

Abstract
Single-cell omics data are growing at an unprecedented rate, whereas effective integration of them remains challenging due to different sequencing methods, quality, and expression pattern of each omics data. In this study, we propose a universal framework for the integration of single-cell multi-omics data based on graph convolutional network (GCN-SC). Among the multiple single-cell data, GCN-SC usually selects one data with the largest number of cells as the reference and the rest as the query dataset. It utilizes mutual nearest neighbor algorithm to identify cell-pairs, which provide connections between cells both within and across the reference and query datasets. A GCN algorithm further takes the mixed graph constructed from these cell-pairs to adjust count matrices from the query datasets. Finally, dimension reduction is performed by using non-negative matrix factorization before visualization. By applying GCN-SC on six datasets, we show that GCN-SC can effectively integrate sequencing data from multiple single-cell sequencing technologies, species or different omics, which outperforms the state-of-the-art methods, including Seurat, LIGER, GLUER and Pamona.
Keywords: single-cell multi-omics, information transfer, integration, graph convolutional neural networks
INTRODUCTION
Recent single-cell sequencing technology has been developed rapidly, covering multiple levels of omics data such as genomics, transcriptomics, epigenomics, proteomics, metabolomics and spatial transcriptomics, etc. Each omics data has unique advantages and provides high-resolution molecular profiles at the cellular level. Among them, single-cell ribonucleic acid sequencing (scRNA-seq) that enables transcriptome profiling of thousands or even millions of cells at the single-cell level [1], has been the most widely used to dissect cellular heterogeneity using RNA expression. It is powerful to identify different cell populations and study cell–cell communications [2, 3]. Single-cell assay for transposase-accessible chromatin with high throughput sequencing (scATAC-seq) is an extensively used method for measurement of genome-wide chromatin open regions [4]. As transcriptional regulatory elements are enriched in the open chromatin regions, scATAC-seq could provide additional information related to gene expression compared to scRNA-seq [5]. In addition to scATAC-seq, many other technologies have also been developed to measure chromatin accessibility [6–9], deoxyribonucleic acid (DNA) methylation [10, 11], and cell surface protein abundance [12]. Each technology of single-cell omics data might capture the unique molecular features of the cells and harbor the information at different layers of gene expression.
Gene expression involves a variety of regulatory mechanisms, such as transcription and post-transcription regulation, translation, and post-translation regulation and so on. Therefore, integration of multi-omics data can establish linkages between data across modalities, providing more comprehensive insights into the gene expression regulation. Indeed, multi-omics analysis is increasingly applied on varies aspects of biology studies, including microbiology, regulatory genomics, pathogen biology and cancer biology. Integrating single-cell multi-omics data are able to unveil the interaction across two or more types of omics data, which obtain more comprehensive characterization of molecular features in each cell [13]. For instance, integration of single-cell transcriptomic and epigenomic data can be used to study the effect of epigenetic genome changes on gene expression. Furthermore, due to high frequency of dropout in single-cell transcriptomic data, effectively integrating it with epigenomics data is useful to determine cell identity [14–17]. Thus, integration of sequencing data from multiple omics has become an urgent need for single-cell studies.
The choice of anchor is important for omics data integration because the correct anchor is more conducive to establish the connection between different data modalities. To date, there are mainly three types of methods based on the selection of anchors for single-cell multi-omics data integration: vertical, horizontal and diagonal-based methods [14, 18]. The horizontal-based methods identify cell-pairs between datasets based on common gene sets, and the vertical-based methods find cell-pairs between datasets based on the common cell sets, and the diagonal methods conduct integration without common genes or cells datasets. Some commonly used omics data integration algorithms such as Seurat [19], LIGER [20], Harmony [21] and GLUER [22] are all diagonal methods. To integrate two single-cell sequencing data, one reference and one query, Seurat performs joint dimension reduction on both datasets using canonical correlation analysis and further identifies cell-pairs between these two datasets by searching mutual nearest neighbors (MNN) [23] on the shared low-dimension representation. The identified cell-pairs are used for further integration analysis. LIGER first employs integrative non-negative matrix factorization (iNMF) to get a low-dimensional space of cellular features. Then, the maximum factor loading matrix is obtained by iNMF. Finally, each cell is assigned a label and a shared factor neighborhood map is constructed to integrate the multi-omics data. GLUER maps cells to low-dimension representations using iNMF and identifies cell-pairs using MNN. Finally, cell-pairs are used for integration by searching nonlinear mapping relationships between two datasets through neural networks. Harmony groups single cells into multiple clusters through soft clustering. Then data integration is performed with the aid of linear correspondences between classes computed from specific cluster centers in each cluster. Pamona [24] and SCIM [25] are vertical-based methods for integrating single-cell multi-omics datasets. Pamona is a partial manifold alignment algorithm for heterogeneous single-cell multi-omics data integration based on the foundation of partial-GW framework. Firstly, a weighted k-nearest neighbor (KNN) [26] graph is constructed using the reference and query datasets. Then the geodesic distances of cells within the same dataset are computed to link the reference data and query data through common cells. Finally, cells in the reference and query datasets are aligned in a common low-dimensional space. SCIM uses the autoencoder [27] to encoder the reference and query datasets to the shared low-dimensional space. Then, use KNN to find the corresponding relationship between the reference and query in the shared low-dimensional space.
