

Abstract
HIV-1 persists as a latent reservoir in people receiving suppressive antiretroviral therapy (ART). When ART is interrupted (treatment interruption/TI), rebound virus reinitiates systemic infection in the lymphoid system. During TI, HIV-1 is also detected in cerebrospinal fluid (CSF), although the source of this rebound virus is unknown. To investigate whether there is a distinct HIV-1 reservoir in the central nervous system (CNS), we compared rebound virus after TI in the blood and CSF of 11 participants. Peak rebound CSF viral loads vary and we show that high viral loads and the appearance of clonally amplified viral lineages in the CSF are correlated with the transient influx of white blood cells. We found no evidence of rebound macrophage-tropic virus in the CSF, even in one individual who had macrophage-tropic HIV-1 in the CSF pre-therapy. We propose a model in which R5 T cell-tropic virus is released from infected T cells that enter the CNS from the blood (or are resident in the CNS during therapy), with clonal amplification of infected T cells and virus replication occurring in the CNS during TI.
HIV-1 predominantly replicates in CD4+ T cells using CCR5 as a co-receptor, and requires the high density of CD4 present on these cells for efficient entry (R5 T cell-tropic)(see1,2, reviewed in3). Antiretroviral therapy (ART) reduces the amount of detectable virus in the blood to low levels, but discontinuation of ART (known as treatment interruption, TI) invariably results in renewed virus replication4–6 usually to pre-therapy levels7. HIV-1 is maintained in a latent state during therapy with much of our understanding of the latent viral reservoir in humans based on analyses of blood cells. Whether there is a reservoir in the central nervous system (CNS), which under certain circumstances can support viral replication, is unclear.
HIV-1 RNA can typically be detected in blood within one to four weeks after TI7,8. The initial rebound virus is typically oligoclonal9–15. Resting CD4+ T cells are considered to be the main source of persisting long-lived reservoirs of HIV-116 and therefore of rebound virus, but other HIV-infected cells in different states of activation have been detected in people on long-term ART17,18. CD4+ T cells circulate between the blood and tissues, or differentiate into tissue-resident cells with limited migratory ability (reviewed in19).
The CNS is isolated by the blood-brain barrier, but can support HIV-1 replication under certain circumstances and thus might be a reservoir for latent virus. There are relatively few T cells in the CNS20–22, and cerebrospinal fluid (CSF) generally has low antibody concentrations (see23 and reviewed by22). CD4+ T cells that enter the CNS crossing the blood-brain barrier release HIV-1 that is genetically similar to populations in the blood, giving rise to a viral load in CSF that is typically 1–10% of the viral load in the blood24. Within the first 2 years of infection approximately 20% of untreated people have genetically distinct viral populations in their CSF, or indirect evidence for viral replication in that compartment24, and rates of compartmentalized replication in the CNS are likely much higher in individuals late in disease with severe neurocognitive impairment, including HIV-associated dementia (HAD)25. Long-term replication of HIV-1 in the CNS compartment often is the result of viral adaptation to replication in myeloid cells with low levels of surface CD4, such as macrophages and microglia (macrophage tropism)26. The ability of infected CD4+ T cells to cross the blood-brain barrier, and the ability of virus to evolve to infect new cell types within the CNS, raise the possibility that there are specific features of latent infection in the CNS that are distinct from those in the lymphoid system27.
Many TI studies have been performed using animal models6,28–33, but characterization of viral rebound in the CNS has been limited by insufficient CSF/brain tissue specimens, use of imaging methods that cannot capture viral dynamics in the CNS and/or use of models that do not efficiently colonize the CNS. Replication competent reservoirs have been reported in the CNS and/or myeloid cells of infected and ART-suppressed NHPs34–36 and humanized mice30,37. In a single study, elevated RNA in the CSF of an ART-treated NHP was detected after treatment with a latency reversing agent34, consistent with the animal having a CNS reservoir of SIV.
Characterization of any CNS HIV-1 reservoir in humans is challenging, but is essential for developing robust cure strategies. One approach has been to analyze brain tissue at autopsy, after adherence to ART up to the time of death has been documented38. Another approach is to examine rebound virus in the CSF which typically trails the appearance of virus in the blood by a few weeks39,40. Current TI protocols usually focus on reinitiating therapy after early detection of HIV-1 in blood (before rebound in the CSF) and do not include lumbar punctures to harvest CSF. Thus it is unlikely a protocol examining rebound virus in the CSF would be acceptable at this time. However, archival samples are available from individuals who chose to stop therapy, or stopped therapy owing to therapy failure41.
We reasoned that detection of a distinct viral population in the CSF after TI would provide indirect evidence of a persistent CNS-specific HIV-1 reservoir. Archived paired blood and CSF specimens were examined for 8 men in one cohort who had discontinued therapy and were experiencing rebound virus especially in the CSF27,42. Similar paired specimens were examined for 3 men in a second cohort where pretherapy specimens were also available to compare to rebound specimens43. Longitudinal viral populations were examined using deep sequencing along with an assessment of entry phenotype to characterize the nature of the rebound virus in the CSF/CNS after TI.
RESULTS AND DISCUSSION
Cohort and TI rebound blood and CSF specimens.
Eight male participants were identified as either failing ART based on detectable plasma viral loads (N=5) or recruited while on suppressive therapy (N=3) and enrolled in a TI protocol; between 3 and 8 longitudinal paired blood and CSF specimens were collected over a period of between 1 to 4 months, with the initial specimens collected at the time of enrollment (Extended Data Table 1)27,42. Here, we examined the archived specimens from these previously described participants27,42,43. The participants ranged in age from 39 to 56 years old, with CD4+ T cell counts between 143 and 834 cells/μl (Table 1). Figure 1 shows blood and CSF viral loads, and CSF white blood cell counts (WBC) counts for five participants in whom the CSF viral load approached that of the blood after TI (Figure 1 a–e). Data for the remaining three participants, whose CSF viral load remained at least 10-fold lower than the blood viral load after TI are also shown in Figure 1 (Figure 1 f–h). All 8 participants experienced viral rebound in the CSF after TI, while 5 of the 8 participants had elevated viral loads in the blood prior to TI but experienced a further increase in blood viral load with TI. Consistent with previous findings40, we observed that the appearance of detectable rebound virus in the CSF lagged behind that in the blood, in our case by approximately 2 weeks.
Table 1.
Participant Characteristics
| Study Sitea | PIDb | Year of TI | Age (years) | Sex | ARVc Before TI | Years From HIV Diagnosis | CD4 Nadir (cells/ul) | Suppressed (S) or Viremic (V) Before TId |
|---|---|---|---|---|---|---|---|---|
| UCSF | 4026 | 1998 | 51 | M | NFV/d4T/ddi | 118 | V | |
| 6004 | 1999 | 51 | M | 3TC/d4T/IDV | 16 | 116 | V | |
| 6005 | 1999 | 53 | M | 3TC/d4T/IDV | 12 | 3 | V | |
| 6006 | 1999 | 39 | M | EFV/NFV/ABC/Adefovir | 17 | 69 | V | |
| 6008 | 2000 | 35 | M | SQV/RTV | 6 | 360 | S | |
| 6011 | 2001 | 43 | M | EFV/ZDV/3TC | 20 | 191 | S | |
| 6012 | 2001 | 51 | M | EFV/d4T/3TC | 23 | 199 | S | |
| 6013 | 2001 | 42 | M | EFV/d4T/3TC | 12 | 275 | V | |
| GOT | 5207 | 2001 | 56 | F | d4T/3TC/IDV/r | 1 | 110 | S |
| 5299 | 2006 | 55 | M | TDF/3TC/AZV/r | 3 | 120 | V | |
| 51126 | 2016 | 53 | M | ABC/3TC/DRV/r | 5 | 60 | V |
Study sites were the University of Californai San Francisco (UCSF) and the University of Gothenburg (GOT)
Personal identification (PID) number for each participant
Antirertiviral (ARV) regimen
Designation for detectable viral load in the blood
Figure 1. HIV-1 rebound populations in participants with high CSF viral load.
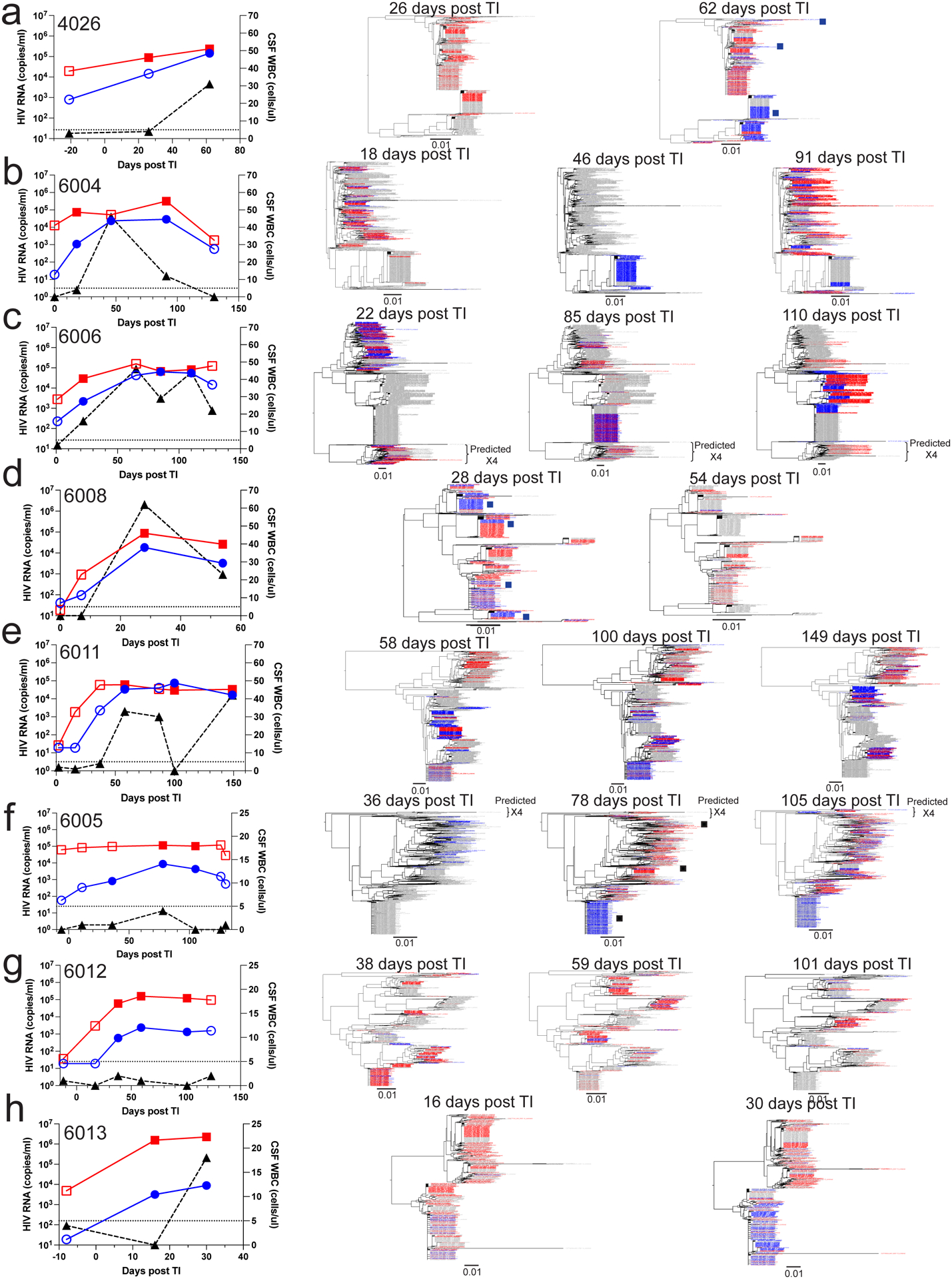
Viral load timelines and phylogenetic trees are shown for 8 participants: 4026 (a), 6004 (b), 6006 (c), 6008 (d), 6011 (e), 6005 (f), 6012 (g), and 6013 (h). The first panel depicts viral loads in the plasma (red squares) and the CSF (blue circles) as well as the CSF WBC count (black triangles) at indicated intervals (in days) after TI. Time points for which sequencing data are available are represented by filled symbols, and time points for which no sequencing data are available are represented by open symbols. Neighbor-joining phylogenetic trees were constructed using all the V1-V3 sequences (about 500 nucleotides in length) generated by Primer ID MiSeq deep sequencing. The trees are shown to the right of the viral load graphs with the time point indicated above each tree. Sequences from the plasma (in red) and CSF (in blue) from each specific time point are colored while the sequences from other time points are in gray. Black squares correspond to the closest positions in the tree of the full length HIV-1 env gene sequences that were used in pseudotyping assays for which entry phenotypes were assessed (Figure 2). In these trees we have limited the number of sequences used to ensure there are similar numbers for the two compartments or to make the trees more visually accessible, with the number of sequences used provided in Extended Data Table 1. In order to validate this approach, we also examined trees for the same participants that included all available sequences and still observed that the CSF was often dominated by clonally amplified sequences and inclusion of more sequences did not provide additional information.
