

Abstract
The critical first step in Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)–associated (CRISPR–Cas) protein-mediated gene editing is recognizing a preferred protospacer adjacent motif (PAM) on target DNAs by the protein’s PAM-interacting amino acids (PIAAs). Thus, accurate computational modeling of PAM recognition is useful in assisting CRISPR–Cas engineering to relax or tighten PAM requirements for subsequent applications. Here, we describe a universal computational protein design framework (UniDesign) for designing protein–nucleic acid interactions. As a proof of concept, we applied UniDesign to decode the PAM–PIAA interactions for eight Cas9 and two Cas12a proteins. We show that, given native PIAAs, the UniDesign-predicted PAMs are largely identical to the natural PAMs of all Cas proteins. In turn, given natural PAMs, the computationally redesigned PIAA residues largely recapitulated the native PIAAs (74% and 86% in terms of identity and similarity, respectively). These results demonstrate that UniDesign faithfully captures the mutual preference between natural PAMs and native PIAAs, suggesting it is a useful tool for engineering CRISPR–Cas and other nucleic acid-interacting proteins. UniDesign is open-sourced at https://github.com/tommyhuangthu/UniDesign.
Keywords: CRISPR–Cas, PAM, gene editing, computational protein design, UniDesign
INTRODUCTION
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)–associated (CRISPR–Cas) protein-mediated genome editing shows great promise in biotechnology and medicine [1–5]. However, the specific recognition of protospacer adjacent motifs (PAMs) before DNA cleavage limits the target range of Cas nucleases [6]. For instance, the most widely used SpCas9 from Streptococcus pyogenes recognizes an NGG (N is A/C/G/T) PAM, allowing it to target about 1/8 (¼ × ¼ × 2 DNA strands) human genomic sequences. Engineering the PAM requirement of a CRISPR–Cas protein has immediate applications [7–10]. For example, relaxing the PAM requirement (e.g. from NGG to NGN) would increase the overall targetable sequences, whereas tightening the PAM requirement (e.g. from NGG to AGG) would increase editing specificity and reduce off-target edits [6].
Directed evolution and structure-guided engineering are the two prevalent strategies to engineer the PAM preference of CRISPR–Cas proteins. In recent years, the latter has gained more popularity and success along with the increased number of solved Cas protein structures. Most of these efforts, however, are limited to utilizing the structural biology knowledge to identify key PAM-interacting amino acids (PIAAs) for mutagenesis engineering.
By contrast, computational protein design (CPD) approaches make use of protein structures as a basis to interrogate the effects of mutagenesis with advanced computer algorithms [11–13] and at a higher level to de novo design a protein sequence with desired function [14, 15]. CPD methods have been widely used in protein engineering and yielded proteins with improved functional characteristics [16–20]. However, the powerful CPD methods were rarely adapted to engineer Cas proteins to modify their PAM requirements. The COMET workflow is by far the only computational approach to model PAM recognition [21]. COMET provided a computational interpretation to the KKH variant of Staphylococcus aureus Cas9 (SaCas9), which was obtained by directed evolution that relaxes the PAM from NNGRRT into NNNRRT [10], followed by COMET-guided design of two new variants SaCas9-NR and SaCas9-RL, both with a relaxed PAM of NNGRRN [21]. COMET leveraged molecular dynamics (MD) simulations and free-energy perturbation (FEP) to calculate mutation-induced binding free energy change, which was subsequently correlated with PAM recognition [21]. One caveat of COMET is that both MD and FEP are computation-intensive and resource-inefficient, limiting their large-scale application and adaptation.
We previously developed two CPD methods, namely EvoDesign and EvoEF2, for monomer protein design and protein–protein interaction (PPI) design [22, 23]. EvoDesign is an evolution-based approach that combines the evolutionary profile [represented as the position-specific scoring matrix (PSSM)] derived from multiple structure/sequence alignment (MSSA) with a physics-based energy function (EvoEF) for protein sequence design [22]. In some cases, the PSSM term may be less reliable due to insufficient structure analogs obtained, whereas the accuracy of EvoEF by itself is moderate for de novo protein design [23]. To increase the physical energy function’s accuracy, we developed EvoEF2 by introducing a few statistical energy terms into EvoEF and reoptimized all the energy weights through extensive de novo sequence design [23]. EvoEF is a linear combination of five weighted energy terms, including van der Waals, hydrogen bonding, electrostatics, solvation and reference energy, and optimized by maximizing the accuracy of predicting the thermodynamic changes upon mutations [22]. Compared with EvoEF, EvoEF2 includes four extra knowledge-based terms, i.e. disulfide bonding, amino acid propensity, Ramachandran and rotamer frequency. Benchmark results showed that all nine terms are important to EvoEF2’s high accuracy, and overall, EvoEF2 improved the sequence design accuracy by about two folds relative to EvoEF [23]. We have successfully used these two methods to design novel protein and peptide binders [24, 25].
Unlike COMET, which relies on resource-heavy algorithms (e.g. MD and FEP), the EvoDesign and EvoEF2 methods comprise resource-friendly components: a rotamer library for efficient amino-acid conformation sampling, an efficient energy function for protein sequence scoring, and a fast optimization algorithm for searching low-energy designer sequences [23, 26, 27]. With more approximations, these two methods are much faster than MD-based approaches yet still very accurate, making them particularly appropriate to efficiently explore the vast sequence space, such as those of the CRISPR–Cas proteins.
Here we report the development of a universal CPD framework (UniDesign) that is built on EvoDesign and EvoEF2 by adding a new capacity to model and design the protein–nucleic acid interaction (PNI), a functionality that is necessary yet underdeveloped for engineering CRISPR–Cas protein’s PAM preference. Our detailed computational analyses show that UniDesign captures the ‘self-consistency’ between PIAAs and PAMs satisfactorily, and provides computational explanation at the molecular and energetic levels, thus suggesting the potential application of UniDesign in assisting CRISPR–Cas protein engineering.
METHODS
Cas structures collection and preprocessing
The Protein Data Bank (PDB) files for eight Cas9 and two Cas12a proteins were downloaded from Research Collaboratory for Structural Bioinformatics (RCSB) PDB [28]. Ions and water molecules were removed except for 5AXW, 5CZZ, 5B43, 5XUS and 6JDV, in which PAM-interacting water molecules were retained. Specifically, HOH1202, HOH1217, HOH1227, HOH201, HOH202 and HOH203 were kept in 5AXW. HOH1207, HOH1233, HOH201, HOH202, HOH203 and HOH204 were reserved in 5CZZ. HOH21 was retained in 5B43. HOH36, HOH65, HOH119, HOH122 and HOH124 was retained in 5XUS. HOH1201 was retained in 6JDV. The missing amino-acid side chains were added using the ‘RepairStructure’ command in UniDesign, and sidechain steric clashes were reduced using the UniDesign ‘Minimization’ command.
Atomic parameters and topologies for nucleotides
The amino-acid atomic parameters and topologies in EvoEF2 [and the UniDesign energy function (UniEF)] are adapted from the united-atom force field CHARMM19 [29]. However, there are no parameters and topologies for nucleotides in CHARMM19. We took nucleotide topologies from the all-atom CHARMM36 force field [30] but removed all nonpolar hydrogen atoms. To be consistent with nucleotide nomenclature in PDB, the nucleotides were renamed as DA, DC, DG and DT for DNA, and A, C, G and U for RNA in the UniEF energy function. The nucleotide atoms were parameterized based on their similarity to atoms in amino acid chemical groups. Our previous study demonstrated the good performance of CHARMM19-based EvoEF2 on protein sequence design with a good balance of accuracy and speed [23], rationalizing the development of UniEF using CHARMM19-like parameters and topologies. The UniEF atomic parameters and residue topologies can be found at https://github.com/tommyhuangthu/UniDesign/blob/master/library/toppar/param_charmm19_lk.prm and https://github.com/tommyhuangthu/UniDesign/blob/master/library/toppar/top_polh19.inp, respectively.
UniEF and modification to the hydrogen-bonding energy term
UniEF inherits the EvoEF2 energy function for protein design [23]. UniEF is the linear combination of nine energy terms:
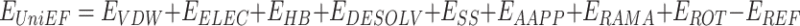 |
(1) |
Here, 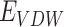 ,
, 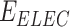 ,
, 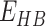 ,
, 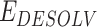 and
and 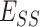 represents the total weighted van der Waals, electrostatic, hydrogen-bonding, de-solvation and disulfide-bonding interaction, respectively; these terms are calculated as the weighted sum of pairwise atomic interaction energy.
represents the total weighted van der Waals, electrostatic, hydrogen-bonding, de-solvation and disulfide-bonding interaction, respectively; these terms are calculated as the weighted sum of pairwise atomic interaction energy. 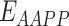 ,
, 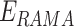 and
and 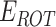 represents the weighted term for calculating amino acid propensity, the Ramachandran term, and the term for modeling rotamer frequency in the rotamer library, respectively; these terms are dependent on protein backbone geometry and are calculated as the sum of residue- or rotamer-wise energy. Finally,
represents the weighted term for calculating amino acid propensity, the Ramachandran term, and the term for modeling rotamer frequency in the rotamer library, respectively; these terms are dependent on protein backbone geometry and are calculated as the sum of residue- or rotamer-wise energy. Finally, 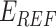 , namely protein reference energy, models the energy of a protein in the unfolded state and is roughly calculated as the sum of amino acid reference energy. Our previous work described the energy weight optimization procedure in detail [23].
