

Abstract
Necroptosis contributes to hepatocyte death in nonalcoholic steatohepatitis (NASH), but the fate and roles of necroptotic hepatocytes (necHCs) in NASH remain unknown. We show here that the accumulation of necHCs in human and mouse NASH liver is associated with an up-regulation of the “don’t-eat-me” ligand CD47 on necHCs, but not on apoptotic hepatocytes, and an increase in the CD47 receptor SIRPα on liver macrophages, consistent with impaired macrophage-mediated clearance of necHCs. In vitro, necHC clearance by primary liver macrophages was enhanced by treatment with either anti-CD47 or anti-SIRPα. In a proof-of-concept mouse model of inducible hepatocyte necroptosis, anti-CD47 antibody treatment increased necHC uptake by liver macrophages and inhibited markers of hepatic stellate cell (HSC) activation, which is responsible for liver fibrogenesis. Treatment of two mouse models of diet-induced NASH with anti-CD47, anti-SIRPα, or AAV8-H1-shCD47 to silence CD47 in hepatocytes increased the uptake of necHC by liver macrophages and decreased markers of HSC activation and liver fibrosis. Anti-SIRPα treatment avoided the adverse effect of anemia found in anti-CD47–treated mice. These findings provide evidence that impaired clearance of necHCs by liver macrophages due to CD47-SIRPα up-regulation contributes to fibrotic NASH, and suggest therapeutic blockade of the CD47-SIRPα axis as a strategy to decrease the accumulation of necHCs in NASH liver and dampen the progression of hepatic fibrosis.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a rapidly growing health issue, affecting ~25% of the adult population worldwide and ~40% of the U.S. adult population (1–3). In 20 to 30% of subjects with NAFLD, nonalcoholic steatohepatitis (NASH) will develop, manifested by liver injury, necroinflammation, and fibrosis (1). NASH fibrosis in particular is associated with an increased risk of cirrhosis, liver failure, and hepatocellular carcinoma, resulting in a higher demand for liver transplantation (4, 5). Although several potential therapeutics are being investigated in clinical trials (6, 7), there are currently no U.S. Food and Drug Administration–approved drugs for NASH treatment due in part to an incomplete understanding of the pathophysiology of NASH, particularly the progression to NASH fibrosis.
The onset and progression of NASH are driven by multiple factors, including insulin resistance, hepatosteatosis, inflammation, oxidative and endoplasmic reticulum stress, and hepatocyte death (8–10). One theory holds that molecules leaking from uncleared dead cells, such as damage-associated molecular patterns, might activate hepatic macrophages and hepatic stellate cells (HSCs), triggering hepatic inflammation and fibrosis (11, 12). However, major gaps remain in our understanding of the roles of hepatocyte death in NASH. There is evidence that both apoptosis and necroptosis, among other types of cell death, are involved in NASH pathogenesis (13, 14). However, a recent clinical trial showed that suppressing hepatocyte apoptosis with the pan-caspase inhibitor, emricasan, failed to improve hepatic fibrosis (15). We therefore turned our attention to necroptosis, which has emerged as a likely pathogenic pathway in many diseases (16, 17), including human and experimental NASH (18–25). In necroptosis, the interaction of tumor necrosis factor (TNF) family death receptors with their ligands, for example, TNFα and FasL, in cells with impaired apoptosis signaling promotes the formation of a complex of receptor-interacting protein (RIP) kinases 1 and 3 (“necrosome”), which phosphorylate the mixed-lineage kinase domain-like (MLKL) pseudokinase. This process promotes MLKL oligomerization and plasma membrane association, causing membrane permeabilization, calcium efflux, and, ultimately, lytic cell death (26).
The consequences of cell death are linked in large part to the efficiency of their phagocytic clearance by macrophages and other cell types (27, 28). When cells die by apoptosis, macrophages carry out the process of efferocytosis to rapidly engulf and degrade dead cells, which prevents post-apoptotic necrosis, dampens inflammation, and activates tissue resolution pathways (27, 28). The inadvertent phagocytosis of live cells is inhibited by the expression of the “don’t-eat-me” signal CD47, which prevents cell uptake by macrophages owing to CD47-mediated activation of the macrophage SIRPα receptor (29, 30). In particular, CD47 binding to SIRPα activates Src homology region 2 domain-containing phosphatase-2 (SHP-2), which inhibits myosin accumulation at the cell surface and subsequent dead cell engulfment (29). In contrast to efferocytosis of apoptotic cells, a recent study indicated that macrophages could only ingest small pieces of necroptotic cells (31). The study showed that necroptotic cells express high amounts of CD47 and that blocking CD47 enabled macrophages to more fully engulf necroptotic cells (31).
Studies focusing on acute liver injury have suggested the beneficial anti-inflammatory and pro-resolving effects of efferocytosis by liver macrophages (11, 32, 33). However, the possible role of impaired efferocytosis in the pathogenesis of NASH remains largely unknown. In considering the importance of necroptosis in NASH and the idea that necroptotic cells may be poorly cleared by macrophages in other settings, we hypothesized that failed uptake of necroptotic hepatocytes (necHCs) by liver macrophages in NASH might promote NASH progression. The findings here support this hypothesis, focusing on the role of the CD47-SIRPα axis and its therapeutic implications.
RESULTS
CD47+ necHCs are increased in human and mouse NASH
To further substantiate previous data implicating necHCs in human NASH (18–21), we showed increases of the necroptosis markers p-MLKL and RIP3 by immunoblot (fig. S1A, first and third blots) and increased p-MLKL+ and RIP3+ hepatocytes by immunofluorescence microscopy in the livers of subjects with NASH compared with normal liver tissue (fig. S1, B and C). Macrophages in these livers did not show increased p-MLKL (fig. S1D). In contrast to p-MLKL, total MLKL was lower in human NASH liver compared with normal liver (fig. S1A, second blot). We then turned to the fructose-palmitate-cholesterol (FPC) diet–induced mouse NASH model, which shows parallels to human NASH, including weight gain, insulin resistance, shared NASH-relevant signaling pathways in hepatocytes and HSCs, and fibrosis (34–41). In this model, steatosis occurs by 8 weeks of diet and features of NASH, including early fibrosis, occur by 16 weeks (34). We found a progressive increase in p-MLKL+ cells in the liver during the 16-week FPC diet feeding period (fig. S1E). Furthermore, RIP3 was higher in the 16-week livers compared with control livers (fig. S1F, first blot). Because we were unable to obtain an anti–p-MLKL antibody for immunoblot analysis of mouse liver, we isolated a pelleted, or “insoluble,” form of MLKL, which has been used as an indicator of necrosome formation and necroptotic signal transmission in NASH liver (19). Immunoblot analysis showed a marked increase in insoluble MLKL (and soluble MLKL) in the 16-week FPC livers compared with control livers (fig. S1F, second and third blots). Furthermore, RIP3+ cells having the morphology of hepatocytes were detected by immunofluorescence staining of 16-week FPC livers, and these cells colocalized with terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL), a marker of cell death (fig. S1G). In contrast, <1% of NASH liver macrophages were p-MLKL positive by immunohistochemical staining (fig. S1H). To evaluate these findings in a different NASH model, we examined mice that were fed a high-fat choline-deficient l-amino–defined diet (HF-CDAA) (42). As in the FPC model, livers from HF-CDAA–fed versus chow-fed mice showed increased expression of p-MLKL, RIP3, and insoluble MLKL in the HF-CDAA cohort, and RIP3+ TUNEL+ cells were observed by immunofluorescence staining (fig. S1, I to K).
On the basis of a previous study showing that expression of the don’t-eat-me signaling molecule CD47 on necroptotic bone marrow–derived macrophages contributed to an impairment in their clearance by macrophages (31), we asked whether necHCs in NASH liver expressed CD47. CD47 protein was markedly increased in human NASH liver versus steatotic or normal liver (Fig. 1A and fig. S1A, fourth blot), and in these NASH livers, CD47 colocalized with p-MLKL+ cells versus p-MLKL− cells (Fig. 1B). In contrast, CD47 expression was low in p-MLKL–negative cells and in macrophages in human NASH liver (Fig. 1B and fig. S2A). To test this finding in experimental NASH, we started with mice expressing ZsGreen in hepatocytes and fed them either the FPC diet for 12 weeks or the HF-CDAA diet for 4 weeks. In both models, CD47 expression was observed on RIP3+ hepatocytes in NASH liver, whereas RIP3+ cells were rarely observed in control liver (Fig. 1, C and D). As with the human study, CD47 expression was not detected in liver macrophages (fig. S2B). We were unable to detect CD47 on hepatocytes that were positive for the apoptosis marker cleaved caspase-3 in human NASH liver (fig. S2C), indicating specificity for necHCs versus apoptotic hepatocytes. To examine these findings in a more controlled, cell-autonomous setting, we used a cross-linkable chimeric RIP3 construct (RIP3-2xFV) that, when dimerized by the compound AP20187, becomes activated and triggers necroptosis (43). We injected mice with AAV8-TBG-mRIP3-2xFV to transduce hepatocytes with the construct and then isolated hepatocytes from these mice. Upon exposure of these hepatocytes to AP20187 ex vivo, necroptosis was induced, and robust CD47 expression on these necHCs was observed (Fig. 1E). We also created an ex vivo model of apoptotic hepatocytes by isolating hepatocytes from anti-Fas (Jo2)–treated mice (44), and consistent with the data in fig. S2C, we saw no expression of CD47 on these cells (fig. S2D). These combined data provide further support for the occurrence of hepatocyte necroptosis in human and experimental NASH and reveal that CD47 expression is induced specifically in necHCs in human and mouse NASH liver and in primary hepatocytes upon induction of necroptosis ex vivo.
Fig. 1. CD47 + necHCs are increased in human and mouse NASH.
