

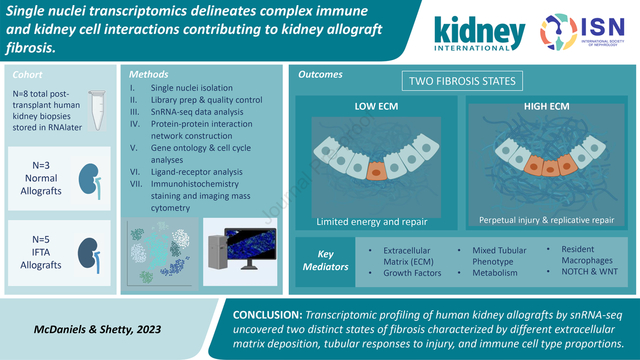
Abstract
Chronic allograft dysfunction (CAD), characterized histologically by interstitial fibrosis and tubular atrophy, is the major cause of kidney allograft loss. Here, using single nuclei RNA sequencing and transcriptome analysis, we identified the origin, functional heterogeneity, and regulation of fibrosis-forming cells in kidney allografts with CAD. A robust technique was used to isolate individual nuclei from kidney allograft biopsies and successfully profiled 23,980 nuclei from five kidney transplant recipients with CAD and 17,913 nuclei from three patients with normal allograft function. Our analysis revealed two distinct states of fibrosis in CAD; low and high extracellular matrix (ECM) with distinct kidney cell subclusters, immune cell types, and transcriptional profiles. Imaging mass cytometry analysis confirmed increased ECM deposition at the protein level. Proximal tubular cells transitioned to an injured mixed tubular (MT1) phenotype comprised of activated fibroblasts and myofibroblast markers, generated provisional ECM which recruited inflammatory cells, and served as the main driver of fibrosis. MT1 cells in the high ECM state achieved replicative repair evidenced by dedifferentiation and nephrogenic transcriptional signatures. MT1 in the low ECM state showed decreased apoptosis, decreased cycling tubular cells, and severe metabolic dysfunction, limiting the potential for repair. Activated B, T and plasma cells were increased in the high ECM state, while macrophage subtypes were increased in the low ECM state. Intercellular communication between kidney parenchymal cells and donor-derived macrophages, detected several years post-transplantation, played a key role in injury propagation. Thus, our study identified novel molecular targets for interventions aimed to ameliorate or prevent allograft fibrogenesis in kidney transplant recipients.
Keywords: Kidney transplantation, chronic kidney injury, snRNA-seq
Graphical Abstract
ONE SENTENCE SUMMARY:
Single nuclei evaluation of human kidney allografts uncovered two distinct types of fibrosis characterized by different ECM levels and compositions, tubular responses to injury, and immune cell types and their associated molecular profiles.
INTRODUCTION
Biological mechanisms underpinning long-term allograft survival after kidney transplantation (KT) remain unknown.1–6 Late graft loss after KT occurs because of chronic allograft dysfunction (CAD), a multifactorial, progressive, and irreversible disease.1,3–11 Interstitial fibrosis and tubular atrophy (IFTA), the final common pathway for CAD,3,7–10,12–14 predicts allograft failure and correlates with increased patient mortality.5–9,11 Little is known about the cellular and molecular interactions between damaged kidney cells in the tubuloinsterstitium and infiltrating host cells.15–17
Single cell transcriptomics have evolved to delineate complex interactions between the immune system and renal cells contributing to CAD.18 Using single nuclei RNA-seq, we evaluated 41,893 nuclei from kidney allograft biopsies, including functioning grafts with IFTA (in patients with CAD) and with normal/nonspecific histopathology (in patients with stable graft function).
Allografts with IFTA segregated into fibrosis with low extracellular matrix (ECM) and high ECM states, based on their ECM composition and deposition level, immune cell types, and associated differential transcriptomics. Trajectory-based pseudotime inferences indicated a proximal tubular to fibroblast phenotypic transition (with a mixed tubular intermediate state) in fibrotic grafts. Ligand receptor analyses revealed dynamic immune and non-immune interactions driven by infiltrating and resident macrophages. Overall, single nuclei transcriptomic analysis of kidney allograft fibrosis yielded mechanistic insights into the role of immune cell responses and maladaptive repair signatures driving fibrosis. This new approach has the potential to better stratify graft injury and identify new targets for personalized interventions.
METHODS
Patients and samples.
Kidney graft biopsies from 8 kidney transplant recipients (KTRs) were studied; 5 patients with CAD whose biopsies were categorized as IFTA and 3 patients with normal/stable graft function were categorized as normal/non-specific. Independent histological evaluations were performed by two kidney transplant pathologists. The Institutional Review Board approved the study and patients signed an informed consent at time of transplantation (HP-00091954). The clinical and research activities being reported are consistent with the Principles of the Declaration of Istanbul as outlined in the ‘Declaration of Istanbul on Organ Trafficking and Transplant Tourism’.
Sample processing and single nuclei isolation.
Kidney allograft tissue was obtained using an 18-gauge biopsy needle and all samples were immersed in RNAlater (Ambion) immediately after collection. Single nuclei isolation from tissue was performed using our recently validated approach.19 See supplementary information for more details.
Quality control, data analyses, and validation reactions.
Detailed analyses and applied tools are provided in supplementary information.
RESULTS
Nuclei isolation using kidney allograft biopsies preserved in RNAlater
The feasibility of using kidney core biopsies preserved in RNAlater for snRNA-seq was assessed (See supplementary methods, Fig. S1A–C). Eight biopsies passed quality control (QC) (Fig. S2A–G). There were no significant differences in QC when compared to snap-frozen kidney graft biopsies (KUT vs. GSE13188220,21) (Figs. S3A–C). RNAlater offers an alternative preservation method prior to snRNA-seq with no evidence of transcripts or genes affected (Tables S1–2).
Identification of cell types in fibrosis and normal graft biopsies
Characteristics of donor, recipients, and samples are provided in Tables 1A–B, S3. Using the Banff criteria,22,23 biopsies were classified as IFTA (fibrosis, N=5, 23,930 nuclei) or normal/non-specific (normal, N=3, 17,913 nuclei) (Table 1B). UMAP integration of 41,893 single nuclei generated 18 main cell clusters (Figs. 1A, S4A, Table S4). Cell marker genes identified distinct clusters (Fig. S4B).
Table 1A.
Characteristics of donor and recipient from kidney grafts graft biopsies included in this study.
| Condition | Recipient characteristics | Donor characteristics | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Age (yo) | Sex | Race | DSA | Creatine at biopsy (mg/dL) | Post-tx time (mo) | More recent creatinine (mg/dL) | Age | Race | Sex | |
| Normal/non-specific | KUT023 | 70 | M | AA | − | 0.80 | 18 | 1.50 | 21 | C | M |
| KUT060 | 32 | F | AA | − | 0.76 | 15 | 1.00 | 17 | C | M | |
| KUT081 | 46 | F | C | − | 1.00 | 24 | 1.00 | 23 | H | F | |
| Fibrotic | KUT076 | 46 | M | AA | − | 1.70 | 16 | 1.80 | 52 | C | F |
| KUT088 | 40 | M | AA | − | 3.00 | 60 | 6.00 | 18 | C | F | |
| KUT040 | 49 | M | AA | − | 2.00 | 48 | 1.25 | 27 | C | F | |
| KUT065 | 60 | M | AA | − | 2.80 | 24 | 10.60 | 52 | C | F | |
| KUT100 | 35 | F | C | + | 1.00 | 26 | 1.33 | 42 | C | F | |
AA, African American; C, Caucasian; DSA, Donor Specific Antibodies; F, Female; H, Hispanic; M, Male; mo, months; Tx, Transplantation; yo, years old; −, absent; +, present.
Table 1B.
Histological classification kidney graft biopsies evaluated using the Banff score.
| Normal/non-specific | Fibrotic | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Banff lesion scores | KUT023 | KUT060 | KUT081 | KUT040 | KUT065 | KUT076 | KUT088 | KUT100 | |
| Acute | Glomerulitis (g) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Interstitial inflammation (i) | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 0 | |
| Peritubular capillaritis (ptc) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| Tubulitis (t) | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 1 | |
| Intimal arteritis (v) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Chronic | Arteriolar hyalinosis (ah) | 0 | 0 | 0 | 2 | 1 | 0 | 1 | 1 |
| Glomerular basement membrane double contours (cg) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Interstitial fibrosis (ci) | 0 | 0 | 0 | 2 | 2 | 2 | 2 | 3 | |
| Peritubular capillary basement membrane multilayering (cptc) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Tubular atrophy (ct) | 0 | 0 | 1 | 2 | 2 | 2 | 2 | 3 | |
| Vascular fibrous intimal thickening (cv) | 0 | 0 | 0 | 2 | 2 | 3 | 2 | 3 | |
| Inflammation in areas of interstitial fibrosis and tubular atrophy (iIFTA) | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | |
| Total inflammation (ti) | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | |
| Mesangial matrix expansion (mm) | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | |
| BK virus (SV40 large T antigen) | - | - | - | - | - | - | - | - | |
| C4d | - | - | - | - | - | - | - | - | |
| Global glomerulosclerosis (%) | <5 | 0 | <5 | 2 | 17 | <5 | 8 | 16 | |
All biopsies were assessed using light (LM), immunofluorescence (IF), and electron microscopy (EM) and reported independently by two transplant pathologists using Banff 2019 update1 of the Banff 97 classification.2 All biopsies were negative for peritubular capillary staining for complement factor 4 fragment d (C4d) tested by immunofluorescence. All biopsies were negative for BK virus (SV40 large T antigen, immunoperoxidase staining).
KUT023: LM: 72 non-sclerotic glomeruli, IF: up to 13 non-sclerotic glomeruli; KUT060: LM: 37 non-sclerotic glomeruli, IF: up to 10 non-sclerotic glomeruli; KUT081: LM: 12 non-sclerotic glomeruli, IF: up to 10 non-sclerotic glomeruli; KUT040: LM: 17 non-sclerotic glomeruli, 2 globally sclerotic, IF: up to 10 non-sclerotic glomeruli; KUT065: LM: 43 non-sclerotic glomeruli, 5 globally sclerotic IF: 15 non-sclerotic glomeruli, 8 globally sclerotic; KUT076: LM: 39 non-sclerotic glomeruli, 4 globally sclerotic IF: up to 10 non-sclerotic glomeruli; KUT088: 39 non-sclerotic glomeruli, 2 globally sclerotic IF: 8 non-sclerotic glomeruli; KUT100: 46 non-sclerotic glomeruli, 14 globally sclerotic IF: up to 10 non-sclerotic glomeruli.
Fig. 1. Single nuclei RNA-seq analysis in normal/nonspecific and fibrotic human kidney grafts.
