

Abstract
Background:
Tissue factor pathway inhibitor (TFPI) is the primary inhibitor of events initiating the blood coagulation pathway. Tfpi−/− mice die during embryonic development. The absence of protease–activated receptor (PAR) 4, the major thrombin receptor on mouse platelets, rescues Tfpi−/−mice to adulthood. Among the 3 TFPI isoforms in mice, TFPIα is the only isoform within platelets (pltTFPIα) and the only isoform that inhibits prothrombinase, the enzymatic complex that converts prothrombin to thrombin.
Objectives:
To determine biological functions of pltTFPIα.
Methods:
Tfpi−/−/Par4−/− mice were irradiated and transplanted with bone marrow from mice lacking or containing pltTFPIα. Thus, PAR4 expression was restored in the recipient mice, which differed selectively by the presence or absence of pltTFPIα and lacked other forms of TFPI.
Results:
Recipient mice lacking pltTFPIα had reduced survival over the 200-day posttransplant period. Necropsy revealed radiation injury associated with large intraventricular platelet-rich thrombi, whereas other organs were not affected. Thrombi were associated with fibrotic presentations, including increased collagen deposition, periostin–positive activated fibroblasts, myofibroblasts, and macrophage infiltrates. Recipient mice containing pltTFPIα showed evidence of radiation injury but lacked heart pathology.
Conclusions:
Tfpi−/−/Par4−/− mice develop severe cardiac fibrosis following irradiation and transplantation with bone marrow lacking pltTFPIα. This pathology is markedly reduced when the mice are transplanted with bone marrow containing pltTFPIα. Thus, in this model system pltTFPIα has an important physiological role in dampening pathological responses mediated by activated platelets within the heart tissue.
Keywords: cardiac, fibrosis, platelet, radiation, TFPI
1 |. INTRODUCTION
Three anticoagulant proteins, tissue factor pathway inhibitor α (TFPIα), protein S (PS), and protease nexin (PN)-1, are released from activated platelets [1]. These 3 proteins inhibit different stages of blood clotting. TFPIα acts during the initiation phase of the procoagulant response by inhibiting tissue factor(TF)-factor VIIa (TF-FVIIa) [2] and nascent prothrombinase [3]. PS acts during the initiation phase by enhancing the ability of TFPI to inhibit FXa [4] and prothrombinase [5–7] and in the propagation phase by acting as a cofactor for activated protein C in the degradation of FVa [8]. PN-1 acts in later stages of coagulation through direct thrombin inhibition [9]. Each protein has been shown to dampen thrombus development under in vivo conditions [10–12]. However, broader physiological roles for these anticoagulant proteins in the procoagulant platelet remain to be explored.
Here, we have focused on defining physiological functions of platelet TFPIα (pltTFPIα). TFPIα is one of the 3 alternatively spliced TFPI isoforms present in mice [13]. All 3 isoforms have identical N-terminal regions containing 2 Kunitz-type serine protease inhibitory domains. The second Kunitz domain directly inhibits FXa, and the first Kunitz domain inhibits TF-FVIIa in an FXa-dependent manner [2,14]. TFPIβ is a glycosylphosphatidylinositol–anchored protein on endothelium [15], whereas TFPIγ is the primary isoform circulating in mouse plasma [13,16]. TFPIα is present in human plasma but not in mouse plasma [15]. There is heparin-releasable TFPIα in humans [17–19] and mice [15]. TFPIα is the only isoform present in human and mouse platelets [20]. It has a C-terminal region with high affinity for partially cleaved forms of FVa that promotes inhibition of nascent forms of prothrombinase [3,21]. In humans, high-affinity binding of the TFPIα C-terminus to FV-short variants produce bleeding disorders [22–25]. Thus, TFPIα inhibition of prothrombinase is a physiologically important anticoagulant activity occurring early in the coagulation cascade when nascent forms of prothrombinase have assembled but before thrombin is generated. Interestingly, saliva from the black fly, Simulium vittatum, has a protein containing a single Kunitz domain that inhibits FXa, followed by a C-terminal region with homology to TFPIα and the FV B-domain [26]. This evolutionary adaptation suggests that the fly anticoagulates its blood meals by inhibiting prothrombinase through the same mechanism used by TFPIα and further amplifies the biological importance of prothrombinase inhibition by TFPIα.
Mice lacking the first Kunitz domain of TFPI (Tfpi−/−) do not survive embryogenesis [27]. We previously found that the embryonic lethality could be rescued by breeding Tfpi−/− mice with mice lacking protease–activated receptor 4 (Par4−/−), the major thrombin receptor on mouse platelets [28]. In addition, our laboratory commissioned the production of a conditional knockout mouse that allowed production of mice with selective removal of TFPIα from platelets. The bone marrow from mice lacking or containing pltTFPIα was transplanted into irradiated Tfpi−/−/Par4−/− recipient mice. This breeding and bone marrow transplantation strategy allowed for functional studies of pltTFPIα in the absence of other sources of TFPI inhibitory activity. The study findings indicate that in the transplant recipient mice, activated platelets without TFPIα drive a thrombotic reaction that promotes pathological fibrosis in the heart that is prevented by pltTFPIα.
2 |. MATERIALS AND METHODS
2.1 |. Production of the floxed total TFPI mouse model
Mutant mice carrying a conditional Tfpi total KO allele (Tfpitm1.1Amast or Tfpifl) were generated at the University of California, Davis, Mouse Biology Program, using a 2-construct approach as described in Supplementary Figure S1. Founders were confirmed by Sanger sequencing of the genomic regions spanning the insertion sites of the 5’ and 3’ constructs. Founders were outcrossed to homozygous ACTB-FLPe mice [29] for recombinase-mediated removal of the Neo selection cassette generating mice carrying the final Tfpi floxed (Tfpifl) conditional allele. Tfpifl genotyping was performed by polymerase chain reaction (PCR) using primers P1 and P2 (Table S1) to confirm presence of the haploblock. The Pf4-Cre transgene (Stock No: 008535; Jackson Laboratory) was bred onto the line to generate Tfpifl/fl Pf4-Cre+ mice. Cre transgene genotyping was performed using primers Cre-1, Cre-2, +PCR-1, and +PCR-2 (Table S1). The Institutional Animal Care and Use Committee of the Medical College of Wisconsin approved all procedures.
2.2 |. Bone marrow transplants
Recipient Tfpi−/−/Par4−/− mice were obtained by breeding heterozygous TFPI K1 knockout mice (Tfpitm1Gjb;Tfpi+/−) to Par4−/−mice (F2rl3tm1.1Cgh; Par4−/−). Bone marrow donor mice were obtained by crossing total TFPIfl/fl mice (Tfpitm1.1Amast; total Tfpi−/−) with Pf4-Cre+ mouse [C57BL/6-Tg(Pf4-icre)Q3Rsko/J]. Tfpi−/−/Par4−/− recipient mice received 11Gy of radiation 24 hours before bone marrow transplantation. Bone marrow was isolated from the iliac crest and long bones of donor mice. Cells were passed through a 70-μm filter, counted for live cells, and 1 × 106 live cells were injected through retro-orbital injection in the anesthetized recipient mouse. Recipient mice were provided trimethoprim/sulfamethoxazole for 1 month after transplantation and maintained in the colony for up to 217 days.
2.3 |. Blood isolation
Blood was collected from anesthetized mice through the inferior vena cava into 3.2% citrate (10% volume/volume). Complete blood counts were determined using an animal blood counter (scil Vet abc Plus). Platelet-rich plasma was collected by 10-minute centrifugation at 3000g and plasma by 9000g centrifugation. Platelet-rich plasma from 2 genetically identical mice was collected from whole blood, pooled, and centrifuged (100g, 5 minutes), the platelets were isolated and then pelleted (700g, 10 minutes), and washed 3 times with phosphate-buffered saline. Platelets were lysed by repeat freeze-thaw cycles (5 times), and then sonicated for a 10-minute cycle of 30 seconds on and 30 seconds off (Bioruptor Pico Sonicator; Diagenode).
2.4 |. TFPI activity assays
TF/FVIIa–dependent FXa generation assays were used to measure TFPI activity in platelet lysates standardized to 750 μg total protein/mL using the bicinchoninic acid assay (Pierce). Samples were incubated for 20 minutes with 1:500 rabbit brain cephalin (Pel-Freez Biologicals), 0.001 nM human FVIIa (Novo Nordisk A/S), and 1:4000 human TF (Siemens), followed by the addition of Spectrozyme FXa (0.5 mM; Sekisui Diagnostics). Finally, the reactions were initiated with 20 nM human FX (Enzyme Research Labs), and FXa generation was measured at 405 nm for 1 hour using SoftMax Pro, version 4.3.1. Platelet TFPI activity was interpolated from standard curves using GraphPad Prism 9.1.0 and sigmoidal 4 parameter logistic regression where X is the recombinant mouse TFPIα concentration. Standard curves for platelet lysate assays were generated using a 2-fold dilution series of murine recombinant TFPIα (Novo Nordisk A/S).
2.5 |. Thrombin-antithrombin complexes
Plasma thrombin-antithrombin (TAT) complexes were measured by ELISA according to the manufacturer’s instructions (Abcam).
2.6 |. Histology
Tissues were stained with trichrome and picrosirius Red (Poly-sciences), for histologic examination. Tissues were evaluated using immunohistochemistry (IHC) and fluorescent microscopy as previously described [30]. Antibodies used for IHC and immunofluorescence included α-smooth muscle actin (SMA) and S100B (GeneTex), fibrinogen (Exalpha Biologicals), platelet CD41 (gift from Richard Aster; Versiti), periostin (Abcam), IBA-1 (Waco Chemicals USA), CD45 (R&D Systems), and Cre recombinase protein (Novus Biologicals). Appropriate fluorochrome conjugated anti-goat, anti-mouse, and anti-rabbit secondary antibodies were used for immunodetection (10 μg/mL) (Jackson ImmunoResearch Laboratories). The Easy Scan imaging system (Motic) scanned IHC slides. A Nikon Eclipse Ti2 inverted microscope with a DS-Ri2 high-speed color camera using a low-magnification 10×/0.45, 20×/0.75, 40×/0.95 or a high-magnification 100× oil/1.45 numerical aperture objective (Nikon Instruments) acquired immunofluorescent images. Images were analyzed with the Nikon NIS-Elements software platform and processed with Imaris multichannel microscopy software (Bitplane). Image formatting was performed in Adobe Photoshop CS-6 and Adobe Illustrator CS-6 (Adobe).
