

Abstract
Peptide epitope tags offer a valuable means for detection and manipulation of protein targets for which high quality detection reagents are not available. Most commonly used epitope tags are bound by conventional, full-size antibodies (Abs). The complex architecture of Abs complicates their application in protein engineering and intracellular applications. To address these shortcomings, single domain antibodies (nanobodies, Nbs) that recognize short peptide epitopes have become increasingly prized. Here we characterize the interaction between a Nb (Nb6E) and a 14-mer peptide epitope. We identify residues in the peptide epitope essential for high affinity binding. Using this information in combination with computational modeling we propose a mode of interaction between Nb6E and this epitope. We apply this nanobody-epitope pair to augment the potency of a ligand at an engineered adenosine A2A receptor. This characterization of the nanobody-epitope pair opens the door to diverse applications including mechanistic studies of G protein-coupled receptor function.
Introduction.
Antibodies (Abs) are prized as reagents for the highly specific recognition of biological molecules of interest, especially for protein targets. These reagents are widely used in basic biomedical research for the detection and manipulation of proteins and in therapeutic applications for blocking the action of soluble proteins in circulation or receptors found on the cell surface. In these settings, the identification of antibodies with high affinity, specificity, and stability can propel research and therapeutic progress1. For some types of targets the identification of antibodies with such desirable properties is difficult. For example, many members of the G protein-coupled receptor (GPCR) superfamily are mostly found embedded in the plasma membrane and have little extracellular surface area exposed for binding by Abs2. In such cases, it is often useful to tag proteins of interest with a short peptide epitope tag to allow for recognition by high quality and well-established anti-epitope tag Abs3. Abs that bind to epitope tags such as myc, HA, and FLAG have been used routinely for decades as immunological detection reagents.
Conventional Abs are comprised of four polypeptide chains (two heavy chains and two light chains) that require glycosylation and disulfide bond formation for assembly, folding, and function. This complex architecture makes the expression of antibodies in the reducing environment of the cellular cytoplasm difficult or impossible. Attempts to create fusions between Abs and bioactive proteins of interest often results in reduced production yields, a loss in folded protein stability, or undesirable proteolytic cleavage4. To circumvent some of these problems, Ab fragments that maintain the high affinity and specificity of full-size immunoglobulins have been developed. Fragments derived directly from conventional Abs include constructs known as single chain-fragment variable (scFv) and fragment antigen-binding (Fab); however, these constructs still possess some of the limitations seen in full-size Abs5. For example, scFvs are commonly used as recognition agents in chimeric antigen receptor T cell (CAR-T) constructs; however, scFv misfolding or mispairing often leads to receptor aggregation or other complications6. Single domain antibodies derived from the variable region of camelid heavy chain only antibodies (VHHs or nanobodies, Nbs) offer an alternative to address some of these issues. Nbs are the smallest antibody fragment that maintains full affinity and they usually do not require disulfide bond formation or glycosylation for function7.
Efforts have been undertaken to identify Nbs that bind to peptide epitope tags7. Nbs that bound to the human immunodeficiency virus protein gp41 were shown to bind to short peptide fragments of this protein, which could be applied as epitope tags (MoonTag, PepTag)8,9. Alternatively, a collection of Nbs specifically raised against a prototypical, synthetic α-helical peptide (Alfa Tag) have been used for a variety of applications10,11. Nanobodies raised against the mammalian proteins α-synuclein12, β-catenin13, CXCR2 (ref. 14,15), and UBC6e16 have been shown to bind to small peptide epitopes taken from these proteins. Previous work has shown that a nanobody (previously called VHH05 or VHH6E, here named Nb6E) binds to a 14-mer peptide derived from the protein UBC6e (6E tag) with low nM affinity16. Subsequent work showed that the 6E tag could be appended via genetic fusion to the extracellular portion of parathyroid hormone receptor-1 (PTHR1), a GPCR, where it was served effectively as an epitope tag without sacrificing receptor function17,18. Success with the Nb6E-6E tag pair spurred us to further characterize this bimolecular interaction. Herein we provide a structure-activity relationship study for the Nb6E-6E tag interaction, generate a computational model to contextualize these experimental data, and apply this nanobody-tag pair to interrogate GPCR signaling.
Results and discussion.
We first sought to identify positions within the 6E tag peptide that are important for interaction with Nb6E. Using solid phase peptide synthesis, we prepared a comprehensive scan library where residues at each position were swapped with Ala except for naturally occurring Ala residues, which were swapped with Glu (Figure 1, Supporting Figure 1 for MS, and Supporting Figure 2 for HPLC purity checks). All peptides also contain a triglycine motif at the N-terminus for use in Sortase A-mediated labeling19. Based on preliminary data, we also prepared an analogue containing the non-natural amino acid aminoisobutyric acid (Aib), which promotes helix formation. We then evaluated the binding of these peptides with Nb6E using a variety of methods.
Figure 1. Sequences of peptides used in this study.

“Obs M” represents the neutral mass corresponding to the most abundant ion peak recorded in characterization using liquid chromatography/mass spectrometry (Supporting Figure 1). Neutral mass M was determined from M = (p*z) – z where z is the charge state of the most abundant ion peak p.
We first developed a competition-based enzyme-linked immunosorbent assay (ELISA) in which each member of the scan library was tested for its ability to inhibit binding of Nb6E to immobilized 6E peptide. To facilitate efficient immobilization on the surface of the ELISA plate for 6E peptide we used sortase-mediated labeling (sortagging) to link 6E with enhanced green fluorescent protein (Figure 2A, Supporting Figure 1). We also labeled Nb6E with biotin using sortagging to permit detection with streptavidin-horse radish peroxidase (SA-HRP) conjugate. Several peptides from the scan library performed similarly to the prototype peptide in this assay (Figure 2B, Supporting Figure 3), indicating maintenance of binding affinity upon Ala substitution. Other members of the scan library exhibited modestly weakened binding. A few analogues (6E-E2, A3, E6, A9, A10) exhibited dramatically reduced binding, suggesting these residues are important for Nb6E association.
Figure 2. Characterization of 6E peptide-Nb6E binding by competition ELISA.