Rebound viral load in CSF is correlated with pleocytosis.
The rebound viral populations in the CSF could be divided into two distinct categories based on the presence or absence of pleocytosis (elevated WBC counts) in the CSF. We define pleocytosis as more than 5 cells per μl CSF. In the absence of CSF pleocytosis, the CSF viral load was much lower than the viral load in the blood (Figure 1 f, g, h). In the presence of CSF pleocytosis, the CSF VL was dramatically increased (Figure 1 a, b, c, d, e). When the ratio of viral load in the CSF to viral load in the blood at all time points with pleocytosis was compared to all time points without pleocytosis, elevation of viral load in the CSF relative to the blood viral load was significantly associated with pleocytosis (Extended Data Figure 1, p=0.002).
Virus clonal amplification is linked to pleocytosis.
Viral RNA was extracted from virus particles pelleted from blood plasma and CSF, converted to cDNA, then an env V1-V3 amplicon was generated by PCR which was sequenced using the Primer ID/unique molecular identifier approach44,45. Phylogenetic analysis of blood-derived HIV-1 sequences revealed that there were multiple viral lineages present in the blood at all time points, including those just after TI (Figure 1, center and right panels), an observation consistent with previous findings9–15. Viral sequences were clustered, indicating rapid expansion of specific variants released from the latent reservoir, and clonal expansion shifted among viral lineages over time.
The rebound virus in the CSF followed two patterns. First, in the absence of pleocytosis the virus in the CSF was genetically diverse and intermixed with virus in the blood (i.e. equilibrated). This can be seen for participants 6005 (first and last time point), 6012, and 6013 (Figure 1 f, g, h) as well as in the initial time points for participants 6004 and 6006 (Figure 1 b and c). The proportionately low viral load in the CSF relative to the blood with equilibrated populations mimics what is seen in chronic infection in the absence of CNS-specific replication and in the absence of pleocytosis24. We attribute low level viremia to the background migration of CD4+ T cells from the blood into the CNS, some of which are infected and release virus similar to that in the blood24, consistent with other recent observations46,47. The virus-transporting infected cells are likely CD4+ T cells rather than monocytes since the equilibrated virus in the blood and CSF requires a high density of CD4 for efficient entry into cells (i.e. T cell-tropic) and is inefficient at infecting cells with a low density of CD4, such as is found on monocytes1,2,26.
The second distinct pattern is the appearance of clonally amplified viral lineages within the CSF that occur in the presence of elevated WBCs/pleocytosis and with higher viral loads that approach the levels in the blood. This result can be seen at one or more time points for participants 4026, 6004, 6006, 6008, 6011 (Figure 1 a, b, c, d, e) and even with a small increase in WBCs in the CSF in participant 6005 (Figure 1 f). The clonal amplification of viral lineages in the CSF linked to pleocytosis was an unexpected observation. The clonally amplified viral lineages in the CSF appear to be disproportionately represented in the CSF versus the blood indicating distinct compartmentalized viral amplification within the CNS. We assessed the blood and CSF viral populations for evidence of compartmentalization. First we examined the phylogenetic trees at peak CSF viral loads for compartmentalization relative to the blood using the Slatkin-Maddison test48. As can be seen in Extended Data Table 1, all eight examples of rebound virus were significantly compartmentalized in the CSF relative to the blood. However, we have previously noted the application of this test is overly sensitive when analyzing deep sequencing data. We therefore employed the Structured Slatkin-Maddison test49 and found that all of the trees remained significantly compartmentalized with the exception of 6012 which did not experience pleocytosis.
The presence of clonally amplified virus is also amenable to another type of analysis for compartmentalization. We examined the proportion of blood virus and CSF virus in the two most abundant clonal lineages at peak CSF viral load and found that these were not randomly distributed between the two compartments, again with the exception of 6012 (Extended Data Table 1, Fisher’s Exact Test). Thus, clonal viral lineages are either uniquely (within the limits of sampling) or disproportionately amplified in the blood or the CSF as a form of compartmentalization. In most cases the virus amplified in the CSF is first observed, or disproportionately observed as a lineage in the CSF. The influx of T cells into the CNS could be playing one of two roles (that are not mutually exclusive) that would account for both the high CSF viral loads and compartmentalization. First, the CD4+ T cells could be supporting the high level of viral replication leading to the high viral loads. Second, antigen-specific CD4+ T cells could define a limited number of infected cells that expand in situ to seed the CNS infection with homogeneous virus giving rise to clonal lineages.
Our results differ from those reported by Gianella et al.40 where they observed diverse compartmentalized rebound virus in the CSF. Their approach did not use UMIs as part of the sequencing strategy which can contribute to two artifacts in the sequencing. First, collapsing identical sequences into “haplotypes” loses information about clonal amplification, which we observe as a dominant feature of the rebound virus in the CSF and is a well-known feature of rebound in the blood9–15. Second, given that deep sequencing platforms are error-prone, the inability to create a consensus sequence for each template to reduce sequencing errors results in these errors being recorded as diversity in the viral population creating the appearance of compartmentalization. We believe the difference in the inclusion of a UMI in the sequencing approach is sufficient to explain our different observations.
Rebound HIV-1 preferentially replicates in CD4+ T cells.
Clonally amplified HIV-1 sequences are well known as part of the low level viremia (LLV) that persists in the blood of people on suppressive ART50–56. Given that LLV occurs in the face of antiviral drugs blocking viral replication, LLV is most likely produced solely by clonally expanded, HIV-infected cells releasing virus rather than replication of genetically similar viruses. However, the fact that the viral load of LLV is typically several logs lower than that of virus rebounding in the CSF (~101-102 RNA cp/ml in LLV blood vs. ~104-105 RNA cp/ml in rebounding CSF) makes it unlikely that virus production from clonally expanded cells alone can generate the high viral loads observed in the CSF during rebound.
In order to explore the mechanisms that generate clonal amplification of virus in the CSF after TI, we analyzed longer sequences from the viral population, specifically the entire viral env gene both to assess whether there was any evidence of ongoing viral replication (in the form of small amounts of sequence diversity) and to determine the entry phenotype as indirect evidence of the cell type that had been harboring the virus. We examined env genes from three CSF specimens chosen at times when clonally amplified virus was apparent. The positions of the V1/V3 region of the env amplicons that were cloned are shown with squares in the figures as indicated: 4026 at 62 days post TI (Figure 1 a); 6008 at 28 days post TI (Figure 1 d); 6005 at 78 days post TI (Figure 1 f). A phylogenetic tree of the full length env gene sequences (left panel) for CSF samples from these three participants is shown in Figure 2 a–c. Again, the sequences designated with squares were those cloned for testing of the entry phenotype, targeting both sequences within the clonally amplified lineages and those from the more diverse portions of the tree.
Figure 2. HIV-1 in CSF is replicating and adapted to growth in T cells.
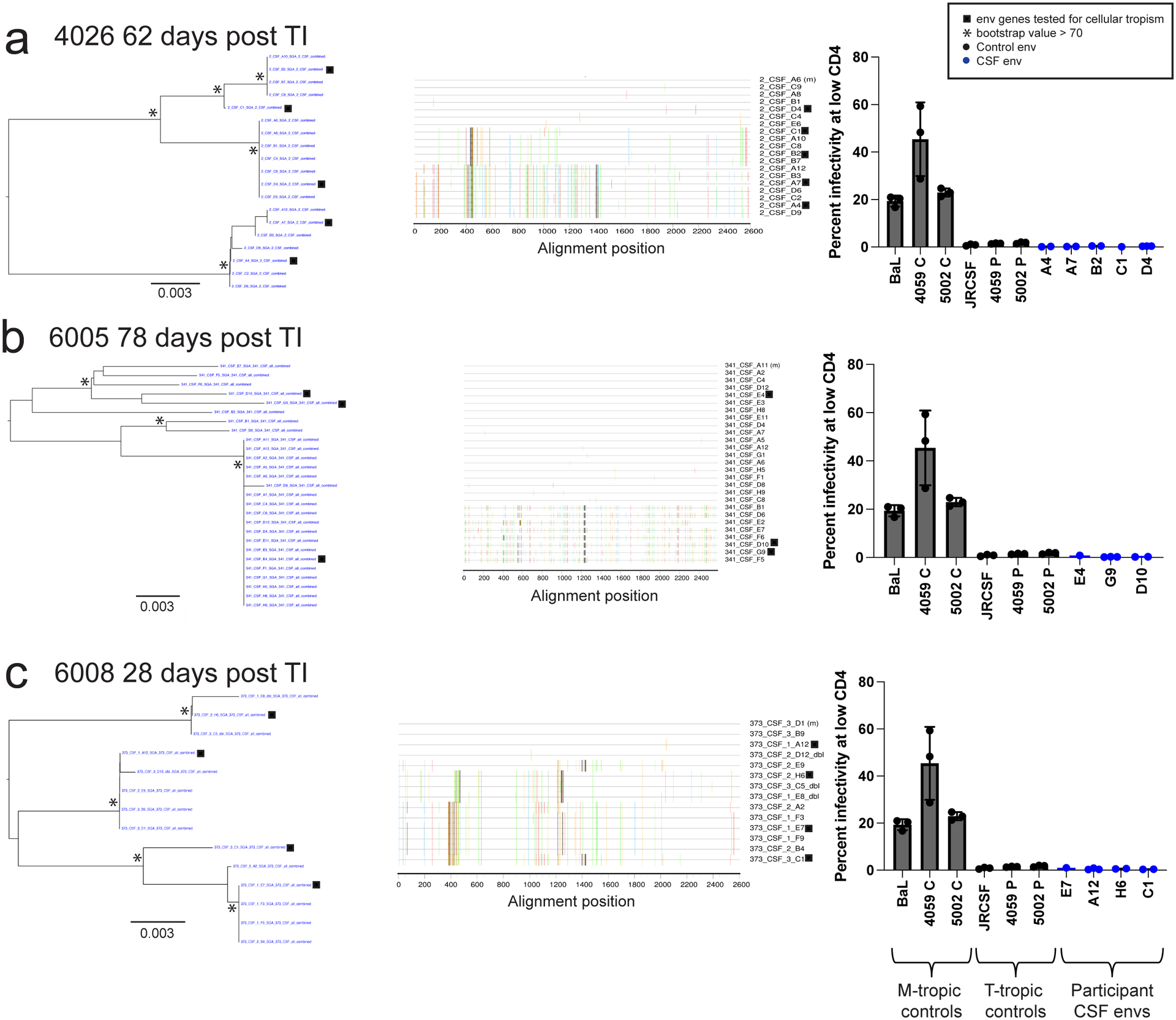
Full length env gene amplicons were generated by end-point dilution PCR of cDNA synthesized using viral RNA in the CSF as template. The entry phenotype was assessed for three representative participants with clonal CSF-specific lineages in the viral rebound populations following TI at the indicated time: 4026 (a), 6005 (b), and 6008 (c). Panel 1 shows neighbor-joining phylogenetic trees of full-length env gene sequences. Panel 2 to the right is a highlighter plot of the full-length env gene sequences depicting the low but detectable sequence diversity within these lineages in each participant. The third panel (far right) graphs the relative infectivity at low levels of CD4 for macrophage-tropic env gene controls used to pseudotype a reporter virus, T cell-tropic env gene controls, as well as the participant-derived env genes shown in the first panel. 3 technical replicates were done for each clone and 1–3 replicate clones were tested for each env. Specific numbers of clones tested for each env are as follows; A4: 2 clones, A7: 2 clones, B2: 2 clones, C1: 1 clones, D4: 3 clones, E4: 1 clones, G9: 3 clones, D10: 2 clones, E7: 1 clone, A12: 3 clones, H6: 2 clones, C1: 2 clones. The infectivity at low CD4 values for each replicate clone are shown with the dots on the bar graph and the mean and standard deviation values for the replicates are shown with the bar and error bars, respectively. All env genes cloned from the CSF virus of the three participants were T cell-tropic when used to pseudotype the reporter virus and tested in the CD4lowCCR5high/CD4highCCR5high Affinofile cell entry assay.