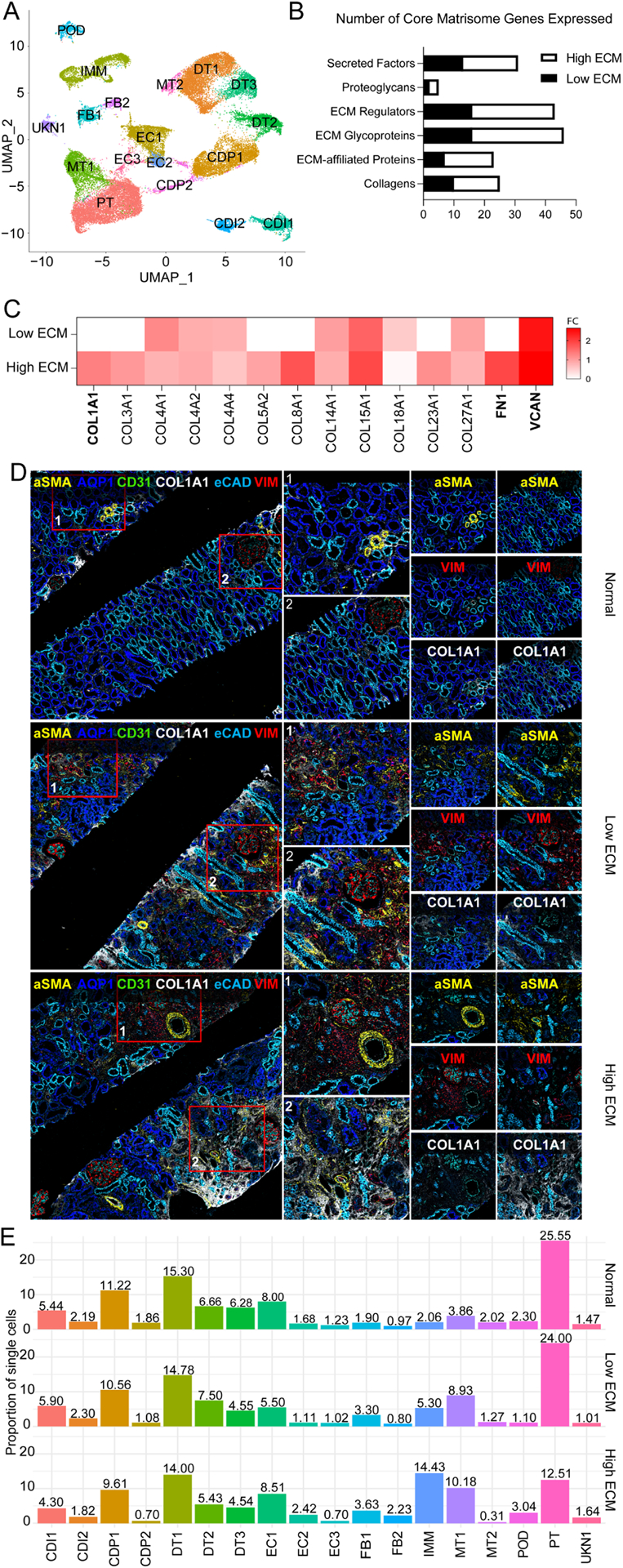
(A) UMAP visualization of 41,893 nuclei from (3 normal and 5 fibrotic grafts) integrated into a single dataset. CDI1, collecting duct intercalated 1; CDI2, collecting duct intercalated 2; CDP1, collecting duct principal 1; CDP2, collecting duct principal 2; DT1, distal tubular 1; DT2, distal tubular 2; DT3, distal tubular 3; EC1, endothelial 1; EC2, endothelial 2; EC3, endothelial 3; FB1, fibroblast 1; FB2; fibroblast 2; IMM, immune; MT1, mixed tubular 1; MT2, mixed tubular 2; POD, podocyte; PT, proximal tubular cells; and UNKN1, unknown 1. (B) Number of matrisome genes expressed by each classification. Black, low ECM; White, high ECM. (C) Heatmap of the expression of 14 selected matrisome genes from fibrotic kidney grafts with low and high ECM expression. Gene expression is represented across all cell clusters. Bolded, critical fibrogenic genes; FC, log2 fold change. (D) Imaging mass cytometry (IMC) staining confirmed ECM deposition levels and localization in matched kidney biopsies. The image shows expression of αSMA (alpha smooth muscle actin cells, AQP1 (proximal tubulars), CD31 (endothelial cells), COL1A1 (collagen 1), eCAD (tubulars), and VIM (fibroblasts). Red boxes denoted by a 1 or 2 is enlarged to the right of the image to show spatial resolution of the glomerulus. The last panels (far right) are enlarged to detail αSMA, VIM, or COL1A1 expression. (E) Proportion of single cells in each cluster per classification. Cell clusters are colored by population.
Among the 1,755 differentially expressed genes (DEGs) (Fig. S5A) between fibrosis and normal, 1,006 DEGs were upregulated in fibrosis and 118 overlapped with the Human Core Matrisome Database.24
Within the fibrosis allograft biopsies, in addition to differential types and transcriptional profiles of immune cells, qualitative and quantitative differences in ECM gene profiles were identified (Figs. 1B, S5B–E). Biopsies with fibrosis were classified into two states, low ECM (8,250 nuclei) and high ECM (15,730 nuclei). Core matrisome genes were more abundant in high ECM (N=109) compared to low ECM (N=64) (with 49% overlapping DEGs between the two states (Figs. 1B, S5C)). Notably, FN1, COL1A1, and VCAN were differentially expressed in high ECM (Fig. 1C). To validate gene expression at the protein level, imaging mass cytometry (IMC) of kidney biopsies confirmed the presence and level of ECM deposition (Fig. 1D). COL1A1, αSMA, and VIM expression increased from normal<low ECM<high ECM. Gene ontology (GO) analyses showed upregulation of inflammatory responses in high ECM (Fig. S5C), and in concordance with higher proportions of immune cells.
Pathway and enrichment analysis of upregulated genes in fibrosis were significant in cell morphogenesis, wound healing, and epithelial cell differentiation (Fig. S5E).
Cell distribution among kidney grafts
Proximal tubular (PT) cells were abundant in normal kidney grafts (25.55%) (Figs. 1E, S5D). PTs significantly decreased in high ECM (12.51%), and slightly diminished in low ECM (24.00%) compared to normal grafts (Fig. 1E). Fibroblasts (FB) subcluster FB1 (PDGFR-α) increased in both fibrosis states compared to normal. Endothelial cell (EC) subcluster, EC2 (EHD3), was more abundant in the high ECM state (2.42%). Mixed tubular (MT) subcluster, MT1, was overrepresented in fibrosis (8.93%−10.18%) compared to normal (3.86%). Immune cells increased significantly from normal (2.10%) to low ECM (5.30%) to high ECM (14.43%) (Fig. 1E). Loop of Henle (LOH) cell markers were identified (UMOD). Following recommendations,18 LOH cells were denoted as “Unknown 1” (UKN1).
Distinct GO enriched terms and pathways are shown for low and high ECM (Fig. S6A–B). A potassium channel-interacting protein 4, KCNIP4, was upregulated in tubular clusters specific to fibrosis states (Figs. S6C–D). KCNIP4 is upregulated in a proinflammatory, profibrotic state that persists after acute kidney injury.25 Low and high ECM states present different cell distributions and transcriptional profiles, supporting heterogeneity within the fibrosis states.
Injured tubular epithelial cells and graft fibrosis
PT physiological pathways were downregulated in both fibrosis states (Figs. S7A–B). Protein-protein interaction enrichment analysis identified 6 Molecular Complex Detection (MCODEs) components (Figs. S7C–D). Low ECM displayed two significantly downregulated modules. MCODE_1 represented fatty acid oxidation, lipid oxidation, and energy metabolism (red, Fig. S7C). MCODE_6 represented negative regulation of extrinsic apoptotic signaling pathway (yellow, Fig. S7C). Shared pathways are shown in Fig. S7E. Lipid and fatty acid metabolism alterations were more significant in low ECM (Fig. S7F), supporting PT metabolic dysfunction.
Interestingly, our MT1–2 cell clusters (Fig. 1A) were transcriptionally similar to the “mixed identities” cluster recently described in a murine model of acute kidney injury.26 Select MT1 cell markers are listed in Table S5.
Cell cycle arrest has been associated with kidney injury and plays an important role in tubular cell protection and maladaptive repair.25,27–29 The identified tubule cell clusters (PT, MT1, MT2) presented differences related to cell cycling proportions among the three groups, slightly decreased in low ECM and increased in high ECM compared to normal reaching statistical significance (Figs. 2A, S8A–C). Critically, low ECM displayed decreased expression of G2M cell cycle regulators compared to high ECM and normal (Fig. S8D).
Fig. 2. Cell cycle and gene expression analysis of the mixed tubular 1 cell cluster.