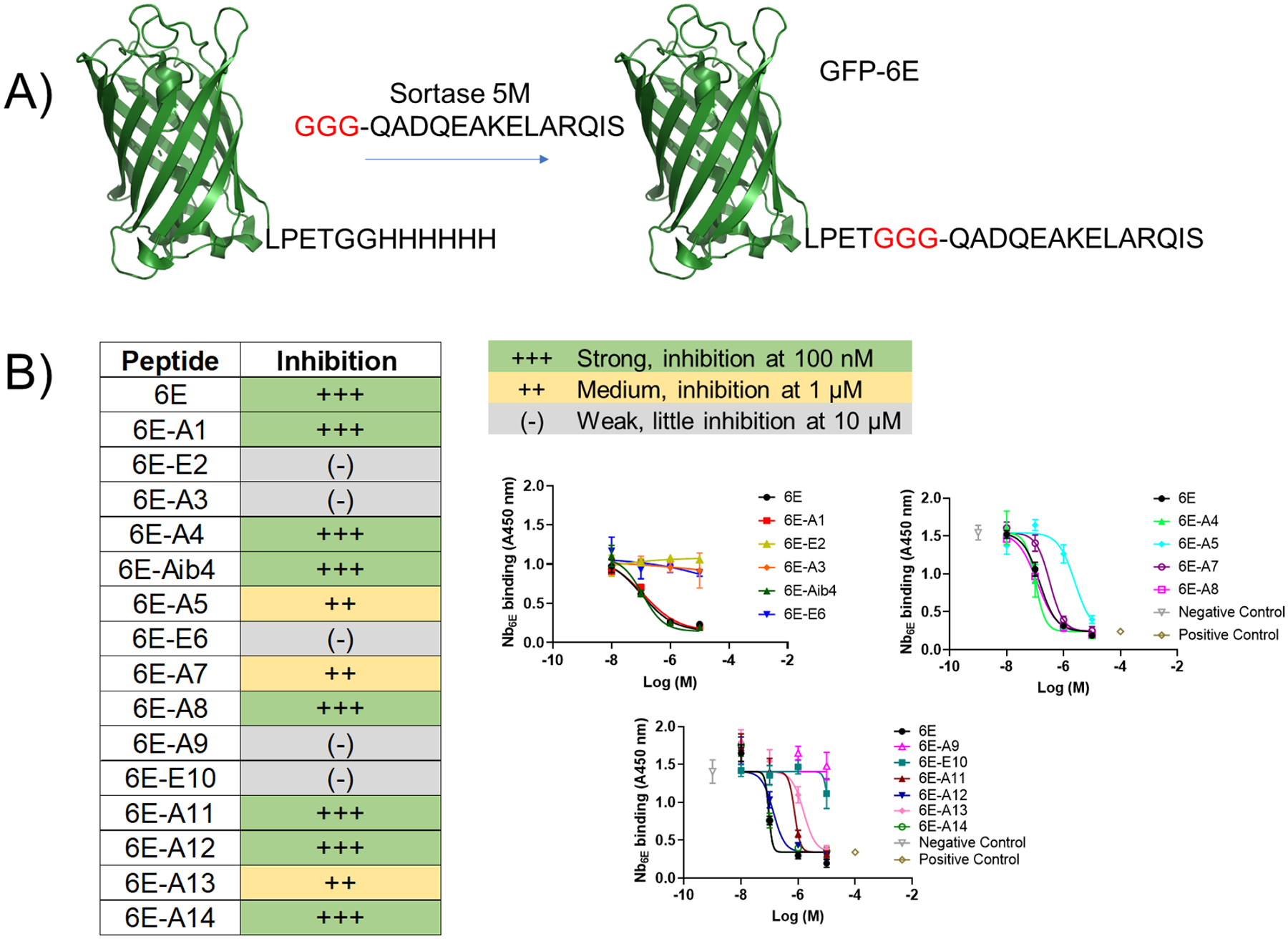
A) Schematic for the preparation of GFP-6E conjugates using sortagging for immobilization in ELISA. See methods for reaction details. B) Summary data for analysis of peptide analogue binding using ELISA. Peptides were categorized into three groups based on their performance in these assays. Representative data from individual assays are shown at right. Data points show mean ± SD from technical replicates in a single experiment. Positive control data points correspond to signal measured in the absence of Nb6E-biotin. Negative control data points correspond to signal observed in the absence of competitor peptide (but with Nb6E-biotin). Curves result from fitting a four-parameter sigmoidal dose-response model to the data (except for weak (−) compounds, which show connected points). Independent replicate data are shown in Supporting Figure 3. Each analogue was tested in ≥ 3 independent experiments. If peptide inhibition categories varied between independent experiments, we categorized compounds in this table based on their modal performance in assays.
To further characterize binding for selected analogues we performed surface plasmon resonance (SPR) experiments (Figure 3). We chose representative peptides from strong, medium, and weak binder categories for these experiments. We focused on modifications in the central portion of the 6E peptide with the reasoning that one or both of the terminal regions of the peptide might project into bulk solution without making direct contact with Nb6E. We immobilized biotinylated Nb6E onto sensor chips coated with streptavidin (SA) and flowed peptides over the coated sensor chips. All peptides demonstrated binding and dissociation in SPR sensorgrams, although the data were not well fit with a simple one-state binding model, requiring the use of a more complex two-state model (see Supporting Methods). The data obtained in SPR experiments mirrored observations from ELISA. 6E peptide bound with the highest affinity, 6E-A5 with modestly reduced affinity, and 6E-A9 with the lowest affinity (Figure 3A–B). Binding association and dissociation parameters were variable among the different analogues tested, with interpretation complicated by use of the two-state model (Supporting Figure 4).
Figure 3. Characterization of Nb6E-6E peptide association and dissociation by surface plasmon resonance assays.
