

Abstract
RNA methylation has emerged recently as an active research domain to study post-transcriptional alteration in gene expression regulation. Various types of RNA methylation, including N6-methyladenosine (m6A), are involved in human disease development. As a newly developed sequencing biotechnology to quantify the m6A level on a transcriptome-wide scale, MeRIP-seq expands RNA epigenetics study in both basic and clinical applications, with an upward trend. One of the fundamental questions in RNA methylation data analysis is to identify the Differentially Methylated Regions (DMRs), by contrasting cases and controls. Multiple statistical approaches have been recently developed for DMR detection, but there is a lack of a comprehensive evaluation for these analytical methods. Here, we thoroughly assess all eight existing methods for DMR calling, using both synthetic and real data. Our simulation adopts a Gamma–Poisson model and logit linear framework, and accommodates various sample sizes and DMR proportions for benchmarking. For all methods, low sensitivities are observed among regions with low input levels, but they can be drastically boosted by an increase in sample size. TRESS and exomePeak2 perform the best using metrics of detection precision, FDR, type I error control and runtime, though hampered by low sensitivity. DRME and exomePeak obtain high sensitivities, at the expense of inflated FDR and type I error. Analyses on three real datasets suggest differential preference on identified DMR length and uniquely discovered regions, between these methods.
Keywords: Epigenomics, RNA Methylation, N6-methyladenosine, MeRIP-seq, Differentially Methylated Regions
INTRODUCTION
RNA chemical modification is a crucial epigenetic mechanism for post-transcriptional regulation. In eukaryotes, over 60% of total RNA modifications are RNA methylations. RNA methylations regulate gene expression through different pathways including RNA stability [1], translation and slicing [2], and have been actively involved in the studies of physiology [3, 4] and oncology [5, 6]. Common methylation modifications include 5-methylcytosine (m5C), 7-methylguanosine (m7G), N1-methyladenosine (m1A) and N6-methyladenosine (m6A). Among these, m6A discovered in 1974 is the major type of methylation in messenger RNA (mRNA) [7]. Over the past few years, studies on m6A modifications have been gaining tremendous popularity in biological research due to its association with mechanisms of complex human diseases and pathogenesis of cancer [2, 8, 9].
As the most prevalent modification type in eukaryotic mRNA, m6A is predominantly enriched in the promoter region, stop codon and DRACH (D=G/A/U, R=G/A, H=A/U/C) motif [10, 11]. As a reversible and dynamic event, m6A is catalyzed by methyltransferases (METTL3, METTL4), read by m6A binding proteins (YTHDF1, IGF2BP1) and demethylated by demethylases (FTO, ALKBH5), which are often referred to as ‘writer’, ‘reader’ and ‘eraser’ [8]. Many human diseases, including cancer, neurological conditions, cardiovascular diseases or delays in embryonic development, have been reported to correlate with abnormal levels of m6A modification enzymes. For example, the overexpression of the m6A demethylase FTO promotes angiogenesis, decreases fibrosis and improves cardiac contractile performance [12]. It has also been discovered that increased m6A methylation level is associated with cardiac hypertrophy [13]. In cancer progression, m6A can play a regulatory role in tumor growth by controlling the expression of oncogenes or antioncogenes [6, 14, 15]. In addition, the aberrant expression of m6A enzymes may serve as a biomarker for cancer diagnosis, prognosis and therapy [5].
The study of m6A has been facilitated by the development of m6A-specific methylated RNA immunoprecipitation with next-generation sequencing (MeRIP-seq), which offers the first opportunity to measure m6A modification on a transcriptome-wide scale. An overview of a typical MeRIP-seq experiment, along with its data analysis workflow, is illustrated in Figure 1. Briefly, RNA samples are first fragmented and then immunoprecipitated (IP) by anti-m6A antibodies. Those antibodies will enrich RNA fragments with m6A methylation. Next, the IP samples are sequenced by Next-Generation Sequencing (NGS) to obtain the mapped reads. To provide a reference for IP samples, input control (pre-IP) mRNA fragments are also subject to high-throughput sequencing [11]. Then, MeRIP-seq generates paired input control and IP profiles for each sample. Here, the IP profile stores the methylated RNA sequencing read counts, while the input control profile stores the basal gene expression read counts. The relative abundance of these two counts reflects the m6A methylation level information, which often serves as the input of computational methods in m6A data analysis. As outlined in Figure 1, an important goal of MeRIP-seq data analysis is to find Differentially Methylated Regions (DMRs) across phenotypes-of-interest (e.g. cases versus controls). DMR identification helps reveal biomarkers, functional regions and pathways associated with the disease or development condition.
Figure 1.

Schematic overview of MeRIP-seq experiment and DMR detection. MeRIP-seq generates paired IP and control input data from RNA samples. Sequencing reads are aligned to the reference genome and Differentially Methylated Regions (DMRs) are then identified by recently developed statistical methods. Their core statistical models and features are listed at the inner circles of the pie chart. Called DMR can then be analyzed in downstream for peak annotation, biomarker discovery, pathway and gene ontology analysis.
With the expanding usage of MeRIP-seq over the last couple of years, several computational methods have been developed to detect DMRs. All available methods, to our best knowledge, have been compiled and presented in Table 1 with methodology details. As the first published tool, exomePeak [16] applies a Fisher’s exact test (FET) on normalized read counts from input control and IP samples between two experimental conditions. It ignores the heterogeneity across biological replicates because it uses the pooled read counts from all replicates. Later, MeTDiff [18] and FET-HMM [17] were introduced as improved tools over exomePeak. MeTDiff assumes beta-binomial distributions and compares methylation levels across conditions by a Likelihood Ratio Test (LRT). However, MeTDiff does not appropriately address the technical variation in sequencing depth. FET-HMM adopts a modified version of FET and uses the binary decisions from FET as the observation of differential methylation states. It then fits a Hidden Markov Model (HMM) on small bins within the detected methylation regions, to incorporate the dependency along the genome. However, FET-HMM merges replicates in each group for testing, neglecting the within-group variability among biological replicates. In 2016, DRME [19] was developed to address this issue, especially under small sample size scenarios. DRME assumes negative binomial models for both IP and input control count data, and uses input control data only for the estimation of background gene expression. DMR are detected by calculating the statistical significance of an observation based on IP data. Authors of DRME later improved their model and published QNB [20]. QNB also utilizes negative binomial distributions. Different from DRME, QNB combines both input control and IP data in the estimation of background expression and the calculation of test statistics. One common limitation of DRME and QNB is that they both take the within-IP and within-input variation as the variation-of-interest. However, in MeRIP-seq, the signal is IP/input ratio, thus it is the variance of that ratio that should be rigorously modeled. Later, authors of exomePeak proposed exomePeak2 [21]. Compared with exomePeak, exomePeak2 accounts for variations from IP efficiency and GC content bias. When multiple replicates exist, exomePeak2 invokes DESeq2 to identify DMRs by treating the IP and input as paired samples.
Table 1.