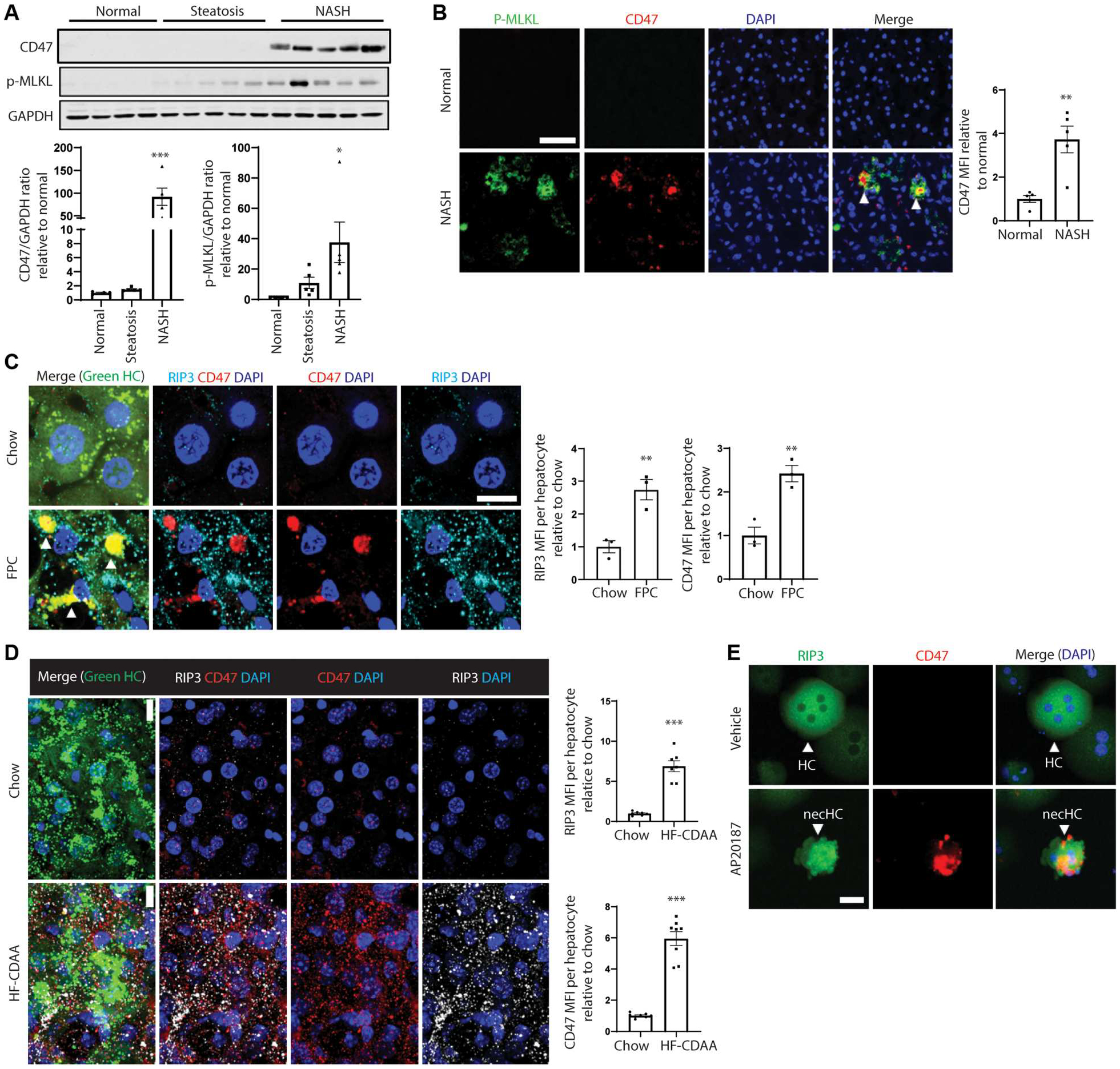
(A) Immunoblots of CD47 and p-MLKL in normal (n = 4), steatotic (n = 5), and NASH (n = 5) human livers, with data quantification (*P < 0.05; **P < 0.01 versus normal). (B) Immunofluorescence staining of human normal and NASH liver sections using anti–p-MLKL (green) and anti-CD47 (red). Arrowheads indicate p-MLKL and CD47 colocalization. Scale bar, 50 μm (**P < 0.01, n = 5 per group). (C) Male ZsGreen-inducible mice were injected with AAV8-TBG-Cre to label hepatocytes and were then fed the FPC NASH diet for 12 weeks. Liver sections (100 μm thickness) were immunostained with anti-CD47 (red) and anti-RIP3 (white). The green channel, which identifies hepatocytes, is shown in the merge image. Arrowheads indicate RIP3-CD47 colocalization. Scale bar, 10 μm. Data were quantified as mean fluorescence intensity (MFI) of CD47 and RIP3 per hepatocyte (**P < 0.01, n = 3 biological replicate hepatocytes per group). (D) Similar to (C), but the mice were fed the HF-CDAA diet for 4 weeks. Scale bar, 10 μm. ***P < 0.001, n = 7 to 8 biological replicate hepatocytes per group. (E) Primary hepatocytes isolated from AAV8-TBG-mRIP3-2xFV–injected mice were treated with vehicle control or AP20187 treatment (10 nM) to induce necroptosis ex vivo and then immunostained for RIP3 (green) and CD47 (red). Scale bar, 25 μm. necHC, necroptotic hepatocyte; HC, live hepatocyte. For all images, nuclei are stained with 4’,6-diamidino-2-phenylindole (DAPI; blue). All data are means ± SEM.
Blocking CD47 increases necHC uptake by liver macrophages and blocks markers of HSC activation in a mouse model of hepatocyte necroptosis
To study the functional implications of increased CD47 expression on necHCs ex vivo, we exposed either primary hepatocytes from AAV8-TBG-mRIP3-2xFV mice or hRIP3-2xFV–transduced human hepatocytes to AP20187 to generate necHCs. We then incubated these necHCs with mouse or human liver macrophages in the presence of anti-CD47 or immunoglobulin G (IgG) control. We used spinning-disk confocal microscopy and image analysis to quantify the volume of necHCs engulfed by the liver macrophages and found that anti-CD47 treatment promoted necHC uptake in both the mouse (Fig. 2A, bars 1 and 2) and human (Fig. 2B) systems. In contrast, the uptake of apoptotic hepatocytes by liver macrophages was initially high and was not further increased by anti-CD47 (Fig. 2A, bars 3 and 4).
Fig. 2. Blocking CD47 increases necHC uptake by liver macrophages and blocks markers of HSC activation in a mouse model of hepatocyte necroptosis.
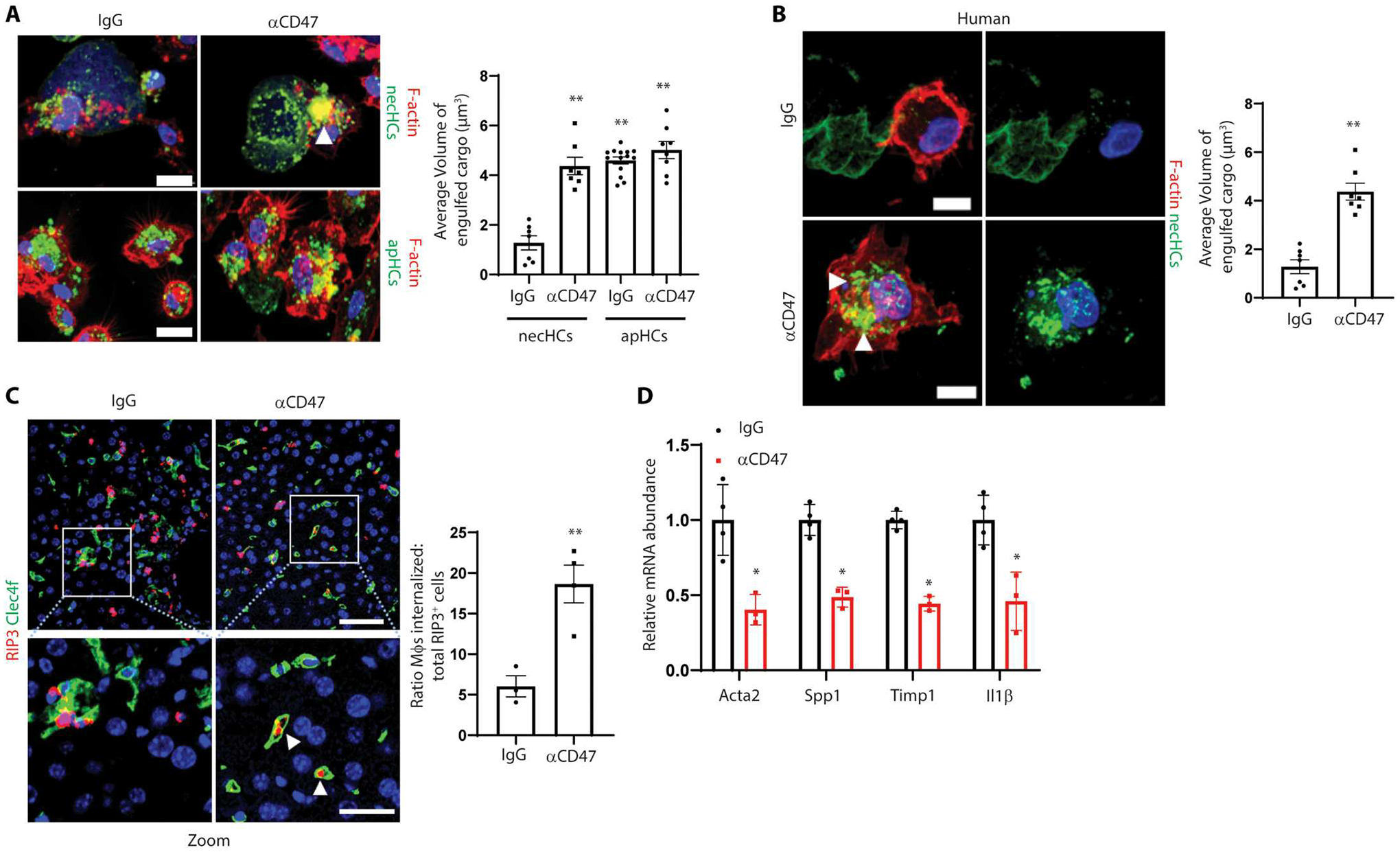
(A) Representative images of primary mouse macrophages stained for F-actin (red) after exposure to mouse necHCs (green) or apoptotic hepatocytes (apHCs; green) in the presence of IgG or anti-CD47 for 5 hours. Scale bars, 10 μm. Arrowheads indicate large volume of engulfed cargo. The data were quantified as the average volume of engulfed cargo (**P < 0.01 versus IgG necHC group, n = 6 biological replicate cells per group). (B) Two representative images of F-actin–labeled primary human macrophages (red) after incubation with human necHCs (green) in the presence of IgG or anti-CD47 for 5 hours. Arrowheads indicate large volume of engulfed cargo. Scale bars, 10 μm. The data were quantified as the average volume of engulfed cargo (**P < 0.01, n = 7 cells per group). (C) Male C57BL/6J mice were injected with AAV8-TBG-mRIP3-2xFV virus, and 10 days later, they were administered AP20187 to induce hepatocyte necroptosis plus either 16 hours of IgG or anti-CD47 treatment. The top images show costaining of Kupffer cells (KCs) using anti-CLEC4F (green) and RIP3 (red) in liver sections. Scale bar, 50 μm. Zoom image areas in the white square are shown in the bottom panel. Arrowheads depict macrophages (Mϕs) with internalized necHCs. Scale bar, 25 μm. The data were quantified as the ratio of macrophage-internalized necHCs to total necHCs (**P < 0.01, n = 3 to 4 mice per group). (D) The livers from (C) were quantified for mRNAs related to HSC activation and liver fibrosis (*P < 0.05, n = 3 to 4 mice per group). For all images, nuclei are stained with DAPI (blue). All data are means ± SEM.