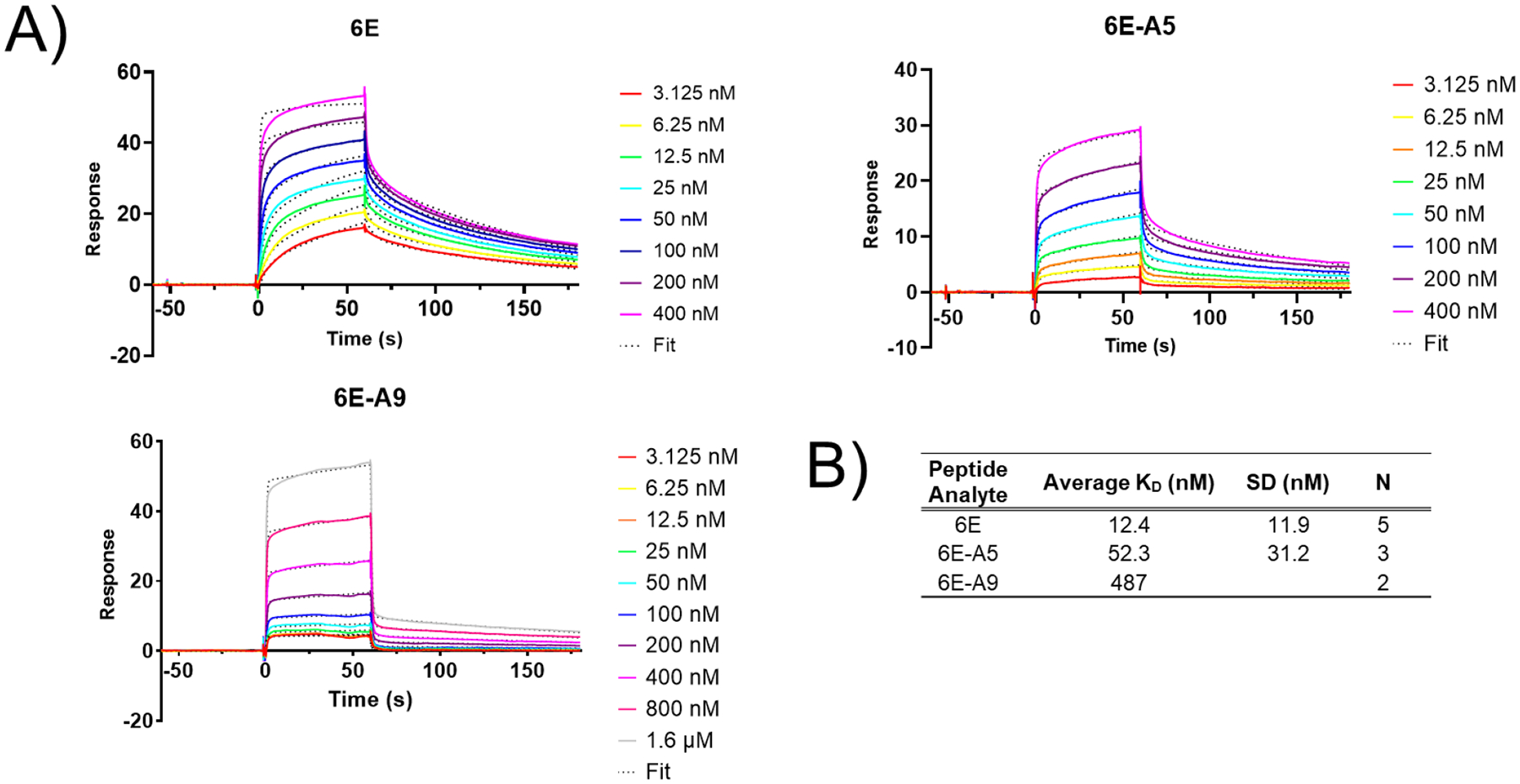
A) Representative sensorgrams showing binding and dissociation of 6E peptides to immobilized Nb6E. Nb6E was conjugated to biotin at its C-terminus using sortagging and immobilized onto sensor chips functionalized with streptavidin. Peptides (at indicated concentrations) were flowed over functionalized sensor chips for 60 s (association) followed by 120 s of buffer flow with no peptide (dissociation). Data were fit a two-state binding model with local R-max with curve fits shown as dotted lines. B) Tabulation of binding parameters from independent replicate experiments. KD averages (mean) and SD were calculated using KD values derived for the indicated number of independent experiments (“N”).
The binding properties of 6E analogues were also tested in a biological milieu. We developed a flow cytometry-based assay using HEK293 cells stably transfected to express the adenosine A2A receptor (a GPCR) fused at its N-terminus with Nb6E [named A2AR(Nb6E)]. This receptor topology places Nb6E at the extracellular face of the plasma membrane, allowing for assessment of binding on live cells expressing this receptor (Figure 4A). We performed a competition assay in which a fluorescein tagged version of 6E peptide (FAM-6E-C14, tracer peptide) was mixed with varying concentrations of unlabeled analogues of 6E (6E, 6E-Aib4, 6E-A5, 6E-A9) and added to cells (Figure 4B–D, Supporting Figure 5). The extent of binding was quantified through use of an Alexa Fluor-647 (AF647) tagged anti-fluorescein secondary antibody, with analysis of labeled cells by flow cytometry. As expected, cells expressing A2AR(Nb6E) showed intense staining with tracer peptide (Supporting Figure 6). Each of the unlabeled peptides inhibited binding of tracer peptide to A2AR(Nb6E) in a concentration-dependent manner. Binding was quantified by measurement of median fluorescence intensity (MFI) in the AF647 channel (Figure 4C–D, Supporting Figure 7). The trends observed mirrored those in previous assays, with 6E and 6E-Aib4 showing strong inhibition of tracer peptide binding, 6E-A5 modest inhibition, and 6E-A9 weak inhibition. To support findings made with flow cytometry, cells expressing A2AR(Nb6E) were also analyzed using fluorescence microscopy (Supporting Figure 8). We observed signal from FAM-6E-C14 on A2AR(Nb6E)-expressing cells. This signal overlapped with that from a tetramethylrhodamine-labeled nanobody (NbAlfa-TMR), which binds to an epitope tag engineered into the extracellular portion of the receptor (see sequence in Supporting Information). We observed negligible staining of cells not transfected with A2AR(Nb6E) via analysis by flow cytometry (Supporting Figure 9), suggesting the observed signals result from interaction of FAM-6E-C14 with receptor.
Figure 4. Assessment of Nb6E-6E peptide interaction on the surface of mammalian cells.