Summary of existing differential RNA methylation analysis methods. Methods are ordered chronologically. TDR: True Discovery Rate, the true positive proportion among top identified regions at a certain cutoff. FDR: False Discovery Rate
| Features | |||||
|---|---|---|---|---|---|
| Package | Input | Call Peaks | Algorithm | Pros | Cons |
| exomePeak [16] (2014) | BAM | Yes; by exomePeak | Fisher’s exact test on averaged normalized counts across all replicates. | High sensitivity and TDR. | 1. Ignores the heterogeneity across replicates by using pooled read counts. 2. Poor FDR/ Type I error control. 3. Long runtime, especially for large sample sizes. |
| FET-HMM [17](2015) | Read count matrix | Yes; by exomePeak | Fisher’s exact test combined with hidden Markov’s model to improve the spatial resolution of DMR detection. | 1. High TDR and sensitivity. 2. Best type I error control. | 1. Ignores the heterogeneity within replicates by using pooled read counts 2. Poor FDR/ Type I error control. |
| MeTDiff [18](2015) | BAM | Yes; by HEPeak | Builds Beta- binomial models for raw IP counts given the total of IP and Input counts. | Low memory consumption. | 1. Does not account for variation in sequencing depth. 2. Poor performance under small sample size. 3. Long runtime, especially for large sample sizes. |
| DRME [19](2016) | Read count matrix | Yes; by exomePeak | Builds negative- binomial models for both raw IP and input counts, with only input counts used to estimate baseline expression. | Highest sensitivity, even for small sample sizes and low expression. | 1. Inappropriate variation modeling. 2. Produces the most ”liberal” p-values, leading to the highest FDR and type I error. |
| QNB [20] (2017) | Read count matrix | Yes; by exomePeak | Builds negative- binomial models for both raw IP and input counts, with both IP and input counts used to estimate baseline expression. | Good FDR control. | Improper variation modeling. |
| exomePeak2 [21] (2019) | BAM | Yes; by exomePeak2 | Applies DESeq2 where the regression adjusts for GC content bias estimated by cubic splines expanded Poisson GLM. | 1. High TDR. 2. One of the Best FDR control. 3. Valid p-value distribution. | 1. Unable to account for additional experimental factors in the model.2. Consumes large amount of memory. |
| RADAR [22] (2019) | BAM | No | Poisson random effect model on preprocessed IP count data. Allows for the inclusion of confounding factors. | First method accounting for confounding factors. | 1. Improper distribution assumption for preprocessed data. 2. Long runtime. |
| TRESS [23] (2022) | BAM | Yes; by TRESS | Negative-binomial models on both raw IP and Input count data. Allows for the inclusion of confounding factors. | 1. High TDR. 2. One of the Best FDR/type I error control. 3. Valid p-value distribution. 4. Least runtime and low memory consumption. | Low sensitivity under small sample size. |
All aforementioned methods are only applicable for two-group comparisons. In real biological experiments, especially large studies, confounding covariates (e.g. age or gender) are often observed, but cannot be properly accounted for in aforementioned approaches. To address this problem, two methods were recently proposed: RADAR [22] and TRESS [23]. Both methods link the methylation level to experimental factors using a linear framework. RADAR adopts a Poisson random effect model, while TRESS utilizes a Gamma–Poisson distribution. TRESS differs from RADAR in two aspects. One is that TRESS assumes that raw read counts follow a negative binomial distribution, which is commonly adopted in modeling various sequencing data types. In contrast, RADAR assumes that the preprocessed (starting from library size normalization and followed by input control adjustment) count data follow a Poisson distribution. After preprocessing, the data are not counts format anymore and thus Poisson assumption is equivocal. Another difference is that TRESS can test the effect of all included factors or any linear combinations among them once the model is fitted. In contrast, to test a different factor using RADAR, one needs to re-provide a design matrix and fit the model again, which is computationally inefficient. Overall, methods described above are compiled and listed in Table 1, exhibiting input data type, brief algorithm description and unique characteristics in usage.
Despite the uprising trend of epitranscriptome studies and the rapid evolution of DMRs detection methods, a rigorous assessment of existing methods is still unavailable. In this paper, we systematically compared and evaluated all eight methods listed above with both synthetic and real data. In the simulation study, we investigated their detection accuracy, consistency and similarity with multiple evaluation metrics under various scenarios, including different sample sizes, input expression levels and true DMR proportions. We applied methods on multiple publicly available m6A datasets, and evaluated methods’ performance using distributions of genomics features, DMR overlapping, methods conservativeness and pathways from consensus DMR. Our comprehensive review will assist researchers in choosing suitable approaches accordingly in differential RNA methylation analysis.
DATA GENERATIVE MODEL AND SIMULATION
The core of our simulation framework is a Gamma–Poisson distribution, with modifications to adapt to the MeRIP-seq data. Suppose there are 10 000 candidate DMRs in total, and 10% of them are differentially methylated between treated and untreated conditions. The number of replicates under each condition varies from 2 to 10 to assess the effect of sample size on DMR calling. For each candidate DMR 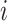 from sample
from sample 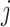 , we first simulate methylation levels
, we first simulate methylation levels 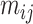 , using
, using 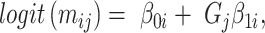 dispersion of methylation
dispersion of methylation 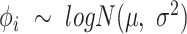 and Gamma scale parameter
and Gamma scale parameter  based on its relationship with
based on its relationship with 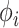 reported and validated previously [23].
reported and validated previously [23]. 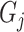 is a binary group (case/control) scalar index.
is a binary group (case/control) scalar index. 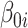 reflects baseline methylation level and
reflects baseline methylation level and 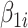 contains the effect of all included factors. For two-group comparison,
contains the effect of all included factors. For two-group comparison, 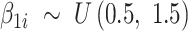 or
or 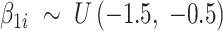 for differential regions (Figure S1), and
for differential regions (Figure S1), and 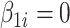 otherwise. The generation of
otherwise. The generation of 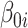 ,
, 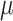 and
and 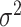 match with their empirical distributions estimated from real data. Given
match with their empirical distributions estimated from real data. Given 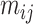 ,
, 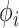 and
and 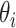 , normalized Poisson rates
, normalized Poisson rates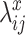 and
and 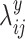 are randomly sampled from Gamma distributions. Then with
are randomly sampled from Gamma distributions. Then with 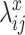 ,
,  and size factors
and size factors  ,
,  , raw read counts are randomly simulated from the Poisson models. A detailed simulation procedure is in Supplementary Materials Section S1.1.1. In addition to the scenario of 10% true DMRs, we also conduct simulations under true DMR proportions of 5 and 15%, and present results in Supplementary Materials Section S1.1.3.
, raw read counts are randomly simulated from the Poisson models. A detailed simulation procedure is in Supplementary Materials Section S1.1.1. In addition to the scenario of 10% true DMRs, we also conduct simulations under true DMR proportions of 5 and 15%, and present results in Supplementary Materials Section S1.1.3.
OVERALL DIFFERENTIAL PEAK COMPARISON
We use simulation procedures described in the previous section to benchmark eight m6A DMR detection methods, with 20 simulation iterations under each scenario. First, we assess all methods under one common experimental design outlined below, to serve as a baseline of the overall comparison before investigating each influential factor separately in later sections.
In each iteration, 1000 out of the 10 000 candidate DMRs are designed to be true DMRs, with three replicates in both case and control groups. We assess the performance of eight DMR detection algorithms using several evaluation metrics. For example, we use the True Discovery Rate (TDR), the proportion of true DMRs among the top regions that are ranked by their adjusted P-values produced by each method. TDR is based on a pragmatic consideration: in the clinical setting, researchers would focus on the top-ranking biomarkers identified by each method; thus, a high TDR indicates a good precision of biomarker discovery. We also examine classic metrics such as Receiver Operating Characteristic curve, sensitivity and False Discovery Rate. To be noted, FETHMM has implemented three strategies: ‘FHB’, ‘FHC’ and ‘FastFHC’. We employ FHC rather than the default setting (FastFHC) because the coding in the default setting involves unusual manipulations of P-values.