Although various integration algorithms have been proposed, the differences in sequencing methods, quality and expression patterns of single-cell multi-omics data are still the challenges for the integration [8, 28–30]. The current methods are mainly confronting two problems. On one hand, they only consider the cells relationship between the reference and query datasets but ignore the relationship among cells within each dataset. On the other hand, since most single cell sequencing techniques are still cell-destructive, multiple datasets of same or different omics often have unpaired cells.
To overcome these two bottlenecks, we propose GCN-SC, a novel framework for single-cell sequencing data integration based on GCN [31]. The main advantage of GCN is that it can handle data with incomplete spatial relationships with the power of convolutional networks. Unlike other machine-learning models, GCN does not encode data using one-dimensional vectors (or two-dimensional matrices). Instead, it uses graph structures to encode relationships among cells. GCN-SC uses both intra-dataset and inter-dataset cell-pairs to construct a mixed graph. The GCN model is further applied on the mixed graph to adjust single-cell data and finally non-negative matrix factorization (NMF) algorithm is applied for dimension reduction. With six datasets from multiple single-cell sequencing methods (Table S1), we show that GCN-SC is efficient to integrate single-cell data across different single-cell sequencing methods, species and omics. Moreover, it outperforms the existing methods, including Seurat, GLUER, LIGER and Pamona, for the integration of single-cell multi-omics data.
MATERIALS AND METHODS
Datasets
To evaluate the performance of the GCN-SC, we collected six single-cell multi-omics datasets from different tissues and organs in humans and mice. All the six datasets used in this study are publically available. Dataset 1 (PCF_2k) contains scRNA-seq data of human pancreatic islets from two single-cell sequencing technologies, Including Cel-seq2 (GSE81076) [32] and Fluidigm C1 (GSE86469) [33], which captured 1,728 and 638 cells, respectively. Dataset 2 (PSG_5k) consists of cells from human pancreas and islet from Smart-seq2 (E-MTAB-5061) [33] and 10X (GSE81608) [33], which have 3,514 and 1,600 cells, respectively. Dataset 3 (HM_10k) [34] consists of scRNA-seq data of 8,569 and 1,886 cells from human and mouse pancreases, respectively. Dataset 4 includes CITE-seq data [35] consisting of 161,764 human bone marrow cells with 228 antibodies, and scRNA-seq data composed of 2,700 cells from PBMC [36]. Dataset 5 consists of scRNA-seq and scATAC-seq of 3,009 human peripheral blood mononuclear cells [35]. Dataset 6 includes scRNA-seq and scATAC-seq of 14,645 cells from human lymphoma [35]. The specific introduction of data preprocess is shown in Supplementary Si1.
scImpute
Single-cell transcriptome sequencing has revolutionized traditional analysis of gene expression, enabling comprehensive characterization of the transcriptomics of individual cells with unprecedented throughput [37]. However, transcriptomic data analysis is limited by its own dropout events and curse of dimensionality, and its high sparsity brings serious challenges to downstream analysis of omics data integration [38–41]. Dropout events occur when gene expression cannot be detected due to sequencing technology limitations or low expression levels of genes [42]. Thus, the zero values in the expression matrix include both true zero and false zero values. Many methods have been developed for the imputation of dropout events in scRNA-seq data, such as DrImputes [43], SAVER [44], scImpute [45] and autoencoder networks [46]. The comparative study found that using scImpute imputation method to process scRNA-seq data showed better performance [47]. It can overcome the sparsity problem of scRNA-seq data and provide a high-quality expression matrix. Thus, we used scImpute in GCN-SC framework for the imputation of dropout events in scRNA-seq data.
Mnn algorithm
In order to integrate multi-omics data, it is necessary to explore the correspondence between cells from each omics data. A cell-pair consists of two similar cells, and some cells may form cell-pairs with multiple cells or may not form cell-pair with any cell. The selection of cell-pairs is mostly based on the characteristics of the cells. In our framework, the MNN algorithm is used to find cell-pairs within the same omics and between different omics data. This algorithm is an optimization of the KNN algorithm. The KNN algorithm is designed to find the 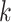 closest examples in the training dataset to the new input instance. Classify the input sample into a class, which most samples in the
closest examples in the training dataset to the new input instance. Classify the input sample into a class, which most samples in the 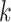 neighbors belong. The typical KNN algorithm is used for classification, while the MNN algorithm adapted the idea to find its
neighbors belong. The typical KNN algorithm is used for classification, while the MNN algorithm adapted the idea to find its 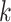 nearest neighbors for a new input instance. The specific introduction of MNN algorithms is shown in Supplementary Si2.
nearest neighbors for a new input instance. The specific introduction of MNN algorithms is shown in Supplementary Si2.
Graph convolutional neural networks
To correlate omics data obtained from different experiments and transfer information between different omics, it is necessary to assume that there is a correspondence between datasets. Previous methods assume that the correspondence between omics data is linear, but this is not true in reality [48–51]. Graph neural networks (GNN) can convert the practical association as a connection and transfer message between nodes in a graph. With the progress of deep learning, more and more types of GNN emerged. Among them, GCN is a kind of graph network that generalizes the convolution operation from traditional data to graph data [52].
Our framework used a spectral-based GCN, which consists of four layers, one input layer, two convolutional layers and one output layer. The use of the two convolutional layers ensures that the method is able to learn undirected graph relationships.
