

Abstract
Background
Serotype coxsackievirus B (CVB) infection has been linked to viral myocarditis, dilated cardiomyopathy, meningitis, and pancreatitis in children and young adults. As of yet, no antiviral drug has been authorized for the treatment of coxsackievirus infection. Therefore, there is perpetual demand for new therapeutic agents and the improvement of existing ones. Benzo[g]quinazolines, the subject of several well-known heterocyclic systems, have risen to prominence and played a significant role in the development of antiviral agents, particularly those for anti-coxsackievirus B4 infection.
Methods
This study investigated the cytotoxicity of the target benzo[g]quinazolines (1–16) in the BGM cells line as well as their anti-coxsackievirus B4 activity. Determination of CVB4 titers using a plaque assay.
Results
Most of the target benzoquinazolines exhibited antiviral activity, however, compounds 1–3 appeared to be the most effective (reduction percentages of 66.7, 70, and 83.3%, respectively). The binding mechanisms and interactions of the three most active 1–3 with the constitutive amino acids in the active site of the multi-target of coxsackievirus B4 (3Clpro and RdRp) targets were also investigated using molecular docking.
Conclusion
The anti coxsackievirus B4 activity has resulted, and the top three active benzoquinazolines (1–3) have bonded to and interacted with the constitutive amino acids in the active region of the multi-target coxsackievirus B4 (RdRp and 3Clpro). Further research is required in the lab. to determine the exact benzoquinazolines mechanism of action.
Supplementary Information
The online version contains supplementary material available at 10.1007/s43440-023-00495-z.
Keywords: Benzo[g]quinazolines, Coxsackievirus B4, Docking study, Cytotoxicity, Molecular dynamic simulation
Introduction
Coxsackievirus B4 (CVB4) is an enterovirus in the Picornaviridae family. It belongs to the six viruses that comprise the B serogroup and has a solitary, positively charged RNA genome of about 7.4 kb [1–3]. Coxsackievirus B4 can cause various diseases ranging from mild upper respiratory illness to severe myocarditis, pericarditis, meningitis, pneumonia, pleurodynia, gastroenteritis, and pancreatitis. CVB4 infection produces a wide spectrum of clinical manifestations including asymptomatic cases, undifferentiated fevers, mild upper respiratory symptoms accompanied by significant symptoms such as fever, chest pain, pleural inflammation, headache, and sore throat [4–6]. Infectious diseases have posed major challenges to modern treatment. This is especially true for viruses whose high mutation rates enable immune evasion and resistance to standard antiviral therapies [7, 8]. The CVB4 infection has been linked to the onset of insulin-dependent diabetic mellitus (IDDM) likely due to virus-induced damage of pancreatic cells. There is currently no cure or vaccination for CVB4 infections. Available treatment options only alleviate symptoms [5, 9, 10]. Multitarget drugs, defined as molecules that modify multiple disease-relevant targets to improve efficacy and/or safety, can unlock opportunities for new therapies [11, 12]. Studying multifunctional compounds can involve extensive experiments or computer-aided rational drug design followed by screening and selecting relevant targets, optimizing leads, and avoiding increased costs [11]. Potential therapies for CVB4 infection were explored, including RNA interference (RNAi). The 19-mer siRNAs reportedly inhibited CVB4 replication in rhabdomyosarcoma (RD) cells with good specificity and no detectable off-target effects according to certain reports [12]. Additionally, the siRNA targeting the 3Cpro region of the CVB4 genome most effectively inhibited CVB4 replication in the RD cell line in a dose-dependent manner [12]. CVB4 expresses a large monocistronic polyprotein comprising one structural polyprotein (P1) and two non-structural polyproteins from its long open reading frame (ORF)[13]. The icosahedral capsid forms from the structural capsid protein P1 (VP1-VP4). Seven non-structural proteins P2 and P3 including peptidase C3 constitute the virus essential for polyprotein processing and replication (Fig. 1) [14]. RNAi targeting the 3Cpro cysteine protease critical for polyprotein processing, shows promise for inhibiting CVB4 without off-target impacts. However, high mutation rates may enable escape, necessitating refined siRNA designs or alternative strategies [12]. A multitarget approach combining RNAi with other inhibitors could limit resistance while providing broader coverage against CVB4 variants. Rational drug design guided by homology modeling could identify optimal target combinations. Pre-clinical studies would evaluate lead candidates before clinical progress to treating CVB4 infections [15, 16]. Despite obstacles, RNAi and rational drug design for multitarget therapy represent promising routes to managing CVB4 and related enteroviruses. Continued progress in understanding viral vulnerabilities, developing new therapeutics and implementing combined treatment-prevention strategies could help address these persistent global health threats [15].
Fig. 1.

CVB4 polyprotein and proteases cleavage diagram. A A single-stranded RNA genome encodes the linear amino acid sequence of the CVB4 polyprotein. Capsid protein and RNA helicase cleavage sites are marked. B The protein, mature peptide AA, and region characteristics of the CVB4 polyprotein are displayed. VP1-VP4 capsid proteins and 2A-3D replication proteins are protein characteristics. Each peptide’s amino acid length following cleavage is shown by its mature AA characteristics. Proteinase, core protein, and RNA polymerase domains are shown in the region features
Our research team previously investigated synthetic heterocyclic compounds for CVB4 inhibition. Certain 2-methylsulfanyl-triazoloquinazolines exhibited the highest activity, reducing CVB4 by 63 and 83% respectively [17]. Extensive studies on benzoquinazolines revealed potent effects against CVB4 (EC50 = 38, 41 and 43 μg/mL vs ribavirin EC50 = 29 μg/mL) and other contexts [18–29]. The insights gained guided designing more active benzo[g]quinazolines as anti-CVB4 agent. In the present work, we evaluated a new series of 2-thioxo-benzo[g]quinazolines (1–16) for their activity against CVB4. The most active benzoquinazolines (1, 2, 3) and co-bound inhibitor were docked into the 3D binding site to establish optimal binding orientation. Molecular docking also studied binding modes and interactions of benzoquinazolines 1, 2 and 3 with 3Clpro and RdRp, CVB4 multi-targets.
Materials and methods
The target benzo[g]quinazolines 1–16
The targets 1–16 were previously synthesized and completely described [21–25, 27]. Benzoquinazolines 1–3 were produced in high yields by reacting ethyl(methyl)isothiocyanate or (benzyl)isothiocyanate with 3-amino-2-naphthaoic acid in DMF for 3–5 h. TEA was added to serve as the basic medium. Thioalkylated benzoquinazolines 4–13 were prepared by treating 1–3 with benzyl halides (benzyl substituted halides) and using K2CO3 as the base in DMF at 80 °C for 20 h. Benzoquinazolines 14–16 formed by hydrazinolyzing 1–3 in boiling DMF for 15–18 h. Details of characterization including IR, 1H and 13C NMR spectroscopy as well as mass spectrometry have been reported, confirming the synthesized 1–16 [21–25, 27]. Purity was further evidenced by TLC and melting point analyses.
Cytotoxicity test
Cytotoxicity of a tested sample was assessed on BGM cells. A 100 mg sample was diluted in 1 mL ethanol (Aldrich, Darmstadt, Germany) for quick analysis. Samples were decontaminated by adding 24 μL of antibiotic–antimycotic [penicillin–streptomycin (antibiotic) amphotericin B (antifungal)] solution (100 × concentration) to 1 mL of the sample. Then, bi-fold dilutions were performed on 100 µL of the original dissolved sample, and 100 µL of each dilution was inoculated onto BGM cells (obtained from the Holding Company for Biological Products & Vaccines VACSERA, Giza, Egypt) that had been cultured in 96 multi well plates (Greiner-Bioone, Frickenhausen, Germany) to determine the sample’s non-toxic dose. For the cytotoxicity assay, an inverted light microscope (Olympus with 100 × magnification) and trypan blue (Sigma Aldrich) dye exclusion cell viability experiments were utilized [30, 31].