Figure 2 shows the DMR calling performance comparison among all methods, in the baseline simulation scenario described above. As displayed in Figure 2A, the TDRs by all methods, TRESS and exomePeak2 have the highest and nearly identical TDR values at each cutoff. exomePeak, FETHMM, DRME and QNB yield TDRs that are comparable but slightly lower than TRESS and exomePeak2. Figure 2B shows that TRESS, exomePeak2, exomePeak, FETHMM and DRME have the highest AUC, whereas MeTDiff has the lowest. It is worth noting that although both TDR and ROC are valid metrics in methods comparison, TDR is more informative because top-ranking ones are more relevant in biomarker discovery. In Figure 2C, P-values from TRESS, exomePeak2, exomePeak, FETHMM and DRME are highly correlated (Spearman correlation 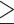 0.93), while MeTDiff produces much more different P-values from other methods. We examine four pairs of methods having the highest and lowest correlations, and demonstrate similarities between FETHMM, exomePeak and exomePeak2 (Figure S3). In Figure 2D, E, Benjamini–Hochberg-adjusted P-values are used to calculate sensitivity and false discovery rate, using 0.05 as the cutoff. Although DRME, FETHMM and exomePeak have high sensitivity, their FDR values are high as well. Combining the two metrics, it indicates an inflated type-I error for DRME, FETHMM and exomePeak. MeTDiff and QNB exhibit unstable performance across simulations. MeTDiff does not perform well in all comparisons, as it has difficulty identifying true positives. TRESS and exomePeak2 achieve nearly identical best overall performance, finding a substantial fraction of true positives while maintaining low FDR. Results under other sample size scenarios are shown in Figure S4 and S5. Overall performance, using the joint distribution of averaged sensitivity and FDR, is also summarized in Figure 2F. Ideally, good methods should have high sensitivity while keeping FDR low, so the methods that reside in the top-left area are the preferred ones.
0.93), while MeTDiff produces much more different P-values from other methods. We examine four pairs of methods having the highest and lowest correlations, and demonstrate similarities between FETHMM, exomePeak and exomePeak2 (Figure S3). In Figure 2D, E, Benjamini–Hochberg-adjusted P-values are used to calculate sensitivity and false discovery rate, using 0.05 as the cutoff. Although DRME, FETHMM and exomePeak have high sensitivity, their FDR values are high as well. Combining the two metrics, it indicates an inflated type-I error for DRME, FETHMM and exomePeak. MeTDiff and QNB exhibit unstable performance across simulations. MeTDiff does not perform well in all comparisons, as it has difficulty identifying true positives. TRESS and exomePeak2 achieve nearly identical best overall performance, finding a substantial fraction of true positives while maintaining low FDR. Results under other sample size scenarios are shown in Figure S4 and S5. Overall performance, using the joint distribution of averaged sensitivity and FDR, is also summarized in Figure 2F. Ideally, good methods should have high sensitivity while keeping FDR low, so the methods that reside in the top-left area are the preferred ones.
Figure 2.

Performance comparison of m6A DMR detection methods. (A) True Discovery Rate (TDR) along top-ranked regions identified by each method. TDR is defined as the proportion of true DMRs among top regions ranked by adjusted P-values. (B) Receiver Operating Characteristic (ROC) curves of DMR detection methods. (C) Heatmap of P-value correlations across eight methods. (D), (E) The violin plots showing sensitivity and FDR distributions of each method, calculated with BH-adjusted P-values. (F) Averaged sensitivity versus FDR of DMRs detection by each method. Here, simulations are conducted under the scenario of three cases, three controls and 10% true DMR. 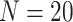 simulations are conducted.
simulations are conducted.
SAMPLE SIZE
We next examine the effect of sample size on DMR calling accuracy as sample size is often the primary parameter-of-interest in experimental designs. Here, simulations are conducted with sample sizes of 2, 3, 5, 7 and 10 in both groups. The sample size values used here in simulations largely reflect the popular choices in current real-world experiments. TDRs under the scenarios of 2, 3, 7 and 10 samples per condition, are shown in Figure 3A–D, respectively. Nearly all methods obtain high TDRs (> 0.8) at very top ranking (e.g. top 100 or 200) regions called, and show decreasing accuracies when moving down the rank. Specifically, TRESS and exomePeak2 maintain the highest accuracies at all cutoffs, whereas MeTDiff performs the worst, with the lowest diminishing accuracies along the rank. As the sample size increases, all methods achieve improved accuracies. This trend is especially evident when  and 10, where methods report similar TDR values. In Figure 3E, TDRs are presented as a heatmap, including results under all simulation scenarios (
and 10, where methods report similar TDR values. In Figure 3E, TDRs are presented as a heatmap, including results under all simulation scenarios (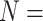 2, 3, 5, 7, 10), stratified by top 400, 700, 1000 and 1500 ranked regions. Overall, TDR values increase as called regions rank higher and sample sizes increase, across all methods. We noticed that a larger sample size can substantially improve detection accuracy, even for mid-ranked regions (e.g. top 1000). RADAR and MeTDiff, compared with other methods, yield lower detection accuracies under small sample size scenarios (i.e.
2, 3, 5, 7, 10), stratified by top 400, 700, 1000 and 1500 ranked regions. Overall, TDR values increase as called regions rank higher and sample sizes increase, across all methods. We noticed that a larger sample size can substantially improve detection accuracy, even for mid-ranked regions (e.g. top 1000). RADAR and MeTDiff, compared with other methods, yield lower detection accuracies under small sample size scenarios (i.e. 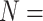 2 and 3), but achieve nearly identical performance as sample size increases. They benefit the most by a boost in sample size. A similar and evident trend is observed in Figure S6, where MeTDiff shows the greatest gains in sensitivity and FDR from large sample sizes. TRESS and exomePeak2 achieve TDRs greater than 0.8 even for an extremely small sample size (N = 2). They outperform other methods in small sample size, due to their implementation of information borrowing across the genome, under an empirical Bayes framework. Such modeling techniques have been shown to be effective statistical frameworks in other genomics studies, especially for small sample sizes [24–27]. In summary, TRESS and exomePeak2 are preferred choices for projects with small sample sizes.
2 and 3), but achieve nearly identical performance as sample size increases. They benefit the most by a boost in sample size. A similar and evident trend is observed in Figure S6, where MeTDiff shows the greatest gains in sensitivity and FDR from large sample sizes. TRESS and exomePeak2 achieve TDRs greater than 0.8 even for an extremely small sample size (N = 2). They outperform other methods in small sample size, due to their implementation of information borrowing across the genome, under an empirical Bayes framework. Such modeling techniques have been shown to be effective statistical frameworks in other genomics studies, especially for small sample sizes [24–27]. In summary, TRESS and exomePeak2 are preferred choices for projects with small sample sizes.
Figure 3.

Comparison of DMR detection accuracy across different sample sizes. (A)–(D) True Discovery Rate (TDR) along top-ranked regions identified by each method, under sample size at 2, 3, 7 and 10 replicates, in each group. (E) Heatmap showing TDR values under combinations of different sample sizes and top-ranked regions cutoffs. Sample sizes are labeled on the right, with 2, 3, 5, 7 and 10, per group. Cutoffs of top-ranked regions are labeled on the left, ranging from top 400, top 700, top 1000 to top 1500. Methods are ordered by column means in heatmap.  simulations are performed under the scenario of 10% true DMRs, and TDR values are averaged across
simulations are performed under the scenario of 10% true DMRs, and TDR values are averaged across 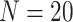 simulations.
simulations.
