

Abstract
Context
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of the COVID-19 infection and responsible for millions of victims worldwide, remains a significant threat to public health. Even after the development of vaccines, research interest in the emergence of new variants is still prominent. Currently, the focus is on the search for effective and safe drugs, given the limitations and side effects observed for the synthetic drugs administered so far. In this sense, bioactive natural products that are widely used in the pharmaceutical industry due to their effectiveness and low toxicity have emerged as potential options in the search for safe drugs against COVID-19. Following this line, we screened 10 bioactive compounds derived from cholesterol for molecules capable of interacting with the receptor-binding domain (RBD) of the spike protein from SARS-CoV-2 (SC2Spike), responsible for the virus’s invasion of human cells. Rounds of docking followed by molecular dynamics simulations and binding energy calculations enabled the selection of three compounds worth being experimentally evaluated against SARS-CoV-2.
Methods
The 3D structures of the cholesterol derivatives were prepared and optimized using the Spartan 08 software with the semi-empirical method PM3. They were then exported to the Molegro Virtual Docking (MVD®) software, where they were docked onto the RBD of a 3D structure of the SC2Spike protein that was imported from the Protein Data Bank (PDB). The best poses obtained from MVD® were subjected to rounds of molecular dynamics simulations using the GROMACS software, with the OPLS/AA force field. Frames from the MD simulation trajectories were used to calculate the ligand’s free binding energies using the molecular mechanics – Poisson-Boltzmann surface area (MM-PBSA) method. All results were analyzed using the xmgrace and Visual Molecular Dynamics (VMD) software.
Supplementary information
The online version contains supplementary material available at 10.1007/s00894-023-05586-5.
Keywords: SARS-CoV-2, Natural products, Molecular dynamics, Spike protein
Introduction
The COVID-19 pandemic, which began in 2020 and has already resulted in millions of victims worldwide, is caused by a viral disease known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Since the outbreak of the pandemic, laboratories from around the world have collaborated to quickly sequence the entire viral genome and understand its pathobiology [1]. However, the development of effective drugs without strong side effects has not yet occurred due to the many time-consuming requirements and steps that must be followed in a typical drug discovery process [2]. One strategy used to accelerate this process during the coronavirus pandemic was to repurpose previously known commercial antivirals, such as lopinavir, ritonavir, and remdesivir. However, this approach has not shown much success [3–5]. Even after the development of vaccines, the interest in a safe and effective drug against COVID-19 is still prominent due to concerns surrounding the emergence of new variants. In light of this, many fundamental aspects of the SARS-CoV-2 infectious cycle remain a focus of scientific interest in order to explore potential drug targets. One commonly used approach is to search for factors that can interfere with the formation of a fusion-competent virus [6].
Many early studies have focused on the entry of SARS-CoV-2 into the cell, where a trimeric glycoprotein known as spike (which gives the viral envelope its crown appearance) and the human angiotensin-converting enzyme-2 (HssACE-2) play an essential role. The association of these two proteins underlies the adhesion of the virus to the cell, which results in conformational changes in the spike protein, triggering the fusion machinery to infiltrate the cell [6]. The SARS-CoV-2 spike protein (SC2Spike) is a homotrimer, with each monomer containing a receptor-binding domain (RBD). It is located on the outer surface of the virus and directly binds to the peptidase domain of HssACE-2, promoting the entry of the virus into the cell [3, 7]. While receptor binding plays an important role, an alternative hypothesis is that these proteins exhibit an intrinsic preference for membrane domains of specific lipid composition. In fact, certain viruses require an association between specific receptor proteins and lipids to trigger endocytosis. So, if fusion is related to the composition of the viral membrane and the host cell, this information suggests the potential therapeutic use of molecules like cholesterol for the treatment of COVID-19 [6]. In fact, vitamin D, a cholesterol-like molecule, has already shown promising results in the treatment of COVID-19 [8–11].
Cholesterol preferentially interacts with certain membrane proteins, especially those modified with specific lipid fractions [12–14]. One study pointed out the key role of lipidic structure and composition in spike-mediated membrane fusion, suggesting that many classes of compounds might be implicated in the disruption of the lipid bilayer. These lipid-targeting drugs are responsible for disrupting spike-mediated membrane fusion. Furthermore, since viral membranes are produced by the membranes of infected host cells, lipid-targeted treatments can disrupt the formation of viral particles required for fusion [6]. One proposed strategy is the use of drugs for hyperlipidemia, such as statins, which have already been evaluated in terms of their effects on COVID-19 patients. These studies have shown significant reductions in mortality associated with low cholesterol levels [15, 16]. This can mainly explain why certain factors related to cholesterol (such as menopause and obesity) are associated with increased risk in COVID-19, as they are similarly involved in cholesterol metabolism. Therefore, for the present study, we selected 10 natural products (listed in Table S1) with chemical features similar to commercially used statins, such as simvastatin, lovastatin, and pravastatin. These compounds have a decalin system with a change in the ring junction stereochemistry and different substituents in a position equivalent to the 3-hydroxy group of cholesterol. This investigation aims to establish the relationship between the reduction of cholesterol and effective treatment of COVID-19 through lipid modification. Therefore, a virtual screening (VS) study was conducted on the bioactive compounds listed in Table S1 to identify molecules capable of targeting SC2Spike. The VS was followed by docking, molecular dynamics (MD) simulations, and binding energy calculations to assess the potential affinities of the ligands for the RBD domain of SC2Spike. This approach involves the structure-based drug design method [17, 18], which is recurrent in the literature and has been successfully applied to the search for potential drug candidates against several SARS-CoV-2 targets [19–24].