To test the role of CD47 in necHCs uptake in vivo, AAV8-TBG-mRIP3-2xFV–transduced and control AAV8-TBG-LacZ–transduced mice were administered AP20187 to promote necroptosis in hepatocytes. As expected, AP20187 administration led to up-regulation of hepatocyte RIP3 and increased plasma alanine transaminase (ALT) in the mRIP3-2xFV–transduced mice but not in the AAV8-TBG-LacZ–transduced mice (fig. S3, A and B). Furthermore, the mRIP3-2xFV group showed increased expression of fibrosis-related mRNAs associated with HSC activation and liver fibrosis (fig. S3C), suggesting that necHCs promote HSC activation. We then injected the AP20187-treated mRIP3-2xFV–transduced mice with anti-CD47 or control IgG and observed an increase in the internalization of RIP3+ necHCs by liver macrophages (CLEC4F+) in the anti-CD47 cohort (Fig. 2C). Anti-CD47 did not alter hepatocyte necroptosis itself as assessed by measuring p-MLKL ex vivo in hepatocytes from mRIP3-2xFV–transduced mice treated with AP20187 to induce necroptosis and then incubated with anti-CD47 or IgG (fig. S3D). Anti-CD47 suppressed the expression of mRNAs associated with activated HSCs and liver fibrosis and the expression of the inflammatory marker Il1b (Fig. 2D). We then linked this finding to HSC activation using the ex vivo model in Fig. 2A, in which anti-CD47 promoted the uptake of necHCs by liver macrophages. The data show that, when conditioned medium (CM) from a coculture of liver macrophages with necHCs was transferred to primary HSCs, two mRNAs associated with HSC activation—Col3a1 and Timp1—and this effect was blocked when the coculture included anti-CD47 (fig. S3E). In summary, in a mouse model of induced hepatocyte necroptosis, antibody-mediated blockade of CD47 increased the uptake of necHCs by liver macrophages and decreased markers of HSC activation, and CM from a liver macrophage-necHC coculture induced HSC activation genes ex vivo in a CD47-dependent manner.
Anti-CD47 treatment and silencing hepatocyte CD47 increases necHC engulfment and mitigates hepatic fibrosis in experimental NASH
We next tested whether anti-CD47 treatment promotes necHC uptake by liver macrophages and suppresses NASH fibrosis in our two complementary mouse models of diet-induced NASH. In the FPC diet model, we intervened with anti-CD47 or IgG control after 8 weeks of diet, that is, after steatosis had developed, to test the hypothesis that anti-CD47 would attenuate the progression to fibrotic NASH. We first documented that anti-CD47 treatment did not affect body weight or liver weight (fig. S4A). As hypothesized, anti-CD47 treatment increased the ratio of macrophage-internalized RIP3+ cells to total RIP3+ cells and decreased total RIP3+ cells (Fig. 3A), consistent with increased uptake of necHCs by liver macrophages. Anti-CD47 lowered fibrosis (as measured by staining with both picrosirius red and aniline blue in Masson’s trichrome), alpha-smooth muscle actin (α-SMA)+ area, and expression of mRNAs associated with activated HSCs and liver fibrosis (Fig. 3, B to E). Furthermore, bulk RNA sequencing analysis of the livers from the two cohorts showed an overall decrease in collagen-related genes (false discovery rate–adjusted P < 0.05, fold change > 1.5) in the anti-CD47 versus IgG-treated cohorts (Fig. 3F). Plasma ALT was also decreased by anti-CD47 treatment (Fig. 3G). These beneficial changes occurred despite no differences between the two groups in steatosis, the expression of key genes associated with NASH inflammation, and NAFLD activity score (NAS) (fig. S4, B to D). The red blood cell number was ~15 to 20% lower in the anti-CD47 cohort (fig. S4E), consistent with human data showing that treatment with anti-CD47 lowers the red blood cell count in patients with cancer (45).
Fig. 3. Anti-CD47 treatment increases necHC engulfment and mitigates hepatic fibrosis in experimental NASH.
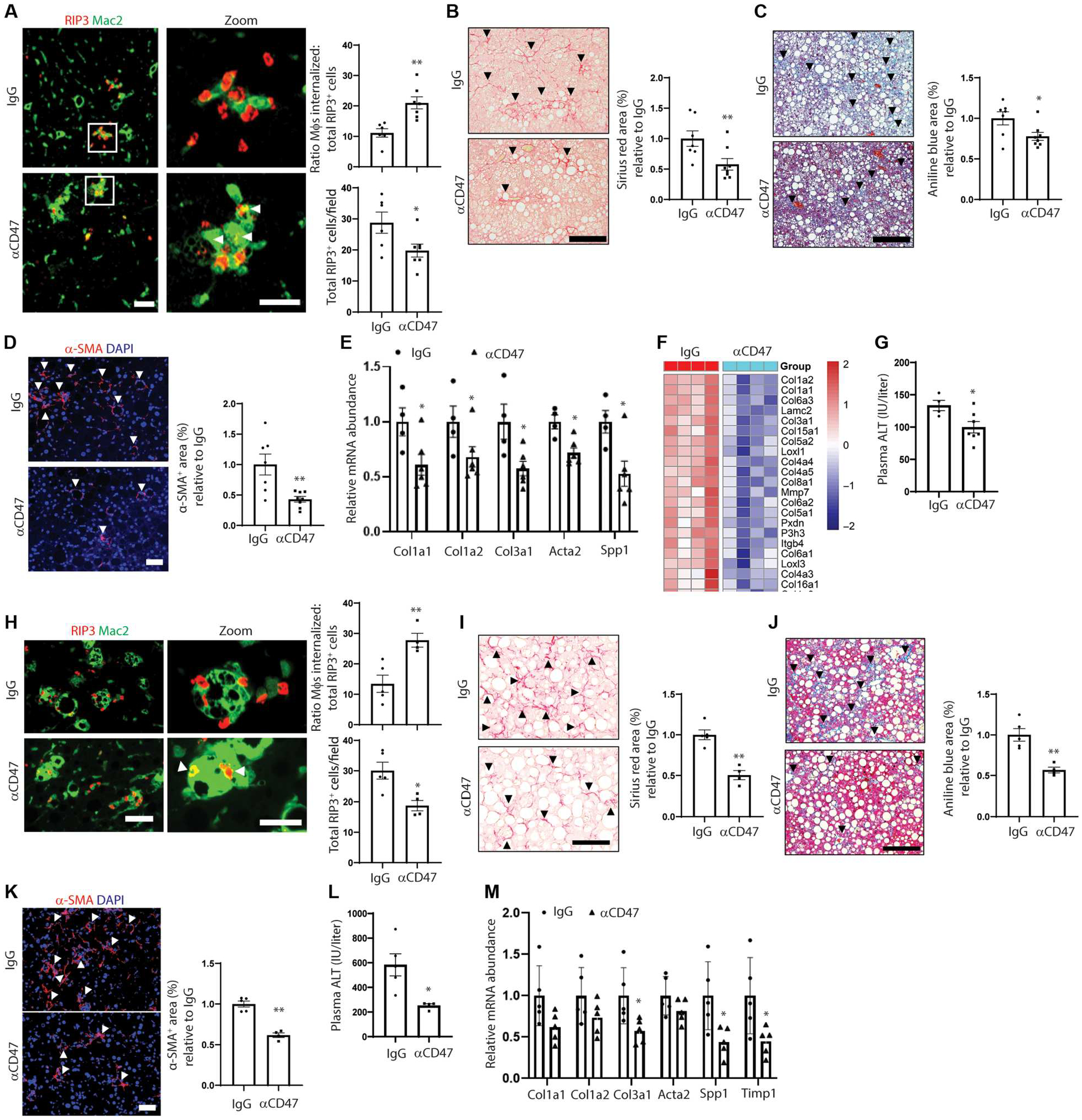
(A to G) Male C57BL/6J mice were fed the FPC NASH diet for 16 weeks and treated with IgG or anti-CD47 between weeks 8 and 16 (n = 7 to 8 mice per group). (A) Representative images of liver sections immunostained with anti-Mac2 to stain macrophages (green) and anti-RIP3 (red). Scale bar, 50 μm. Zoom images of the areas in the white square are shown in the right panel. Arrowheads depict macrophages with internalized necHCs. Scale bar, 25 μm. The data were quantified as the ratio of macrophage-internalized necHCs to total necHCs and total RIP3+ cells (*P < 0.05; **P < 0.01). (B) Staining and quantification of picrosirius red–positive area (arrowheads) (*P < 0.05). Scale bar, 100 μm. (C) Trichrome-stained images, with quantification of aniline blue–positive area, indicated by arrowheads (*P < 0.05). Scale bar, 100 μm. (D) Immunofluorescence staining and quantification of α-SMA–positive area (arrowheads) (**P < 0.01). Scale bar, 50 μm. (E) Quantification of mRNAs related to HSC activation and liver fibrosis (*P < 0.05) (n=4 to 7 mice). (F) Heatmap of genes involved in collagen formation in IgG versus anti-CD47 cohorts (n = 4 mice per group). (G) Plasma ALT activity (*P < 0.05) (n=4 to 7 mice). (H to L) Male C57BL/6J mice were fed the HF-CDAA NASH diet for 8 weeks and treated with IgG or anti-CD47 between 2 and 8 weeks (n = 4 to 5 mice per group). (H) Representative images of liver sections immunostained with anti-Mac2 to stain macrophages (green) and RIP3 (red) in liver sections. Scale bar, 100 μm. Zoom images of the areas in the white square are shown in the right panel. Arrowheads depict macrophages with internalized necHCs. Scale bar, 25 μm. The data were quantified as the ratio of macrophage-internalized necHCs to total necHCs and total RIP3+ cells (*P < 0.05; **P < 0.01). (I) Staining and quantification of picrosirius red–positive area (arrowheads) (*P < 0.05). Scale bar, 100 μm. (J) Trichrome-stained images, with quantification of aniline blue–positive area, indicated by arrowheads (**P < 0.01). Scale bar, 100 μm. (K) Immunofluorescence staining and quantification of α-SMA–positive area (arrowheads) (**P < 0.01). Scale bar, 50 μm. (L) Plasma ALT activity (*P < 0.05). (M) Mice were fed the HF-CDAA NASHdiet for 4 weeks and treated with IgG or anti-CD47 between weeks 2 and 4, followed by quantification of mRNAs related to HSC activation and liver fibrosis (*P < 0.05, n = 5 mice per group). All data are means ± SEM.
In the HF-CDAA diet–fed NASH model, anti-CD47 antibody treatment between weeks 2 to 8 did not affect body weight but slightly decreased liver weight (fig. S4F). Similar to the findings with the FPC NASH model, anti-CD47 treatment enhanced RIP3+ cell uptake by liver macrophages and decreased total RIP3+ cells (Fig. 3H). In this model, we could accurately assess apoptotic hepatocyte clearance, because cleaved caspase-3+ (apoptotic) cells are more prominent than in the FPC model. Consistent with the ex vivo data in Fig. 2A, we found that anti-CD47 treatment did not increase apoptotic cell clearance by liver macrophages (fig. S4G). Anti-CD47 decreased fibrosis in this model as assessed by staining for picrosirius red, aniline blue, and α-SMA+ area, and plasma ALT was also decreased by anti-CD47 treatment (Fig. 3, I to L). In mice fed the diet for 4 weeks, with IgG or anti-CD47 given between weeks 2 and 4, the expression of mRNAs associated with activated HSCs and liver fibrosis was decreased by anti-CD47 treatment (Fig. 3M). As with the FPC model, steatosis, inflammatory parameters, and NAS score were similar between the two cohorts (fig. S4, H to J). In this model, the red blood cell number was not changed by anti-CD47 cohort (fig. S4K), which may be due to the shorter period of treatment in this model versus the FPC model.