We first examined the level of sequence diversity within the clonally amplified sequences (Figure 2 middle panels) to assess whether observed low levels of diversity were due to sequencing errors or virus replication. The synthesis of the cDNA product by reverse transcriptase in vitro is reported to have an error rate of 1 in 10,000 nucleotides by the manufacturer, and we previously examined this directly and found an error rate of 1 in 20,000 nucleotides in this PCR end-point dilution protocol45. While diversity in the amplified sequences is low (Figure 2, three highlighter plots, middle panels), it is greater than this expected error rate. Based on the length of the sequenced regions across the three participants, we would expect to see 4 sequencing errors if the sequences were actually identical (i.e. clonal). In contrast, when the sequences are compared to the designated master sequence we observe 24 mutations. The accumulation of this low level sequence diversity is most easily explained by the clonally amplified virus undergoing a brief period of viral replication to attain the high viral loads. Thus, while proliferation of an infected cell could play a role in the appearance of clonal lineages, the high level of virus in the CSF and the accumulation of some genetic diversity suggests ongoing viral replication. Shorter amplicons can underestimate overall diversity, especially when recombinants are not recorded within the amplicon57–59. However, in this case the short amplicons evaluated in the deep sequencing analysis in Figure 1 (500 bp) give similar interpretations when evaluated with less sampling depth using a 2.5 kbp amplicon as shown in Figure 2.
HIV-1 typically requires a high density of CD4 for efficient entry, thus it preferentially targets and replicates in CD4+ T cells26. Under circumstances where CD4+ T cells are limiting, the virus can adapt to enter cells with a low density of CD4, a phenotype termed macrophage tropism (reviewed by3). By comparing viral infectivity at high and low density of CD4, an assay conveniently done using Affinofile cells where the level of CD4 can be regulated60, it is possible to identify cells that have significant infectivity at a low density of CD4 (macrophage tropism) versus those that do not (T cell tropism). We used this assay to assess the entry phenotype encoded by env gene clones from both the clonally amplified sequences and from the more diverse sequences (Figure 2 a–c, right panels). As can be seen, all of the env gene clones tested encoded Env proteins that had very poor infectivity at a low density of CD4. Thus we infer that the major populations of rebound virus in the CSF, including the variants in clonally amplified lineages, were previously selected for replication in CD4+ T cells. One explanation of our results is that viruses that had been replicating in CD4+ T cells entered the latent reservoir in T cells and these were the source of the rebound virus in both the blood and the CNS/CSF.
Viral lineages in the CSF contribute to rebound populations.
We examined three participants from a second cohort who underwent unsupervised TI with less frequent blood and CSF sampling during the rebound period but with pre-therapy plasma and CSF specimens available. As can be seen in Figure 3, participants 5207, 5299, and 51126 (Figure 3 a–c) all experienced rebound with viral loads in the CSF approaching those in the blood, with low to moderate CSF WBC influx at or above 5 cells/μl (Figure 3 left panels) and with largely clonal rebound virus in the CSF from one or several variants (middle/right panels, phylogenetic trees); these patterns are consistent with what was observed in the first cohort (Figure 1).
Figure 3. HIV-1 rebound populations compared to pre-therapy viruses.
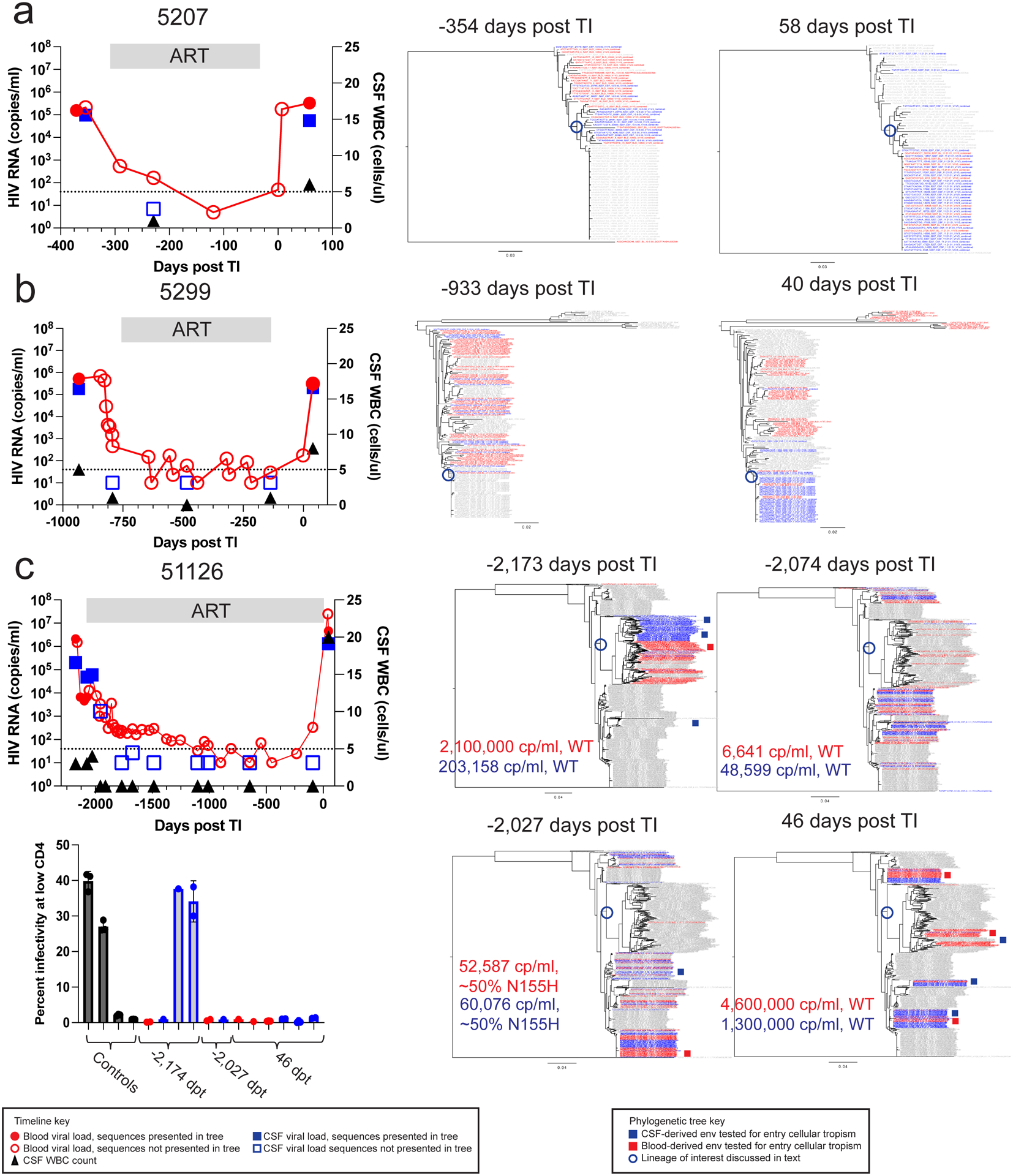
Timelines and phylogenetic trees are shown for 3 participants: 5207 (a), 5299 (b), and 51126 (c). The first panel depicts viral loads in the plasma (red circles) and the CSF (blue squares) as well as the CSF WBC count (black triangles) before ART, during ART, and after TI. Time points prior to TI are shown as negative days. Neighbor-joining phylogenetic trees were constructed using up to 50 V1-V3 sequences (or all that were available if less) generated by Primer ID MiSeq deep sequencing at the indicated for each participant. Sequences from the plasma (in red) and CSF (in blue) from each specific time point are colored while the sequences the from other time points are gray. Full length env amplicons were generated from blood and CSF lineages for 51126 and their shorter sequence equivalents are shown in the deep sequencing trees as red and blue squares, respectively. In these trees we have limited the number of sequences used to ensure there are similar numbers for the two compartments or to make the trees more visually accessible, with the number of sequences used provided in Extended Data Table 1. In order to validate this approach, we also examined trees for the same participants that included all available sequences and still observed that the CSF was often dominated by clonally amplified sequences and inclusion of more sequences did not provide additional information. The full length amplicons were tested for entry phenotype in the Affinofile assay with the results shown in the lower left panel with the clones grouped by day post TI of the sample. Positive and negative controls for the M-tropic entry phenotype were included and shown in black, while clones generated from CSF are shown in blue and from blood in red. 3 technical replicates were done for each clone and 1–3 replicate clones were tested for each env. Specific numbers of clones tested for each env are as follows from left to right: 2 clones, 1 clone, 1 clone, 2 clones, 2 clones, 1 clone, 1 clone, 1 clone, 3 clones, 2 clones, 3 clones, 2 clones. The infectivity at low CD4 values for each replicate clone are shown with the dots on the bar graph and the mean and standard deviation values for the replicates are shown with the bar and error bars, respectively.
In this cohort we had the opportunity to sequence the virus present in both the blood and CSF at the time point just before the initiation of ART and preceding TI. For participant 5207 (Figure 3 a) the clonal rebound virus in the blood and the CSF was from a lineage that pre-therapy had been over-represented in the CSF compared to the blood (lineage branch point circled blue). Similarly, for participant 5299 (Figure 3 b) the largely clonal TI virus in the CSF also appears to be from a lineage (branch point circled blue) that was rare pre-therapy, but observed at a higher frequency in the CSF during TI; in contrast, rebound virus in the blood for this participant branched with pretherapy blood virus lineages. One explanation for these patterns is that virus most similar to lineages in the CNS pre-therapy gave rise to at least some of the rebound virus. Also, the predominant virus in the CSF can appear at a low level in the blood, consistent with transport of the predominant virus out of the CNS into the blood. This observation emphasizes the potential of a CNS viral reservoir to reseed the periphery during treatment interruption.
For participant 51126 (Figure 3 c; diagnosed with HAD) samples were available at therapy initiation and rebound as well as at two intermediate time points. The entry time point (−2,173 days post TI) consisted of major lineages (branch point circled) that represented most of the virus in the blood and a compartmentalized population in the CSF; this compartmentalized population in the CSF consisted of macrophage-tropic virus (Figure 3, lower left panel). The rebound virus in the blood (46 days post TI) represented this major lineage in the blood as well as several other lineages that were all R5 T cell-tropic (Figure 3, lower left panel). However, the rebound virus in the CSF did not include virus from this macrophage-tropic virus lineage. When we sampled the rebound virus in the CSF to a high level (2200 templates sequenced) we failed to observe any virus from the M-tropic lineage (Extended Data Figure 2). In this one case we failed to detect any contribution of a CNS infection with a macrophage-tropic virus lineage contributing to virus rebound after TI even with deep sampling. The rebound virus in the CSF (46 days post TI) represents several lineages of virus with a T cell-tropic entry phenotype within lineages not detected prior to therapy (−2173 days from TI).
Viral RNA declined slowly after ART initiation for participant 51126 and sequencing of residual viral populations present after 8 weeks of ART (−2074 days from TI) revealed the presence of virus more similar to the rebound lineage that was drug sensitive and genetically distinct from the variants detected in the blood and CSF before ART (−2173 days from TI). By 14 weeks of ART (−2027 days from TI), approximately 50% of variants in this newly detected lineage were resistant to raltegravir (IN N155H), and this virus contributed to an increasing viral load in the blood; two viruses sampled from these lineages were R5 T cell-tropic. A subsequent change in therapy improved suppression in the blood and CSF. After treatment interruption the rebound virus in the CSF represented drug sensitive variants, largely related to the lineage that appeared during therapy initiation. Overall, these phylogenetic patterns suggest that viral lineages over-represented in the CSF pre-ART (or revealed during viral load decay post-ART) can contribute to rebound of virus in the CSF and blood. It seems likely that inadequate regimen potency and penetration into the CNS allowed localized viral replication in the CNS when therapy was initiated (i.e. before it was changed which then led to suppression).
Recently Fisher et al.12 reported the detection of rebound virus in the blood with the same sequence as the pretherapy blood virus. This suggests that the cells producing virus at the time of ART initiation may form the portion of the reservoir that gives rise to rebound virus. This is consistent with the observation that cells infected near the time of ART initiation comprise the majority of the inducible, replication-competent reservoir in the blood61. Similarly, a study of three participants who initiated ART during acute infection12 observed that all participants had identical RNA sequences (with sequencing of either an 8 or 5 kbp region) in their blood plasma pre-ART and post-TI. However, the lack of genetic diversity pre-ART in this case limits the inferences that can be made about the source of the rebound virus. When smaller amplicons were examined in people treated during chronic infection, a subset of individuals were identified as having identical (or very similar) variants in their blood plasma pre-ART and post-TI13,62,63. However, as can be seen in Figure 3, a subset of rebound viruses in the blood may come from sources that have a distinct genetic composition compared to virus in the blood pre-therapy.
CSF biomarkers of inflammation and neuronal damage during TI.