Cell morphology evaluation by inverted light microscopy
BGM cell (Vaccines VACSERA, Giza, Egypt) monolayers were seeded at 2 × 105 cells/mL in separate 96-well plates (Greiner-Bioone, Frickenhausen, Germany) and incubated until confluent (24 h). The media was removed from each well and replaced with 100 µL of dulbecco’s modified eagle medium (DMEM) (GIBCO BRL, Darmstadt, Germany)-prepared bi-fold dilutions of the sample being examined. For cell control goals, 100 µL of DMEM containing no samples was added. All cultures were maintained in a humidified atmosphere containing 5% (v/v) CO2 and incubated at 37 C for 72 h. Observations done on a daily basis revealed that the cells had lost their confluence, were rounding and contracting, and that the cytoplasm was granulating and vacuolizing. Morphological alterations were observed using an inverted light microscope (Olympus with 100 × magnification) and assessed [30].
Cell viability assay
The sample toxicity on BGM cells was assessed using the trypan blue dye exclusion method [31]. BGM cells cultures (2 × 105 cells/mL) were developed in 12-well plates. After 24 h incubation, 100 μL of sample dilutions (bi-fold dilutions) were applied to each well. After 72 h, the medium was removed, cells trypsinized and an equal volume of 0.4% trypan blue dye (Sigma Aldrich) solution added. The number of viable cells was counted under a phase contrast microscope. Trypan blue only stains dead cells blue, allowing distinguishing viable (unstained) cells. Counting unstained (viable) cells under a phase contrast microscope determined safe, minimally cytotoxic and lethal concentration ranges of the sample. Comparing viable cell counts in controls versus sample-treated wells showed the concentration-dependent effects on cell viability.
Determination of coxsackievirus B4 titers using plaque assay
Coxsackievirus B4 (106 PFU/mL, 100 μL) was mixed with 100 μL non-toxic sample dilutions. Incubating for 30 min at 37 °C allowed the sample to interact with CVB4. In 12-well plates, BGM cells (10 × dilution, 100 μL) received treated/untreated CVB4 (100 μL). After 1 h adsorption incubation at 37 °C in 5% CO2, plates were shaken occasionally to prevent drying. After adsorption, 1 mL DMEM media and 1 mL 1% agarose were added per well. Incubating enabled plaque formation which was stopped by fixation in formalin, staining with crystal violet and counting plaques. Viral titers (PFU/mL) were determined based on plaque counts. The described plaque assay enabled determining concentration-dependent anti-CVB4 activity of the test sample. It demonstrated the ability to inhibit plaque formation, representing virus replication, before and after cell exposure [32].
Homology modeling
In homology modeling, a protein sequence with a known 3D structure (template) is used to generate a structural model of a homologous protein sequence (target) that lacks an experimental 3D structure. The template selection, target-template alignment, model construction, and model evaluation are the main steps in homology modeling [33]. RNA-dependent RNA polymerase (RdRp) polyprotein [coxsackievirus B4] protein sequences with uniprot entry code UWQ56810.1 were used as targets for homology modeling using the SWISS-MODEL. The FASTA sequences were extracted and searched for similarity patterns automatically. The resulting alignments were scored to determine high quality predicted models. The SWISS-MODEL server performed target-template sequence alignment to generate 3D models for all target sequences by searching the PDB for proteins that function as X-ray templates [34]. The top homologs were selected using quantitative global model quality estimation (GMQE) and qualitative model energy analysis (QMEAN) data. The quality estimate ranges from 0 to 1, with higher values indicating better models. The QMEAN4 scoring function is a linear combination of four structural descriptors [35, 36].
Model quality estimation, structure validation and energy minimization
The models, including the backbone torsion angles, were uploaded to the SAVES server Version 5, where PROCHECK was used to assess the stereochemical quality of each model [37]. The QMEAN Z-score, which was calculated by subtracting the mean and dividing by the standard deviation of the normalized QMEAN score distribution, was used to evaluate the model quality. The normalized QMEAN score was plotted against the number of residues to compare the quality of different models. Energy minimization was performed in the Swiss-PDB viewer with the GROMOS96 force field to fix any distorted geometry in our model [38].
Molecular docking
The structures of each substance were saved in molecular operating environment files (MOE -2015). Triangle matcher was used as a placement method to dock the database of chemicals at the active site of the target protein, using the London dG score as a scoring function. The London dG score estimates the free binding energy (kcal/mol) between the chemical and the protein based on their intermolecular interactions. Only the top ten positions with the lowest London dG scores were recorded for each chemical.
Molecular dynamic simulation
To examine the atomic behavior and molecular structural stability of the top-ranked compounds and the crystal structures of the 3C-protease, 3CLpro, and RdRp enzymes, as well as to analyze their conformational changes during binding, the molecular dynamic (MD) approach is frequently employed. The top-ranked compounds were selected based on their docking scores and binding modes. The nanoscale molecular dynamics (NAMD) program [39] was used to perform the simulation. The necessary configuration files for MD simulation were generated by accessing the CHARMM-GUI website (http://www.charmm-gui.org/) [40]. Ligands were parameterized using the CHARMM general force field (CGenFF) tool, which is available online (https://cgenff.umaryland.edu/). The entire system was solvated using the water-molecule transferable intermolecular potential (TIP3P) model [41]. The NVT ensemble was used to minimize the energy of the systems across 10,000 steps during equilibration on a 5 ns timescale and subsequent production on a 50 ns timescale. The conformational changes of the molecules were analyzed by measuring their root mean square deviation (RMSD) and root mean square fluctuation (RMSF) values.
Binding free energy calculations
The binding free energy (ΔGbind) was determined by employing the widely used molecular mechanics poisson boltzmann surface area mechanics (MM-PBSA) method [42, 43]. Using the latest 10 ns of MD trajectories and a total of 1000 snapshots, all energy components were calculated. This includes the van der waals, electrostatic, polar solvation, and nonpolar solvation contributions. The reported studies have shown that the trend of inhibitor binding affinities in physiological circumstances may be reproduced using MD simulation coupled with MM-PBSA [43].
Results and discussion
Biological evaluation
The antiviral properties of the synthesized 2-thioxo-benzo[g]quinazolines (1–16, Fig. 2) were tested in vitro against CVB4. Their cytotoxicity were initially on the BGM cells line tested, as detailed in the supporting information (Table 1). There was no significant difference in the levels of the nontoxic doses for the targets 1–16, which ranged from 25 to 65 µg/mL. Most benzoquinazolines showed antiviral activity, however, effectiveness varied among compounds. This is typical due to coxsackievirus B4’s unique nature, structure, size, and genomic structure (RNA). Even though CVB4 has an RNA genomic structure, various responses to the investigated benzoquinazolines are observed. Antiviral bioassays reveal distinct differences in the reactions of viruses carrying RNA with diverse subsets of components. It’s possible that the compounds’ effect on viral infectivity was assessed exclusively by the compounds’ direct action on the virus, and not by their ability to block viral adsorption or disrupt the viral replication cycle. This could be due to the nature of the tests, which solely target the virus with non-toxic concentrations of the investigated benzoquinazolines. This study’s findings might have resulted from any of the aforementioned mechanisms, or they could have been influenced by the substances investigated at non-toxic doses that affected the genome. Another possible explanation is that increasing the dose may increase the probability of obtaining an antiviral impact, as evidenced by the fact that different doses demonstrated no toxicity on different cell lines. All of the investigated compounds 1–16 have shown varied levels of activity against coxsackievirus B4, ranging from low to high; however, compounds 5 and 13 appeared inactive (Table 1). Antiviral efficiency against coxsackievirus B4 is promising for compounds 1, 2, 3, 9, and 16, with reduction percentages ranging from 60 to 83%. However, benzoquinazoline 14 has shown good efficacy (56%) in comparison to its parent and 16. Benzoquinazolines 4, 6, 7, 10–12, and 15 exhibited minimal antiviral activity against CVB4. Therefore, more research is required to identify the precise fraction responsible for the antiviral potency of the many promising materials or to confirm the synergism of the fractions that contribute to the antiviral potency of the studied materials.