, namely protein reference energy, models the energy of a protein in the unfolded state and is roughly calculated as the sum of amino acid reference energy. Our previous work described the energy weight optimization procedure in detail [23].
Compared to EvoEF2, we updated the hydrogen-bonding energy term in UniEF to modeling water-mediated hydrogen bonds, which are commonly seen in biological systems. For a regular hydrogen-bonding interaction, 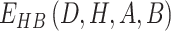 is defined by four atoms: the hydrogen atom (H), the hydrogen acceptor (A), the hydrogen donor (D) and the base atom to which
is defined by four atoms: the hydrogen atom (H), the hydrogen acceptor (A), the hydrogen donor (D) and the base atom to which 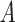 is attached (B).
is attached (B). 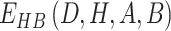 is a linear combination of three terms that depend on the distance between H and A (
is a linear combination of three terms that depend on the distance between H and A (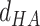 ), the angle between D, H and A (
), the angle between D, H and A (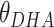 ) and the angle between H, A and B (
) and the angle between H, A and B (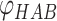 ):
):
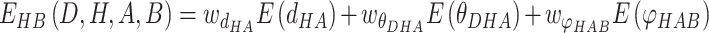 |
(2) |
where:
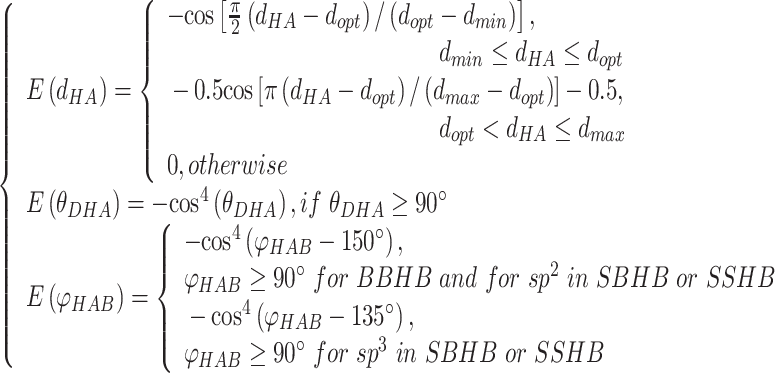 |
(3) |
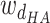 ,
, 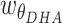 and
and 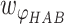 are weights for the three terms. The optimal distance between H and A,
are weights for the three terms. The optimal distance between H and A, 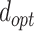 , is set to 1.9 Å. Additionally,
, is set to 1.9 Å. Additionally, 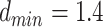 Å and
Å and 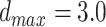 Å are the lower and upper bounds of the distance between the hydrogen–acceptor pair. The optimal
Å are the lower and upper bounds of the distance between the hydrogen–acceptor pair. The optimal 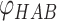 value is set to either 150° or 135°, depending on the acceptor hybridization (sp2 or sp3) and the locations of the donor and acceptor atoms (BBHB: backbone–backbone hydrogen bond; SBHB: sidechain–backbone hydrogen bond; SSHB: sidechain–sidechain hydrogen bond).
value is set to either 150° or 135°, depending on the acceptor hybridization (sp2 or sp3) and the locations of the donor and acceptor atoms (BBHB: backbone–backbone hydrogen bond; SBHB: sidechain–backbone hydrogen bond; SSHB: sidechain–sidechain hydrogen bond).
Water molecules can be hydrogen bond donors, acceptors, or both. In UniDesign, the water hydrogen atoms are not explicitly modeled due to the difficulty to determine their positions. Water-mediated hydrogen bonding interactions are treated in three cases:
Case 1: the hydrogen bond acceptor (A) is water while the donor is normal. In this case, the base atom B does not exist, and hence 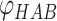 related terms in Equations (2) and (3) are omitted. The other terms are the same.
related terms in Equations (2) and (3) are omitted. The other terms are the same.
Case 2: the hydrogen bond donor (D) is water while the acceptor is normal. In this case, the hydrogen atom (H) is omitted, and 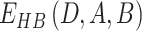 is used to calculate water-mediated hydrogen-bonding energy.
is used to calculate water-mediated hydrogen-bonding energy.
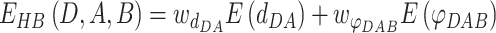 |
(4) |
where:
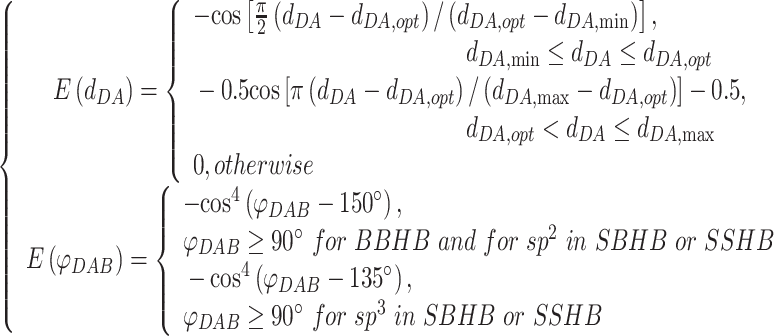 |
(5) |
The optimal distance between D and A, 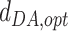 , is set to 2.8 Å. The minimum and maximum distances for considering a water-mediated hydrogen bond are:
, is set to 2.8 Å. The minimum and maximum distances for considering a water-mediated hydrogen bond are: 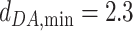 Å and
Å and 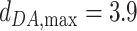 Å.
Å.
Case 3: both D and A are water. In this case, H and A do not exist, and only the 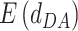 term in Equations (4) and (5) is used to calculate water-mediated hydrogen bonding energy.
term in Equations (4) and (5) is used to calculate water-mediated hydrogen bonding energy.
Generation of PAM variant models with UniDesign
The ‘BuildMutant’ command, implemented in EvoEF2 to build amino-acid mutant models, was extended to build models for nucleotide mutations in UniDesign. When mutating a nucleotide, the torsional angle centered on the bond that connects the backbone and sidechain is kept unchanged, and the coordinates of sidechain atoms are calculated based on the nucleotide topology described above. For instance, for the DA → DC mutation, the value of the torsional angle O4'–C1'–N1–C2 in nucleotide DC will be taken from that of O4'–C1'–N9–C4 in nucleotide DA. For a double-stranded DNA, if one nucleotide is mutated on one strand, the paired nucleotide on the other strand will be automatically mutated to ensure reverse complementarity. Below is the command line to build PAM mutants using 4UN3 as an example:
path_to_UniDesign/UniDesign --command=BuildMutant --pdb=4UN3.pdb --mutant_file=mutants.txt
The ‘mutants.txt’ file contains one or more lines like ‘tD5a,gD6a,gD7a;’ Each line ended with ‘;’ represents one mutant for which mutations are separated by ‘,’. In this example, the TGG PAM will be mutated into AAA. All other PAM variants can be built similarly.
Computational repacking and redesign of PIAAs
The ‘ProteinDesign’ command in UniDesign was used to repack or redesign the PIAA residues. Below is the command line using 4UN3 as an example:
path_to_UniDesign/UniDesign --command=ProteinDesign --ppint --pdb=4UN3.pdb --design_chains=B --resfile=RESFILE_4UN3_UniDesign.txt
The ‘--ppint’ option specifies a PPI or PNI design task, which is equally treated in the prototype UniDesign. The ‘--design_chains=B’ option means that design is carried out on chain B, i.e. the SpCas9 protein chain. The residues to be repacked and/or redesigned were controlled using a restraint file (RESFILE) named ‘RESFILE_4UN3_UniDesign.txt’. The RESFILE format is explained in detail at https://github.com/tommyhuangthu/UniDesign/blob/master/manual.docx.
As a comparison to UniDesign, the Rosetta FixBB protocol (version 3.15) [31, 32] was also applied to redesign PIAA residues for each native Cas protein and its related PAM variants generated by UniDesign. Below is the command line using 4UN3 as an example:
path_to_rosetta/main/source/bin/fixbb.static.linuxgccrelease -in:file:s 4UN3.pdb -in:file:fullatom -resfile RESFILE_4UN3_Rosetta.txt -nstruct 1
Similar to that in UniDesign, the residues to be redesigned were restricted by a RESFILE following Rosetta’s syntax.