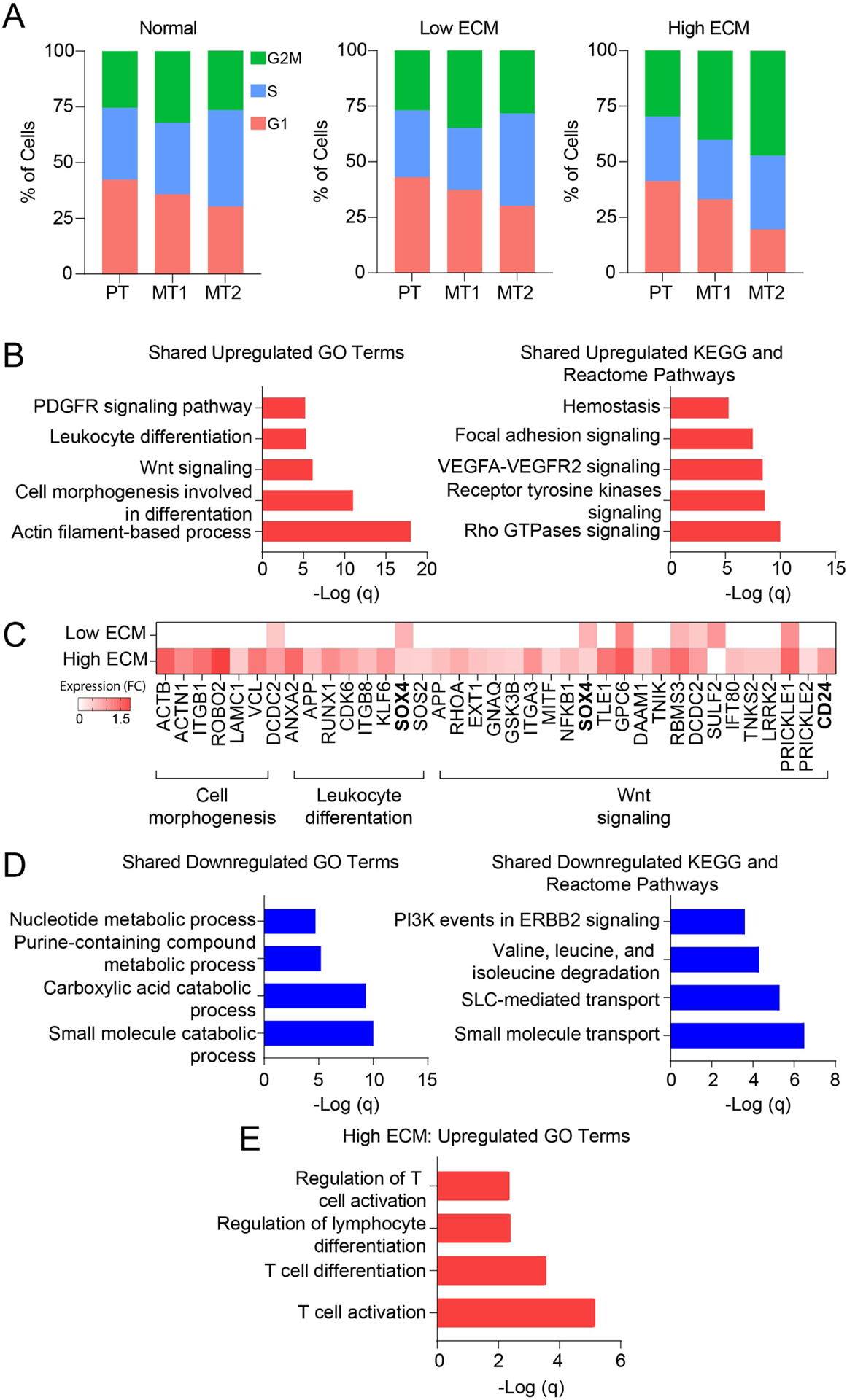
(A) Proportion of epithelial tubular cells at different cell cycle stages using average transcriptional expression data. Orange, G1 phase; green, S phase; and blue, G2M phase. MT1, mixed tubular 1; MT2, mixed tubular 2; and PT, proximal tubular cells. (B) Gene enrichment analysis showing the top upregulated GO terms and pathways shared in fibrotic grafts. (C) Heatmap of upregulated gene expression using selected genes panel B. Expression is represented as log2 FC values. (D) Gene ontology pathway analysis showing the top downregulated GO terms and pathways shared in fibrotic grafts. (E) Gene ontology pathway analysis showing the top upregulated pathways unique to high ECM.
DEG analysis of fibrotic allografts showed that MT1 overexpressed genes enriched in actin filament-based process, cell morphogenesis, receptor tyrosine kinases, and VEGF signaling (Fig. 2B). High ECM was characterized by a strong signature of cell morphogenesis, leukocyte differentiation, and Wnt signaling (Fig. 2C, Table S6). Conversely, low ECM was marked by negative regulation of cell proliferation and apoptosis (Table S6). High ECM showed enrichment of a senescence-gene signature compared to low ECM (Table S6).
In fibrotic allografts, MT1 downregulated GO terms and pathways included amino acid degradation and small molecule transport (Fig. 2D). T-cell activation and differentiation were enriched in high ECM (Fig. 2E).
Trajectory analysis confirmed MT1 epithelial origin (Fig. 3A). Changes in gene expression along a trajectory illustrated a cellular transition from PT to MT1 to a FB-like state (Fig. 3B). Over time, expression of a PT marker (CUBN) decreased while a FB marker (C7) increased as cells moved toward the MT1 transition signature (Fig. 3C). MT1 also showed increased αSMA and VIM expression and reduced E-CADHERIN expression. Together, MT1 is an intermediate transcriptional state in the epithelial to mesenchymal transition (EMT).
Fig. 3. Pseudotime and trajectory analysis of gene expression displaying dynamic changes in proximal tubular cells and mixed tubular cell clusters.
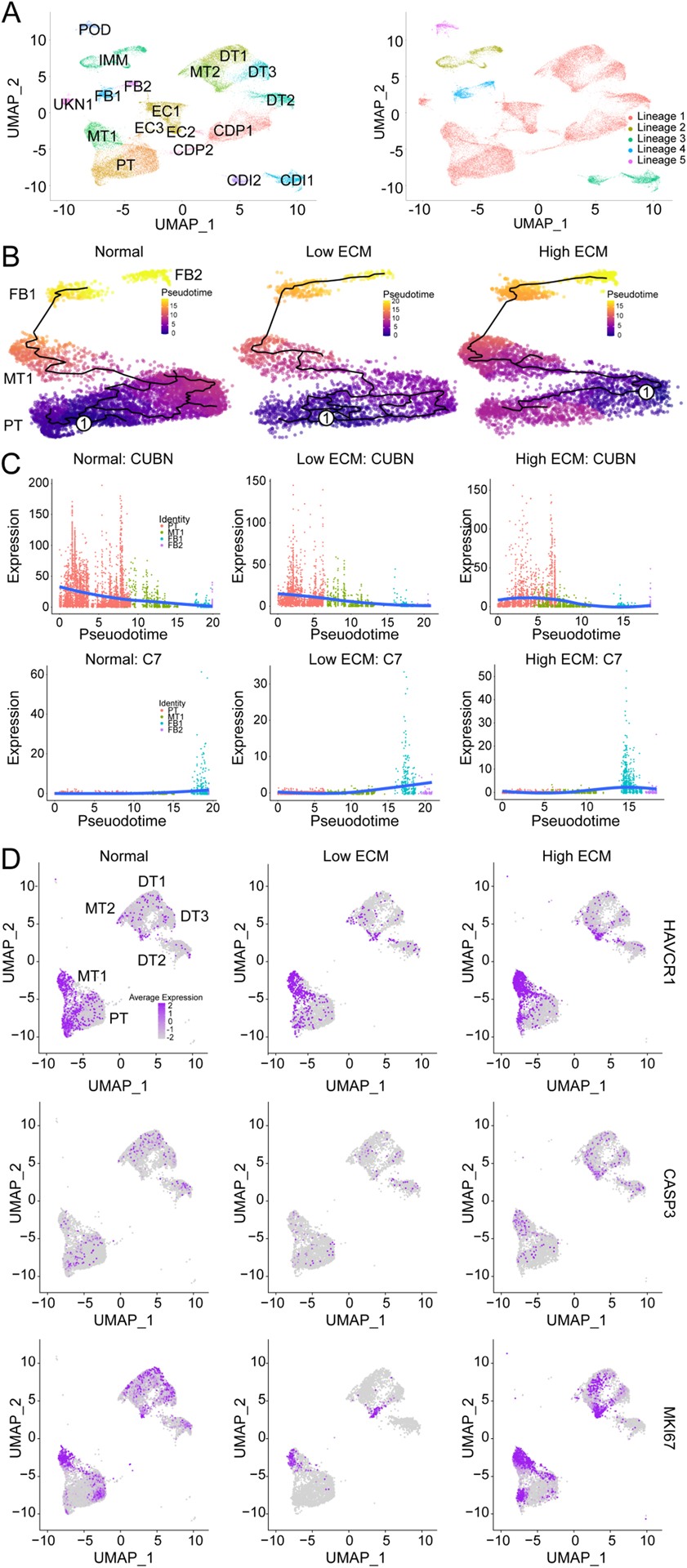
(A) UMAP of combined classes with 18 cell clusters (right) giving rise to a total of five unsupervised partitions (right). CDI1, collecting duct intercalated 1; CDI2, collecting duct intercalated 2; CDP1, collecting duct principal 1; CDP2, collecting duct principal 2; DT1, distal tubular 1; DT2, distal tubular 2; DT3, distal tubular 3; EC1, endothelial 1; EC2, endothelial 2; EC3, endothelial 3; FB1, fibroblast 1; FB2; fibroblast 2; IMM, immune; MT1, mixed tubular 1; MT2, mixed tubular 2; POD, podocyte; PT, proximal tubular cells; and UNKN1, unknown 1. (B) The single-cell trajectory reconstructed by Monocle 2 displaying normal (left), low ECM (middle), and high ECM (right). Each point represents a cell state at a specific time. Cells start at the root, marked by encircled one, and progress to one of three alternative reprogramming outcomes, denoted by PT, MT, FB1, and FB2. Cells must pass through the intermediate cluster MT1. Trajectory curve is highlighted by a solid black line. (C) Expression dynamics of CUBN (top) and C7 (bottom) along pseudotime that supports functional transition from PT to MT1 to FB1 and FB2 cell clusters. Cells are colored by their cluster. Orange, PT; green, MT1; blue, FB1; and purple, FB2. Solid blue line denotes average expression along pseudotime. (D) UMAP of epithelial cells with quantification of HAVCR1, CASP3, and MKI67 expression across each classification.
For normal grafts, cell trajectories ended at the FB1 cluster, whereas fibrotic grafts trajectories extended to the FB2 cluster. These discrete pseudotime trajectories suggest that changes in gene expression illustrated two distinct FB lineages: i) originated and ended in the PT cluster and ii) led to a functional transition from the PT to FB2 cluster (Fig. 3B).
Transcriptomic analyses revealed that the MT1 co-expressed PT (LRP, CUBN) and injury (HAVCR1, VCAM1)25 markers (Fig. 3D). MT1 in the high ECM state expressed a higher level of injury markers. Normal graft expression of injury and normal PT markers are likely a consequence of ongoing subclinical immune responses and chronic exposure to calcineurin inhibitors.
As PT cells regenerate after injury,30 the injured MT1 population was evaluated for replication- or impaired reparation-specific markers. Co-upregulation of a proliferation marker (MKI67) and an apoptotic marker (CASP3) in high ECM MT1 showed a subset of injured PT cells that underwent injury-induced replication. MT1 in high ECM was also characterized by reduced SLC34A1 expression compared to low ECM and normal, indicating dedifferentiation of injured PT cells. Critically, a nephrogenic signature26 normally expressed during early kidney development was associated with high ECM evidenced by high SOX4 and CDC24 expression (Fig. 2C) while not observed in low ECM. MT2 cells were not as abundant as MT1 and were mainly present in normal allografts (Fig. 1E).
Fibroblast subtypes in the kidney grafts
Two distinct fibroblasts clusters were identified (FB1, FB2) (Fig. 4A). FB1 was more abundant in fibrotic kidneys (Fig. 1E), defined by PDGFR-α and COL1A1 expression (Fig. 4B), and enriched in myofibroblasts markers (COL1A1, COL3A1, COL5A1, COL5A2, COL6A3, COL8A1, FBN1).17,27 GO analysis for FB1 in low ECM showed upregulation of cell junction, focal adhesion, and VEGF signaling pathways (Fig. 4C), while in high ECM there was an upregulation of ECM organization, smooth muscle contraction, and wound healing (Fig. 4D).