Methodology
The three-dimensional structure of SC2Spike used in this study was that complexed with the LCB1 polypeptide inhibitor available in the Protein Data Bank (PDB) online server (www.rcsb.org), under the code 7JZU [3]. This crystallographic structure was downloaded and optimized for docking studies using the Molegro Virtual Docker (MVD®) program [25]. The three-dimensional structures of the 10 potential ligands (listed in Table S1) were built and optimized using the Spartan 08 software with the semi-empirical method PM3 [26]. After calculating the atoms’ partial charges using the natural population analysis method [27], the ligands’ three-dimensional structures were exported to MVD® [25].
Molecular docking
As in previous studies [22, 23], the search space for molecular docking was defined to include all residues of the RBD of SC2Spike that interact with HssACE-2: Lys417, Gly446, Tyr449, Tyr453, Leu455, Phe456, Ala475, Phe486, Asn487, Tyr489, Gln493, Gly496, Gln498, Thr500, Asn501, Gly502, and Tyr505 [3, 7]. The search space was a sphere with a radius of 23 Å and its center located at coordinates x = 215, y = 211, and z = 217.25. Each of the 10 ligands shown in Table S1 was docked into this search space using the same docking protocol as before [22], with 6 molecular docking runs conducted and the best 30 poses per run returned, resulting in 180 poses per ligand to be analyzed. The best-ranked poses, based on their MolDock scores (which estimate protein–ligand interaction energy) and the establishment of intermolecular interactions with residues of the RBD, were selected for molecular dynamics (MD) simulations. Figures were created using the MVD® [25] and MOE® (https://www.chemcomp.com/Products.htm) software, and the results were analyzed as reported in previous literature [28–30].
Molecular dynamics simulations
The best poses selected from the docking runs were processed using the Open Babel [31] and the Antechamber Python Parser Interface (ACPYPE) [32] programs to generate the corresponding topology and coordinate files in the.gro format, which could be recognized by the OPLS/AA force field [33, 34] for conducting the MD simulations. Similarly, the topology and coordinate files of the protein were generated using the pdb2gmx routine of GROMACS 2019.4 [35, 36] that were compatible with the OPLS/AA force field [33, 34]. Each protein–ligand complex was then centered in a dodecahedron box with a volume of nearly 400 nm3, which was filled with approximately 12,000 TIP3P water molecules [37], under periodic boundary conditions [38], while maintaining a minimum distance of 1 nm between the solute and the box wall. The systems were then subjected to two energy minimization steps of 100 ps each to relax them. The first step was performed with position restraint (PR) of protein and ligand to accommodate the water molecules around the complex, while the second step was performed without PR to reach a local minimum of the potential energy surface. Both energy minimization steps were carried out using the steepest descent algorithm with a convergence criterion of 100.0 kJ mol−1 nm−1. Two stages of equilibration, each of 100 ps, were then carried out to bring the system to 310 K and 1 bar, simulating physiological conditions. The first equilibration was performed in the NVT ensemble (constant number of particles, volume, and temperature), while the second was performed in the NPT ensemble (constant number of particles, pressure, and temperature). Temperature and pressure stability were maintained using the velocity-rescale thermostat [39] and the Parrinello-Rahman pressure coupling method [40], respectively. Finally, the MD simulation production step was conducted with a simulation time of 100 ns at 310 K and 1 bar using an integration time of 2 fs. Complex coordinates and energy data were saved every 10 ps to enable further analysis of properties, such as total energy, root mean square deviation (RMSD), and the average number of hydrogen bonds formed during the simulation. The stability of temperature and pressure was maintained using the velocity-rescale thermostat and the Parrinello-Rahman pressure coupling method, respectively. Results of the MD simulations were analyzed through plots of RMSD, root mean square deviation fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and H-bonds, similar to works already reported in the literature [22, 23, 41, 42]. Graphs and figures were generated using xmgrace 5.1.25 and Visual Molecular Dynamics 1.9.3 (VMD) [43].