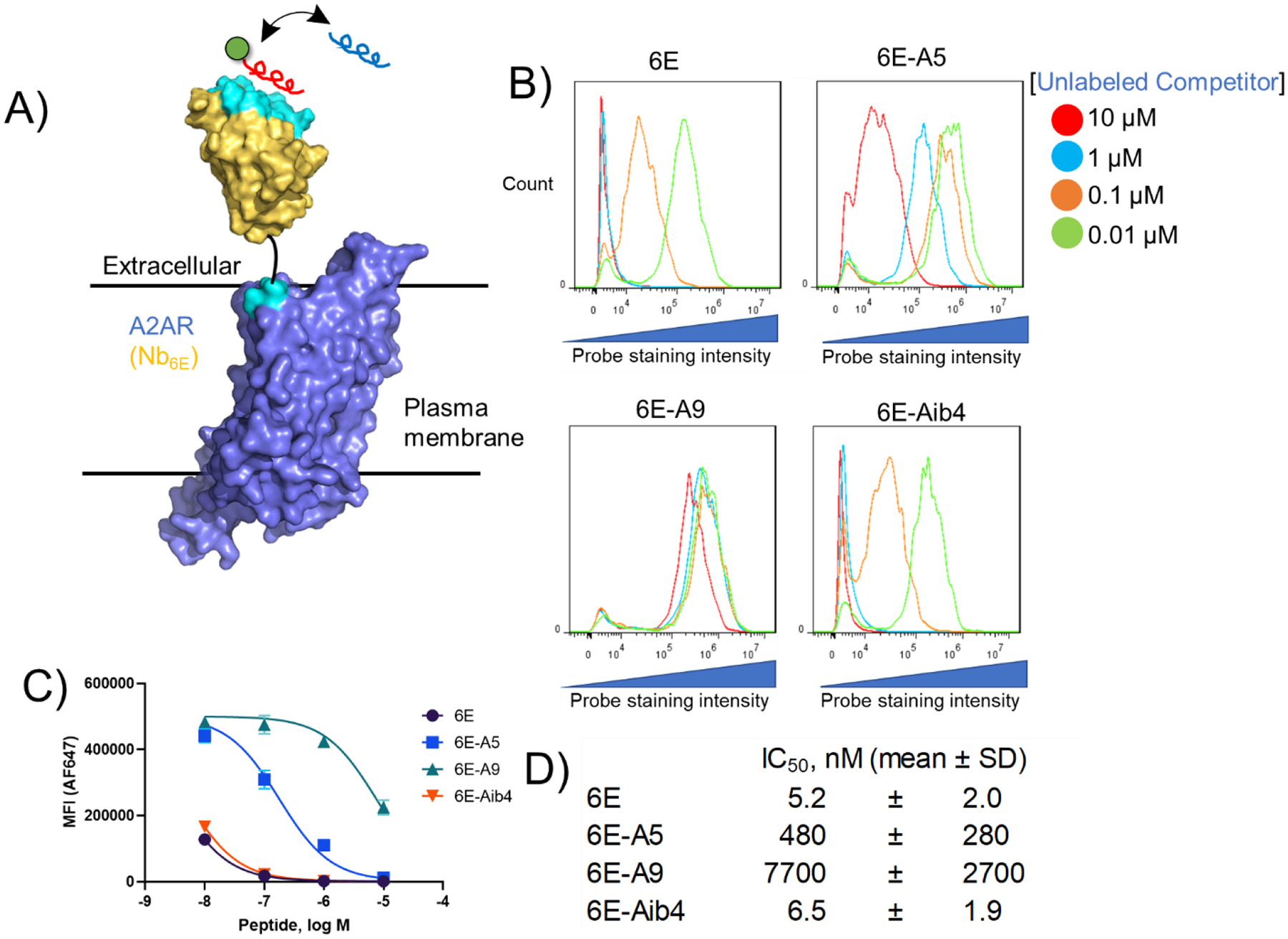
A) Schematic of a competition binding assay involving cells expressing A2AR(Nb6E), a fluorescein-labeled tracer peptide (red), and unlabeled competitor peptides (blue). B) Representative data for flow cytometry analysis of the inhibition of fluorescein-labelled tracer peptide binding by indicated concentrations of unlabeled competitor peptides. Tracer peptide (10 nM) and unlabeled competitor peptides were incubated with cells expressing A2AR(Nb6E), followed by washing, and detection with anti-fluorescein secondary antibody labeled with AF647. Data is presented as a histogram of the intensity of AF647 staining of live cells. Data from independent replicate experiments are shown in Supporting Figure 5. C) Median fluorescence intensity (MFI) quantification of the histograms presented in panel 4B. Curves correspond to application of a sigmoidal dose-response response model to the data. Data points and error bars correspond to mean ± SD from triplicate measurements. Data from independent replicate experiments are shown in Supporting Figure 7. D) Mean IC50 values for inhibition of FAM-6E-C14 binding for each unlabeled peptide tested. IC50 values are reported as mean ± SD from three independent experiments.
Based on these binding data we sought to develop a model to describe the interaction between Nb6E and 6E peptide. Efforts to characterize the Nb6E-6E complex by crystallography failed to yield suitable crystals (data not shown). Recent advances in protein structure prediction methods have enabled rapid and accurate predictions of protein structure20,21. Since high resolution structural information for UBC6e is currently not available, we elected to model the structure of full-length human UBC6e using Alphafold2. The output of our query suggested that the 6E epitope region adopts an α-helical conformation that makes contact with the folded core of the protein (Figure 5A). Alphafold2 has also been used previously to model peptide-protein complex structure22. Using a similar approach, we generated a model of the Nb6E-6E interaction in which the 6E peptide forms a helical structure docked near portions of Nb6E that include the complementarity determining regions (CDRs) (Figure 5B–D), in line with previously published nanobody-peptide epitope crystal structures10,12,13. This model of binding is supported by structure-activity relationship data above, in which substitutions at residues predicted to occur at the binding interface with Nb6E cause substantial losses in binding (Figure 1, Figure 2B). Further evidence for a helical binding conformation for 6E comes from the observation that the incorporation of a non-natural amino acid that promotes helix formation (Aib, Figure 1)23 supports high affinity binding.