When we use the model for integration between different omics datasets the activation function of the convolutional layer selects ReLu. The input of the GCN includes both the query omics data expression matrix 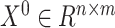 , and the hybrid graph. In this paper, the matrix
, and the hybrid graph. In this paper, the matrix 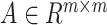 is constructed based on the cell-pairs matrix
is constructed based on the cell-pairs matrix 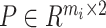 obtained by the MNN algorithm. When
obtained by the MNN algorithm. When 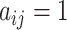 , it means that cell
, it means that cell 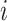 and cell
and cell 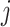 are the nearest neighbor to each other. When
are the nearest neighbor to each other. When 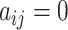 , the situation is opposite, and the diagonal elements of matrix
, the situation is opposite, and the diagonal elements of matrix 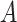 are all zero. Since the diagonal elements are set to zero, the cell’s own characteristics are ignored, so it is necessary to add a self-loop to it to obtain the matrix
are all zero. Since the diagonal elements are set to zero, the cell’s own characteristics are ignored, so it is necessary to add a self-loop to it to obtain the matrix 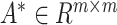 , and the degree matrix
, and the degree matrix 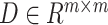 is obtained from the matrix
is obtained from the matrix 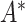 .
. 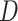 is a diagonal matrix, and the value of
is a diagonal matrix, and the value of 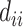 represents the number of MNNs of cell
represents the number of MNNs of cell 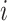 in the query dataset. To prevent the neighbor information of a certain cell from excessively affecting the cell characteristics, we use the degree matrix to normalize the matrix
in the query dataset. To prevent the neighbor information of a certain cell from excessively affecting the cell characteristics, we use the degree matrix to normalize the matrix 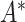 to obtain the adjacency matrix
to obtain the adjacency matrix 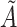 , which is:
, which is:
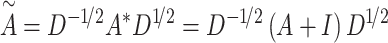 |
(1) |
where 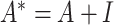 ,
, 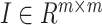 is the identity matrix.
is the identity matrix.
When we use the model for label transfer between omics data, the activation function of the convolutional layers selects ReLu and Softmax, respectively. The Softmax activation function is defined as follows:
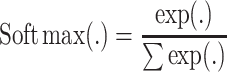 |
(2) |
At this time, the input of the GCN is the expression matrix 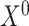 of the query omics data, the cell label
of the query omics data, the cell label 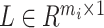 in the reference omics data corresponding to the cell-pairs index between the omics data and the adjacency matrix
in the reference omics data corresponding to the cell-pairs index between the omics data and the adjacency matrix 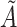 constructed by the cell-pairs inside the query omics data. The reference dataset cell label in the cell-pairs is selected as the corresponding query dataset cell label, where
constructed by the cell-pairs inside the query omics data. The reference dataset cell label in the cell-pairs is selected as the corresponding query dataset cell label, where 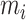 is the number of cell-pairs between omics. Since it is not guaranteed that every cell in the query dataset can form a cell-pair with cell in the reference dataset during cell-pairs selection, so the label transfer in this paper is a semi-supervised process.
is the number of cell-pairs between omics. Since it is not guaranteed that every cell in the query dataset can form a cell-pair with cell in the reference dataset during cell-pairs selection, so the label transfer in this paper is a semi-supervised process.
The layer weight matrix and the bias matrix are introduced inside the GCN model, and the initialization of 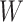 and
and 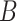 consists of data randomly taken from
consists of data randomly taken from 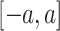 , where:
, where:
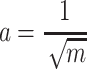 |
(3) |
the output of each layer of the GCN is denoted as 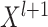 , where:
, where:
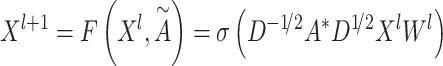 |
(4) |
when the gap between the output 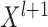 and the target matrix
and the target matrix 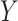 is large, the model is optimized by adding the bias matrix
is large, the model is optimized by adding the bias matrix 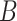 , that is:
, that is:
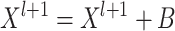 |
(5) |
In the model, the mean square loss function is used to feedback the model performance, and we choose Adam optimizer as the model optimizer. The objective function of the GCN model is:
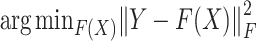 |
(6) |
where 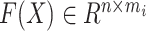 is the expression matrix extracted from the model output data by the cell index number in the second column of cell-pairs matrix
is the expression matrix extracted from the model output data by the cell index number in the second column of cell-pairs matrix 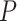 , and
, and 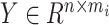 is the expression matrix extracted from the reference dataset by the cell index number in the first column of
is the expression matrix extracted from the reference dataset by the cell index number in the first column of 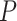 .
.
Non-negative matrix factorization
Single-cell sequencing generates high-dimensional data and a basic step in single-cell data analysis is to cluster and visualize each cell after using dimensionality reduction techniques [53]. In GCN-SC, we used NMF for the dimensionality reduction because of two reasons. On one hand, the processed single-cell sequencing data is non-negative, which is suitable for NMF [54]. On the other hand, the NMF algorithm has many advantages compared with traditional algorithms, such as the simplicity of implementation, the interpretability of the decomposition form and decomposition result. Since the activation functions in the GCN are Relu and Softmax, the expression values of the omics data after being processed by the GCN are still non-negative numbers, which ensures that each expression value has biological significance. The specific introduction of NMF algorithm is shown in Supplementary Si3.