2.7 |. Quantification of collagen and periostin
Collagen and periostin signals in tissue sections were quantified using ImageJ software. After the region of interest was assigned and a monochromatic image produced, the tissue sections were outlined and any space outside the outlined tissue was obscured to avoid quantitation. The outlined tissue was analyzed to quantify the percentage area with positive pixels.
2.8 |. Statistical analyses
Statistical analyses were performed using GraphPad Prism v9.1.0. Log-rank test was used to estimate significant differences in Kaplan–Meier survival curves. For other quantitative assays, Kruskal–Wallis test and post hoc analyses using the Wilcoxon signed-rank test were performed with an α level of 0.05; p values were adjusted for multiple comparisons using the Benjamini and Hochberg false discovery rate with significance reported as the adjusted q value when applicable. Data are presented as the mean ± standard deviation.
3 |. RESULTS
3.1 |. Generation of mice lacking pltTFPIα
The pltTFPIα–deficient mice serving as transplant donors were generated by targeted genetic deletion of Tfpi within the megakaryocytic lineage by crossing the Pf4 promoter–driven Cre transgene onto the conditional total Tfpifl/fl mice (Supplementary Figure S1). Tfpifl/fl Pf4-Cre+ mice had normal survival. Bone marrow from Tfpifl/fl Pf4-Cre− or Tfpifl/fl Pf4-Cre+ donor mice (both PAR4+/+) was transplanted into Tfpi−/−/PAR4−/− recipients. The transplant restored platelet PAR4 activity, and the transplanted mice differed by the presence (Tx-pltTFPIαpos) or absence (Tx-pltTFPIαneg) of pltTFPIα but lacking other sources of TFPI. Moreover, TFPI expression may have differed in other hematopoietic cells that express Pf4 and TFPI, such as macrophages. Hearts from Tfpifl/fl Pf4-Cre+ donor mice and Tfpi−/−/Par4−/− recipient mice that did not have radiation exposure were histologically evaluated and appeared normal on trichrome stain. Immunohistochemical staining revealed only background collagen deposition and the absence of periostin (Supplementary Figures S2 and S3). Thus, there was no baseline pathology in the hearts of the recipient mice before transplant.
3.2 |. Characterization of transplanted mice
Bone marrow from pltTFPIαneg and pltTFPIαpos mice engrafted equally well in Tfpi−/−/Par4−/− mice, as assessed by complete blood counts of peripheral whole blood. Blood cell counts after transplant were comparable with those previously reported for similar aged mice (Table S2). Platelet TFPI activity after transplant was 0.084 ± 0.16 fmoles TFPI/mg total platelet protein in Tx-pltTFPIαneg mice compared with 4.10 ± 1.3 fmoles TFPI/mg total platelet protein in Tx-pltTFPIαpos mice (p < .0001) (Figure 1A). Plasma TAT complex levels were not different between Tx-pltTFPIαneg and Tx-pltTFPIαpos mice (2.2 ± 1.4 vs 1.8 ± 0.9 ng/mL; p = .6541) (Figure 1B) indicating that platelets lacking TFPIα did not produce a generalized prothrombotic condition. However, Tx-pltTFPIαneg mice had reduced survival as 5 of 12 Tx-pltTFPIαneg mice died before day 217 posttransplantation. The Tx-pltTFPIαpos mice had 100% survival until day 203 posttransplantation when 1 of the 6 mice died (Figure 1C). Necropsy with detailed histologic analyses was performed to determine the cause of death.
FIGURE 1.
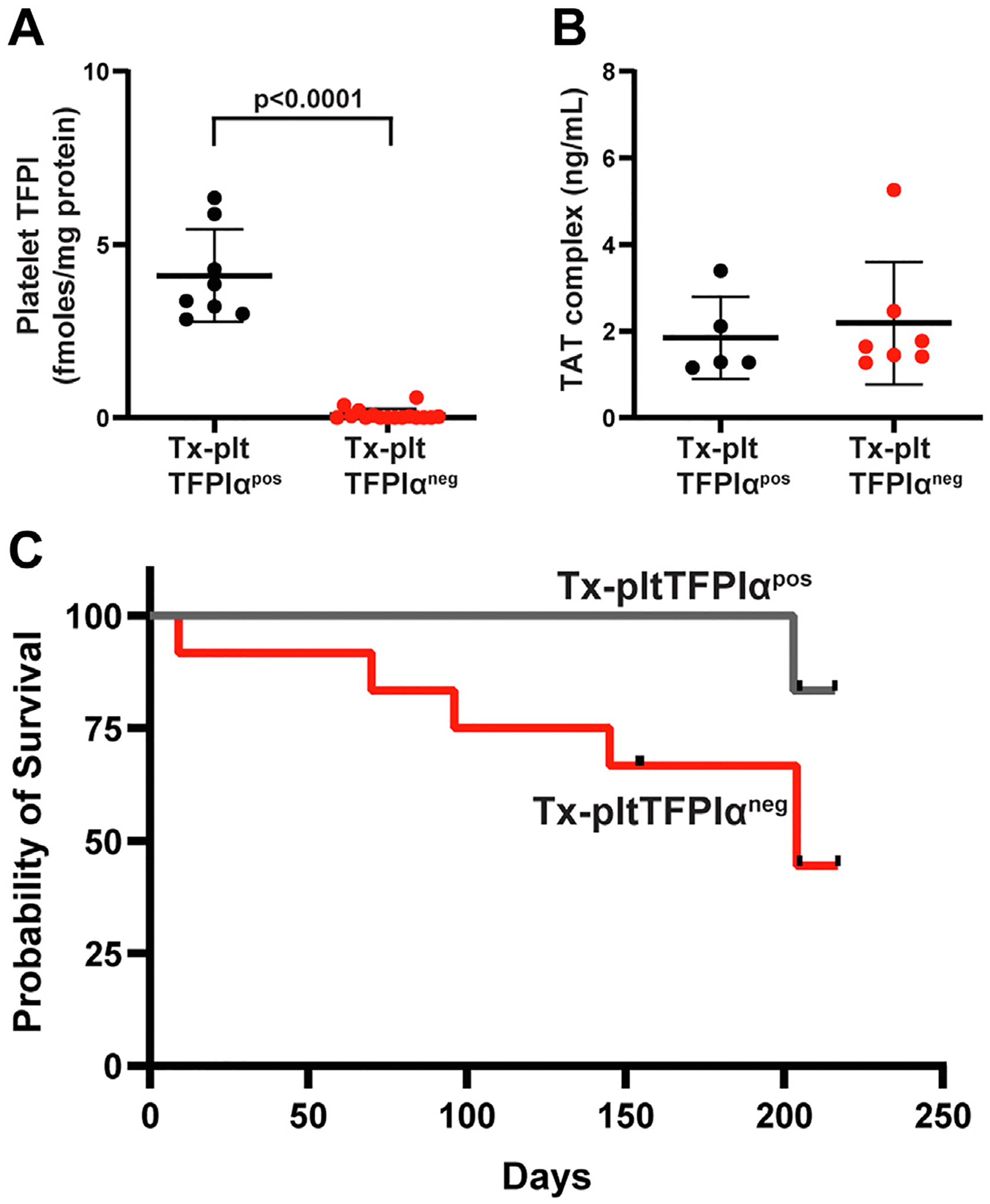
Tx-pltTFPIαneg mice did not have a global coagulopathy but had decreased survival. (A) TF/FVIIa–dependent FXa generation assays of platelet lysates confirm the absence of TFPIα inhibitory activity in platelets from Tx-pltTFPIαneg mice. Platelets from 2 mice of the same genotype were pooled and lysed. Each point represents a technical replicate from 2 platelet lysates of each genotype. (B) Plasma thrombin-antithrombin (TAT) complex levels were not different between Tx-pltTFPIαneg and Tx-pltTFPIαpos mice, indicating the absence of a global coagulopathy produced by platelets lacking TFPIα. (C) A Kaplan–Meier survival analysis of Tx-pltTFPIαneg (n = 12) and Tx-pltTFPIαpos (n = 6) mice. Five Tx-pltTFPIαneg mice died before posttransplant day 217. By contrast, Tx-pltTFPIαpos mice had 100% survival until posttransplant day 203, when 1 mouse died. The black tick marks indicate censored mice harvested before natural death. The difference between groups was not significant (p = .1359).
3.3 |. Both groups of mice had radiation–induced tissue damage
Radiation is a known risk factor for cardiac fibrosis in patients receiving chest or thoracic radiotherapy for treatment of left-sided breast or lung cancer [31,32]. Cardiomyocytes are thought to be resistant to irradiation. This contrasts with endothelial cells that become apoptotic or have increased cellular senescence following irradiation [33]. The endothelial cell damage results in leaky vessels and perivascular β-amyloid deposition, increased adhesion of inflammatory cells, and interstitial fibrosis [34,35]. Two predominant phenotypes were observed in the transplanted mice: 1) radiation–induced vascular leakage with low level interstitial fibrosis present in the Tx-pltTFPIαneg and Tx-pltTFPIαpos mice; and 2) excessive cardiac thrombosis with extensive fibrosis present only in Tx-pltTFPIαneg mice. For the former, typical radiation–induced cardiomyopathy changes were observed in both groups of mice including β-amyloid deposition adjacent to individual vessels (Supplementary Figure S4A) and mild interstitial fibrosis as shown by collagen deposition in perivascular areas and between the ventricular cardiomyocytes (Supplementary Figure S4B). However, as described further, the thrombotic phenotype and cardiac tissue responses were more severe in mice lacking pltTFPIα and likely contributed to their death.