Fig. 2.

Chemical structures of the investigated benzo[g]quinazolines 1–16
Table 1.
Non-toxic doses of tested benzoquinazolines (1–16) on the BGM cells line; Percentages of reduction done by the non-toxic doses of 1–16 against coxsackievirus B4
| Comp | Non-toxic doses (µg/mL) On the BMG cell line |
% of Coxsackievirus B4 reduction |
|---|---|---|
| 1 | 60 | 66.7 ± 4 |
| 2 | 60 | 70 ± 4 |
| 3 | 60 | 83.3 ± 4 |
| 4 | 50 | 23.3 ± 2 |
| 5 | 55 | 0 |
| 6 | 65 | 26.7 ± 2 |
| 7 | 60 | 20 ± 4 |
| 8 | 50 | 50 ± 2 |
| 9 | 65 | 60 ± 3 |
| 10 | 50 | 20 ± 1 |
| 11 | 65 | 33.3 ± 2 |
| 12 | 25 | 20 ± 3 |
| 13 | 40 | 0% |
| 14 | 55 | 56.7 ± 3 |
| 15 | 55 | 10 ± 1 |
| 16 | 60 | 60 ± 1 |
Results are mean ± SD of 3 results against 3 doses of virus (1X106) PFU/ml
Based on the aforementioned findings, preliminary structural activity relationships (SAR) have been constructed. Table 1 shows that the parent compounds (1–3) showed the greatest activity against CVB4 (66.7, 70, and 83.3%); with compound 3, featuring a benzyl substitution at position 3 in the benzo[g]quinazoline scaffold, showing the highest reduction percent, followed by 2, and then 1, which featured by ethyl and methyl groups, respectively. This could be attributable to the target molecule being more lipophilic. The activity profile is not improved by chemically transforming the thioxo group (1–3) into the thioether group (4–13). The anti-CVB4 activity, however, was shown to vary depending on the type of substitution introduced to the thioether moiety. It is obvious that compound 8, which contains a chloride substituent at the para position of the benzyl moiety, is more active against CBV4 than compound 7, which contains the same substituent, providing further validity to the aforementioned fact dealing with lipophilicity features. Substituted CN compounds 9–11 had varying biological effects; compound 11 with N–benzyl substitution was more active than compound 10 with N–ethyl substitution, whereas compound 9 with CN in the meta position on the benzyl moiety was most active. Derivatives (14–16) were developed by performing further chemical transformations on the thioxo group via inserting of the hydrazine functional group; nevertheless, only derivatives (14) and (16) showed increased activity (56.7 and 60%, respectively). It is probable that the hydrazine functionality may have low intrinsic reactivity with the CVB4 active site or may have the capacity to bind to active site residues. Therefore, the addition of an electron-donating or electron-withdrawing group such as methoxy, chloride, or cyano to the benzyl group, or the insertion of a rich hydrogen group, produced numerous different types of activity profiles.
Molecular and homology modeling
Homology modeling
Coxsackievirus major protease 3C and B3 3C proteases, which correspond to PDB entries 5iyt and 4y2a, respectively, have 97.78% and 96.10% sequence identity (Table 2). The top three models in terms of GMQE and QMEAN4 quality scores indicate reliable models with accurate tertiary structures. The model quality estimation plot shows that the main protease 3C and B3 3C protease models are within the expected range for proteins of similar size (Fig. 3A and C). PROCHECK analysis showed that 90.1% and 92.4% of the amino acid residues in the main 3C and B3 3C protease models, respectively, were within the most favored region, while 9.3% and 7.1% were in the additionally allowed region (Fig. 3B and D).
Table 2.
Summary of coxsackievirus 3CLpro and RdRp homology models and validations
| Coxsackie proteins | Swiss-model server results | Procheck analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Name | PDB ID | Sequence identity | GMQE | QMEAN4 | Most favoured regions | Additionally allowed regions | Generously allowed regions | Disallowed regions |
| 3CLpro | 5iyt | 97.78% | 0.80 | 0.80 ± 0.05 | 90.1% | 9.3% | 0.7% | 0.0% |
| RdRp | 4y2a | 96.10% | 0.80 | 0.88 ± 0.05 | 92.4% | 7.1% | 0.2% | 0.2% |
Fig. 3.

Ramachandran plots illustrate modelled amino acid residue dihedral angles Psi and Phi. Red color indicates the favored angles, yellow color indicates acceptable angles, beige color indicates generous angles, and white color indicates banned angles. Panel A shows a model quality comparison plot for coxsackievirus’s primary protease 3C, whereas panel B shows its ramachandran plots. Panel C shows a coxsackievirus B3 3C protease model quality comparison graphic. Panel D displays coxsackievirus B3’s 3C protease ramachandran maps
Molecular modeling study
Docking simulations are used to study the molecular structure and structure–activity relationship of various compounds. In this study, we used the MOE 2015.10 software from chemical computing group inc. (Montreal, Quebec, Canada) for the docking procedure. The target compounds and the co-bound inhibitor were docked onto the putative binding site of the protein to obtain an optimal binding orientation. Molecular docking was performed on the three most active compounds 1–3 to investigate their binding modes and interactions with the key amino acids in the active site of the multi-target coxsackievirus B4 (3Clpro and RdRp targets). The crystal structures of 3Clpro, RdRp receptors, and capsid binder in complex with NZN, 1FS, and QFW were obtained from the RCSB protein data bank (PDB ID: homology model for 3Clpro, RdRp, and 6ZCK for capsid binder) and used them to generate the initial docking model of 3Clpro, RdRp receptors, and capsid binder.
Coxsackievirus B4 3C protease (picornain 3C)
To verify and validate the docking procedure, the NZN was subjected to one run of docking into the binding site. NZN inhibitor exhibited a covalent interaction with Cys147 and a hydrogen bond with Cys147 via the carbonyl group (Fig. 4). Docking studies for 1, 2, and 3 revealed that all three molecules occupy almost the same location in the enzyme active site as NZN.
Fig. 4.

NZN binding to the active site of 3C protease for coxsackievirus B4 (homology model). A Three-dimensional view of the complex, B Two-dimensional representation of the interactions
Benzoquinazoline scaffold’s two nitrogen atoms are important for the interaction with enzymes. One of the nitrogen atoms in compounds 1, 2, and 3 (Score = –8.10, –8.4, and –9.6 kcal/mol, respectively) (Table 3) forms a hydrogen bond with the essential amino acid Cys147 in the active site of coxsackievirus 3C protease. The other nitrogen atom interacts with the amino acid residue Thr142, while the aromatic ring has a pi-H interaction with Ala144 and Gly145, as shown in Fig. 5 (A–D), Figures S1, S2 and Table 3.
Table 3.