Evaluation indicators
In this paper, we used two different evaluation indicators to assess the model performance. In evaluating the performance of integration of scRNA-seq and scATAC-seq data, we used ‘alignment score’, an evaluation index proposed by Seurat. The better the integration effect, the more comprehensive the information transfer, and the higher the ‘alignment score’ obtained. The alignment score is a value from 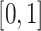 . The solution process of the ‘alignment score’ is as follows: find the
. The solution process of the ‘alignment score’ is as follows: find the 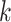 nearest neighbors for each cell in the data, observe how many cells in the
nearest neighbors for each cell in the data, observe how many cells in the 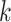 cells belong to the same omics, and compare the
cells belong to the same omics, and compare the 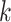 nearest neighbors of all cells with the search for
nearest neighbors of all cells with the search for 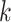 nearest neighbors. Cells belonging to the same omics are summed and averaged to get
nearest neighbors. Cells belonging to the same omics are summed and averaged to get 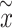 . If the two omics data are well integrated and the information transfer is sufficiently comprehensive, it should be ensured that the number of cells of the own omics and the other omics cells in the
. If the two omics data are well integrated and the information transfer is sufficiently comprehensive, it should be ensured that the number of cells of the own omics and the other omics cells in the 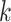 nearest neighbors of each cell is almost equal. This process is to ensure that the two sets of data are evenly mixed and indistinguishable. Calculated as follows:
nearest neighbors of each cell is almost equal. This process is to ensure that the two sets of data are evenly mixed and indistinguishable. Calculated as follows:
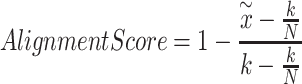 |
(7) |
where 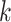 is 0.01 of the total number of cells in the integrated dataset, and
is 0.01 of the total number of cells in the integrated dataset, and 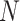 is the number of omics types included in the integrated dataset.
is the number of omics types included in the integrated dataset.
In the process of label transfer that map the scRNA-seq data to CITE-seq data, a custom ‘transfer accuracy rate’ is used to evaluate the model’s label transfer performance. The better the label transfer effect, the higher the evaluation index, and its value range is 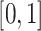 . When evaluating the label transfer accuracy, let
. When evaluating the label transfer accuracy, let 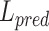 be the cell label predicted by GCN-SC,
be the cell label predicted by GCN-SC, 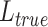 be the accurately predicted label, and the calculation formula is:
be the accurately predicted label, and the calculation formula is:
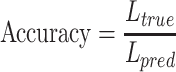 |
(8) |
Illustration of the GCN-SC workflow
In the paper, we put forward a novel method GCN-SC for single-cell multi-omics data integration, and the flowchart is shown in Figure 1. The source codes and datasets are available at https://github.com/YuBinLab-QUST/GCN-SC/.
Figure 1.

Overview of the GCN-SC framework on single-cell multi-omics data integration. GCN-SC can take single-cell multi-omics data as input, including data of gene expression (scRNA-seq), chromatin accessibility (scATAC-seq) and protein expression (CITE-seq). Each omics data was pre-processed to a uniform count matrix, in which each row represents one feature and each column represents one cell. As gene expression has much higher frequency of dropout compared to other omics data, scImpute was conducted on the matrix to reduce the sparsity of the matrix. Next, one scRNA-seq data were used as the reference, while the rest were considered as the query. By using MNN algorithm, cell-pairs with the closest Euclidean distance were identified, and then a mixed graph was constructed based on cell–cell relationships, which includes both inter-dataset edges and intra-dataset edges. A GCN that includes four layers further took this mixed graph to adjust matrices from the query dataset. Finally, NMF was implemented for dimension reduction on the adjusted matrices from the query and the matrix from the reference.
The steps of GCN-SC method are:
Step1. Single-cell multi-omics data as input, including data of gene expression (scRNA-seq), chromatin accessibility (scATAC-seq) and protein expression (CITE-seq).
Step2. The scImpute algorithms were used to reduce potential bias caused by high sparsity of scRNA-seq data.
Step3. Using MNN algorithm to find cell-pairs within pre-processed data.
Step4. After cell-pair selection, input the mixed graph into graph convolutional neural network (GCN) for omics data integration and information transfer.
Step5. NMF was implemented for dimension reduction on the adjusted matrices from the query and the matrices from reference for clustering analysis.
Step6. Six single-cell datasets across different single-cell sequencing technologies, species and omics were used to assess GCN-SC model.
RESULTS AND DISCUSSION
Integration of scRNA-seq data from different single-cell sequencing technologies
Due to the difference in sequencing depth of RNA from single cell and heterogeneities of biological samples, as well as efficiency of single-cell experiments, RNA expression of each gene could be quite variable across scRNA-seq data. Particularly, the variations might be even more dramatic between scRNA-seq data from experiments with different single-cell sequencing technologies, such as Fluidigm, Cel-seq2 and Smart-seq. This kind of systematic bias in gene expression is known as the batch effect. A strong batch effect would complicate downstream analysis and lead to potential misinterpretation of results. To test the ability of GCN-SC to remove such kind of batch effect, we applied GCN-SC to integrate scRNA-seq data from different experimental batches with two datasets. Dataset 1 (PCF_2k) contains scRNA-seq data of human pancreatic islets from two single cell sequencing technologies, including Cel-seq2 and Fluidigm C1. Dataset 2 (PSG_5k) consists of cells from the human pancreas and islet from Smart-seq2 and 10X.