3.4 |. Tx-pltTFPIαneg mice had thrombotic lesions in the heart
Trichrome staining revealed mural thrombi in the heart ventricles from 11 of 12 Tx-pltTFPIαneg mice, including 1 harvested at day 154, 3 at day 155, 1 at day 205, and 1 at day 217 posttransplant, and 5 that died as indicated in Figure 1C. One mouse harvested at day 154 did not have cardiac thrombi. Because there were equal numbers of male and female Tx-pltTFPIαneg mice, a sex-specific difference in development of heart pathology was not present. None of the Tx-pltTFPIαpos mice had cardiac thrombi. In Figure 2, hearts from 3 Tx-pltTFPIαpos (A–C) and 3 Tx-pltTFPIαneg (D–F) mice that were harvested before natural death are shown to highlight the pathology observed. The Tx-pltTFPIαneg mice had ventricular thrombi that varied in size and number. The thrombi stained blue and were located either adjacent to the ventricular wall or within the ventricular chamber. Some mice had multiple small to medium mural thrombi (Figure 2D, F), whereas others had large thrombi that filled a sizable portion of the ventricle (Figure 2E). Thrombi were also present in the atria of 4 of 12 Tx-pltTFPIαneg mice (Figure 2E), and extensive pericardial fibrosis was observed in a single mouse (Figure 2F). Overall, among the 11 Tx-pltTFPIαneg mice with thrombi, 6 had clots in both ventricles, 4 had only right ventricle clots, and 1 had only a left ventricular clot (Table S3). Thrombi were not present in the brain, liver, kidneys, spleen, or lungs from Tx-pltTFPIαneg mice. Because thrombi were present within the right ventricle of several mice, lungs were examined for emboli, but none were observed.
FIGURE 2.
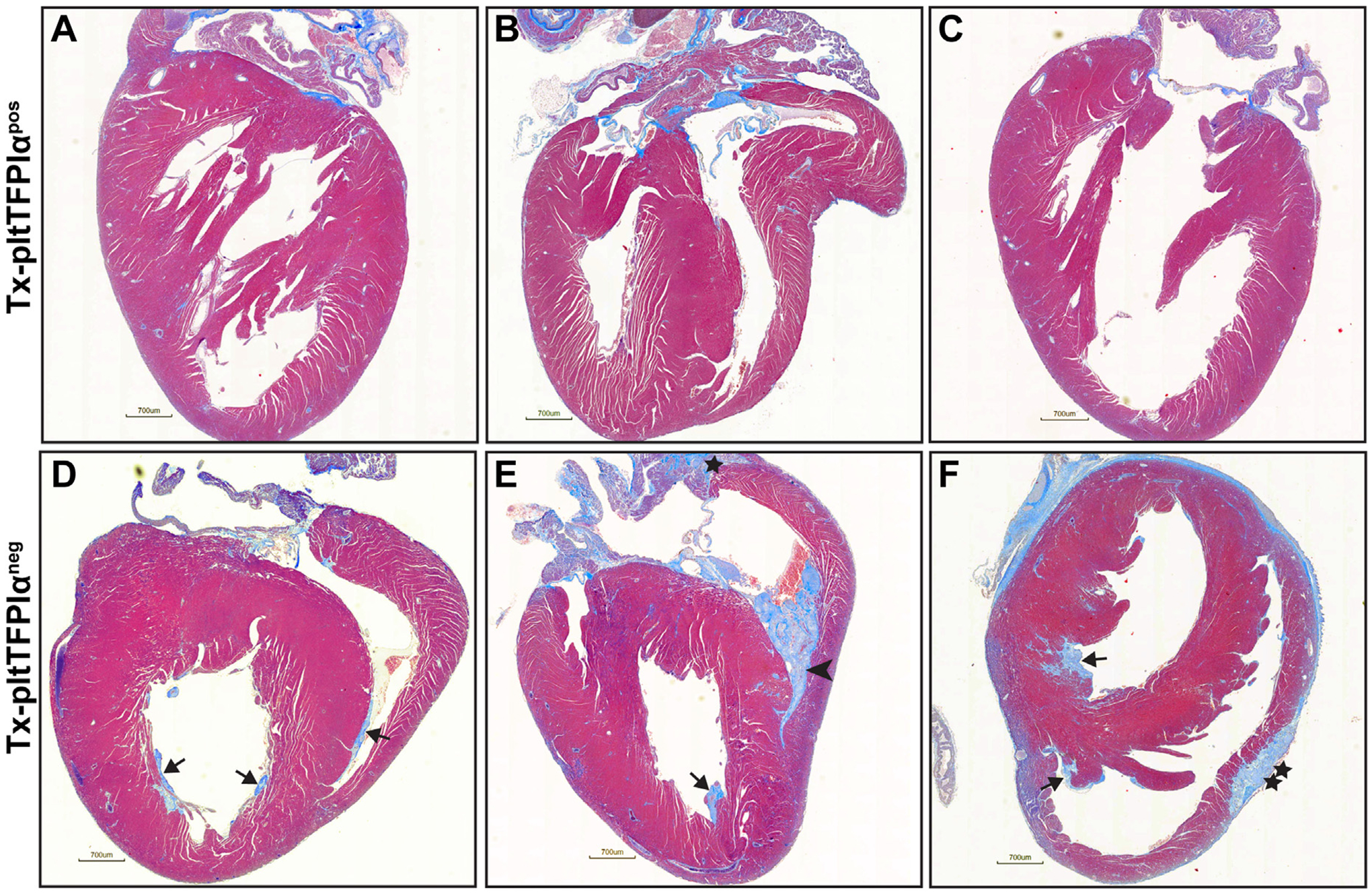
Hearts from Tx-pltTFPIαneg mice had extensive thrombotic disease. Hearts from Tx-pltTFPIαpos and Tx-pltTFPIαneg mice were stained with trichrome. (A–C) Three representative Tx-pltTFPIαpos hearts harvested on days 205 (2 hearts) and 216 (1 heart) posttransplant. No cardiac thrombi were present. (D–F) Three representative Tx-pltTFPIαneg hearts harvested on days 154, 155, and 205 posttransplant with different patterns of thrombus formation. In D–F, arrows indicate mural ventricular thrombi. In E, the arrowhead indicates a large thrombus that filled the ventricle, and the star indicates an atrial thrombus. In F, the double star indicates a region of pericardial fibrosis (bar = 700 μm).
3.5 |. Thrombi in Tx-pltTFPIαneg hearts were platelet and fibrin rich
Selected thrombi from the Tx-pltTFPIαneg hearts shown in Figure 2 were enlarged and are presented in Figure 3 with hearts from Tx-pltTFPIαpos mice displayed in Supplementary Figure S5. In the trichrome–stained images (Figure 3A), the blue ventricular thrombi have invaded into the cardiac tissue. Immunostaining of the cardiac thrombi characterized them as fibrin and platelet rich (Figure 3B and Supplementary Figure S6). Similar staining of Tx-pltTFPIαpos hearts confirmed the absence of thrombi (Supplementary Figure S5A, B).
FIGURE 3.
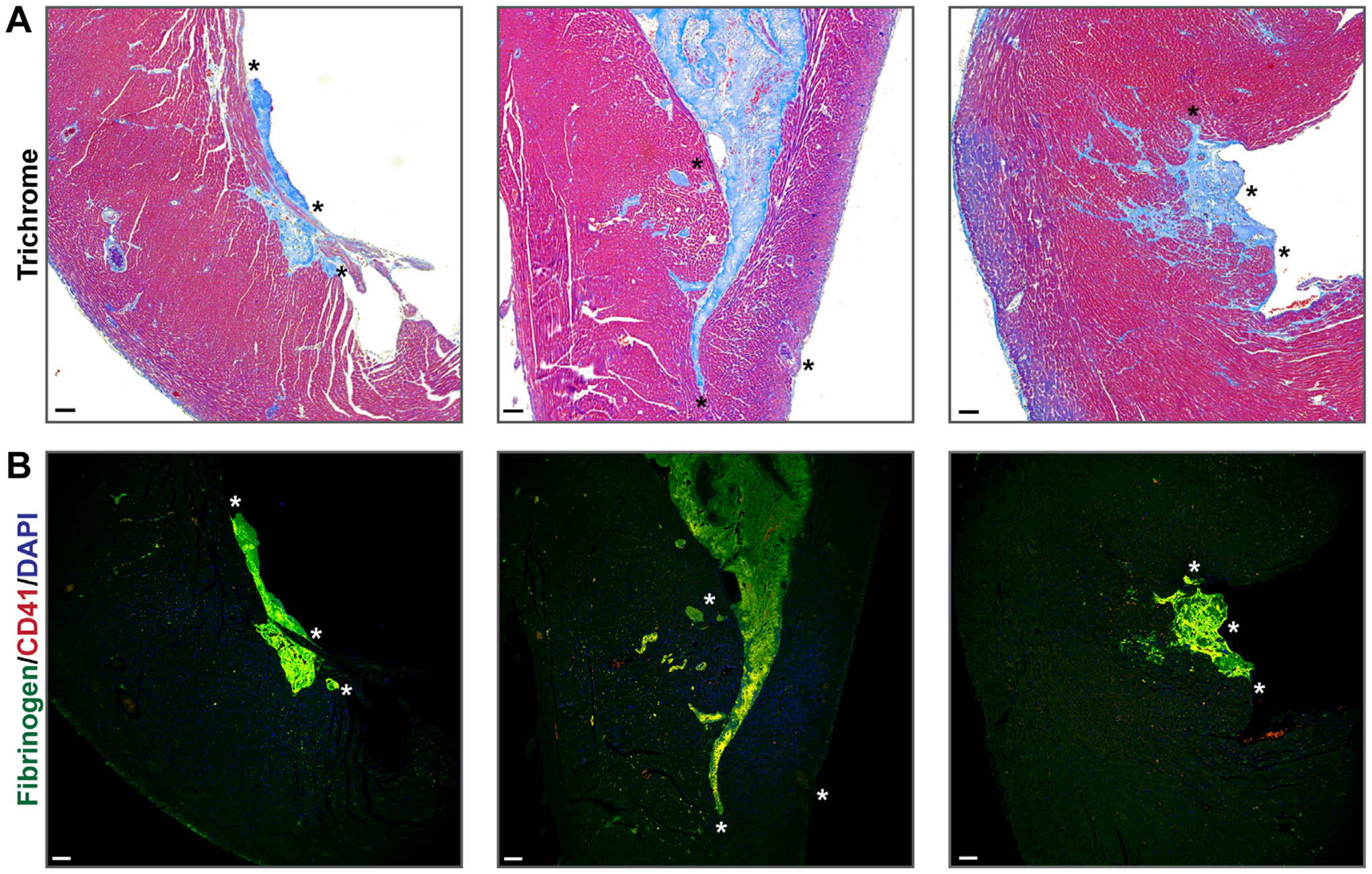
Tx-pltTFPIαneg cardiac thrombi were platelet and fibrin rich. (A) Enlarged images of thrombi from the Tx-pltTFPIαneg hearts presented in Figure 2D–F. The left and right panels display ventricular mural thrombi. The thrombi and associated interstitial collagen (blue) extend into the heart wall. The middle panel shows a large mural thrombus filling the right ventricle. (B) The thrombi in all 3 panels were fibrin and platelet rich. Tissue sections were probed with anti-fibrinogen (green) and anti–platelet CD41 (red) with DAPI nuclear counterstain (blue). Although platelets were stained red, they appeared yellow because they colocalize with the green fibrinogen. Asterisks are landmarks for images in A and B (bar = 100 μm).
