

Abstract
Transcriptional profiling studies have identified several protective genes upregulated in tubular epithelial cells during acute kidney injury (AKI). Identifying upstream transcriptional regulators could lead to the development of therapeutic strategies augmenting the repair processes. SOX9 is a transcription factor controlling cell-fate during embryonic development and adult tissue homeostasis in multiple organs including the kidneys. SOX9 expression is low in adult kidneys, however, stress conditions can trigger its transcriptional upregulation in tubular epithelial cells. SOX9 plays a protective role during the early phase of AKI and facilitates repair during the recovery phase. To identify the upstream transcriptional regulators that drive SOX9 upregulation in tubular epithelial cells, we used an unbiased transcription factor screening approach. Preliminary screening and validation studies show that zinc finger protein 24 (ZFP24) governs SOX9 upregulation in tubular epithelial cells. ZFP24, a Cys2-His2 (C2H2) zinc finger protein is essential for oligodendrocyte maturation and myelination, however, its role in the kidneys or in SOX9 regulation remains unknown. Here, we found that tubular epithelial ZFP24 gene ablation exacerbated ischemia, rhabdomyolysis, and cisplatin-associated AKI. Importantly, ZFP24 gene deletion resulted in suppression of SOX9 upregulation in injured tubular epithelial cells. Chromatin immunoprecipitation and promoter luciferase assays confirmed that ZFP24 bound to a specific site in both murine and human SOX9 promoters. Importantly, CRISPR/Cas9 mediated mutation in the ZFP24 binding site in the SOX9 promoter in vivo led to suppression of SOX9 upregulation during AKI. Thus, our findings identify ZFP24 as a critical stress-responsive transcription factor protecting tubular epithelial cells through SOX9 upregulation.
Keywords: Acute kidney injury (AKI), renal tubular epithelial cells (RTECs), Sox9 (sex-determining region Y (SRY) box 9), Zinc finger protein (ZFP24), ischemia, cisplatin
Graphical Abstract
Translational Statement
The transcription factor SOX9 is upregulated in injured tubular epithelial cells and plays a protective role during AKI. Identifying the upstream factors could lead to the development of therapeutic strategies that increase the expression of protective genes such as SOX9, reduce injury, and augment the repair processes. To this end, the present study describes the identification of zinc factor protein ZFP24 as the transcriptional regulator that controls the upregulation of a protective SOX9-mediated transcriptional program in tubular epithelial cells during AKI.
Introduction
Sox (Sex-determining region-Y related HMG-box) proteins are a conserved family of transcription factors and cell fate regulators with widespread roles in embryonic development and adult tissue homeostasis1. The SOX family, which encompasses about 20 members with highly divergent functions, is defined by the presence of a conserved high-mobility group (HMG) domain that mediates DNA binding2. SOX9 is a key member of the SOX family and was initially identified as the gene underlying the haploinsufficiency disorder campomelic dysplasia (CD), a human syndrome characterized by defective chondrogenesis and sex reversal3–5. Notwithstanding its critical role in skeletal development and male sex determination, studies with Sox9-deficient mice have revealed the importance of this transcription factor in cell fate determination in multiple organs such as the pancreas, intestine, kidney, heart, and brain5.
Sox9 is widely expressed during embryonic development, including in the kidneys. However, in most adult tissues its expression is low, except in certain cell types such as astrocytes6 in the brain and adult stem cells in tissues with high turnovers, such as in the intestine7 and hair follicles8. In hair follicles, which undergo cyclical periods of growth, Sox9 is highly expressed and is functionally critical for stem cells that reside at the base of the resting follicle8. Sox9 also drives postnatal injury repair9 as well as fibrosis10 in multiple organs. Thus, in adult tissues, Sox9 seems to be part of a tightly regulated transcriptional program that ensures proper tissue architecture during normal cell turnover and post injury repair.
Within the adult kidneys, Sox9 expression is low in the renal tubular epithelial cells (RTECs). However, under conditions of cellular stress and injury, Sox9 is transcriptionally upregulated11. Functionally, Sox9 plays a cytoprotective role during the early phase of ischemia reperfusion injury (IRI)12, cisplatin nephrotoxicity12, and rhabdomyolysis-associated AKI13 and facilitates repair during the recovery phase11, 14. We have recently identified posttranslational regulatory mechanisms that suppress Sox9 protein function during AKI12. However, the mechanisms that control Sox9 mRNA upregulation in RTECs during AKI remains unknown.
In the current study, we have utilized an unbiased RNAi mediated functional screening approach to identify transcription factors that regulate stress induced Sox9 upregulation in RTECs. Our studies have identified Zinc finger protein Zfp2415, also known as Zfp191 and Znf24, as a crucial upstream regulator of Sox9 in RTECs. Zfp24 is a Cys2-His2 (C2H2) zinc finger protein that contains four putative DNA-binding zinc finger domains15, 16. Zfp24 plays crucial roles in oligodendrocyte maturation and CNS myelination15, 17, however its role in Sox9 regulation or in the pathogenesis of AKI has not been previously explored. Our results show that Zfp24 drives Sox9 upregulation in RTECs during AKI.
Methods
Primary RNAi screen to identify SOX9 transcriptional regulators.
Boston University mouse proximal tubule cells (BUMPT, generated by Drs. Wilfred Lieberthal and John Schwartz18 and obtained from Dr. Zheng Dong, Augusta University, Augusta, GA) were grown at 37 °C in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum. We used a promoter-reporter lentiviral firefly luciferase vector (Applied Biological Materials, Catalog numbers: LV653 and LV655) to generate two stable BUMPT cell lines expressing empty vector (promoter-less) and murine Sox9 promoter (−2500 bp from the TSS) driven firefly luciferase. BUMPT stable cells were then reverse transfected with control siRNA or siRNAs targeting transcriptional regulators using methods described in our previous work12, 19. Briefly, the Dharmacon mouse siRNA library targeting transcriptional regulators (1887 genes) containing four pooled siRNAs for each gene was utilized in the primary screen. Stable BUMPT cells were plated in 96-well plates and reverse-transfected with 25 nM siRNA using Lipofectamine RNAiMAX reagent (Life Technologies). At 48 h post transfection, cells were treated with 15 μM cisplatin in fresh media. Subsequently, 8 h post treatment, firefly luciferase assay (Thermo Fischer, Catalog number 16176) was performed. The siRNAs that reduced luciferase activity in stable cells that expressed the Sox9 promoter driven luciferase were selected for secondary screening. The primary screen was carried out in triplicate samples, and data analysis was performed according to established and previously reported methods12, 19. The details of the secondary screening are provided in the supplementary methods section.
Mice breeding.
All animals were housed, handled, and animal studies were conducted in accordance with approved Institutional Animal Care and Use Committee procedures. Mice were housed in a temperature-controlled environment with a 12-h light cycle and given a standard diet and water ad libitum. Conditional gene knockout in RTECs was achieved through breeding of Zfp24-floxed mice (Jackson Laboratory, stock no. 029023) and Sox9-floxed mice (Jackson Laboratory, stock no. 013106) with Ggt1-Cre mice (Jackson Laboratory, stock no. 012841). For all mouse colonies, the pups were ear-tagged and genotyped at 3 weeks of age as described in our previous studies. Methods used for generating mice where CRISPR-Cas9 mediated knock-in mutation was introduced in the SOX9 promoter (Prommut mice) is described in detail in the supplementary methods section. The Zfp24 floxed and Prommut strains were obtained and maintained on C57BL/6J background. The Ggt1-Cre mice have a mix of BALB/cJ and C57BL/6 background.
Animal models of acute kidney injury.
Since AKI is associated with distinct disease conditions, multiple mouse models have been developed20. Ischemia, cisplatin, and rhabdomyolysis associated AKI was triggered in male mice at 8–12 weeks of age using methods described in our recent studies12, 13, 21, 22. Littermate controls were used in all studies. Experiments were carried out in a blinded fashion where the researchers evaluating, quantifying, or analyzing experimental outcomes were blinded to the genotype of the mice. Briefly, to cause ischemia-reperfusion injury, mice were anesthetized by isoflurane and placed on a surgical platform where the body temperature was monitored during the procedure. The skin was disinfected, kidneys were exposed, and bilateral renal pedicles were clamped for 30 minutes, followed by clamp removal, and suturing to close the muscle and skin around the incision. To compensate for the fluid loss, 0.5 ml of warm sterile saline was administered via intraperitoneal injection. The sham (mock) groups underwent similar procedure except without bilateral clamping. Nephrotoxicity was trigged by a single cisplatin (30 mg/kg) intraperitoneal injection as described previously21. The sham (vehicle) groups were injected with equal volumes of normal saline. To induce rhabdomyolysis, mice were injected with 7.5 ml/kg 50% glycerol intramuscularly to the two hind-legs or injected with saline as a control. Following injury, blood was collected at 24 h (IRI and rhabdomyolysis) or 72 h (cisplatin) via cardiac puncture after carbon dioxide asphyxiation. Renal tissues were collected and processed for immunoblot, qPCR, and histological analysis as described previously23, 24 and in supplementary methods.
Cell culture, promoter mutagenesis, protein and gene expression analysis.