In PCF_2k, 10 clusters and 5 clusters of cells were identified in Cel-seq2 and Fluidigm C1 data by using UMAP [55], respectively (Figure 2A and B). Even though these scRNA-seq data are from the same types of human tissue, using UMAP algorithm directly on the raw expression matrix, results in almost fully separation of cells from different technologies in a two-dimensional space (Figure 2C). This batch effect was almost unchanged after imputation by using scImpute algorithm (Figure 2D). Next, Cel-seq2 data as the reference and Fluidigm C1 data as the query, and then applied MNN algorithm to find three MNNs for each cell in the query dataset. The cell-pairs were used to construct the internal cell graph of the query dataset. Similarly, MNN algorithm was applied to find five MNNs between cells from the reference and the query dataset, and a cross-dataset graph was further constructed. By using the mixture of internal cell graph and the cross-dataset graph as the input of GCN, the batch effect between two scRNA-seq data was greatly reduced (Figure 2E). Finally, we used NMF to reduce the dimension of integrated data and followed by UMAP for two-dimensional embedding (Figure 2F). Notably, the results maintain a similar cell cluster structures as the original reference data, whereas the query data was successfully integrated (Figure 2A and F). Similar results could be observed by applying the analysis on the dataset 2 (Figure S1), demonstrate that GCN-SC is efficient to integrate scRNA-seq data from various single cell sequencing technologies.
Figure 2.

Integration of scRNA-seq data from multiple experimental batches. (A) Raw cluster map of Cel-seq2 transcriptome sequencing data with UMAP. (B) Raw cluster map of Fluidigm C1 transcriptome sequencing data with UMAP. (C) The UMAP embedding of the original expression matrix from Cel-seq2 and Fluidigm, which contain 2000 hypervariable genes. (D) UMAP embedding of the expression matrix processed by scImpute algorithm. (E) UMAP embedding of integrated expression matrix processed by the GCN-SC framework. (F) Clustering diagram of the integrated data processed by GCN-SC.
GCN-SC enables cross-species cell alignment based on scRNA-seq data
Single cell sequencing data has been widely used for the identification of cell types of different species, such as humans and mice. However, association of different cell types across species is poorly understood, which is at least partially due to the lack of appropriate methods to align each type of cell between different species. To examine the feasibility of the GCN-SC for cross-species cell alignment, we selected dataset 3 (HM_10k) from the previous study [56], which consists transcriptomic data of 8,569 and 1,886 cells from human and mouse pancreases, respectively (Figure S2).
Firstly, we retained the top 2,000 hypervariable genes to obtain the gene expression matrix in human and mouse. Similar as the integration of scRNA-seq data from different single-cell sequencing technologies, directly applying UMAP on the gene expression matrix of human and mouse cells to project them into two-dimensional space, almost did not find any cells from these two species are clustered together (Figure 3A), even though there are common types of cells. These results indicate the huge batch effect of scRNA-seq data across different species. In contrast, applying GCN-SC on this dataset by using human data as the reference and mouse data as the query, the cells from human and mouse were able to cluster together (Figure 3B). We also using the mouse data as the reference and the human data as the query, the results were not impacted (Figure S3). The effective integration of the data enables to cluster cells from different cell types, rather than different species, which identified 8 main types of cells, including alpha, beta, delta, ductal, endothelial, gamma, mast, quienscent_stellate and each class was present in both species (Figure 3C and Table S2). For each cell type, proportion of the misaligned cells are very low and almost not found in some cell types (Figure 3D). Taken together, our results suggested that GCN-SC is not only able to remove batch effect between different batches of scRNA-seq data, but also able to align the same type of cells across different species with scRNA-seq data.
Figure 3.

Cell alignment of cross-species transcriptome sequencing data. (A) The UMAP embedding of the original expression matrix of cells from human and mouse. (B) The UMAP embedding the integrated data processed by GCN-SC. (C) Identification of cell types on the UMAP embedding of the results from GCN-SC. (D) Number of cells from human and mouse in each cell type in the original data (upper) and in the processed data from GCN-SC (bottom).
Label transfer between scRNA-seq and CITE-seq data with GCN-SC
CITE-seq enables the detection of surface proteins and transcriptome profiling simultaneously in each single cell, which provide association between RNA expression pattern and different surface proteins [57, 58]. Using the surface proteins, different types of cells could be identified. Due to different sequencing technologies, the feasibility to directly map scRNA-seq data to CITE-seq data remains unclear. To this aim, we collected dataset 4, which includes CITE-seq data consisting of 161,764 human bone marrow cells with 228 antibodies (CITE161k), and scRNA-seq data composed of 2,700 cells (PBMC3k). Based on the surface proteins, the 161,764 cells were classified into 5 types including B cell, Progenitor cell, NK, Mono/DC and T cell (Figure 4A). On the other hand, the 2,700 cells were annotated into 6 cell types including B cells, Platelet cells, NK cells, T cells, Mono/DC cells and a few unlabeled cells (Figure 4B).
Figure 4.

Mapping cell types between scRNA-seq and CITE-seq data. (A) The UMAP visualization diagram of the CITE-seq data. (B) The UMAP visualization diagram of the scRNA-seq data. (C) The UMAP visualization of the predicted cell type in scRNA-seq data based on cell labels from CITE-seq data. (D) Proportion of the correctly predicted cells in each cell type with different numbers of anchors.