3.6 |. Localized areas of myofibroblast transformation were present in Tx-pltTFPIαneg hearts
Activated platelets release transforming growth factor (TGF)-β, a potent activator of fibroblasts that stimulates their differentiation into myofibroblasts [36,37]. α-SMA is a marker of myofibroblast differentiation and induced by TGF-β1. The Tx-pltTFPIαpos hearts did not express detectable levels of α-SMA (Figure 4A), whereas in 3 Tx-pltTFPIαneg hearts containing 7 thrombi, 4 of the thrombi had associated α-SMA interstitial expression, indicating the presence of myofibroblasts (Figure 4B).
FIGURE 4.
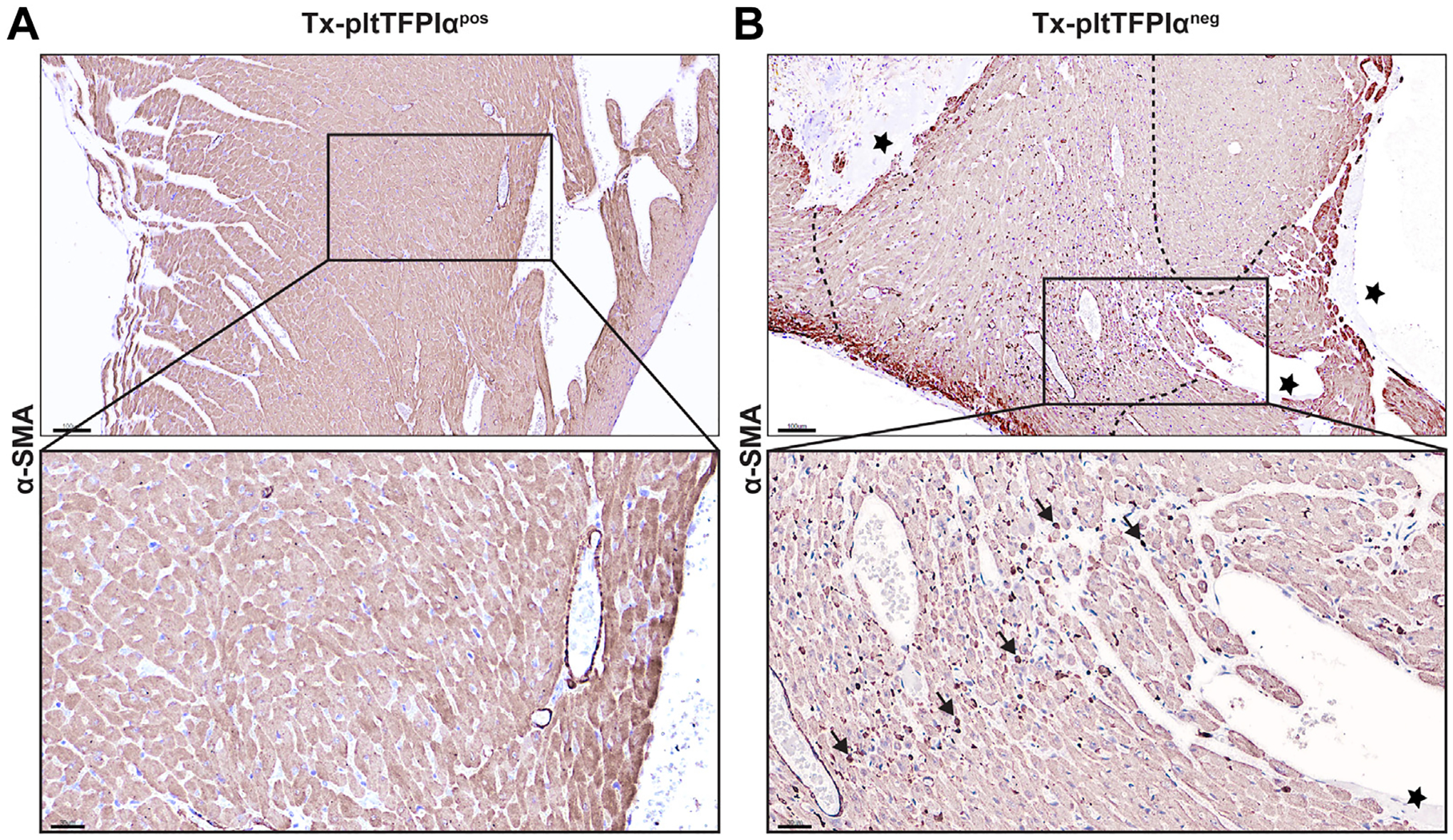
Tx-pltTFPIαneg hearts had myofibroblasts in localized areas adjacent to thrombi. Tx-pltTFPIαpos and Tx-pltTFPIαneg heart tissue was stained with α-smooth muscle actin (α-SMA). (A) α-SMA staining was not present in Tx-pltTFPIαpos hearts, indicating the absence of myofibroblasts. (B) In the upper panel, stars indicate mural thrombi, and dashed lines delineate the tissue areas surrounding mural thrombi in Tx-pltTFPIαneg hearts in which myofibroblasts were present. Bar = 100 μm. The insets are enlarged areas. In (B), the star indicates the mural thrombi. The arrows point out 5 of the many myofibroblasts present throughout the region surrounding the mural thrombi (bar = 30 μm).
3.7 |. Abundant periostin was present in areas surrounding thrombi in Tx-pltTFPIαneg hearts
Periostin is a TGF-β superfamily-responsive glycoprotein secreted into the extracellular matrix (ECM) by activated fibroblasts and myofibroblasts [38,39]. Tx-pltTFPIαneg heart tissue had increased periostin expression radiating outwardly from the thrombus into the surrounding ECM (Figure 5A). Lower levels of periostin were present in pltTFPIαpos hearts, most likely related to background radiation–induced activation of fibroblasts (Supplementary Figure S5C). Periostin immunoreactivity was quantified in Tx-pltTFPIαneg and Tx-pltTFPIαpos hearts (Supplementary Figure S5E). Within the Tx-pltTFPIαneg hearts, periostin in areas adjacent to a thrombus increased over background areas of the heart (16.77 ± 7.07% vs 0.81 ± 0.85% periostin–positive pixels, respectively; q = 0.0009). There was no difference in background periostin staining between Tx-pltTFPIαneg and the Tx-pltTFPIαpos hearts (0.81 ± 0.85% vs 0.85 ± 0.37% periostin–positive pixels, respectively; q = 0.4318).
FIGURE 5.
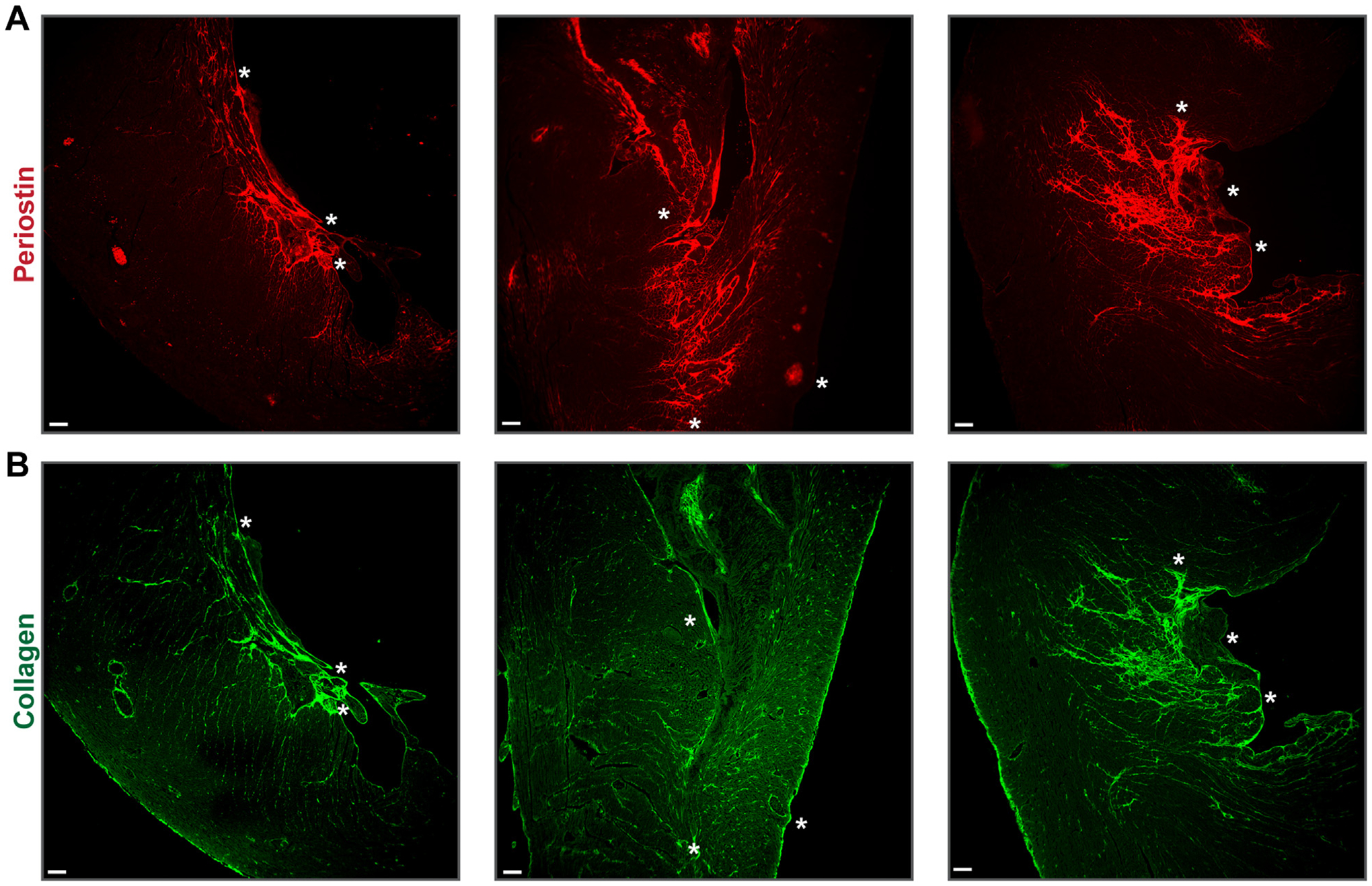
Tx-pltTFPIαneg hearts had periostin and collagen adjacent to mural thrombi. Sections from 3 Tx-pltTFPIαneg hearts are shown. Asterisks are landmarks within the periostin stained thrombus in A and the same thrombus stained with picrosirius red directly below in B and in the thrombi shown in Figure 3. (A) Immunolabeling with rabbit antiperiostin antibody (red). In the left and right panels, periostin was adjacent to the mural ventricular thrombi and extended into the myocardium. In the middle panel, an area with extensive periostin penetrating the myocardium can be seen immediately adjacent to a region of the ventricular thrombus that had many platelets at its narrow end (see Figure 3B; indicated by yellow colocalization of CD41 and fibrinogen). (B) Picrosirius red immunofluorescence (green) shows collagen deposition in an outwardly radiating pattern like periostin. In the middle panel, collagen extended from the platelet-rich area at the narrow end of the large ventricular clot (Figure 3B) across the entire ventricular wall (bar = 100 μm).