We conducted another experiment in which we treated steatotic mice with AAV8-H1-shCD47 (shCD47) to silence CD47 in hepatocytes (34, 46). Mice were fed the FPC diet for 8 weeks to develop simple steatosis and then treated with AAV8-H1-shCD47 (shCD47), or AAV8-H1-shControl (shCtr), for an additional 8 weeks. We first documented that AAV8-H1-shCD47 treatment caused an about 50% decrease in hepatocyte CD47 expression (Fig. 4A), without affecting body or liver weight (fig. S5A). As hypothesized, shCD47 increased the ratio of macrophage-internalized RIP3+ cells to total RIP3+ cells and decreased total RIP3+ cells (Fig. 4B), consistent with increased uptake of necHCs by liver macrophages. shCD47 lowered fibrosis (as measured by staining with both picrosirius red and aniline blue in Masson’s trichrome), α-SMA+ area, and expression of mRNAs associated with activated HSCs and liver fibrosis (Fig. 4, C to F). Plasma ALT was also decreased by shCD47 treatment (Fig. 4G). These beneficial changes occurred despite no differences between the two groups in either steatosis or the expression of key genes associated with NASH inflammation and NAS score (fig. S5, B to D), and AAV8-H1-shCD47 did not affect red blood cell number compared to the control group (fig. S5E). Together, these data demonstrate that neutralization of CD47 or silencing of hepatocyte CD47 in mice with diet-induced NASH facilitates necHC uptake by liver macrophages and suppresses progression to hepatic fibrosis.
Fig. 4. Hepatocyte CD47 silencing promotes necHC engulfment and attenuates hepatic fibrosis in experimental NASH.
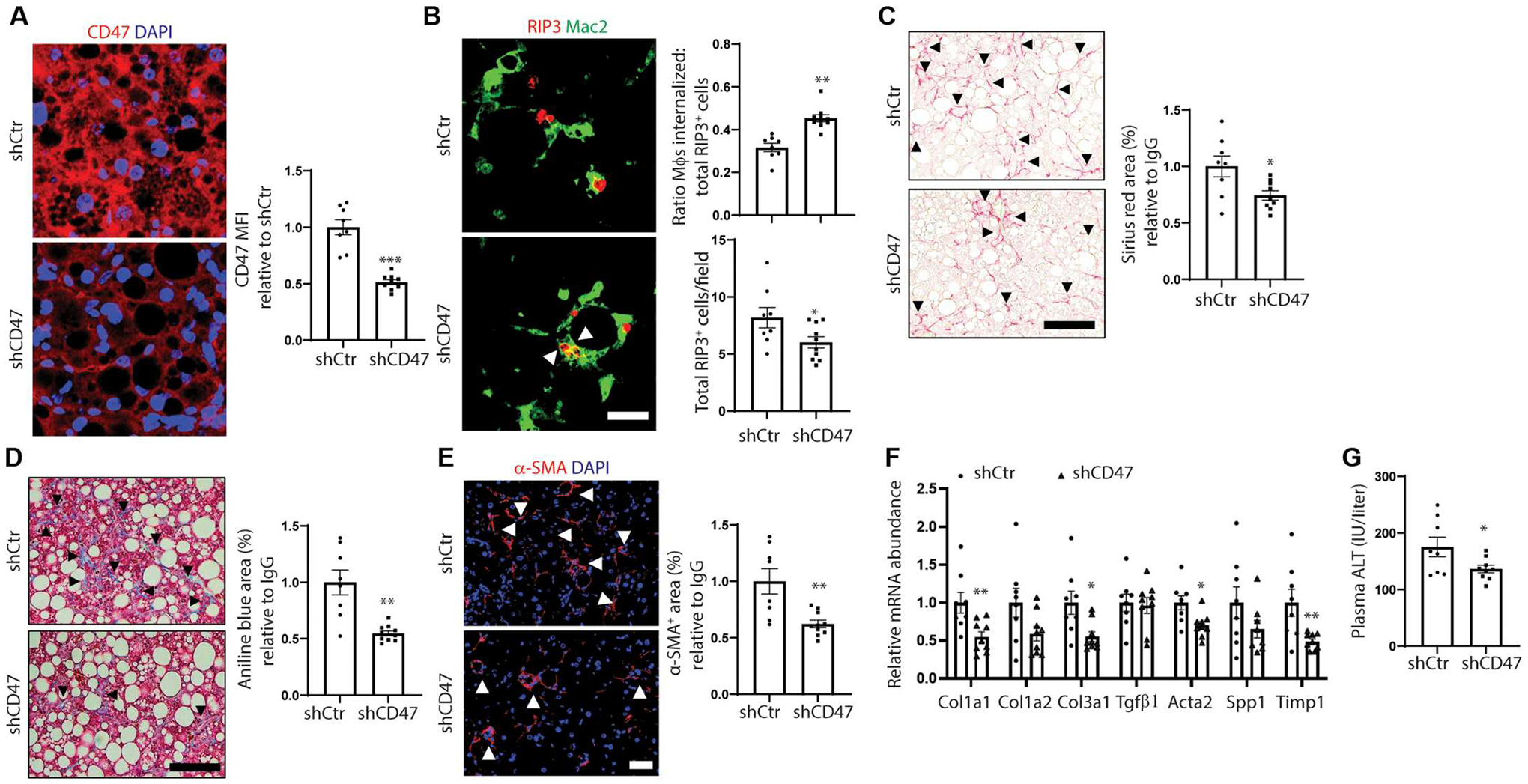
Male C57BL/6J mice were fed the FPC NASH diet for 16 weeks and treated with AAV8-H1-shControl (shCtr) or AAV8-H1-shCD47 (shCD47) between weeks 8 and 16 (n = 8 to 10 mice per group). (A) Relative hepatocyte CD47 MFI. Scale bar, 25 μm. (B) Representative images of liver sections immunostained with anti-Mac2 to stain macrophages (green) and anti-RIP3 (red). Scale bar, 50 μm. Arrowheads depict macrophages with internalized necHCs. The data were quantified as the ratio of macrophage-internalized necHCs to total necHCs and total RIP3+ cells (*P < 0.05; **P < 0.01). (C) Staining and quantification of picrosirius red–positive area (arrowheads) (*P < 0.05). Scale bar, 50 μm. (D) Trichrome-stained images, with quantification of aniline blue–positive area, indicated by arrowheads (*P < 0.05). Scale bar, 50 μm. (E) Immunofluorescence staining and quantification of α-SMA–positive area (arrowheads) (**P < 0.01). Scale bar, 50 μm. (F) Quantification of mRNAs related to HSC activation and liver fibrosis (*P < 0.05). (G) Plasma ALT activity (*P < 0.05).
SIRPα+ macrophages are increased in human and mouse NASH, and anti-SIRPα increases necHC engulfment and decreases hepatic fibrosis in experimental NASH
CD47 blocks the engulfment of apoptotic cells by interacting with macrophage SIRPα (29), but little is known about its regulation or role in impaired necroptotic cell uptake. In this context, we found that SIRPα expression in liver macrophages was markedly up-regulated in human NASH liver (Fig. 5, A and B), but not in human steatotic liver (fig. S6A). SIRPα expression was also increased in the livers of 16-week FPC diet–fed NASH mice compared with the normal livers of chow diet–fed mice or the steatotic livers of 4-week FPC diet–fed mice (Fig. 5, C and D, and fig. S6, B and C). The increase in SIRPα seen in FPC-NASH liver was found in both liver-resident Kupffer cells (KCs) (Mac2+Clec4f+) and infiltrated liver macrophages (Mac2+Clec4f−) (fig. S6D). Similar findings were also observed in the HF-CDAA mouse models of NASH (Fig. 5, E and F, and fig. S6, E to G).
Fig. 5. SIRPα+ macrophages are increased in human and mouse NASH.
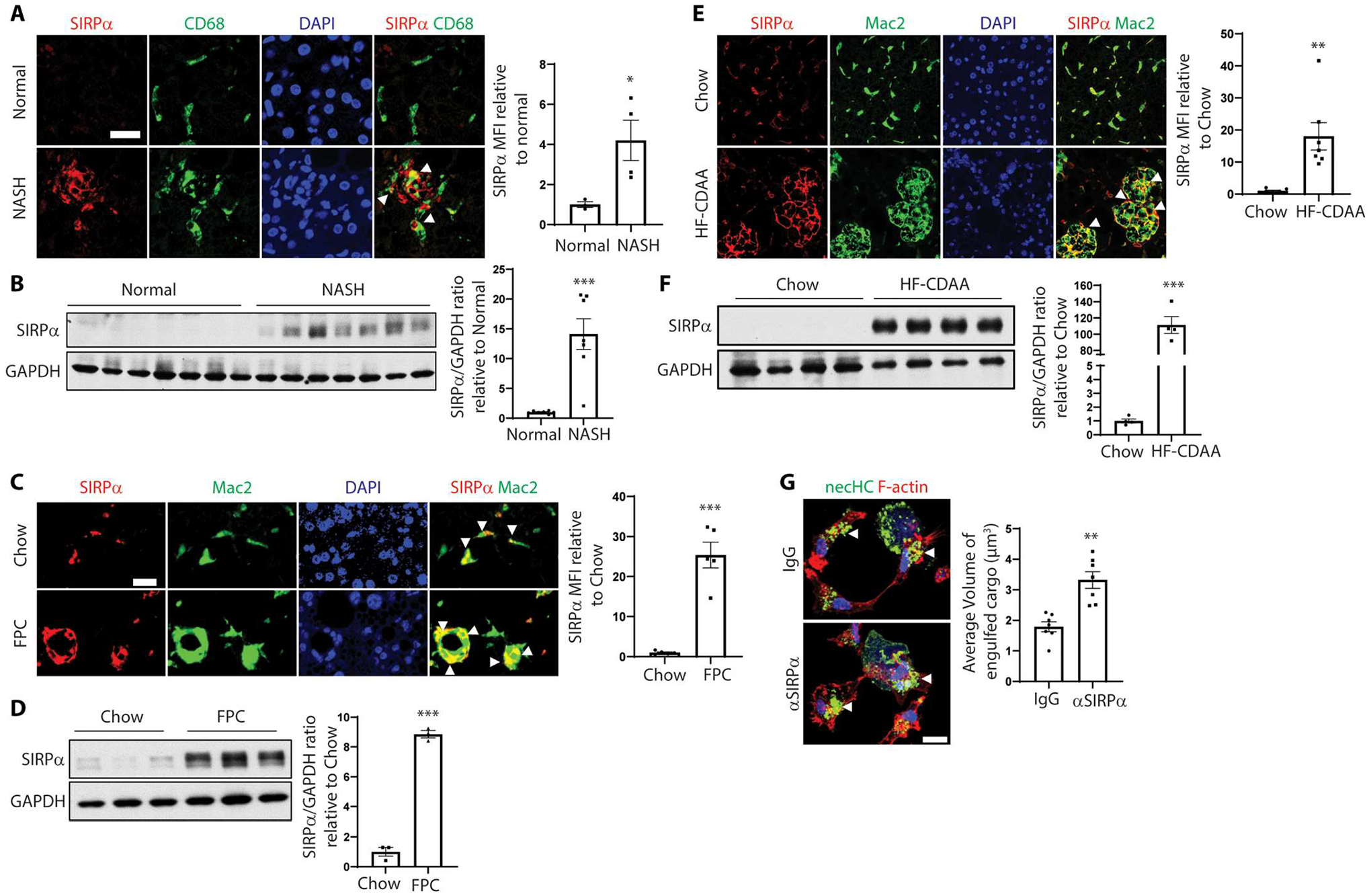
(A) Immunofluorescence staining of SIRPα (red) and CD68+ macrophage (green) in human normal or NASH liver sections. Arrowheads indicate SIRPα-macrophage colocalization. Scale bar, 25 μm. The data were quantified as MFI of SIRPα relative to the normal liver group (*P < 0.05, n = 4 per group). (B) Immunoblots of SIRPα in human livers of normal or patients with NASH, with data quantification (***P < 0.001, n = 7 livers per group). (C) Immunofluorescence staining of SIRPα (red) and Mac2+ macrophages (green) in liver sections from 16-week FPC-fed mice. Arrowheads indicate SIRPα-macrophage colocalization. Scale bar, 25 μm. The data were quantified as relative SIRPα MFI (***P < 0.001, n = 5 mice per group). (D) Immunoblots of SIRPα in livers from chow-fed or 16-week FPC-fed mice, with data quantification (***P < 0.001, n = 3 mice per group). (E) Immunofluorescence staining of SIRPα (red) and Mac2+ macrophages (green) in liver sections of 12-week HF-CDAA–fed mice. Arrowheads indicate SIRPα-macrophage colocalization. Scale bar, 50 μm. The data were quantified as SIRPα MFI (**P < 0.01, n = 5 mice per group). (F) Immunoblots of SIRPα in livers from chow-fed or 12-week HF-CDAA–fed mice, with data quantification (***P < 0.001, n = 4 mice per group). (G) Representative images of primary mouse macrophages stained for F-actin (red) after exposure to mouse necHCs (green) in the presence of IgG or anti-SIRPα. Scale bar, 10 μm. Arrowheads indicate large volume of engulfed cargo. The data were quantified as the average volume of engulfed cargo (**P < 0.01 versus IgG necHC group; n = 7 biological replicate cells per group). For all images, nuclei are stained with DAPI (blue). All data are means ± SEM.