Free energy calculations (MM-PBSA)
Molecular mechanics – Poisson-Boltzmann surface area (MM-PBSA) studies were carried out for each ligand submitted to MD simulation using the g_mmpbsa tool [44], which is compatible with GROMACS 4.6.5 [45]. To perform the analysis, 250 frames of the MD trajectory were extracted at regular intervals of 400 ps. A binary file with the extension.tpr was then generated using the grompp tool (GROMACS preprocessor). The.tpr file contained the initial structure for the simulation, the molecular topology, and all the simulation parameters necessary for the use of g_mmpbsa and was compatible with the previous version of GROMACS. The protein–ligand interaction energy was estimated by considering the potential energy variation in vacuum, the energy related to polar solvation, and the energy related to nonpolar solvation, with the latter using SASA. Additionally, decomposition analysis, similar to works reported in the literature [21, 42, 45], was performed to identify the most relevant residues contributing to the ligand’s binding to the RBD of SC2Spike after stabilization from the MD simulation.
Results and discussion
Molecular docking
Analysis of the poses obtained from the docking studies resulted in the selection of three ligands listed in Table 1. Besides being the best-ranked (showing poses with more negative energy values), these ligands also showed multiple hydrogen bonds (H-bonds) with RBD residues. The MolDock score values estimated by MVD® [25] (Table 1) suggest that ligands A and B are the most promising, as they scored the most negative, suggesting more stable interactions with the protein. Three poses were selected for ligand A because, despite being located in distinct regions of the SC2Spike (see Fig. 1), they ranked well in terms of energy and, at the same time, showed interactions with RBD residues. This suggests that this ligand has the potential to bind to the RBD domain in three distinct orientations, increasing the chances of disrupting the binding between SC2Spike and HssACE-2.
Table 1.
Docking results

Fig. 1.
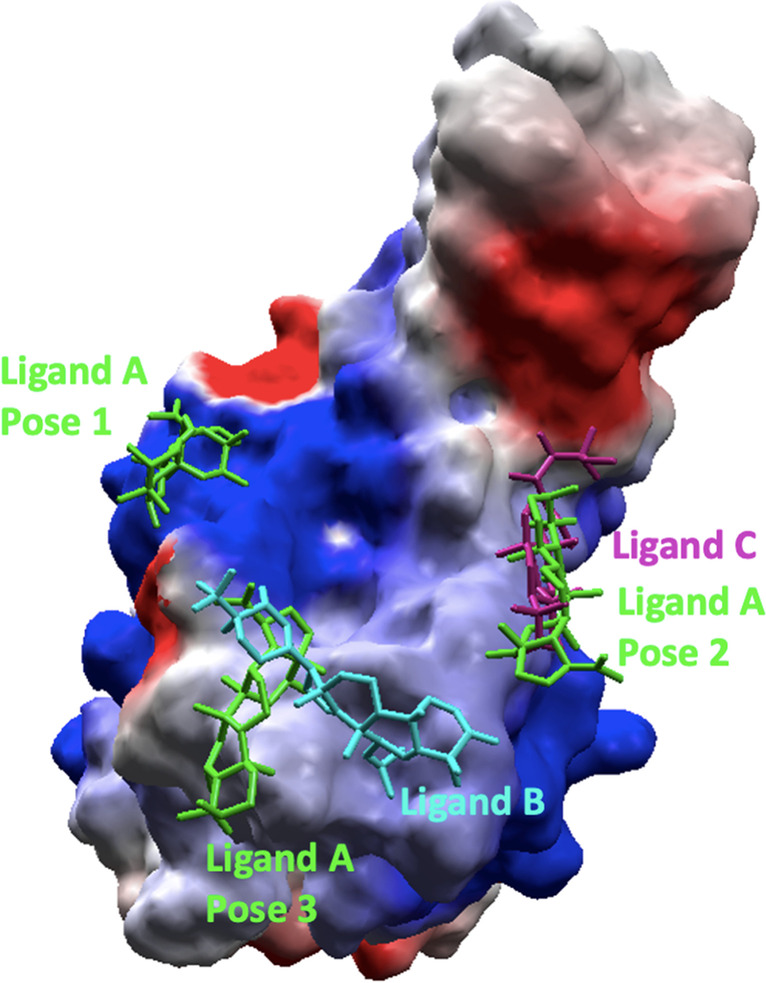
Location of the best poses from the docking over SC2Spike. Ligand A is shown in green, ligand B in blue, and ligand C in pink. The protein is represented as an electrostatic surface
Figure 1 shows the binding pockets in SC2Spike for the poses listed in Table 1, while Fig. 2 details the interactions observed for these poses. As can be seen, the poses of ligands B, together with the best-ranked pose of ligand A (pose 3), are in the same pocket where the RBD residues Gly446, Gln498, Thr500, and Asn501 can be found (see Fig. 2). This result suggests that this pocket is more favorable for interactions of those ligands with SC2Spike. Other possible binding pockets were identified by ligand C and poses 1 and 2 of ligand A. These regions contain the RBD residues Lys417, Tyr449, Gln493, and Tyr505 (see Fig. 2).