Docking data for compounds 1- 3 against 3C protease of coxsackievirus B4 and using (PDB ID: 5iyt) as template crystal
| Ligand | Receptor | Interaction | Distance | E (kcal/mol) | Scoring |
|---|---|---|---|---|---|
| Co-crystal (NZN) | –10.0 | ||||
| S 30 | CG1 VAL 210 (A) | H-acceptor | 4.26 | −0.2 | |
| S 30 | CG2 VAL 210 (A) | H-acceptor | 4.12 | –0.2 | |
| S 30 | OH TYR 327 (A) | H-acceptor | 3.18 | –2.6 | |
| C 4 | 6-ring TYR 327 (A) | H-pi | 4.79 | –0.2 | |
| 6-ring | CE1 HIS 199 (A) | pi-H | 3.54 | –0.5 | |
| 6-ring | CE1 HIS 199 (A) | pi-H | 4.56 | –0.5 | |
| 6-ring | NE2 HIS 199 (A) | pi-H | 3.29 | –0.3 | |
| 6-ring | CG1 VAL 210 (A) | pi-H | 4.46 | –0.7 | |
| Comp. 1 | –8.1 | ||||
| N 9 | SG CYS 147 (A) | H-donor | 3.22 | –3.9 | |
| 6-ring | CA ALA 144 (A) | pi-H | 3.47 | –0.3 | |
| 6-ring | N GLY 145 (A) | pi-H | 4.04 | –0.8 | |
| Comp. 2 | –8.4 | ||||
| N 12 | SG CYS 147 (A) | H-donor | 3.19 | –3.6 | |
| S 30 | CB THR 142 (A) | H-acceptor | 4.48 | –0.4 | |
| 6-ring | CA ALA 144 (A) | pi-H | 3.44 | –0.3 | |
| 6-ring | N GLY 145 (A) | pi-H | 4.02 | –0.7 | |
| Comp. 3 | –9.6 | ||||
| C17 23 | OG1 THR 142 (A) | H-donor | 3.01 | –1.1 | |
| O28 1 | N GLY 145 (A) | H-acceptor | 2.75 | –2.9 | |
| O28 1 | N GLN 146 (A) | H-acceptor | 3.04 | –1.7 | |
| O28 1 | N CYS 147 (A) | H-acceptor | 3.21 | –0.6 | |
Fig. 5.

A Three-dimensional view of the active site of the 3C protease with the three most active ligands (1–3) overlaid; B, C, and D Two-dimensional representation of the interactions between the active site of the 3C protease and each of the three most active ligands (1–3) after docking and minimization
The RNA-dependent RNA polymerase for coxsackievirus B4
All information regarding the active site can be found in the homology model. This model sheds light on the hydrophobicity of the binding site, which is surrounded by multiple hydrophobic residues, including Ile296, Asp111, Leu110, Val210, Ser295, and Tyr327. The antiviral effect of these benzoquinazolines against coxsackievirus could be due to their hydrophobic nature (Fig. 6, Figs. S3 and S4). To ensure the accuracy and reliability of the docking procedure, it was validated on the 1FS by performing a single docking run into the binding site. The hydrogen connection between 1FS inhibitor and Tyr327 was observed. Docking studies with compounds 1–3 showed that they all occupied similar locations in the enzyme active site to the 1FS (Table 4, Fig. 6D). The docking analysis revealed a hydrogen connection between the thioxo moiety (C = S) of compounds 1–3 and Tyr327 in the catalytic site (Fig. 6A-C). Hydrogen bonds with Gly293 and Tyr327 were observed in benzoquinazolines 1–3, whereas hydrophobic interactions with Arg188 and Asp111 were seen in compounds 1 and 2 (Fig. 6 A and B, Fig. S3 A and B and Table 4). The N-benzyl ring in compound 3 is crucial for its ability to form a hydrophobic connection with Ser292 and bridge through water molecule to Gly293 as well as the phenyl ring bind via a π-π interaction with the catalytic site residue Tyr327. (Fig. 6C and Fig. S3C). Hydrogen bonds were observed between the carbonyl (C = O) group of compound 3 and Thr294. This may help to explain why compound 3 is the most potent inhibitor and why its percent reduction value is so much higher than that of compounds 1 and 2.
Fig. 6.

Two-dimensional representation of the interactions between the active site of B4 (RNA-dependent RNA polymerase) and the three most active ligands (1, 2, and 3) in (A), (B), and (C), respectively, and the co-crystal in (D)
Table 4.
Docking data for compounds 1–3, and IFS, against (RNA-dependent RNA polymerase of coxsackievirus B4) ( template (PDB ID: 4y2a)
| Ligand | Receptor | Interaction | Distance | E (kcal/mol) | Scoring |
|---|---|---|---|---|---|
| Co-crystal (IFS) | – 8.3 | ||||
| S 30 | CG1 VAL 210 (A) | H-acceptor | 4.26 | – 0.2 | |
| S 30 | CG2 VAL 210 (A) | H-acceptor | 4.12 | – 0.2 | |
| S 30 | OH TYR 327 (A) | H-acceptor | 3.18 | – 2.6 | |
| C 4 | 6-ring TYR 327 (A) | H-pi | 4.79 | – 0.2 | |
| 6-ring | CE1 HIS 199 (A) | pi-H | 3.54 | – 0.5 | |
| 6-ring | CE1 HIS 199 (A) | pi-H | 4.56 | – 0.5 | |
| 6-ring | NE2 HIS 199 (A) | pi-H | 3.29 | – 0.3 | |
| 6-ring | CG1 VAL 210 (A) | pi-H | 4.46 | – 0.7 | |
| Comp. 1 | – 6.3 | ||||
| S 27 | CG1 VAL 210 (A) | H-acceptor | 4.05 | – 0.2 | |
| S 27 | CG2 VAL 210 (A) | H-acceptor | 4.09 | – 0.2 | |
| S 27 | OH TYR 327 (A) | H-acceptor | 3.27 | – 2.4 | |
| 6-ring | CE1 HIS 199 (A) | pi-H | 3.5 | – 0.3 | |
| 6-ring | CE1 HIS 199 (A) | pi-H | 4.47 | – 0.4 | |
| 6-ring | NE2 HIS 199 (A) | pi-H | 3.36 | – 1 | |
| 6-ring | CG1 VAL 210 (A) | pi-H | 4.42 | – 0.6 | |
| Comp. 2 | –6.5 | ||||
| S 30 | NZ LYS 127 (A) | H-acceptor | 3.6 | – 8.4 | |
| S 30 | OG SER 184 (A) | H-acceptor | 3.25 | – 0.9 | |
| 6-ring | CA SER 292 (A) | pi-H | 4.04 | – 0.8 | |
| 6-ring | CA SER 292 (A) | pi-H | 4.36 | – 0.8 | |
| 6-ring | N GLY 293 (A) | pi-H | 3.7 | – 0.6 | |
| 6-ring | CA GLY 293 (A) | pi-H | 3.74 | – 0.6 | |
| Comp. 3 | – 6.9 | ||||
| O 1 | CB SER 295 (A) | H-acceptor | 3.36 | – 0.5 | |
| S 37 | CG1 VAL 210 (A) | H-acceptor | 4.14 | – 0.2 | |
| S 37 | OH TYR 327 (A) | H-acceptor | 4.09 | – 1.3 | |
| 6-ring | CG2 VAL 210 (A) | pi-H | 3.42 | – 0.3 | |
The capsid protein of coxsackievirus B4
The docking method was tested by performing a single docking run on the QFW into the binding site. The carboxylic group of the QFW inhibitor formed a hydrogen bond with Arg A228. Identical to QFW, compounds 1–3 docked in the same general area of the enzyme active site (Fig. 7, Figs. S5 and S6). The two ArgA228 residues on the pocket’s inner surface and the Gln B233 residue at the entrance were found to play a crucial function in the model (Table 5). As with QFW’s anchoring to CVB4, the investigated compounds are retained in place by stacking interactions between two hydrophobic residues (TyrA67, CysA64 in compounds 1 and 2, beside thioxo group in both compounds form hydrogen bond with GlnB233 and ValA91 (Fig. 7 B and C). Four hydrogen bonds formed by 3 including two H-bond of thioxo group with TyrA67 and ArgA228, the carbonyl group with PheB236, as well as phenyl group with GlnB233 (Fig. 7d, and Fig. S5C). The benzoquinazolines 1–3 scoring are around −7.93, −7.96, and −9.53, respectively, vs −10.78 kcal/mol for QFW with CVB4.