To test whether anti-SIRPα could improve necHC uptake ex vivo, we incubated primary mouse liver macrophages with necHCs plus either anti-SIRPα or control IgG and observed an about twofold increase in necHC engulfment by anti-SIRPα (Fig. 5G). To test the role of anti-SIRPα in NASH, mice on the FPC diet for 8 weeks were treated with anti-SIRPα or IgG for an additional 8 weeks. Body weight was similar between the two cohorts, but liver weights were ~20% lower in anti-SIRPα–treated mice (fig. S7A). As with anti-CD47, anti-SIRPα increased RIP3+ cell internalization by liver macrophages and decreased total RIP3+ cells (Fig. 6A). Anti-SIRPα decreased hepatic fibrosis and α-SMA+ area (Fig. 6, B to D) and lowered the expression of mRNAs associated with HSC activation and liver fibrosis (Fig. 6E). Plasma ALT and hepatic Il1b and Mcp1 were also decreased by anti-SIRPα treatment (Fig. 6F and fig. S7B), but there was no change in hepatic steatosis or NAS score (fig. S7, C and D). Last, in contrast to the finding with anti-CD47, anti-SIRPα treatment did not decrease the red blood cell count (fig. S7E), which is consistent with the idea that this adverse effect can be avoided with anti-SIRPα versus anti-CD47 therapy (47).
Fig. 6. Anti-SIRPα increases necHC engulfment and decreases hepatic fibrosis in experimental NASH.
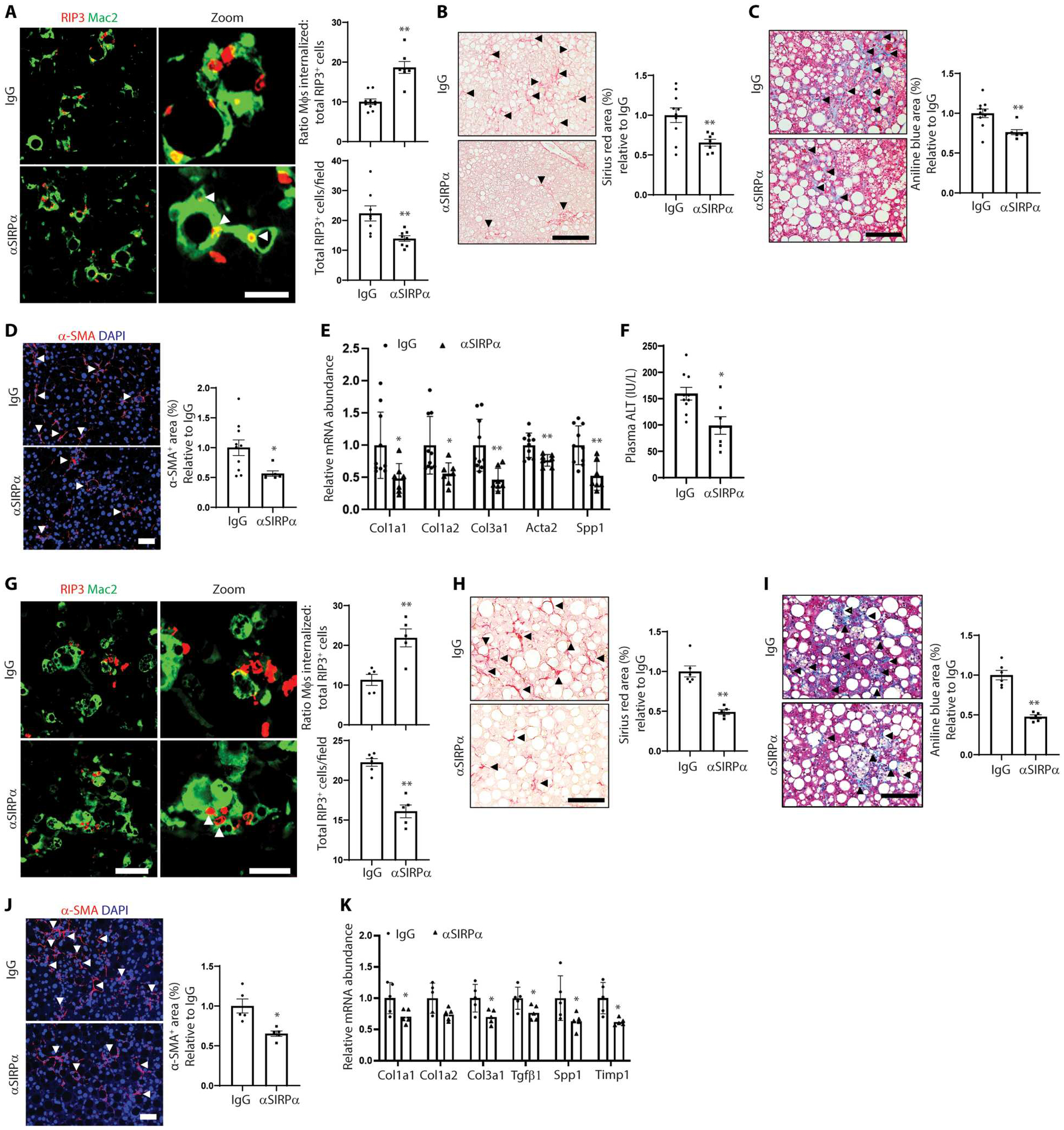
(A to F) Male C57BL/6J mice were fed the FPC NASH diet for 16 weeks and treated with IgG or anti-SIRPα between weeks 8 and 16 (n = 7 to 10 mice per group). (A) Representative images of liver sections immunostained with anti-Mac2 to stain macrophages (green) and RIP3 (red) in liver sections. Scale bar, 50 μm. Zoom images of the areas in the white square are shown in the right panel. Arrowheads depict macrophages with internalized necHCs. Scale bar, 25 μm. The data were quantified as the ratio of macrophage-internalized necHCs to total necHCs and total RIP3+ cells (**P < 0.01). (B) Staining and quantification of picrosirius red–positive area (arrowheads) (**P < 0.01). Scale bar, 100 μm. (C) Trichrome-stained images, with quantification of aniline blue–positive area, indicated by arrowheads (*P < 0.05). Scale bar, 100 μm. (D) Immunofluorescence staining and quantification of α-SMA–positive area (arrowheads) (*P < 0.05). Scale bar, 50 μm. (E) Quantification of mRNAs related to HSC activation and liver fibrosis (*P < 0.05; **P < 0.01). (F) Plasma ALT activity (**P < 0.01). (G to J) Male C57BL/6J mice were fed the HF-CDAA NASH diet for 8 weeks and treated with IgG or anti-SIRPα between 2 and 8 weeks (n = 5 to 6 mice per group). (G) Representative images of liver sections immunostained with anti-Mac2 to stain macrophages (green) and RIP3 (red) in liver sections. Scale bar, 50 μm. Zoom images of the areas in the white square are shown in the right panel. Arrowheads depict macrophages with internalized necHCs. Scale bar, 25 μm. The data were quantified as the ratio of macrophage-internalized necHCs to total necHCs and total RIP3+ cells (**P < 0.01). (H) Staining and quantification of picrosirius red–positive area (arrowheads) (**P < 0.01). Scale bar, 100 μm. (I) Trichrome-stained images, with quantification of aniline blue–positive area, indicated by arrowheads (**P < 0.01). Scale bar, 100 μm. (J) Immunofluorescence staining and quantification of α-SMA–positive area (arrowheads) (*P < 0.05). Scale bar, 50 μm. (K) Mice were fed the HF-CDAA NASH diet for 4 weeks and treated with IgG or anti-SIRPα between weeks 2 and 4, followed by quantification of mRNAs related to HSC activation and liver fibrosis (*P < 0.05, n = 5 mice per group). All data are means ± SEM.
Similar findings were observed in the HF-CDAA model. Anti-SIRPα treatment between weeks 2 and 8 did not affect body or liver weight (fig. S7F) but increased RIP3+ cell internalization by liver macrophages and decreased total RIP3+ cells (Fig. 6G), decreased hepatic fibrosis and α-SMA+ area (Fig. 6, H to J), and reduced mRNAs associated with HSC activation and liver fibrosis (Fig. 6K), all without affecting plasma ALT (fig. S7G). As with the anti-SIRPα–treated FPC NASH model, we found reductions in Il1b and Mcp1 expression without a change in steatosis or NAS score (fig. S7, H to J), and red blood cell number was not lowered by anti-SIRPα treatment (fig. S7K).