Details regarding the generation of CRISPR/Cas9 mediated knockout cells is provided in the supplementary methods sections. Site directed mutagenesis was carried out using methods described in our previous studies23, 25, 26 and as described in the supplementary methods. Methods used to analyze protein and gene expression in cell culture and renal tissues have been described previously27–29 and in the supplementary methods.
Statistical analysis.
Data in all the graphs are presented as mean with S.D. Statistical analysis were carried out using GraphPad Prism. p < 0.05 was considered statistically significant. To calculate statistical significance between two groups, two-tailed unpaired Student’s t test was performed. For comparisons among three or more groups one-way analysis of variance (ANOVA) followed by Tukey’s or Dunnett’s multiple-comparison test was used. No outliers were excluded, and all experiments were repeated at least three times.
Results
RNAi screen to identify upstream transcriptional regulators of Sox9 in RTECs.
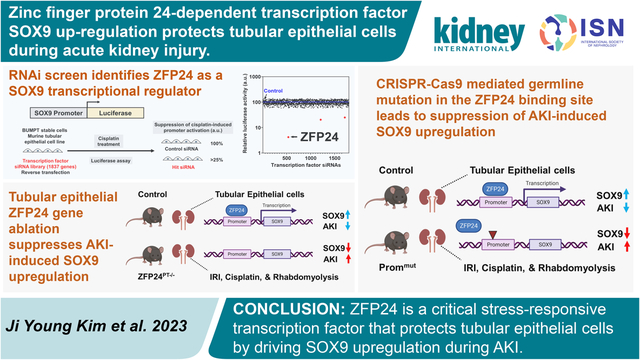
To identify the transcriptional factors that mediate Sox9 upregulation in RTECs under stress conditions associated with kidney injury, we used a modified version of a previously developed RNAi based screening method12. To this end, we used BUMPT cells18 because high transient transfection efficiency (~95%) of this murine RTEC cell line makes it a suitable model for high-throughput (siRNA) screening assays12. We generated stable BUMPT cells in which a luciferase-based reporter plasmid encoding the Sox9 promoter was introduced. Treatment of these cells with cisplatin results in a robust induction of Sox9 promoter activity (Supplementary Figure S1 and Figure 1a). We then transiently transfected these cells with control siRNAs (non-specific) or transcription factor specific siRNAs (set of four pooled per target, 1837 targets), followed by cisplatin treatment, and assessment of Sox9 promoter activity using luciferase reporter assay (Figure 1b). The primary screen was carried out in triplicate, and subsequent data analysis identified three hit candidates, namely Interferon regulatory factor 1 (Irf1), SMAD family member 4 (Smad4), and Zinc finger protein 24 (Zfp24) (Figure 1c–d). Our secondary screening in a human RTEC cell line, HK-2 cells, transfected with a plasmid encoding human SOX9 promoter luciferase plasmid, showed that among these candidates, knockdown of ZFP24 had the highest effect in suppressing stress-induced SOX9 promoter activity (Figure 1e). For final validation, we generated HK-2 cells in which ZFP24 gene was knocked out using CRISPR/Cas9 lentiviral construct. We then performed add-back experiments where empty or wild-type human ZFP24 expression vectors were introduced in the knockout cells (Figure 1f). Subsequent, functional assays showed that ZFP24 knockout suppresses stress induced SOX9 promoter activity, which is reversed by re-introduction of ZFP24 (Figure 1g).
Figure 1. RNAi screen identifies Zfp24 as a Sox9 transcriptional regulator in tubular epithelial cells.
(a) Stable BUMPT cells expressing either promoter-less luciferase (empty) or Sox9-promoter driven luciferase were treated with vehicle or 25 μM cisplatin for 8 h followed by measurement of luciferase activity. Cisplatin treatment induced a robust induction of luciferase activity that was dependent on the Sox9 promoter. (b) Schematic representation of the primary screen. Stable BUMPT cells were reverse transfected with control or transcription factor siRNAs (1837 genes) followed by treatment with cisplatin and measurement of luciferase activity. The luciferase activity in the cisplatin treated control siRNA group was set as 100% and any transcription factor siRNAs that reduced luciferase activity to 25% or lower, were considered as hits. (c) Graphical representation of the data obtained in the primary screen. (d) Secondary screens were performed in BUMPT cells for the top three hits, using distinct siRNAs. (e) Stable HK-2 cells that express either promoter-less luciferase (empty) or human SOX9 promoter driven luciferase. These cells were transfected with siRNA for the top three hits, followed by treatment with vehicle or 50 μM cisplatin for 8 h and measurement of luciferase activity. (f) CRISPR/Cas9 mediated ZFP24 gene knockout was carried out in HK-2 cells. Control (WT) or ZFP24 knockout cells were then transiently transfected with empty vector or ZFP24 expression plasmid. Representative immunoblot confirming both gene knockout and successful addback. (g) Control, ZFP24 knockout, and add-back cells were transfected with SOX9-promoter driven luciferase construct, followed by treatment with cisplatin, and measurement of luciferase activity. The results showed that ZFP24 knockout suppresses cisplatin-mediated activation of the SOX9-promoter driven luciferase activation and this phenotype was reversed by ZFP24 add-back. In all the bar graphs (n = 8 biologically independent samples), experimental values are presented as mean ± S.D. The height of error bar = 1 S.D., and p < 0.05 was indicated as statistically significant. Student’s t test was carried out and statistical significance is indicated by *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar: 100 μm.
Renal Zfp24 expression during acute kidney injury.
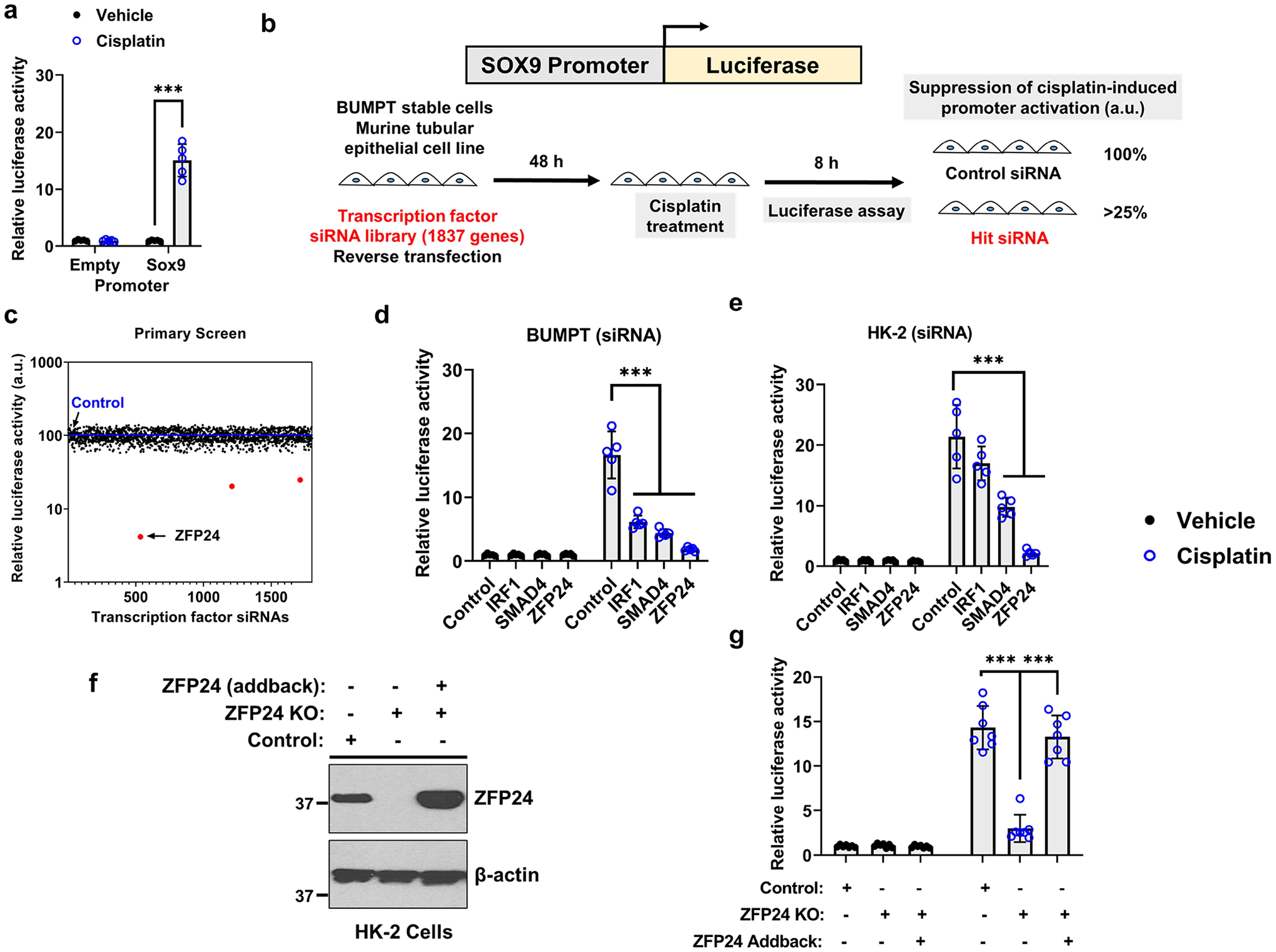
Since the role of Zfp24 in kidneys remains unknown, we initially examined its expression under normal and disease conditions. Moreover, Sox9 expression is undetectable in the RTECs of the adult murine kidneys; however, under conditions of stress such as ischemia-reperfusion injury (IRI), cisplatin nephrotoxicity, and rhabdomyolysis associated AKI, there is a profound increase in the mRNA11, 30 and protein12, 13, 23 levels of Sox9. So, we first sought to examine the expression of Zfp24 under normal and AKI conditions. To induce AKI, C57BL/6J mice underwent bilateral ischemic surgery or were injected with a single 30 mg/kg intraperitoneal dose of cisplatin or rhabdomyolysis was induced by glycerol injection (7.5 ml/kg 50% glycerol) in the hind-leg muscles. The extent of renal damage was determined by measurement of blood urea nitrogen and serum creatinine (Figure 2a–b). In case of ischemia-reperfusion and rhabdomyolysis, renal impairment occurs 24 h post-surgery, whereas in the cisplatin-associated kidney injury models, renal impairment is observed 72 h post-injection. Gene and protein analysis of kidney tissues at these time-points showed that Zfp24 expression does not appreciably change during AKI (Supplementary Figure S2 and Figure 2c–d).