We examined the longitudinal specimens from five participants (6004, 6005, 6006, 6011, and 6012) for a set of inflammatory markers (Figure 4 a–e). Three of these participants had strong evidence of clonal amplification of viral lineages with WBCs migrating into the CNS (6004, 6006, and 6011, Figure 1) and two did not (6005 and 6012, Figure 1). Some of these data (including CSF NfL and neopterin) have been reported previously43 but are included here with the other inflammatory biomarkers to allow a more comprehensive comparison. Our results suggest that the initial increase in the CSF viral load, and the concomitant influx of WBCs, are associated with several inflammatory markers. As can be seen in Figure 4, rising CXCL10/IP-10 levels were closely linked to the peaking of WBC counts during rebound as seen for participants 6004, 6006, 6011, and to a lesser extent for participant 6005 (Figure 4 a–c, e) where there was only a slight increase in CSF WBCs; this was not the case for participant 6012 (Figure 4 d) where there was no influx of cells into the CSF. MCP-1 (CXCL2), TNFα, and MMP9 were more variable in their appearance, sometimes tracking with CXCL 10/IP-10 and in some cases lagging; neopterin also increased in concentration but always lagged CXCL10/IP-10 (Figure 4). By contrast, in the two cases where an increase in NfL was detected after TI (participants 6011 and 6012; Extended Data Figure 3 b and d) this increase occurred either well after the initial rise in inflammatory markers or in their absence. We tested these associations statistically by comparing the biomarker level in the CSF for all time points with CSF WBC counts greater than 5 cells/μl to those with 5 or fewer cells/μl. As can be seen in Extended Data Figure 4, both IP10 and MMP9 were significantly elevated during pleocytosis (Extended Data Figure 4 a and b) while neopterin was not significantly elevated (Extended Data Figure 4 c). The neurocognitive score QNPZ-4 was not significantly different between the two groups (Extended Data Figure 4 e), while the NfL marker was higher in the group with low CSF white blood cells (Extended Data Figure 4 d), perhaps driven by the increase in the CSF of participant 6012. However, the direction of significant difference is the opposite from expected based on the hypothesis that acute inflammation would result in increased neuronal damage as measured by the release of NfL. Thus, while pleocytosis is linked to several inflammatory markers it is not linked to acute neuronal damage.
Figure 4. Inflammatory biomarkers in treatment interruption.
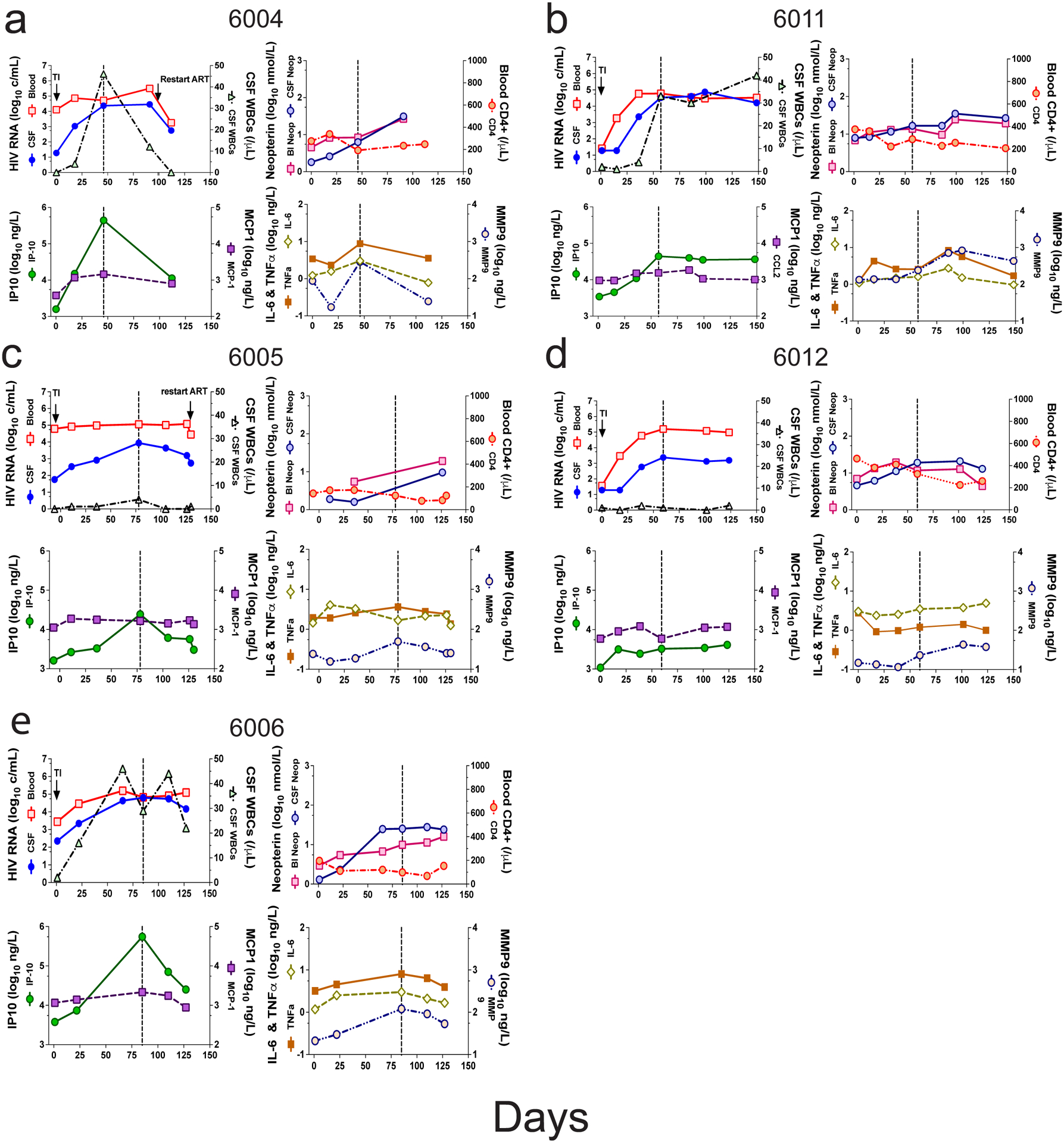
Biomarkers are shown for five participants in a group of four panels per participant tracking the biomarkers over the course of the TI: 6004 (a), 6011 (b), 6005 (c), 6012 (d), 6006 (e). The upper left panel in each group reproduces the viral load from blood (red squares) and CSF (blue circles), and CSF WBC counts (black triangles) shown in Figure 1. The upper right panel in each group shows blood (red square) and CSF (blue circles) levels of neopterin, and the blood CD4+ T cell count (red circles). The lower left panel of each group shows the levels of IP10 (green circles) and MCP1 (purple squares). The lower right panel of each group shows the levels of IL-6 (open olive diamonds), TNFα (filled orange squares), and MMP9 (open tan circles).
Conclusions
We found that the majority of the viral populations that rebounds in the CSF during TI represents clonally amplified virus with a T cell-tropic entry phenotype (requiring a high density of CD4 for efficient entry). This suggests that the main rebounding populations in the blood and CNS compartments are likely produced from T cell reservoirs.
The primary difference observed between rebounding populations in these two compartments is that rebound virus in the CSF is strongly influenced by transient pleocytosis in the CSF that often accompanies TI. The influx of WBCs into the CNS compartment during TI is closely linked to the clonal amplification of virus, elevated viral loads, and elevated inflammatory markers in the CSF. Our results did not identify any rebound virus from HIV-infected myeloid cells in the CNS (even when documented to be present pre-therapy) as assessed by virus entry phenotype. This observation can not exclude the possibility of a myeloid cell HIV-1 reservoir, but it does reveal that if such a reservoir exists it is likely to be a very minor population in the CSF during TI. We observed T cell-tropic viral lineages enriched in the CSF prior to ART that were also observed in the rebounding virus population in the CSF and blood during TI; in addition, many of the viral lineages present in the CSF were disproportionately represented in the CSF relative to the blood. This raises the possibility that latently infected CD4+ T cells present in the CNS during ART contribute to rebound virus in the CSF/CNS, and that a portion of these viral lineages migrate to the blood. Additionally antigen-specific T cells may enter the CNS compartment after TI and expand, with some of them being infected and thus able to release homogeneous virus disproportionately represented in the CSF. At present it is unclear why WBCs transit into the CNS during TI and whether this by itself is indicative of a CNS reservoir. Understanding the nature of the putative antigen (locally produced HIV-1 for example), the extent of T cell clonality, and the antigen specificity of the T cells in the CSF during TI pleocytosis will help give further insight into virus rebound in the CNS compartment.
Online Methods
Study design.
Participant specimens were retrospectively identified at the University of California at San Francisco (UCSF) and at the University of Gothenburg (Gothenburg, Sweden) to identify those who had undergone serial lumbar punctures before and after ART interruption with accompanying blood sampling (Table 1, Extended Data Table 1). The decision to interrupt treatment was made by each participant and their primary caregivers. In some cases, participants from a UCSF cohort41 were on failing antiretroviral therapy (i.e. were viremic with drug resistance in virus in the blood), but all participants showed increases in viral load in the blood with TI and all had dramatic increases in the CSF viral load with TI (Extended Data Table 1). Before and after ART TI, participants underwent site-specific protocols to evaluate responses in the CSF to ART modifications, although in some cases TI was inferred by the return of viremia (Extended Data Table 2). Site-specific study protocols were approved by the IRB at UCSF and the University of Gothenburg, respectively. All participants provided informed consent for the use of their stored specimens.
Specimen collection and viral load analysis.
Whole blood was collected in EDTA. Blood plasma was separated from whole blood by centrifugation, and the plasma was stored at - 80°C. CSF was collected in uncoated tubes and cells were removed by centrifugation. CSF supernatant was then stored at - 80°C. HIV-1 RNA levels in blood plasma and CSF were quantified with the Amplicor HIV-1 Monitor assay, version 1.5 (Roche Diagnostic Systems, Hoffman-La Roche, Basel, Switzerland) with a dynamic range down to 50 copies/mL and a detection limit of approximately 20 copies/mL. Each center used their standard clinical laboratory methods to measure CSF WBC counts and peripheral blood CD4+ T lymphocyte counts.
Biomarker analysis.
Neopterin was measured by a commercially available enzyme-linked immunosorbent assay (ELISA) (BRAHMS GmbH, Hennigsdorf, Germany) with an upper normal reference value of ≤ 5.8 nmol/L in CSF64 . CSF neurofilament light protein (NfL) concentration was measured by means of a sensitive ELISA assay (NF-light ELISA kit; UmanDiagnostics AB, Umeå, Sweden) as previously described65,66. Upper reference values are age dependent67. Seven additional soluble inflammatory biomarkers were tested at Vitalant Research Institute, San Francisco (formerly Blood Systems Research Institute) both in plasma and CSF, including TNFα, MMP9, CXCL10 (IP-10), sCD14, sCD163, CCL2 (MCP-1), and IL6. Five different assays were used: MILLIPLEX MAP (MilliporeSigma, Burlington, MA) Human High Sensitivity T Cell Panel (TNFα and IL6), MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel (IP-10 and MCP-1), MILLIPLEX MAP Human MMP Magnetic Bead Panel 2 (MMP9), sCD163 and sCD14 R&D Human Quantikine ELISA Kit (R&D Systems, Minneapolis MN). Data were collected by Luminex 200, analyzed by Bioplex 6.1 and compiled using Bioplex DataPro (Bio-Rad, Hercules CA).
Sequence analysis.
Most sequences were generated by Illumina MiSeq deep sequencing with Primer ID45, however a subset of sequences was generated by single genome amplification (SGA) in order to facilitate analyses of viral entry. Deep sequencing was performed as follows: viral RNA was extracted from blood plasma or CSF using the QIAmp Viral RNA Mini Kit (Qiagen), and cDNA was generated using a single primer for the V1-V3 region of env or a pool of 4 primers for the V1-V3 region and 3 regions in pro/pol (partial RT, IN, and PR) for synthesis using reverse transcriptase. Each cDNA primer included a random 11 base Primer ID. cDNAs were amplified by PCR and sequenced using the Illumina MiSeq 300 base paired-end multiplex library preparation protocol. Pimer sequences are listed in Extended Data Table 3. A template consensus sequence (TCS) was generated using the multiple reads for each Primer ID. TCSs were aligned using multiple sequence comparison by log-expression (MUSCLE)68, and neighbor-joining phylogenetic trees were generated and visualized using FigTree v1.4.4.
Single genome amplification (SGA) was performed as previously described1. Briefly, viral RNA was extracted from blood plasma or CSF using the QIAmp Viral RNA Mini Kit (Qiagen) and cDNA was generated using an Oligo(dT)20 primer (ThermoFisher Scientific, 18418020) using reverse transcriptase. End-point dilution PCR was used to generate full-length env genes that were bidirectionally sequenced by Sanger sequencing to yield 2X or greater coverage. Primer sequences are listed in Extended Data Table 3. The env genes were aligned using MUSCLE and neighbor-joining phylogenetic trees were generated and visualized using FigTree (v1.4.4). Highlighter plots were generated using the Highlighter tool in the Los Alamos National Laboratory’s HIV sequence database (hiv.lanl.gov).