Fig. 4. Gene expression and trajectory analysis of fibroblast clusters.
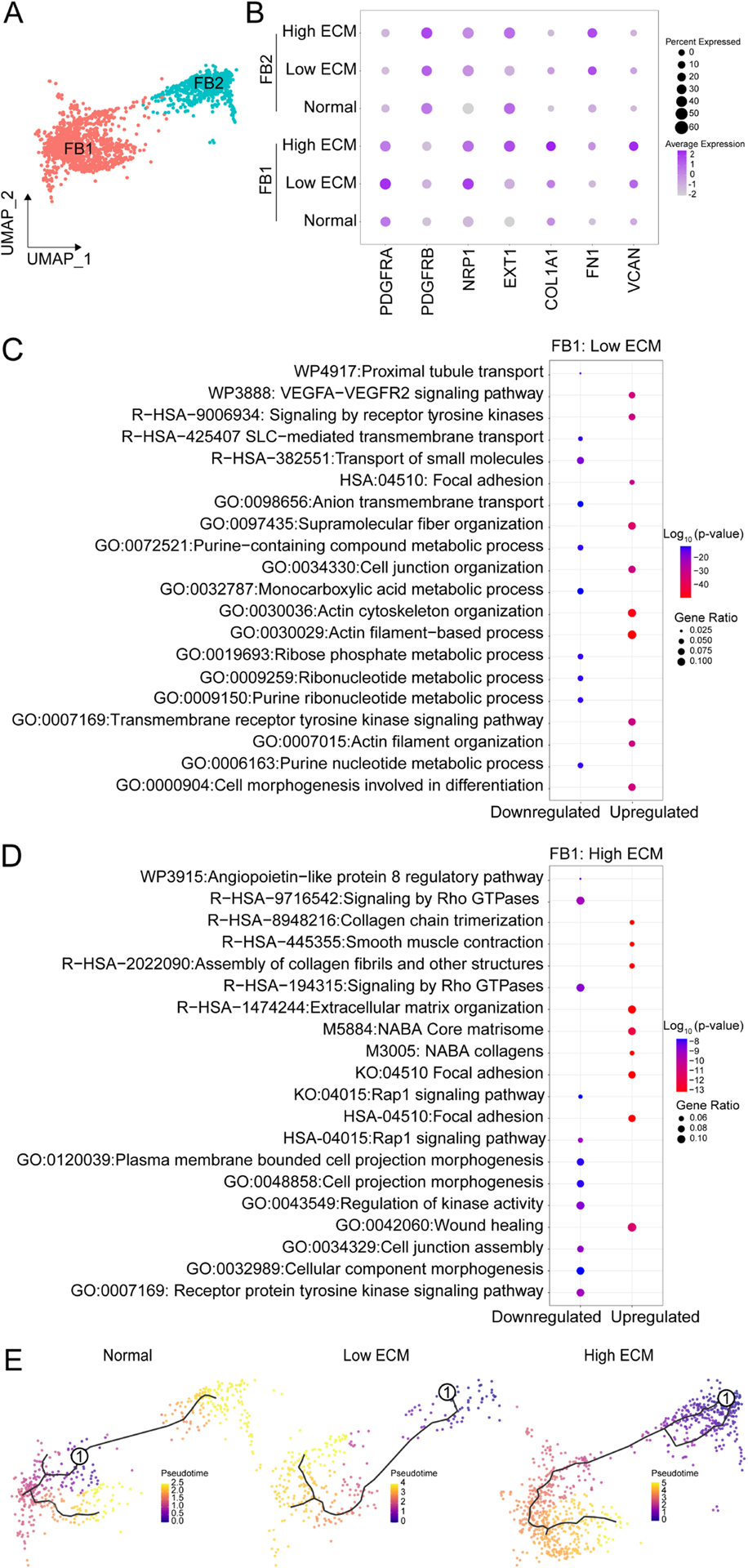
(A) UMAP of FB1 and FB2. (B) Dot plot of DEGs to distinguish clusters. FB1 GO enrichment analysis for (C) low and (D) high ECM. Comparison of the GO terms by p-value and gene ratio (defines the number of DEGs in associated with the GO term). Analysis was performed with ggplot2 in R. (E) Individual trajectories along pseudotime for normal (left), low ECM (middle), and high ECM (right). Solid black line depicts the expression curves for each branch over pseudotime. Encircled one, origin of trajectory.
FB2 was predominant in high ECM (Fig. 1E), defined by PDGFR-β expression (Fig. 4B), and enriched in pericyte markers (PDGFR-β, CALD1, COL4A2, NOTCH3). FB2-associated pathways included RhoA, NOTCH, and integrin-linked kinase signaling. In high ECM, FB2 pathways were associated with actin filament-based process, focal adhesion, and positive regulation of cell motility and adhesion.
Independent of cell proportions, FB1–2 were more transcriptionally active in high ECM compared to low ECM, as indicated by unique DEGs and global expression levels; FBs in normal allografts were significantly less transcriptionally active than in fibrotic grafts.
Lineage mapping analysis, as previously described,31 determined FB reprogramming trajectories across the normal and fibrosis states. FBs in normal grafts were homogenous along pseudotime trajectories whereas FBs in fibrotic grafts were heterogeneous (Fig. 4E). This data supports a FB functional phenotypic transition in fibrotic grafts.
The immune landscape of the failing kidney
A total of 2,655 immune cells were analyzed and 12 immune subclusters were identified amongst the studied groups (Figs. 5A–B, S9A–B). Normal allografts presented the fewest immune cells (Figs. 5A, S9B). Low ECM had the highest myeloid cell proportions containing diverse macrophages (MΦ1, MΦ2), classical and plasmacytoid dendritic cells (cDC, pDC), and mast cells. High ECM had the highest proportions of B, plasma, T, and Treg cells (Figs. 5A–C, S9B). The relative proportion and spatial distribution of immune cells (T (CD4+, CD8+), B (CD20+), and MΦ (CD68+)) were evaluated using IMC staining32 from formalin-fixed paraffin embedded biopsies (Fig. 5D). The panel also consisted of normal kidney architectural markers (endothelial (CD31+), tubular (AQP1+)) to validate spatial morphological cells for each group. For high ECM, the combined panel showed increased abundance of T and B cells, located in close proximity to the endothelium and to each other.
Fig. 5. Immune cells in the human kidney grafts.
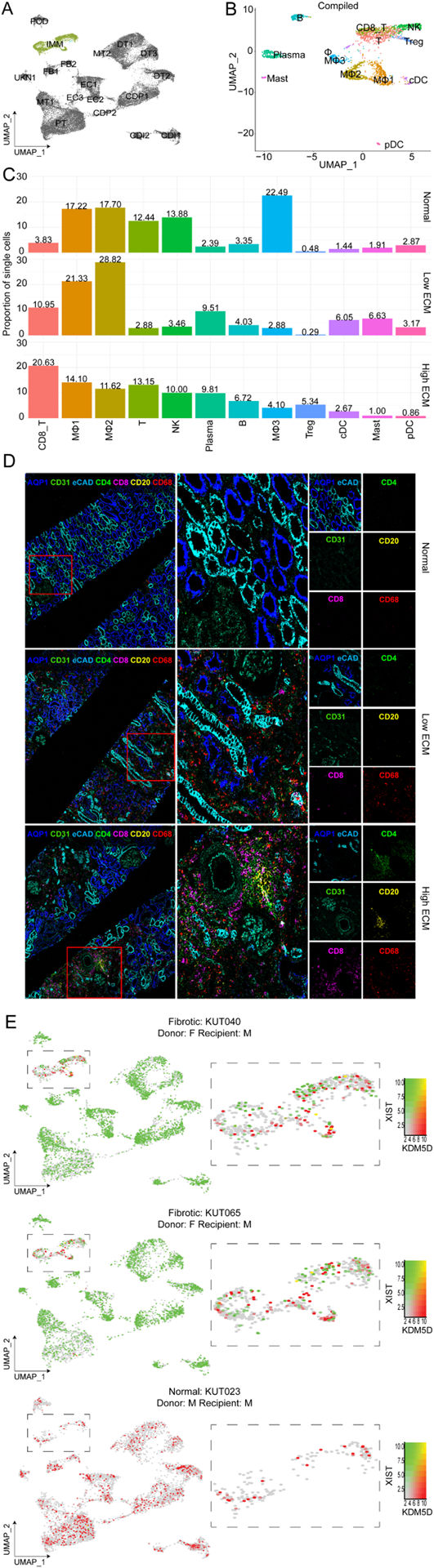
(A) UMAP visualization of the immune cell cluster, highlighted in green. (B) Compiled immune cell subclustering of over 2,000 cells integrated into a single dataset and by each classification. B, B cells; CD8 T, CD8+ T cells, cDC, conventional dendritic cells; Mast, mast cells; MΦ1, macrophage subcluster 1; MΦ2, macrophage subcluster 2; MΦ3, macrophage subcluster 3; NK, natural killer T cells; pDC; plasmacytoid dendritic cells; Plasma, plasma cells; T, T cells; and Tregs, regulatory T cells. (C) Proportion of single immune cells. Cell clusters are represented with the same colors as in A. (D) Imaging mass cytometry (IMC) staining confirmed immune cell presence and localization in matched kidney biopsies. The image shows expression of AQP1 (proximal tubules), CD31 (endothelial cells), CD4+ (T cells), CD8+ (T cells), CD20 (B cells), and CD68 (macrophages). Red boxes denoted by a 1 or 2 is enlarged to the right of the image to show spatial resolution of the glomerulus. The last panels (far right) are enlarged to detail kidney architecture (AQP1, CD31, eCAD), CD4, CD8, CD29, CD68 expression. (E) UMAPs of XY chromosome linked gene expression analysis of immune cells in sex-matched and -mismatched kidney transplants. Red, KDM5D expression (Y chromosome linked gene); and green, XIST expression (X chromosome linked gene). F, female; and M, male.
The T cell cluster in high ECM was enriched in DEGs associated with B and T cell activation, NF-kB signaling, TCR signaling, and Th17 cell differentiation, which matched its increased activity enriched in the MT1 cluster (Fig. 2E). The B cell cluster (enriched in B memory cells) (Table S7) pathway-associated genes included regulation of B cell activation, leukocyte chemotaxis, endocytosis, and NF-kB and FC receptor-mediated stimulatory signaling pathways. Plasma cells in high ECM had an active transcriptional profile although the biopsies lacked histological evidence for antibody-mediated rejection (Table S7).