Figure 2. Renal Zfp24 expression during acute kidney injury.
Ischemia reperfusion (IRI), cisplatin nephrotoxicity, and rhabdomyolysis-associated acute kidney injury was induced in 8–12-week-old male C57BL/6J mice. Bilateral renal ischemia was induced for 30 min, cisplatin nephrotoxicity was induced by a single intraperitoneal cisplatin injection (30 mg/kg), and rhabdomyolysis was induced by glycerol injection (7.5 ml/kg 50% glycerol) in the hind-leg muscles. Sham groups represent either mock surgery or vehicle injections. At 24 h (IRI and rhabdomyolysis) and 72 h (cisplatin), serum and kidneys were collected for further analysis. Blood urea nitrogen (a) and serum creatinine (b) measurements were carried out to examine kidney function and damage. (c-d) Western blot analysis showed that Zfp24 expression remains unaltered during AKI. Representative blots show Zfp24 expression in the renal tissues, while the graph depict densitometric analysis (Zfp24 expression normalized to β-actin). (e-f) Renal cortical tissues were used for Zfp24 immunoprecipitation followed by immunoblot analysis of total and phosphorylated Zfp24 using a phospho-linker antibody. Zfp24 was predominantly present in the dephosphorylated state in the injured kidneys. Representative blots show Zfp24 phosphorylation in the renal tissues, while the graph depict densitometric analysis (Phospho-Zfp24 expression normalized to total Zfp24 levels). In all the bar graphs (n = 7 biologically independent samples from three independent experiments), experimental values are presented as mean ± S.D. The height of error bar = 1 S.D., and p < 0.05 was indicated as statistically significant. Student’s t test was carried out and statistical significance is indicated by *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar: 100 μm.
Zfp24 contains a SCAN domain and four DNA-binding C2H2 zinc finger domains that are interspersed by linkers that contain phosphorylation sites15. Linker phosphorylation is a mechanism that can result in the deactivation of the C2H2 proteins because phosphorylation introduces a negative charge to the DNA-binding domain of the protein and hence reduces its affinity to DNA. To examine the phosphorylation status of Zfp24, we performed immunoprecipitation studies in normal and injured kidneys. Our results (Figure 2e–f) show that Zfp24 is present in the phosphorylated state in normal renal tubular epithelial cells. However, Zfp24 is de-phosphorylated and activated early during the development of AKI. Time-course experiments in the IRI model showed that Zfp24 dephosphorylation occurs within 1-hour post-reperfusion and precedes Sox9 upregulation (Supplementary Figure S3).
RTEC-specific Zfp24 gene deletion aggravates the severity of kidney injury.
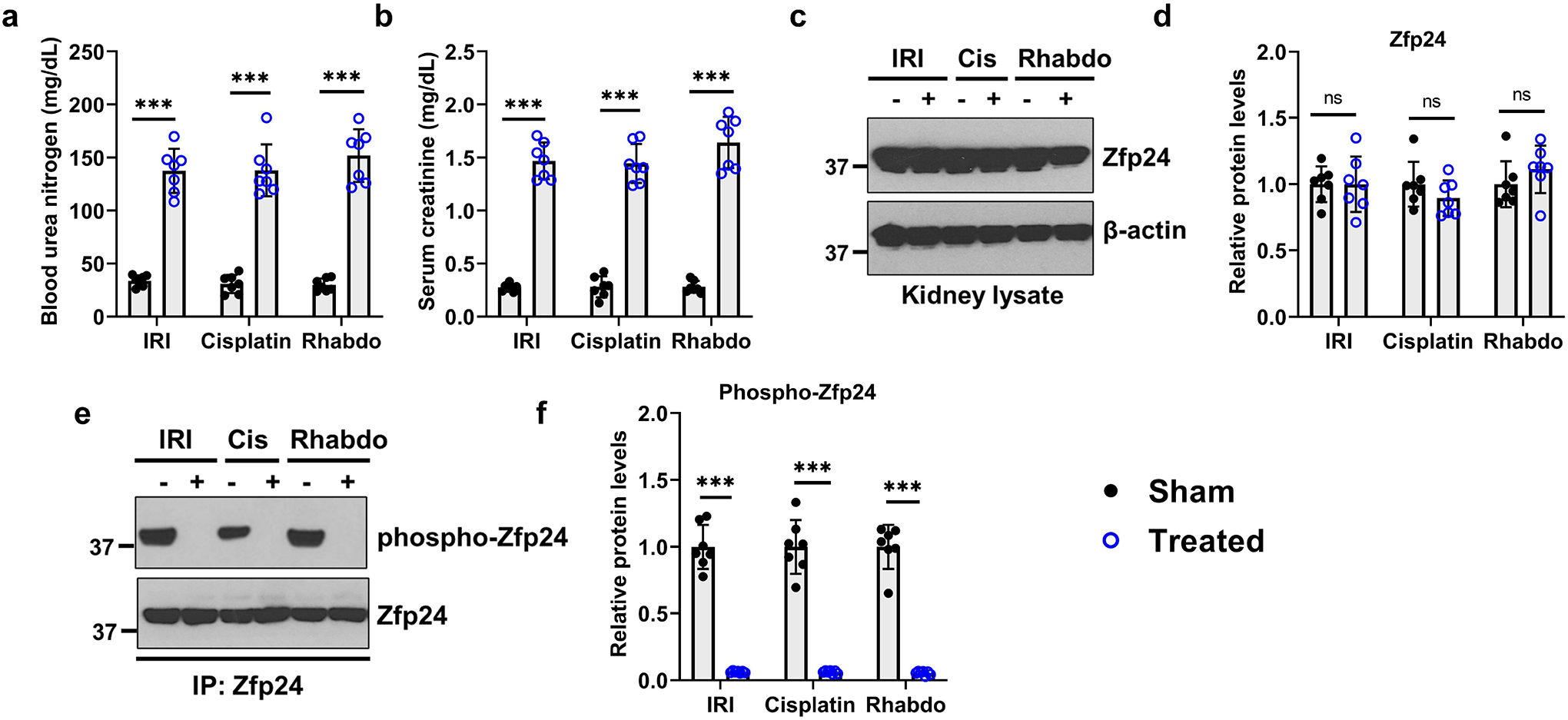
We next sought to examine the consequence of RTEC-specific Zfp24 gene deletion on normal renal function and the severity of AKI. To this end, we generated Zfp24 conditional knockout mice (Zfp24PT−/−) by crossing the Zfp24-floxed15 mice with the Ggt1-Cre31 mice. The Ggt1-driven Cre is expressed in RTECs 7–10 days after birth, post completion of renal development31. Initial studies showed that normal renal function (Supplementary Figure S4) is not affected by Zfp24 deficiency. Remarkably, Zfp24 gene ablation in RTECs (Figure 3a) markedly increased kidney damage during IRI, cisplatin, and rhabdomyolysis associated AKI (Figure 3b–f). Zfp24 deficiency also resulted in increased cell death activation as measured by Caspase-3 activation (Supplementary Figure S5). To determine the effect of RTEC-specific Zfp24 gene ablation on overall survival post-injury, we used the rhabdomyolysis model for long-term studies. As shown in the Supplementary Figure S6, rhabdomyolysis-associated AKI was not severely fatal in the control mice and 90% mice survived until the end of the observation period of 4 weeks. However, all the Zfp24PT−/− mice and 50% of the Sox9PT−/− mice died within the first week. These results indicate that Zfp24 and Sox9 are critical survival factors in RTECs during AKI. Next, we cultured primary RTECs from the control and Zfp24PT−/− mice. These cells were then treated with cisplatin, and subsequent viability and caspase assays showed that Zfp24 knockout sensitizes RTECs to cisplatin-mediated cell death (Supplementary Figure S7). Collectively, these studies suggested that Zfp24 plays a protective role in RTECs during AKI.
Figure 3. Zfp24 gene ablation in tubular epithelial cells exacerbates the severity of AKI.