Figure 5. Models of 6E peptide structure and interactions.
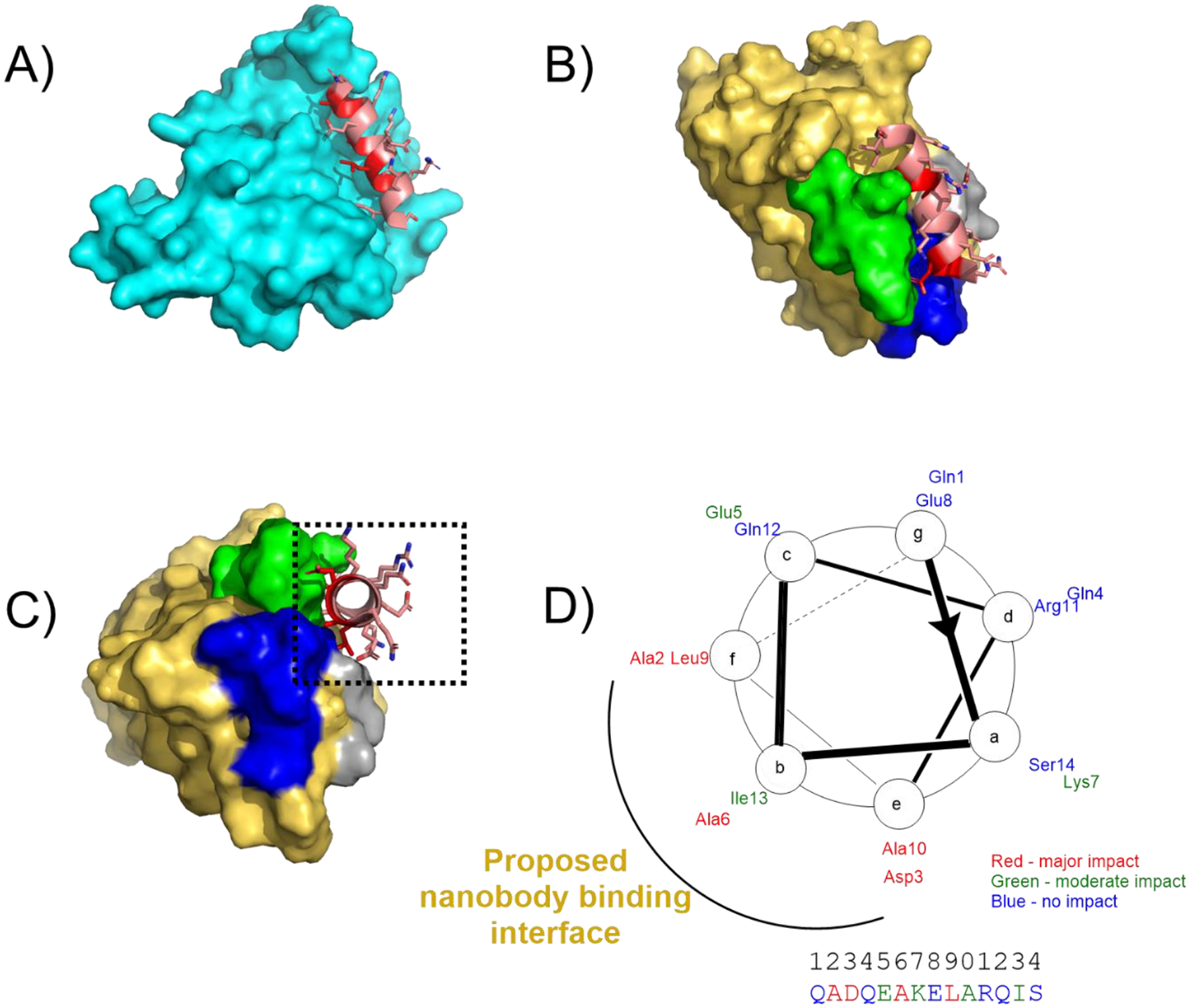
A) Model of the 6E peptide portion of UBC6e (cartoon, shown in salmon with residues important for binding to Nb6E in red) in the context of UBC6e (remainder of the modeled protein shown in cyan surface). The structural model was generated using Alphafold 220. The structure of the C-terminal portion (residues 183 onward) of UBC6e was predicted with low confidence and is omitted from this structure. The color-coded sequence is shown in Supporting Figure 10. B) Model of 6E peptide (salmon/red cartoon) bound to Nb6E (gold surface) with Nb complementary determining regions (CDRs) highlighted in contrasting colors (CDR1: gray, CDR2: green, CDR3: blue). The structure was generated by inputting a sequence consisting of Nb6E fused to 6E peptide by a flexible linker peptide. The color-coded input sequence is shown in Supporting Figure 10. Note that the 6E peptide used for modeling does not contain the GGG extension at the N-terminus. C) Reoriented perspective of hypothetical complex between 6E (red/salmon) and Nb6E (gold). D) Helical wheel diagram of 6E peptide with ELISA data summarized using color coding. The putative nanobody binding interface is proposed based on the structural model shown in panel C and structure-activity relationship data.
We then explored whether this nanobody-peptide tag pair could be used to generate a ligand that specifically activates an engineered receptor. There is broad interest in the development of designer ligands that activate engineered receptors, such as GPCR variants, but not naturally occurring receptors24. Such engineered receptor-designer ligand pairs have proven highly valuable to characterize the biological consequences of receptor activation without interference from activation of endogenous receptors. Because most designer receptors possess mutations in their transmembrane signaling domains, they may not reproduce subtle features of naturally occurring receptor-specific signaling outputs, such as signaling duration and subcellular localization25, which are now appreciated to be important determinants of the type and strength of biological responses induced26. We hypothesized that appending Nb6E to receptor (A2AR) as an artificial ligand binding domain would facilitate the design of an engineered receptor-specific ligand without altering the domain of the receptor responsible for signal generation.