Three macrophage clusters (MΦ1–3) were identified (Figs. 5BC, S10A–D) and characterized by common macrophage markers (CSF1R, MANBA, PLXDC2, RTN1, GRK3). MΦ1s were categorized by upregulation of antigen processing and presentation, HLA class II genes, and Th1/Th2/Th17 cell differentiation (Fig. S10B, Table S7). MΦ2s (characterized by STAB1, F13A1, CD163, NRP1) showed MHC class II-mediated antigen presentation and macrophage M2-related phagocytosis (Fig. S10C, Table S7). MΦ1–2 also overexpressed CD74 in high ECM kidney grafts, recently described together with C1QA, CD81, and APOE as a potential marker of resident macrophages across species33 (Fig. S10A, Table S7).
Low ECM had the largest MΦ1 and MΦ2 populations (21.33%, 28.82%) when compared to high ECM (14.10%, 11.62%) and normal (17.22%, 17.70%), respectively (Fig. 5C). Notably, in both fibrosis states, MΦ2 expression profiles were associated with cell adhesion and endocytosis regulation, whereas IFN-α and-γ responses were unique to high ECM. MΦ3 (characterized by CD86, FCN1, CSFR1) was identified as phagocytic macrophages (Table S7).
cDC and pDC were predominant in low ECM (6.05%, 3.17%) compared to high ECM (2.67%, 0.86%), respectively (Fig. 5C). cDCs and pDCs were distinguished by their selective cell marker expression (e.g., BTLA, ITGAX, NRP1, IRF7) (Fig. S9D). Importantly, donor vs recipient immune cells contribution to the allograft was evaluated using XY chromosome linked gene expression signatures for sex-mismatched cases across the three groups (Table 1A). Immune cells were mainly from the recipient with a small proportion of immune cells from the donor (Figs. 5E). There was no evidence of recipient non-immune cells. A sex-matched case (KUT100) was evaluated showing specificity of the signature. For some samples, we categorized immune cell populations into donor/recipient specific immune cells based on the expression of sex-specific genes (Fig. S11). Donor immune cells were identified in the kidney graft up to 60-months posttransplant (KUT088) (Fig. S11).
Evaluation of DEGs between donor and recipient-labelled immune cells in a particular cell type within sample KUT040 was done (Fig. S12). This proof-of-principle analyses showed the differential contribution of immune cells based on donor/recipient origin, supporting an important role of resident macrophages in mediating, and exacerbating immune-mediated inflammation in kidney grafts.
Paracrine signaling during fibrosis
Ligand-receptor (LR) analyses focused on clusters with significant differences in cell proportions and strong implications in kidney fibrogenesis. Figs. 6A–B depict signaling interactions between macrophages and fibroblasts in low and high ECM, respectively. Their fibroblast receptors shared functional roles including fibroblast activation and ECM expansion evidenced by several interactions with collagens, fibronectins, and integrins. PDGFC, a potent profibrotic mitogen,34 was secreted in fibrotic grafts, supporting the fibroblast-to-myofibroblast transdifferentiation.35 Upregulation of NOTCH signaling pathways was also shared. Toll-like receptors (TLRs) triggered NOTCH signaling in macrophages via DLL1, a main NOTCH ligand36 secreted in high ECM. For low ECM, FB1-MΦ3 LRs were also involved in PI3K-Akt-mTOR-signaling.
Fig. 6. Cell-cell interactions and stained tissue spatial validation.
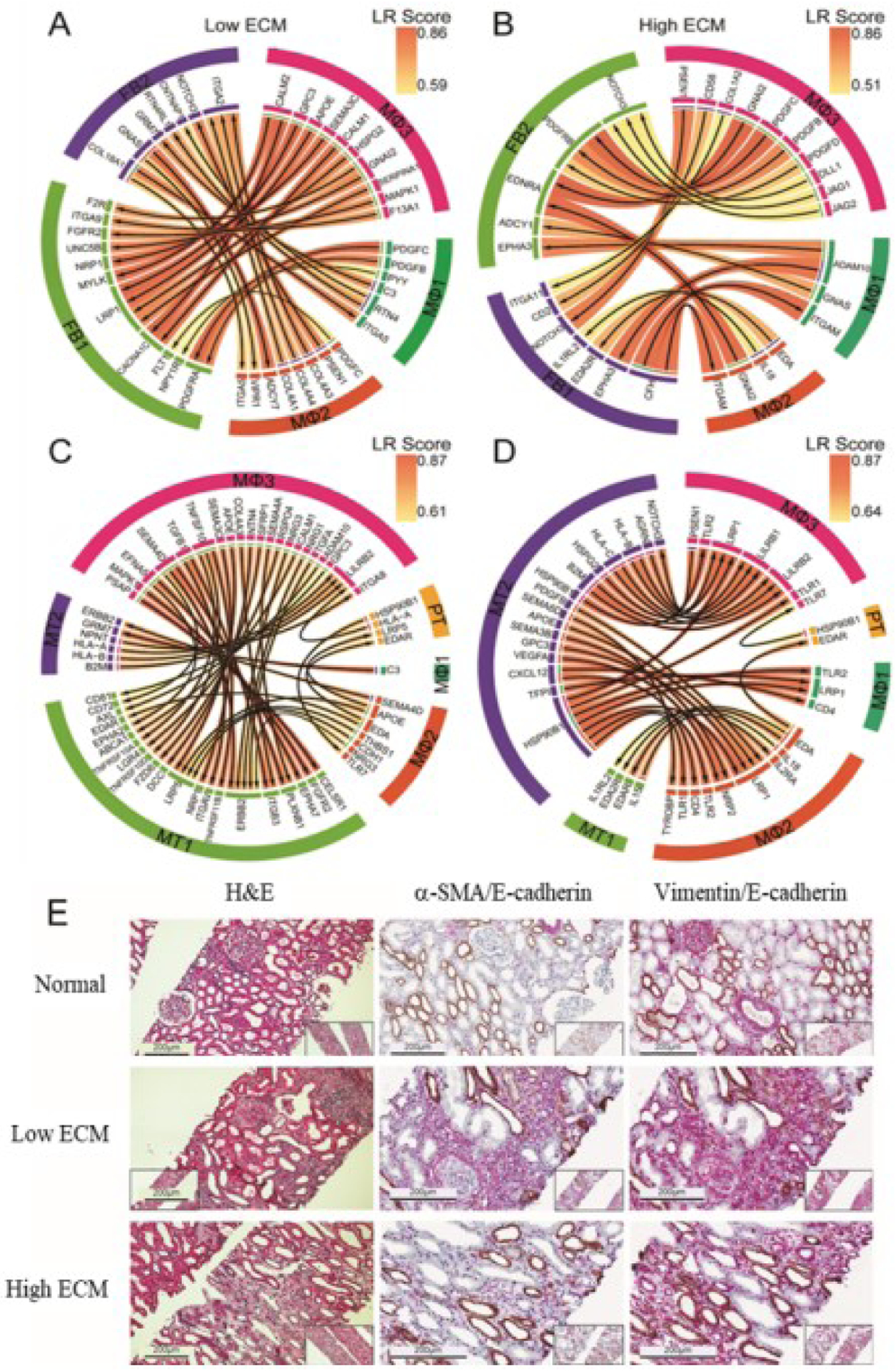
(A-B) Cell-cell interactions and stained tissue spatial validation. (A-B) Ligand receptor (LR) interactions between tissue macrophages (MΦ1–3) and fibroblasts (FB1–2) subclusters. (C-D) LR interactions between tissue macrophages (MΦ1–3) and tubular epithelial cells (PT, MT1, and MT2). Arrows point towards receptors; left panels: Low ECM and right panels: high ECM. (E) Representative images of H&E staining and immunohistochemistry staining of normal (KUT060), low ECM (KUT040), and high ECM (KUT076). Histopathological abnormalities in low and high ECM states are shown. Scale bars, 200μm.
Figs. 6C–D depict signaling interactions between macrophages and tubulars in low and high ECM. For low ECM, MT1-MΦ3 and MT1-MΦ1 LRs were involved in chemotaxis and tubular/tissue morphogenesis. Interactions like CELSR1-PSAP, a regulator of cell polarity,37 and FGFR2-MAPK1, a regulator of fibroblast proliferation and activation,38 further supported EMT in low ECM. In high ECM, MT1-MΦ1 (and FB1-MΦ1) interactions reflected cytokine-mediated and NIK/NF-kB signaling pathways, in concordance with the increased immune cell abundance and activation.
In high ECM, MT2-MΦ3 LRs stressed interactions between immune and non-immune cells. HSP90B1-LRP1 and HSP90-TLR1 were shown to stimulate inflammation and apoptosis.39 HSP90, a functioning damage-associated molecular pattern (DAMP),40 also interacted with TLR2 and TLR7, both expressed by MΦ3. This signaling cascade is predicted to mediate tissue repair.40
H&E and immunohistochemistry (IHC) staining further corroborates LR interactions with infiltrating cells and damaged kidney architecture compared to normal (Fig. 6E). Low ECM kidney parenchyma displayed patches of fibrosis whereas high ECM showed more compact areas of fibrosis. E-CADHERIN expression marked abnormal and atrophying tubulars in fibrotic tissues. αSMA marked a higher proportion of myofibroblasts in high ECM relative to low ECM. Interstitial presence and relative abundance of myofibroblasts plays a key role in fibrosis pathogenesis—supported by our molecular findings and IMC evaluations (Fig. 5D).
DISCUSSION
The pathophysiology of fibrosis in the kidney graft is characterized by multidimensional regulation of complex networks influenced by various factors unique to the human transplant model, such as the alloimmune response and kidney parenchymal toxicity, resulting in impaired reparation.
Our study identified high heterogeneity in the composition of fibrosis, diverse cell types, cell-cell interactions, and differential transcriptional profiles, providing a deeper insight into deleterious processes leading to graft loss. We propose a model of fibrogenesis that integrates our experimental results (Fig. 7).
Fig. 7. Molecular and cellular landscape of human kidney graft fibrogenesis.