Ggt1-Cre mice were crossed with Zfp24-floxed mice to generate tubular epithelial conditional knockout mice (Zfp24PT−/−). (a) Immunoblot analysis of Zfp24 protein in renal cortical tissues showed successful gene knockout. Blots are representative of three independent experiments. Littermate control and Zfp24 conditional knockout mice were then challenged with bilateral renal ischemia (30 min), cisplatin (30 mg/kg, single intraperitoneal injection) nephrotoxicity, or glycerol-induced rhabdomyolysis (7.5 ml/kg 50% glycerol in the hind-leg muscles) followed by examination of the severity of AKI using biochemical and histological analysis. (b-f) Blood urea nitrogen, serum creatinine, renal Ngal gene expression (qPCR), and histological analysis (H&E) showed that tubular Zfp24 deficiency results in significantly worsened renal impairment in the IRI, cisplatin, and rhabdomyolysis-associated mouse models of AKI. The asterisk in the histological images depicts damaged tubules. Histopathologic analysis and tubular damage were scored by calculation of the percentage of tubules that showed dilation, epithelium flattening, cast formation, loss of brush border and nuclei, and denudation of the basement membrane. In all the bar graphs (n = 8 biologically independent samples from three independent experiments), experimental values are presented as mean ± S.D. The height of error bar = 1 S.D., and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparison test was carried out, and statistical significance is indicated by *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar: 100 μm.
RTEC-specific Zfp24 gene deletion suppresses stress-induced Sox9 upregulation.
To determine the role of Zfp24 in stress induced Sox9 upregulation, we carried out gene expression analysis of renal cortical tissues under normal and AKI conditions. Strikingly, our studies showed that AKI-dependent upregulation of Sox9 is suppressed in the Zfp24 conditional knockout mice (Figure 4a and Supplementary Figure S8). More than 90% reduction in Sox9 mRNA upregulation was observed in the Zfp24 conditional knockout mice. Immunoblot analysis (Figure 4b–c) also corroborated the results obtained with the gene expression analysis. Our previous studies12, 23 have identified several downstream target genes of Sox9 in RTECs. So, we examined the expression of these Sox9 target genes and found that Zfp24 gene deletion suppresses the injury-induced upregulation of Sox9 target genes in RTECs (Supplementary Figure S9). These results provide compelling evidence that Zfp24 drives Sox9 upregulation in RTECs during AKI.
Figure 4. Essential role of Zfp24 in AKI-induced Sox9 upregulation.
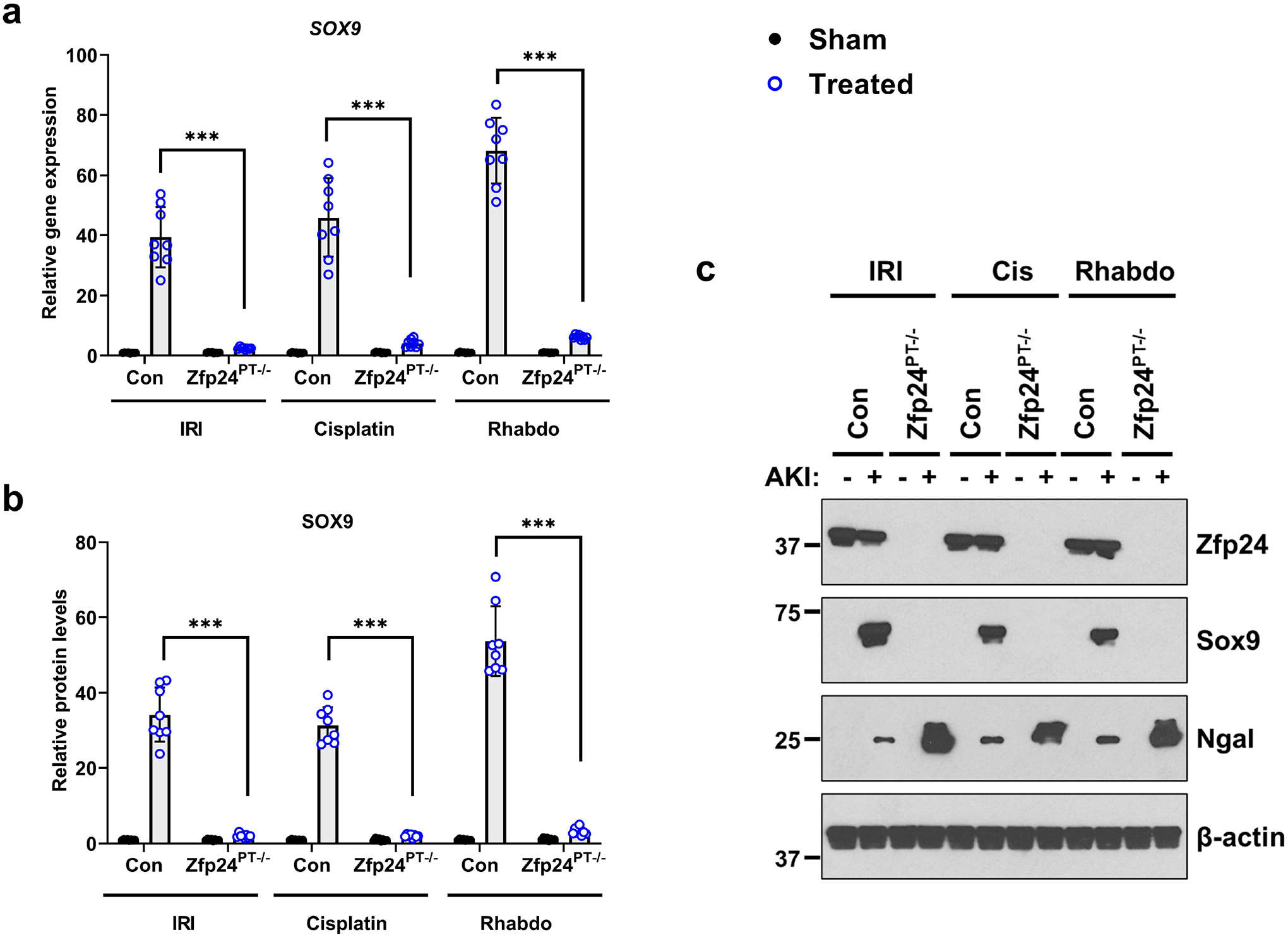
Littermate control and Zfp24 conditional knockout mice were challenged with bilateral renal ischemia (30 min), cisplatin (30 mg/kg, single intraperitoneal injection) nephrotoxicity, or glycerol-induced rhabdomyolysis (7.5 ml/kg 50% glycerol in the hind-leg muscles) followed by examination of Sox9 protein and gene expression in renal tissues. (a) qPCR-based gene expression analysis was carried out in the renal tissues from littermate control and knockout mice 24–72 h after induction of kidney injury. The AKI-induced Sox9 mRNA up-regulation was suppressed in the Zfp24 conditional knockout mice. (b-c) Western blot analysis showed that AKI-induced Sox9 protein induction is dependent on Zfp24. Blots are representative of four independent experiments. Densitometric analysis was performed for Zfp24 protein expression using Image J and normalization was carried out using β-actin as loading control. In all the bar graphs (n = 8 biologically independent samples from 3–4 independent experiments), experimental values are presented as mean ± S.D. The height of error bar = 1 S.D., and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparison test was carried out, and statistical significance is indicated by *P < 0.05, **P < 0.01, ***P < 0.001.
Stress-induced recruitment of Zfp24 to the Sox9 promoter.
To test if Zfp24 binds to the Sox9 promoter under normal and AKI conditions, we performed chromatin immunoprecipitation studies in normal and injured renal cortical tissues. We then used Sox9 promoter specific primers and qPCR analysis to examine Zfp24 binding to Sox9 promoter. Our results showed that under normal conditions, Zfp24 binding to the Sox9 promoter is very limited. However, during AKI, Zfp24 is recruited to the Sox9 promoter (Figure 5a). Bioinformatics analysis of murine and human SOX9 promoter identified a putative binding site for ZFP24 (Figure 5b). In promoter luciferase assays, site-directed mutagenesis of this putative ZFP24 binding site in the murine (Figure 5c–d and Supplementary Figure S10) and human (Figure 5c and 5e) SOX9 promoter, significantly abrogated stress-induced promoter activation. Electrophoresis mobility shift assay (EMSA) further confirmed the direct binding of murine Zfp24 to the TCAT site in the Sox9 promoter (Supplementary Figure S11). Collectively, these results suggest that Zfp24 binds to the Sox9 promoter under stress conditions and drives Sox9 upregulation during AKI.
Figure 5. ZFP24 binds to human and murine SOX9 promoter under stress conditions.