To test whether anti-SIRPα treatment was beneficial after NASH developed, we fed mice the HF-CDAA diet for 6 weeks, which results in steatosis and early fibrosis (fig. S8, A and B). The mice were continued on the diet for an additional 6 weeks, during which they were treated with anti-SIRPα or IgG for additional 6 weeks (Fig. 7A). As in the NASH prevention experiment, anti-SIRPα treatment did not affect body or liver weight, liver steatosis, NAS score, plasma ALT (fig. S7, C to F), or red blood cell number (fig. S7G). However, the treatment increased RIP3+ cell internalization by liver macrophages and decreased total RIP3+ cells (Fig. 7B). Anti-SIRPα decreased hepatic fibrosis and collagen deposition (Fig. 7, C and D) and reduced both α-SMA+ area and Opn+ area (Fig. 7, E and F). Thus, antibody-mediated neutralization of SIRPα administered either before or after early NASH develops promotes necHC engulfment by liver macrophages and dampens progression of NASH fibrosis.
Fig. 7. Anti-SIRPα increases necHC engulfment and ameliorates hepatic fibrosis in mice with established early NASH.
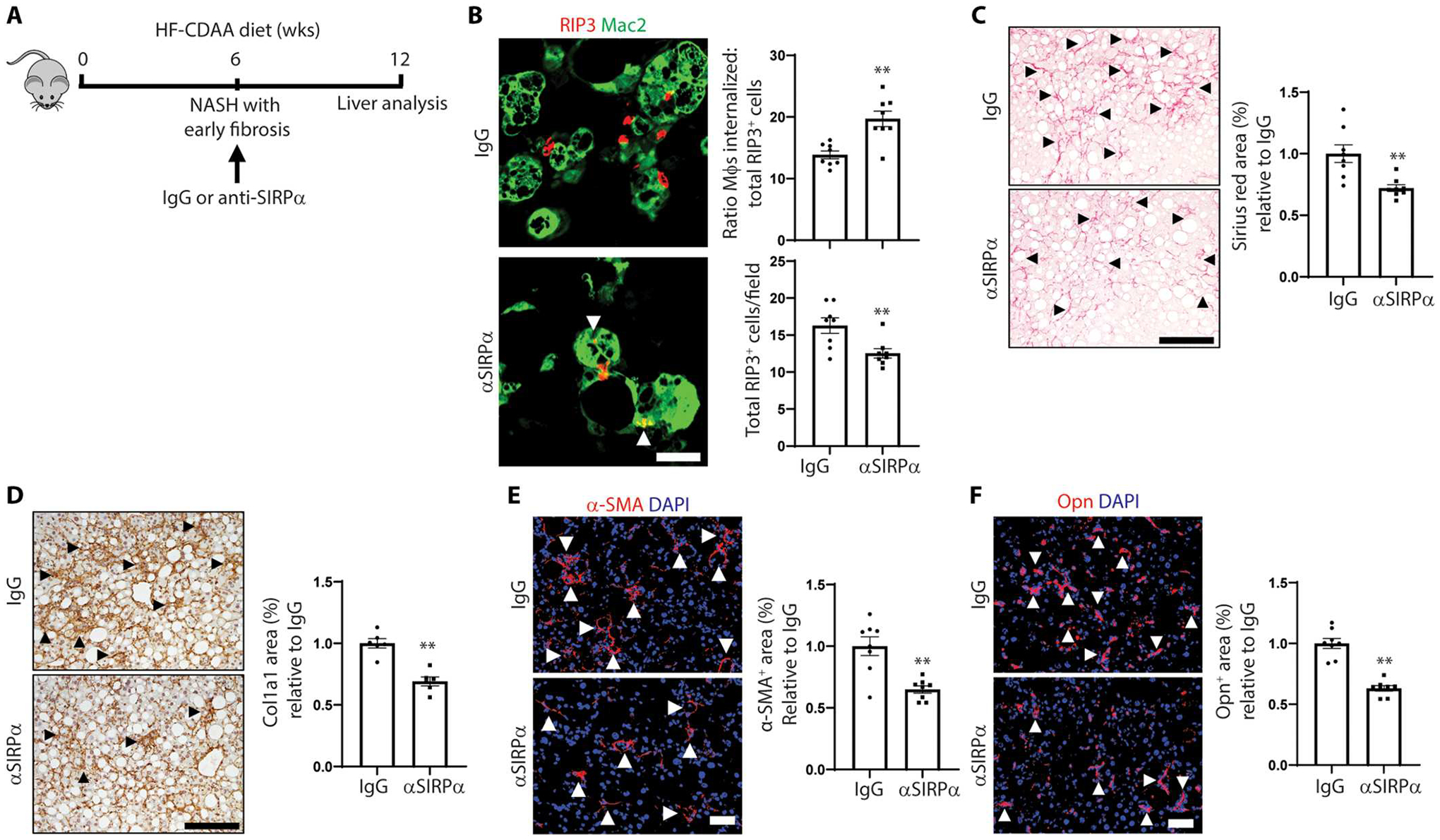
Male C57BL/6J mice were fed the HF-CDAA NASH diet for 12 weeks and treated with anti-SIRPα or IgG between weeks 6 and 12 (n = 6 to 8 mice per group). (A) Illustration of the experimental design. (B) Representative images of liver sections immunostained with anti-Mac2 to stain macrophages (green) and RIP3 (red) in liver sections. Arrowheads depict macrophages with internalized necHCs. Scale bar, 25 μm. The data were quantified as the ratio of macrophage-internalized necHCs to total necHCs and total RIP3+ cells (**P < 0.01). (C) Staining and quantification of picrosirius red–positive area (arrowheads) (**P < 0.01). Scale bar, 100 μm. (D) Liver sections immunostained with anti–collagen 1a1 (Col1a1), with quantification of Col1a1 area, indicated by arrowheads (**P < 0.01). Scale bar, 100 μm. (E) Immunofluorescence staining and quantification of α-SMA–positive area (arrowheads) (**P < 0.05). Scale bar, 50 μm. (F) Immunofluorescence staining and quantification of osteopontin (Opn)–positive area (arrowheads) (**P < 0.05). Scale bar, 50 μm. All data are means ± SEM.
DISCUSSION
Different types of cell death, such as apoptosis and necroptosis, have been implicated in NASH development and progression, and inhibition of cell death has been shown to ameliorate NASH in various murine experimental models (25, 48–51). However, when an anti-apoptosis strategy was tested in a human clinical trial through the administration of the caspase inhibitor emricasan, NASH end points, particularly fibrosis, were not improved (48). An alternative approach is to promote the clearance of dead hepatocytes by liver macrophages, which, based on general concepts (27, 28), would be predicted to prevent hepatocyte necrosis and possibly promote inflammation resolution. There is a paucity of information on the role of dead hepatocyte clearance in the setting of NASH. We have shown here that CD47 expression by necHCs and SIRPα expression by liver macrophages are up-regulated in human and mouse NASH liver and that blocking the CD47-SIRPα axis ex vivo and in experimental NASH promotes necHC uptake by liver macrophages, which is associated with decreases in markers of HSC activation and liver fibrosis during NASH progression.
Two recent reports deserve comment. First, an interesting study suggested that hepatocytes have an epigenetic mechanism to prevent the expression of RIP3 (52). However, using the techniques described in Materials and Methods, we, like others (18–20, 22–25), were able to show expression of RIP3 and p-MLKL in human and mouse NASH liver, and we documented specific expression in hepatocytes (18). Second, we showed that necHCs are the major cell type in NASH liver expressing CD47 and that silencing CD47 specifically in hepatocytes mitigated NASH. Nonetheless, it is possible that certain macrophage populations in NASH liver express lower levels of CD47 (53), which could, in theory, contribute to some of the effects of anti-CD47 on NASH progression.
Several studies have shown that genetic deficiency of RIP3 (19) or MLKL (54), or inhibition of necroptosis (25), improves experimental NASH. However, another study showed that RIP3 deficiency exacerbates high-fat diet (HFD)–induced liver inflammation, and the authors suggested that fibrosis may be exacerbated owing to a shift in hepatocyte death to apoptosis (55). Our work was not designed to address this question directly, but the associations of hepatocyte necroptosis with worsening NASH and of necHC clearance with NASH improvement are consistent with a pro-NASH effect of hepatocyte necroptosis. Nonetheless, we do not yet know how much of the improvement in NASH with CD47-SIRPα blockade is due to the ridding of necHCs versus a possible beneficial process activated in macrophages when they are stimulated to engulf necHCs.
Despite our analyses of human NASH liver and our ex vivo experiments with human necHCs and human macrophages, it is uncertain whether our anti-CD47/anti-SIRPα treatment studies in mouse NASH will translate to human NASH. In terms of previous studies supporting translational significance, CD47 deficiency was shown to protect against HFD-induced obesity and liver steatosis (56), and a recent preprint reported a beneficial effect of anti-CD47 on NASH in mice fed the so-called Amylin liver NASH diet (57), which induces NASH-like features. In contrast, another study showed that CD47 deficiency exacerbates chronic HFD-induced NASH (58). The reason for the differences among these studies remains to be determined, but none of them reported on the effects of the absence or neutralization of CD47 on dead cell clearance by liver macrophages or tested the effect of anti-SIRPα. This latter point is key for several reasons. First, the similar effects of anti-CD47 and anti-SIRPα on liver fibrosis implicate the CD47-SIRPα axis per se in the mechanism, for example, by enhancing dead cell engulfment versus some other effect of blocking CD47. Second, whereas systemic blocking of CD47 can result in anemia (45), as shown here, we did not observe anemia in mice treated with anti-SIRPα, which is consistent with previous reports (47, 59), or with specifically silencing of hepatocyte CD47. Third, anti-SIRPα but not anti-CD47 or hepatocyte CD47 silencing lowered markers of liver inflammation in NASH. Thus, although hepatocyte-specific CD47 silencing using small interfering RNA (siRNA) platforms in current clinical use (60–62) may have promise, these data suggest a possible benefit of targeting SIRPα for NASH treatment in the future. An anti-SIRPα monoclonal antibody (GS-0189) designed to treat non-Hodgkin’s lymphoma is undergoing a phase 1 clinical trial (NCT04502706), which could provide further information for clinical applications of targeting SIRPα by antibody treatment.
MATERIALS AND METHODS
Study design
The overall objective of this study was to investigate the role of liver macrophages and their interactions with necHCs in NASH progression, focusing on the hypothesis that increased CD47-SIRPα axis signaling prevents liver macrophages from efficiently engulfing necHCs, contributing to NASH progression. After confirming that the hepatocyte necroptosis occurred in both human and mouse NASH liver, we found the increased CD47 expression in necHCs in these livers. We then tested whether blocking CD47 by anti-CD47 antibody or using AAV8-H1-shCD47 could promote necHC engulfment in vitro and in vivo, using initially an AAV8-TBG-mRIP3-2xFV model, in which hepatocyte necroptosis can be induced by a cross-linker. We then tested anti-CD47 in NASH progression using two well-known NASH models, the FPC model and the HF-CDAA model (34, 42). Given the unfavorable effects of systemic anti-CD47 treatment and the therapeutic potential of using anti-SIRPα to target the CD47-SIRPα axis, we turned our attention to SIRPα and found increased SIRPα expression in NASH liver macrophages, which led us to test whether anti-SIRPα treatment could also enhance necHC uptake in vitro and in vivo and dampen NASH progression in the aforementioned NASH models.