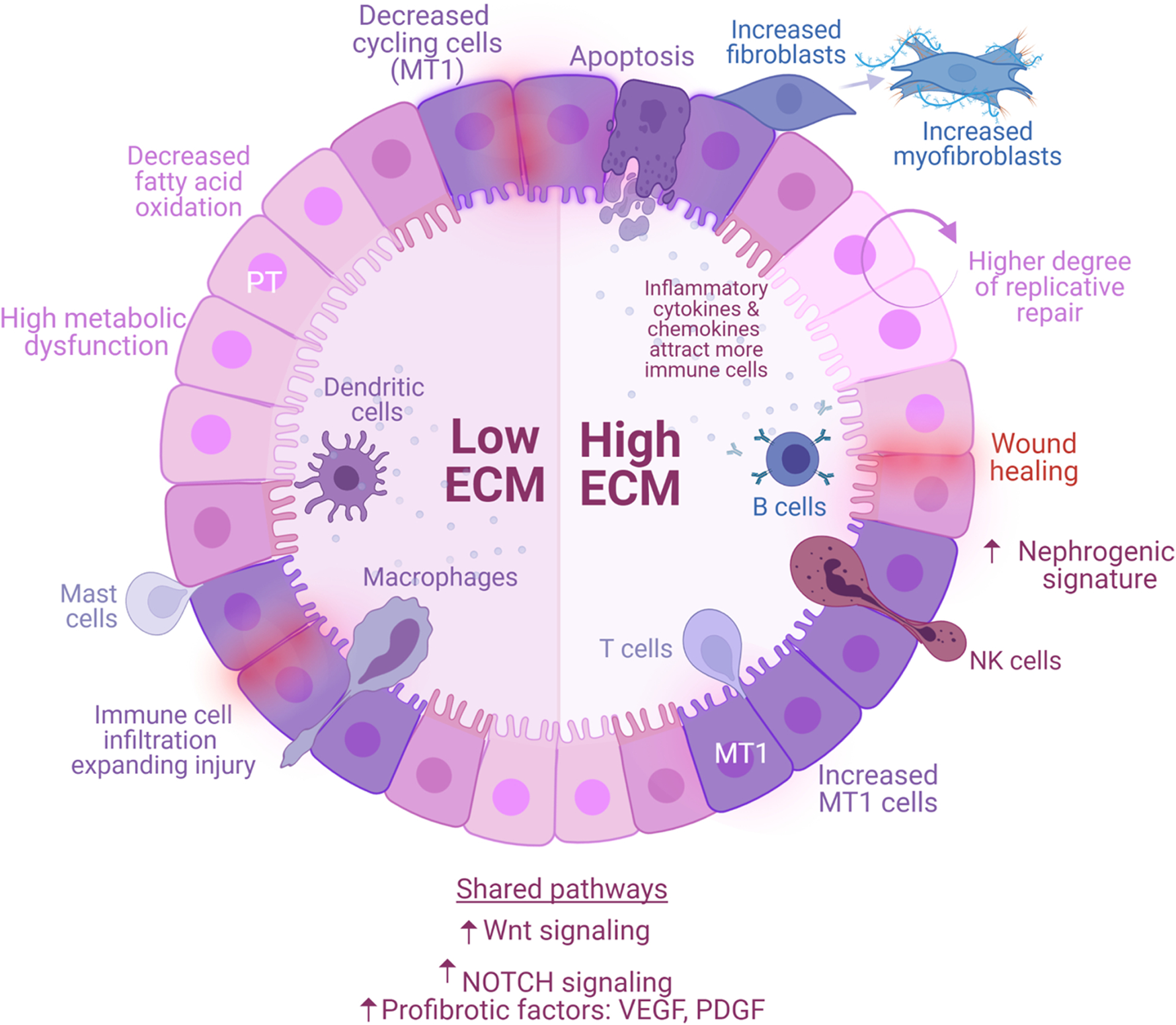
Proximal tubular (PT) epithelial cells are the first responders to kidney insults, such as ischemia reperfusion injury (early stressors) or sustained subclinical injury (late stressors). Healthy PT cells may resolve the injury and stabilize graft function (normal allografts), while injured PT cells (MT1) generate a provisional ECM that attracts inflammatory cells, facilitating their infiltration and propagating injury (fibrotic allografts). Fibrotic signaling is enhanced by secretion of growth factors (PDGF, VEGF) and upregulation of signaling pathways (NOTCH, Wnt). The processes are heterogeneous and lead to different molecular states—low ECM and high ECM. Low ECM is characterized by a decreased of cycling cells, severe PT metabolic dysfunction, and decreased apoptosis. High ECM activates maladaptive replicative repair and enters a perpetual state of tissue regeneration, scarring, and ECM accumulation. These two conditions are characterized by different proportions of immune cells. Low ECM is marked by increased dendritic (cDCs, pDCs), mast, and macrophages (MΦ1, MΦ2) cells whereas high ECM is marked by increased B, T (CD8+ T cells, NKs, and Tregs), and plasma cells. Resident macrophages and kidney cells interactions demonstrate the critical role of these cells and their cross talk in the propagation of injury. Low and high ECM are distinguishable by the secretion of different cytokines and ECM-related factors. In response to injury, MT1 cells transition into transcriptionally active fibroblasts enriched in myofibroblast markers. MT1 in high ECM was characterized by dedifferentiation and nephrogenic signatures, supporting a high degree of replicative repair, that was not observed in low ECM. Although both conditions progress to fibrosis, the severity of metabolic dysfunction of PT cells in low ECM limits repair, whereas the degree of immune cell activation in high ECM promotes a positive feedback loop inducing maladaptive repair. Interventions aimed at graft fibrogenesis likely require a more targeted approach based on the unique molecular pathways characterizing each condition.
This study has several unique features including: i) the inclusion of surveillance allograft biopsies obtained at similar posttransplant times (>15-months post-transplantation), ii) samples obtained from functioning grafts, and iii) patients on similar immunosuppression regimens. Previous reports did not include normal allografts as a control group20,21,25 and, consequently, did not illustrate normal kidney graft biology in an alloimmune environment.
Our unsupervised analysis identified 18 unique clusters with well-represented kidney and immune cell populations. Based on the qualitative and quantitative molecular differences in ECM production and immune cell types and associated transcriptomics, two sub-states of Banff histological IFTA categories were revealed. The two different fibrotic states were further validated by showing higher ECM-related protein expression and immune cells in high ECM compared to low ECM using IMC.
In response to injury, provisional ECM (cross-linked fibrin, fibronectin, fibrinogen, proteoglycans) serves as a migration scaffold for inflammatory, fibroblastic, and vascular cells to enter and repopulate sites of injury.41 We aimed to discern the main cellular subtypes affected by provisional ECM deposition, propagating further injury.
PT cells play a central role in sensing injury and mediating a response,42 but their role in kidney graft fibrogenesis has not been fully explored. Our results demonstrate that PT cells in the kidney graft are responsible for initiating the reparation process in response to injury. One critical characteristic of normal tissue repair is controlled apoptosis of inflammatory cells and myofibroblasts that attenuates and/or terminates the healing process.43 This was reflected in normal allografts, where PT cells displayed some level of injury, apoptotic markers, and fibroblasts with low transcriptional activity. Moreover, primary biological PT functions were also preserved in normal grafts. Conversely, PT cells in low ECM were significantly compromised due to metabolic dysfunction and a slight decrease in cycling tubular cells. These cells had decreased expression of G2/M regulators, leading to inhibition of apoptosis and necrosis.
Healthy PT cells rely primarily on FAO as their energy source.44 Our data showed fibrotic allografts with significantly downregulated FAO, which contributes to IFTA development in CKD.44,45 These findings support that PT cells undergo a PT “renal hibernation”42 or reduction in energy metabolism, which was not observed in normal kidney allografts. Accordingly, the low ECM state with altered metabolism is in prolonged renal hibernation whereas high ECM can exit hibernation supported by our epithelial cell proliferation data.
The decline of PT cells in high ECM is likely a consequence of a phenotypic transition to a fibroblast-like state. A newly identified MT1 cell cluster (injured tubular cell intermediate between PT and FB cells) was increased in fibrotic grafts. These transitional cells have not been reported in chronic kidney conditions. MT1 expressed profibrotic growth factors (VEGF, PDGF) and injury markers (HAVCR1, VCAM1) as reported in acute kidney injury.21,25–27,46,47 Although cellular senescence was enriched in both states, a nephrogenic signature was observed in the high ECM MT1 cluster, supporting regeneration of PT cells.
Pseudotime trajectory analyses showed dynamic transcriptional transitions from PT to MT1 to FB. These cells expressed collagens, integrins, PDGFR, and other growth factors perpetuating MT1 transformation, immune cell recruitment, and active production of ECM. Continuous expression of these signals resulted in a positive feedback loop of fibroblast activation. These fibroblasts expressing PDGFR-α/-β may give rise to a myofibroblast lineage as demonstrated by high expression of αSMA.17 The final FB1 cluster, unique to fibrosis states, was enriched in myofibroblasts markers (COL1A1, FN1, VCAN, PDGFR-α/-β).
Through paracrine signaling, PTs can communicate damage by recruiting immune cells to the injury site. An activated inflammatory response was observed in both fibrosis states but not in normal allografts. Critically, low and high ECM states were characterized by distinct immune cell landscapes. Myeloid-derived immune cells were increased in low ECM while B, T, and plasma cells were increased in high ECM. However, immune cells were transcriptionally more active in high ECM.
PT and MT1 cells in high ECM were characterized by their genes associated with transendothelial cell migration and T cell activation. B memory and B cells (also acting as antigen presenting cells) propagate and heighten these responses. B memory cells are characterized by production of inflammatory cytokines and chemokines that attract more macrophages, NK, and inflammatory cells48,49 as observed in the high ECM state. Likely, upregulation of Th17 cells (evidenced by pathway analysis) supported B cell expansion. Antigen-driven activation of B memory cells results in their rapid proliferation and differentiation into plasma cells that produce large amounts of higher-affinity antibodies,48,49 explaining the higher population of plasma cells in high ECM.
Differential immune infiltrates associated with kidney graft fibrogenesis deserve further evaluation. Using single-cell analysis, tissue-resident macrophages are abundant in healthy kidneys.50 We report a similar observation in our MΦ3 cluster where tissue-resident macrophages were dominant in normal compared to fibrotic grafts. Tregs assist in repair and prevent inflammation by modulating macrophage phenotype and function.51–54 Our data complements this finding by showing that the nephrogenic, high ECM state have a 5-fold increase in Tregs relative to the low ECM state. B cell activity is strongly associated with maladaptive kidney repair and immune-mediated injury hypothesized to drive the adaptive immune response.55 Overall, our data extends these published findings to chronic injury models in humans not yet reported.
Interestingly, a study evaluating human kidney allograft samples using bulk gene expression-based approaches demonstrated that IFTA biopsies without alternative explanations for pathogenesis (like the samples included in our study) showed differential gene expression evidence of ongoing cellular immune-mediated injury.56 Moreover, an IFTA group without histological evidence of inflammation showed a molecular profile consistent with immune-mediated inflammation largely overlapping with gene expression findings in acute rejection biopsies,56 similar to our high ECM samples. The resolution of sn/scRNA-seq to individual cell types improve our understanding of the cell-specific pathways of injuries and provide for better disease subclassification.