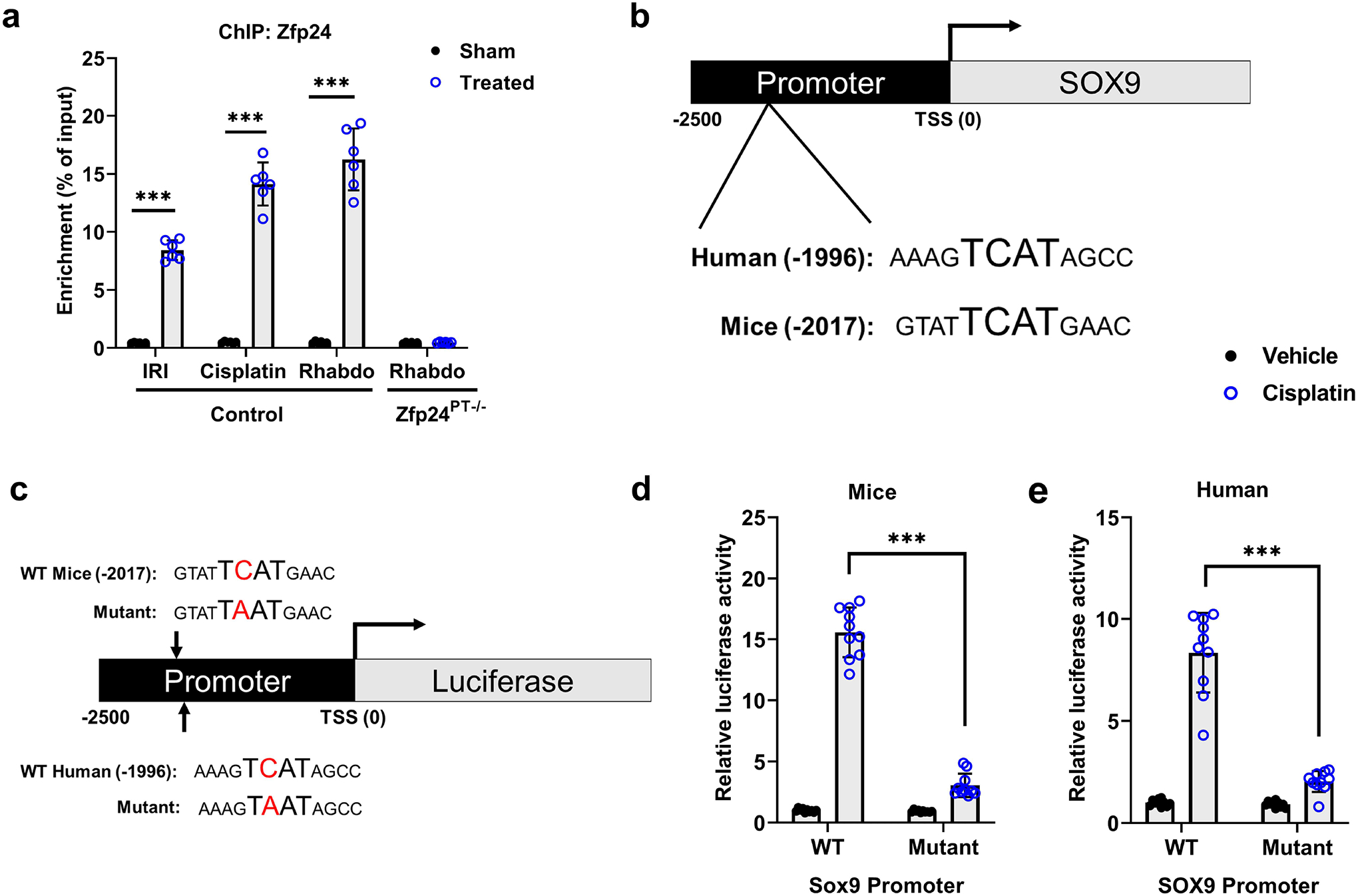
(a) Zfp24 chromatin immunoprecipitation (ChIP) was carried out using renal tissues from sham and ischemia-, cisplatin nephrotoxicity-, and rhabdomyolysis treated mice. Subsequent qPCR analysis using primers specific for the murine Sox9 promoter region showed that Zfp24 can bind to Sox9 promoter in vivo. Renal tissues from Zfp24PT−/− mice were used as negative control. (b) Bioinformatic analysis was carried out to identify potential ZFP24 binding sites within the human and murine SOX9 promoter. Figure shows schematic representation of SOX9 promoter, highlighting the putative ZFP24 binding site in humans and mice. TSS indicates the transcription start site. (c) The SOX9 promoter sequences (−2500bp from transcription start site (TSS)) was cloned in a luciferase reporter construct. A single point mutation was introduced in the putative ZFP24 binding site (TCAT to TAAT) in both human and murine SOX9 promoter sequences. (d) BUMPT cells were transfected with WT or mutant murine Sox9 promoter driven Renilla luciferase reporter construct, followed by cisplatin treatment, and luciferase activity measurement. These cells were also co-transfected with a plasmid encoding a constitutively expressed Cypridina luciferase, which was used as the normalizing control. (e) HK-2 cells were transfected with WT or mutant human SOX9 promoter driven Renilla luciferase reporter construct, followed by cisplatin treatment, and luciferase activity measurement. These cells were also co-transfected with a plasmid encoding a constitutively expressed Cypridina luciferase, which was used as the normalizing control. Results indicate that cisplatin associated increased in SOX9 promoter activity is suppressed by mutations in the ZFP24 binding site. In all the bar graphs (n = 6–10 biologically independent samples), experimental values are presented as mean ± S.D. The height of error bar = 1 S.D., and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparison test was carried out, and statistical significance is indicated by *P < 0.05, **P < 0.01, ***P < 0.001.
Mutation in the Zfp24 binding site in the SOX9 promoter exacerbates AKI.
To functionally test our hypothesis that Zfp24 binding to the putative site in the promoter region drives Sox9 upregulation during AKI, we used a CRISPR-Cas9 driven method to generate a germline mutant mouse in which the Zfp24 binding site was mutated from TCAT to TAAT (Figure 6 a). When the heterozygous mice were used for breeding, WT and homozygous mutant mice (Prommut) were born at expected mendelian ratios. The adult homozygous mutant mice appeared normal and had no functional renal abnormalities (Supplementary Figure S12). We next investigated how this mutation in the Zfp24 binding site influences the severity of AKI in IRI, cisplatin, and rhabdomyolysis-associated mouse models of AKI. Our results showed that when challenged with injury, the Prommut mice had significantly higher renal impairment as compared to the WT littermates (Figure 6 b–f). Prommut mice also had elevated cell death activation as measured by caspase-3 activation (Supplementary Figure S13).
Figure 6. CRISPR/Cas9 mediated point mutation in the Zfp24 binding site in the Sox9 promoter abrogates AKI-induced Sox9 upregulation.
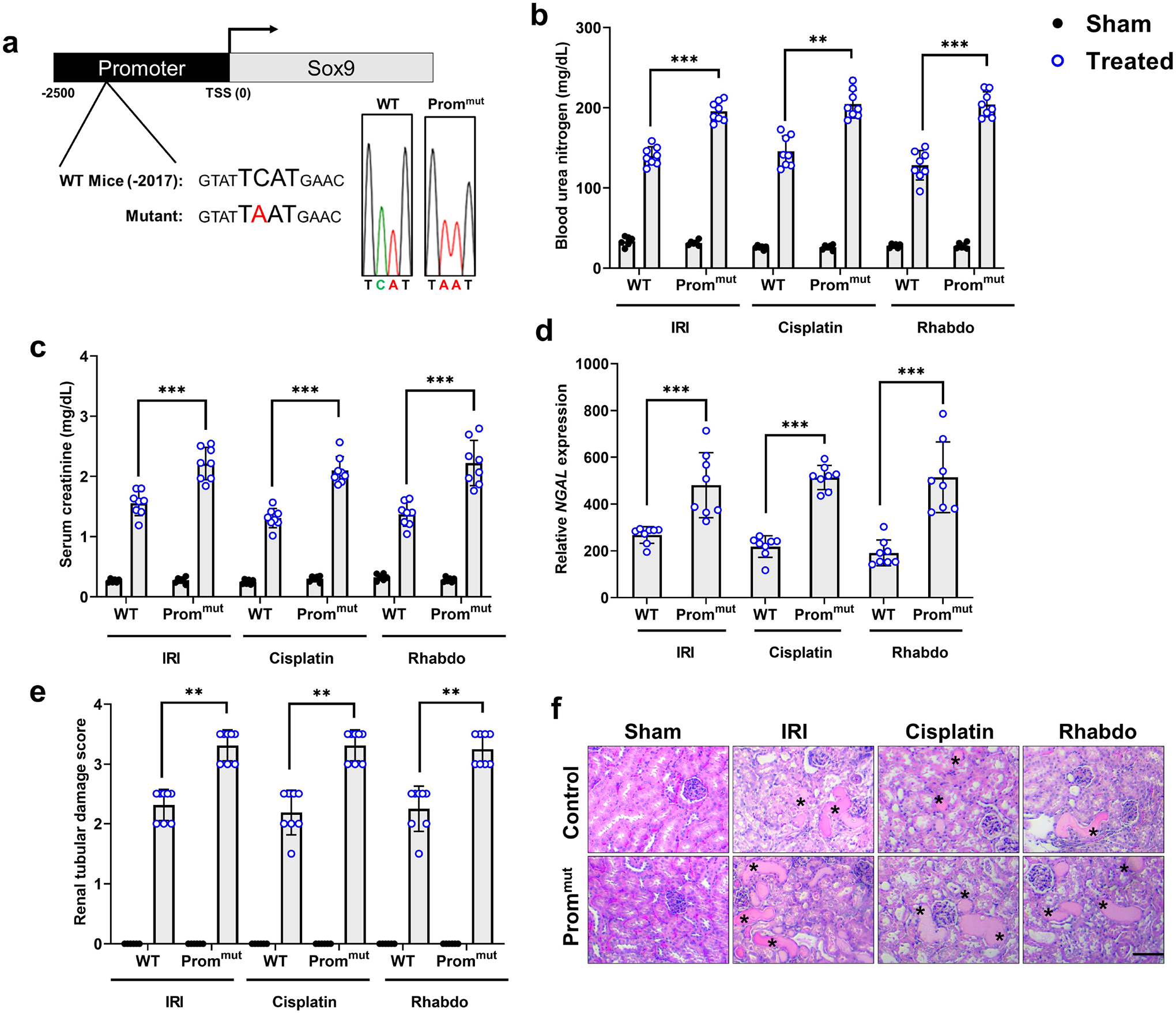
A transgenic knock-in mouse was generated using CRISPR/Cas9 and a point mutation was introduced in the Zfp24 binding site within the murine Sox9 promoter. (a) Schematic representation of the single point mutation in the Zfp24 binding site within the murine Sox9 promoter. Heterozygous mutant mice were crossed to generate littermate control (WT) and promoter mutant (Prommut) mice. Representative DNA sequencing data confirming the mutation is shown here. These mice were then challenged with bilateral renal ischemia (30 min), cisplatin (30 mg/kg, single intraperitoneal injection) nephrotoxicity, or glycerol-induced rhabdomyolysis (7.5 ml/kg 50% glycerol in the hind-leg muscles) followed by examination of the severity of AKI using biochemical and histological analysis. (b-f) Blood urea nitrogen, serum creatinine, renal Ngal gene expression (qPCR), and histological analysis (H&E) showed that mutations in the Zfp24 binding site within the Sox9 promoter results in significantly worsened renal impairment in the IRI, cisplatin, and rhabdomyolysis-associated mouse models of AKI. The asterisk in the histological images depicts damaged tubules. Histopathologic analysis and tubular damage were scored by calculation of the percentage of tubules that showed dilation, epithelium flattening, cast formation, loss of brush border and nuclei, and denudation of the basement membrane. In all the bar graphs (n = 8 biologically independent samples from3–4 independent experiments), experimental values are presented as mean ± S.D. The height of error bar = 1 S.D., and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparison test was carried out, and statistical significance is indicated by *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar: 100 μm.