Fig. 7.

Two-dimensional representation of the interactions between the active site of the capsid protein and the co-crystal in (A) and the three most active ligands 1–3 in (B–D), respectively
Table 5.
Docking data for compounds 1–3 against the capsid protein of coxsackievirus B4; protein receptor (PDB ID: 6ZCK)
| Ligand | Receptor | Interaction | Distance | E (kcal/mol) | scoring |
|---|---|---|---|---|---|
| Co-crystal (QFW) | –10.78 | ||||
| O4 26 | NH2 ARG 228 (A) | H-acceptor | 3.17 | –4.7 | |
| O5 27 | NE ARG 228 (A) | H-acceptor | 3.16 | –4.6 | |
| O4 26 | NE ARG 228 (A) | Ionic | 3.57 | –2.9 | |
| O4 26 | NH2 ARG 228 (A) | Ionic | 3.17 | –5 | |
| O5 27 | NE ARG 228 (A) | Ionic | 3.16 | –5.1 | |
| C16 19 | 6-ring PHE 236 (B) | H-pi | 3.94 | –0.9 | |
| 6-ring | CE2 TYR 67 (A) | pi-H | 4.12 | –0.5 | |
| 6-ring | CA THR 235 (B) | pi-H | 3.87 | –0.5 | |
| Comp. 1 | –7.93 | ||||
| N 9 | OE1 GLN 233 (B) | H-donor | 3.11 | –3.2 | |
| S 27 | CB VAL 91 (A) | H-acceptor | 3.84 | –0.4 | |
| S 27 | N GLN 93 (A) | H-acceptor | 4.1 | –2.5 | |
| S 27 | CB GLN 93 (A) | H-acceptor | 3.77 | –0.7 | |
| 6-ring | CB CYS 64 (A) | pi-H | 4.86 | –0.7 | |
| 6-ring | OH TYR 67 (A) | pi-H | 4.01 | –0.3 | |
| Comp. 2 | –7.96 | ||||
| C 16 | OE1 GLN 233 (B) | H-donor | 3.49 | –0.2 | |
| S 30 | N CYS 64 (A) | H-acceptor | 4.68 | –0.8 | |
| S 30 | OH TYR 230 (A) | H-acceptor | 4.61 | –0.6 | |
| 6-ring | CE2 TYR 67 (A) | pi-H | 4.72 | –0.2 | |
| Comp. 3 | |||||
| S 37 | CE2 TYR 67 (A) | H-acceptor | 3.77 | –0.3 | –9.53 |
| S 37 | NH2 ARG 228 (A) | H-acceptor | 4.65 | –1.4 | |
| C 4 | 6-ring PHE 236 (B) | H-pi | 4.39 | –0.6 | |
| 6-ring | N CYS 64 (A) | pi-H | 3.64 | –0.3 | |
| 6-ring | CB CYS 64 (A) | pi-H | 3.72 | –0.4 | |
Molecular dynamic simulation
The most active compound 3, the crystal structures of the 3C-protease, 3CLpro, and RdRp enzymes, were simulated using the nanoscale molecular dynamics (NAMD) software. Generation of the configuration files needed for MD simulation was done by using the CHARMM-GUI website (http://www.charmm-gui.org/).
To examine the structural stability and mobility of the enzyme-ligand complex during simulation, the fluctuations of the complex and the movements of the associated ligand within a hydrated environment were analyzed. These movements ensure the reliability and efficiency of the system in a biological context. As shown in Fig. 8 (A, B, C), the system was overall stable. The RMSD of the docked enzyme-ligand pair did not change significantly during the 50 ns simulation. Table 6 showed the mean RMSD and maximum deviation for three targets with benzoquinazoline 3. The lack of major structural changes along the RMSD plot indicates the stability and strong ligand binding in the active site pocket of the protein.
Fig. 8.
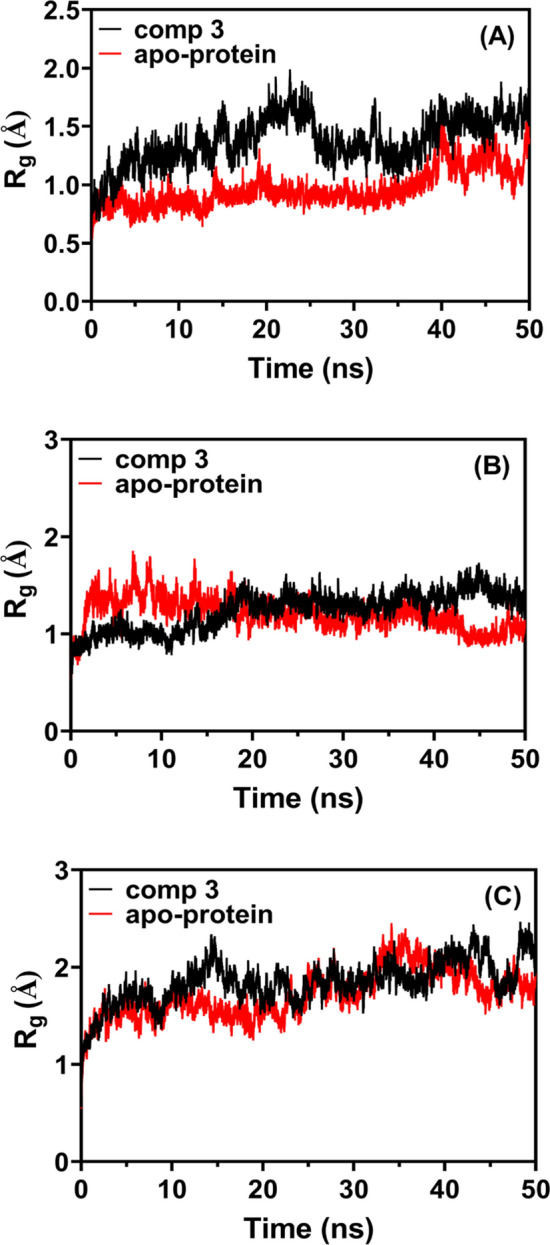
The root means square deviation (RMSD) of compound 3 with A 3C-protease, B 3CLpro, and C RdRp
Table 6.
Average, maximum values, and time of the maximum fluctuations in RMSD results
| Targets | Average value of RMSD (Å) | Maximum value of RMSD (Å) | Time of the maximum value (ns) |
|---|---|---|---|
| 3C_protease | 1.24 | 1.722 | 44.93 |
| 3CLpro | 1.38 | 1.98 | 22.68 |
| RdRp | 1.85 | 2.457 | 48.32 |
The relative mobility of the flexible Cα atoms in each residue is an important metric that can be determined using the RMSF. As can be shown in Fig. 9 (A, B, C), the average C fluctuations of 3C-protease, 3CLpro, and RdRp were 0.686, 0.639, and 0.825 Å, respectively. Considering that loops and coils function as bridges between β-sheets and α-helices, it makes sense that these locations would show the most pronounced fluctuations. These sheets and helices are typically oriented over simulation time to facilitate ligand interactions and maintain ligand localization inside the active site. Within the first 50 ns after a ligand enters the active site pocket of an enzyme, β-sheets and α-helices frequently extend and reduce.