Fig. 2.

Interactions of the best poses over SC2Spike
Molecular dynamics
The 5 poses shown in Figs. 1 and 2 were subjected to rounds of MD simulations, following the protocol described in the “Methodology” section, to evaluate their behavior over time. Figures S1 and 3 show the plots of total energy and RMSD over time for each simulated system, while the averaged RMSDs for protein and ligand can be found in Table 2. Analysis of these graphs and the table reveals energy stabilization since the beginning of the simulation at very negative values, around − 4.0 × 105 kJ/mol (see Figure S1), and average fluctuations below 4.2 Å and 2.0 Å for protein and ligand, respectively. These results indicate stable dynamic behavior of all systems over time, with the ligands stabilizing over the RBD domain of SC2Spike.
Table 2.
Average values of RMSD (protein and ligand), Rg, SASA, and intra- and inter-H-bond numbers of the spike-ligand complexes during the MD simulations
| Compound | RMSD (nm) | Rg (nm) | SASA (nm2) | H-bonda | H-bondb | |
|---|---|---|---|---|---|---|
| Protein | Ligand | |||||
|
Ligand A Pose 1 |
3.6 ± 0.5 | 0.49 ± 0.07 | 1.87 ± 0.02 | 108.8 ± 2.5 | 119.2 ± 5.3 | 1.2 ± 0.8 |
|
Ligand A Pose 2 |
3.5 ± 0.4 | 0.48 ± 0.08 | 1.82 ± 0.01 | 109.5 ± 2.0 | 117.4 ± 5.2 | 1.2 ± 1.2 |
|
Ligand A Pose 3 |
3.5 ± 0.7 | 0.50 ± 0.10 | 1.87 ± 0.02 | 110.8 ± 2.2 | 113.2 ± 5.2 | 0.7 ± 0.9 |
| Ligand B | 3.0 ± 0.3 | 1.87 ± 0.12 | 1.85 ± 0.01 | 110.2 ± 3.2 | 115.8 ± 6.8 | 0.7 ± 0.9 |
| Ligand C | 4.1 ± 0.5 | 1.02 ± 0.30 | 1.81 ± 0.01 | 109.5 ± 1.7 | 118.5 ± 5.2 | 2.1 ± 1.5 |
aAverage number of intra-protein H-bonds observed during the MD simulation
bAverage number of protein–ligand H-bonds observed during the MD simulation
Figure 3 and Table 2 also show that ligands A and C are the ones presenting the lowest fluctuations during the simulations, with values around 0.5 Å for all poses of ligand A and 1.0 Å for ligand C. Ligand B, on the other hand, showed larger fluctuations ranging around 2.0 Å. This suggests a better dynamic behavior and stabilization of ligands A and C over the RBD domain of SC2Spike compared to ligand B.
Fig. 3.
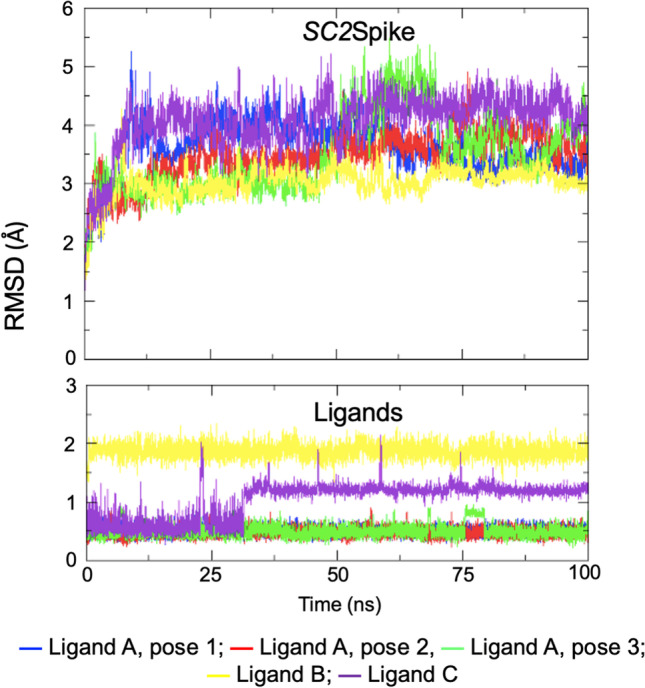
Plots of RMSD for SC2Spike and the ligands during the MD simulations
The Rg values were calculated to verify the compactness of the protein–ligand complexes during the MD simulations (Table 2). The complexes with ligand C and ligand A (pose 2) showed lower Rg values, while the complexes with ligand B and the other poses of ligand A showed greater Rg values. Additionally, the average SASA values calculated for the complexes (Table 2) show that smaller values were observed for ligand A (pose 2) and ligand C. Furthermore, the plots of RMSF variation shown in Figure S2 reveal higher fluctuations of the SC2Spike/Ligand B complex compared to the complexes with ligands A (pose 2) and C. These results align with the RMSD and total energy results, indicating that the complexes with ligands A and C are more stable.