To this end, we prepared a chimeric A2AR ligand that binds to both the receptor orthosteric site and the high affinity, non-orthosteric binding (Nb6E) site. First, we developed chemistry to link an A2AR agonist small molecule to 6E peptide (Figure 6A, Supporting Figures 1–2 for synthetic characterization). For the A2AR agonist we used CGS21680, which has been used previously to synthesize bitopic ligands targeting A2AR and GPCR dimers27. The binding of CGS21680 to human A2AR has been characterized by X-ray crystallography28. This structure shows that the carboxylate on CGS21680 projects from the extracellular opening of A2AR, offering a logical site for linking to other ligands. To the carboxylate on CGS21680 we coupled an alkyne group connected by a short polyethylene glycol (PEG4) linker. To facilitate linkage between 6E and CGS21680 we synthesized an analogue of 6E with an azide moiety appended at the peptide N-terminus (Azide-6E, Figure 6A). Using standard copper-catalyzed azide-alkyne cycloaddition (“click”) chemistry conditions29 we prepared a CGS21680–6E conjugate (CGS-6E, Figure 6A). Because structural information is not available for A2AR(Nb6E) it is not straightforward to predict the optimal length of linker between the orthosteric agonist (CGS21680) and the Nb6E binding peptide. Although the modestly sized PEG4 linker used here is effective, future empirical optimization of linker length might yield further improvements in conjugate biological activity.
Figure 6. Synthesis and application of a GPCR ligand-6E peptide conjugate for enhancing ligand potency.

A) Synthetic scheme for the preparation of conjugates consisting of an A2AR agonist (CGS21680) and 6E peptide using “click” chemistry. Detailed synthetic methodology and characterization is provided in the methods section and Supporting Figures 1–2. B) Evaluation of pharmacological properties of CGS-6E. (left) A representative dose-response curve for the action of indicated compounds for inducing cAMP responses on cells expressing A2AR(Nb6E). Data points represent readings from individual wells in a single representative experiment. Additional dose-response data sets are shown in Supporting Figure 11. (right) Tabulation of compound agonist potency parameters. EC50 values are reported as mean ± SD from three independent experiments.
We then compared CGS-6E to CGS21680 for inducing activation of A2AR(Nb6E) by monitoring HEK293 cells stably transfected with receptor and a cAMP-sensitive luciferase variant30. We found that CGS-6E was as efficacious as CGS21680 for inducing a maximal cyclic adenosine monophosphate (cAMP) response and that it was >25-fold more potent (see EC50 in Figure 6B, Supporting Figure 11). In contrast, CGS-6E was very weakly active (EC50 > 1,000 nM) on cells expressing either wild-type human A2AR or A2AR fused to enhanced green fluorescent protein (A2AREGFP, Supporting Figure 12). The difference in biological activity for CGS-6E between A2AR(Nb6E) and wild-type A2AR (>1,000-fold difference in EC50 values) offers a large selectivity window for biological applications. Addition of 6E peptide to cells expressing A2AR(Nb6E) did not cause an increase in cAMP signal (Supporting Figure 12). Based on this finding, we suggest that the installation of a nanobody as an artificial ligand binding domain into GPCRs might offer a straightforward path to generate receptors effectively activated by modular, chimeric ligands.
Collectively, these experiments have identified 6E peptide features important for its interaction with Nb6E. These data support the structural model generated here (Figure 5) for this interaction. Given the relative paucity of nanobody-peptide epitope pairs with corresponding structure-activity relationship studies13, these findings should empower others to adapt these tools for studies in their system of choice. Worth noting is the use of the Nb6E-6E tag pair for applications as diverse as the study of Drosophila and intracellular enzyme engineering15,31. We demonstrate the power of these tools for studying GPCR pharmacology through engineering a receptor engrafted with an artificial epitope recognizing module (Nb6E), which functions to augment the potency of an engineered ligand. The engraftment of Nbs as artificial recognition domains has been previously reported for cytokine receptors32 and for a phosphatase33. More work has been extended to adapt Nbs as recognition domains for chimeric antigen receptor T cell (CAR-T) constructs for targeted immunotherapy34,35. This work represents the first example of the engraftment of a Nb into a GPCR, which may be useful for the design of bitopic ligands36. Our success in leveraging Nb-epitope interaction for improving the potency of an already high affinity ligand suggests this approach should be widely useful for basic biomedical research. The expanding collection of nanobodies that target GPCRs bodes favorably for these efforts37.
Methods.
Synthesis.
Peptides were synthesized using solid-phase synthesis with Fmoc protection of backbone amines. All compounds were purified with reverse-phase high performance liquid chromatography and compound identity was confirmed by mass spectrometry. Synthetic details are described in the supporting methods section. Mass spectrometry characterization of compounds is shown in Supporting Figure 1.
Protein expression and labeling.
Nanobodies were expressed in the E. coli (BL21 or WK6 strains) periplasm and purified as previously described17 and detailed in Supporting Methods. The sequence of Nb6E has been published previously16. Purified nanobodies were site specifically labeled using sortagging as previously described38 and detailed in the Supporting Methods.
Data calculations and display.
Data were analyzed and prepared for display using GraphPad Prism, FlowJo, and Fiji (ImageJ). Flow cytometry data were quantified by measuring median fluorescence of intensity (MFI) measurements.
Fluorescence Microscopy.
HEK293 cells were analyzed as described in Supporting Methods.
Cell culture and cell-based assays.
Cell-based experiments were run with clonal cell lines derived from HEK293 (ATCC CRL-1573) stably transfected with a biosensor for cAMP30 and receptors of interest. Receptor sequences are found in Supporting Methods. Biological response assays were run in white walled 96-well plates and luminescence responses were recorded on a plate reader as described in Supporting Methods. Cell-based binding assays were recorded using flow cytometry as described in Supporting Methods.
Computational protein modeling.
Structure prediction was performed using the online tool ColabFold39 implementing Alphafold2.
Supplementary Material
Acknowledgments:
We acknowledge D. Appella, C. Bewley, S. Gellman, and K. Jacobson for helpful discussions. We acknowledge the NIDDK mass spectrometry core (J. Lloyd and E. Anderson) for assistance. This work was supported by the NIH Intramural Research Program (NIDDK) and funding from the NIH Director’s Award.