RESULTS
UniDesign for protein–nucleic acid interaction modeling and design
EvoDesign and EvoEF2 lack the capacity for PNI design or other functional protein design tasks like protein–ligand interaction (PLI) design and enzyme design since they cannot model non-protein molecules. To overcome these limitations, we have developed a universal CPD approach named UniDesign to deal with these four kinds of functional protein design tasks (i.e. PPI, PNI, PLI and enzyme design) (Figure 1A).
Figure 1.

The UniDesign workflow for protein–nucleic acid interaction (PNI) design. (A) UniDesign can work for protein–protein interaction design, protein–ligand interaction design, enzyme design and PNI design. (B) The UniDesign pipeline for PNI design comprises four stages (denoted as i, ii, iii and iv).
UniDesign inherits the overall methodology of EvoEF2 while adopting the evolutionary component of EvoDesign. Figure 1B illustrates the pipeline for PNI design. First, design sites of interest can be defined based on the input PNI complex structure. Second, UniDesign searches for structure analogs to the scaffold protein and constructs a PSSM using the MSSA obtained from the pairwise structure alignment. Third, building on the PNI scaffold, UniDesign performs sequence redesign for the predefined design sites, using a composite energy function by combining the 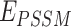 and
and 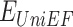 energy terms, where UniEF is an energy function extended from EvoEF2 [23] and is used to model the physical interactions between protein and nucleic acid (see Methods). An efficient simulated annealing Monte Carlo (SAMC) simulation procedure is employed to search the sequence space. Finally, after the SAMC simulation, the sequence with the lowest total energy is taken as the best design. Due to SAMC’s stochasticity, the lowest-energy designs from multiple independent simulations will be collected for analysis. In UniDesign, the evolutionary module is set as optional; UniEF alone is used for design when evolution is disabled.
energy terms, where UniEF is an energy function extended from EvoEF2 [23] and is used to model the physical interactions between protein and nucleic acid (see Methods). An efficient simulated annealing Monte Carlo (SAMC) simulation procedure is employed to search the sequence space. Finally, after the SAMC simulation, the sequence with the lowest total energy is taken as the best design. Due to SAMC’s stochasticity, the lowest-energy designs from multiple independent simulations will be collected for analysis. In UniDesign, the evolutionary module is set as optional; UniEF alone is used for design when evolution is disabled.
Summary of CRISPR–Cas proteins in this study
We selected eight CRISPR–Cas9 proteins, namely SpCas9, SaCas9, St1Cas9, FnCas9, Nme1Cas9, Nme2Cas9, CdCas9 and AceCas9, and two CRISPR–Cas12a proteins, namely AsCas12a and LbCas12a (Table 1), to study their PAM recognition because (1) their experimentally determined consensus PAMs are consistent in different studies, providing a ground truth to computational modeling; (2) their Cas/gRNA (guide RNA)/DNA complex structures are available with PDB PAM being a part of the consensus PAMs (Table 1) [PAM sequence is 5′ to 3′ on the non-target strand (NTS)]. The ‘consensus’ PAMs refer to the preferred PAMs that are associated with high cleavage efficiency.
Table 1.
PAMs for eight Cas9 and two Cas12a proteins
| Cas | Type | Species | Length | Consensus PAM | PDB (resolution) | PDB PAM |
|---|---|---|---|---|---|---|
| SpCas9 | II-A | Streptococcus pyogenes | 1368 | NGG | 4UN3 (2.59 Å) 5F9R (3.40 Å) |
TGG TGG |
| SaCas9 | II-A | Staphylococcus aureus | 1053 | NNGRRT | 5AXW (2.70 Å) 5CZZ (2.60 Å) |
TTGGGT TTGAAT |
| St1Cas9 | II-A | Streptococcus thermophilus | 1121 | NNRGAA | 6M0W (2.76 Å) | AAAGAA |
| FnCas9 | II-B | Francisella novicida | 1629 | NGG | 5B2O (1.70 Å) | TGG |
| Nme1Cas9 | II-C | Neisseria meningitidis | 1082 | NNNNGATT | 6JDV (3.10 Å) | ATATGATT |
| Nme2Cas9 | II-C | Neisseria meningitidis | 1082 | NNNNCC | 6JE3 (2.93 Å) | AGGCCC |
| CdCas9 | II-C | Corynebacterium diphtheriae | 1084 | NNRHHHY | 6JOO (2.90 Å) | GGGTAAT |
| AceCas9 | II-C | Acidothermus cellulolyticus | 1138 | NNNCC | 6WBR (2.91 Å) | ATACC |
| AsCas12a | V-A | Acidaminococcus sp. | 1307 | TTTV | 5B43 (2.80 Å) | TTTA |
| LbCas12a | V-A | Lachnospiraceae bacterium ND2006 | 1228 | TTTV | 5XUS (2.50 Å) | TTTA |
For SpCas9, two structures determined at different resolutions, which represent the catalytically inactive and active states, respectively, were chosen (PDB ID: 4UN3 and 5F9R). For SaCas9, two structures (5AXW and 5CZZ) with different PDB PAMs were considered. One structure was used for each of the remaining six Cas9 and two Cas12a proteins (i.e. FnCas9, Nme1Cas9, Nme2Cas9, CdCas9, St1Cas9, AceCas9, AsCas12a and LbCas12a). We did not consider the minimal Cas9 from Campylobacter jejuni (CjCas9, 984 amino acids) because its PAM determined in different studies are not consistent [33–36].
Protein structures provide direct insights to understand how Cas amino acids recognize PAMs. Based on the degree of homology between Cas9 proteins and the presence/absence of an additional Cas protein besides Cas1, Cas2 and Cas9, the type II CRISPR–Cas9 systems are subdivided into three subtypes (II-A, II-B and II-C) [37]. Below we summarize the PAM-involved interactions in the order of II-A, II-B and II-C Cas9 proteins, followed by the two Cas12a proteins.
In the two structures of the type II-A SpCas9 [38, 39], whose PAM preference is NGG, it is noted that while the second G and third G of the PDB PAM form bidentate hydrogen bonds with Arg1333 and Arg1335, respectively, the first T does not form direct contacts with amino acids (Figure 2A and B).
Figure 2.

Visualization of the interactions between PAM nucleotides and PAM-recognizing amino acids. (A) 4UN3 (SpCas9) with TGG PAM; (B) 5F9R (SpCas9) with TGG PAM; (C) 5AXW (SaCas9) with TTGGGT PAM; (D) 5CZZ (SaCas9) with TTGAAT PAM; (E) 6M0W (St1Cas9) with AAAGAA PAM; (F) 5B2O (FnCas9) with TGG PAM; (G) 6JDV (Nme1Cas9) with ATATGATT PAM; (H) 6JE3 (Nme2Cas9) with AGGCCC PAM; (I) 6JOO (CdCas9) with GGGTAAT PAM; (J) 6WBR (AceCas9) with ATACC PAM; (K) 5B43 (AsCas12a) with TTTA PAM and (L) 5XUS (LbCas12a) with TTTA PAM. Nucleotides on nontarget and target strands are shown as yellow and magenta sticks, respectively; nontarget-stranded nucleotides are marked with asterisks. Amino acids are shown in green sticks. Hydrogen bonds are shown as green dashed lines.
Similarly, in the two structures of the type II-A SaCas9 (Figure 2C and D) [40], which has a PAM preference of NNGRRT, the first two T’s of the PDB PAM do not form direct interactions or water-mediated interactions with amino acids, whereas the third G forms bidentate hydrogen bonds with Arg1015, the fourth G or A forms one hydrogen bond with Asn985, the fifth G or A forms two water-mediated hydrogen bonds with Asn985 and Asn986, respectively, and the sixth T forms a hydrogen bond with Arg991. Besides, Asn986 forms a water-mediated hydrogen bond with the phosphate oxygen atoms of the fifth G or A.
In the structure of the type II-A St1Cas9 (Figure 2E) [41], whose PAM preference is NNRGAA, the third A forms bidentate hydrogen bonds with Gln1084, which is positioned by Glu1057. The fourth G forms two hydrogen bonds with Lys1086. The fifth A and the T in pair with the sixth A form van der Waals interactions with Met1049.
FnCas9, a type II-B Cas9 protein, was reported to recognize both NGG and NGA with a much higher preference for NGG [42]. In the structure of FnCas9 (Figure 2F), the second G forms coplanar bidentate hydrogen bonds with Arg1585. The third G or A can form distorted bidentate hydrogen bonds or a single hydrogen bond with Arg1556.