STRATIFIED ASSESSMENT
Motivated by the fact that the accuracy of differential expression analyses of high-throughput sequencing data, such as bulk RNA-seq, is highly dependent on expression levels [25], we proceed to investigate the DMRs detection accuracies stratified by input value ranges. Based on the distribution of input control values (Figure S7), candidate regions are divided into five strata based on their averaged input counts: stratum 1 [0, 10), stratum 2 [10, 20), stratum 3 [20, 40), stratum 4 [40, 80) and stratum 5 [80, +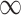 ). At a nominal significance level of 0.05, the sensitivity and FDR of all methods at five strata are shown in Figure 4. Methods are ordered by mean values across strata. All methods have increased sensitivities when moving from a low stratum to a higher one (Figure 4A–C). This is within our expectation, as low-input regions are typically the most susceptible to simulation noise. DRME generates high sensitivities (
). At a nominal significance level of 0.05, the sensitivity and FDR of all methods at five strata are shown in Figure 4. Methods are ordered by mean values across strata. All methods have increased sensitivities when moving from a low stratum to a higher one (Figure 4A–C). This is within our expectation, as low-input regions are typically the most susceptible to simulation noise. DRME generates high sensitivities (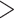 0.75) even at the lowest stratum, and relatively favorable performance in all regions. Sensitivity of DRME still improves for each stratum as sample size increases, but the overall performance gain of DRME over other methods reduces at larger sample sizes. As the sample size increases, all methods exhibit increased and less variable sensitivities, and such performance gain is substantial for lower strata, suggesting that large sample size contributes to more reliable inferences especially for regions affected more by high background noise. Among all, exomePeak2 benefits the most from an increase in sample size, moving from seventh to fourth place. For results on FDR, the benefit of a larger sample size is not as profound as in sensitivity (Figure 4D–F). TRESS and exomePeak2 show small and the most consistent FDRs across all strata and sample sizes. MeTDiff provides drastically poorer FDRs in lower input regions under a small sample scenario (N = 3), whereas it receives a major boost with elevated sample sizes. exomePeak, FETHMM and DRME remain to suffer from poor FDR control, even in a large sample scenario (N = 10).
0.75) even at the lowest stratum, and relatively favorable performance in all regions. Sensitivity of DRME still improves for each stratum as sample size increases, but the overall performance gain of DRME over other methods reduces at larger sample sizes. As the sample size increases, all methods exhibit increased and less variable sensitivities, and such performance gain is substantial for lower strata, suggesting that large sample size contributes to more reliable inferences especially for regions affected more by high background noise. Among all, exomePeak2 benefits the most from an increase in sample size, moving from seventh to fourth place. For results on FDR, the benefit of a larger sample size is not as profound as in sensitivity (Figure 4D–F). TRESS and exomePeak2 show small and the most consistent FDRs across all strata and sample sizes. MeTDiff provides drastically poorer FDRs in lower input regions under a small sample scenario (N = 3), whereas it receives a major boost with elevated sample sizes. exomePeak, FETHMM and DRME remain to suffer from poor FDR control, even in a large sample scenario (N = 10).
Figure 4.

Sensitivity and FDR stratified by mean input count values. Five strata are defined: Stratum 1 [0, 10), Stratum 2 [10, 20), Stratum 3 [20, 40), Stratum 4 [40, 80) and Stratum 5 [80,+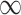 ). Sensitivity and FDR are calculated with BH-adjusted P-values, with a cutoff of 0.05 to define significance. (A)–(C) Stratified sensitivity with 3, 5 and 10 replicates per group, respectively. (D)–(F) Stratified FDR with 3, 5 and 10 replicates per group, respectively.
). Sensitivity and FDR are calculated with BH-adjusted P-values, with a cutoff of 0.05 to define significance. (A)–(C) Stratified sensitivity with 3, 5 and 10 replicates per group, respectively. (D)–(F) Stratified FDR with 3, 5 and 10 replicates per group, respectively. 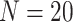 simulations are conducted with 10% DMRs.
simulations are conducted with 10% DMRs.
TYPE I ERROR AND VALIDITY OF P-VALUES
To investigate the type I errors of these methods and the validity of P-values, we conduct simulations under the null hypothesis where none (0%) of candidate regions are differentially methylated. We then obtain the DMR identified by each method, at a nominal significance level of 0.05 using BH-adjusted P-values. Under the scenarios of 2, 3, 5, 7 and 10 replicates per group, empirical type I error rates are computed and shown in Table 2. TRESS and FETHMM achieve type I error rates close to 0.05 in all circumstances, demonstrating that their type I error rates are close to the nominal value. exomePeak2 tends to be more ‘conservative’ with a smaller sample size (N = 2, 3), leading to the best FDR control at the expense of low sensitivity (Figure 2D, E). DRME is the most ‘liberal’ method, and this matches its high sensitivity and FDR, as shown in Figure 2D, E. We also examine the validity of P-values, checking if P-values under the null are uniformly distributed between 0 and 1, and illustrate the results using three replicates per group in Figure 5. TRESS and exomePeak2 yield the most aligned P-values to the expected in the QQ plot (Figure 5A, fall on or close to the diagonal reference line). Most methods generate liberal P-values (fall in the bottom-right area), whereas FETHMM is being too conservative for most regions (fall in the top-left area). Since small P-values are more informative in the DMRs detection, we also apply a -log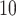 transformation and focus on the distributions of small P-values in Figure 5B. TRESS, exomePeak2 and RADAR perform the best, while other methods provide excessively small P-values, indicating inflated type I error. The results are insensitive to sample size (Figure S9), and consistent with Figure 2E, where TRESS, exomePeak2 and RADAR produce the most well-controlled and stable FDRs.
transformation and focus on the distributions of small P-values in Figure 5B. TRESS, exomePeak2 and RADAR perform the best, while other methods provide excessively small P-values, indicating inflated type I error. The results are insensitive to sample size (Figure S9), and consistent with Figure 2E, where TRESS, exomePeak2 and RADAR produce the most well-controlled and stable FDRs.
Table 2.
Type I error of DMRs detection by eight methods, under the null hypothesis where there is no true DMR. Type I errors are calculated at a nominal significance level of 0.05, and averaged over 20 simulations.
| Methods | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sample Size | FETHMM | TRESS | exomePeak2 | MeTDiff | QNB | RADAR | exomePeak | DRME |
| 2 versus 2 | 0.052 | 0.054 | 0.020 | 0.075 | 0.102 | 0.169 | 0.169 | 0.363 |
| 3 versus 3 | 0.054 | 0.048 | 0.038 | 0.085 | 0.094 | 0.130 | 0.173 | 0.366 |
| 5 versus 5 | 0.055 | 0.043 | 0.053 | 0.077 | 0.081 | 0.104 | 0.173 | 0.365 |
| 7 versus 7 | 0.055 | 0.040 | 0.057 | 0.093 | 0.093 | 0.095 | 0.175 | 0.360 |
| 10 versus 10 | 0.057 | 0.039 | 0.060 | 0.096 | 0.094 | 0.091 | 0.174 | 0.360 |
Figure 5.

Examination of validity of the observed P-values from simulations under the null. (A) Quantile–quantile plot (QQ plot) comparing the distribution of observed P-values against the expected distribution under the null. (B) QQ plot with a
under the null. (B) QQ plot with a  log
log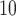 transformation, focusing on small P-values. 20 simulations are conducted under the null hypothesis where there is no DMR. Sample size N=3 per group.
transformation, focusing on small P-values. 20 simulations are conducted under the null hypothesis where there is no DMR. Sample size N=3 per group.