Donor-derived immune cells are limited and decreased over time after transplantation.55,57 However, the analysis of sex-mismatched donor/recipient pairs detected donor resident immune cells present in allograft tissue (up to 60-months post-transplantation), demonstrating long-term immune cell chimerism. To our knowledge, immune cell chimerism in human kidney allografts—several years after transplantation—has not been reported. LR analyses showed that these resident macrophages were strongly associated with a proinflammatory environment, likely activating myofibroblasts and perpetuating injury and/or impaired reparation. Furthermore, non-immune cells were predominantly donor derived. This is in contrast with a previous study by Suryawanshi et al., which identified recipient-derived fibroblasts.58
A major factor driving kidney injury vs tissue restoration is the activation state of macrophages within local tissues rather than the degree of monocyte infiltration.59–62 MΦ1–2 were more transcriptionally active in the high ECM tissues although present in lower proportions. MΦ1 was characterized by overexpression of genes regulating B cell proliferation, lymphocyte activation, and IL2-production. These findings also validated increased leukocyte transendothelial migration in the high ECM state.
Low ECM activated a less regulated immune response via expression of ABCA1, ADAM10, AXL, CALM1/2, ERBB2, F2R, GNAI2, HSPG2, LGR4, LRP1/5, MAPK1, and TNFSF10 based on the Innatedb.63 Also, sustained injury precluded tubular regeneration and activated macrophages responded to local tissue injury, inducing a hostile environment and an overall failed repair state.64
A limitation of our study is its cross-sectional design. Despite careful matching of normal and fibrosis states, the cross-sectional design precludes longitudinal evaluation of dynamic mechanisms leading to IFTA. Future studies are needed to examine the sequential events that associate with onset, propagation, and resolution of injury.
The best practices for sample size determination relies on the hypothesis and number of cells.65 Our study of normal vs. IFTA kidney grafts profiled 41,893 cells, which was sufficient to capture transcriptional heterogeneity, subcluster populations, and generated an average of 28,057 unique genes derived from human biopsies. However, as all the single cell studies using human clinical biopsies,16,58,66 our study is limited by the small sample size, and therefore, results cannot be generalized without further additional evaluations.
New methods and tools will be necessary to scale up single-cell analysis for applications that require higher throughput, including in-depth clinical characterization. The optimal approach will be not only scalable and affordable but also efficient to equally capture all the cells contained in the original sample.
In conclusion, this is the first study describing the cellular and molecular heterogeneity of human graft fibrosis at single nuclei resolution. Critically, our findings underscore the molecular immunological heterogeneity observed in allograft fibrosis despite being on a similar immunosuppression regimen. Interventions aimed at ameliorating allograft fibrosis require a more targeted approach based on the unique molecular pathways characterizing each state. Such findings emphasize the need for complementary molecular approaches to the standard transplant pathology, allowing for better patient stratification and personalized CAD treatment.
Supplementary Material
Table S1. Summary of NovaSeq results.
Table S2. Summary of Q30 scores.
Table S3. Donor and recipient HLA typing for patients included in the study.
Table S4. Counts of isolated cells in each cell cluster.
Table S5. Fibrosis MT1 shared cell type markers.
Table S6. Functional enrichment analysis of low and high ECM MT1 upregulated genes compared to PT.
Table S7. Immune cell subset gene expression signatures.
Fig. S1. Isolation of nuclei from kidney allograft biopsies stored in RNAlater and validation of cDNA libraries prior to snRNA-seq.
Fig. S2. Sequencing quality of allografts stored in RNAlater.
Fig. S3. Quality control assessment of RNAlater-stored allografts to and frozen allografts.
Fig. S4. UMAP visualization of each individual biopsy sample and cell cluster identity.
Fig. S5. Distinct cellular and molecular profile of fibrotic grafts.
Fig. S6. Enrichment pathway analysis and volcano plots of fibrotic human grafts.
Fig. S7. Pathway enrichment analyses for proximal tubular cells.
Fig. S8. Cell cycle analysis of tubular cells.
Fig. S9. Immune cell subcluster analysis.
Fig. S10. Macrophage subcluster analyses.
Fig. S11. XY chromosome linked gene expression analysis of immune and nonimmune subclusters in sex-mismatched kidney transplants.
TRANSLATIONAL STATEMENT:
After the first year of transplantation, approximately 4–5% of kidneys are lost due to chronic allograft dysfunction, characterized by interstitial fibrosis and tubular atrophy (IFTA). Preventing kidney graft dysfunction and loss is a critical unmet need. In this study, single nuclei RNA-seq identified genes and molecular pathways that were differentially expressed between normal and IFTA clinical biopsies. We provide a web-accessible database that delineated cellular pathways driving beneficial or deleterious tissue repair after chronic injury. These novel fibrosis-driving pathways, cell types, and cell-cell interactions can be leveraged to design new individualized therapeutics to avoid IFTA and graft dysfunction. 99/100
ACKNOWLEDGEMENTS:
We would also like to thank members of the oncology tissue service core at the Johns Hopkins University for their assistance with the immunostaining assays. We would like to acknowledge members of the Jonathan Bromberg, Alexander Krupnick, and Christine Lau labs for their invaluable input and advice on the study design and critical evaluation of this study. We would also like to thank the members of the Institute for Genome Sciences at the University of Maryland, Baltimore for their invaluable contribution and assistance using the 10x Genomics Chromium.
FUNDING:
National Institute of Diabetes and Digestive and Kidney Diseases grant R21DK100678 (DM)
National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK080074 (VRM)
National Institute of Diabetes and Digestive and Kidney Diseases grant 3R01DK122682 (VRM)
National Institutes of Health grant R01DK109581 (VRM)
National Institutes of Health grant R01DK122682 (VRM)
DISCLOSURE:
All the authors declared no competing interests. The research reported in this publication is supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health under awards numbers: R21DK100678 (DGM), R01DK109581 (VRM), and R01DK122682 (VRM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTARY MATERIAL:
Supplemental methods and materials.
Supplemental results. Quality control metrics performed on RNAlater kidney biopsies.
Supplemental references.
Supplementary information is available on Kidney International’s website.
DATA AVAILABILITY:
The data that supports the findings of this study will be publicly available (GEO ID: GSE195718).
REFERENCES:
- 1.Srivastava A, Palsson R, Kaze AD, et al. The Prognostic Value of Histopathologic Lesions in Native Kidney Biopsy Specimens: Results from the Boston Kidney Biopsy Cohort Study. J Am Soc Nephrol. 2018;29(8):2213–2224. doi: 10.1681/ASN.2017121260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nankivell BJ, Borrows RJ, Fung CLS, et al. The natural history of chronic allograft nephropathy. N Engl J Med. 2003;349(24):2326–2333. doi: 10.1056/NEJMoa020009 [DOI] [PubMed] [Google Scholar]
- 3.Najafian B, Kasiske BL. Chronic allograft nephropathy. Curr Opin Nephrol Hypertens. 2008;17(2):149–155. doi: 10.1097/MNH.0b013e3282f4e514 [DOI] [PubMed] [Google Scholar]
- 4.El-Zoghby ZM, Stegall MD, Lager DJ, et al. Identifying specific causes of kidney allograft loss. Am J Transplant. 2009;9(3):527–535. doi: 10.1111/j.1600-6143.2008.02519.x [DOI] [PubMed] [Google Scholar]
- 5.Nicholson ML, McCulloch TA, Harper SJ, et al. Early measurement of interstitial fibrosis predicts long-term renal function and graft survival in renal transplantation. Br J Surg. 1996;83(8):1082–1085. doi: 10.1002/bjs.1800830813 [DOI] [PubMed] [Google Scholar]
- 6.Khan H, Mubarak M, Aziz T, et al. Prevalence and risk factors for early chronic allograft nephropathy in a live related renal transplant program. J Nephropathol. 2014;3(2):69–79. doi: 10.12860/jnp.2014.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreso F, Lopez M, Vallejos A, et al. Serial protocol biopsies to quantify the progression of chronic transplant nephropathy in stable renal allografts. Am J Transplant. 2001;1(1):82–88. doi: 10.1034/j.1600-6143.2001.010115.x [DOI] [PubMed] [Google Scholar]
- 8.Boor P, Floege J. Renal allograft fibrosis: biology and therapeutic targets. Am J Transplant. 2015;15(4):863–886. doi: 10.1111/ajt.13180 [DOI] [PubMed] [Google Scholar]
- 9.Ishida H, Ogura G, Uehara S, et al. Preventive effect of early introduction of everolimus and reduced-exposure tacrolimus on renal interstitial fibrosis in de novo living-donor renal transplant recipients. Clin Exp Nephrol. 2020;24(3):268–276. doi: 10.1007/s10157-019-01822-6 [DOI] [PubMed] [Google Scholar]
- 10.Naesens M, Khatri P, Li L, et al. Progressive histological damage in renal allografts is associated with expression of innate and adaptive immunity genes. Kidney Int. 2011;80(12):1364–1376. doi: 10.1038/ki.2011.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lv W, Booz GW, Wang Y, et al. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur J Pharmacol. 2018;820:65–76. doi: 10.1016/j.ejphar.2017.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mengel M, Loupy A, Haas M, et al. Banff 2019 Meeting Report: Molecular diagnostics in solid organ transplantation-Consensus for the Banff Human Organ Transplant (B-HOT) gene panel and open source multicenter validation. Am J Transplant. 2020;20(9):2305–2317. doi: 10.1111/ajt.16059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin Q, Liu H. Connective Tissue Growth Factor and Renal Fibrosis. Adv Exp Med Biol. 2019;1165:365–380. doi: 10.1007/978-981-13-8871-2_17 [DOI] [PubMed] [Google Scholar]
- 14.Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012;5(1):S24. doi: 10.1186/1755-1536-5-S1-S24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halloran PF, Famulski KS, Reeve J. Molecular assessment of disease states in kidney transplant biopsy samples. Nat Rev Nephrol. 2016;12(9):534–548. doi: 10.1038/nrneph.2016.85 [DOI] [PubMed] [Google Scholar]
- 16.Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. JASN. 2019;30(1):23–32. doi: 10.1681/ASN.2018090912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuppe C, Ibrahim MM, Kranz J, et al. Decoding myofibroblast origins in human kidney fibrosis. Nature. 2021;589(7841):281–286. doi: 10.1038/s41586-020-2941-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Clark JZ, Nelson JW, et al. Renal-Tubule Epithelial Cell Nomenclature for Single-Cell RNA-Sequencing Studies. J Am Soc Nephrol. 2019;30(8):1358–1364. doi: 10.1681/ASN.2019040415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rousselle TV, McDaniels JM, Shetty AC, et al. An optimized protocol for single nuclei isolation from clinical biopsies for RNA-seq. Sci Rep. 2022;12(1):9851. doi: 10.1038/s41598-022-14099-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson PC, Wu H, Kirita Y, et al. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci U S A. 2019;116(39):19619–19625. doi: 10.1073/pnas.1908706116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muto Y, Wilson PC, Ledru N, et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat Commun. 2021;12(1):2190. doi: 10.1038/s41467-021-22368-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loupy A, Haas M, Roufosse C, et al. The Banff 2019 Kidney Meeting Report (I): Updates on and clarification of criteria for T cell- and antibody-mediated rejection. Am J Transplant. 2020;20(9):2318–2331. doi: 10.1111/ajt.15898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Racusen LC, Solez K, Colvin RB, et al. The Banff 97 working classification of renal allograft pathology. Kidney Int. 1999;55(2):713–723. doi: 10.1046/j.1523-1755.1999.00299.x [DOI] [PubMed] [Google Scholar]