3.8 |. Collagen deposition was accentuated in areas surrounding thrombi in Tx-pltTFPIαneg hearts
Cardiac fibrosis is characterized by excess collagen deposition in ECM observed by picrosirius red staining. Extensive collagen deposition was present in a radiating pattern extending from the mural thrombi into the ECM (Figure 5B) similar to periostin staining. Collagen deposition was limited to radiation–induced background levels in the Tx-pltTFPIαpos hearts (Supplementary Figures S4B and S5D).
3.9 |. Fibrosis was present in the Tx-pltTFPIαneg heart but not in other organs
Collagen deposition in the heart parenchyma was examined using ImageJ software to quantify histologic observations. There was no difference in collagen deposition between nonirradiated Tfpi−/−/Par4−/− mice and Tx-pltTFPIαpos mice (Figure 6A). However, there were different fibrotic patterns in picrosirius red–stained heart tissue from the Tx-pltTFPIαpos and Tx-pltTFPIαneg hearts (Supplementary Figure S7). The Tx-pltTFPIαneg hearts had significantly more collagen deposition than the Tx-pltTFPIαpos hearts (1.42 ± 0.85% vs 0.27 ± 0.11% picrosirius red–positive pixels; q = 0.0012) (Figure 6A), and there was an increased amount of collagen associated with the thrombi. Interestingly, there were high and low collagen deposition groups among the Tx-pltTFPIαneg hearts. The high collagen group had 8.4-fold more collagen deposition than the Tx-pltTFPIαneg hearts (2.28 ± 0.28 vs 0.27 ± 0.11, q = 0.0095), and the low collagen group had 2.7-fold more collagen in the pltTFPIαneg hearts (0.73 ± 0.18 vs 0.27 ± 0.11, q = 0.0065). The differences in collagen deposition in the Tx-pltTFPIαneg hearts were not related to the sex of the recipient mouse because approximately equal numbers of male and female mice were in each collagen group. Collagen deposition was quantified in other tissues with no differences found between the Tx-pltTFPIαneg and Tx-pltTFPIαpos mice in the kidney (0.17 ± 0.10 vs 0.18 ± 0.07; p = .61), liver (0.38 ± 0.21 vs 0.27 ± 0.12; p = .33), or lung (0.52 ± 0.17 vs 0.50 ± 0.10; p = .86) (Figure 6B–D).
FIGURE 6.
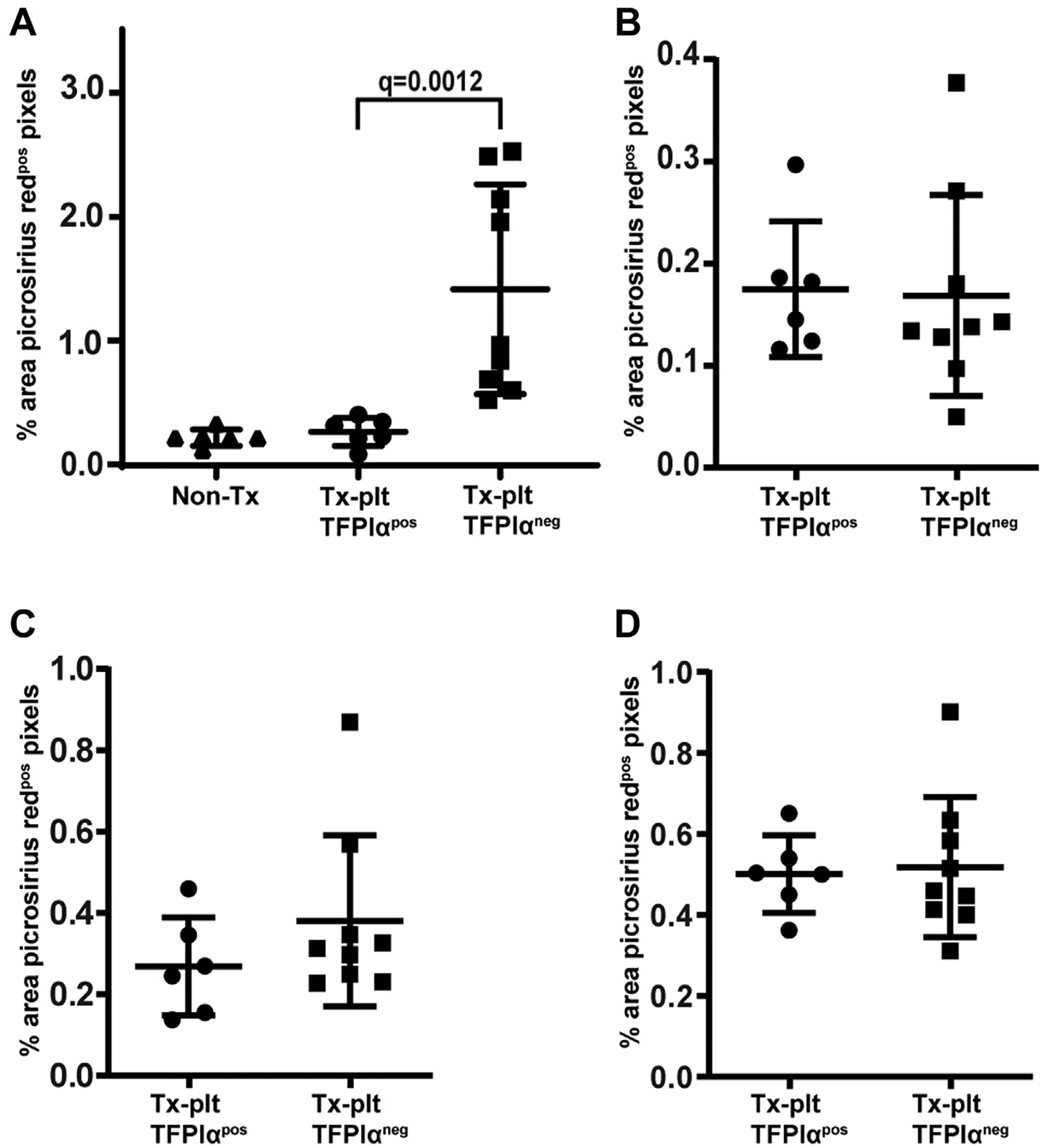
Tx-pltTFPIαneg mice had fibrosis only in the heart. ImageJ software was used to quantify organ fibrosis in picrosirius red–stained tissues. Data are presented as the percent area of picrosirius red–positive pixels for each image analyzed. (A) Tx-pltTFPIαneg hearts had greater collagen deposition than Tx-pltTFPIαpos hearts (q = 0.0012), which separated into low and high fibrotic groups. The low and high groups showed more fibrosis than Tx-pltTFPIαpos hearts (low q = 0.0095 and high q = 0.0065). Collagen deposition was not different between the hearts from TFPI−/−/Par4−/− mice that did not have radiation exposure and the hearts from the Tx-pltTFPIαpos mice (p = .407). (B–D) There were no differences in fibrosis between Tx-pltTFPIαpos and Tx-pltTFPIαneg mice in other tissues analyzed. Collagen deposition in (B) the kidney (p = .6070); (C) the liver (p = .328); and (D) the lung (p = .8639).
3.10 |. Tx-pltTFPIαneg cardiomyocytes secreted S100B with associated macrophage infiltrates
Myofibroblasts secrete ECM proteins and cytokines that promote macrophage tissue infiltration and activation [40,41]. S100B is secreted by myofibroblasts and activated macrophages [42–44]. S100B was not detected in Tx-pltTFPIαpos hearts (Figure 7A). Abundant S100B was present in Tx-pltTFPIαneg hearts, particularly in areas surrounding mural thrombi, where expression extended through the ventricular wall (Figure 7B). Hearts were stained for ionized calcium binding adaptor molecule (IBA)-1, a pan-specific marker of activated macrophages. There are other pan-specific markers of activated macrophages, such as F4/80 and CD68, which differentially detect macrophages in various anatomical sites. IBA-1 was used in this study because it is a marker of membrane ruffling associated with phagocytosis, a key feature of macrophage activation. Although macrophages were not observed in the Tx-pltTFPIαpos hearts (Figure 7C), they were abundant in the mural clot and the adjacent myocardium within Tx-pltTFPIαneg mouse hearts (Figure 7D). In addition, immunofluorescence imaging revealed CD45+ inflammatory cell infiltration into the ECM, secondary to mural clot formation in Tx-pltTFPIαneg hearts but not in Tx-pltTFPIαpos hearts (Supplementary Figure S8), which strongly indicates these clots were not postmortem artifacts.
FIGURE 7.