RUNTIME AND MEMORY CONSUMPTION
We assess the software runtime and computing memory consumption for each method, using aligned BAM files as the default input. Under different sample sizes, the runtime for five methods is shown in Figure 6A. As the sample size increases, all methods show longer runtime. Both TRESS and exomePeak2 take shorter runtime compared with other approaches, and this is increasingly evident with larger sample sizes. exomePeak and MeTDiff have similar runtime across all sample sizes. RADAR is the slowest among all, in our benchmark. Results above are from assessments on the High-Performance Computing (HPC) with 1 node, 1 core and 200 GBs of memory. Because most methods take BAM files as the standard input, we further benchmark computing memory consumption and show the results in Figure 6B. MeTDiff and exomePeak consume the least amount of memory (3.81 and 4.62 GB, respectively). TRESS consumes slightly more memory than MeTDiff and exomePeak. exomePeak2 utilizes the most memory (170.28 GB) than the remaining methods. Here, simulations are conducted in HPC with 1 node, 40 cores and 200 GBs of memory available for each method to invoke.
Figure 6.

Runtime and memory consumption comparison of m6A DMR detection methods. (A) Runtime comparison of five different methods under various sample size scenarios. Units in hours. (B) Computing memory consumption, in units of GBs, for five different methods.
REAL DATA ANALYSIS
We first obtain a real dataset (GSE46705) [1, 28, 29] from a study investigating METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation, and denote it as ‘RD1’. In this study, there are four sample types from human HeLa cell line: one wild type (WT) sample and three treated samples. The treatments correspond to the knockdown (KD) of complex METTL3, METLL14 and WTAP [28]. Each sample contains two replicates. We apply TRESS, exomePeak, exomePeak2, MeTDiff and RADAR on this real data, to identify the differential m6A methylation. Here we also incorporate the method MACS3 [30], due to its applicability in analyzing MeRIP-seq data. MACS3 has been adopted by several previous studies [31–33], indicating its potential as an effective tool for the differential analysis of MeRIP-seq data. We only include methods taking BAM file as input for comparison and thus exclude methods taking reads count matrix, like QNB and DRME.
Raw FASTQ files are mapped to human reference genome hg18, using the standard pipeline of STAR with default parameters. Post-aligned BAM files serve as the input for all five methods for comparison. We are interested in the differential methylation between WT and METTL3 samples. DMR calling is conducted at the significance level of FDR < 0.05. After filtering out short (width < 150) and overlapping regions, TRESS, exomePeak, exomePeak2, MeTDiff and RADAR identify 1,413, 1,397, 5,272, 161 and 2,924 DMRs, respectively. exomePeak2 identifies the most amount of DMRs, while MeTDiff finds the least.
The performance comparison of five methods using WT versus METTL3 group real data is shown in Figure 7. We annotate the DMRs using ChIPseeker [34]. The distributions of annotated genomic features of the DMRs are shown in Figure 7A. Here, most methods except RADAR favor DMRs at 3’ UTR. RADAR favors exons located toward the downstream of genes (i.e. non-first exon). For all methods, promoters and downstream exons contribute a considerable amount of constituting genomic features. Figure 7B shows the counts of overlapping regions among them. Five overlapping regions are found by all five methods. 3,348 unique DMRs are found by exomePeak2, which is the highest among all. The highest number of common regions between two methods is 1038 overlaps called both by exomePeak and exomePeak2, while the least count of overlapping regions between two methods is 15 overlaps called by TRESS and MeTDiff. For peak width comparison, the peak width distributions of DMRs, in log scale, are shown in Figure 7C. TRESS favors mid-length regions of 150–400bp. RADAR has a bimodal distribution covering both mid-length and long regions. The FDRs of 1038 common regions are shown in Figure 7D. Here, we observe that exomePeak is a more conservative method compared with exomePeak2. Figure 7E shows the two random examples of consensus DMRs between WT and METTL3 samples, called by TRESS, exomePeak, exomePeak2, MeTDiff and RADAR simultaneously. These two regions cover protein-coding genes TEX264(chr3), PRICKLE4, TOMM6 and USP49(chr6). Previous studies indicate that TEX264 enables signaling receptor activity and also involves in protein-DNA covalent cross-linking repair [35]. USP46 involves in cysteine-type endopeptidase activity, histone H2B conserved C-terminal lysine deubiquitination and mRNA splicing, via spliceosome [36]. Pathway analyses are conducted for exomePeak2 and RADAR, the top two methods identifying the most DMRs. As demonstrated in Figure 7F, the top three enrichment terms are ‘Diseases of signal transduction by growth factor receptors and second messengers’, ‘Transcriptional regulation by TP53’ and ‘Class I MHC mediated antigen processing & presentation’, in exomePeak2’s DMRs. The result for RADAR is summarized in Figure S24.
Figure 7.

Differential m6A methylation methods on real data. (A) Barplots showing distributions of various genomic feature at identified DMRs. Same FDR cutoff of 0.05 was adopted by TRESS, exomePeak, exomePeak2, MeTDiff, RADAR and MACS3 to call significance. (B) Venn diagram showing the overlaps of DMRs identified by five methods. (C) Density plot of distributions of peak width, in log scale, for six methods. (D) Scatterplot of pairwise FDR values of 1,038 consensus region called both by exomePeak and exomePeak2. (E) Two examples of differential peak visualization from consensus DMRs. All differential peaks analyses are between wild-type (WT) group and METTL3 group. (F) Gene Ontology (GO) analysis for DMR genes from exomePeak2, which called the highest number of DMRs. (G) Ranking order based on DMR counts in three real datasets. (H) Ranking order based on 3’UTR percentage in three real datasets.
We also test on two other real datasets (GSE94613 and GSE115105) [37–39] for comparison, and denote them as ‘RD2’ and ‘RD3’. Here, ‘RD2’ contains 12 human samples of METTL3 knockdown cell lines and controls. ‘RD3’ contains bone marrow-derived dendritic cells (BMDCs) from two Ythdf1 knockdown and paired wild-type mice. We perform the same sets of analyses for these two datasets, and present additional results in the Supplementary Materials (S2.2, S2.3). We order five methods by the number of DMRs and the percentage of 3’UTR across three real datasets (Figure 7G). exomePeak2 calls the most DMRs in three real datasets, followed by RADAR. The relative rank stays the same except for TRESS. In terms of the 3’UTR, a consistent relative rank across datasets is observed again, except for a minor difference between exomePeak2 and MeTDiff (Figure 7H).
DISCUSSION
We conduct comprehensive assessments of eight differential epitranscriptome methods, using both synthetic and real data. Leveraging on key metrics including sensitivity, TDR, FDR, AUC and type I error rate, we investigated these methods’ DMR detection precision, false positive rate, consistency and similarity. Additionally, analyses are conducted to explore the effect of sample size and input expression level.
Our simulations adopt the modeling from TRESS, incorporating the Gamma–Poisson distribution and logit linear model for data generation. Parameters are estimated from publicly available datasets to best mimic characteristics of m6A data. In order to facilitate a broad analysis, data are also simulated under various sample size and true DMR proportion situations.
TRESS and exomePeak2 have the highest TDR and AUC values, as well as the lowest FDRs, indicating they are able to generate an accurate order of prediction, and will not produce an excessive number of false positives when evaluated at a specific threshold. Although DRME, FETHMM and exomePeak have high sensitivity, they did a rather poor job in FDR control. MeTDiff, one of the earliest-developed methods, performs poorly in nearly all comparisons, evidenced by the lowest TDR, AUC and sensitivity. Overall, TRESS and exomePeak2 should be favored due to their well-controlled FDR and high precision in differential epitranscriptome analysis.