The stability of each SC2Spike-ligand complex was also assessed through the plot of intra-protein H-bonds shown in Fig. 4 and averaged in Table 2. As can be seen, the complexes with ligand A (poses 1 and 2) and ligand C were the ones showing the highest numbers of H-bonds. This is aligned with the intermolecular H-bonds formed by the ligands (see Fig. 5, Tables 2 and 3) where one can see that ligand A (poses 1 and 2) and ligand C showed more H-bonds with an average number of 2.1 and 1.2, respectively, and also a higher % of occupancy, forming more H-bonds over time with residues of the RBD domain of SC2Spike compared to ligand B.
Fig. 4.
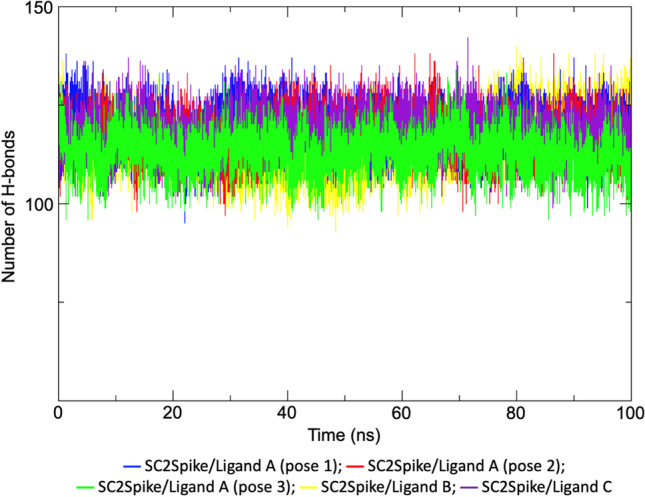
Plots of variation of intra-protein H-bonds during the MD simulations
Fig. 5.
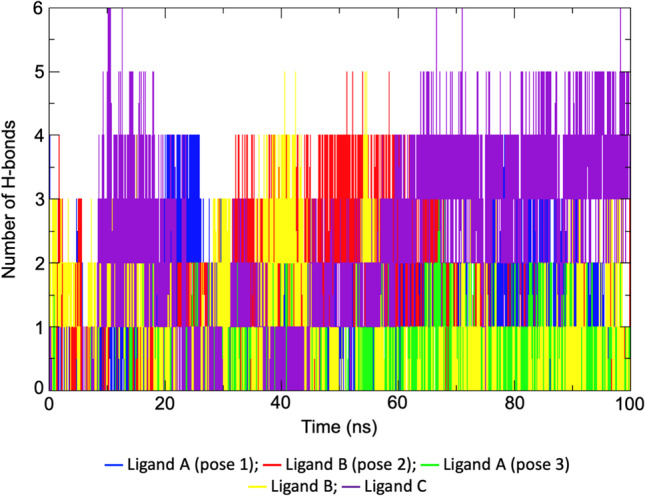
Plot of the variation of protein–ligand H-bonds formed during the MD simulations
Table 3.
% occupancy of H-bonds between the ligands and SC2Spike during the MD simulations. Only residues forming H-bonds for more than 10% of the simulation time are listed
| Ligand | Interacting residues | |
|---|---|---|
| Acceptor | Donor | |
| Ligand A (pose 1) | Thr376 (12.8%), Val503 (19.5%) | Thr376 (25.5%) |
| Ligand A (pose 2) | Tyr449 (13.2%), Phe490 (25.1%), Ser494 (25.1%), Gly482 (20.3%), Val483 (10.4%), Cys480 (11.6%) | Ser494 (10.4%), Gly482 (19.2%) |
| Ligand A (pose 3) | Tyr489 (26.1%), Leu492 (15.0%) | –– |
| Ligand B | Gly404 (33.5%), Tyr508 (17.9%), Val503 (12.0%) | Arg408 (14.3%) |
| Ligand C | Phe490 (10.8%), Tyr449 (21.1%), Gly482 (37.5%), Asn481 (11.6%), Val483 (10.4%) | Arg346 (24.7%), Gly482 (27.1%), Lys444 (74.9%), Tyr449 (12.8%) |
Free energy calculations (MM-PBSA)
Figure 6 shows the plots of binding energy calculated by the MM-PBSA method for all ligands during the MD simulation. The very negative values below − 60 kJ/mol observed for all ligands corroborate the docking and MD simulation results and point to those compounds as good binders to SC2Spike. Comparison among the compounds reveals that ligand C was the one showing the lowest value of binding energy, followed by poses 1 and 2 of ligand A, ligand B, and, finally, pose 3 of ligand A. Despite showing the worse energy result in the docking study (see Table 1), this ligand was the one showing the most H-bonds during the MD simulation studies. Also, it showed RMSD values similar to the poses of ligand A, after accommodating in a new position, around 30 ns of simulation (see RMSD plot in Fig. 3). Regarding ligand A, the MM-PBSA and MD simulation results corroborate and suggest that the binding pockets of poses 1 and 2 are more favorable for the binding of this ligand to SC2Spike, despite the better docking results shown for pose 3. The decomposition analysis of the residues’ individual contributions to the binding energy (Table 4) reveals that ligand A (pose 2) and ligand C were the ones with the most contributions with the lowest energetic values being observed for the RBD residue Tyr449 in both cases (− 17.1 and − 21.5 kJ/mol, respectively).