HIV-1 entry phenotype assessment.
Full-length env genes were cloned into the pcDNA3.1D/V5-His-TOPO expression vector (Invitrogen) using the pcDNA3.1 directional TOPO cloning kit (Invitrogen). We used our established protocol previously described69 to determine the ability of the env genes to facilitate entry of pseudotyped reporter viruses into Affinofile cells expressing a low density of surface CD4, a marker of macrophage tropism.
Statistical analyses.
All statistical analyses were performed using R statistical software (version 3.6.3) or with Prism (version 9.3.1) using tests as indicated in the text and/or figure legends.
Extended Data
Extended Data Figure 1. Statistical analysis of the link between CSF viral load and pleocytosis.
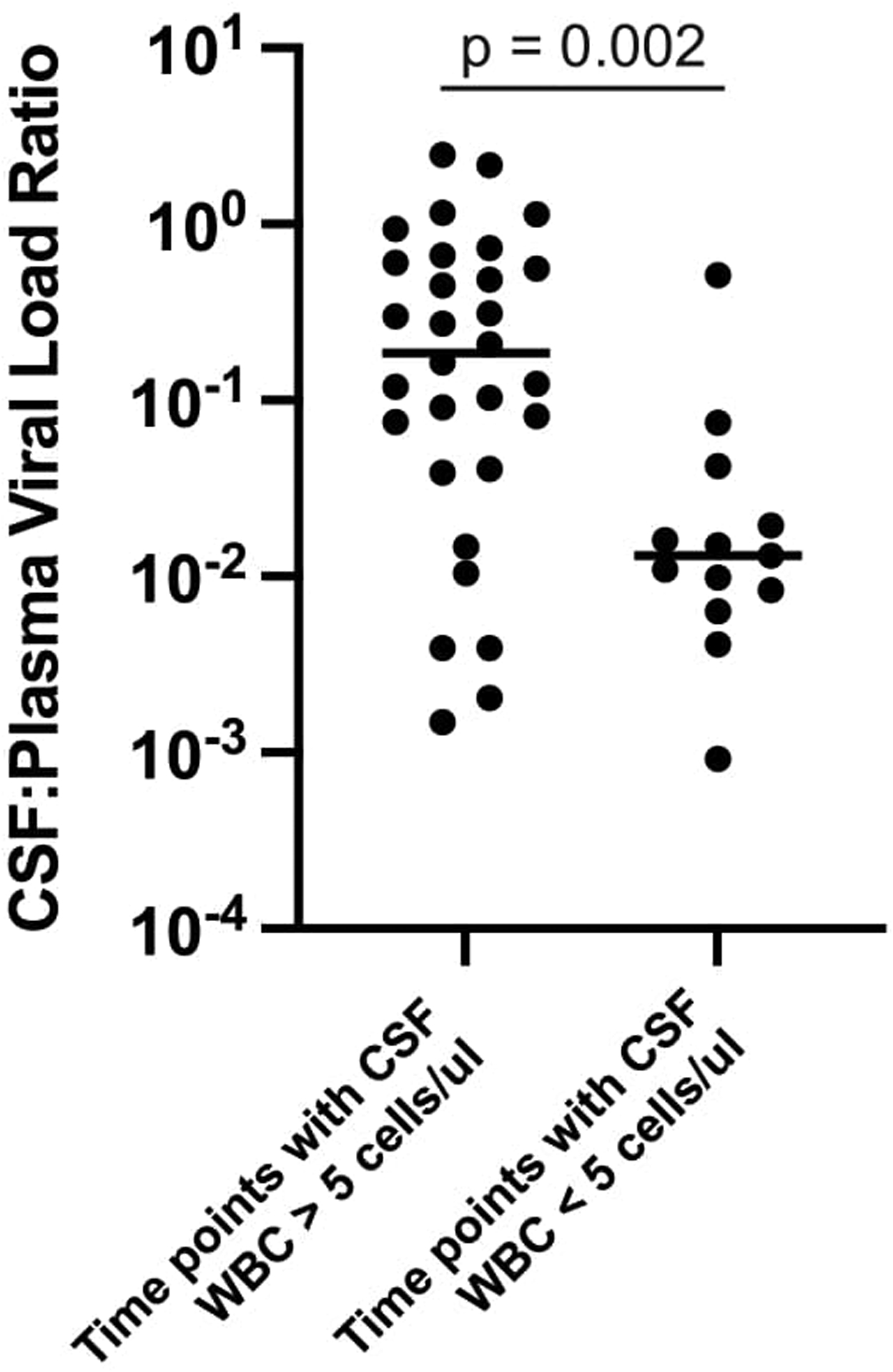
The ratio of CSF viral load to blood viral load was calculated for each time point shown in Figure 1. This was done to normalize the CSF viral load as a fraction of the blood viral load thus reducing the variability in blood set point viral load; in this analysis the larger the value the closer the CSF viral load approached that of the blood. These values were then grouped as coming from time points where the white blood cell count was greater than 5/μl or less than or equal to 5/μl. The values of these two groups were compared using the two-sided Mann-Whitney test, with the p value included in the graph. In this analysis we did not correct for the fact that multiple values were collected for each participant.
Extended Data Figure 2. Deep sequencing of HIV-1 populations from participant 51126.
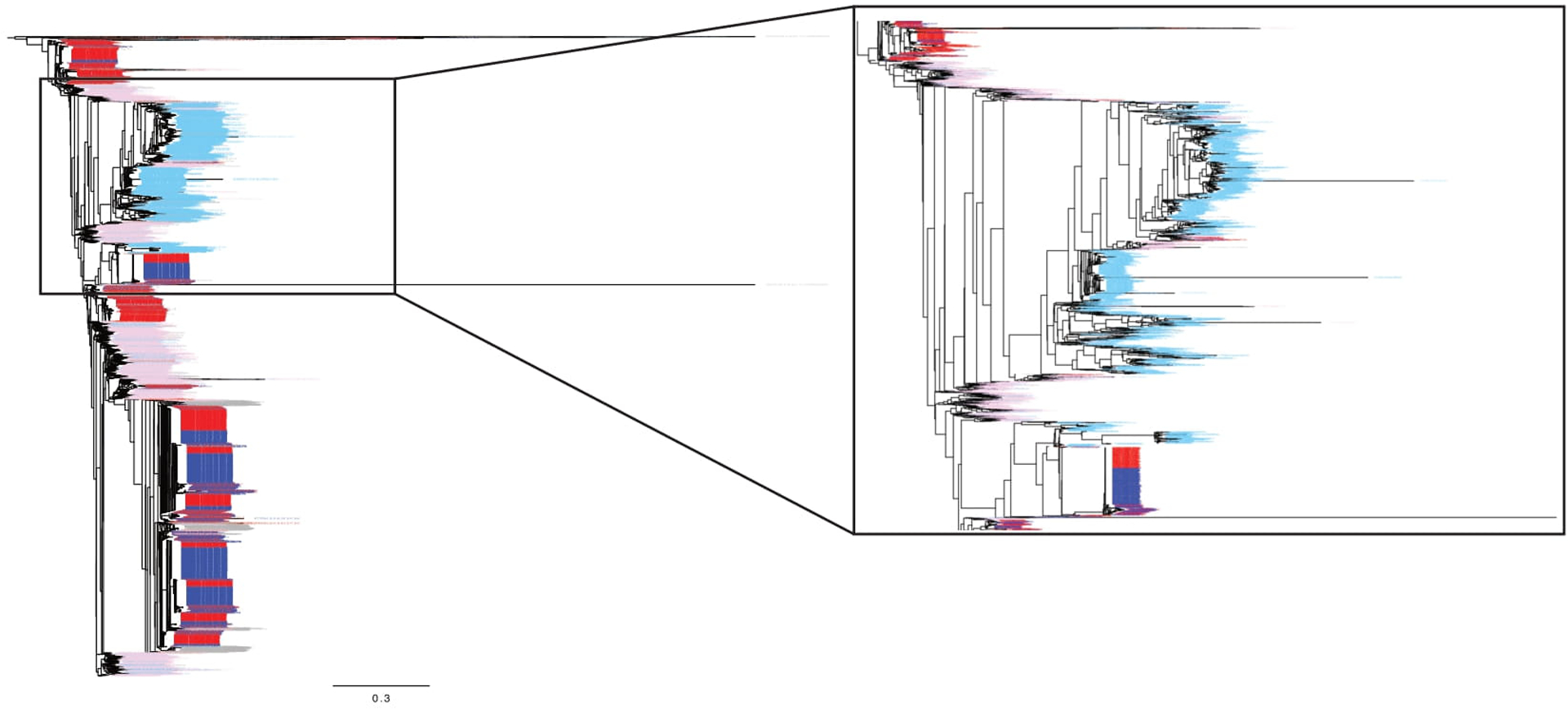
Neighbor-joining phylogenetic tree containing a large sampling of template consensus sequences (TCS) from MiSeq/Primer ID sequencing from the pretherapy and TI time points. Sequences from the blood plasma pretherapy are shown in pink (1,734 TCS). Sequences from the CSF pretherapy are shown in light blue (1,734 TCS). Sequences from the blood plasma post TI are shown in red (2,233 TCS). Sequences from the CSF post TI are shown in dark blue (2,233 TCS). Also included in gray are 50 sequences from the blood and CSF from each of the two intermediate decay time points. On the right the tree is expanded to show the portion where the macrophage-tropic virus lineage was found in the CSF pretherapy.
Extended Data Figure 3. Marker analysis during TI.
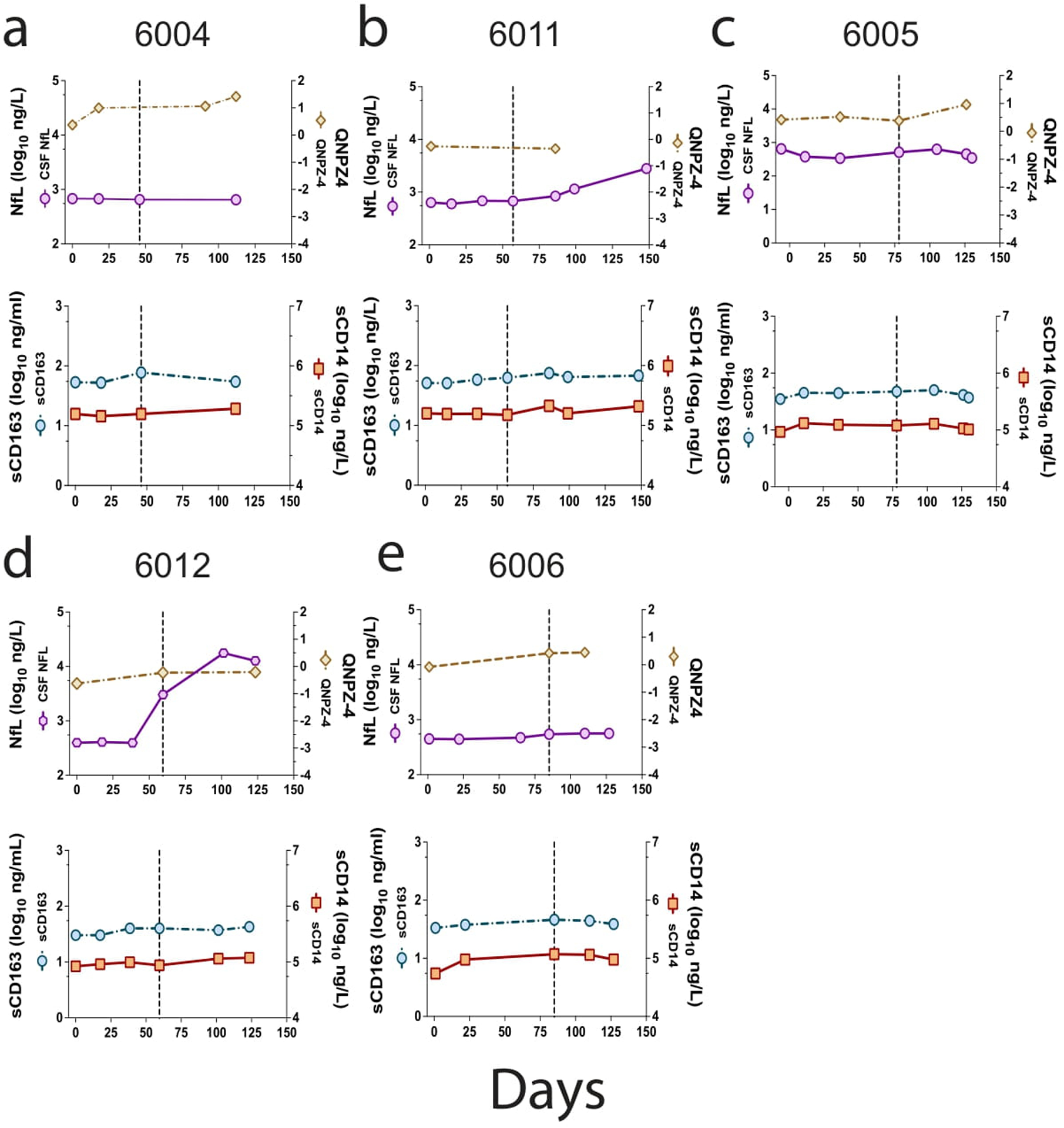
Further analysis of markers was done for the five participants shown in Figure 4. Two additional graphs are presented in vertical columns for each of the participants. In the top graph is shown the level of NfL as a function of time post TI/enrollment (purple circles). Also included is the QNPZ4 score test of neurocognition (tan diamonds). The vertical dashed line shows the peak viral load associated with pleocytosis (Figure 1). In the lower graph of the pair, the values of sCD163 are shown (light blue circles), and sCD14 (orange squares).