Human liver specimens
Deidentified normal and NASH human liver specimens were acquired from the Liver Tissue Cell Distribution System at the University of Minnesota, and deidentified steatosis human liver specimens were acquired from the Cooperative Human Tissue Network at the University of Pennsylvania. The specimens were collected postmortem on the date of liver transplantation and preserved as frozen samples. The diagnostic information is included in table S1. Phenotypic and pathological characterizations were conducted by physicians and pathologists associated with the Liver Tissue Cell Distribution System. All human studies (protocol AAAU1906) were approved by the Columbia University Institutional Review Board and were conducted in accordance with National Institutes of Health and institutional guidelines for human subject research.
Experimental models
Male wild-type C57/BL6J mice (10 to 12 weeks old) were obtained from The Jackson Laboratory (Bar Harbor, ME, #000664) and were allowed to adjust to the housing environment in the Columbia University Irving Medical Center for 1 week before initiation of experiments. We randomly assigned mice to the experimental groups and were blinded for the NASH studies. The mice were fed either an FPC diet (Envigo, #TD. 160785 PWD) (34) for 16 weeks or HF-CDAA (Research Diets, #A06071302) (42) for 12 weeks to induce fibrotic NASH. Age-matched mice were fed a control diet (PicoLab Rodent Diet 20, #5053). For the FPC diet–induced NASH model, the mice were fed the diet for 8 weeks before intraperitoneal injection with either anti-CD47 antibody (200 μg per mouse; Bio X Cell, #BE0283, RRID: AB_2687806) or anti-SIRPα antibody (100 μg per mouse; Bio X Cell, #BE0322, RRID: AB_2819049) three times a week for an additional 8 weeks. Mice in the control group were treated with the same dose of isotype IgG. To silence CD47 in hepatocytes in the FPC model, the mice were fed the FPC diet for 8 weeks, followed by intravenous injection with AAV8-H1-shCD47 [3.0 × 1011 genome copies (gc) per mouse, 5’-CACCAgcagaactacttggattagttCTCGAGAACTAATCCAAGTAGTTCTGC-3’] or AAV8-H1-shControl virus as previously published (34). The mice were continued on the diet for an additional 8 weeks before analysis. For the HF-CDAA diet–induced NASH model, the mice were fed on HF-CDAA diet for 2 weeks, followed by intraperitoneal injection with either anti-CD47 or anti-SIRPα antibody (same dose as above) three times a week for an additional 6 weeks. Mice in the control group were treated with isotype-matched IgG. To investigate the treatment effects of anti-SIRPα in the HF-CDAA diet–induced NASH model, the mice were fed the diet for 6 weeks and then injected intraperitoneally with anti-SIRPα antibody (100 μg per mouse; Bio X Cell, #BE0322) or the same dose of isotype IgG three times a week for an additional 6 weeks. For one experiment, we treated inducible ZsGreen mice [B6.Cg-Gt(ROSA)26Sortm6(CAG-ZsGreen1)Hze/J; The Jackson Laboratory, #007906] with AAV8-TBG-Cre virus (2 × 1011 gc per mouse; Addgene, #107787-AAV8) and then fed the mice either the FPC or HF-CDAA NASH diet for 12 or 4 weeks, respectively. Livers were collected and fixed in 10% formalin for 24 hours before cutting into 100-μm sections using a vibratome. To establish necHCs in vivo, we generated AAV8-TBG-mRIP3-2XFv based on previous research (43). Male wild-type C57/BL6J mice were treated with the virus (2 × 1011 gc permouse) via intravenous injection (RIP3-transduced mice) before administering a homodimerizer (10 mg/kg; AP20187) to induce necroptosis in hepatocytes. In the anti-CD47 treatment experiment, RIP3-transduced mice were treated with anti-CD47 (200 μg per mouse; Bio X Cell, #BE0283, RRID: AB_2687806) or IgG 30 min before injection with AP20187, and the livers were treated 16 hours later. All in vivo data were biological replicates. All animals were housed in standard cages at 22°C in a 12-hour–12-hour light-dark cycle in a barrier facility, and experiments were performed by following the Guide for the Care and Use of Laboratory Animals. The protocol AABL-5573 was approved by the Institutional Animal Care and Use Committee at Columbia University.
Statistical analysis
All quantitative data are presented as means ± SEM. Statistical significance was determined using GraphPad Prism software (version 9.3). Shapiro-Wilk test was used to test normality. Statistical significance between two groups was analyzed using the Student’s t test if the data followed a normal distribution. Otherwise, a non-parametric Mann-Whitney U test was used. Multiple groups were analyzed using one-way analysis of variance (ANOVA) with Tukey post hoc testing. P values of <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments:
We thank C. Troy, C. Collier, and C. Chen for conducting vibratome sectioning of liver and R. Schwabe for helpful discussions. We thank the Liver Tissue Cell Distribution System at the University of Minnesota and University of Pittsburgh for providing human normal and NASH liver tissues. We acknowledge W. Liu (Columbia University) for excellent technical assistance and D. Ngai and M. Schilperoort for proofreading and editing the manuscript.
Funding:
This work was supported by NIH grants T32 5T32HL007343-42 (to B.D.G.); R00HL145131 (to A.Y.); R00HL130574, R01HL151611, and the Irving Scholars Program through UL1TR001873 (to H.Z.); R00DK115778 (to B.C.); and R01DK116620 (to I.T.). Other support included an American Liver Foundation Liver Scholar Award (to X.W.); an American Heart Association Postdoctoral Fellowship 20POST35130003 (to F.L.); an Irma T. Hirschl/Monique Weill-Caulier Trust Research Award (to B.C.); a PhRMA Foundation Research Starter Grant in Translational Medicine (to B.C.); an American Society of Hematology Fellow Scholar Award (to Z.Z.); an American Heart Association Career Development Award 19CDA34660043 (to Z.Z.); the Columbia University Digestive and Liver Diseases Research Center P30 grant supported by NIH (1P30DK132710-01); Italian Ministry of Health (Ministero della Salute), Ricerca Finalizzata RF-2016-02364358, Fondazione IRCCS Ca’ Granda “Liver BIBLE” (PR-0391) (to L.V.); Innovative Medicines Initiative 2 joint undertaking of European Union’s (EU) Horizon 2020 research and innovation programme and EFPIA EU Programme Horizon 2020 (under grant agreement no. 777377) for the Project LITMUS (to L.V.); and the EU programme “Photonics” under grant agreement 101016726 (to L.V.). Human liver samples were obtained from the Liver Tissue Cell Distribution System (University of Minnesota), which was funded by NIH contract HHSN276201200017C. Samples for histological analysis were prepared in the Molecular Pathology Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by NIH/NCI grant no. P30 CA013696. The microscopy work in this study was conducted in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by NIH grants P30CA013696 and S10RR025686.
Footnotes
Supplementary Materials
This PDF file includes:
Other Supplementary Material for this manuscript includes the following:
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. The raw RNA-sequencing data have been deposited at the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) under accession number GSE197111.
REFERENCES AND NOTES
- 1.Younossi ZM, Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol 70, 531–544 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE, Swain MG, Congly SE, Kaplan GG, Shaheen AA, The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol 7, 851–861 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Harrison SA, Gawrieh S, Roberts K, Lisanti CJ, Schwope RB, Cebe KM, Paradis V, Bedossa P, Aldridge Whitehead JM, Labourdette A, Miette V, Neubauer S, Fournier C, Paredes AH, Alkhouri N, Prospective evaluation of the prevalence of non-alcoholic fatty liver disease and steatohepatitis in a large middle-aged US cohort. J. Hepatol 75, 284–291 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Sanyal AJ, Van Natta ML, Clark J, Neuschwander-Tetri BA, Diehl A, Dasarathy S, Loomba R, Chalasani N, Kowdley K, Hameed B, Wilson LA, Yates KP, Belt P, Lazo M, Kleiner DE, Behling C, Tonascia J, Prospective study of outcomes in adults with nonalcoholic fatty liver disease. N. Engl. J. Med 385, 1559–1569 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, Haflidadottir S, Bendtsen F, Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149, 389–397.e10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qureshi K, Neuschwander-Tetri BA, The molecular basis for current targets of NASH therapies. Expert Opin. Investig. Drugs 29, 151–161 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Cardoso A-C, de Figueiredo-Mendes C, Villela-Nogueira CA, Sanyal AJ, New drugs for non-alcoholic steatohepatitis. Liver Int 40, 96–101 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Loomba R, Friedman SL, Shulman GI, Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwabe RF, Tabas I, Pajvani UB, Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158, 1913–1928 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buzzetti E, Pinzani M, Tsochatzis EA, The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65, 1038–1048 (2016). [DOI] [PubMed] [Google Scholar]
- 11.An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M, Kuramitsu K, Lai M, Popov YV, Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun 11, 2362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gautheron J, Gores GJ, Rodrigues CMP, Lytic cell death in metabolic liver disease. J. Hepatol 73, 394–408 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwabe RF, Luedde T, Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol 15, 738–752 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirsova P, Ibrabim SH, Gores GJ, Malhi H, Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res 57, 1758–1770 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, Sheikh MY, Schattenberg JM, Kayali Z, Zivony A, Sheikh A, Garcia-Samaniego J, Satapathy SK, Therapondos G, Mena E, Schuppan D, Robinson J, Chan JL, Hagerty DT, Sanyal AJ, A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J. Hepatol 72, 816–827 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Choi ME, Price DR, Ryter SW, Choi AMK, Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 4, e128834 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, Schadt EE, Quertermous T, Betancur P, Maegdefessel L, Matic LP, Hedin U, Weissman IL, Leeper NJ, CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, Kreggenwinkel K, Schneider AT, Bartneck M, Neumann UP, Canbay A, Reeves HL, Luedde M, Tacke F, Trautwein C, Heikenwalder M, Luedde T, A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med 6, 1062–1074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H, Castro RE, Rodrigues CM, Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci 129, 721–739 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Guo L, Zhang P, Chen Z, Xia H, Li S, Zhang Y, Kobberup S, Zou W, Lin JD, Hepatic neuregulin 4 signaling defines an endocrine checkpoint for steatosis-to-NASH progression. J. Clin. Invest 127, 4449–4461 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FK-M, Wu H, The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luedde T, Kaplowitz N, Schwabe RF, Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 147, 765–783.e4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gautheron J, Vucur M, Luedde T, Necroptosis in nonalcoholic steatohepatitis. Cell. Mol. Gastroenterol. Hepatol 1, 264–265 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyata T, Wu X, Fan X, Huang E, Sanz-Garcia C, Ross CKC, Roychowdhury S, Bellar A, McMullen MR, Dasarathy J, Allende DS, Caballeria J, Sancho-Bru P, McClain CJ, Mitchell M, McCullough AJ, Radaeva S, Barton B, Szabo G, Dasarathy S, Nagy LE, Differential role of MLKL in alcohol-associated and non-alcohol-associated fatty liver diseases in mice and humans. JCI Insight 6, e140180 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majdi A, Aoudjehane L, Ratziu V, Islam T, Afonso MB, Conti F, Mestiri T, Lagouge M, Foufelle F, Ballenghien F, Ledent T, Moldes M, Cadoret A, Fouassier L, Delaunay JL, Aït-Slimane T, Courtois G, Fève B, Scatton O, Prip-Buus C, Rodrigues CMP, Housset C, Gautheron J, Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J. Hepatol 72, 627–635 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Cai Z, Jitkaew S, Zhao J, Chiang H-C, Choksi S, Liu J, Ward Y, Wu L.