Mutation in the Zfp24 binding site leads to suppression of AKI-induced SOX9 upregulation.
We next carried out gene expression analysis of renal cortical tissues from wild type and Prommut littermate mice under normal and AKI conditions. Remarkably, our studies showed that Sox9 mRNA upregulation is impaired in the Prommut mice during IRI, cisplatin nephrotoxicity, and rhabdomyolysis associated AKI (Figure 7a). Furthermore, immunoblot and densitometric analysis (Figure 7b–c) confirmed the results obtained in the gene expression analysis and showed a significant impairment in the induction of Sox9 proteins levels in the injured kidneys of the Prommut mice. These results provide strong evidence that Zfp24 binds to the Sox9 promoter during AKI and drives stress induced upregulation of an essential and protective transcriptional program.
Figure 7. Zfp24-binding site in the Sox9 promoter is essential for AKI induced Sox9 upregulation.
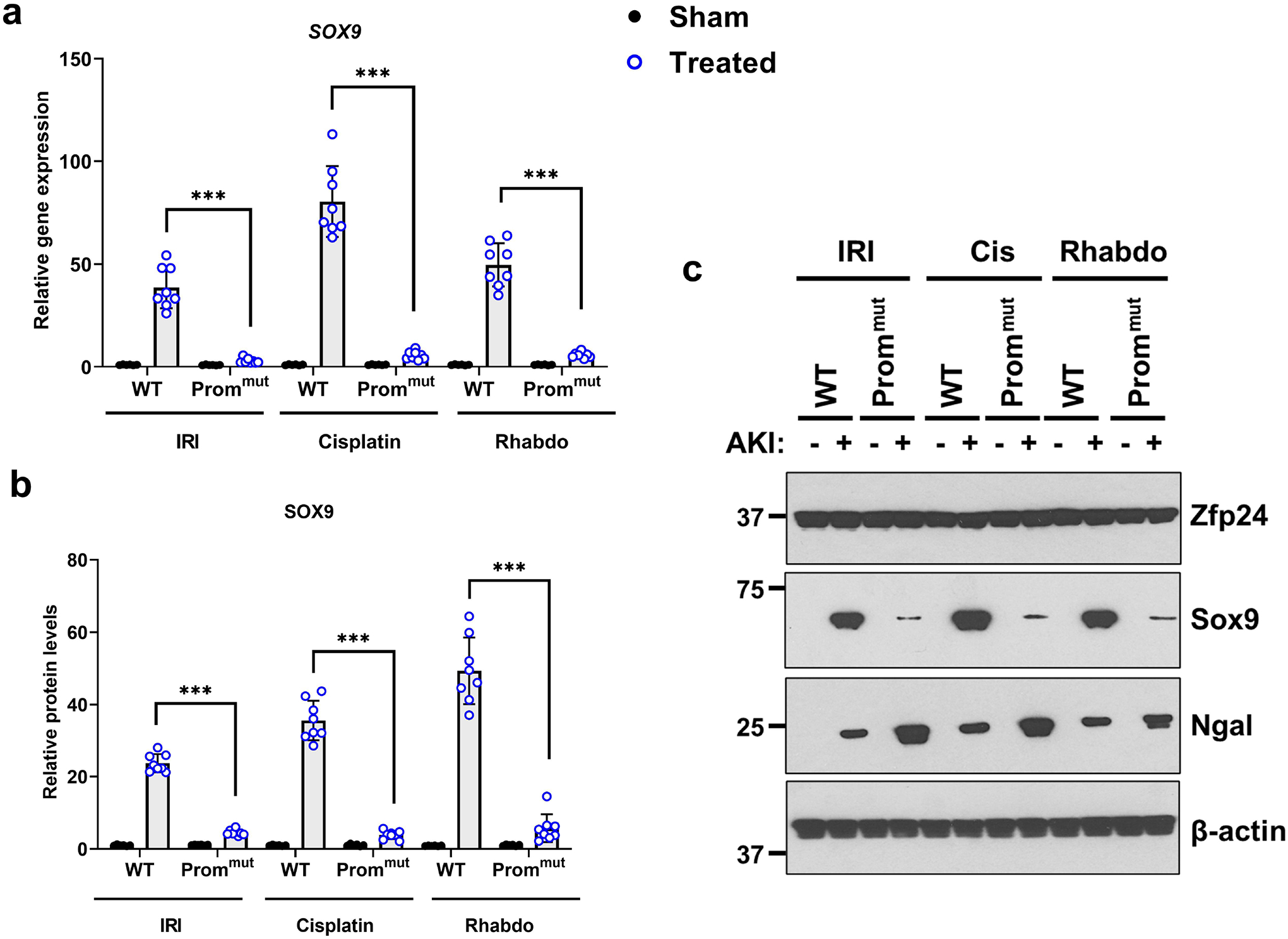
Littermate WT and promoter mutant (Prommut) mice were challenged with bilateral renal ischemia (30 min), cisplatin (30 mg/kg, single intraperitoneal injection) nephrotoxicity, or glycerol-induced rhabdomyolysis (7.5 ml/kg 50% glycerol in the hind-leg muscles) followed by examination of Sox9 protein and gene expression in renal tissues. (a) qPCR-based gene expression analysis was carried out in the renal tissues from WT and Prommut mice 24–72 h after induction of kidney injury. The AKI-induced Sox9 mRNA up-regulation was suppressed in the Prommut mice. (b-c) Western blot analysis showed that AKI-induced Sox9 protein induction is suppressed in the Prommut mice. Blots are representative of four independent experiments. Densitometric analysis was performed for Sox9 protein expression using Image J and normalization was carried out using β-actin as loading control. In all the bar graphs (n = 8 biologically independent samples from 3–4 independent experiments), experimental values are presented as mean ± S.D. The height of error bar = 1 S.D., and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparison test was carried out, and statistical significance is indicated by *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Acute kidney injury is associated with a precipitous decline in renal function due to inflammatory, hypoxic, and nephrotoxic insults to tubular epithelial cells32–36. The initial injury phase is characterized by RTEC cell death37, 38, inflammation39–42, endothelial dysfunction43–45, and decline in glomerular filtration rate. The subsequent phase involves tissue repair and recovery46. Finally, depending on the extent of injury and ensuing repair, AKI can be fatal, result in complete recovery or transition into chronic kidney disease46–50. Sox9 is one of the highly upregulated genes in the RTECs11, 51, 52 and plays protective roles during the injury12 and repair phases11, 14. Due to its consistent expression in RTECs within tubules with incomplete repair53, it has also been speculated that Sox9 might drive fibrosis in the later phases. Pro-fibrotic role of Sox9 has also been demonstrated in other cell types such as myofibroblasts54. Due to the clear importance of Sox9 in renal response to injury, further studies are required to decipher its spatial and temporal regulation in various cell types. In the current study, we have focused on understanding the mechanisms that drive Sox9 upregulation in RTECs during the early phase of AKI.
