediatr. 2018;85(8):699. [DOI] [PubMed] [Google Scholar]
- 40.Schuch LA, Forstner M, Rapp CK, Li Y, Smith DEC, Mendes MI, et al. FARS1-related disorders caused by bi-allelic mutations in cytosolic phenylalanyl-tRNA synthetase genes: Look beyond the lungs! Clin Genet. 2021;99(6):789–801. [DOI] [PubMed] [Google Scholar]
- 41.Boussouar S, Benattia A, Escudie JB, Gibault L, Capron F, Legrand A, et al. Vascular Ehlers-Danlos syndrome (vEDS): CT and histologic findings of pleural and lung parenchymal damage. Eur Radiol. 2021;31(8):6275–85. [DOI] [PubMed] [Google Scholar]
- 42.Pelizzo G, Collura M, Puglisi A, Pappalardo MP, Agolini E, Novelli A, et al. Congenital emphysematous lung disease associated with a novel Filamin A mutation. Case report and literature review. BMC Pediatr. 2019;19(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alimi A, Taytard J, Abou Taam R, Houdouin V, Forgeron A, Lubrano Lavadera M, et al. Pulmonary hemosiderosis in children with Down syndrome: a national experience. Orphanet J Rare Dis. 2018;13(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bush A, Cunningham S, de Blic J, Barbato A, Clement A, Epaud R, et al. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax. 2015;70(11):1078–84. [DOI] [PubMed] [Google Scholar]
- 45.Asano K, Ueki S, Tamari M, Imoto Y, Fujieda S, Taniguchi M. Adult-onset eosinophilic airway diseases. Allergy. 2020;75(12):3087–99. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki Y, Suda T. Eosinophilic pneumonia: A review of the previous literature, causes, diagnosis, and management. Allergol Int. 2019;68(4):413–9. [DOI] [PubMed] [Google Scholar]
- 47.Papiris SA, Manali ED, Kalomenidis I, Kapotsis GE, Karakatsani A, Roussos C. Bench-to-bedside review: pulmonary-renal syndromes--an update for the intensivist. Crit Care. 2007;11(3):213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.von Vigier RO, Trummler SA, Laux-End R, Sauvain MJ, Truttmann AC, Bianchetti MG. Pulmonary renal syndrome in childhood: a report of twenty-one cases and a review of the literature. Pediatr Pulmonol. 2000;29(5):382–8. [DOI] [PubMed] [Google Scholar]
- 49.Kohler S, Gargano M, Matentzoglu N, Carmody LC, Lewis-Smith D, Vasilevsky NA, et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021;49(D1):D1207–D17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haimel M, Pazmandi J, Heredia RJ, Dmytrus J, Bal SK, Zoghi S, et al. Curation and expansion of Human Phenotype Ontology for defined groups of inborn errors of immunity. J Allergy Clin Immunol. 2022;149(1):369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cetin Gedik K, Lamot L, Romano M, Demirkaya E, Piskin D, Torreggiani S, et al. The 2021 European Alliance of Associations for Rheumatology/American College of Rheumatology Points to Consider for Diagnosis and Management of Autoinflammatory Type I Interferonopathies: CANDLE/PRAAS, SAVI, and AGS. Arthritis Rheumatol. 2022;74(5):735–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.