We also investigate the impact of sample size on the accuracy of DMR detection, as sample size is critical for the majority of study designs. We observe a rise in TDR and AUC as sample size increases, across all methods. TRESS and exomePeak2 continue to be the optimal options. Because of the incorporation of empirical Bayes framework, they can borrow information across the regions along the genome and thus have high precision even at small sample sizes. For example, they attain high TDRs (> 0.8) with only two replicates per group. MeTDiff and RADAR gain the most from an increase in sample size, obtaining significantly enhanced TDRs, sensitivities and FDRs under scenarios of larger sample sizes.
Because biological difference is frequently obscured by background noise, the accuracy of differential expression analysis of high-throughput sequencing data often depends on expression levels. Therefore, we stratify candidate regions by their input expression levels and assess their sensitivity and FDR along the strata. Reduced sensitivities are reported in regions of lower input levels, although they improve substantially as sample sizes grow. DRME manages to achieve a high sensitivity (> 0.75) even at the lowest stratum, but at the cost of a high FDR (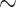 0.75), across all sample sizes. Compared with other methods, the sensitivity of TRESS is most affected by input level, particularly when the sample size is small.
0.75), across all sample sizes. Compared with other methods, the sensitivity of TRESS is most affected by input level, particularly when the sample size is small.
To minimize potential bias in the statistical modeling for simulation, we further propose a real-data-based strategy adopting the Beta-Binomial distribution for data generation (Section S1.2.1). Specifically, IP counts are modeled with a Binomial distribution where the probability parameter is assumed to follow the Beta distribution. This time, we estimate relevant parameters from a publicly available dataset for read counts sampling, using a beta-binomial regression. We conduct a same set of analyses and obtain comparable results to the simulation described in the main manuscript (Section S1.2.2).
Throughout the examination of type I error and P-value distribution under the null hypothesis when there is no DMR, both TRESS and exomePeak2 achieve type I errors close to the nominal value of 0.05 and P-value distributions most aligned with the expected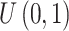 distribution. MeTDiff, QNB, RADAR, exomePeak and DRME have been demonstrated to be liberal, especially for small P-values, which matches their inflated FDRs as observed in Figure 2E. Regarding the computing performance, TRESS yields the shortest runtime across sample sizes, while consuming a relatively light amount of memory. MeTDiff and exomePeak have the least amount of memory consumption, but have considerably longer runtime.
distribution. MeTDiff, QNB, RADAR, exomePeak and DRME have been demonstrated to be liberal, especially for small P-values, which matches their inflated FDRs as observed in Figure 2E. Regarding the computing performance, TRESS yields the shortest runtime across sample sizes, while consuming a relatively light amount of memory. MeTDiff and exomePeak have the least amount of memory consumption, but have considerably longer runtime.
Nanopore sequencing, a cutting-edge third-generation sequencing technology, has also become increasingly valuable in the analysis of gene regulation and disease mechanisms due to its sensitivity and accuracy in detecting RNA modifications. To support the detection of RNA modifications, several bioinformatics techniques have been developed, including Nanocompore [40], a model-free comparative approach, ELIGOS [41] for direct detection of RNA base modifications, xPore [42] for identifying differences in RNA modifications and nanoDoc [43] which uses convolutional neural networks and Deep One-Class Classification. Moreover, various supervised learning methods, such as EpiNano [44], Nanom6A [45] and DENA [46], have been developed to accurately measure m6A modifications with single-nucleotide resolution using synthetic training data. Furthermore, tModBase [47] and directRMDB [48] have been established as databases to understand the landscape of tRNA modification profiles and to integrate quantitative modification profiles obtained from direct RNA sequencing. As nanopore sequencing technology advances, we can anticipate the emergence of more precise methods, leading to new discoveries in the field of RNA modification research.
SOFTWARE AND DATA AVAILABILITY
In this study, FET-HMM, exomePeak (version 2.16.0), MeTDiff (version 1.1.0), DRME, QNB (version 1.0), exomePeak2 (version 1.9.1), RADAR (version 0.2.4) and TRESS (version 1.4.0) were adopted. In exomePeak2, the test method was set to ‘DESeq2’, rather than the default setting. In FET-HMM, we employ FHC rather than the default setting (FastFHC) because the coding in the default setting involves unusual manipulations of P-values. All other methods were tested under the default settings. The software were obtained from Bioconductor or their respective GitHub repositories. Source code of simulations have been uploaded to GitHub at: https://github.com/dxd429/m6A_Benchmark_simulation. The latest version (3.0.0b1) of ‘MACS3’ was used to implement the workflow for the differential enrichment analysis of the real data.
Key Points
Novel epitranscriptomic sequencing technology enables the evaluation of RNA modifications using a data-driven approach.
Differential epitranscriptome analysis requires proper modeling of paired input and IP samples, accommodating technical and biological noise, peak detection and addressing the small sample size issue.
TRESS and exomePeak2 achieve high TDR, low FDR and outstanding sensitivity in benchmark studies.
Detection accuracy can be undermined by low input expression but benefits from an increase in sample size.
RADAR, TRESS and exomePeak2 show top-tier rigorous type I error control and valid P-value distribution under the null. MeTDiff has the least computing memory consumption, and TRESS has the fastest runtime.
AUTHOR CONTRIBUTIONS STATEMENT
Z.G. and H.F. conceived the experiments, D.D. conducted the simulations and compiled results. W.T. conducted the real data analysis. R.W compiled the figures. D.D., Z.G. and H.F. wrote the manuscript. All authors reviewed the manuscript.
Supplementary Material
FUNDING
This work was supported by the American Cancer Society Institutional Research Grant (ACS IRG) [#IRG-16-186-21 to H.F.] through Case Comprehensive Cancer Center; and the Corinne L. Dodero Foundation for the Arts and Sciences and the Case Western Reserve University (CWRU) Program for Autism Education and Research to H.F.
Daoyu Duan is a PhD student in Epidemiology and Biostatistics at the Department of Population and Quantitative Health Sciences in Case Western Reserve University School of Medicine. He is interested in methodology development in -omics data.
Wen Tang is a biostatistician in the Department of Population and Quantitative Health Sciences at Case Western Reserve University School of Medicine. She is interested in applied biostatistics and bioinformatics methods in team-science research projects.
Runshu Wang is a master student in Biostatistics at University of Michigan School of Public Health. He is interested in applied biostatistical methods.
Zhenxing Guo is an assistant professor in the School of Data Science at The Chinese University of Hong Kong, Shenzhen. Her research interest lies in the development of statistically principled and computationally efficient method and tools for the analysis of large biomedical data, in particular -omics data.
Hao Feng is an assistant professor in the Department of Population and Quantitative Health Sciences at Case Western Reserve University School of Medicine. His main research interest is to develop statistical methods and computational tools for high-throughput bioinformatics data.
Contributor Information
Daoyu Duan, Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, 44106, Ohio, USA.
Wen Tang, Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, 44106, Ohio, USA.
Runshu Wang, Department of Biostatistics, University of Michigan, Ann Arbor, 48109, Michigan, USA.
Zhenxing Guo, School of Data Science, The Chinese University of Hong Kong - Shenzhen, Shenzhen, 518172, Guangdong, China.
Hao Feng, Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, 44106, Ohio, USA.