Extended Data Figure 4. Statistical analysis of links between biomarkers and pleocytosis during TI.
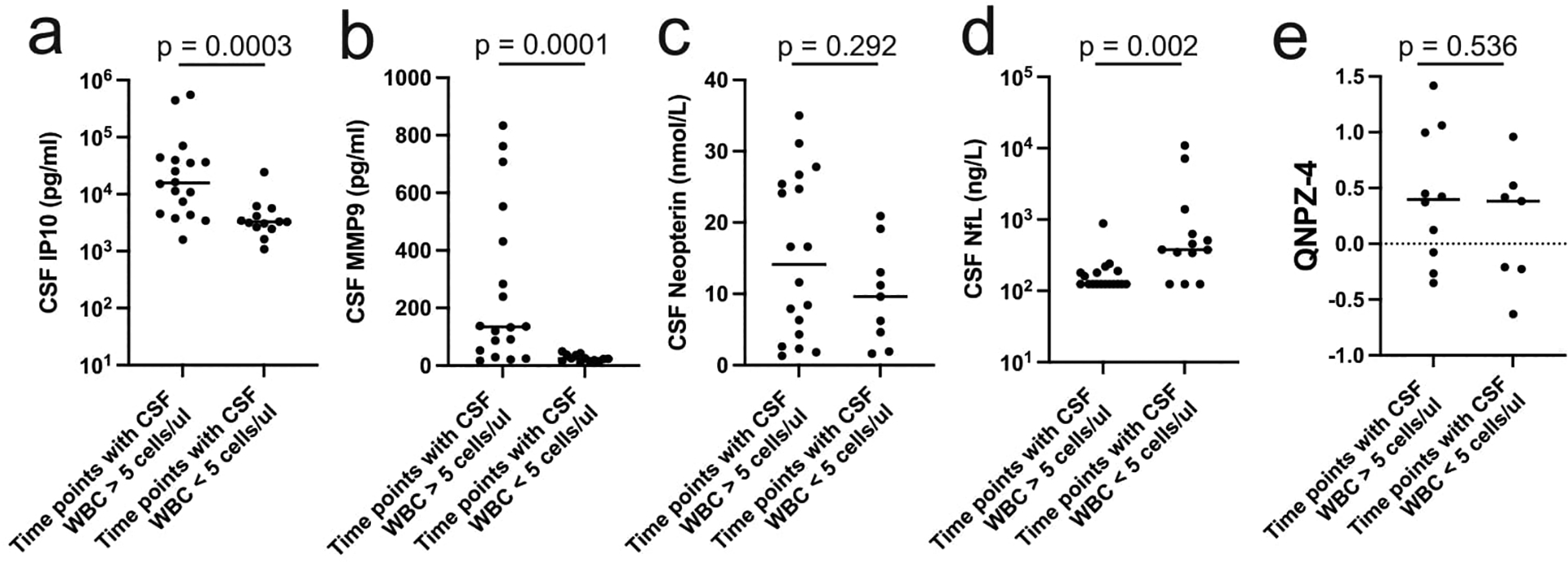
Selected biomarker data presented in Figure 4 and Extended Data Figure 3 were pooled based on the presence or absence of pleocytosis (WBC count greater than or less than 5/μl, respectively) in the CSF. These two groups were compared for IP10 (a), MMP9 (b), neopterin (c), NfL (d), or the neurocognitive score QNPZ4 (e). The groups were compared using the two-sided Mann-Whitney test, with the p values indicated on each graph. A Bonferroni correction for multiple comparisons indicates a significant p value cutoff of 0.01. No corrections were made for the fact that multiple values were included for each participant.
Extended Data Table 1.
Extended information about sample collection, viral loads, cell counts, sequencing results, and compartmentalization tests.
| Site | PID | VisitDate | Days post interruption | Plasma Viral Load | CSF Viral Load | ART | CSF WBC | CD4 | Sequences in figure? | Total BLD TCS (TCS in tree) | Total CSF TCS (TCS in tree) | Total SGA envs | Panmictic Slatkin-Maddison p value | Structured Slatkin-Maddison p value | Fisher’s exact test p value | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UCSF | 4026 | On ART, viremic | 12/23/1998 | −21 | 19500 | 798 | NFV/d4T/ddi | 3 | 163 | N | ||||||
| 4026 | Interruption | 1/13/1999 | ||||||||||||||
| 4026 | Rebound | 2/8/1999 | 26 | 88800 | 14600 | 4 | 174 | Y | 1520 (200) | 0 | ||||||
| 4026 | Rebound | 3/16/1999 | 62 | 237000 | 143000 | 31 | 98 | Y | 630 (200) | 1248 (200) | 19 CSF | <0.0001 | <0.0001 | <0.0001 | ||
| 6004 | Interruption | 8/2/1999 | 3TC/d4T/IDV | |||||||||||||
| 6004 | On ART, viremic | 8/2/1999 | 0 | 12800 | <20 | 0 | 275 | N | ||||||||
| 6004 | Rebound | 8/20/1999 | 18 | 73600 | 1090 | 4 | 339 | Y | 1293 (200) | 65 (65) | ||||||
| 6004 | Rebound | 9/17/1999 | 46 | 52700 | 23600 | 46 | 192 | Y | 0 | 692 (200) | ||||||
| 6004 | Rebound | 11/1/1999 | 91 | 318000 | 28900 | 12 | 234 | Y | 744 (200) | 1888 (200) | <0.0001 | <0.0001 | 0.0002 | |||
| 6004 | Rebound | 12/10/1999 | 130 | 1815 | 564 | 0 | 247 | N | ||||||||
| 6005 | On ART, viremic | 8/27/1999 | −6 | 62000 | 57 | 3TC/d4T/IDV | 0 | 143 | N | 1896 (0) | 0 | |||||
| 6005 | Interruption | 9/2/1999 | ||||||||||||||
| 6005 | Rebound | 9/13/1999 | 11 | 82900 | 340 | 1 | 170 | N | 0 | 25 (0) | ||||||
| 6005 | Rebound | 10/8/1999 | 36 | 97800 | 818 | 1 | 173 | Y | 0 | 62 (62) | ||||||
| 6005 | Rebound | 11/19/1999 | 78 | 115583 | 8660 | 4 | 124 | Y | 1079 (200) | 1340 (200) | 27 CSF | <0.0001 | <0.0001 | <0.0001 | ||
| 6005 | Rebound | 12/16/1999 | 105 | 103177 | 4360 | 0 | 78 | Y | 2277 (200) | 318 (200) | ||||||
| 6005 | Rebound | 1/6/2000 | 126 | 120000 | 1570 | 0 | 83 | N | 1684 (0) | 79 (0) | ||||||
| 6005 | Rebound | 1/10/2000 | 130 | 28052 | 548 | 1 | 124 | N | ||||||||
| 6006 | Interruption | 9/15/1999 | EFV/NFV/ABC/Adefovir | |||||||||||||
| 6006 | On ART, viremic | 9/16/1999 | 1 | 2790 | 227 | 2 | 196 | N | 76 (0) | 21 (0) | ||||||
| 6006 | Rebound | 10/7/1999 | 22 | 29200 | 2210 | 16 | 112 | Y | 299 (200) | 276 (200) | ||||||
| 6006 | Rebound | 11/19/1999 | 65 | 156735 | 42830 | 46 | 120 | N | 6527 (0) | 1392 (0) | ||||||
| 6006 | Rebound | 12/9/1999 | 85 | 67578 | 63381 | 29 | 99 | Y | 2089 (200) | 821 (200) | <0.0001 | <0.0001 | <0.0001 | |||
| 6006 | Rebound | 1/3/2000 | 110 | 82100 | 54800 | 44 | 69 | Y | 1130 (200) | 348 (200) | ||||||
| 6006 | Rebound | 1/20/2000 | 127 | 123886 | 14747 | 22 | 154 | N | 179 (0) | 13 (0) | ||||||
| 6008 | Interruption | 9/6/2000 | SQV/RTV | |||||||||||||
| 6008 | Rebound | 9/6/2000 | 0 | <20 | 41 | 0 | 834 | N | 0 | |||||||
| 6008 | Rebound | 9/13/2000 | 7 | 929 | 96 | 0 | 793 | N | 4 (0) | 2 (0) | ||||||
| 6008 | Rebound | 10/4/2000 | 28 | 87851 | 18323 | 62 | 798 | Y | 232 (200) | 149 (149) | 14 CSF | <0.0001 | <0.0001 | <0.0001 | ||
| 6008 | Rebound | 10/30/2000 | 54 | 26242 | 3265 | 23 | 738 | Y | 95 (95) | 12 (12) | ||||||
| 6011 | Interruption | 1/9/2001 | EFV/ZDV/3TC | |||||||||||||
| 6011 | Rebound | 1/11/2001 | 2 | 26 | <20 | 2 | 375 | N | ||||||||
| 6011 | Rebound | 1/25/2001 | 16 | 1827 | <20 | 1 | 359 | N | ||||||||
| 6011 | Rebound | 2/15/2001 | 37 | 59052 | 2286 | 4 | 223 | N | 0 | 36 (0) | ||||||
| 6011 | Rebound | 3/8/2001 | 58 | 60609 | 34016 | 33 | 288 | Y | 224 (200) | 690 (2\00) | <0.0001 | <0.0001 | <0.0001 | |||
| 6011 | Rebound | 4/6/2001 | 87 | 34566 | 40156 | 30 | 229 | N | 24 (0) | 399 (0) | ||||||
| 6011 | Rebound | 4/19/2001 | 100 | 30130 | 74650 | 0 | 256 | Y | 344 (200) | 343 (200) | ||||||
| 6011 | Rebound | 6/7/2001 | 149 | 33380 | 16202 | 42 | 208 | Y | 378 (200) | 938 (200) | ||||||
| 6011 | Rebound | 8/24/2001 | 227 | 20887 | 23867 | 46 | 266 | N | 141 (0) | 235 (0) | ||||||
| 6011 | Rebound | 10/23/2001 | 287 | 41600 | 12500 | 31 | 253 | N | 116 (0) | 450 (0) | ||||||
| 6012 | On ART, suppressed | 3/7/2001 | −12 | 37 | <20 | EFV/d4T/3TC | 1 | 464 | N | |||||||
| 6012 | Interruption | 3/19/2001 | ||||||||||||||
| 6012 | Rebound | 4/5/2001 | 17 | 2994 | <20 | 0 | 382 | N | 3 (0) | 0 | ||||||
| 6012 | Rebound | 4/26/2001 | 38 | 59369 | 586 | 2 | 410 | Y | 1818 (200) | 31 (31) | ||||||
| 6012 | Rebound | 5/17/2001 | 59 | 158418 | 2382 | 1 | 325 | Y | 252 (200) | 36 (36) | <0.0001 | 0.454 | 0.27 | |||
| 6012 | Rebound | 6/28/2001 | 101 | 121119 | 1328 | 0 | 225 | Y | 85 (85) | 26 (26) | ||||||
| 6012 | Rebound | 7/20/2001 | 123 | 96583 | 1552 | 2 | 260 | N | 703 (0) | 35 (0) | ||||||
| 6013 | On ART, viremic | 4/9/2001 | −8 | 4843 | <20 | EFV/d4T/3TC | 4 | 618 | N | 23 (0) | 0 | |||||
| 6013 | Interruption | 4/17/2001 | ||||||||||||||
| 6013 | Rebound | 5/3/2001 | 16 | 1558556 | 3193 | 0 | 369 | Y | 224 (200) | 22 (22) | ||||||
| 6013 | Rebound | 5/17/2001 | 30 | 2254433 | 8835 | 18 | 275 | Y | 130 (130) | 117 (117) | <0.0001 | <0.0001 | <0.0001 | |||
| GOT | 5207 | Pre therapy | 9/18/2000 | −371 | 155000 | 110 | N | |||||||||
| 5207 | Pre therapy | 10/5/2000 | −354 | 209000 | 95500 | 147 | Y | 23 (23) | 13 (13) | |||||||
| 5207 | On ART, decay | 12/7/2000 | −291 | 534 | ZDV/3TC+IDV/r | N | ||||||||||
| 5207 | On ART, decay | 2/7/2001 | −229 | 165 | <20 | ZDV/3TC+IDV/r | 1 | 365 | N | |||||||
| 5207 | On ART, suppressed | 5/28/2001 | −119 | <20 | d4T+3TC+IDV/r | 240 | N | |||||||||
| 5207 | On ART, suppressed | 9/24/2001 | 0 | 49 | d4T+3TC+IDV/r | 330 | N | |||||||||
| 5207 | Interruption | 9/24/2001 | ||||||||||||||
| 5207 | Rebound | 10/1/2001 | 7 | 178000 | 230 | N | ||||||||||
| 5207 | Rebound | 11/21/2001 | 58 | 330000 | 55400 | 6 | 244 | Y | 9 (9) | 31 (31) | ||||||
| 5299 | Pre therapy | 5/19/2004 | −933 | 520000 | 181000 | 5 | 240 | Y | 274 (50) | 16 (16) | ||||||
| 5299 | Pre therapy | 8/16/2004 | −844 | 686000 | 120 | N | ||||||||||
| 5299 | Pre therapy | 9/2/2004 | −827 | 443000 | N | |||||||||||