Nme1Cas9 and Nme2Cas9 are type II-C Cas9 proteins in which both DNA strands are involved in PAM recognition [43]. Nme1Cas9 was reported to recognize promiscuous PAMs with a consensus on NNNNGATT [44–46]. Nme2Cas9 has a PAM preference for NNNNCC [47]. In the structure of Nme1Cas9 (Figure 2G) [43], the fifth G forms two hydrogen bonds with His1024. The sixth A forms one hydrogen bond with Thr1027, whereas its paired T can form a water-mediated hydrogen bond with Thr1027. The seventh and eighth T’s of PAM do not have contact with amino acids, but the paired A’s on the target strand (TS) form one and two hydrogen bonds with Asn1029 and Gln981, respectively. In the structure of Nme2Cas9 (Figure 2H) [43], the fifth C forms a hydrogen bond with Asp1028. The sixth C does not form contact with amino acids but the paired G forms bidentate hydrogen bonds with Arg1033.
CdCas9 is another type II-C Cas9 protein with a PAM preference for NNRHHHY [48]. In its reported structure (Figure 2I) [48], the third G forms a hydrogen bond with Arg1017. The fourth to seventh PAM nucleotides (TAAT) do not form any hydrogen bond with amino acids. The fourth T and the fifth–sixth A’s paired partners (two T’s) form van der Waals contacts with Phe1011, Pro1043, Leu1046 and Lys1015. The seventh T’s paired A forms a hydrogen bond with Lys1015.
In the structure of the type II-C AceCas9 (Figure 2J) [49], whose PAM preference is NNNCC, the fourth C forms a hydrogen bond with Glu1044, which is positioned by Arg1091, and the paired G forms a hydrogen bond with Arg1088. The fifth C’s paired G on the TS forms bidentate hydrogen bonds with Arg1091.
Compared to the Cas9 proteins, which usually recognize G-rich PAMs, the type V-A Cas12a proteins usually prefer T-rich PAMs [37, 50]. Both AsCas12a and LbCas12a prefer a canonical PAM of TTTV (V = A, G or C) [50–52]. In the structure of AsCas12a (Figure 2K) [53], the first T is surrounded by the side-chain methyl groups of Thr167 and Thr539, whereas the paired A forms a hydrogen bond with the side chain of Lys607. The second T forms a van der Waals interaction with the side-chain methyl group of Thr167, whereas the paired A forms hydrogen bonds with Lys607 and Lys548, respectively. The third T forms a hydrogen bond with Lys607, whereas the nucleobase and deoxyribose moieties of the paired A form van der Waals interactions with the side chain of Lys607 and Pro599/Met604, respectively. The fourth A and its paired T do not form base-specific contacts with the Cas12a protein, consistent with the lack of specificity at the fourth position of the TTTV PAM.
In the structure of LbCas12a (Figure 2L) [52], the first T forms van der Waals contacts with the side chains of Thr149 and Gln529, whereas the paired A forms a hydrogen bond with Lys595. The 5-methyl group of the second T is in the vicinity of the side-chain methyl group of Thr149, whereas the paired A forms hydrogen bonds with Lys595 and Lys538. The third T forms a hydrogen bond with Lys595, whereas the paired A forms a hydrogen bond with Tyr542. Similar to that in AsCas12a, the fourth A and its paired T do not form base-specific contacts with the protein, supporting the overall lack of specificity at the fourth position.
In sum, these reported CRISPR–Cas structures provide direct visual clues to the key interactions, commonly through hydrogen bonds or van der Waals forces, between PIAAs and PAMs.
Computational modeling reveals that native PIAAs prefer consensus PAMs
We proceeded to develop a CPD method to study the interaction between the CRISPR–Cas protein’s PIAAs and their preferred PAMs on the target DNAs. We defined the amino acids that are in direct contact (<4.5 Å) with the side chains of PAMs or the paired nucleotides as PIAAs (Supplementary Table 1). Of note, PIAAs defined by this simple rule contained all PAM-recognizing residues described in the above section.
For each PDB, its PAM nucleotides were mutated to generate all 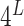 PAM variants, where
PAM variants, where 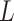 is PAM length. For example, the consensus PAM of SpCas9 is NGG, and we generated all 64 (
is PAM length. For example, the consensus PAM of SpCas9 is NGG, and we generated all 64 (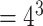 ) variants from AAA to TTT. The number of variants increases exponentially as
) variants from AAA to TTT. The number of variants increases exponentially as 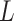 grows. Too many structures need to be generated for very long PAMs, e.g. 16 384 and 65 536 structures for CdCas9 and Nme1Cas9 that have seven and eight PAM nucleotides, respectively. Thus, only the last six PAM positions were varied if
grows. Too many structures need to be generated for very long PAMs, e.g. 16 384 and 65 536 structures for CdCas9 and Nme1Cas9 that have seven and eight PAM nucleotides, respectively. Thus, only the last six PAM positions were varied if 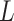 > 6, resulting in a handleable maximum of 4096 structures (this is reasonable because the first two nucleotides form no contact with amino acids in CdCas9 or Nme1Cas9). When PAM nucleotides on NTS were mutated, the paired nucleotides on TS were correspondingly substituted to ensure complementarity.
> 6, resulting in a handleable maximum of 4096 structures (this is reasonable because the first two nucleotides form no contact with amino acids in CdCas9 or Nme1Cas9). When PAM nucleotides on NTS were mutated, the paired nucleotides on TS were correspondingly substituted to ensure complementarity.
We first computationally examined the preference of native Cas proteins for all PAM variants. This is to validate whether UniDesign can recapitulate such PAM preference for each of the ten Cas proteins. To do this, we used UniDesign to repack the PIAAs in the Cas/gRNA/DNA complex model of each PAM variant and calculate the total binding energy. The ‘total energy’ represents the energy of the entire system including protein, gRNA, DNA and water molecules (if applicable). The ‘binding energy’ refers to the interaction between protein/gRNA and DNA, which is important to PAM recognition.
We reasoned that the binding energy is a good indicator for quantifying to what extent a Cas protein prefers a PAM (lower binding energy indicates a higher preference). Interestingly, for each Cas, the energy associated with variants with consensus PAMs distributed in a relatively narrow window to the minimum binding energy (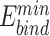 ) and/or the minimum total energy (
) and/or the minimum total energy (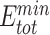 ) (Figure 3). Through structure inspection, we noticed that some variants scored low binding energy at the cost of high total energy (LbHt) because of inter-residue steric clashes. Mathematically, we defined a PAM as LbHt if its associated total energy is greater than a threshold (
) (Figure 3). Through structure inspection, we noticed that some variants scored low binding energy at the cost of high total energy (LbHt) because of inter-residue steric clashes. Mathematically, we defined a PAM as LbHt if its associated total energy is greater than a threshold (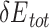 ) of the minimum total energy (
) of the minimum total energy (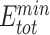 ). Because the PNI interactions between these LbHt PAMs and the corresponding Cas9s may not be physically stable/feasible, we filtered them out for subsequent computations.
). Because the PNI interactions between these LbHt PAMs and the corresponding Cas9s may not be physically stable/feasible, we filtered them out for subsequent computations.
Figure 3.

UniDesign computed total versus binding energy for all PAM variants on different Cas protein scaffolds. (A) 4UN3 (SpCas9); (B) 5F9R (SpCas9); (C) 5AXW (SaCas9); (D) 5CZZ (SaCas9); (E) 6M0W (St1Cas9); (F) 5B2O (FnCas9); (G) 6JDV (Nme1Cas9); (H) 6JE3 (Nme2Cas9); (I) 6JOO (CdCas9); (J) 6WBR (AceCas9); (K) 5B43 (AsCas12a) and (L) 5XUS (LbCas12a). The variants with consensus PAMs are colored in red while the others are in gray. UEU, UniDesign energy units.
Only PAM variants that satisfy 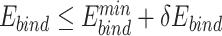 and
and 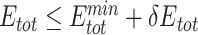 were subjected to sequence logo analysis using WebLogo [54] with appropriate
were subjected to sequence logo analysis using WebLogo [54] with appropriate 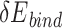 and
and 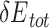 parameters;
parameters; 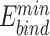 and
and 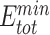 were yielded from UniDesign calculations (Supplementary Table 2). For comparison, sequence logos were also plotted with
were yielded from UniDesign calculations (Supplementary Table 2). For comparison, sequence logos were also plotted with 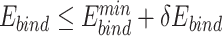 or
or 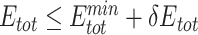 (Supplementary Figures 1 and 2).
(Supplementary Figures 1 and 2).
For both SpCas9 structures, the computationally determined preferred PAMs are NGG (Figure 4A and B), consistent with the experimentally determined NGG consensus.
Figure 4.