References
- 1. Wang X, Zhike L, Gomez A, et al. m6A-dependent regulation of messenger RNA stability. Nature 2014. ISSN 00280836.; 505(7481): 117. 10.1038/NATURE12730. URL. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3877715/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Geula S, Moshitch-Moshkovitz S, Dominissini D, et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015; 347(6225): 1002–6. ISSN 10959203. 10.1126/science.1261417. URL. https://www.science.org/doi/10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 3. Dermentzaki G, Lotti F. New insights on the role of N6-Methyladenosine RNA methylation in the physiology and pathology of the nervous system. Front Mol Biosci 2020; 7:229. ISSN 2296889X. 10.3389/fmolb.2020.555372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu J, Frazier K, Zhang J, et al. Emerging role of m6A RNA methylation in nutritional physiology and metabolism. Obes Rev 2020; 21(1): e12942. ISSN 1467-789X. 10.1111/OBR.12942. URL. https://onlinelibrary.wiley.com/doi/10.1111/obr.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pinello N, Sun S, Wong JJL. Aberrant expression of enzymes regulating m6A mRNA methylation: implication in cancer. Cancer Biol Med 2018; 15(4): 323. ISSN 20953941. 10.20892/J.ISSN.2095-3941.2018.0365. URL. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6372906/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Uddin MB, Wang Z, Yang C. The m6A RNA methylation regulates oncogenic signaling pathways driving cell malignant transformation and carcinogenesis. Mol Cancer 2021; 20(1): 61–18, ISSN 1476-4598. 10.1186/S12943-021-01356-0. URL. https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-021-01356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A 1974; 71(10): 3971–5. ISSN 00278424. 10.1073/pnas.71.10.3971.URL. https://www.pnas.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen XY, Zhang J, Zhu JS. The role of m6A RNA methylation in human cancer. Mol Cancer 2019; 18(1): 103–9. ISSN 14764598. 10.1186/s12943-019-1033-z. URL. https://link.springer.com/article/10.1186/s12943-019-1033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lan Q, Liu PY, Haase J, et al. The critical role of RNA M6A methylation in cancer. Cancer Res 2019; 79(7): 1285–92. ISSN 15387445. 10.1158/0008-5472.CAN-18-2965/661324/P/THE-CRITICAL-ROLE-OF-RNA-M6A-METHYLATION-IN. URL. https://aacrjournals.org/cancerres/article/79/7/1285/640506/The-Critical-Role-of-RNA-m6A-Methylation-in. [DOI] [PubMed] [Google Scholar]
- 10. Ke S, Alemu EA, Mertens C, et al. A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation. Genes Dev 2015; 29(19): 2037–53. ISSN 1549-5477. 10.1101/GAD.269415.115. https://pubmed.ncbi.nlm.nih.gov/26404942/?dopt=Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012; 149(7): 1635. ISSN 10974172. 10.1016/J.CELL.2012.05.003. URL. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3383396/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mathiyalagan P, Adamiak M, Mayourian J, et al. FTO-dependent N 6-Methyladenosine regulates cardiac function during Remodeling and repair. Circulation 2019; 139(4): 518–32. ISSN 1524–4539. 10.1161/CIRCULATIONAHA.118.033794. URL. https://pubmed.ncbi.nlm.nih.gov/29997116/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dorn LE, Lasman L, Chen J, et al. The N-Methyladenosine mRNA Methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation 2019; 139(4): 533–45. ISSN 15244539. 10.1161/CIRCULATIONAHA.118.036146. URL. https://pubmed.ncbi.nlm.nih.gov/30586742/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hui Y, Zhang Z. ALKBH5-mediated m6A demethylation of lncRNA RMRP plays an oncogenic role in lung adenocarcinoma. Mamm Genome 2021; 32(3): 195–203. ISSN 14321777. 10.1007/s00335-021-09872-6. URL. https://link.springer.com/article/10.1186/s12935-020-1105-6. [DOI] [PubMed] [Google Scholar]
- 15. Nishizawa Y, Konno M, Asai A, et al. Oncogene c-Myc promotes epitranscriptome m6A reader YTHDF1 expression in colorectal cancer. Oncotarget 2018; 9(7): 7476. ISSN 19492553. 10.18632/ONCOTARGET.23554. URL. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5800917/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meng J, Cui X, Rao MK, et al. Exome-based analysis for RNA epigenome sequencing data. Bioinformatics 2013; 29(12): 1565. ISSN 13674803. 10.1093/BIOINFORMATICS/BTT171. URL. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3673212/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang YC, Zhang SW, Liu L, et al. Spatially enhanced differential RNA methylation analysis from affinity-based sequencing data with hidden Markov model. Biomed Res Int 2015; 2015:852070. ISSN 23146141. 10.1155/2015/852070. URL. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4537718/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cui X, Lin Z, Meng J, et al. MeTDiff: a novel differential RNA methylation analysis for MeRIP-Seq data. IEEE/ACM Trans Comput Biol Bioinform 2018ISSN 15579964; 15(2): 526–34. 10.1109/TCBB.2015.2403355. [DOI] [PubMed] [Google Scholar]
- 19. Liu L, Zhang SW, Gao F, et al. DRME: count-based differential RNA methylation analysis at small sample size scenario. Anal Biochem 2016ISSN 0003-2697; 499:15–23. 10.1016/J.AB.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 20. Liu L, Zhang SW, Huang Y, et al. QNB: differential RNA methylation analysis for count-based small-sample sequencing data with a quad-negative binomial model. BMC Bioinform 2017; 18(1):1–12. ISSN 14712105. 10.1186/S12859-017-1808-4/FIGURES/10. URL. https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tang Y, Chen K, Song B, et al. m6A-atlas: a comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res 2021; 49(D1): D134–43. ISSN 1362-4962. 10.1093/NAR/GKAA692. URL. https://pubmed.ncbi.nlm.nih.gov/32821938/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Z, Zhan Q, Eckert M, et al. RADAR: differential analysis of MeRIP-seq data with a random effect model. Genome Biol 2019; 20(1): 1–17. ISSN 1474760X. 10.1186/S13059-019-1915-9/FIGURES/7. URL. https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1915-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo Z, Shafik AM, Jin P, et al. Differential RNA methylation analysis for MeRIP-seq data under general experimental design. Bioinformatics (Oxford, England) 2022; 38:4705–12. ISSN 1367-4811. 10.1093/BIOINFORMATICS/BTAC601. URL. https://pubmed.ncbi.nlm.nih.gov/36063045/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu H, Wang C, Wu Z. A new shrinkage estimator for dispersion improves differential expression detection in RNA-seq data. Biostatistics (Oxford, England) 2013; 14(2): 232–43. ISSN 1468-4357. 10.1093/BIOSTATISTICS/KXS033. URL. https://pubmed.ncbi.nlm.nih.gov/23001152/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hao W, Wang C, Zhijin W. PROPER: comprehensive power evaluation for differential expression using RNA-seq. Bioinformatics 2015; 31(2): 233. ISSN 14602059. 10.1093/BIOINFORMATICS/BTU640. URL. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4287952/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng H, Conneely KN, Hao W. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res 2014; 42(8): e69. ISSN 13624962. 10.1093/nar/gku154. URL. https://pubmed.ncbi.nlm.nih.gov/24561809/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15(12) ISSN 1474-760X:550. 10.1186/S13059-014-0550-8. URL. https://pubmed.ncbi.nlm.nih.gov/25516281/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 2014; 10(2): 93–5. ISSN 1552-4469. 