Fig. 9.

Root mean square fluctuations (RMSF) plot for compound 3 with A 3C-protease, B 3CLpro, and C RdRp
The compactness of a molecular’s structure can be used to calculate its radius of gyration (Rg), which is defined as the root mean square distance between all atoms in the molecule and the centroid. Rg of compound 3, a delivery agent, varies during the simulation with respect to its three targets (3C-protease, 3CLpro, and RdRp; 15.250.0492, 15.490.0722, and 23.460.0815, respectively) as illustrated in Fig. 10 (A, B, C). Up to 10 ns, a high Rg value indicated its bioavailability, and this behavior showed that the target benzoquinazoline might sustain its native structure in MD and its connections.
Fig. 10.
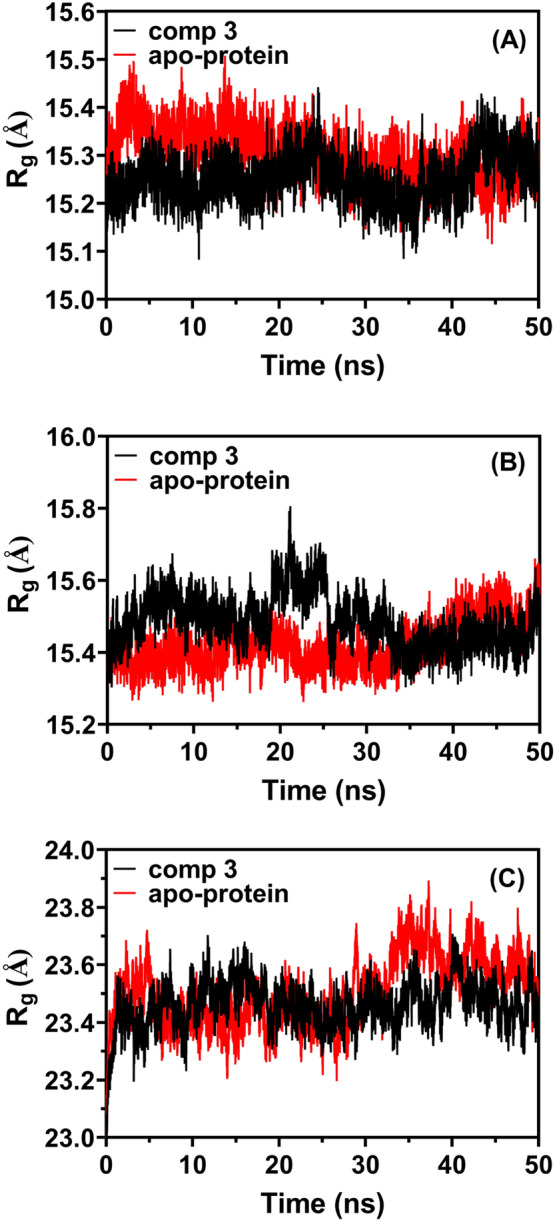
The radius of gyration (Rg) of compound 3 with A 3C-protease, B 3CLpro, and C RdRp over MD time
There is a significant function for hydrogen bonding in all interactions. Measuring the intermolecular H-bond formed between compound 3 and three targets (3C-protease, 3CLpro, and RdRp) allowed us to assess the MD simulation’s robustness. Using a minimum donor–acceptor distance of 3.2 and a maximum angle of 30°, the hydrogen-bonding pattern was calculated for the top-ranked complexes. The intermolecular hydrogen bonds formed between compound 3 and the hydrophobically-charged proteins (3C-protease, 3CLpro, and RdRp) in all of the systems have been checked and are listed in Fig. 11 (A, B, C). The cut-off distance and cut-off angle of hydrogen-donor–acceptor are the hydrogen bonding criterion for the donor–acceptor. If compound 3 was in close proximity to the proteins, then hydrogen bonding between the molecules would be possible. Furthermore, the amount of intermolecular hydrogen bonds is highly correlated with the drug-protein interaction in the last 30 ns period in 3C-protease and 3CLpro, whereas in RdRp the number of H-bonds was consistently high throughout.
Fig. 11.

Inter-molecular hydrogen bonds formed between compound 3 with A 3C-protease, B 3CLpro and C RdRp
Binding energy calculation
Each ligand’s participation in the ligand–protein interaction was determined by estimating the binding free energy using MD simulation trajectories [44]. The MD directed molecular mechanics poisson boltzmann surface area (MM/PBSA) approach was used to determine binding free energy using the ambertool20 mmpbsa tool. A bigger negative binding energy indicates a better ligand affinity for its target protein. The method accounts for more ligand–protein affinity than static or the most advanced flexible molecular docking methods. MM/PBSA is as accurate as free-energy perturbation but cheaper to process [45]. Sample frames were extracted/saved from the final 20 ns of MD simulation trajectories using the SASA (solvent accessible surface area) only model of the free binding energy computation and the single trajectory technique to calculate each energy term and their average values over all three MD simulation runs. Binding free energy may be calculated with great accuracy using the mechanics/poisson boltzmann surface area (MM/PBSA) approach. By analyzing the last 20 ns of MD trajectories with the MM/PBSA method, the binding free energies of compound 3 with 3C-protease, 3CLpro, and RdRp were calculated. Complexes of 3 with 3C-protease, 3 with 3CLpro, and 3 with RdRp have different binding free energies (ΔGbinding) of −73.13, −76.71, and −82.64 kJ/mol, respectively (Table 7). Compound 3 with 3C-protease complex has a lower computed van der waals contribution (ΔEvdw) to the binding free energy (−143.52 kJ/mol) than the 3 with 3CLpro complex (−153.21 kJ/mol) or the 3 with RdRp complex (−170.081 kJ/mol). The binding of compound 3 with RdRp active site was favored by a sum of solvation energy (ΔEsolv), van der waals energy (ΔEvdw), and electrostatic energy (ΔEelect), indicating that compound 3 is highly selective to the RdRp target.
Table 7.
Molecular energy data of compound 3: 3C-protease, 3: 3CLpro and 3: RdRp complexes
| Energy components (kJ/mol) | Comp.3:3C-protease | Comp.3:3CLpro | Comp.3:RdRp |
|---|---|---|---|
| van der waal energy (ΔGvdW) | –143.52 | – 153.21 | – 170.081 |
| Electrostatic energy (ΔGele) | –33.25 | – 35.21 | – 42.52 |
| Polar solvation energy (ΔGps) | 117.06 | 124.06 | 148.261 |
| SASA energy (ΔGsasa) | –13.42 | – 12.35 | – 18.3 |
| Binding energy (ΔGbinding) | –73.13 | – 76.71 | – 82.64 |
Conclusion
The antiviral activity of benzo[g]quinazolines (1–16) against coxsackievirus B4 was investigated, as well as their cytotoxicity, utilizing the BGM cells line. The results demonstrated that benzoquinazolines 1–3 have the highest antiviral effects against CVB4. The activity was influenced by the type and position of the substituents on the benzoquinazoline scaffold as shown by benzoquiazolines (7–11). The docking results performed the binding and interactions of the top three active benzoquinazolines 1–3 with the constitutive amino acids in the active region of the multi-target coxsackievirus B4 (RdRp and 3Clpro) targets. The structural stability and mobility of the enzyme-ligand complex were analyzed by measuring the RMSD and RMSF of the system. The MD simulation results verified benzoquinazoline 3’s intrinsic action. A combination of solvation energy (ΔEsolv), van der waals energy (ΔEvdw), and electrostatic energy (ΔEelect) preferred compound 3 binding to the RdRp active site, demonstrating that compound 3 is highly selective to the RdRp target. Further laboratory investigation is warranted to better understand the benzoquinazolines mechanism action.