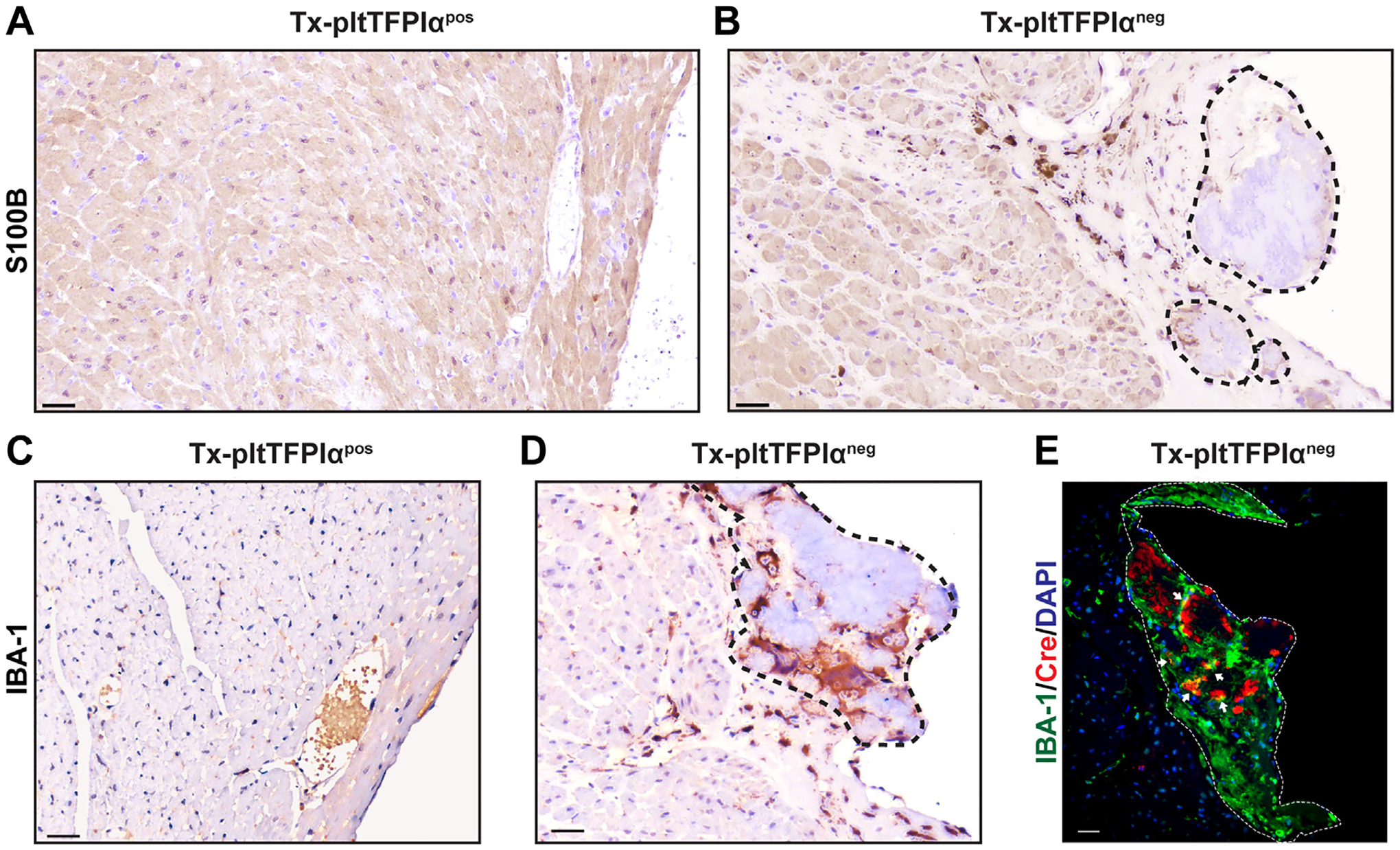
Tx-pltTFPIαneg hearts had cardiomyocytes adjacent to mural thrombi producing S100B and activated macrophages localized primarily within the mural thrombus. Immunohistochemistry of Tx-pltTFPIαpos and Tx-pltTFPIαneg hearts using anti-S100B (A and B) and anti–IBA-1 (C and D) antibodies. Dashed lines demarcate mural thrombi. (A) The Tx-pltTFPIαpos hearts did not show S100B expression. (B) The Tx-pltTFPIαneg hearts had S100B expression at the periphery of the mural thrombi and within the adjacent myocardium. (C) The Tx-pltTFPIαpos heart did not have IBA-1 expression, indicating the absence of activated macrophages. (D) The Tx-pltTFPIαneg hearts had numerous macrophages within the mural thrombus, with some also present in the adjacent myocardium (bar = 30 μm). (E) The Tx-pltTFPIαneg hearts were immunostained for macrophage IBA-1 expression (750 nm, green) and Cre recombinase protein (647 nm, red). Abundant macrophages and areas with Cre expression were present within the platelet-rich thrombus. However, Cre+ macrophages that stained yellow were rare (arrows). The Cre+ cells may be platelets or CD45+ cells within thrombus (bar = 10 μm). The images shown in (B), (D,) and (E) are sections from the same heart tissue, but the image in B is from a deeper sequential section than D and E.
3.11 |. Rare IBA-1–positive bone marrow–derived macrophages were present in Tx-pltTFPIαneg heart after transplant
Two macrophage subsets are found in the heart, resident macrophages that are derived from the yolk sac and nonresident macrophages that originate from bone marrow monocytes [45]. Because TFPI [46] and PF4 [47] are present in various macrophage populations, the contribution of macrophage TFPI to the thrombotic phenotype was examined. Heart macrophages in the recipient mice lacked the TFPI-K1 domain before transplantation, but after transplant the hearts of Tx-pltTFPIαpos mice may have had bone marrow–derived macrophages expressing TFPI that contributed to the protective phenotype. However, macrophages were not readily detectable in the hearts from Tx-pltTFPIαpos mice. Therefore, macrophages associated with clots in the Tx-pltTFPIαneg mice were examined for Cre expression. If they expressed Cre, it would suggest that bone marrow–derived macrophages were within the hearts of Tx-pltTFPIαneg and Tx-pltTFPIαpos mice. However, only rare IBA-1 and PF4 Cre+ macrophages were associated with the heart thrombi in the Tx-pltTFPIαneg mice (Figure 7E), indicating that most macrophages within the hearts of both groups of mice were resident macrophages that did not produce TFPI activity.
4 |. DISCUSSION
TFPIα is an anticoagulant protein present within the procoagulant platelet [10,20,48]. We developed and characterized a mouse model to examine how pltTFPIα anticoagulant activity affects platelet function. Tfpi−/−/Par4−/− recipient mice [28] were transplanted with PAR4+ (F2rl3+/+) bone marrow either with or without pltTFPIα to produce 2 groups of mice that differed in whether they produced pltTFPIα. Because the recipient mice lacked the first Kunitz domain of TFPI, effects of pltTFPIα activity were observed without complication from TFPI activity derived from other sources. Hearts from both groups of mice displayed a low level of background cardiac fibrosis from the whole-body irradiation. However, pltTFPIαneg mice had large intraventricular mural thrombi and associated cardiac fibrosis radiating from the thrombus into the ventricular wall. This was a localized, heart-specific process because thrombi and associated tissue fibrosis were not present in other organs. The process was related to the absence of TFPIα within platelets because cardiac thrombi and associated fibrosis were not observed in the pltTFPIαpos mice.
A mouse with the entire TFPI gene floxed was bred to the Pf4-Cre+ mouse to generate bone marrow donors for these studies. Recipient mice had equal engraftment blood count profiles, and the Tx-pltTFPIαneg mice had greatly reduced pltTFPIα activity. The plasma TAT levels were not different between the 2 recipient groups indicating that the lack of pltTFPIα did not induce generalized hypercoagulability. However, Tx-pltTFPIαneg mice had reduced survival compared with the Tx-pltTFPIαpos mice that was likely caused by localized hypercoagulability in the heart.
The Tx-pltTFPIαneg hearts had atrial and ventricular thrombi, as well as pericardial fibrosis, whereas the Tx-pltTFPIαpos hearts did not display this pathology. Some Tx-pltTFPIαneg hearts had large platelet–and fibrin–rich thrombi that filled most of the ventricle and incorporated valvular tissue. The thrombi penetrated surrounding cardiac tissue where they were associated with activated fibroblasts secreting periostin and collagen, the central effectors of cardiac fibrosis [49,50].
We propose that the cardiac insult to the Tx-pltTFPIαneg mice was 2-fold. The first insult was the full body irradiation used for the bone marrow transplant procedure. The Tx-pltTFPIαpos hearts had radiation damage evidenced by arterioles surrounded by amyloid and the low background amounts of periostin and collagen. These findings were equally present in Tx-pltTFPIαpos hearts and areas of the Tx-pltTFPIαneg hearts not affected by thrombosis-associated fibrosis. The second insult was the platelet-rich thrombi. TGF-β is 40- to 100-fold more abundant in platelets than in other cells and is exclusively the profibrotic β1 isoform that promotes differentiation of cardiac fibroblasts into myofibroblasts and strongly stimulates collagen deposition within the ECM [51–53]. TGF-β1 is released from activated platelets and becomes trapped within thrombi. Then, it is slowly released and activated as the clot undergoes plasmin-mediated lysis [53,54]. Thus, TGF-β is likely the primary driver of the cardiac fibrosis observed in the pltTFPIαneg mice. The profibrotic effects may have been further enhanced by platelet polyphosphate, which induces in vitro differentiation of murine fibroblasts into myofibroblasts [55], and by platelet factor 4, which promotes disease in mouse models of skin, lung and heart fibrosis [56]. Furthermore, thrombin cleavage of PARs located on endothelial cells, myocytes, and fibroblasts may contribute to cardiac fibrosis [57].
The ventricular mural thrombi were associated with small, localized areas of myofibroblasts expressing α-SMA, whereas periostin was abundant in areas surrounding thrombi. This finding is consistent with periostin being a TGF-β responsive matricellular protein secreted by activated fibroblasts and myofibroblasts [41,49]. Activated fibroblasts proliferate independently from their differentiation into myofibroblasts. However, differentiation into myofibroblasts robustly promotes inflammation because myofibroblasts secrete additional ECM proteins and cytokines that recruit macrophages and other immune cells to the injured tissue [58]. S100B IHC was used to examine the inflammatory response in the Tx-pltTFPIαneg hearts. Adult cardiomyocytes repress S100B expression, but it becomes upregulated in many pathological states, including fibrosis [59]. Once upregulated, S100B engages the receptor for advanced glycation end products expressed on cardiomyocytes and recruits additional macrophages to the lesion [42]. In the Tx-pltTFPIαneg mice, S100B was adjacent to mural clots and radiated into the surrounding heart tissue. This histological pattern is consistent with the presence of a cycle of macrophage and cardiomyocyte activation contributing to the fibrosis observed.