We used the transcriptomic data of CITE161k as the reference and applied GCN-SC to map the 2,700 cells from PBMC3k to the reference. In this way, we were able to predict the cell types of each cell in the query by transferring the label in CITE161k to PBMC3k. The predicted label that is identical to the annotated one was recorded as an accurate prediction. Among the four common cell types between the two datasets, NK cell has the lowest transfer accuracy and many of them were predicted as T cells, probably due to relative similar expression profiles of these two cell types (Figure 4C). We also test the robustness of the prediction by changing the value of the MNN coefficient between groups. Interestingly, with the increasing of the coefficient, the transfer accuracy also increased (Figure 4D and Table S3). In addition, we also explores the change of prediction accuracy of cells in common cell types with the number of anchor pairs (Table S4). Eventually, we used the nearest neighbor parameter 24, which achieves 97% accuracy after 100 iterations. Therefore, GCN-SC is an efficient tool to predict cell types by mapping them to a reference with labels, even if the cell types in the reference and query dataset are not fully matched.
GCN-SC outperforms the existing methods on integration of single-cell multi-omics data
Single-cell transcriptomic data can quantify RNA expression of thousands of genes in parallel in each cell, whereas single-cell epigenomics data is able to characterize DNA methylation or chromatin accessibility in nearby genomic regions, which is essential to determine the transcriptional activity of each gene [59]. Therefore, these two omics data should be highly correlated. Integrating these two is not only able to reveal gene regulatory relationships associated with cell heterogeneity but can also increase the power for classification of subcellular population comparing to using only single omics data [4, 60].
Therefore, we explored the performance of GCN-SC in integrating transcriptomics and epigenomics data from the same group of cells using two datasets, including dataset 5 (PBMC_3k) and dataset 6 (HL_11k). Both datasets have transcriptomics data and epigenomics data (Figure S4). As single-cell transcriptomics data has been more extensively used for the clustering, we chose it as the reference, and epigenomics data as the query. We retained the top 2,000 hypervariable genes in the two omics data, and then selected the common ones to obtain a gene expression and a gene activity matrix with the identical dimension.
Using UMAP, we projected the cells from scRNA-seq and scATAC-seq data into a two-dimension plot based on gene expression and gene activity matrix. As expected, the cells from scRNA-seq are almost fully separated from the cells from scATAC-seq on both PBMC_3K and HL_11k, suggesting that different omics data have distinct batch effect (Figure 5A). However, by applying UMAP on the output of GCN, we found that the batch effect between scRNA-seq and scATAC-seq was greatly removed, for which the cells from two omics data could be clustered together (Figure 5B). Interestingly, some clusters of cells were only observed from one omics data, suggesting a compensation effect when integrating these two omics data.
Figure 5.

Integration of single-cell multi-omics data. (A) The UMAP visualization of the scRNA-seq and scATAC-seq data from the same group of cells (dataset 5). (B) The UMAP visualization of the integrated data processed by GCN-SC. The UMAP visualization of the integration data by Seurat (C), GLUER (D) and GCNSC (E). (F) The alignment score map of the three integration algorithms Seurat, GLUER and GCN-SC on dataset 5.
We further compared GCN-SC with four existing integration algorithms, Seurat, LIGER, GLUER and Pamona. By using UMAP on the data integrated by these four methods on PBMC_3k, we classified the cells into different clusters. Seven clusters that were identified on data from Seurat and LIGER, while the later one is more compacted (Figure 5C and Figure S5A). Both these two methods use the low-dimensional embedding vector of the data in the integration process, so that the information carried by each omics data might be lost, and the feature information carried in each omics dataset cannot be fully taken into account. GLUER obtained 10 clusters, while the boundary of each cluster is not clear (Figure 5D). Surprisingly, GCN-SC was able to identify 14 clusters and each cluster was clearly separated from each other (Figure 5E). The similar trend could also be observed when applying the four methods on dataset 6 (HL11k), in which GCN-SC obtained the most and clearest cell clusters (Figure S5B–G).
To compare the performance quantitatively, we calculated the alignment scores (Methods) of the data processed by Seurat, GLUER, Pamona and GCN-SC on PBMC_3k and HL_11k. Since LIGER costs dramatically more computational resources, it is not feasible to use 2,000 features for integration. Therefore, we only included Seurat, GLUER and Pamona for a fair comparison. The alignment score of Pamona on datasets 5 and 6 is 0.242 and 0.309, respectively, which is very different from the other three methods. As expected, GCN-SC achieved much higher alignment score than Seurat, Pamona and GLUER on both datasets (Figure 5F and Figure S5H). Taken together, our results demonstrated that GCN-SC is more accurate to integrate single-cell transcriptomic and epigenomic data compared to the existing methods, indicating that it could be powerful to integrate single-cell multi-omics data.
CONCLUSION
In this paper, we proposed a GCN-based framework for integrating single-cell multi-omics data (GCN-SC). GCN-SC uses multiple ways to address the challenges across different steps of the integration. First, the use of imputation algorithms addresses the challenge about sparsity of transcriptomic data. Second, the MNN algorithm captures one-to-many and many-to-many relationships between cells in a biological sense. Then, the GCN takes into account both reference data and query data. It uses mixed graph to explore the nonlinear functional relationship between different omics datasets and realize the information transfer between omics data. Finally, NMF can obtain low-dimensional representation of single-cell omics data therefore overcomes the issue of high-dimensional data. In summary, GCN-SC is an effective framework for single-cell omics data integration. As single-cell multi-omics data become more and more abundant, integrating data promises to deepen our recognition of the role of cellular heterogeneity in the context of development and pathogenesis.