Supplementary Information
Below is the link to the electronic supplementary material.
{kind=link}
Supplementary file1 (PNG 6689 KB) Molecular modeling of the catalytic active site of coxsackievirus 3CLpro. (i)Surface analysis using three most active ligands (1-3 superimposed); (ii) Magnified view ofthe active site and the ligands.
{kind=link}
Supplementary file2 (PNG 2320 KB) Interactions of amino acids in the coxsackievirus 3CLpro homology model andthe overlay of co-crystalline ligand in (A), and ligands 1-3 (B), (C) and (D), respectively.
{kind=link}
Supplementary file3 (PNG 4082 KB) Interactions between amino acids of the coxsackievirus B4 3C protease (PDBcode: 6ZCK) and the three most active ligands 1-3 in (A-C), respectively; Figure S5.Interactions between amino acids of the coxsackievirus RdRp homology model and the threemost active ligands 1-3 in (D-F), respectively.
{kind=link}
Supplementary file4 (PNG 4791 KB) Molecular modeling of the catalytic active site of coxsackievirus RdRp and itsinteraction with the tested ligand (1). (i) Surface representation of the active site and theligand; (ii) Close-up view of the active site and the ligand showing the binding mode andinteractions.
{kind=link}
Supplementary file5 (PNG 5837 KB) Molecular modeling of the catalytic active site of Coxsackievirus B4 3C protease(PDB code: 6ZCK) and its interaction with the tested ligand (1). (i) Surface representation ofthe active site and the ligands; (ii) Close-up view of the active site and the ligands showingthe binding mode and interactions.
Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project, King Saud University, Riyadh, Saudi Arabia for funding this work through grant no. RSP-2023R353
Authors contribution
HAA: writing original draft, investigation and data curation; ABH: software and validation; MM.: writing-reviewing; WMA methodology and biological experiments MMA: writing-reviewing; GAEM writing reviewing, QS software and validation, SBH software and validation, RA-S writing original draft, investigation, data curation and supervision. All authors have read the final version and approved the manuscript for publication.
Funding
This research was funded by Researchers Supporting Project, King Saud University, through grant no. RSP-2023R353.
Data availability
Original data are available upon request.
Declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Flodström M, Horwitz MS, Maday A, Balakrishna D, Rodriguez E, Sarvetnick N. A critical role for inducible nitric oxide synthase in host survival following coxsackievirus B4 infection. Virology. 2001;281:205–215. doi: 10.1006/viro.2000.0801. [DOI] [PubMed] [Google Scholar]
- 2.Chapman NM, Ramsingh AI, Tracy S. Genetics of coxsackievirus virulence. Coxsackie B Viruses 1997:227–58. [DOI] [PubMed]
- 3.Honkimaa A, Kimura B, Sioofy-Khojine A-B, Lin J, Laiho J, Oikarinen S, et al. Genetic adaptation of coxsackievirus B1 during persistent infection in pancreatic cells. Microorganisms. 2020;8:1790. doi: 10.3390/microorganisms8111790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMinn PC. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol Rev. 2002;26:91–107. doi: 10.1111/j.1574-6976.2002.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 5.Azzam RA, Osman RR, Elgemeie GH. Efficient synthesis and docking studies of novel benzothiazole-based pyrimidinesulfonamide scaffolds as new antiviral agents and Hsp90α inhibitors. ACS Omega. 2020;5:1640–1655. doi: 10.1021/acsomega.9b03706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nekoua MP, Bertin A, Sane F, Gimeno J-P, Fournier I, Salzet M, et al. Persistence of coxsackievirus B4 in pancreatic β cells disturbs insulin maturation, pattern of cellular proteins, and DNA methylation. Microorganisms. 2021;9:1125. doi: 10.3390/microorganisms9061125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strasfeld L, Chou S. Antiviral drug resistance: mechanisms and clinical implications. Infect Dis Clin. 2010;24:809–833. doi: 10.1016/j.idc.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Sun L, Meijer A, Froeyen M, Zhang L, Thibaut HJ, Baggen J, et al. Antiviral activity of broad-spectrum and enterovirus-specific inhibitors against clinical isolates of enterovirus D68. Antimicrob Agents Chemother. 2015;59:7782–7785. doi: 10.1128/AAC.01375-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brilot F, Jaïdane H, Geenen V, Hober D. Coxsackievirus B4 infection of murine foetal thymus organ cultures. J Med Virol. 2008;80:659–666. doi: 10.1002/jmv.21016. [DOI] [PubMed] [Google Scholar]
- 10.Dotta F, Censini S, van Halteren AGS, Marselli L, Masini M, Dionisi S, et al. Coxsackie B4 virus infection of β cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci. 2007;104:5115–5120. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 12.Tan EL, Wong APY, Poh CL. Development of potential antiviral strategy against coxsackievirus B4. Virus Res. 2010;150:85–92. doi: 10.1016/j.virusres.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 13.Bixing H, Harrower B, Burtonclay P, Constantino T, Warrilow D. Genome sequences of Coxsackievirus B5 isolates from two children with meningitis in Australia. Microbiol Resour Announc. 2017 doi: 10.1128/genomeA.01125-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tuthill TJ, Groppelli E, Hogle JM, Rowlands DJ. Picornaviruses. Cell entry by non-enveloped viruses. Berlin: Springer; 2010. pp. 43–89. [Google Scholar]
- 15.Olasunkanmi OI, Chen S, Mageto J, Zhong Z. Virus-induced cytoplasmic aggregates and inclusions are critical cellular regulatory and antiviral factors. Viruses. 2020;12:399. doi: 10.3390/v12040399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Al-Shakliah NS, Attwa MW, AlRabiah H, Kadi AA. Identification and characterization of in vitro, in vivo, and reactive metabolites of tandutinib using liquid chromatography ion trap mass spectrometry. Anal Methods. 2021;13:399–410. doi: 10.1039/D0AY02106G. [DOI] [PubMed] [Google Scholar]
- 17.Al-Salahi RA, Al-Omar MA, Alswaidan I, Marzouk M, El-Senousy WM, Amr AE-GE. Antiviral activities of some synthesized methylsulfanyltriazoloquinazoline derivatives. Res Chem Intermed. 2015;41:151–161. doi: 10.1007/s11164-013-1177-1. [DOI] [Google Scholar]
- 18.Abuelizz HA, Marzouk M, Bakhiet A, Abdel-Aziz MM, Ezzeldin E, Rashid H, et al. In silico study and biological screening of benzoquinazolines as potential antimicrobial agents against methicillin-resistant Staphylococcus aureus, carbapenem-resistant Klebsiella pneumoniae, and fluconazole-resistant Candida albicans. Microb Pathog. 2021;160:105157. doi: 10.1016/j.micpath.2021.105157. [DOI] [PubMed] [Google Scholar]