| 5299 | On ART, decay | 9/9/2004 | −820 | 28900 | TDF+3TC+AZV/r | N | ||||||||||
| 5299 | On ART, decay | 9/16/2004 | −813 | 4290 | TDF+3TC+AZV/r | N | ||||||||||
| 5299 | On ART, decay | 9/23/2004 | −806 | 3510 | TDF+3TC+AZV/r | N | ||||||||||
| 5299 | On ART, decay | 10/4/2004 | −795 | 1520 | TDF+3TC+AZV/r | 230 | N | |||||||||
| 5299 | On ART, decay | 11/29/2004 | −739 | 464 | <20 | TDF+3TC+AZV/r | 1 | 390 | N | |||||||
| 5299 | On ART, suppressed | 3/4/2005 | −644 | 150 | TDF+3TC+AZV/r | 220 | N | |||||||||
| 5299 | On ART, suppressed | 3/16/2005 | −632 | <20 | TDF+3TC+AZV/r | N | ||||||||||
| 5299 | On ART, suppressed | 5/31/2005 | −556 | 172 | TDF+3TC+AZV/r | 260 | N | |||||||||
| 5299 | On ART, suppressed | 6/14/2005 | −542 | 23 | TDF+3TC+AZV/r | N | ||||||||||
| 5299 | On ART, suppressed | 8/11/2005 | −484 | 61 | <20 | TDF+3TC+AZV/r | 0 | 330 | N | |||||||
| 5299 | On ART, suppressed | 9/23/2005 | −441 | <20 | TDF+3TC+AZV/r | N | ||||||||||
| 5299 | On ART, suppressed | 1/23/2006 | −319 | 124 | TDF+3TC+AZV/r | 410 | N | |||||||||
| 5299 | On ART, suppressed | 2/2/2006 | −309 | 24 | TDF+3TC+AZV/r | N | ||||||||||
| 5299 | On ART, suppressed | 4/18/2006 | −234 | 86 | TDF+3TC+AZV/r | N | ||||||||||
| 5299 | On ART, suppressed | 5/5/2006 | −217 | <20 | TDF+3TC+AZV/r | N | ||||||||||
| 5299 | On ART, suppressed | 7/24/2006 | −137 | 29 | <20 | TDF+3TC+AZV/r | 1 | 380 | N | |||||||
| 5299 | On ART, suppressed | 12/7/2006 | −1 | 175 | TDF+3TC+AZV/r | 360 | N | |||||||||
| 5299 | Interruption | 12/8/2006 | ||||||||||||||
| 5299 | Rebound | 1/17/2007 | 40 | 316000 | 205000 | 8 | 410 | Y | 768 (50) | 34 (34) | ||||||
| 51126 | Pre therapy | 2/22/2011 | −2173 | 2100000 | 203158 | 3 | 60 | Y | 4948 (50) | 1734 (50) | 21 BL, 14 CSF | |||||
| 51126 | Pre therapy | 3/7/2011 | −2160 | 1500000 | 70 | N | ||||||||||
| 51126 | On ART, decay | 4/4/2011 | −2132 | 6628 | DRV/r+RGV | 320 | N | |||||||||
| 51126 | On ART, decay | 5/9/2011 | −2097 | 4594 | DRV/r+RGV | 280 | N | |||||||||
| 51126 | On ART, viremic | 6/1/2011 | −2074 | 6641 | 48599 | DRV/r+RGV | 3 | 230 | Y | 112 (50) | 51 (50) | |||||
| 51126 | On ART, viremic | 6/20/2011 | −2055 | 12825 | DRV/r+RGV | N | ||||||||||
| 51126 | On ART, viremic | 7/18/2011 | −2027 | 52587 | 60076 | DRV/r+RGV | 4 | 340 | Y | 800 (50) | 174 (50) | 16 BL, 9 CSF | ||||
| 51126 | On ART, decay | 8/22/2011 | −1992 | 7692 | ZDV+3TC+NVP+DRV/r | N | ||||||||||
| 51126 | On ART, decay | 9/28/2011 | −1955 | 3401 | 1645 | ZDV+3TC+NVP+DRV/r | 0 | 140 | N | |||||||
| 51126 | On ART, decay | 10/17/2011 | −1936 | 1006 | ZDV+3TC+NVP+DRV/r | N | ||||||||||
| 51126 | On ART, decay | 11/11/2011 | −1911 | 874 | ZDV+3TC+NVP+DRV/r | 0 | 280 | N | ||||||||
| 51126 | On ART, decay | 12/5/2011 | −1887 | 306 | ZDV+3TC+NVP+DRV/r | N | ||||||||||
| 51126 | On ART, decay | 1/3/2012 | −1858 | 3626 | ZDV+3TC+NVP+DRV/r | 440 | N | |||||||||
| 51126 | On ART, decay | 1/17/2012 | −1844 | 438 | ZDV+3TC+NVP+DRV/r | N | ||||||||||
| 51126 | On ART, decay | 2/2/2012 | −1828 | 319 | ZDV+3TC+NVP+DRV/r | N | ||||||||||
| 51126 | On ART, decay | 2/20/2012 | −1810 | 216 | ZDV+3TC+NVP+DRV/r | N | ||||||||||
| 51126 | On ART, decay | 3/6/2012 | −1795 | 256 | ZDV+3TC+NVP+DRV/r | N | ||||||||||
| 51126 | On ART, decay | 3/20/2012 | −1781 | 189 | ZDV+3TC+NVP+DRV/r | N | ||||||||||
| 51126 | On ART, decay | 4/2/2012 | −1768 | 243 | <20 | ZDV+3TC+NVP+DRV/r | 0 | 400 | N | |||||||
| 51126 | On ART, decay | 5/21/2012 | −1719 | 185 | ZDV+3TC+NVP+DRV/r | 470 | N | |||||||||
| 51126 | On ART, decay | 7/5/2012 | −1674 | 286 | 27 | ZDV+3TC+NVP+DRV/r | 0 | 440 | N | |||||||
| 51126 | On ART, decay | 8/6/2012 | −1642 | 162 | ZDV+3TC+NVP+DRV/r | 460 | N | |||||||||
| 51126 | On ART, decay | 9/26/2012 | −1591 | 270 | ZDV+3TC+NVP+DRV/r | 580 | N | |||||||||
| 51126 | On ART, decay | 11/29/2012 | −1527 | 250 | ZDV+3TC+NVP+DRV/r | N | ||||||||||
| 51126 | On ART, decay | 1/9/2013 | −1486 | 300 | <20 | ZDV+3TC+NVP+DRV/r | 0 | 680 | N | |||||||
| 51126 | On ART, suppressed | 4/29/2013 | −1376 | 106 | ABC+3TC+NVP+DrV/r | 510 | N | |||||||||
| 51126 | On ART, suppressed | 6/11/2013 | −1333 | 87 | ABC+3TC+NVP+DrV/r | N | ||||||||||
| 51126 | On ART, suppressed | 8/30/2013 | −1253 | 95 | ABC+3TC+NVP+DrV/r | N | ||||||||||
| 51126 | On ART, suppressed | 1/28/2014 | −1102 | 33 | <20 | ABC+3TC+DRV/r | 0 | 520 | N | |||||||
| 51126 | On ART, suppressed | 3/28/2014 | −1043 | 81 | ABC+3TC+DRV/r | 640 | N | |||||||||
| 51126 | On ART, suppressed | 4/30/2014 | −1010 | 56 | <20 | ABC+3TC+DRV/r | 0 | 840 | N | |||||||
| 51126 | On ART, suppressed | 8/19/2014 | −899 | <20 | ABC+3TC+DRV/r | N | ||||||||||
| 51126 | On ART, suppressed | 11/17/2014 | −809 | 41 | ABC+3TC+DRV/r | N | ||||||||||
| 51126 | On ART, suppressed | 4/30/2015 | −645 | <20 | <20 | ABC+3TC+DRV/r | 0 | 830 | N | |||||||
| 51126 | On ART, suppressed | 8/7/2015 | −546 | 72 | ABC+3TC+DRV/r | N | ||||||||||
| 51126 | On ART, suppressed | 11/9/2015 | −452 | <20 | ABC+3TC+DRV/r | N | ||||||||||
| 51126 | On ART, suppressed | 6/14/2016 | −234 | 25 | ABC+3TC+DRV/r | 730 | N | |||||||||
| 51126 | On ART, suppressed | 11/3/2016 | −92 | 334 | <20 | ABC+3TC+DRV/r | 0 | 740 | N | |||||||
| 51126 | Interruption | 2/3/2017 | ||||||||||||||
| 51126 | Rebound | 3/16/2017 | 41 | 25000000 | 380 | N | ||||||||||
| 51126 | Rebound | 3/21/2017 | 46 | 4600000 | 1300000 | 20 | 250 | Y | 7675 (50) | 2233 (50) | 11 BL, CSF 20 |
Extended Data Table 2.
Diagnosis and treatment summary
| 4026: Estimate infected for 10 years. Started monotherapy in 1994. Triple drug therapy in 1996. Documented suppression on 6/98, 7/98, and 11/98 but viremic on 10/6/98 (blood VL 11,241) and 12/23/98, with TI on 1/16/99. |
| 6004: Treatment started more than 3 years before TI. Documented suppression 1/31/97–12/23/98. Viremic 1/29/99 on therapy; TI 8/2/99. |
| 6005: No information. |
| 6006: Prior treatment greater than 12 months. |
| 6008: Estimate infected 10 years before TI. Treatment started in 1996. TI on 9/6/00. |
| 6011: Suppressed (with one blip) from 3/20/00 until TI on 1/9/01. |
| 6012: Suppressed from 12/7/99 until TI on 3/19/01 |
| 6013: On treatment from 3/20/97 to TI on 4/17/01 with viral load reduced but not suppressed. |
| 5207: Diagnosed 9/18/2000 with CD4+ T cell count of 110/ul. Started treatment on 10/14/2000 and was treated until 9/24/2001 when TI occurred. |
| 5299: Diagnosed 4/25/2003, estimated 12 weeks post infection. Treated 9/4/2004 until 12/8/2006 when found to be viremic, then started on a new regimen on 1/17/2007. |
| 51126: Diagnosed 2/1/2011 with CD4+ T cell count of 70/ul and HIV-associated dementia. Started treatment on 3/7/2011 but developed INSTI resistance (N155H, V2011, T206S) leading to an early change in the drug regimen, with treatment interruption on 2/4/2017. |
Available information about the HIV-1 diagnosis and antiretroviral (ARV) treatment history of all 11 study participants.
Extended Data Table 3.