Surface marker profiling studies of cardiac macrophages have delineated at least 4 subsets. Three subsets populate the heart during embryogenesis, and the fourth results from migration and differentiation of circulating monocytes into the heart tissue [60]. PF4 expression has been reported in resident aorta, peritoneal, and perivascular macrophages and within atherosclerotic lesions [61–64]. We considered the possibility that decreased expression of TFPI in monocyte–derived macrophages in the Tx-pltTFPIαneg mice contributed to the thrombotic and fibrotic pathology that was not observed in the Tx-pltTFPIαpos hearts. Macrophages were present within and surrounding thrombi in the Tx-pltTFPIαneg hearts, but we were not able to reliably determine if they expressed TFPI using IHC. Pf4-Cre transgene-mediated recombination events occur in 48% of mouse splenic macrophages and in 45% of peripheral blood leukocytes [65,66], but heart macrophages have not been previously examined. An anti-Cre recombinase antibody was used to examine heart tissue for transplanted bone marrow lineage macrophages. There were only rare Cre+ macrophages present suggesting low or no contribution of macrophage TFPI expression to the phenotype.
In addition to macrophages, there were large numbers of CD45+ cells in the Tx-pltTFPIαneg hearts that localized adjacent to the mural thrombi and extended outwardly, whereas the Tx-pltTFPIαpos hearts had substantially fewer CD45+ cells. We propose that leukocytes and macrophages induce further production of TGF-β by the activated fibroblasts with loops of activation and release of fibrogenic factors and cytokines leading to continuous TGF-β release and supporting a model where the fibrosis in the Tx-pltTFPIαneg hearts is initiated by TGF-β released from activated platelets and propagated by the lack of pltTFPIα, which is not available to dampen cardiac thrombi formation. Chronic induction and release of fibroblast periostin and collagen, along with myofibroblast activation and inflammatory cell infiltration, further propagates this response producing severe clot-associated fibrosis and lethality of some of the Tx-pltTFPIαneg mice.
Previous studies have implicated platelets in the pathophysiology of pulmonary [67], hepatic [68], and cardiac fibrosis, [69] as well as in vessel wall fibrosis following venous thrombosis [70]. The work presented in this study illustrated that pltTFPIα has a key role modulating platelet reactivity to prevent thrombus production and fibrosis within the cardiac tissue of irradiated mice that lack other sources of TFPI activity. Dabigatran, a direct thrombin inhibitor, attenuates murine cardiac fibrosis in the setting of pressure overload [71]. It is likely that pltTFPIα prevented cardiac fibrosis by reducing thrombin generation and thrombin-mediated platelet activation in the studies described in this work. TFPI is one of the 3 anticoagulant proteins in platelets [1]. The large effect of its absence from platelets in the development of cardiac thrombi in the Tx-pltTFPIαneg mice despite the presence of PS and PN-1 in the platelets may be related to the ability of TFPIα to dampen early responses to procoagulant stimuli [2,3] before bursts of thrombin are generated that cannot be controlled by other platelet inhibitors of later stages of the coagulation cascade.
Supplementary Material
Essentials.
Tissue factor pathway inhibitor α (TFPIα) is the only TFPI isoform in platelets (pltTFPIα) and inhibits prothrombinase.
Mice differing in the presence or absence of pltTFPIα and lacking other TFPIs were produced.
Mice without pltTFPIα developed cardiac thrombi and severe cardiac fibrosis.
Platelets promoted cardiac fibrosis after irradiation in a manner modulated by pltTFPIα.
Funding information
This work was supported by the National Institutes of Health (HL068835 to A.E.M. and U01AI133561 to H.W.) and by Advancing a Healthier Wisconsin Project (5520519 to J.L.).
Footnotes
DECLARATION OF COMPETING INTERESTS
A.E.M. receives research funding from Novo Nordisk and Pharma-cosmos and has received honoraria for serving on Novo Nordisk Advisory Boards. The other authors have no competing interests to disclose.
SUPPLEMENTARY MATERIAL
The online version contains supplementary material available at https://doi.org/10.1016/j.jtha.2022.11.034
DATA SHARING STATEMENT
For original data, please contact the corresponding author at aemast@versiti.org.
REFERENCES
- [1].Siebert AE, Mast AE. Platelet anticoagulant proteins: modulators of thrombosis propensity within a procoagulant cell. J Thromb Haemost. 2020;18:2083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rao LV, Rapaport SI. Studies of a mechanism inhibiting the initiation of the extrinsic pathway of coagulation. Blood. 1987;69:645–51. [PubMed] [Google Scholar]
- [3].Wood JP, Bunce MW, Maroney SA, Tracy PB, Camire RM, Mast AE. Tissue factor pathway inhibitor-alpha inhibits prothrombinase during the initiation of blood coagulation. Proc Natl Acad Sci U S A. 2013;110:17838–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hackeng TM, Sere KM, Tans G, Rosing J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc Natl Acad Sci U S A. 2006;103:3106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dahlback B Novel insights into the regulation of coagulation by factor V isoforms, tissue factor pathway inhibitoralpha, and protein S. J Thromb Haemost. 2017;15:1241–50. [DOI] [PubMed] [Google Scholar]
- [6].Dahlback B, Tran S. A hydrophobic patch (PLVIVG; 1481–1486) in the B-domain of factor V-short is crucial for its synergistic TFPIalpha-cofactor activity with protein S and for the formation of the FXa-inhibitory complex comprising FV-short, TFPIalpha, and protein S. J Thromb Haemost. 2022;20:1146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dahlback B, Tran S. The preAR2 region (1458–1492) in factor V-Short is crucial for the synergistic TFPIalpha-cofactor activity with protein S and the assembly of a trimolecular factor Xa-inhibitory complex comprising FV-Short, protein S, and TFPIalpha. J Thromb Haemost. 2022;20:58–68. [DOI] [PubMed] [Google Scholar]
- [8].Walker FJ. Regulation of activated protein C by a new protein. A possible function for bovine protein S. J Biol Chem. 1980;255: 5521–4. [PubMed] [Google Scholar]
- [9].Baker JB, Low DA, Simmer RL, Cunningham DD. Protease-nexin: a cellular component that links thrombin and plasminogen activator and mediates their binding to cells. Cell. 1980;21:37–45. [DOI] [PubMed] [Google Scholar]
- [10].Maroney SA, Cooley BC, Ferrel JP, Bonesho CE, Mast AE. Murine hematopoietic cell tissue factor pathway inhibitor limits thrombus growth. Arterioscler Thromb Vasc Biol. 2011;31:821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Boulaftali Y, Adam F, Venisse L, Ollivier V, Richard B, Taieb S, Monard D, Favier, Marie-Christine Alessi R, Bryckaert M, Arocas V, M-Perrus Jandrot, Bouton M-C. Anticoagulant and antithrombotic properties of platelet protease nexin-1. Blood. 2010;115:97–106. [DOI] [PubMed] [Google Scholar]
- [12].Calzavarini S, Prince-Eladnani R, Saller F, Bologna L, Burnier L, Brisset AC, Quarroz C, Caro MDR, Ermolayev V, Matsumura Y, Fernández JA, Hackeng TM, Griffin JH, Angelillo-Scherrer A. Platelet protein S limits venous but not arterial thrombosis propensity by controlling coagulation in the thrombus. Blood. 2020;135:1969–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Maroney SA, Ferrel JP, Collins ML, Mast AE. Tissue factor pathway inhibitor-gamma is an active alternatively spliced form of tissue factor pathway inhibitor present in mice but not in humans. J Thromb Haemost. 2008;6:1344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Girard TJ, Warren LA, Novotny WF, Likert KM, Brown SG, Miletich JP, Broze GJ Jr. Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature. 1989;338:518–20. [DOI] [PubMed] [Google Scholar]
- [15].Maroney SA, Ferrel JP, Pan S, White TA, Simari RD, McVey JH, Mast AE. Temporal expression of alternatively spliced forms of tissue factor pathway inhibitor in mice. J Thromb Haemost. 2009;7: 1106–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Girard TJ, Grunz K, Lasky NM, Malone JP, Broze GJ Jr. Re-evaluation of mouse tissue factor pathway inhibitor and comparison of mouse and human tissue factor pathway inhibitor physiology. J Thromb Haemost. 2018;16:2246–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sandset PM, Abildgaard U, Larsen ML. Heparin induces release of extrinsic coagulation pathway inhibitor (EPI). Thromb Res. 1988;50: 803–13. [DOI] [PubMed] [Google Scholar]
- [18].Novotny WF, Palmier M, Wun TC, Broze GJ Jr, Miletich JP. Purification and properties of heparin-releasable lipoprotein-associated coagulation inhibitor. Blood. 1991;78:394–400. [PubMed] [Google Scholar]
- [19].Peterson JA, Maroney SA, Martinez ND, Mast AE. Major reservoir for heparin-releasable TFPIalpha (tissue factor pathway inhibitor alpha) is extracellular matrix. Arterioscler Thromb Vasc Biol. 2021;41, 1942–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Maroney SA, Haberichter SL, Friese P, Collins ML, Ferrel JP, Dale GL, Mast AE. Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood. 2007;109:1931–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mast AE, Ruf W. Regulation of coagulation by tissue factor pathway inhibitor: implications for hemophilia therapy. J Thromb Haemost. 2022;20:1290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vincent LM, Tran S, Livaja R, Bensend TA, Milewicz DM, Dahlbäck B. Coagulation factor V(A2440G) causes east Texas bleeding disorder via TFPIalpha. J Clin Invest. 2013;123:3777–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cunha ML, Bakhtiari K, Peter J, Marquart JA, Meijers JCM, Middeldorp S. A novel mutation in the F5 gene (factor V Amsterdam) associated with bleeding independent of factor V procoagulant function. Blood. 2015;125:1822–5. [DOI] [PubMed] [Google Scholar]