- 19.Al-Salahi R, Taie HAA, Bakheit AH, Marzouk M, Almehizia AA, Herqash R, et al. Antioxidant activities and molecular docking of 2-thioxobenzo[g]quinazoline derivatives. Pharmacol Reports. 2019;71:695–700. doi: 10.1016/j.pharep.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Almehizia AA, Abuelizz HA, Taie HAA, ElHassane A, Marzouk M, Al-Salahi R. Investigation the antioxidant activity of benzo[g]triazoloquinazolines correlated with a DFT study. Saudi Pharm J. 2019 doi: 10.1016/j.jsps.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Salahi R, Marzouk M, Abuelizz HA. Anti-HAV evaluation and molecular docking of newly synthesized 3-benzyl (phenethyl) benzo [g] quinazolines. Bioorg Med Chem Lett. 2019;29:1614–1619. doi: 10.1016/j.bmcl.2019.04.043. [DOI] [PubMed] [Google Scholar]
- 22.Al-Salahi R, Ahmad R, Anouar E, Iwana Nor Azman NI, Marzouk M, Abuelizz HA. 3-Benzyl(phenethyl)-2-thioxobenzo[g]quinazolines as a new class of potent α-glucosidase inhibitors: Synthesis and molecular docking study. Future Med Chem. 2018;10:1889–1905. doi: 10.4155/fmc-2018-0141. [DOI] [PubMed] [Google Scholar]
- 23.Al-Salahi R, Moustapha ME, Abuelizz HA, Alharthi AI, Alburikan KA, Ibrahim IT, et al. Radioiodination and biodistribution of newly synthesized 3-benzyl-2-([3-methoxybenzyl] thio) benzo [g] quinazolin-4-(3H)-one in tumor bearing mice. Saudi Pharm J. 2018;26:1120–1126. doi: 10.1016/j.jsps.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abuelizz HA, Marzouk M, Bakheit AH, Al-Salahi R. Investigation of some benzoquinazoline and quinazoline derivatives as novel inhibitors of HCV-NS3/4A protease: Biological, molecular docking and QSAR studies. RSC Adv 2020; doi: 10. 10.1039/d0ra05604a. [DOI] [PMC free article] [PubMed]
- 25.Abuelizz HA, Marzouk M, Bakheit AH, Awad HM, Soltan MM, Naglah AM, et al. Antiproliferative and antiangiogenic properties of new VEGFR-2-targeting 2-thioxobenzo [g] quinazoline derivatives (In Vitro) Molecules. 2020;25:5944. doi: 10.3390/molecules25245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Salahi R, Abuelizz HA, El Dib R, Marzouk M, Alshammari MB. Antimicrobial activity of new 2-thioxo-benzo[g]quinazolin-4(3H)-one derivatives. Med Chem (Los Angeles) 2017; doi: 10.2174/1573406412666160610095706. [DOI] [PubMed]
- 27.Al-Salahi R, El Dib RA, Marzouk M. Synthesis and in vitro cytotoxicity evaluation of new 2-thioxo-benzo[g]quinazolin-4(3h)-one derivatives. Heterocycles. 2015;91:1735–1751. doi: 10.3987/COM-15-13282. [DOI] [Google Scholar]
- 28.Al-Salahi R, A Abuelizz H, Wadi M, A El Dib R, A Alotaibi M, Marzouk M. Antimicrobial activity of synthesized 2-methylthiobenzo [g][1, 2, 4]-triazolo [1, 5-a] quinazoline derivatives. Med Chem 2016;12:760–766. [DOI] [PubMed]
- 29.Al-Salahi R, Abuelizz HA, Ghabbour HA, El-Dib R, Marzouk M. Molecular docking study and antiviral evaluation of 2-thioxo-benzo[g]quinazolin-4(3H)-one derivatives. Chem Cent J 2016; doi: 10.1186/s13065-016-0168-x. [DOI] [PMC free article] [PubMed]
- 30.Simões CMO, Amoros M, Girre L. Mechanism of antiviral activity of triterpenoid saponins. Phyther Res An Int J Devoted to Pharmacol Toxicol Eval Nat Prod Deriv. 1999;13:323–328. doi: 10.1002/(SICI)1099-1573(199906)13:4<323::AID-PTR448>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 31.Walum E, Stenberg K, Jenssen D. Understanding cell toxicology. E. Horwood; 1990.
- 32.Schmidtke M, Knorre C, Blei L, Stelzner A, Birch-Hirschfeld E. Penetration and antiviral activity of Coxsackievirus B3 (CVB3)-specific phosphorothioate oligodeoxynucleotides (PS-ODN) Nucleosides Nucleotides. 1998;17:1557–1566. doi: 10.1080/07328319808004686. [DOI] [Google Scholar]
- 33.Krieger E, Nabuurs SB, Vriend G. Homology modeling. Methods Biochem Anal. 2003;44:509–524. doi: 10.1002/0471721204.ch25. [DOI] [PubMed] [Google Scholar]
- 34.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benkert P, Tosatto SCE, Schomburg D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins Struct Funct Bioinforma. 2008;71:261–277. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- 36.Benkert P, Künzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37:W510–W514. doi: 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 38.van Gunsteren WF, Billeter SR, Eising AA, Hünenberger PH, Krüger P, Mark AE, et al. Biomolecular simulation: the GROMOS96 manual and user guide. Vdf Hochschulverlag AG an Der ETH Zürich, Zürich. 1996;86:1–1044. [Google Scholar]
- 39.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 41.Boonstra S, Onck PR, van der Giessen E. CHARMM TIP3P water model suppresses peptide folding by solvating the unfolded state. J Phys Chem B. 2016;120:3692–3698. doi: 10.1021/acs.jpcb.6b01316. [DOI] [PubMed] [Google Scholar]
- 42.Hakami AR, Bakheit AH, Almehizia AA, Ghazwani MY. Selection of SARS-CoV-2 main protease inhibitor using structure-based virtual screening. Future Med Chem. 2022;14:61–79. doi: 10.4155/fmc-2020-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji B, Liu S, He X, Man VH, Xie X-Q, Wang J. Prediction of the binding affinities and selectivity for CB1 and CB2 ligands using homology modeling, molecular docking, molecular dynamics simulations, and MM-PBSA binding free energy calculations. ACS Chem Neurosci. 2020;11:1139–1158. doi: 10.1021/acschemneuro.9b00696. [DOI] [PubMed] [Google Scholar]
- 44.Cavasotto CN. Binding free energy calculation using quantum mechanics aimed for drug lead optimization. Quantum Mech Drug Discov 2020:257–268. [DOI] [PubMed]
- 45.Miller III BR, McGee Jr TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA. py: an efficient program for end-state free energy calculations. J Chem Theory Comput 2012;8:3314–3321. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file1 (PNG 6689 KB) Molecular modeling of the catalytic active site of coxsackievirus 3CLpro. (i)Surface analysis using three most active ligands (1-3 superimposed); (ii) Magnified view ofthe active site and the ligands.
Supplementary file2 (PNG 2320 KB) Interactions of amino acids in the coxsackievirus 3CLpro homology model andthe overlay of co-crystalline ligand in (A), and ligands 1-3 (B), (C) and (D), respectively.
Supplementary file3 (PNG 4082 KB) Interactions between amino acids of the coxsackievirus B4 3C protease (PDBcode: 6ZCK) and the three most active ligands 1-3 in (A-C), respectively; Figure S5.Interactions between amino acids of the coxsackievirus RdRp homology model and the threemost active ligands 1-3 in (D-F), respectively.
Supplementary file4 (PNG 4791 KB) Molecular modeling of the catalytic active site of coxsackievirus RdRp and itsinteraction with the tested ligand (1). (i) Surface representation of the active site and theligand; (ii) Close-up view of the active site and the ligand showing the binding mode andinteractions.
Supplementary file5 (PNG 5837 KB) Molecular modeling of the catalytic active site of Coxsackievirus B4 3C protease(PDB code: 6ZCK) and its interaction with the tested ligand (1). (i) Surface representation ofthe active site and the ligands; (ii) Close-up view of the active site and the ligands showingthe binding mode and interactions.
Data Availability Statement
Original data are available upon request.