Fig. 6.
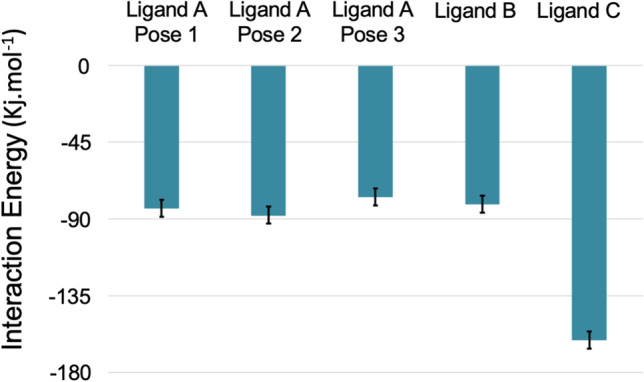
Plots of binding energy calculated based on the MM-PBSA method
Table 4.
Decomposition analysis of the binding energy (kJ/mol) per residue
| Ligand A (pose 1) | Ligand A (pose 2) | Ligand A (pose 3) | Ligand B | Ligand C | |
|---|---|---|---|---|---|
| Thr376 | − 17.6 ± 0.3 | ||||
| Gly404 | − 7.7 ± 0.3 | ||||
| Arg408 | − 15.7 ± 0.7 | ||||
| Tyr449 | − 17.1 ± 0.5 | − 21.5 ± 0.4 | |||
| Cys480 | − 7.4 ± 0.2 | ||||
| Asn481 | − 11.4 ± 0.3 | ||||
| Gly482 | − 11.2 ± 0.2 | − 13.4 ± 0.4 | |||
| Val483 | − 10.6 ± 0.2 | − 4.8 ± 0.4 | |||
| Tyr489 | − 20.4 ± 0.5 | ||||
| Phe490 | − 15.4 ± 0.3 | − 14.3 ± 0.4 | |||
| Leu492 | − 10.6 ± 0.6 | ||||
| Ser494 | − 12.0 ± 0.3 | ||||
| Val503 | − 8.7 ± 0.3 | − 9.4 ± 0.3 | |||
| Tyr508 | − 11.8 ± 0.5 |
Conclusion
Computational methods used to study protein–ligand interactions cannot replace experimental studies, but they have the potential to filter results and guide further experiments, saving time and resources. In this study, molecular docking, MD simulations, and MM-PBSA calculations were used to assess the binding potential of cholesterol-derived molecules to the RBD domain of the SC2Spike protein. The results showed that three of the 10 evaluated molecules had good docking results and could adopt stable conformations and interact with RBD domain residues. The MD simulations corroborated the binding potential of these three compounds and ranked ligands A and C as the most promising candidates for further experimental evaluation against the binding between SC2Spike and HssACE-2 to prevent SARS-CoV-2 entry into human cells. The study also suggests that ligand A has the potential to bind to three distinct parts of the RBD domain, increasing the likelihood of disrupting interactions between SC2Spike and HssACE-2.
Supplementary information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors wish to thank the Instituto Militar de Engenharia (IME) for the infrastructure and overall support and the Federal University of Lavras (UFLA) – Brazil for software availability. This was also supported by the university of Hradec Kralové. S.R.L and T.C.C.F also wish to thank CQDM - Canada for support.
Author contribution
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Rayssa Ribeiro, Fernanda D. Botelho, Amanda M. V. Pinto, Antonia M. A. La Torre, and Joyce S. F. D. Almeida. The first draft of the manuscript was written by Rayssa Ribeiro, Fernanda D. Botelho, and Marcelo C. Santos. The final version was written by Tanos C. C. Franca and Valdir F. Veiga Jr. The project was supported by Tanos C. C. Franca, Steven R. LaPlante, Valdir F. Veiga Jr., and Marcelo C. Santos. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
Funding was provided by the Conselho Nacional de Pesquisa (CNPq) (Grant Numbers: 305432/2022–2-PQ2 and 310782/2022-8-PQ1) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (Grant Numbers: E-02/202.961/2017 - CNE; E-26/200.512/2023-CNE, and E-26/211.315/2021-TEMATICOS).