- [24].Zimowski KL, Petrillo T, Ho MD, Wechsler J, Shields JE, Denning G, Jhita N, Rivera AA, Escobar MA, Kempton CL, Camire RM, Doering CB. F5-Atlanta: a novel mutation in F5 associated with enhanced East Texas splicing and FV-short production. J Thromb Haemost. 2021;19:1653–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Peterson JA, Gupta S, Martinez ND, Brandon Hardesty B, Maroney SA, Mast AE. Factor V east Texas variant causes bleeding in a three-generation family. J Thromb Haemost. 2021;20:565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tsujimoto H, Kotsyfakis M, Francischetti IMB, Eum JH, Strand MR, Champagne DE. Simukunin from the salivary glands of the black fly Simulium vittatum inhibits enzymes that regulate clotting and inflammatory responses. PLoS One. 2012;7:e29964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huang ZF, Higuchi D, Lasky N, Broze GJ Jr. Tissue factor pathway inhibitor gene disruption produces intrauterine lethality in mice. Blood. 1997;90:944–51. [PubMed] [Google Scholar]
- [28].Ellery PE, Maroney SA, Cooley BC, Luyendyk JP, Zogg M, Weiler H, Mast AE. A balance between TFPI and thrombin-mediated platelet activation is required for murine embryonic development. Blood. 2015;125:4078–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Marar TT, Martinez ND, Maroney SA, Siebert AE, Wu J, Stalker TJ, Tomaiuolo M, Delacroix S, Simari RD, Mast AE, Brass LF. The contribution of TFPIalpha to the hemostatic response to injury in mice. J Thromb Haemost. 2021;19:2182–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dye BK, Butler C, Lincoln J. Smooth muscle alpha-actin expression in mitral valve interstitial cells is important for mediating extracellular matrix remodeling. J Cardiovasc Dev Dis. 2020;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Totzeck M, Schuler M, Stuschke M, Heusch G, Rassaf T. Cardiooncology—strategies for management of cancer-therapy related cardiovascular disease. Int J Cardiol. 2019;280:163–75. [DOI] [PubMed] [Google Scholar]
- [32].Atkins KM, Rawal B, Chaunzwa TL, Lamba N, Bitterman DS, Williams CL, Kozono DE, Baldini EH, Chen AB, Nguyen PL, D’Amico AV, Nohria A, Hoffmann U, Aerts HJWL, Mak RH. Cardiac radiation dose, cardiac disease, and mortality in patients with lung cancer. J Am Coll Cardiol. 2019;73:2976–87. [DOI] [PubMed] [Google Scholar]
- [33].Saiki H, Moulay G, Guenzel AJ, Liu W, Decklever TD, Classic KL, Pham L, Chen HH, Burnett JC, Russell SJ, Redfield MM. Experimental cardiac radiation exposure induces ventricular diastolic dysfunction with preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2017;313:H392–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Venkatesulu BP, Mahadevan LS, Aliru ML, Yang X, Bodd MH, Singh PK, Yusuf SW, Abe JI, Krishnan S. Radiation-induced endothelial vascular injury: a review of possible mechanisms. JACC Basic Transl Sci. 2018;3:563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mrotzek SM, Rassaf T, Totzeck M. Cardiovascular damage associated with chest irradiation. Front Cardiovasc Med. 2020;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Czubryt MP. Cardiac fibroblast to myofibroblast phenotype conversion—an unexploited therapeutic target. J Cardiovasc Dev Dis. 2019;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. 2018;128:2127–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tallquist MD, Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol. 2017;14:484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tallquist MD. Cardiac fibroblast diversity. Annu Rev Physiol. 2020;82: 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. 2021;117: 1450–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JW, Nordon RE, Harvey RP. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. 2019;8:e43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sorci G, Bianchi R, Riuzzi F, Tubaro C, Arcuri C, Giambanco I, Donato R. S100B protein, a damage-associated molecular pattern protein in the brain and heart, and beyond. Cardiovasc Psychiatry Neurol. 2010;2010:656481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sorci G, Riuzzi F, Arcuri C, Tubaro C, Bianchi R, Giambanco I, Donato R. S100B protein in tissue development, repair and regeneration. World J Biol Chem. 2013;4:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang Y, Gao H, Shi C, Erhardt PW, Pavlovsky A, A Soloviev D, Bledzka K, Ustinov V, Zhu L, Qin J, Munday AD, Lopez J, Plow E, Simon DI. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbalpha. Nat Commun. 2017;8: 15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bozzi M, Parisi V, Poggio P. Macrophages in the heart: active players or simple bystanders? Int Rev Cell Mol Biol. 2022;368:109–41. [DOI] [PubMed] [Google Scholar]
- [46].Muller-Calleja N, Hollerbach A, Ritter S, Pedrosa DG, Strand D, Graf C, Reinhardt C, Strand S, Poncelet P, Griffin JH, Lackner KJ, Ruf W. Tissue factor pathway inhibitor primes monocytes for anti-phospholipid antibody-induced thrombosis. Blood. 2019;134: 1119–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kasper B, Petersen F. Molecular pathways of platelet factor 4/CXCL4 signaling. Eur J Cell Biol. 2011;90:521–6. [DOI] [PubMed] [Google Scholar]
- [48].Novotny WF, Girard TJ, Miletich JP, Broze GJ Jr. Platelets secrete a coagulation inhibitor functionally and antigenically similar to the lipoprotein associated coagulation inhibitor. Blood. 1988;72:2020–5. [PubMed] [Google Scholar]
- [49].Ivey MJ, Tallquist MD. Defining the cardiac fibroblast. Circ J. 2016;80:2269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].de Oliveira Camargo R, Abual’anaz B, Rattan SG, Filomeno KL, Dixon IMC. Novel factors that activate and deactivate cardiac fibroblasts: a new perspective for treatment of cardiac fibrosis. Wound Repair Regen. 2021;29:667–77. [DOI] [PubMed] [Google Scholar]
- [51].Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. 1983;258:7155–60. [PubMed] [Google Scholar]
- [52].Wilson SE. TGF beta-1, -2 and -3 in the modulation of fibrosis in the cornea and other organs. Exp Eye Res. 2021;207:108594. [DOI] [PubMed] [Google Scholar]
- [53].Grainger DJ, Wakefield L, Bethell HW, Farndale RW, Metcalfe JC. Release and activation of platelet latent TGF-beta in blood clots during dissolution with plasmin. Nat Med. 1995;1:932–7. [DOI] [PubMed] [Google Scholar]
- [54].Karolczak K, Watala C. Blood platelets as an important but underrated circulating source of TGFbeta. Int J Mol Sci. 2021;22:4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Suess PM, Smith SA, Morrissey JH. Platelet polyphosphate induces fibroblast chemotaxis and myofibroblast differentiation. J Thromb Haemost. 2020;18:3043–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Affandi AJ, Carvalheiro T, Ottria A, de Haan JJ, Brans MAD, Brandt MM, Tieland RG, Lopes AP, Fernández BM, Bekker CPJ, van der Linden M, Zimmermann M, Giovannone B, Wichers CGK, Garcia S, de Kok M, Stifano G, Xu YJ, Kowalska MA, Waasdorp M, et al. CXCL4 drives fibrosis by promoting several key cellular and molecular processes. Cell Rep. 2022;38:110189. [DOI] [PubMed] [Google Scholar]
- [57].Spronk HM, De Jong AM, Verheule S, De Boer HC, Maass AH, Lau DH, Rienstra M, van Hunnik A, Kuiper M, Lumeij S, Zeemering S, Linz D, Kamphuisen PW, Cate HT, Crijns HJ, Van Gelder IC, van Zonneveld AJ, Schotten U. Hypercoagulability causes atrial fibrosis and promotes atrial fibrillation. Eur Heart J. 2017;38:38–50. [DOI] [PubMed] [Google Scholar]
- [58].Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. 2019;65:70–99. [DOI] [PubMed] [Google Scholar]
- [59].Tsoporis JN, Marks A, Kahn HJ, Butany JW, Liu PP, O’Hanlon D, Parker TG. Inhibition of norepinephrine-induced cardiac hypertrophy in s100beta transgenic mice. J Clin Invest. 1998;102:1609–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18: 733–44. [DOI] [PubMed] [Google Scholar]
- [61].Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, Robbins CS, Monaco C, Park I, McNamara CA, Binder CJ, Cybulsky MI, Scipione CA, Hedrick CC, Galkina EV, Kyaw T, Ghosheh Y, Dinh HQ, Ley K. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. 2020;127: 402–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods. 2014;408:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].McKinsey GL, Lizama CO, Keown-Lang AE, Niu A, Santander N, Larpthaveesarp A, Chee E, Gonzalez FF, Arnold TD. A new genetic strategy for targeting microglia in development and disease. Elife. 2020;9:e54590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Domschke G, Gleissner CA. CXCL4-induced macrophages in human atherosclerosis. Cytokine. 2019;122:154141. [DOI] [PubMed] [Google Scholar]
- [65].Nagy Z, Vögtle T, Geer MJ, Mori J, Heising S, Di Nunzio G, Gareus R, Tarakhovsky A, Weiss A, Neel BG, Desanti GE, Mazharian A, Senis YA. The Gp1ba-Cre transgenic mouse: a new model to delineate platelet and leukocyte functions. Blood. 2019;133:331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pertuy F, Aguilar A, Strassel C, Eckly A, Freund J-N, Duluc I, Gachet C, Lanza F, Léon C. Broader expression of the mouse platelet factor 4-cre transgene beyond the megakaryocyte lineage. J Thromb Haemost. 2015;13:115–25. [DOI] [PubMed] [Google Scholar]
- [67].Carrington R, Jordan S, Wong YJ, Pitchford SC, Page CP. A novel murine model of pulmonary fibrosis: the role of platelets in chronic changes induced by bleomycin. J Pharmacol Toxicol Methods. 2021;109:107057. [DOI] [PubMed] [Google Scholar]
- [68].Ghafoory S, Varshney R, Robison T, Kouzbari K, Woolington S, Murphy B, Xia L, Ahamed J. Platelet TGF-beta1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018;2:470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Meyer A, Wang W, Qu J, Croft L, Degen JL, Coller BS, Ahamed J. Platelet TGF-beta1 contributions to plasma TGF-beta1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood. 2012;119:1064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].DeRoo E, Martinod K, Cherpokova D, Fuchs T, Cifuni S, Chu L, Staudinger C, Wagner DD. The role of platelets in thrombus fibrosis and vessel wall remodeling after venous thrombosis. J Thromb Haemost. 2021;19:387–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dong A, Mueller P, Yang F, Yang L, Morris A, Smyth SS. Direct thrombin inhibition with dabigatran attenuates pressure overload-induced cardiac fibrosis and dysfunction in mice. Thromb Res. 2017;159:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For original data, please contact the corresponding author at aemast@versiti.org.