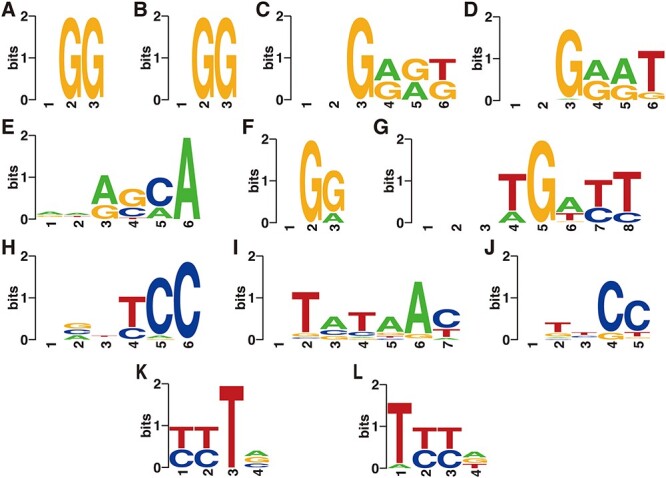
UniDesign predicted consensus PAMs for native Cas proteins. (A) 4UN3 (SpCas9); (B) 5F9R (SpCas9); (C) 5AXW (SaCas9); (D) 5CZZ (SaCas9); (E) 6M0W (St1Cas9); (F) 5B2O (FnCas9); (G) 6JDV (Nme1Cas9); (H) 6JE3 (Nme2Cas9); (I) 6JOO (CdCas9); (J) 6WBR (AceCas9); (K) 5B43 (AsCas12a) and (L) 5XUS (LbCas12a). Note that for Nme1Cas9 and CdCas9, the first two and one positions were excluded from PAM variant generation, respectively, and the ‘-’ symbols were manually added at these positions for plotting sequence logos.
For both SaCas9 structures, the computationally determined PAM preference is NNGRR[T > G] (Figure 4C and D), also matching the experimental consensus NNGRRT and the report that the sixth position tolerates other nucleotides albeit it prefers T most [55, 56].
For St1Cas9, the UniDesign computed PAM is NNR[G ≈ C][C ≈ A]A (Figure 4E), covering the experimentally determined consensus NNRGAA [41]. In the computational model with a UniDesign suggested PAM ATACCA, the fourth C- and the fifth C-paired G’s formed hydrogen bonds with Ser1082 and K1086, respectively. Of note, the conformations of Ser1082 and K1086 in our UniDesign modeling by using the ATACCA PAM were quite different from those in the reported PDB model with a PAM of AAAGAA.
For FnCas9, UniDesign obtains a computational preference for the NG[G > A] PAM (Figure 4F), consistent with the experimentally determined PAM NG[G > A] [42].
For Nme1Cas9, the UniDesign-computed preferred PAM is NNNTGATT (Figure 4G), which is a subset of the experimentally determined consensus NNNNGATT [44–46, 57]. UniDesign modeling computationally confirmed that Nme1Cas9 prefers only G at the fifth position. Further, UniDesign predicted T, C and C as the second favorable nucleotides at the sixth to eighth positions, respectively. Besides, UniDesign suggested NNNNGACT, NNNNGCTT, NNNNGTCT and NNNNGTTT as alternative PAMs for Nem1Cas9 (Figure 4G), which corroborates with the findings by Amrani et al [44]. We note that there is a discrepancy at the fourth PAM position where UniDesign favors T and A but the experimental studies do not indicate any preference. Interestingly, when the restraint on total energy was removed (e.g. 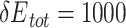 ) in UniDesign, the preference at the fourth PAM position is gone and the modeled consensus PAM becomes NNNNGNNT (Supplementary Figure 1), the same as reported by Esvelt et al. in their study [57] (see Table 1 in the ref.).
) in UniDesign, the preference at the fourth PAM position is gone and the modeled consensus PAM becomes NNNNGNNT (Supplementary Figure 1), the same as reported by Esvelt et al. in their study [57] (see Table 1 in the ref.).
For Nme2Cas9, the UniDesign-modeled PAM is N[G/C/A]N[T/C]CC with dominating C’s at the fifth and sixth positions (Figure 4H), largely consistent with the experimental PAM NNNNCC [43, 47]. UniDesign modeling suggests that the second position prefers equally G, C and A but not T, in part due to the steric clashes between the 5-methyl group on T and Lys1044. Besides, computational modeling suggests that the fourth position prefers T or C probably because the paired A or G on the TS can form a potential hydrogen bond with Tyr1035.
For CdCas9, the UniDesign-modeled PAM is NTATAAY (Figure 4I), a subset of the experimental consensus NNRHHHY [48]. Computational modeling indicates that the third position prefers A > C ≈ G but not T, compared to the preference for G ≈ A > C but not T determined experimentally [48]. The most discrepancy takes place at the second PAM position, where computation suggests a preference of T > G while experimental assay shows that all nucleotides are acceptable with a slight preference of G over others [48].
For AceCas9, UniDesign predicted the PAM preference as NTNCC (Figure 4J), similar to the experimentally determined PAM NNNCC [49]. Computational modeling shows that the fourth and fifth positions strongly prefer C’s while the second position marginally favors T.
For AsCas12a, the UniDesign predicted consensus PAM sequence is [T ≈ C][T ≈ C]TV (Figure 4K), covering the experimentally determined TTTV PAM. Interestingly, it has been shown that besides the TTTV PAM, AsCas12a also recognizes non-canonical C-containing PAMs, especially when the C nucleotides appear at the first two positions [52].
For LbCas12a, the UniDesign preferred consensus PAM is T[T ≈ C][T ≈ C][A ≈ G ≈ T], largely recapitulating the experimentally confirmed TTTV PAM. In addition to the canonical TTTV PAM, previous experimental studies showed that the second and third positions tolerate the C nucleotides very well [50, 52], and as shown, these characteristics were successfully recapitulated by UniDesign.
Compared with the sequence logos with 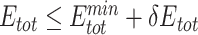 alone, the plots with
alone, the plots with 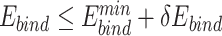 alone better recapitulated those with both
alone better recapitulated those with both 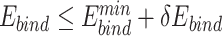 and
and 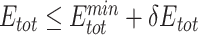 (Figure 4, Supplementary Figures 1 and 2), suggesting that UniDesign binding energy is a better indicator than total energy for PAM preference modeling.
(Figure 4, Supplementary Figures 1 and 2), suggesting that UniDesign binding energy is a better indicator than total energy for PAM preference modeling.
In sum, the computationally predicted consensus PAM profiles by UniDesign largely recapitulated the experimentally determined consensus PAMs (Figure 4 and Table 1). Our data demonstrate that UniDesign is a useful tool to quantitatively interrogate the molecular level integrations between the native PIAAs and different PAM variants.
Computational redesign of Cas proteins reveals that PDB PAMs prefer native PIAAs
On the other hand, we ask the question if the native PIAAs are the most preferred amino acids by PDB PAMs and more generally, the consensus PAMs. We first used UniDesign to redesign the PIAAs for each Cas protein with a fixed PDB PAM. In the redesign process, the evolutionary module was disabled and only UniEF was used for energy calculation. The results are summarized in Table 2 and described below.
Table 2.
Recapitulation of PAM-interacting amino acids by UniDesign with PDB PAMs
| Cas | PDB | Redesigned PIAA residue types | Recovery | Similarity |
|---|---|---|---|---|
| SpCas9 | 4UN3 | K1107K, E1219E, R1333R, R1335R | 4/4 | 4/4 |
| 5F9R | K1107K, E1219E, R1333R, R1335R | 4/4 | 4/4 | |
| SaCas9 | 5AXW | N985N, N986N, L989L, R991R, R1002K, R1015R | 5/6 | 6/6 |
| 5CZZ | N985N, N986N, L989H, R991R, R1002K, R1015R | 4/6 | 5/6 | |
| St1Cas9 | 6M0W | F673Y, T1048T, M1049Q, Y1055Y, S1082S, Q1084Q, K1086Q | 4/7 | 5/7 |
| FnCas9 | 5B2O | R1241R, E1449N, D1472D, S1473S, R1474R, S1555S, R1556R, R1585R, Y1586Y | 8/9 | 8/9 |
| Nme1Cas9 | 6JDV | Q981Q, H1024H, T1027S, N1029T, E1048E, G1049G | 4/6 | 5/6 |
| Nme2Cas9 | 6JE3 | K843K, D1028E, R1033R, Y1035W, K1044K | 3/5 | 5/5 |
| CdCas9 | 6JOO | I47R, K818K, F1011F, K1015Q, R1017R, R1042K, P1043E, L1046L | 4/8 | 5/8 |
| AceCas9 | 6WBR | R55R, E1044E, R1048R, R1088R, R1091R | 5/5 | 5/5 |
| AsCas12a | 5B43 | T167S, T539Q, K548R, P599P, K603K, M604M, K607K | 4/7 | 6/7 |
| LbCas12a | 5XUS | T149T, Q529Q, K538K, Y542Y, P587P, K591Q, M592M, K595K | 7/8 | 7/8 |
| Total | N/A | N/A | 48(47)/65 | 56(55)/65 |
Only one scaffold for SpCas9 and SaCas9 was counted for calculating the total recovery and similarity rates. The values outside and inside the parentheses were obtained with scaffold 5AXW or 5CZZ counted, respectively.