Footnotes
The supporting information is available free of charge at http://pubs.acs.org: mass spectrometry characterization and HPLC purity checks of compounds used in this study; ELISA, flow cytometry and cell-based assay replicates; tabulation of SPR results; fluorescence microscopy data; plasmid and amino acid sequence information; and detailed methodology are provided
References.
- (1).Carter PJ; Lazar GA Next Generation Antibody Drugs: Pursuit of the “High-Hanging Fruit.” Nat. Rev. Drug Discov 2018, 17 (3), 197–223. 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- (2).Hutchings CJ A Review of Antibody-Based Therapeutics Targeting G Protein-Coupled Receptors: An Update. Expert Opin. Biol. Ther 2020, 20 (8), 925–935. 10.1080/14712598.2020.1745770. [DOI] [PubMed] [Google Scholar]
- (3).Vandemoortele G; Eyckerman S; Gevaert K Pick a Tag and Explore the Functions of Your Pet Protein. Trends Biotechnol. 2019, 37 (10), 1078–1090. 10.1016/j.tibtech.2019.03.016. [DOI] [PubMed] [Google Scholar]
- (4).Chodorge M; Celeste AJ; Grimsby J; Konkar A; Davidsson P; Fairman D; Jenkinson L; Naylor J; White N; Seaman JC; et al. Engineering of a GLP-1 Analogue Peptide/Anti-PCSK9 Antibody Fusion for Type 2 Diabetes Treatment. Sci. Rep 2018, 8 (1), 17545. 10.1038/s41598-018-35869-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nelson AL Antibody Fragments: Hope and Hype. mAbs 2010, 2 (1), 77–83. 10.4161/mabs.2.1.10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fujiwara K; Masutani M; Tachibana M; Okada N Impact of ScFv Structure in Chimeric Antigen Receptor on Receptor Expression Efficiency and Antigen Recognition Properties. Biochem. Biophys. Res. Commun 2020, 527 (2), 350–357. 10.1016/j.bbrc.2020.03.071. [DOI] [PubMed] [Google Scholar]
- (7).Cheloha RW; Harmand TJ; Wijne C; Schwartz TU; Ploegh HL Exploring Cellular Biochemistry with Nanobodies. J. Biol. Chem 2020, 295 (45), 15307–15327. 10.1074/jbc.REV120.012960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Boersma S; Khuperkar D; Verhagen BMP; Sonneveld S; Grimm JB; Lavis LD; Tanenbaum ME Multi-Color Single-Molecule Imaging Uncovers Extensive Heterogeneity in MRNA Decoding. Cell 2019, 178 (2), 458–472.e19. 10.1016/j.cell.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Traenkle B; Segan S; Fagbadebo FO; Kaiser PD; Rothbauer U A Novel Epitope Tagging System to Visualize and Monitor Antigens in Live Cells with Chromobodies. Sci. Rep 2020, 10 (1), 14267. 10.1038/s41598-020-71091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Götzke H; Kilisch M; Martínez-Carranza M; Sograte-Idrissi S; Rajavel A; Schlichthaerle T; Engels N; Jungmann R; Stenmark P; Opazo F; et al. The ALFA-Tag Is a Highly Versatile Tool for Nanobody-Based Bioscience Applications. Nat. Commun 2019, 10 (1), 4403. 10.1038/s41467-019-12301-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jedlitzke B; Mootz HD A Light-Activatable Photocaged Variant of the Ultra-High Affinity ALFA-Tag Nanobody. ChemBioChem 2022. 10.1002/cbic.202200079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).De Genst EJ; Guilliams T; Wellens J; O’Day EM; Waudby CA; Meehan S; Dumoulin M; Hsu S-TD; Cremades N; Verschueren KHG; et al. Structure and Properties of a Complex of α-Synuclein and a Single-Domain Camelid Antibody. J. Mol. Biol 2010, 402 (2), 326–343. 10.1016/j.jmb.2010.07.001. [DOI] [PubMed] [Google Scholar]
- (13).Braun MB; Traenkle B; Koch PA; Emele F; Weiss F; Poetz O; Stehle T; Rothbauer U Peptides in Headlock--a Novel High-Affinity and Versatile Peptide-Binding Nanobody for Proteomics and Microscopy. Sci. Rep 2016, 6, 19211. 10.1038/srep19211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bradley ME; Dombrecht B; Manini J; Willis J; Vlerick D; De Taeye S; Van den Heede K; Roobrouck A; Grot E; Kent TC; et al. Potent and Efficacious Inhibition of CXCR2 Signaling by Biparatopic Nanobodies Combining Two Distinct Modes of Action. Mol. Pharmacol 2015, 87 (2), 251–262. 10.1124/mol.114.094821. [DOI] [PubMed] [Google Scholar]
- (15).Xu J; Kim A-R; Cheloha RW; Fischer FA; Li JSS; Feng Y; Stoneburner E; Binari R; Mohr SE; Zirin J; Ploegh HL; Perrimon N Protein Visualization and Manipulation in Drosophila through the Use of Epitope Tags Recognized by Nanobodies. eLife 2022, 11, e74326. 10.7554/eLife.74326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ling J; Cheloha RW; McCaul N; Sun Z-YJ; Wagner G; Ploegh HL A Nanobody That Recognizes a 14-Residue Peptide Epitope in the E2 Ubiquitin-Conjugating Enzyme UBC6e Modulates Its Activity. Mol. Immunol 2019, 114, 513–523. 10.1016/j.molimm.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cheloha RW; Fischer FA; Woodham AW; Daley E; Suminski N; Gardella TJ; Ploegh HL Improved GPCR Ligands from Nanobody Tethering. Nat. Commun 2020, 11 (1), 2087. 10.1038/s41467-020-15884-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cheloha RW; Fischer FA; Gardella TJ; Ploegh HL Activation of a G Protein-Coupled Receptor through Indirect Antibody-Mediated Tethering of Ligands. RSC Chem. Biol 2021, 2 (6), 1692–1700. 10.1039/D1CB00118C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Pishesha N; Ingram JR; Ploegh HL Sortase A: A Model for Transpeptidation and Its Biological Applications. Annu. Rev. Cell Dev. Biol 2018, 34, 163–188. 10.1146/annurev-cellbio-100617-062527. [DOI] [PubMed] [Google Scholar]