-g., Liu Z-G, Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol 16, 55–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morioka S, Maueroder C, Ravichandran KS, Living on the edge: Efferocytosis at the interface of homeostasis and pathology. Immunity 50, 1149–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doran AC, Yurdagul A Jr., I. Tabas, Efferocytosis in health and disease. Nat. Rev. Immunol 20, 254–267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barclay AN, Van den Berg TK, The interaction between signal regulatory protein alpha (SIRPα) and CD47: Structure, function, and therapeutic target. Annu. Rev. Immunol 32, 25–50 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Tsai RK, Discher DE, Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol 180, 989–1003 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerlach BD, Marinello M, Heinz J, Rymut N, Sansbury BE, Riley CO, Sadhu S, Hosseini Z, Kojima Y, Tang DD, Leeper NJ, Spite M, Barroso M, Rayner KJ, Fredman G, Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ 27, 525–539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popov Y, Sverdlov DY, Bhaskar KR, Sharma AK, Millonig G, Patsenker E, Krahenbuhl S, Krahenbuhl L, Schuppan D, Macrophage-mediated phagocytosis of apoptotic cholan-giocytes contributes to reversal of experimental biliary fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol 298, G323–G334 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campana L, Starkey Lewis PJ, Pellicoro A, Aucott RL, Man J, O’Duibhir E, Mok SE, Ferreira-Gonzalez S, Livingstone E, Greenhalgh SN, Hull KL, Kendall TJ, Vernimmen D, Henderson NC, Boulter L, Gregory CD, Feng Y, Anderton SM, Forbes SJ, Iredale JP, The STAT3-IL-10-IL-6 pathway is a novel regulator of macrophage efferocytosis and phenotypic conversion in sterile liver injury. J. Immunol 200, 1169–1187 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, Chung RT, Lefkowitch JH, Schwabe RF, Tabas I, Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab 24, 848–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, Zheng Z, Kasikara C, Davra V, Meroni M, Chung RT, Rothlin CV, Schwabe RF, Blaner WS, Birge RB, Valenti L, Tabas I, Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab 31, 406–421.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, Shi H, Valenti L, Pajvani UB, Sandhu J, Infante RE, Radhakrishnan A, Covey DF, Guan KL, Buck J, Levin LR, Tontonoz P, Schwabe RF, Tabas I, Cholesterol stabilizes TAZ in hepatocytes to promote experimental non-alcoholic steatohepatitis. Cell Metab 31, 969–986.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Sommerfeld MR, Jahn-Hofmann K, Cai B, Filliol A, Remotti HE, Schwabe RF, Kannt A, Tabas I, A therapeutic silencing RNA targeting hepatocyte TAZ prevents and reverses fibrosis in nonalcoholic steatohepatitis in mice. Hepatol. Commun 3, 1221–1234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu J, Zhu C, Wang X, Kim K, Bartolome A, Dongiovanni P, Yates KP, Valenti L, Carrer M, Sadowski T, Qiang L, Tabas I, Lavine JE, Pajvani UB, Hepatocyte TLR4 triggers inter-hepatocyte Jagged1/Notch signaling to determine NASH-induced fibrosis. Sci. Transl. Med 13, eabe1692 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, Meroni M, Graham MJ, Yates KP, Diehl AM, Schwabe RF, Tabas I, Valenti L, Lavine JE, Pajvani UB, Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med 10, eaat0344 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang H-M, Fan S-J, Zhou X-R, Liu Y-J, Li X, Liao L-P, Huang J, Shi C-C, Yu L, Fu R, Fan J-G, Zhang Y-Y, Luo C, Li G-M, Histone deacetylase inhibitor givinostat attenuates nonalcoholic steatohepatitis and liver fibrosis. Acta Pharmacol. Sin 43, 941–953 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu W, Ye C, Cheng Q, Zhang X, Yao L, Li Q, Huang J, Liu Y, Zou Z, Wang H, Yan J, Zhu Y, Wang C, Ai D, Macrophage raptor deficiency-induced lysosome dysfunction exacerbates nonalcoholic steatohepatitis. Cell. Mol. Gastroenterol. Hepatol 7, 211–231 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei G, An P, Vaid KA, Nasser I, Huang P, Tan L, Zhao S, Schuppan D, Popov YV, Comparison of murine steatohepatitis models identifies a dietary intervention with robust fibrosis, ductular reaction, and rapid progression to cirrhosis and cancer. Am. J. Physiol. Gastrointest. Liver Physiol 318, G174–G188 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez DA, Green DR, Generation and use of chimeric rip kinase molecules to study necroptosis. Methods Mol. Biol 1857, 71–83 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Bajt ML, Lawson JA, Vonderfecht SL, Gujral JS, Jaeschke H, Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: Evidence for a postmitochondrial processing of caspase-8. Toxicol. Sci 58, 109–117 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, Colevas AD, O’Rourke T, Narayanan S, Papadopoulos K, Fisher GA, Villalobos V, Prohaska SS, Howard M, Beeram M, Chao MP, Agoram B, Chen JY, Huang J, Axt M, Liu J, Volkmer JP, Majeti R, Weissman IL, Takimoto CH, Supan D, Wakelee HA, Aoki R, Pegram MD, Padda SK, First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol 37, 946–953 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lisowski L, Dane AP, Chu K, Zhang Y, Cunningham SC, Wilson EM, Nygaard S, Grompe M, Alexander IE, Kay MA, Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 506, 382–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu J, Xavy S, Mihardja S, Chen S, Sompalli K, Feng D, Choi T, Agoram B, Majeti R, Weissman IL, Volkmer J-P, Targeting macrophage checkpoint inhibitor SIRPα for anti-cancer therapy. JCI Insight 5, e134728 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Witek RP, Stone WC, Karaca FG, Syn WK, Pereira TA, Agboola KM, Omenetti A, Jung Y, Teaberry V, Choi SS, Guy CD, Pollard J, Charlton P, Diehl AM, Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 50, 1421–1430 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, Zhang Z, Zhang D, Fan D, Nie Y, Shao F, Wu K, Liang J, Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol 68, 773–782 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M, Imai H, Yuet-Yin Kok C, Okochi H, Nakano H, Miyajima A, Tanaka M, Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis 10, 449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qi J, Kim JW, Zhou Z, Lim CW, Kim B, Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am. J. Pathol 190, 68–81 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Preston SP, Stutz MD, Allison CC, Nachbur U, Gouil Q, Tran BM, Duvivier V, Arandjelovic P, Cooney JP, Mackiewicz L, Meng Y, Schaefer J, Bader SM, Peng H, Valaydon Z, Rajasekaran P, Jennison C, Lopaticki S, Farrell A, Ryan M, Howell J, Croagh C, Karunakaran D, Schuster-Klein C, Murphy JM, Fifis T, Christophi C, Vincan E, Blewitt ME, Thompson A, Boddey JA, Doerflinger M, Pellegrini M, Epigenetic silencing of RIPK3 in hepatocytes prevents MLKL-mediated necroptosis from contributing to liver pathologies. Gastroenterology, (2022). [DOI] [PubMed] [Google Scholar]
- 53.Hayes BH, Tsai RK, Dooling LJ, Kadu S, Lee JY, Pantano D, Rodriguez PL, Subramanian S, Shin JW, Discher DE, Macrophages show higher levels of engulfment after disruption of cis interactions between CD47 and the checkpoint receptor SIRPα. J. Cell Sci 133, jcs237800 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu X, Poulsen KL, Sanz-Garcia C, Huang E, McMullen MR, Roychowdhury S, Dasarathy S, Nagy LE, MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J. Hepatol 73, 616–627 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roychowdhury S, McCullough RL, Sanz-Garcia C, Saikia P, Alkhouri N, Matloob A, Pollard KA, McMullen MR, Croniger CM, Nagy LE, Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology 64, 1518–1533 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maimaitiyiming H, Norman H, Zhou Q, Wang S, CD47 deficiency protects mice from diet-induced obesity and improves whole body glucose tolerance and insulin sensitivity. Sci. Rep 5, 8846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gwag T, Ma E, Zhou C, Wang S, Anti-CD47 antibody treatment attenuates liver inflammation and fibrosis in experimental non-alcoholic steatohepatitis models. Liver Int 42, 829–841 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tao HC, Chen KX, Wang X, Chen B, Zhao WO, Zheng Y, Yang YG, CD47 deficiency in mice exacerbates chronic fatty diet-induced steatohepatitis through Its role in regulating hepatic inflammation and lipid Metabolism. Front. Immunol 11, 148 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yanagita T, Murata Y, Tanaka D, Motegi S-I, Arai E, Daniwijaya EW, Hazama D, Washio K, Saito Y, Kotani T, Ohnishi H, Oldenborg P-A, Garcia NV, Miyasaka M, Ishikawa O, Kanai Y, Komori T, Matozaki T, Anti-SIRPα antibodies as a potential new tool for cancer immunotherapy. JCI insight 2, e89140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, Perez J, Chiesa J, Warrington S, Tranter E, Munisamy M, Falzone R, Harrop J, Cehelsky J, Bettencourt BR, Geissler M, Butler JS, Sehgal A, Meyers RE, Chen Q, Borland T, Hutabarat RM, Clausen VA, Alvarez R, Fitzgerald K, Gamba-Vitalo C, Nochur SV, Vaishnaw AK, Sah DWY, Gollob JA, Suhr OB, Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med 369, 819–829 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Zhang X, Goel V, Robbie GJ, Pharmacokinetics of patisiran, the first approved RNA interference therapy in patients with hereditary transthyretin-mediated amyloidosis. J. Clin. Pharmacol 60, 573–585 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ray KK, Wright RS, Kallend D, Koenig W, Leiter LA, Raal FJ, Bisch JA, Richardson T, Jaros M, Wijngaard PLJ, Kastelein JJP; ORION-10 and ORION-11 Investigators, Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N. Engl. J. Med 382, 1507–1519 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ, Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Hasui N, Sakaguchi K, Ogawa T, Sakamoto Y, Shimizu T, In vitro ballooned hepatocytes can be produced by primary human hepatocytes and hepatic stellate cell sheets. Sci. Rep 12, 5341 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Charni-Natan M, Goldstein I, Protocol for primary mouse hepatocyte isolation. STAR Protoc 1, 100086 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T, Green DR, Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol. Cell 61, 589–601 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C, Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Soneson C, Love MI, Robinson MD, Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu A, Ibrahim JG, Love MI, Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 35, 2084–2092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, Wadi L, Meyer M, Wong J, Xu C, Merico D, Bader GD, Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc 14, 482–517 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. The raw RNA-sequencing data have been deposited at the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) under accession number GSE197111.