All PIAAs for SpCas9 (4UN3 and 5F9R) and AceCas9 (6WBR) were successfully recovered. UniDesign confirmed that the native PIAAs in both Cas9s led to the lowest energy scores.
For SaCas9, four PIAAs, i.e. Asn985, Asn986, Arg991 and Arg1015 were chosen as native types, whereas Arg1002 was designed into the physiochemically similar Lys on both scaffolds (5AXW and 5CZZ). An extra mutation, i.e. L989 → H, was chosen on 5CZZ.
For St1Cas9 (6M0W), four out of seven native PIAAs, i.e. Thr1048, Tyr1055, Ser1082, Gln1084 and one physiochemically similar mutation F673 → Y were suggested by UniDesign. Computational modeling suggested that the mutation M1049 → Q formed a 2.9 Å hydrogen bond with the PAM’s sixth A, and K1086 → Q formed two hydrogen bonds (3.0 and 3.2 Å, respectively) with the fourth G of PAM, whereas in the experimental structure, the native Lys1086 also forms two hydrogen bonds (2.8 and 2.4 Å, respectively) with the PAM’s fourth G.
For FnCas9 (5B2O), eight out of nine native PIAAs were picked by UniDesign, except residue Glu1449. UniDesign suggested Asn for this position.
For Nme1Cas9 (6JDV), four out of the six native PIAAs were recapitulated by UniDesign. At the other two PIAAs (Thr1027 and Asn1029), UniDesign suggested Ser and Thr, respectively, both of which do not change amino-acid polarity.
Of the five native PIAAs in Nme2Cas9 (6JE3), three were recapitulated by UniDesign; the two differences were at D1028 → E and Y1035 → W, where UniDesign suggested residues with similar properties.
For CdCas9 (6JOO), four of the eight native PIAAs were suggested by UniDesign. For the other four PIAAs, UniDesign suggested Lys instead of the native Arg at position 1042, Gln instead of the native Lys at position 1015 and Arg instead of the native Ile at position 47. The UniDesign suggested Arg47 is located at a solvent-exposed position that can form π–π stacking interaction with PAM nucleotide G1.
For AsCas12a (5B43), four out of the seven native PIAAs were recovered by UniDesign. The remaining three mutations were T167 → S, T539 → Q and K548 → R, two of which chose similar amino acids through UniDesign modeling.
For LbCas12a (5XUS), only one of the eight native PIAAs was missed, i.e. K591 → Q. Structure shows that K591 protrudes into the bulky solvent and does not form any contact with the PAM nucleotides. Thus, this residue can be difficult to predict due to fewer geometrical restraints.
Overall, 48/65 (74%) native PIAAs from all eight Cas9s were recapitulated by UniDesign. Considering the similarity of the amino acids (e.g. R ↔ K, F ↔ Y ↔ W, D ↔ E and S ↔ T), 56/65 (86%) of the PIAAs could be regarded as recovered (see Table 2). Such high recapitulation rates indicate that PDB PAMs show a high preference for native or native-like PIAAs and that UniDesign is a reliable tool to predict PIAAs given a fixed PAM.
UniDesign analysis suggests that native PIAAs are overall sufficient for consensus PAMs and that in some cases PIAAs need to change to accommodate different PAM sequences
For practical reasons, not all the PAMs will be used in structural biology experiments to determine their integrations with the PIAAs. In this regard, a CPD method, such as UniDesign, serves as an alternative to structural biology experiments to scan all consensus PAMs and provide computational insights into their interactions with the PIAAs. In particular, we wonder if, for each different PAM (e.g. AGG versus CGG), a different combination of PIAAs will be required for favorable Cas–DNA binding to achieve high gene-editing efficiency.
Here for each Cas structure, the models bearing all possible consensus PAM variants were used as a scaffold to interrogate the effects of mutations of the PIAAs on the energy scores in UniDesign. The results from different PAM variants were combined for sequence logo analysis. For instance, four PAM variants of SpCas9 (PAM: NGG) with AGG, CGG, GGG or TGG PAMs were used for PIAA redesign individually. Similarly, 64 (= 4 × 4 × 2 × 2) PAM variants for SaCas9 (PAM: NNGRRT) were fed into UniDesign to redesign the corresponding PIAAs.
The UniDesign results were then used to generate sequence logo plots. It is shown that in general, for most Cas proteins, especially SpCas9, SaCas9, FnCas9, Nme1Cas9 and LbCas12a, all or a very high ratio of native PIAAs were computationally recapitulated based on the UniDesign computed energy scores, indicating that native or native-like PIAAs are overall sufficient for all consensus PAM variants (Figure 5). Compared with the PIAAs recapitulation results with PDB PAMs (Table 2), identical design results were obtained for SpCas9 (Figure 5A and B), SaCas9 (Figure 5C and D), Nme1Cas9 (Figure 5G) and AsCas12a (Figure 5K) with non-PDB, consensus PAMs.
Figure 5.

UniDesign predicted PAM-interacting amino acids based on consensus PAM-bearing Cas protein variants. (A) 4UN3 (SpCas9) with four consensus PAM variants; (B) 5F9R (SpCas9) with four consensus PAM variants; (C) 5AXW (SaCas9) with 64 consensus PAM variants; (D) 5CZZ (SaCas9) with 64 consensus PAM variants; (E) 6M0W (St1Cas9) with 32 consensus PAM variants; (F) 5B2O (FnCas9) with four consensus PAM variants; (G) 6JDV (Nme1Cas9) with 16 consensus PAM variants; (H) 6JE3 (Nme2Cas9) with 256 consensus PAM variants; (I) 6JOO (CdCas9) with 432 consensus PAM variants; (J) 6WBR (AceCas9) with 64 consensus PAM variants; (K) 5B43 (AsCas12a) with three consensus PAM variants and (L) 5XUS (LbCas12a) with three consensus PAM variants. Note that 16 and 432 consensus PAM variants were obtained for Nme1Cas9 and CdCas9 because their first two or one PAM positions were not varied, respectively.
It should also be noted that for some Cas proteins, e.g. Nme2Cas9 (Figure 5H), CdCas9 (Figure 5I), AceCas9 (Figure 5J) and LbCas12a (Figure 5L), UniDesign suggested specific PIAAs changes upon PAM variations although native or native-like amino acids were still dominating at most positions. This means that native PIAAs may not always best fit all PAMs equally even though they are still considered consensus PAMs.
Collectively, these results indicate that, although non-PDB, consensus PAMs still highly prefer native or native-like PIAAs, a single sequence of PIAAs may not tolerate all consensus PAM variants equally, thus suggesting mutation studies on PIAAs to accommodate different PAM variations. UniDesign can serve as a useful computational tool for this purpose.
Comparison with Rosetta on redesigning PIAA residues
As a comparison, we carried out the same PIAA redesign study with Rosetta [31, 32], another widely used CPD method, based on either PDB PAMs or more generally, the non-PDB, consensus PAMs. The results showed that building on PDB PAMs, Rosetta predicted only 19/65 (29%) or 22/65 (34%) of the PIAA residues in terms of amino acid identity or similarity (Supplementary Table 3), much lower than those achieved by UniDesign (Table 2). Similar to its poor performance in recovering native PIAAs with PDB PAMs, Rosetta also largely failed to recapitulate the native or native-like PIAAs given the non-PDB, canonical PAM variants (Supplementary Figure 3). Regarding computation speed, the two programs, UniDesign and Rosetta, were similarly fast on the PIAA redesign tasks, with UniDesign slightly faster (Supplementary Table 4).
DISCUSSION
Understanding the PAM recognition process at the structural biology level is critical to engineering CRISPR–Cas proteins toward relaxed or altered PAM requirements, which have direct impacts on their applications in biotechnology and medicine. To this end, current studies are typically conducted as follows: first, determining the consensus PAMs; second, solving the Cas/gRNA/DNA complex structure(s) to elucidate Cas–PAM interactions; and third, structure-guided engineering of PAM requirement. In the second stage, the DNA substrate usually contained the most preferred PAM (e.g. PDB PAM as defined in this study). Despite these useful and insightful studies, a comprehensive and quantitative understanding of the relationship between a Cas protein and its consensus PAMs is still missing, hindering the systematic computational design of Cas variants with modified PAM requirements. To our knowledge, COMET is the only computational workflow designed for CRISPR–Cas PAM engineering to date [21]. But this method has not been widely used at least partially because it is highly resource-costing.