Zinc finger proteins are among the largest families in the eukaryotic genomes55. The human genome contains ~700 C2H2-containing zinc finger proteins; however, the biological functions of the vast majority of these proteins remain obscure55. The Cys2His2 zinc finger domain is involved in DNA binding and many of these proteins are transcription factors. Among these, the biological functions of Zfp24 which has four DNA-binding zinc finger domains has remained relatively underexplored15, 16. Zfp24 belongs to a subset of the zinc finger protein family whose members also contain SCAN domain56, which is a leucine-rich domain involved in protein–protein interactions. Initial studies showed that Zfp24 is widely expressed during development and in adult tissues17, including the kidneys. Two germline Zfp24 knockout mice have been generated, one showed an embryonic lethal phenotype57, while the other showed postnatal lethality17. While the reasons for these discrepancies are not known, these severe phenotypes highlight the critical non-redundant biological role of Zfp24, especially during development. The most well characterized function of Zfp24 has been its demonstrated role in oligodendrocyte maturation and developmental CNS myelination15, 17.
The role of Zfp24 in the kidneys has remained unexplored. Previous gene expression studies showed high expression of Zfp24 in the developing and adult murine kidneys17. In the current study, we have generated a RTEC-specific Zfp24 knockout mice, which shows that Zfp24 is not required for normal renal function in adult mice. However, under conditions of stress, Zfp24 plays a protective role and gene deletion under these conditions results in aggravation of renal injury. Notably, here we have used a Ggt1-driven Cre mice where gene deletion occurs 7–10 days post birth31, at a time when renal development is already completed. Hence, the lack of adverse phenotype under baseline conditions does not rule out the role of Zfp24 in renal development. However, our studies clearly establish Zfp24 as a stress-protective gene in adult RTECs.
Only a single previous study has explored the transcriptional targets of Zfp2415. Given the role of Zfp24 in oligodendrocyte maturation, chromatin immunoprecipitation (ChIP) sequencing (ChIP-seq) analysis of murine oligodendrocyte progenitor cells and mature oligodendrocytes showed that Zfp24 binds to the regulatory regions of genes important for CNS myelination15. Interestingly, these target genes included Myrf (Myelin Regulatory Factor) and Sox10. In the current study, multiple lines of evidence support the notion that Sox9 is a downstream target of Zfp24 in RTECs. Firstly, gene ablation studies show that AKI-associated Sox9 upregulation is Zfp24-dependent. Secondly, mutagenesis of the putative Zfp24 binding site in the murine and human promoter suppress stress induced Sox9 promoter activity in vivo and in vitro. Thirdly, ChIP experiments demonstrate stress induced binding of Zfp24 to the Sox9 promoter. Our studies combined with the recent work15 raise an intriguing possibility that Zfp24 might be a previously unrecognized regulator of Sox family of transcription factors. Given the severe developmental phenotypes of the Zfp24 germline knockout mice and the role of Sox proteins in development, it would be interesting to examine the expression of Sox family genes in the Zfp24 knockout mice. Also, it would be important to determine if additional Zfp24 binding sites are present within the Sox9 promoter.
Upstream regulators that drive Sox9 transcription during AKI have not been systematically studied. However, a recent study58 has proposed that the transcription factor, early growth response-1 (Egr1), contributes to Sox9 upregulation during IRI associated AKI. Intriguingly, the authors examined gene and protein expression of Sox9 in the WT mice under normal and IRI conditions, but not in mice with Egr1 gene knockout or overexpression. Hence, the relative contribution of Egr1 in Sox9 upregulation in vivo is difficult to decipher from this study. Our examination of Egr1 levels in the WT and Zfp24 conditional knockout mice showed that Egr1 induction during AKI is not influenced by Zfp24 deletion (Supplementary Figure S14). Since, Zfp24 gene deletion reduces Sox9 upregulation by more than 90%, our studies suggest that Zfp24 is the predominant regulator of stress induced Sox9 upregulation during AKI.
Our study provides the first evidence for the functional role of Zfp24 in AKI and Sox9 transcriptional regulation and raises several questions that warrant future investigations. Notably, Zfp24 protein expression seems to not be altered during AKI. However, ChIP experiments show that Zfp24 binds to the Sox9 promoter only under stress conditions. Several possible mechanisms could underlie Zfp24 binding to Sox9 promoter during AKI. One possibility is that Zfp24 binding to Sox9 promoter might be dependent on a hitherto unknown stress-regulated binding partner protein(s). It is also possible that stress-induced post-translational regulation of Zfp24 might drive promoter binding and Sox9 upregulation. Interestingly, studies15 have shown that a phosphorylated form of Zfp24 which does not bind to the DNA is the predominant form in oligodendrocyte progenitor cells. During maturation into oligodendrocytes, the non-phosphorylated, DNA-binding form accumulates and binds to promoter regions of target genes. Our studies show that a similar mechanism might be involved in kidneys since Zfp24 is dephosphorylated early during the development of AKI. Future studies are required to test if functional modulation of Zfp24 phosphorylation status influences Sox9 upregulation during AKI and to identify linked kinases and or phosphatases. Given that survival studies show that Zfp24 gene deletion cause higher injury than Sox9 gene deletion (Supplementary Figure S6), systemic analysis of Zfp24 target genes in RTECs is also required to identify other Sox9-independent genes. Furthermore, studies in inducible knockout mice strains could delineate the role of Zfp24 in the injury, repair, and AKI-to-CKD transition phases. Finally, Zfp24 is a widely expressed gene during development and in adult tissues and hence it would be interesting to examine if the Zfp24-Sox9 axis is also relevant in non-renal tissues.
Collectively, our studies have identified Zfp24 as a critical regulator of Sox9 upregulation during AKI. We propose that Zfp24-Sox9 axis might play a critical role in kidney tissue remodeling in response to stress and injury. Future studies into understanding how protective factors such as Zfp24 and Sox9 drive tissue repair and regeneration could lead to identification of therapeutic strategies to augment recovery and mitigate short- and long-term adverse sequelae of kidney injury.
Disclosure statement
All the authors declared no competing interests.
Supplementary Material
Acknowledgments
This work was supported by funds from the Ohio State University Cancer Center (NSP) and National Institutes of Health grants R03TR003686 (NSP), R01DK132230 (NSP and AB), R01DK117183 (AB), and K08DK123411 (SMM). JAS was supported by predoctoral fellowship from the American Heart Association (900765). AVLR was supported by the OSU College of Pharmacy Summer Undergraduate Research Fellowship (SURF). We sincerely thank Dr. Joseph C. Gigliotti (Liberty University, College of Osteopathic Medicine) for critical reading of the manuscript. The authors thank The Ohio State University Comprehensive Cancer Center (OSUCCC) for use of the following shared resources: genomics shared resource, microscopy shared resource, and the OSU University Laboratory Animal Resources (ULAR) for housing and care of animals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary materials
Supplementary Figure S1: Development of a promoter assay to examine Sox9 transcriptional regulation.
Supplementary Figure S2: Zfp24 gene expression during acute kidney injury.
Supplementary Figure S3: Zfp24 dephosphorylation is an early event during IRI-associated AKI.
Supplementary Figure S4: Effect of Zfp24 gene knockout on kidney function.
Supplementary Figure S5: Zfp24 gene deletion results in increased cell death during AKI.
Supplementary Figure S6: Zfp24 gene deletion in tubular epithelial cells reduces overall survival from rhabdomyolysis.
Supplementary Figure S7: Zfp24 gene deletion sensitizes primary tubular epithelial cells to cisplatin-induced cell death.
Supplementary Figure S8: Time-course analysis of Sox9 gene induction during rhabdomyolysis-associated AKI.
Supplementary Figure S9: Gene expression analysis of Sox9 target genes.
Supplementary Figure S10: Functional effect of Zfp24 binding site mutations on Sox9 promoter activation.
Supplementary Figure S11: Examination of Zfp24 binding to Sox9 promoter by electrophoresis mobility shift assay (EMSA).
Supplementary Figure S12: Effect of Sox9 promoter mutation on baseline kidney function.
Supplementary Figure S13: Prommut mice have increased tubular cell death during AKI.
Supplementary Figure S14: Egr1 expression is not altered in Zfp24 conditional knockout mice.
Supplementary Figure S15: Uncropped images of immunoblot data.
Supplementary methods.
Supplementary Table S1: Primer and probe sequences used in the current study.
Supplementary references.