Data availability
Additional data from this work will be made available upon request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jo S, Kim S, Shin DH, et al. Inhibition of SARS-CoV 3CL protease by flavonoids. J Enzym Inhib Med Ch. 2020;35(1):145–151. doi: 10.1080/14756366.2019.1690480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bambini S, Rappuoli R. The use of genomics in microbial vaccine development. Drug Discov Today. 2009;14(5–6):252–260. doi: 10.1016/j.drudis.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao L, Goreshnik I, Coventry B, et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science. 2020;370(6515):426–431. doi: 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.da Silva AA, Wiedemann LSM, Veiga-Junior VF. Natural products’ role against COVID-19. RSC Adv. 2020;10(39):23379–23393. doi: 10.1039/D0RA03774E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sepay N, Sekar A, Halder UC, et al. Anti-COVID-19 terpenoid from marine sources: a docking, admet and molecular dynamics study. J Mol Struct. 2021;1228:129433. doi: 10.1016/j.molstruc.2020.129433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanders DW, Jumper CC, Ackerman PJ, et al. SARS-CoV-2 requires cholesterol for viral entry and pathological syncytia formation. Elife. 2021;10:e65962. doi: 10.7554/eLife.65962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan R, Zhang Y, Li Y, et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camargo CA, Jr, Martineau AR. Vitamin D to prevent COVID-19: recommendations for the design of clinical trials. FEBS J. 2020;287(17):3689–3692. doi: 10.1111/febs.15534. [DOI] [PubMed] [Google Scholar]
- 9.Speeckaert M, Speeckaert R, Delanghe J. Vitamin D and vitamin D binding protein: the inseparable duo in COVID-19. J Endocrinol Invest. 2021;44:2323–2324. doi: 10.1007/s40618-021-01573-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alshahawey M. A genetic insight into vitamin D binding protein and COVID-19. Med Hypotheses. 2021;149:110531. doi: 10.1016/j.mehy.2021.110531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meltzer DO, Best TJ, Zhang H, et al. Solway, association of vitamin D status and other clinical characteristics with COVID-19 test results. JAMA Netw open. 2020;3(9):e2019722–e2019722. doi: 10.1001/jamanetworkopen.2020.19722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levental I, Lingwood D, Grzybek M, et al. Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc Natl Acad Sci. 2010;107(51):22050–22054. doi: 10.1073/pnas.1016184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin BR. Chemical approaches for profiling dynamic palmitoylation. Biochem Soc Trans. 2013;41(1):43–49. doi: 10.1042/BST20120271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sobocińska J, Roszczenko-Jasińska P, Ciesielska A, et al. Protein palmitoylation and its role in bacterial and viral infections. Front Immunol. 2018;8:2003. doi: 10.3389/fimmu.2017.02003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daniels LB, Sitapati AM, Zhang J, et al. Relation of statin use prior to admission to severity and recovery among COVID-19 inpatients. Am J Card. 2020;136:149–155. doi: 10.1016/j.amjcard.2020.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang XJ, Qin JJ, Cheng X, et al. In-hospital use of statins is associated with a reduced risk of mortality among individuals with COVID-19. Cell Metab. 2020;32(2):176–187. e4. doi: 10.1016/j.cmet.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andricopulo AD, Salum LB, Abraham DJ. Structure-based drug design strategies in medicinal chemistry. Curr Top Med Chem. 2009;9(9):771–790. doi: 10.2174/156802609789207127. [DOI] [PubMed] [Google Scholar]
- 18.Reynolds CH. Impact of computational structure-based methods on drug discovery. Curr Pharm Des. 2014;20(20):3380–3386. doi: 10.2174/138161282020140528105532. [DOI] [PubMed] [Google Scholar]
- 19.Dhankhar P, Dalal V, Kumar V. Screening of severe acute respiratory syndrome coronavirus 2 RNA-dependent RNA polymerase inhibitors using computational approach. J Comput Biol. 2021;28(12):1228–1247. doi: 10.1089/cmb.2020.0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar KA, Sharma M, Dalal V, et al. Multifunctional inhibitors of SARS-CoV-2 by MM/PBSA, essential dynamics, and molecular dynamic investigations. J Mol GraphModel. 