In this study, we aim to develop a universal, easy-to-use CPD approach to model PNI accurately and efficiently. To test UniDesign’s effectiveness, we used it to model PAM recognition for eight Cas9 and two Cas12a proteins. We noted that, given native PIAAs, the whole Cas/gRNA/DNA system with a consensus PAM, in general, had relatively lower predicted UniDesign binding energy (and total energy) (Figure 3), suggesting that PIAAs have a preference for consensus PAMs. This is consistent with experiments that the Cas protein initiates DNA interrogation through specific recognition of preferred PAMs [58, 59]. By setting 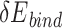 and
and 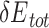 thresholds appropriately, we found that the predicted low-energy PAM variants to a large extent recapitulated the preferred consensus PAM profiles as determined by experiments (Figure 4). Also, we showed that the UniDesign binding energy (together with
thresholds appropriately, we found that the predicted low-energy PAM variants to a large extent recapitulated the preferred consensus PAM profiles as determined by experiments (Figure 4). Also, we showed that the UniDesign binding energy (together with 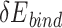 ) was a better descriptor for PAM preference than the total energy (together with
) was a better descriptor for PAM preference than the total energy (together with 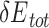 ) (Figure 4, Supplementary Figures 1 and 2). Conversely, we noted that, given PDB PAMs or consensus PAMs, computational redesign of PIAAs by UniDesign largely recapitulated the naturally occurring amino acids at these positions (Table 2 and Figure 5), suggesting that consensus PAMs also strongly favor native PIAA residues. Thus, computational modeling of the Cas–PAM recognition by the UniDesign reveals the inherent mutual preference between consensus PAMs and native PIAAs resulting from long-time evolution. It should be noted that although the UniEF was trained only on protein monomers and PPIs, the Cas–PAM recognition modeling results suggested that the prototype UniDesign generally works on PNIs.
) (Figure 4, Supplementary Figures 1 and 2). Conversely, we noted that, given PDB PAMs or consensus PAMs, computational redesign of PIAAs by UniDesign largely recapitulated the naturally occurring amino acids at these positions (Table 2 and Figure 5), suggesting that consensus PAMs also strongly favor native PIAA residues. Thus, computational modeling of the Cas–PAM recognition by the UniDesign reveals the inherent mutual preference between consensus PAMs and native PIAAs resulting from long-time evolution. It should be noted that although the UniEF was trained only on protein monomers and PPIs, the Cas–PAM recognition modeling results suggested that the prototype UniDesign generally works on PNIs.
Another point we would like to discuss is how to use UniDesign to engineer Cas proteins with relaxed or altered PAMs. Based on the above analysis, we speculate that sufficient binding interaction between the Cas protein and the PAM is a prerequisite for DNA interrogation initiation. As a consequence, the UniDesign binding and total energy of Cas/gRNA/DNA with PDB PAM and native PIAAs can be used as a reference/baseline. When PAM is altered (even though it is still a consensus PAM), native PIAAs may not fit the modified PAM best, resulting in a binding loss (e.g. reduced binding energy). Thus, the PIAA residues may need to be redesigned to accommodate the changed PAM, and meanwhile, other PAM-surrounding amino acids may also need to be redesigned to enhance nonspecific interactions between Cas and PAM to make up for the lost binding affinity. UniDesign can automate this design process to generate variants for further analysis. Also, it should be noted that UniDesign requires only minimal computational resources and can run efficiently on a personal computer and different operating systems. For instance, for the PIAA redesign tasks of each Cas ortholog, only a single CPU (Intel(R) Xeon(R) Gold 6140 CPU @ 2.30 GHz) with a 768 MB memory was used by UniDesign, and the average computation time ranged from about 3 to 6 s, depending on a Cas protein’s length and the number of PIAA residues to be calculated (Supplementary Table 4).
We also want to point out some possible limitations of UniDesign and this study. First, the PAM variant modeling and PIAA repacking/redesign are based on fixed protein and/or nucleic acid backbone(s). Certain variations of PAMs or PIAAs may result in large backbone conformational changes that are not captured by UniDesign, which may have an unexpected impact on PAM modeling and Cas protein engineering. One possible solution is to combine UniDesign with MD approaches by using UniDesign to narrow down a list of the most promising Cas variants followed by MD simulations to elucidate their effects on desired functions. Second, we note that there are some variations between the experimentally determined PAMs and the UniDesign predicted consensus PAMs. For instance, there is a discrepancy at the fourth PAM position of Nme1Cas9 between the experimental data (e.g. N) and predicted results (e.g. T and A). One possible reason is that we used the polar-hydrogen CHARMM19 force field in UniEF/UniDesign to achieve a very high computation speed, but its accuracy may not be as good as the all-atom force fields such as AMBER [60] or CHARMM36 [30]. We are incorporating these force fields into UniDesign and will benchmark them in the follow-up studies.
In sum, we report UniDesign as a universal CPD approach for PNI modeling and design. We demonstrate UniDesign’s effectiveness by applying it to decode Cas–PAM recognition quantitatively for eight Cas9 and two Cas12a proteins. This work represents the first systematic computational modeling on PAM recognition that can provide new insights for PAM engineering. We expect that UniDesign will serve as an important tool for CRISPR–Cas protein engineering in the field.
Key Points
We report UniDesign as a new universal computational protein design (CPD) framework for protein–nucleic acid interaction modeling and design.
UniDesign is the first systematic CPD method for engineering CRISPR–Cas protein’s PAM requirements and achieved good performance on diverse Cas proteins.
UniDesign accurately modeled the mutual preference between natural PAMs and native PAM-interacting amino acids caused by long-term evolution.
UniDesign is fully open-sourced, computationally efficient and inexpensive, and can run on personal computers and different operating systems.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Y.E.C. laboratory for their insightful discussions and suggestions for this study.
Author Biographies
Xiaoqiang Huang received his BS, MS, and PhD degrees from Tsinghua University. He is currently a Research Investigator at the University of Michigan. He is working in the field of bioinformatics and computational biology and his research is primarily focused on computational protein design and engineering.
Jun Zhou received the dual BSC degrees from the University of Michigan and China Pharmaceutical University in 2021. He is currently a PhD student in pharmacology at the University of Michigan. His research area focuses on gene editing therapy for cardiovascular diseases.
Dongshan Yang is an Assistant Professor at University of Michigan. His research focuses on development of genetic engineered large animal models for human diseases and development of novel gene editing therapy.
Jifeng Zhang received his PhD degree in Cardiovascular Pathophysiology from the University of Yamanashi. He is an Associate Professor in the Frankel Cardiovascular Center at the University of Michigan. His research aims to understand the molecular mechanisms of cardiovascular diseases, including atherosclerosis and abdominal aortic aneurysm.
Xiaofeng Xia received his BS and PhD degrees from Tsinghua University. He is the CEO of ATGC Inc. His research focuses on novel gene editing techniques, embryonic stem cell technologies and antibody therapeutics.
Yuqing Eugene Chen is the Frederick Huetwell Professor of Cardiovascular Medicine at the University of Michigan. His research is focused on using human genetic discoveries and gene-editing techniques to improve the diagnosis and treatment of cardiovascular disease through translational research.
Jie Xu received his BS degree from Tsinghua University and PhD degree from the University of Connecticut. He is an Associate Professor of Internal Medicineat the University of Michigan. His research focuses on the development of gene edited animal models for biomedical research, as well as the improvement of the gene editing tools.
Contributor Information
Xiaoqiang Huang, Center for Advanced Models for Translational Sciences and Therapeutics, Department of Internal Medicine, University of Michigan Medical School, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Jun Zhou, Center for Advanced Models for Translational Sciences and Therapeutics, Department of Internal Medicine, University of Michigan Medical School, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Dongshan Yang, Center for Advanced Models for Translational Sciences and Therapeutics, Department of Internal Medicine, University of Michigan Medical School, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Jifeng Zhang, Center for Advanced Models for Translational Sciences and Therapeutics, Department of Internal Medicine, University of Michigan Medical School, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Xiaofeng Xia, Research & Development, ATGC Inc., 100 E Lancaster Avenue, LIMR Building Lab 129, Wynnewood, PA 19096, USA.
Yuqing Eugene Chen, Center for Advanced Models for Translational Sciences and Therapeutics, Department of Internal Medicine, University of Michigan Medical School, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
Jie Xu, Center for Advanced Models for Translational Sciences and Therapeutics, Department of Internal Medicine, University of Michigan Medical School, 2800 Plymouth Road, Ann Arbor, MI 48109, USA.
FUNDING
This research was funded by the Cystic Fibrosis Foundation (Grant No. XU19XX0 to J.X.) and the National Institutes of Health (Grant No. GM149016 to X.H. and X.X., HL159900, HL147527, HL159871 to Y.E.C. and GM122181 to J.Z.). This research was supported in part through computational resources and services provided by Advanced Research Computing at the University of Michigan, Ann Arbor.
CODE AVAILABILITY
UniDesign is freely available at https://github.com/tommyhuangthu/UniDesign. All Perl scripts for this work are available at https://doi.org/10.5281/zenodo.7426026.