Amplification and sequencing primers
| Primer | Sequence |
|---|---|
| SGA1 Forward | TGCCAAGAAAAGCAAAGATCATTAG |
| SGA1 Reverse | GACTCTCGAGAAGCACTCAAGGCAAGCTTTATTGAG |
| SGA2 Forward | GATCAAGCTTTAGGCATCTCCTATGGCAGGAAGAAG |
| SGA2 Reverse | GACTCTCGAGAAGCACTCAAGGCAAGCTTTATTGAG |
| MiSeq Subtype B V3 cDNA* | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNCAGTCCATTTTGCTYTAYTRABVTTACAATRTGC |
| MiSeq Subtype C V3 cDNA* | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNGCTATGTGTTGTAATTTCTAGGTCCCCT |
| MiSeq PR cDNA* | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNCAGTTTAACTTTTGGGCCATCCATTCC |
| MiSeq IN cDNA* | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNATCGAATACTGCCATTTGTACTGC |
| MiSeq RT cDNA* | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNCAGTCACTATAGGCTGTACTGTCCATTTATC |
| MiSeq V3 PCR1 | GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNNTTATGGGATCAAAGCCTAAAGCCATGTGTA |
| MiSeq PR PCR1 | GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNNTCAGAGCAGACCAGAGCCAACAGCCCCA |
| MiSeq IN PCR1 | GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNNAAAAGGAGAAGCCATGCATG |
| MiSeq RT PCR1 | GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNNGGCCATTGACAGAAGAAAAAATAAAAGC |
| MiSeq PCR1 reverse primer | GTGACTGGAGTTCAGACGTGTGCTC |
| MiSeq PCR2 primer 1 | AATGATACGGCGACCACCGAGATCTACACGCCTCCCTCGCGCCATCAGAGATGTG |
| MiSeq PCR2 primer 2** | CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTC |
| Sequencing primer for MiSeq runs | GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAG |
Primers used to amplify and sequence HIV-1 RNA for both Illumina MiSeq with Primer ID and single genome amplification (SGA).
Ns in the cDNA primer sequences are random primers
In the index primer the 6-N is not a random primer but a pre-designed index sequence. We use a set of 24 indices
Acknowledgement
First, we would like to thank the many participants who donated the specimens that were analyzed in this study. This work was supported by NIH grant R01 NS094067 (R.W.P.). The work was also supported by the UNC Center For AIDS Research (NIH award P30 AI050410) (R.S.), the UNC Lineberger Comprehensive Cancer Center (NIH award P30 CA16068) (R.S.) and by the Swedish state, under an agreement between the Swedish government and the county councils (ALF agreement ALFGBG-717531) (M.G.). HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931). We thank the UNC High Throughput Sequencing Facility for their assistance in generating the sequence data.
Competing Interests
UNC is pursuing IP protection for Primer ID, and R.S. has received nominal royalties.
Footnotes
Code availability. Illumina MiSeq data was analyzed using TCS pipeline version 2.5.1 (https://www.primer-id.org/?from_old).
Data availability.
The sequences of the full length env amplicons are available in GenBank (accession numbers ON599411-ON59572). The deep sequencing data is available in the Short Read Archive (https://www.ncbi.nlm.nih.gov/sra/PRJNA880316). Remaining source data are available with this paper.
References
- 1.Ping LH, et al. Comparison of viral Env proteins from acute and chronic infections with subtype C human immunodeficiency virus type 1 identifies differences in glycosylation and CCR5 utilization and suggests a new strategy for immunogen design. J. Virol 87, 7218–7233 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parrish NF, et al. Transmitted/founder and chronic subtype C HIV-1 use CD4 and CCR5 receptors with equal efficiency and are not inhibited by blocking the integrin α4β7. PLoS Pathog. 8, e1002686 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joseph SB & Swanstrom R The evolution of HIV-1 entry phenotypes as a guide to changing target cells. J. Leuk. Biol 103, 421–431 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Colby DJ, et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat. Med 24, 923–926 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henrich TJ, et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PLoS Med. 14, e1002417 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitney JB, et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 512, 74–77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Treasure GC, et al. Relationship among viral load outcomes in HIV treatment interruption trials. JAIDS 72, 310–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li JZ, et al. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 30, 343–353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bar KJ, et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N. Eng.l J. Med 375, 2037–2050 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bednar MM, et al. Diversity and tropism of HIV-1 rebound virus populations in plasma level after treatment discontinuation. J. Infect. Dis 214, 403–407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Scheerder MA, et al. HIV rebound Is predominantly fueled by genetically identical viral expansions from diverse reservoirs. Cell Host Microbe 26, 347–358 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisher K, et al. Plasma-derived HIV-1 virions contain considerablelevels of defective genomes. J. Virol 96, e0201121 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kearney MF, et al. Origin of rebound plasma HIV includes cells with identical proviruses that are transcriptionally active before stopping of antiretroviral therapy. J. Virol 90, 1369–1376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rothenberger MK, et al. Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc. Natl. Acad. Sci. U. S. A 112, E1126–1134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrade VM, et al. A minor population of macrophage-tropic HIV-1 variants is identified in recrudescing viremia following analytic treatment interruption. Proc. Natl. Acad. Sci. U. S. A 117, 9981–9990 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chun TW, et al. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med 1, 1284–1290 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Hiener B, et al. Identification of genetically intact HIV-1 proviruses in specific CD4(+) T cells from effectively treated participants. Cell Reports 21, 813–822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chomont N, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med 15, 893–900 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shacklett BL, Ferre AL & Kiniry BE Defining T cell tissue residency in humans: Implications for HIV pathogenesis and vaccine design. Current HIV/AIDS Reports 17, 109–117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louveau A, Harris TH & Kipnis J Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 36, 569–577 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engelhardt B, Vajkoczy P & Weller RO The movers and shapers in immune privilege of the CNS. Nat. Immunol 18, 123–131 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Iwasaki A Immune regulation of antibody access to neuronal tissues. Trends Mol. Med 23, 227–245 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burbelo PD, et al. Anti-Human Immunodeficiency Virus antibodies in the cerebrospinal fluid: Evidence of early treatment impact on central nervous system reservoir? The J. Infect. Dis 217, 1024–1032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sturdevant CB, et al. Compartmentalized replication of R5 T cell-tropic HIV-1 in the central nervous system early in the course of infection. PLoS Pathog. 11, e1004720 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnell G, et al. HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog. 7, e1002286 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joseph SB, et al. Quantification of entry phenotypes of macrophage-tropic HIV-1 across a wide range of CD4 densities. J. Virol 88, 1858–1869 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price RW & Deeks SG Antiretroviral drug treatment interruption in human immunodeficiency virus-infected adults: Clinical and pathogenetic implications for the central nervous system. J. NeuroVirol 10 Suppl 1, 44–51 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Santangelo PJ, et al. Whole-body immunoPET reveals active SIV dynamics in viremic and antiretroviral therapy-treated macaques. Na.t Methods 12, 427–432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Honeycutt JB, et al. T cells establish and maintain CNS viral infection in HIV-infected humanized mice. J. Clin. Invest 128, 2862–2876 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Honeycutt JB, et al. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med 23, 638–643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitney JB, et al. Prevention of SIVmac251 reservoir seeding in rhesus monkeys by early antiretroviral therapy. Nat. Commun 9, 5429 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fennessey CM, et al. Genetically-barcoded SIV facilitates enumeration of rebound variants and estimation of reactivation rates in nonhuman primates following interruption of suppressive antiretroviral therapy. PLoS Pathog. 13, e1006359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Obregon-Perko V, et al. Dynamics and origin of rebound viremia in SHIV-infected infant macaques following interruption of long-term ART. JCI Insight 6, e152526 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gama L, et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS 31, 5–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Avalos CR, et al. Brain macrophages in Simian Immunodeficiency Virus-infected, antiretroviral-suppressed macaques: a functional latent reservoir. mBio 8, e01186–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abreu C, et al. Brain macrophages harbor latent, infectious simian immunodeficiency virus. AIDS 33 Suppl 2, S181–s188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su H, et al. Recovery of latent HIV-1 from brain tissue by adoptive cell transfer in Virally suppressed humanized mice. J. Neuroimmune Pharmacol 16, 796–805 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dubé K, et al. Ethical considerations for HIV cure-related research at the end of life. BMC Med. Ethics 19, 83 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Almeida SN, et al. Dynamics of monocyte chemoattractant protein type one (MCP-1) and HIV viral load in human cerebrospinal fluid and plasma. J. Neuroimmunol 169, 144–152 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Gianella S, et al. Compartmentalized HIV rebound in the central nervous system after interruption of antiretroviral therapy. Virus Evolution 2, vew020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deeks SG, et al. Virologic and immunologic consequences of discontinuing combination antiretroviral-drug therapy in HIV-infected patients with detectable viremia. N. Engl. J. Med 344, 472–480 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Price RW, et al. Cerebrospinal fluid response to structured treatment interruption after virological failure. AIDS 15, 1251–1259 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Gisslen M, et al. Cerebrospinal fluid signs of neuronal damage after antiretroviral treatment interruption in HIV-1 infection. AIDS Res. Ther 2, 6 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou S, et al. Deep Sequencing of the HIV-1 env gene reveals discrete X4 lineages and linkage disequilibrium between X4 and R5 viruses in the V1/V2 and V3 variable regions. J. Virol 90, 7142–7158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou S, et al. Primer ID validates template sampling depth and greatly reduces the error rate of next-generation sequencing of HIV-1 denomic RNA populations. J. Virol 89, 8540–8555 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lustig G, et al. T cell derived HIV-1 is present in the CSF in the face of suppressive antiretroviral therapy. PLoS Pathog. 17, e1009871 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma V, et al. Cerebrospinal fluid CD4+ T cell infection in humans and macaques during acute HIV-1. PLoS Pathog. 17(12), e1010105 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slatkin M & Maddison WP A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 123, 603–613 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adewumi OM, et al. HIV-1 Central Nervous System Compartmentalization and Cytokine Interplay in Non-Subtype B HIV-1 Infections in Nigeria and Malawi. AIDS Res. Hum. Retroviruses 36, 490–500 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aamer HA, et al. Cells producing residual viremia during antiretroviral treatment appear to contribute to rebound viremia following interruption of treatment. PLoS Pathog. 16, e1008791 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bailey JR, et al. Residual human immunodeficiency virus type 1 viremia in some patients on Antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4(+) T cells. J. Virol 80, 6441–6457 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cole B, et al. In-depth single-cell analysis of translation-competent HIV-1 reservoirs identifies cellular sources of plasma viremia. Nat. Commun 12, 3727 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halvas EK, et al. HIV-1 viremia not suppressible by antiretroviral therapy can originate from large T cell clones producing infectious virus. J. Clin. Invest 130, 5847–5857 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rassler S, et al. Prolonged persistence of a novel replication-defective HIV-1 variant in plasma of a patient on suppressive therapy. Virol. J 13, 157 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sahu GK, Sarria JC & Cloyd MW Recovery of replication-competent residual HIV-1 from plasma of a patient receiving prolonged, suppressive highly active antiretroviral therapy. J. Virol 84, 8348–8352 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simonetti FR, et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc. Natl. Acad. Sci. U. S. A 113, 1883–1888 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu CL, et al. Relationship between intact HIV-1 proviruses in circulating CD4(+) T cells and rebound viruses emerging during treatment interruption. Proc. Natl. Acad. Sci. U. S. A 115, E11341–E11348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cohen YZ, et al. Relationship between latent and rebound viruses in a clinical trial of anti-HIV-1 antibody 3BNC117. J. Exp. Med 215, 2311–2324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu PT, et al. Origin of rebound virus in chronically SIV-infected Rhesus monkeys following treatment discontinuation. Nat. Commun 11, 5412 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnston SH, et al. A quantitative affinity-profiling system that reveals distinct CD4/CCR5 usage patterns among human immunodeficiency virus type 1 and simian immunodeficiency virus strains. J. Virol 83, 11016–11026 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abrahams MR, et al. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med 11, eaaw5589 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imamichi H, et al. Human immunodeficiency virus type 1 quasi species that rebound after discontinuation of highly active antiretroviral therapy are similar to the viral quasi species present before initiation of therapy. J. Infect. Dis 183, 36–50 (2001). [DOI] [PubMed] [Google Scholar]
- 63.Kearney MF, et al. Lack of detectable HIV-1 molecular evolution during suppressive antiretroviral therapy. PLoS Pathog. 10, e1004010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hagberg L, et al. Cerebrospinal fluid neopterin: an informative biomarker of central nervous system immune activation in HIV-1 infection. AIDS Res. Ther 7, 15 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jessen Krut J, et al. Biomarker evidence of axonal injury in neuroasymptomatic HIV-1 patients. PLoS One 9, e88591 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Norgren N, Rosengren L & Stigbrand T Elevated neurofilament levels in neurological diseases. Brain Res. 987, 25–31 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Yilmaz A, et al. Neurofilament light chain protein as a marker of neuronal injury: review of its use in HIV-1 infection and reference values for HIV-negative controls. Expert Rev. Mol. Diagn 17, 761–770 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Edgar RC MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Joseph SB, Lee B & Swanstrom R Affinofile assay for identifying macrophage-tropic HIV-1. Bio-Protocol 4, e1184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequences of the full length env amplicons are available in GenBank (accession numbers ON599411-ON59572). The deep sequencing data is available in the Short Read Archive (https://www.ncbi.nlm.nih.gov/sra/PRJNA880316). Remaining source data are available with this paper.