- 24.Naba A, Clauser KR, Hoersch S, et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012;11(4):M111.014647. doi: 10.1074/mcp.M111.014647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerhardt LMS, Liu J, Koppitch K, et al. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc Natl Acad Sci U S A. 2021;118(27):e2026684118. doi: 10.1073/pnas.2026684118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudman-Melnick V, Adam M, Potter A, et al. Single-Cell Profiling of AKI in a Murine Model Reveals Novel Transcriptional Signatures, Profibrotic Phenotype, and Epithelial-to-Stromal Crosstalk. J Am Soc Nephrol. 2020;31(12):2793–2814. doi: 10.1681/ASN.2020010052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirita Y, Wu H, Uchimura K, et al. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci U S A. 2020;117(27):15874–15883. doi: 10.1073/pnas.2005477117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonventre JV. Primary proximal tubule injury leads to epithelial cell cycle arrest, fibrosis, vascular rarefaction, and glomerulosclerosis. Kidney Int Suppl (2011). 2014;4(1):39–44. doi: 10.1038/kisup.2014.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535–543, 1p following 143. doi: 10.1038/nm.2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Humphreys BD, Czerniak S, DiRocco DP, et al. Repair of injured proximal tubule does not involve specialized progenitors. Proc Natl Acad Sci U S A. 2011;108(22):9226–9231. doi: 10.1073/pnas.1100629108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao J, Spielmann M, Qiu X, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566(7745):496–502. doi: 10.1038/s41586-019-0969-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kakade VR, Weiss M, Cantley LG. Using Imaging Mass Cytometry to Define Cell Identities and Interactions in Human Tissues. Front Physiol. 2021;12:817181. doi: 10.3389/fphys.2021.817181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zimmerman KA, Bentley MR, Lever JM, et al. Single-Cell RNA Sequencing Identifies Candidate Renal Resident Macrophage Gene Expression Signatures across Species. J Am Soc Nephrol. 2019;30(5):767–781. doi: 10.1681/ASN.2018090931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eitner F, Bücher E, van Roeyen C, et al. PDGF-C is a proinflammatory cytokine that mediates renal interstitial fibrosis. J Am Soc Nephrol. 2008;19(2):281–289. doi: 10.1681/ASN.2007030290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin L, Hu K. LRP-1: Functions, Signaling and Implications in Kidney and Other Diseases. Int J Mol Sci. 2014;15(12):22887–22901. doi: 10.3390/ijms151222887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.López-López S, Monsalve EM, Romero de Ávila MJ, et al. NOTCH3 signaling is essential for NF-κB activation in TLR-activated macrophages. Sci Rep. 2020;10(1):14839. doi: 10.1038/s41598-020-71810-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brzóska HŁ, d’Esposito AM, Kolatsi-Joannou M, et al. Planar cell polarity genes Celsr1 and Vangl2 are necessary for kidney growth, differentiation, and rostrocaudal patterning. Kidney Int. 2016;90(6):1274–1284. doi: 10.1016/j.kint.2016.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Z, Dai C. Ablation of FGFR2 in Fibroblasts Ameliorates Kidney Fibrosis after Ischemia/Reperfusion Injury in Mice. Kidney Dis (Basel). 2017;3(4):160–170. doi: 10.1159/000484604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoter A, El-Sabban ME, Naim HY. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int J Mol Sci. 2018;19(9):E2560. doi: 10.3390/ijms19092560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010:672395. doi: 10.1155/2010/672395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barker TH, Engler AJ. The provisional matrix: setting the stage for tissue repair outcomes. Matrix Biol. 2017;60–61: 1–4. doi: 10.1016/j.matbio.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chevalier RL. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol. 2016;311(1):F145–161. doi: 10.1152/ajprenal.00164.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hinz B, Lagares D. Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol. 2020;16(1):11–31. doi: 10.1038/s41584-019-0324-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Console L, Scalise M, Giangregorio N, et al. The Link Between the Mitochondrial Fatty Acid Oxidation Derangement and Kidney Injury. Front Physiol. 2020;11:794. doi: 10.3389/fphys.2020.00794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21(1):37–46. doi: 10.1038/nm.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu YA, Liao CT, Raybould R, et al. Single-Nucleus RNA Sequencing Identifies New Classes of Proximal Tubular Epithelial Cells in Kidney Fibrosis. J Am Soc Nephrol. 2021;32(10):2501–2516. doi: 10.1681/ASN.2020081143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu H, Malone AF, Donnelly EL, et al. Single-Cell Transcriptomics of a Human Kidney Allograft Biopsy Specimen Defines a Diverse Inflammatory Response. JASN. 2018;29(8):2069–2080. doi: 10.1681/ASN.2018020125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vazquez MI, Catalan-Dibene J, Zlotnik A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine. 2015;74(2):318–326. doi: 10.1016/j.cyto.2015.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oleinika K, Mauri C, Salama AD. Effector and regulatory B cells in immune-mediated kidney disease. Nat Rev Nephrol. 2019;15(1):11–26. doi: 10.1038/s41581-018-0074-7 [DOI] [PubMed] [Google Scholar]
- 50.Bell RMB, Denby L. Myeloid Heterogeneity in Kidney Disease as Revealed through Single-Cell RNA Sequencing. Kidney360. 2021;2(11):1844–1851. doi: 10.34067/KID.0003682021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.do Valle Duraes F, Lafont A, Beibel M, et al. Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight. 2020;5(3):130651. doi: 10.1172/jci.insight.130651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuswanto W, Burzyn D, Panduro M, et al. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity. 2016;44(2):355–367. doi: 10.1016/j.immuni.2016.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panduro M, Benoist C, Mathis D. Treg cells limit IFN-γ production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc Natl Acad Sci U S A. 2018;115(11):E2585–E2593. doi: 10.1073/pnas.1800618115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Q, Dwyer GK, Zhao Y, et al. IL-33-mediated IL-13 secretion by ST2+ Tregs controls inflammation after lung injury. JCI Insight. 2019;4(6):123919. doi: 10.1172/jci.insight.123919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cippà PE, Liu J, Sun B, et al. A late B lymphocyte action in dysfunctional tissue repair following kidney injury and transplantation. Nat Commun. 2019;10(1):1157. doi: 10.1038/s41467-019-09092-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Modena BD, Kurian SM, Gaber LW, et al. Gene Expression in Biopsies of Acute Rejection and Interstitial Fibrosis/Tubular Atrophy Reveals Highly Shared Mechanisms That Correlate With Worse Long-Term Outcomes. Am J Transplant. 2016;16(7):1982–1998. doi: 10.1111/ajt.13728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malone AF, Wu H, Fronick C, et al. Harnessing expressed single nucleotide variation and single cell RNA sequencing to define immune cell chimerism in the rejecting kidney transplant. J Am Soc Nephrol. 2020;31(9):1977–1986. doi: 10.1681/ASN.2020030326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suryawanshi H, Yang H, Lubetzky M, et al. Detection of Infiltrating Fibroblasts by Single-Cell Transcriptomics in Human Kidney Allografts.; 2021:2020.09.03.281733. doi: 10.1101/2020.09.03.281733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han HI, Skvarca LB, Espiritu EB, et al. The role of macrophages during acute kidney injury: destruction and repair. Pediatr Nephrol. 2019;34(4):561–569. doi: 10.1007/s00467-017-3883-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guiteras R, Flaquer M, Cruzado JM. Macrophage in chronic kidney disease. Clin Kidney J. 2016;9(6):765–771. doi: 10.1093/ckj/sfw096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kluth DC, Erwig LP, Rees AJ. Multiple facets of macrophages in renal injury. Kidney Int. 2004;66(2):542–557. doi: 10.1111/j.1523-1755.2004.00773.x [DOI] [PubMed] [Google Scholar]
- 62.Meng XM, Tang PMK, Li J, Lan HY. Macrophage Phenotype in Kidney Injury and Repair. Kidney Dis (Basel). 2015;1(2):138–146. doi: 10.1159/000431214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Breuer K, Foroushani AK, Laird MR, et al. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013;41(Database issue):D1228–1233. doi: 10.1093/nar/gks1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duffield JS. Macrophages and immunologic inflammation of the kidney. Semin Nephrol. 2010;30(3):234–254. doi: 10.1016/j.semnephrol.2010.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goldman SL, MacKay M, Afshinnekoo E, et al. The Impact of Heterogeneity on Single-Cell Sequencing. Front Genet. 2019;10:8. doi: 10.3389/fgene.2019.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malone AF, Humphreys BD. Single Cell Transcriptomics and Solid Organ Transplantation. Transplantation. 2019;103(9):1776–1782. doi: 10.1097/TP.0000000000002725 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of NovaSeq results.
Table S2. Summary of Q30 scores.
Table S3. Donor and recipient HLA typing for patients included in the study.
Table S4. Counts of isolated cells in each cell cluster.
Table S5. Fibrosis MT1 shared cell type markers.
Table S6. Functional enrichment analysis of low and high ECM MT1 upregulated genes compared to PT.
Table S7. Immune cell subset gene expression signatures.
Fig. S1. Isolation of nuclei from kidney allograft biopsies stored in RNAlater and validation of cDNA libraries prior to snRNA-seq.
Fig. S2. Sequencing quality of allografts stored in RNAlater.
Fig. S3. Quality control assessment of RNAlater-stored allografts to and frozen allografts.
Fig. S4. UMAP visualization of each individual biopsy sample and cell cluster identity.
Fig. S5. Distinct cellular and molecular profile of fibrotic grafts.
Fig. S6. Enrichment pathway analysis and volcano plots of fibrotic human grafts.
Fig. S7. Pathway enrichment analyses for proximal tubular cells.
Fig. S8. Cell cycle analysis of tubular cells.
Fig. S9. Immune cell subcluster analysis.
Fig. S10. Macrophage subcluster analyses.
Fig. S11. XY chromosome linked gene expression analysis of immune and nonimmune subclusters in sex-mismatched kidney transplants.
Data Availability Statement
The data that supports the findings of this study will be publicly available (GEO ID: GSE195718).