DATA AVAILABILITY
All computational data, including preprocessed Cas structure PDB files, PAM variant models, PIAAs repacked and redesigned models, RESFILEs, a summary of PAM recognition and PIAA redesign results are available at https://doi.org/10.5281/zenodo.7426026.
References
- 1. Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods 2013;10:957–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014;157:1262–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Komor AC, Badran AH, Liu DR. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell 2017;168:20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang X, Yang D, Zhang J, et al. . Recent advances in improving gene-editing specificity through CRISPR–Cas9 nuclease engineering. Cell 2022;11:2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cong L, Ran FA, Cox D, et al. . Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collias D, Beisel CL. CRISPR technologies and the search for the PAM-free nuclease. Nat Commun 2021;12:555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walton RT, Christie KA, Whittaker MN, et al. . Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020;368:290–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishimasu H, Shi X, Ishiguro S, et al. . Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 2018;361:1259–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu JH, Miller SM, Geurts MH, et al. . Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018;556:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kleinstiver BP, Prew MS, Tsai SQ, et al. . Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 2015;33:1293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tian Y, Huang X, Li Q, et al. . Computational design of variants for cephalosporin C acylase from pseudomonas strain N176 with improved stability and activity. Appl Microbiol Biotechnol 2017;101:621–32. [DOI] [PubMed] [Google Scholar]
- 12. He J, Huang X, Xue J, et al. . Computational redesign of penicillin acylase for cephradine synthesis with high kinetic selectivity. Green Chem 2018;20:5484–90. [Google Scholar]
- 13. Hettiaratchi MH, O’Meara MJ, O’Meara TR, et al. . Reengineering biocatalysts: computational redesign of chondroitinase ABC improves efficacy and stability. Sci Adv 2020;6:eabc6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cao L, Coventry B, Goreshnik I, et al. . Design of protein-binding proteins from the target structure alone. Nature 2022;605:551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cao L, Goreshnik I, Coventry B, et al. . De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020;370:426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goldenzweig A, Goldsmith M, Hill SE, et al. . Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol Cell 2016;63:337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khersonsky O, Lipsh R, Avizemer Z, et al. . Automated design of efficient and functionally diverse enzyme repertoires. Mol Cell 2018;72:178–186.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ashworth J, Havranek JJ, Duarte CM, et al. . Computational redesign of endonuclease DNA binding and cleavage specificity. Nature 2006;441:656–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang X, Xue J, Zhu Y. Computational design of cephradine synthase in a new scaffold identified from structural databases. Chem Commun 2017;53:7604–7. [DOI] [PubMed] [Google Scholar]
- 20. Tian Y, Xu Z, Huang X, et al. . Computational design to improve catalytic activity of cephalosporin C acylase from pseudomonas strain N176. RSC Adv 2017;7:30370–5. [DOI] [PubMed] [Google Scholar]
- 21. Luan B, Xu G, Feng M, et al. . Combined computational-experimental approach to explore the molecular mechanism of SaCas9 with a broadened DNA targeting range. J Am Chem Soc 2019;141:6545–52. [DOI] [PubMed] [Google Scholar]
- 22. Pearce R, Huang X, Setiawan D, et al. . EvoDesign: designing protein-protein binding interactions using evolutionary interface profiles in conjunction with an optimized physical energy function. J Mol Biol 2019;431:2467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang X, Pearce R, Zhang Y. EvoEF2: accurate and fast energy function for computational protein design. Bioinformatics 2020;36:1135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shultis D, Mitra P, Huang X, et al. . Changing the apoptosis pathway through evolutionary protein design. J Mol Biol 2019;431:825–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang X, Pearce R, Zhang Y. De novo design of protein peptides to block association of the SARS-CoV-2 spike protein with human ACE2. Aging 2020;12:11263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang X, Pearce R, Zhang Y. FASPR: an open-source tool for fast and accurate protein side-chain packing. Bioinformatics 2020;36:3758–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang X, Pearce R, Zhang Y. Toward the accuracy and speed of protein side-chain packing: a systematic study on Rotamer libraries. J Chem Inf Model 2020;60:410–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berman HM, Battistuz T, Bhat TN, et al. . The Protein Data Bank. Acta Crystallogr Sec D Biol Crystallogr 2002;58:899–907. [DOI] [PubMed] [Google Scholar]
- 29. Brooks BR, Bruccoleri RE, Olafson BD, et al. . CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 1983;4:187–217. [Google Scholar]
- 30. Huang J, MacKerellAD, Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 2013;34:2135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alford RF, Leaver-Fay A, Jeliazkov JR, et al. . The Rosetta all-atom energy function for macromolecular modeling and design. J Chem Theory Comput 2017;13:3031–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leaver-Fay A, Tyka M, Lewis SM, et al. . ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 2011;487:545–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fonfara I, le Rhun A, Chylinski K, et al. . Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res 2014;42:2577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim E, Koo T, Park SW, et al. . In vivo genome editing with a small Cas9 orthologue derived from campylobacter jejuni. Nat Commun 2017;8:14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamada M, Watanabe Y, Gootenberg JS, et al. . Crystal structure of the minimal Cas9 from campylobacter jejuni reveals the molecular diversity in the CRISPR-Cas9 systems. Mol Cell 2017;65:1109–1121.e3. [DOI] [PubMed] [Google Scholar]
- 36. Nakagawa R, Ishiguro S, Okazaki S, et al. . Engineered campylobacter jejuni Cas9 variant with enhanced activity and broader targeting range. Commun Biol 2022;5:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shmakov S, Smargon A, Scott D, et al. . Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol 2017;15:169–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Anders C, Niewoehner O, Duerst A, et al. . Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014;513:569–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jiang F, Taylor DW, Chen JS, et al. . Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 2016;351:867–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nishimasu H, Cong L, Yan WX, et al. . Crystal structure of Staphylococcus aureus Cas9. Cell 2015;162:1113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Y, Zhang H, Xu X, et al. . Catalytic-state structure and engineering of Streptococcus thermophilus Cas9. Nature Catalysis 2020;3:813–23. [Google Scholar]
- 42. Hirano H, Gootenberg JS, Horii T, et al. . Structure and engineering of Francisella novicida Cas9. Cell 2016;164:950–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun W, Yang J, Cheng Z, et al. . Structures of Neisseria meningitidis Cas9 complexes in catalytically poised and anti-CRISPR-inhibited states. Mol Cell 2019;76:938–952.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amrani N, Gao XD, Liu P, et al. . NmeCas9 is an intrinsically high-fidelity genome-editing platform. Genome Biol 2018;19:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hou Z, Zhang Y, Propson NE, et al. . Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA 2013;110:15644–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee CM, Cradick TJ, Bao G. The Neisseria meningitidis CRISPR-Cas9 system enables specific genome editing in mammalian cells. Mol Ther 2016;24:645–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Edraki A, Mir A, Ibraheim R, et al. . A compact, high-accuracy Cas9 with a dinucleotide PAM for in vivo genome editing. Mol Cell 2019;73:714–726.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hirano S, Abudayyeh OO, Gootenberg JS, et al. . Structural basis for the promiscuous PAM recognition by Corynebacterium diphtheriae Cas9. Nat Commun 2019;10:1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das A, Hand TH, Smith CL, et al. . The molecular basis for recognition of 5’-NNNCC-3’ PAM and its methylation state by Acidothermus cellulolyticus Cas9. Nat Commun 2020;11:6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zetsche B, Gootenberg JS, Abudayyeh OO, et al. . Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015;163:759–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim HK, Song M, Lee J, et al. . In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat Methods 2017;14:153–9. [DOI] [PubMed] [Google Scholar]
- 52. Yamano T, Zetsche B, Ishitani R, et al. . Structural basis for the canonical and non-canonical PAM recognition by CRISPR-Cpf1. Mol Cell 2017;67:633–645.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yamano T, Nishimasu H, Zetsche B, et al. . Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell 2016;165:949–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Crooks GE, Hon G, Chandonia JM, et al. . WebLogo: a sequence logo generator. Genome Res 2004;14:1188–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Friedland AE, Baral R, Singhal P, et al. . Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol 2015;16:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ran FA, Cong L, Yan WX, et al. . In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015;520:186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Esvelt KM, Mali P, Braff JL, et al. . Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods 2013;10:1116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sternberg SH, Redding S, Jinek M, et al. . DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014;507:62–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Globyte V, Lee SH, Bae T, et al. . CRISPR/Cas9 searches for a protospacer adjacent motif by lateral diffusion. EMBO J 2019;38:e99466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Case DA, Cheatham TE, Darden T, et al. . The Amber biomolecular simulation programs. J Comput Chem 2005;26:1668–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All computational data, including preprocessed Cas structure PDB files, PAM variant models, PIAAs repacked and redesigned models, RESFILEs, a summary of PAM recognition and PIAA redesign results are available at https://doi.org/10.5281/zenodo.7426026.