- (20).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yang J; Anishchenko I; Park H; Peng Z; Ovchinnikov S; Baker D Improved Protein Structure Prediction Using Predicted Interresidue Orientations. Proc. Natl. Acad. Sci 2020, 117 (3), 1496–1503. 10.1073/pnas.1914677117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tsaban T; Varga JK; Avraham O; Ben-Aharon Z; Khramushin A; Schueler-Furman O Harnessing Protein Folding Neural Networks for Peptide–Protein Docking. Nat. Commun 2022, 13 (1), 176. 10.1038/s41467-021-27838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Karle IL Controls Exerted by the Aib Residue: Helix Formation and Helix Reversal This Article Is a US Government Work and, as Such, Is in the Public Domain in the United States of America. Biopolymers 2001, 60 (5), 351. 10.1002/1097-0282(2001)60:5<351::AID-BIP10174>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- (24).Urban DJ; Roth BL DREADDs (Designer Receptors Exclusively Activated by Designer Drugs): Chemogenetic Tools with Therapeutic Utility. Annu. Rev. Pharmacol. Toxicol 2015, 55, 399–417. 10.1146/annurev-pharmtox-010814-124803. [DOI] [PubMed] [Google Scholar]
- (25).Alvarez-Curto E; Milligan G Defining the Functional Equivalence of Wild-Type and Chemically Engineered G Protein-Coupled Receptors. In Designer Receptors Exclusively Activated by Designer Drugs; Thiel G, Ed.; Neuromethods; Springer New York: New York, NY, 2015; Vol. 108, pp 1–28. 10.1007/978-1-4939-2944-3_1. [DOI] [Google Scholar]
- (26).Irannejad R; Pessino V; Mika D; Huang B; Wedegaertner PB; Conti M; von Zastrow M Functional Selectivity of GPCR-Directed Drug Action through Location Bias. Nat. Chem. Biol 2017, 13 (7), 799–806. 10.1038/nchembio.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Das A; Sanjayan GJ; Kecskés M; Yoo L; Gao Z-G; Jacobson KA Nucleoside Conjugates of Quantum Dots for Characterization of G Protein-Coupled Receptors: Strategies for Immobilizing A2A Adenosine Receptor Agonists. J. Nanobiotechnology 2010, 8, 11. 10.1186/1477-3155-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lebon G; Edwards PC; Leslie AGW; Tate CG Molecular Determinants of CGS21680 Binding to the Human Adenosine A 2A Receptor. Mol. Pharmacol 2015, 87 (6), 907–915. 10.1124/mol.114.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hong V; Presolski SI; Ma C; Finn MG Analysis and Optimization of Copper-Catalyzed Azide-Alkyne Cycloaddition for Bioconjugation. Angew. Chem. Int. Ed Engl 2009, 48 (52), 9879–9883. 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Binkowski BF; Butler BL; Stecha PF; Eggers CT; Otto P; Zimmerman K; Vidugiris G; Wood MG; Encell LP; Fan F; Wood KV A Luminescent Biosensor with Increased Dynamic Range for Intracellular CAMP. ACS Chem. Biol 2011, 6 (11), 1193–1197. 10.1021/cb200248h. [DOI] [PubMed] [Google Scholar]
- (31).Ramirez DH; Aonbangkhen C; Wu H-Y; Naftaly JA; Tang S; O’Meara TR; Woo CM Engineering a Proximity-Directed O-GlcNAc Transferase for Selective Protein O-GlcNAcylation in Cells. ACS Chem. Biol 2020, 15 (4), 1059–1066. 10.1021/acschembio.0c00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Mossner S; Phan HT; Triller S; Moll JM; Conrad U; Scheller J Multimerization Strategies for Efficient Production and Purification of Highly Active Synthetic Cytokine Receptor Ligands. PLOS ONE 2020, 15 (4), e0230804. 10.1371/journal.pone.0230804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Fernandes RA; Su L; Nishiga Y; Ren J; Bhuiyan AM; Cheng N; Kuo CJ; Picton LK; Ohtsuki S; Majzner, et al. Immune Receptor Inhibition through Enforced Phosphatase Recruitment. Nature 2020, 586 (7831), 779–784. 10.1038/s41586-020-2851-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Xie YJ; Dougan M; Jailkhani N; Ingram J; Fang T; Kummer L; Momin N; Pishesha N; Rickelt S; Hynes RO; et al. Nanobody-Based CAR T Cells That Target the Tumor Microenvironment Inhibit the Growth of Solid Tumors in Immunocompetent Mice. Proc. Natl. Acad. Sci 2019, 116 (16), 7624–7631. 10.1073/pnas.1817147116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mo F; Duan S; Jiang X; Yang X; Hou X; Shi W; Carlos CJJ; Liu A; Yin S; Wang W; et al. Nanobody-Based Chimeric Antigen Receptor T Cells Designed by CRISPR/Cas9 Technology for Solid Tumor Immunotherapy. Signal Transduct. Target. Ther 2021, 6 (1), 80. 10.1038/s41392-021-00462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sachdev S; Cabalteja CC; Cheloha RW Strategies for Targeting Cell Surface Proteins Using Multivalent Conjugates and Chemical Biology. Methods Cell Biol. 2021, 166, 205–222. 10.1016/bs.mcb.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Laeremans T; Sands ZA; Claes P; De Blieck A; De Cesco S; Triest S; Busch A; Felix D; Kumar A; Jaakola V-P; Menet C Accelerating GPCR Drug Discovery With Conformation-Stabilizing VHHs. Front. Mol. Biosci 2022, 9, 863099. 10.3389/fmolb.2022.863099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Cheloha RW; Li Z; Bousbaine D; Woodham AW; Perrin P; Volarić J; Ploegh HL Internalization of Influenza Virus and Cell Surface Proteins Monitored by Site-Specific Conjugation of Protease-Sensitive Probes. ACS Chem. Biol 2019, 14 (8), 1836–1844. 10.1021/acschembio.9b00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Mirdita M; Schütze K; Moriwaki Y; Heo L; Ovchinnikov S; Steinegger M ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19 (6), 679–682. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.