References
- 1.Kamachi Y, Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development 2013; 140: 4129–4144. [DOI] [PubMed] [Google Scholar]
- 2.Bowles J, Schepers G, Koopman P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev Biol 2000; 227: 239–255. [DOI] [PubMed] [Google Scholar]
- 3.Lee YH, Saint-Jeannet JP. Sox9 function in craniofacial development and disease. Genesis 2011; 49: 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pritchett J, Athwal V, Roberts N, et al. Understanding the role of SOX9 in acquired diseases: lessons from development. Trends Mol Med 2011; 17: 166–174. [DOI] [PubMed] [Google Scholar]
- 5.Jo A, Denduluri S, Zhang B, et al. The versatile functions of Sox9 in development, stem cells, and human diseases. Genes Dis 2014; 1: 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun W, Cornwell A, Li J, et al. SOX9 Is an Astrocyte-Specific Nuclear Marker in the Adult Brain Outside the Neurogenic Regions. J Neurosci 2017; 37: 4493–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blache P, van de Wetering M, Duluc I, et al. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J Cell Biol 2004; 166: 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadaja M, Keyes BE, Lin M, et al. SOX9: a stem cell transcriptional regulator of secreted niche signaling factors. Genes Dev 2014; 28: 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao J, Patel J, Kaur S, et al. Sox9 and Rbpj differentially regulate endothelial to mesenchymal transition and wound scarring in murine endovascular progenitors. Nat Commun 2021; 12: 2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacraz GPA, Junker JP, Gladka MM, et al. Tomo-Seq Identifies SOX9 as a Key Regulator of Cardiac Fibrosis During Ischemic Injury. Circulation 2017; 136: 1396–1409. [DOI] [PubMed] [Google Scholar]
- 11.Kumar S, Liu J, Pang P, et al. Sox9 Activation Highlights a Cellular Pathway of Renal Repair in the Acutely Injured Mammalian Kidney. Cell Rep 2015; 12: 1325–1338. [DOI] [PubMed] [Google Scholar]
- 12.Kim JY, Bai Y, Jayne LA, et al. A kinome-wide screen identifies a CDKL5-SOX9 regulatory axis in epithelial cell death and kidney injury. Nat Commun 2020; 11: 1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JY, Bai Y, Jayne LA, et al. Involvement of the CDKL5-SOX9 signaling axis in rhabdomyolysis-associated acute kidney injury. Am J Physiol Renal Physiol 2020; 319: F920–F929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang HM, Huang S, Reidy K, et al. Sox9-Positive Progenitor Cells Play a Key Role in Renal Tubule Epithelial Regeneration in Mice. Cell Rep 2016; 14: 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elbaz B, Aaker JD, Isaac S, et al. Phosphorylation State of ZFP24 Controls Oligodendrocyte Differentiation. Cell Rep 2018; 23: 2254–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harper J, Yan L, Loureiro RM, et al. Repression of vascular endothelial growth factor expression by the zinc finger transcription factor ZNF24. Cancer Res 2007; 67: 8736–8741. [DOI] [PubMed] [Google Scholar]
- 17.Howng SY, Avila RL, Emery B, et al. ZFP191 is required by oligodendrocytes for CNS myelination. Genes Dev 2010; 24: 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinha D, Wang Z, Price VR, et al. Chemical anoxia of tubular cells induces activation of c-Src and its translocation to the zonula adherens. Am J Physiol Renal Physiol 2003; 284: F488–497. [DOI] [PubMed] [Google Scholar]
- 19.Sprowl JA, Ong SS, Gibson AA, et al. A phosphotyrosine switch regulates organic cation transporters. Nat Commun 2016; 7: 10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hukriede NA, Soranno DE, Sander V, et al. Experimental models of acute kidney injury for translational research. Nat Rev Nephrol 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JY, Jayne LA, Bai Y, et al. Ribociclib mitigates cisplatin-associated kidney injury through retinoblastoma-1 dependent mechanisms. Biochem Pharmacol 2020; 177: 113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pabla N, Gibson AA, Buege M, et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc Natl Acad Sci U S A 2015; 112: 5231–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JY, Bai Y, Jayne LA, et al. SOX9 promotes stress-responsive transcription of VGF nerve growth factor inducible gene in renal tubular epithelial cells. J Biol Chem 2020; 295: 16328–16341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bai Y, Kim JY, Bisunke B, et al. Kidney toxicity of the BRAF-kinase inhibitor vemurafenib is driven by off-target ferrochelatase inhibition. Kidney Int 2021; 100: 1214–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zavorka Thomas ME, Jeon JY, Talebi Z, et al. Gilteritinib-induced upregulation of S100A9 is mediated through BCL6 in acute myeloid leukemia. Blood Adv 2021; 5: 5041–5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Oosterwijk JG, Buelow DR, Drenberg CD, et al. Hypoxia-induced upregulation of BMX kinase mediates therapeutic resistance in acute myeloid leukemia. J Clin Invest 2018; 128: 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pabla N, Bhatt K, Dong Z. Checkpoint kinase 1 (Chk1)-short is a splice variant and endogenous inhibitor of Chk1 that regulates cell cycle and DNA damage checkpoints. Proc Natl Acad Sci U S A 2012; 109: 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pabla N, Huang S, Mi QS, et al. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J Biol Chem 2008; 283: 6572–6583. [DOI] [PubMed] [Google Scholar]
- 29.Pabla N, Dong G, Jiang M, et al. Inhibition of PKCdelta reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J Clin Invest 2011; 121: 2709–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirita Y, Wu H, Uchimura K, et al. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci U S A 2020; 117: 15874–15883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwano M, Plieth D, Danoff TM, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002; 110: 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kellum JA, Romagnani P, Ashuntantang G, et al. Acute kidney injury. Nat Rev Dis Primers 2021; 7: 52. [DOI] [PubMed] [Google Scholar]
- 33.Zuk A, Bonventre JV. Acute Kidney Injury. Annu Rev Med 2016; 67: 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoue T, Tanaka S, Okusa MD. Neuroimmune Interactions in Inflammation and Acute Kidney Injury. Front Immunol 2017; 8: 945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reeves WB. Innate Immunity in Nephrotoxic Acute Kidney Injury. Trans Am Clin Climatol Assoc 2019; 130: 33–40. [PMC free article] [PubMed] [Google Scholar]
- 36.Ikizler TA, Parikh CR, Himmelfarb J, et al. A prospective cohort study of acute kidney injury and kidney outcomes, cardiovascular events, and death. Kidney Int 2021; 99: 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linkermann A, Chen G, Dong G, et al. Regulated cell death in AKI. J Am Soc Nephrol 2014; 25: 2689–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cummings BS, Schnellmann RG. Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways. J Pharmacol Exp Ther 2002; 302: 8–17. [DOI] [PubMed] [Google Scholar]
- 39.Bajwa A, Kinsey GR, Okusa MD. Immune mechanisms and novel pharmacological therapies of acute kidney injury. Curr Drug Targets 2009; 10: 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Namwanje M, Bisunke B, Rousselle TV, et al. Rapamycin Alternatively Modifies Mitochondrial Dynamics in Dendritic Cells to Reduce Kidney Ischemic Reperfusion Injury. Int J Mol Sci 2021; 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 2002; 110: 835–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pan Y, Cao S, Terker AS, et al. Myeloid cyclooxygenase-2/prostaglandin E2/E-type prostanoid receptor 4 promotes transcription factor MafB-dependent inflammatory resolution in acute kidney injury. Kidney Int 2022; 101: 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int 2007; 72: 151–156. [DOI] [PubMed] [Google Scholar]
- 44.Singh P, Ricksten SE, Bragadottir G, et al. Renal oxygenation and haemodynamics in acute kidney injury and chronic kidney disease. Clin Exp Pharmacol Physiol 2013; 40: 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tiwari R, Kapitsinou PP. Role of Endothelial Prolyl-4-Hydroxylase Domain Protein/Hypoxia-Inducible Factor Axis in Acute Kidney Injury. Nephron 2021: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naved BA, Bonventre JV, Hubbell JA, et al. Kidney repair and regeneration: perspectives of the NIDDK (Re)Building a Kidney consortium. Kidney Int 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chawla LS, Eggers PW, Star RA, et al. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 2014; 371: 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sears S, Siskind L. Potential Therapeutic Targets for Cisplatin-Induced Kidney Injury: Lessons from Other Models of AKI and Fibrosis. J Am Soc Nephrol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Docherty MH, O’Sullivan ED, Bonventre JV, et al. Cellular Senescence in the Kidney. J Am Soc Nephrol 2019; 30: 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 2015; 11: 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerhardt LMS, Liu J, Koppitch K, et al. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc Natl Acad Sci U S A 2021; 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu J, Kumar S, Dolzhenko E, et al. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2017; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar S. Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int 2018; 93: 27–40. [DOI] [PubMed] [Google Scholar]
- 54.Raza S, Jokl E, Pritchett J, et al. SOX9 is required for kidney fibrosis and activates NAV3 to drive renal myofibroblast function. Sci Signal 2021; 14. [DOI] [PubMed] [Google Scholar]
- 55.Cassandri M, Smirnov A, Novelli F, et al. Zinc-finger proteins in health and disease. Cell Death Discov 2017; 3: 17071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Edelstein LC, Collins T. The SCAN domain family of zinc finger transcription factors. Gene 2005; 359: 1–17. [DOI] [PubMed] [Google Scholar]
- 57.Li J, Chen X, Yang H, et al. The zinc finger transcription factor 191 is required for early embryonic development and cell proliferation. Exp Cell Res 2006; 312: 3990–3998. [DOI] [PubMed] [Google Scholar]
- 58.Chen JW, Huang MJ, Chen XN, et al. Transient upregulation of EGR1 signaling enhances kidney repair by activating SOX9(+) renal tubular cells. Theranostics 2022; 12: 5434–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.