10.1038/NCHEMBIO.1432. URL. https://pubmed.ncbi.nlm.nih.gov/24316715/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Niu Y, Xu Z, Wu YS, et al. N6-methyl-adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Genom Proteom Bioinform 2013; 11(1): 8–17. ISSN 2210-3244. 10.1016/J.GPB.2012.12.002. URL. https://pubmed.ncbi.nlm.nih.gov/23453015/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008; 9(9): R137. ISSN 1474-760X. 10.1186/GB-2008-9-9-R137. URL. https://pubmed.ncbi.nlm.nih.gov/18798982/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McIntyre ABR, Gokhale NS, Cerchietti L, et al. Limits in the detection of m6A changes using MeRIP/m6A-seq. Sci Rep 2020; 10(1): 6590. ISSN 2045-2322. 10.1038/S41598-020-63355-3. URL. https://pubmed.ncbi.nlm.nih.gov/32313079/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu H, Flores MA, Meng J, et al. MeT-DB: a database of transcriptome methylation in mammalian cells. Nucleic Acids Res 2015; 43(Database issue): D197–203. ISSN 1362-4962. 10.1093/NAR/GKU1024. URL. https://pubmed.ncbi.nlm.nih.gov/25378335/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Antanaviciute A, Baquero-Perez B, Watson CM, et al. m6aViewer: software for the detection, analysis, and visualization of N6-methyladenosine peaks from m6A-seq/ME-RIP sequencing data. RNA (New York, NY) 2017; 23(10): 1493–501. ISSN 1469-9001. 10.1261/RNA.058206.116. URL. https://pubmed.ncbi.nlm.nih.gov/28724534/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics (Oxford, England) 2015; 31(14): 2382–3. ISSN 1367-4811. 10.1093/BIOINFORMATICS/BTV145. URL. https://pubmed.ncbi.nlm.nih.gov/25765347/. [DOI] [PubMed] [Google Scholar]
- 35. Fielden J, Wiseman K, Torrecilla I, et al. TEX264 coordinates p97- and SPRTN-mediated resolution of topoisomerase 1-DNA adducts. Nat Commun 2020; 11(1): 1–16. ISSN 20411723. 10.1038/s41467-020-15000-w. URL. https://www.nature.com/articles/s41467-020-15000-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi L, Shen X, Shen Y. USP49-mediated histone H2B Deubiquitination regulates HCT116 cell proliferation through MDM2-p53 Axis. Mol Cell Biol 2022; 42(3): e0043421. ISSN 1098-5549. 10.1128/MCB.00434-21. https://pubmed.ncbi.nlm.nih.gov/35072515/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barbieri I, Tzelepis K, Pandolfini L, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017; 552(7683): 126–31. ISSN 14764687. 10.1038/nature24678. URL. https://pubmed.ncbi.nlm.nih.gov/29186125/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yankova E, Blackaby W, Albertella M, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021; 593(7860): 597–601. ISSN 1476-4687. 10.1038/S41586-021-03536-W. URL. https://pubmed.ncbi.nlm.nih.gov/33902106/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han D, Liu J, Chen C, et al. Anti-tumour immunity controlled through mRNA m 6 a methylation and YTHDF1 in dendritic cells. Nature 2019; 566(7743): 270–4. ISSN 1476-4687. 10.1038/S41586-019-0916-X. URL. https://pubmed.ncbi.nlm.nih.gov/30728504/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leger A, Amaral PP, Pandolfini L, et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat Commun 2021; 12(1): 7198. ISSN 2041-1723. 10.1038/S41467-021-27393-3. URL. https://pubmed.ncbi.nlm.nih.gov/34893601/, https://pubmed.ncbi.nlm.nih.gov/34893601/?dopt=Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jenjaroenpun P, Wongsurawat T, Wadley TD, et al. Decoding the epitranscriptional landscape from native RNA sequences. Nucleic Acids Res 2021; 49(2): e7. ISSN 1362-4962. 10.1093/NAR/GKAA620. URL. https://pubmed.ncbi.nlm.nih.gov/32710622/, https://pubmed.ncbi.nlm.nih.gov/32710622/?dopt=Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pratanwanich PN, Yao F, Chen Y, et al. Identification of differential RNA modifications from nanopore direct RNA sequencing with xPore. Nat Biotechnol 2021; 39(11): 1394–402, ISSN 1546-1696. 10.1038/s41587-021-00949-w. URL. https://www.nature.com/articles/s41587-021-00949-w. [DOI] [PubMed] [Google Scholar]
- 43. Ueda H. nanoDoc: RNA modification detection using Nanopore raw reads with deep one-class classification. bioRxiv. 2021;2020.09.13.295089. 10.1101/2020.09.13.295089. URL. https://www.biorxiv.org/content/10.1101/2020.09.13.295089v2. [DOI]
- 44. Liu H, Begik O, Novoa EM. EpiNano: detection of m6A RNA modifications using Oxford Nanopore direct RNA sequencing. Methods in molecular biology (Clifton, NJ) 2021; 2298:31–52. ISSN 1940-6029. 10.1007/978-1-0716-1374-0_3. URL. https://pubmed.ncbi.nlm.nih.gov/34085237/, https://pubmed.ncbi.nlm.nih.gov/34085237/?dopt=Abstract. [DOI] [PubMed] [Google Scholar]
- 45. Gao Y, Liu X, Bizhi W, et al. Quantitative profiling of N6-methyladenosine at single-base resolution in stem-differentiating xylem of Populus trichocarpa using Nanopore direct RNA sequencing. Genome Biol 2021; 22(1): 22. ISSN 1474-760X. 10.1186/S13059-020-02241-7. URL. https://pubmed.ncbi.nlm.nih.gov/33413586/, https://pubmed.ncbi.nlm.nih.gov/33413586/?dopt=Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qin H, Liang O, Gao J, et al. DENA: training an authentic neural network model using Nanopore sequencing data of Arabidopsis transcripts for detection and quantification of N 6-methyladenosine on RNA. Genome Biol 2022; 23(1): 1–23. ISSN 1474760X. 10.1186/S13059-021-02598-3/FIGURES/6. URL. https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02598-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lei HT, Wang ZH, Li B, et al. tModBase: deciphering the landscape of tRNA modifications and their dynamic changes from epitranscriptome data. Nucleic Acids Res 2023; 51(D1): D315–27. ISSN 1362-4962. 10.1093/NAR/GKAC1087. URL. https://pubmed.ncbi.nlm.nih.gov/36408909/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Y, Jiang J, Ma J, et al. DirectRMDB: a database of post-transcriptional RNA modifications unveiled from direct RNA sequencing technology. Nucleic Acids Res 2023; 51(D1): D106–16. ISSN 1362-4962. 10.1093/NAR/GKAC1061. URL. https://pubmed.ncbi.nlm.nih.gov/36382409/. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
In this study, FET-HMM, exomePeak (version 2.16.0), MeTDiff (version 1.1.0), DRME, QNB (version 1.0), exomePeak2 (version 1.9.1), RADAR (version 0.2.4) and TRESS (version 1.4.0) were adopted. In exomePeak2, the test method was set to ‘DESeq2’, rather than the default setting. In FET-HMM, we employ FHC rather than the default setting (FastFHC) because the coding in the default setting involves unusual manipulations of P-values. All other methods were tested under the default settings. The software were obtained from Bioconductor or their respective GitHub repositories. Source code of simulations have been uploaded to GitHub at: https://github.com/dxd429/m6A_Benchmark_simulation. The latest version (3.0.0b1) of ‘MACS3’ was used to implement the workflow for the differential enrichment analysis of the real data.
Key Points
Novel epitranscriptomic sequencing technology enables the evaluation of RNA modifications using a data-driven approach.
Differential epitranscriptome analysis requires proper modeling of paired input and IP samples, accommodating technical and biological noise, peak detection and addressing the small sample size issue.
TRESS and exomePeak2 achieve high TDR, low FDR and outstanding sensitivity in benchmark studies.
Detection accuracy can be undermined by low input expression but benefits from an increase in sample size.
RADAR, TRESS and exomePeak2 show top-tier rigorous type I error control and valid P-value distribution under the null. MeTDiff has the least computing memory consumption, and TRESS has the fastest runtime.