Although GCN-SC could effectively integrate single-cell multi-omics data, there might be many modifications to improve its performance in the future. First, it is reasonable to flexibly adjust the number of convolutional layers according to the size and characteristics of different datasets. Second, by adding weights to the linkages between different omics, cell-pairs across different types of the cells could be discriminatory, which might be helpful to dissect the cell heterogeneities. Hopefully, these and other issues will be addressed in the updated version of the framework.
Key Points
We propose a new method to integrate sing-cell data from different sequencing technologies, species and omics.
The model can transfer labels across different single-cell datasets, which enables to predict cell labels in unlabeled datasets with a labeled dataset as reference.
The results of the six datasets show that GCN-SC achieves robust and good performance for the integration of single-cell omics datasets.
Supplementary Material
Author Biographies
Hongli Gao is a master student at the Qingdao University of Science and Technology, China. Her research interests are bioinformatics and machine learning.
Bin Zhang is a postdoctoral fellow at Computational Bioscience Research Center (CBRC), King Abdullah University of Science and Technology, Thuwal, Saudi Arabia. His research interests are computational biology, cancer biology and RNA biology.
Long Liu is a master student at the Qingdao University of Science and Technology, China. His research interests are bioinformatics and machine learning.
Shan Li is a PhD student at the School of Mathematics and Statistics, Central South University, China. Her research interests are bioinformatics and machine learning.
Xin Gao is a professor at the King Abdullah University of Science and Technology (KAUST), Saudi Arabia. His research interests include bioinformatics, computational biology, artificial intelligence and machine learning.
Bin Yu is a professor at the Qingdao University of Science and Technology, China. His research interests include bioinformatics, artificial intelligence and biomedical image processing.
Contributor Information
Hongli Gao, Qingdao University of Science and Technology, China.
Bin Zhang, Computational Bioscience Research Center (CBRC), King Abdullah University of Science and Technology, Thuwal, Saudi Arabia.
Long Liu, Qingdao University of Science and Technology, China.
Shan Li, School of Mathematics and Statistics, Central South University, China.
Xin Gao, King Abdullah University of Science and Technology (KAUST), Saudi Arabia.
Bin Yu, Qingdao University of Science and Technology, China.
FUNDING
National Natural Science Foundation of China (62172248); Natural Science Foundation of Shandong Province of China (ZR2021MF098); King Abdullah University of Science and Technology (FCC/1/1976-44-01, FCC/1/1976-45-01, URF/1/4379-01-01 and REI/1/4742-01-01).
References
- 1. Rosenberg AB, Roco CM, Muscat RA, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018;360:176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Forcato M, Romano O, Bicciato S. Computational methods for the integrative analysis of single-cell data. Brief Bioinform 2020;22:bbaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zheng L-L, Xiong J-H, Zheng W-J, et al. ColorCells: a database of expression, classification and functions of lncRNAs in single cells. Brief Bioinform 2020;22:bbaa325. [DOI] [PubMed] [Google Scholar]
- 4. Cusanovich DA, Hill AJ, Aghamirzaie D, et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 2018;174:1309–1324.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muto Y, Wilson PC, Ledru N, et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat Commun 2021;12:2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramanathan M, Porter DF, Khavari PA. Methods to study RNA-protein interactions. Nat Methods 2019;16:225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cao J, Cusanovich DA, Ramani V, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 2018;361:1380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lake BB, Chen S, Sos BC, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 2018;36:70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma A, McDermaid A, Xu J, et al. Integrative methods and practical challenges for single-cell multi-omics. Trends Biotechnol 2020;38:1007–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luo C, Keown CL, Kurihara L, et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 2017;357:600–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mulqueen RM, Pokholok D, Norberg SJ, et al. Highly scalable generation of DNA methylation profiles in single cells. Nat Biotechnol 2018;36:428–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu F, Wang S, Dai X, et al. Ensemble learning models that predict surface protein abundance from single-cell multimodal omics data. Methods 2021;189:65–73. [DOI] [PubMed] [Google Scholar]
- 13. Jin S, Zhang L, Nie Q. scAI: an unsupervised approach for the integrative analysis of parallel single-cell transcriptomic and epigenomic profiles. Genome Biol 2020;21:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Argelaguet R, Arnol D, Bredikhin D, et al. MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol 2020;21:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang L, Zhang S. Learning common and specific patterns from data of multiple interrelated biological scenarios with matrix factorization. Nucleic Acids Res 2019;47:6606–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stuart T, Satija R. Integrative single-cell analysis. Nat Rev Genet 2019;20:257–72. [DOI] [PubMed] [Google Scholar]
- 17. Dou J, Liang S, Mohanty V, et al. Bi-order multimodal integration of single-cell data. Genome Biol 2022;23:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Argelaguet R, Cuomo ASE, Stegle O, et al. Computational principles and challenges in single-cell data integration. Nat Biotechnol 2021;39:1202–15. [DOI] [PubMed] [Google Scholar]