2021;107:107969. doi: 10.1016/j.jmgm.2021.107969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhankhar P, Dalal V, Singh V, et al. Computational guided identification of novel potent inhibitors of N-terminal domain of nucleocapsid protein of severe acute respiratory syndrome coronavirus 2. J Biomol Struct Dyn. 2022;40(9):4084–4099. doi: 10.1080/07391102.2020.1852968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almeida JSFD, Botelho FD, de Souza FR, et al. Searching for potential drugs against SARS-CoV-2 through virtual screening on several molecular targets. J Biomol Struct Dyn. 2022;40(11):5229–5242. doi: 10.1080/07391102.2020.1869096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aghamohammadi M, Sirouspour M, Goncalves AS, et al. Modeling studies on the role of vitamins B1 (thiamin), B3 (nicotinamide), B6 (pyridoxamine), and caffeine as potential leads for the drug design against COVID-19. J Mol Model. 2022;28(12):380. doi: 10.1007/s00894-022-05356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farahani MD, França TCC, Alapour S, et al. Jumping from fragment to drug via smart scaffolds. ChemMedChem. 2022;17(10):e202200092. doi: 10.1002/cmdc.202200092. [DOI] [PubMed] [Google Scholar]
- 25.Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem. 2006;49(11):3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 26.Stewart JJ. Optimization of parameters for semiempirical methods IV: extension of MNDO, AM1, and PM3 to more main group elements. J Mol Model. 2004;10(2):155–164. doi: 10.1007/s00894-004-0183-z. [DOI] [PubMed] [Google Scholar]
- 27.Reed AE, Weinstock RB, Weinhold F. Natural population analysis. J Chem Phys. 1985;83:735–746. doi: 10.1063/1.449486. [DOI] [Google Scholar]
- 28.Singh V, Dhankhar P, Dalal V, et al. Drug-repurposing approach to combat Staphylococcus aureus: biomolecular and binding interaction study. ACS Omega. 2022;7(43):38448–38458. doi: 10.1021/acsomega.2c03671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh V, Dhankhar P, Dalal V, et al. In-silico functional and structural annotation of hypothetical protein from Klebsiella pneumonia: a potential drug target. J Mol Graph Model. 2022;116:108262. doi: 10.1016/j.jmgm.2022.108262. [DOI] [PubMed] [Google Scholar]
- 30.Kumari R, Rathi R, Pathak SR, et al. Computational investigation of potent inhibitors against YsxC: structure-based pharmacophore modeling, molecular docking, molecular dynamics, and binding free energy. J Biomol Struct Dyn. 2023;41(3):930–941. doi: 10.1080/07391102.2021.2015446. [DOI] [PubMed] [Google Scholar]
- 31.O’Boyle NM, Banck M, James CA, et al. Open Babel: an open chemical toolbox. J Cheminformatics. 2011;3(1):1–14. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Da Silva AWS, Vranken WF. ACPYPE-Antechamber python parser interface. BMC Res Notes. 2012;5(1):1–8. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236. doi: 10.1021/ja9621760. [DOI] [Google Scholar]
- 34.Kaminski GA, Friesner RA, Tirado-Rives J, et al. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105(28):6474–6487. doi: 10.1021/jp003919d. [DOI] [Google Scholar]
- 35.Abraham MJ, Murtola T, Schulz R. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 36.Pronk S, Páll S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 38.Van Gunsteren WF, Berendsen HJ. Computer simulation of molecular dynamics: methodology, applications, and perspectives in chemistry. Angew Chem Int Ed English. 1990;29(9):992–1023. doi: 10.1002/anie.199009921. [DOI] [Google Scholar]
- 39.Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126:014101 [DOI] [PubMed]
- 40.Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52(12):7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 41.Gupta DN, Dalal V, Savita BK, et al. In-silico screening and identification of potential inhibitors against 2Cys peroxiredoxin of Candidatus Liberibacter asiaticus. J Biomol Struct Dyn. 2022;40(19):8725–8739. doi: 10.1080/07391102.2021.1916597. [DOI] [PubMed] [Google Scholar]
- 42.Kumari R, Dalal V. Identification of potential inhibitors for LLM of Staphylococcus aureus: structure-based pharmacophore modeling, molecular dynamics, and binding free energy studies. J Biomol Struct Dyn. 2022;40(20):9833–9847. doi: 10.1080/07391102.2021.1936179. [DOI] [PubMed] [Google Scholar]
- 43.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 44.Kumari R, Kumar R; O.S.D.D. Consortium et al (2014) g _ mmpbsa - a GROMACS tool for MM-PBSA and its optimization for high-throughput binding energy calculations. J Chem Inf Model 54(7):1951–1962 [DOI] [PubMed]
- 45.Kumari R, Dhankhar P, Dalal V. Structure-based mimicking of hydroxylated biphenyl congeners (OHPCBs) for human transthyretin, an important enzyme of thyroid hormone system. J Mol Graph Model. 2021;105:107870. doi: 10.1016/j.jmgm.2021.107870. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional data from this work will be made available upon request.