- 19. Butler A, Hoffman P, Smibert P, et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Welch JD, Kozareva V, Ferreira A, et al. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 2019;177:1873–1887.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods 2019;16:1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peng T, Chen GM, Tan KJB. GLUER: integrative analysis of single-cell omics and imaging data by deep neural network. bioRxiv preprint bioRxiv:2021. 10.1101/2021.01.25.427845. [DOI]
- 23. Haghverdi L, Lun ATL, Morgan MD, et al. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 2018;36:421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cao K, Hong Y, Wan L. Manifold alignment for heterogeneous single-cell multi-omics data integration using Pamona. Bioinformatics 2021;38:211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stark SG, Ficek J, Locatello F, et al. SCIM: universal single-cell matching with unpaired feature sets. Bioinformatics 2020;36:i919–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang X, Gao H, Qi R, et al. scBKAP: a clustering model for single-cell RNA-Seq data based on bisecting K-means. IEEE ACM T Comput Biol Bioinform 2022;1–10. [DOI] [PubMed] [Google Scholar]
- 27. Yu B, Chen C, Qi R, et al. scGMAI: a Gaussian mixture model for clustering single-cell RNA-Seq data based on deep autoencoder. Brief Bioinform 2021;22:bbaa316. [DOI] [PubMed] [Google Scholar]
- 28. Xu Y, Das P, McCord RP. SMILE: mutual information learning for integration of single-cell omics data. Bioinformatics 2022;38:476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Misra BB, Langefeld CD, Olivier M, et al. Integrated omics: tools, advances, and future approaches. J Mol Endocrinol 2018;62:R21–45. [DOI] [PubMed] [Google Scholar]
- 30. Chen H, Lareau C, Andreani T, et al. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol 2019;20:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shang J, Jiang J, Sun Y. Bacteriophage classification for assembled contigs using graph convolutional network. Bioinformatics 2021;37:i25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Muraro MJ, Dharmadhikari G, Grun D, et al. A single-cell transcriptome atlas of the human pancreas. Cell Syst 2016;3:385–394.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lawlor N, George J, Bolisetty M, et al. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res 2017;27:208–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baron M, Veres A, Wolock SL, et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst 2016;3:346–360.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zheng GX, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu Y, Zhang Z, You L, et al. scIGANs: single-cell RNA-seq imputation using generative adversarial networks. Nucleic Acids Res 2020;48:e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dijk D, Sharma R, Nainys J, et al. Recovering gene interactions from single-cell data using data diffusion. Cell 2018;174:716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Elyanow R, Dumitrascu B, Engelhardt BE, et al. netNMF-sc: leveraging gene-gene interactions for imputation and dimensionality reduction in single-cell expression analysis. Genome Res 2020;30:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zuo C, Dai H, Chen L. Deep cross-omics cycle attention model for joint analysis of single-cell multi-omics data. Bioinformatics 2021;37:4091–9. [DOI] [PubMed] [Google Scholar]
- 41. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bacher R, Kendziorski C. Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biol 2016;17:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gong W, Kwak IY, Pota P, et al. DrImpute: imputing dropout events in single cell RNA sequencing data. BMC Bioinformatics 2018;19:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang M, Wang J, Torre E, et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods 2018;15:539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li WV, Li JJ. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat Commun 2018;9:997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hinton GE, Salakhutdinov RR. Reducing the dimensionality of data with neural networks. Science 2006;313:504–7. [DOI] [PubMed] [Google Scholar]
- 47. Jiang R, Li WV, Li JJ. mbImpute: an accurate and robust imputation method for microbiome data. Genome Biol 2021;22:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cao K, Bai X, Hong Y, et al. Unsupervised topological alignment for single-cell multi-omics integration. Bioinformatics 2020;36:i48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kang M, Ko E, Mersha TB. A roadmap for multi-omics data integration using deep learning. Brief Bioinform 2022;23:bbab454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Welch JD, Hartemink AJ, Prins JF. MATCHER: manifold alignment reveals correspondence between single cell transcriptome and epigenome dynamics. Genome Biol 2017;18:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rautenstrauch P, Vlot AHC, Saran S, et al. Intricacies of single-cell multi-omics data integration. Trends Genet 2022;38:128–39. [DOI] [PubMed] [Google Scholar]
- 52. Song Q, Su J, Zhang W. scGCN is a graph convolutional networks algorithm for knowledge transfer in single cell omics. Nat Commun 2021;12:3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Do VH, Canzar S. A generalization of t-SNE and UMAP to single-cell multimodal omics. Genome Biol 2021;22:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nadif M, Role F. Unsupervised and self-supervised deep learning approaches for biomedical text mining. Brief Bioinform 2021;22:1592–603. [DOI] [PubMed] [Google Scholar]
- 55. Becht E, McInnes L, Healy J, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 2018;37:38–44. [DOI] [PubMed] [Google Scholar]
- 56. Avila Cobos F, Alquicira-Hernandez J, Powell JE, et al. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat Commun 2020;11:5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 2017;14:865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gibney E, Nolan CJH. Epigenetics and gene expression. Heredity 2010;105:4–13. [DOI] [PubMed] [Google Scholar]
- 59. Furlan M, Pretis S, Pelizzola M. Dynamics of transcriptional and post-transcriptional regulation. Brief Bioinform 2020;22:bbaa389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhu C, Preissl S, Ren B. Single-cell multimodal omics: the power of many. Nat Methods 2020;17:11–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.